ES2871001T3 - Conjugados de pirrolobenzodiazepinas y anticuerpos - Google Patents
Conjugados de pirrolobenzodiazepinas y anticuerpos Download PDFInfo
- Publication number
- ES2871001T3 ES2871001T3 ES18704956T ES18704956T ES2871001T3 ES 2871001 T3 ES2871001 T3 ES 2871001T3 ES 18704956 T ES18704956 T ES 18704956T ES 18704956 T ES18704956 T ES 18704956T ES 2871001 T3 ES2871001 T3 ES 2871001T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- alkyl
- saturated
- antibody
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
- A61K31/5517—1,4-Benzodiazepines, e.g. diazepam or clozapine condensed with five-membered rings having nitrogen as a ring hetero atom, e.g. imidazobenzodiazepines, triazolam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/51—Complete heavy chain or Fd fragment, i.e. VH + CH1
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/515—Complete light chain, i.e. VL + CL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cell Biology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Un conjugado de la fórmula (I): Ab - (DL)p (I) en la que: Ab es un anticuerpo que se une a AXL; DL es **(Ver fórmula)** en la que: X se selecciona del grupo que comprende: un enlace simple, -CH2- y -C2H4-; n es de 1 a 8; m es 0 o 1; R7 es metilo o fenilo; cuando existe un enlace doble entre C2 y C3, R2 se selecciona del grupo que consiste en: (ia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3; (ib) un alquilo alifático C1-5 saturado; (ic) un cicloalquilo C3-6 saturado; (id) **(Ver fórmula)** en la que cada uno de R21, R22 y R23 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R2 no es mayor de 5; (ie) **(Ver fórmula)** en la que uno de R25a y R25b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y (if) **(Ver fórmula)** donde R24 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; cuando existe un enlace simple entre C2 y C3, R2 es **(Ver fórmula)** donde R26a y R26b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos 15 alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R26a y R26b es H, el otro se selecciona de nitrilo y un alquiléster C1-4; cuando existe un enlace doble entre C2' y C3', R12 se selecciona del grupo que consiste en: (iia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3; (iib) un alquilo alifático C1-5 saturado; (iic) un cicloalquilo C3-6 saturado; (iid) **(Ver fórmula)** en la que cada uno de R31, R32 y R33 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R12 no es mayor de 5; (iie) **(Ver fórmula)** en la que uno de R35a y R35b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y (iif) **(Ver fórmula)** donde R34 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo, dicho fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; cuando existe un enlace simple entre C2' y C3', R12 es **(Ver fórmula)** donde R36a y R36b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R36a y R36b es H, el otro se selecciona de nitrilo y un alquiléster C1-4; 5 en donde en (ia) y (iia), alquilo C1-7 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un átomo de carbono de un compuesto de hidrocarburo que tiene de 1 a 7 átomos de carbono, que puede ser alifático o alicíclico, y que puede ser saturado o insaturado; y en (if) y (iif), alquilo C1-4 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un compuesto de hidrocarburo que tiene de 1 a 4 átomos de carbono, que puede ser alifático o alicíclico, y que puede 10 ser saturado o insaturado; y p es de 1 a 8.
Description
DESCRIPCIÓN
Conjugados de pirrolobenzodiazepinas y anticuerpos
Referencia cruzada a solicitudes relacionadas
La presente solicitud reivindica el beneficio de GB1702029.8 y GB1702031.4, presentadas el 8 de febrero de 2017 y GB1719906.8 presentada el 30 de noviembre de 2017.
Campo de la invención
La presente invención se refiere a pirrolobenzodiazepinas (PBD) que tienen un grupo protector lábil en la forma de un enlazador a un anticuerpo.
Antecedentes de la invención
Pirrolobenzodiazepinas
Algunas pirrolobenzodiazepinas (PBD) tienen la capacidad de reconocer secuencias de ADN específicas y unirse a estas. La secuencia preferida es PuGPu. El primer antibiótico antitumoral de PBD, antramicina, se descubrió en 1965 (Leimgruber, et ál., J. Am. Chem. Soc., 87, 5793-5795 (1965); Leimgruber, et ál., J. Am. Chem. Soc., 87, 5791-5793 (1965)). Desde entonces, se han informado una cantidad de PBD de origen natural, y se han desarrollado más de 10 rutas de síntesispara varios análogos (Thurston, et ál., Chem. Rev. 1994, 433-465 (1994); Antonow, D. y Thurston, D.E., Chem. Rev. 2011 111 (4), 2815-2864). Los miembros de la familia incluyen abeimicina (Hochlowski, et ál., J. Antibiotics, 40, 145-148 (1987)), chicamicina (Konishi, et ál., J. Antibiotics, 37, 200-206 (1984)), DC-81 (patente japonesa 58-180487; Thurston, et ál., Chem. Brit., 26, 767-772 (1990); Bose, et ál., Tetrahedron, 48 , 751-758 (1992)), mazetramicina (Kuminoto, et ál., J. Antibiotics, 33, 665-667 (1980)), neotramicinas A y B (Takeuchi, et ál., J. Antibiotics, 29, 93-96 (1976)), porotramicina (Tsunakawa, et ál., J. Antibiotics, 41, 1366-1373 (1988)), protracarcina (Shimizu, et al, J. Antibiotics, 29, 2492-2503 (1982); Langley y Thurston, J. Org. Chem., 52, 91-97 (1987)), sibanomicina (DC-102)(Hara, et ál., J. Antibiotics, 41, 702-704 (1988); Itoh, et ál., J. Antibiotics, 41, 1281-1284 (1988)), sibiromicina (Leber, et ál., J. Am. Chem. Soc., 110, 2992-2993 (1988)) y tomamicina (Arima, et ál., J. Antibiotics, 25, 437-444 (1972)). Las PBD tienen la estructura general:

Difieren en cantidad, tipo y posición de los sustituyentes, tanto en sus anillos A aromáticos como en sus anillos C pirrolo, y en el grado de saturación del anillo C. En el anillo B hay una imina (N=C), una carbinolamina(NH-CH(OH)) o un éter metílico de carbinolamina (NH-CH(OMe)) en la posición N10-C11, el cual es el centro electrofílico responsable de la alquilación del ADN. Todos los productos de origen natural conocidos tienen una configuración (S) en la posición quiral C11a que les provee un giro hacia la derecha cuando se observan desde el anillo C en dirección al anillo A. Esto les provee la forma tridimensional adecuada para isohelicidad con la hendidura menor del ADN de forma B, lo que produce un ajuste adecuado en el sitio de unión (Kohn, In Antibiotics III. Springer-Verlag, Nueva York, pp. 3-11 (1975); Hurley y Needham-VanDevanter, Acc. Chem. Res., 19, 230-237 (1986)). Su capacidad de formar un aducto en la ranura menor les permite interferir en el procesamiento de ADN y, por lo tanto, se utilizan como agentes antitumorales.
Un compuesto de pirrolobenzodiazepina se describe en Gregson et ál. (Chem. Commun. 1999, 797-798) como el compuesto 1, y por Gregson et ál. (J. Med. Chem. 2001, 44, 1161-1174) como el compuesto 4a. Este compuesto, también conocido como SG2000, se muestra a continuación:

WO 2007/085930 describe la preparación de compuestos diméricos de PBD con grupos enlazadores para la conexión a un agente de unión celular, tal como un anticuerpo. El enlazador se encuentra presente en el puente que enlaza las unidades monoméricas de PBD del dímero.
Los compuestos diméricos de PBD que tienen grupos enlazadores para la conexión a un agente de unión celular, tal como un anticuerpo, se han descrito en WO 2011/130613 y WO 2011/130616. El enlazador en estos compuestos se une al núcleo de PBD a través de la posición C2 y, en general, se escinde mediante la acción de una enzima en el grupo enlazador. En WO 2011/130598, el enlazador en estos compuestos se une a una de las posiciones N10 disponibles en el núcleo de PBD y, en general, se escinde mediante la acción de una enzima en el grupo enlazador.
Conjugados de anticuerpos y fármacos
Se ha establecido la terapia de anticuerpos para el tratamiento dirigido de pacientes con cáncer, trastornos inmunológicos y angiogénicos (Carter, P. (2006) Nature Reviews Immunology 6:343-357). El uso de conjugados de anticuerpos y fármacos (ADC), es decir, inmunoconjugados, para la administración local de agentes citotóxicos o citostáticos, es decir, fármacos para destruir o inhibir células tumorales en el tratamiento de cáncer, selecciona como diana la administración del resto de fármaco a los tumores, y la acumulación intracelular en ellos, mientras que la administración sistémica de estos agentes de fármacos sin conjugar puede generar niveles inaceptables de toxicidad para células normales (Xie et ál (2006) Expert. Opin. Biol. Ther. 6(3):281-291; Kovtun et ál (2006) Cáncer Res.
66(6):3214-3121; Law et al (2006) Cancer Res. 66(4):2328-2337; Wu et al (2005) Nature Biotech. 23(9):1137-1145; Lambert J. (2005) Current Opin. in Pharmacol. 5:543-549; Hamann P. (2005) Expert Opin. Ther. Patents 15(9):1087-1103; Payne, G. (2003) Cancer Cell 3:207-212; Trail et al (2003) Cancer Immunol. Immunother. 52:328-337; Syrigos and Epenetos (1999) Anticancer Research 19:605-614).
De este modo se busca la máxima eficacia con la mínima toxicidad. Los esfuerzos para diseñar y refinar los ADC se han enfocado en la selectividad de los anticuerpos monoclonales (mAbs) así como también en el mecanismo de acción del fármaco, el enlace con el fármaco, la relación fármaco a anticuerpo (carga) y las propiedades de liberación del fármaco(Junutula, et ál., 2008b Nature Biotech., 26(8):925-932; Dornan et al (2009) Blood 114(13):2721-2729; US 7521541; US 7723485; WO2009/052249; McDonagh (2006) Protein Eng. Design & Sel. 19(7): 299-307; Doronina et ál (2006) Bioconj. Chem. 17:114-124; Erickson et al (2006) Cancer Res. 66(8):1-8; Sanderson et al (2005) Clin. Cancer Res. 11:843-852; Jeffrey et al (2005) J. Med. Chem. 48:1344-1358; Hamblett et al (2004) Clin. Cancer Res. 10:7063-7070). Los restos de fármacos pueden ejercer sus efectos citotóxicos y citostáticos mediante mecanismos que incluyen la unión a tubilina, la unión a ADN o la inhibición de la proteasoma y/o topoisomerasa. Algunos fármacos citotóxicos tienden a ser inactivos o menos activos cuando se conjugan con anticuerpos grandes o ligandos de receptores de proteínas.
El documento WO 2016/166302 A1 desvela anticuerpos anti-AXL humanizados y conjugados de los mismos. Específicamente, se describen conjugados que comprenden PBD que tienen un grupo protector lábil en forma de tipos particulares de enlazadores al anticuerpo.
Estos inventores han desarrollado conjugados de anticuerpos dímeros de PBD particulares.
Compendio de la invención
Un primer aspecto de la presente invención proporciona un conjugado de la fórmula (I):
Ab -(D L)p(I)
donde:
Ab es un anticuerpo que se une a AXL;
DL es

donde:
X se selecciona del grupo que comprende: un enlace simple, -CH2- y -C2H4-;
n es de 1 a 8;
m es 0 o 1;
R7 es metilo o fenilo;
cuando existe un enlace doble entre C2 y C3, R2 se selecciona del grupo que consiste en:
(ia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3; (ib) un alquilo alifático C1-5 saturado;
(ic) un cicloalquilo C3-6 saturado;
(id)
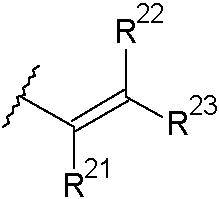
donde cada uno de R21, R22 y R23 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R12 no es mayor de 5;
(ie)

donde uno de R25a y R25b es H y el otro se selecciona de: fenilo, el fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y
(if)

donde R24 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo;
cuando existe un enlace simple entre C2 y C3, R2 es

donde R26a y R26b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R26a y R26b es H, el otro se selecciona de nitrilo y un alquiléster C1-4;
cuando existe un enlace doble entre C2' y C3', R12 se selecciona del grupo que consiste en:
(iia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3;
(iib) un alquilo alifático C1-5 saturado;
(iic) un cicloalquilo C3-6 saturado;
(iid)

donde cada uno de R31, R32 y R33 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R12 no es mayor de 5;
(iie)

donde uno de R35a y R35b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi, piridilo y tiofenilo, y
(iif)

donde R34 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3;
ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo;
cuando existe un enlace simple entre C2' y C3', R12 es

donde R36a y R36b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R36a y R36b es H, el otro se selecciona de nitrilo y un alquiléster C1-4;
donde en (ia) y (iia), alquilo C1-7 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un átomo de carbono de un compuesto de hidrocarburo que tiene de 1 a 7 átomos de carbono, que puede ser
alifático o alicíclico, y que puede ser saturado o insaturado; y en (if) y (iif), alquilo C1-4 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un compuesto de hidrocarburo que tiene de 1 a 4 átomos de carbono, que puede ser alifático o alicíclico, y que puede ser saturado o insaturado;
y p es de 1 a 8.
Se ha descubierto que estos conjugados tienen una buena actividad y una tolerabilidad sorprendente en comparación con los conjugados análogos que no contienen el resto de sulfonamido.
Breve descripción de las figuras
La Figura 1 muestra la unión de un conjugado a AXL;
La Figura 2 muestra la eficacia in vivo de un conjugado;
La Figura 3 muestra la eficacia in vivo de conjugados;
La Figura 4 muestra la eficacia in vivo de conjugados;
La Figura 5 muestra la eficacia in vivo de conjugados en un xenoinjerto derivado de un paciente; y
La Figura 6 muestra la eficacia in vivo de conjugados en otro xenoinjerto derivado de un paciente.
Descripción Detallada de la Invención
La presente invención proporciona un dímero de PBD con un enlazador conectado a través de la posición N10 en uno de los restos de PBD conjugados a un anticuerpo como se define a continuación.
La presente invención es adecuada para usar para proporcionar un compuesto de PBD a un sitio preferido en un sujeto. El conjugado permite la liberación de un compuesto de PBD activo que no retiene ninguna parte del enlazador. No hay tope presente que pueda afectar la reactividad del compuesto de PBD. Por lo tanto, el conjugado de la fórmula (I) liberaría el compuesto RelA:

En la presente invención, el enlace especificado entre el dímero de PBD y el anticuerpo es preferentemente estable de forma extracelular. Antes del transporte o la administración a una célula, el conjugado de anticuerpo y fármaco (ADC) es preferentemente estable y permanece intacto, es decir, el anticuerpo permanece enlazado al resto de fármaco. Los enlazadores son estables fuera de la célula diana y se pueden escindir a una tasa eficaz en el interior de la célula. Un enlazador eficaz: (i) mantendrá las propiedades de unión específicas del anticuerpo, (ii) permitirá la administración intracelular del conjugado o resto de fármaco, (iii) permanecerá estable e intacto, es decir, no se escindirá, hasta que se haya transportado o administrado el conjugado a su sitio seleccionado como diana y (iv) mantendrá un efecto citotóxico destructor de células o un efecto citostático del resto de fármaco de PBD. Es posible medir la estabilidad del ADC mediante técnicas analíticas estándar, tales como espectroscopía de masas, HPLC y la técnica de separación/análisis LC/MS.
La administración de los compuestos de las fórmulas RelA se logra en el sitio de activación deseado del conjugado de la fórmula (I) mediante la acción de una enzima, tal como catepsina, en el grupo de enlace y en particular en el resto dipéptido valina-alanina.
Definición
Sustituyentes
La expresión “opcionalmente sustituido”, tal como se usa en la presente, hace referencia a un grupo original que puede estar no sustituido o que puede estar sustituido.
A menos que se especifique lo contrario, el término “sustituido”, tal como se usa en la presente, hace referencia a un grupo original que contiene uno o más sustituyentes. El término “sustituyente” se utiliza en la presente en el sentido convencional y hace referencia a un resto químico que se encuentra unido de manera covalente, o si corresponde, fusionado, a un grupo original. Se conoce una amplia variedad de sustituyentes y también se conocen métodos para su formación e introducción en varios grupos originales.
A continuación se describen en más detalle ejemplos de sustituyentes.
Alquilo C1-12: El término “alquilo C1-12”, tal como se usa en la presente, hace referencia a un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo de carbono de un compuesto hidrocarburo que tiene de 1 a 12 átomos de carbono, el que puede ser alifático o alicíclico, y que se puede encontrar saturado o insaturado (por ejemplo, parcialmente insaturado, totalmente insaturado). El término “alquilo C1-4”, tal como se usa en la presente, hace referencia a un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo de carbono de un compuesto hidrocarburo que tiene de 1 a 4 átomos de carbono, el que puede ser alifático o alicíclico, y que se puede encontrar saturado o insaturado (por ejemplo, parcialmente insaturado, totalmente insaturado). Por lo tanto, el término “alquilo” incluye las subclases alquenilo, alquinilo, cicloalquilo, etc., las que se describen más adelante.
Los ejemplos de grupos alquilo saturados incluyen, de modo no taxativo, metilo (C1), etilo (C2), propilo (C3), butilo (C4), pentilo (C5), hexilo (Ce) y heptilo (C7).
Los ejemplos de grupos alquilo lineales saturados incluyen, de modo no taxativo, metilo (C1 ), etilo (C2), n-propilo (C3), n-butilo (C4), n-pentilo (amilo) (C5), n-hexilo (Ce) y n-heptilo (C7).
Los ejemplos de grupos alquilo ramificados saturados incluyen, iso-propilo (C3), iso-butilo (C4), sec-butilo (C4), terc-butilo (C4), iso-pentilo (C5) y neo-pentilo (C5).
Alquenilo C2-12: El término “alquenilo C2-12”, tal como se usa en la presente, se refiere a un grupo alquilo que tiene uno o más enlaces dobles carbono-carbono.
Los ejemplos de grupos alquenilo insaturados incluyen, de modo no taxativo, etenilo (vinilo, -CH=CH2), 1-propenilo (-CH=CH-CH3), 2-propenilo (alilo, -CH-CH=CH2), isopropenilo (1-metilvinilo, -C(CH3)=CH2), butenilo (C4), pentenilo (C5) y hexenilo (Ce).
Alquinilo C2-12: El término “alquinilo C2-12”, tal como se usa en la presente, se refiere a un grupo alquilo que tiene uno o más enlaces triples carbono-carbono.
Los ejemplos de grupos alquinilo insaturados incluyen, de modo no taxativo, etinilo (-C=CH) y 2-propinilo (propargilo, -CH2CECH).
Cicloalquilo C3-12: El término “cicloalquilo C3-12” tal como se usa en la presente, se refiere a un grupo alquilo que es también un grupo ciclilo; es decir, un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo del anillo alicíclico de un compuesto de hidrocarburo cíclico (carbocíclico), cuyo resto tiene de 3 a 12 átomos de carbono, incluyendo de 3 a 12 átomos del anillo.
Los ejemplos de grupos cicloalquilo incluyen, de modo no taxativo, aquellos derivados de:
compuestos de hidrocarburos monocíclicos saturados:
ciclopropano (C3), ciclobutano (C4), ciclopentano (C5), ciclohexano (Ce), cicloheptano (C7), metilciclopropano (C4), dimetilciclopropano (C5), metilciclobutano (C5), dimetilciclobutano (Ce), metilciclopentano (Ce), dimetilciclopentano (C7) y metilciclohexano (C7);
compuestos de hidrocarburos monocíclicos insaturados:
ciclopropeno (C3), ciclobuteno (C4), ciclopenteno (C5), ciclohexeno (Ce), metilciclopropeno (C4), dimetilciclopropeno (C5), metilciclobuteno (C5), dimetilciclobuteno (Ce), metilciclopenteno (Ce), dimetilciclopenteno (C7) y metilciclohexeno (C7); y
compuestos de hidrocarburos policíclicos saturados:
norcarano (C7), norpinano (C7), norbornano (C7).
Heterociclilo C3-20: El término “heterociclilo C3-20”, tal como se usa en la presente, se refiere un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo del anillo de un compuesto heterocíclico, cuyo resto tiene de 3 a 20 átomos del anillo, de los cuales de 1 a 10 son heteroátomos del anillo. Preferentemente, cada anillo tiene de 3 a 7 átomos del anillo, de los cuales de 1 a 4 son heteroátomos del anillo.
En este contexto, los prefijos (por ejemplo, C3-20, C3-7, C5-e, etc.) indican la cantidad de átomos del anillo o intervalo de números de átomos del anillo, ya sean átomos de carbono o heteroátomos. Por ejemplo, el término “heterociclilo C5-e”, tal como se usa en la presente, se refiere a un grupo heterociclilo que tiene 5 o e átomos del anillo.
Los ejemplos de grupos heterociclilo monocíclicos incluyen, de modo no taxativo, aquellos derivados de:
N1 : aziridina (C3), azetidina (C4), pirrolidina (tetrahidropirrol) (C5), pirrolina (por ejemplo, 3-pirrolina, 2,5-dihidropirrol) (C5), 2H-pirrol o 3H-pirrol (isopirrol, isoazol) (C5), piperidina (Ce), dihidropiridina (Ce), tetrahidropiridina (Ce), azepina (C7);
O1 : oxirano (C3), oxetano (C4), oxolano (tetrahidrofurano) (C5), oxol (dihidrofurano) (C5), oxano (tetrahidropirano) (Ce), dihidropirano (Ce), pirano (Ce), oxepin (C7);
S1 : tiirano (C3), tietano (C4), tiolano (tetrahidrotiofeno) (C5), tiano (tetrahidrotiopiran) (Ce), tiepano (C7);
O2: dioxolano (C5), dioxano (Ca) y dioxepano (C7);
O3: trioxano (Ca);
N2: imidazolidina (C5), pirazolidina (diazolidina) (C5), imidazolina (C5), pirazolina (dihidropirazol) (C5), piperazina (Ca);
N1O1 : tetrahidrooxazol (C5), dihidrooxazol (C5), tetrahidroisoxazol (C5), dihidroisoxazol (C5), morfolina (Ca), tetrahidrooxazina (Ca), dihidrooxazina (Ca), oxazina (Ca);
N1S1 : tiazolina (C5), tiazolidina (C5), tiomorfolina (Ca);
N2O1 : oxadiazina (Ca);
O1S1 : oxatiol (C5) y oxatiano (tioxano) (Ca); y
N1O1S1 : oxatiazina (Ca).
Los ejemplos de grupos heterociclilo monocíclicos sustituidos incluyen aquellos derivados de sacáridos, en forma cíclica, por ejemplo, furanosas (C5), tales como arabinofuranosa, lixofuranosa, ribofuranosa y xilofuransa, y piranosas (Ca), tales como alopiranosa, altropiranosa, glucopiranosa, manopiranosa, gulopiranosa, idopiranosa, galactopiranosa y talopiranosa.
Arilo C5-20: El término “arilo C5-20”, tal como se usa en la presente, se refiere un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo de un anillo aromático de un compuesto aromático, cuyo resto tiene de 3 a 20 átomos del anillo. El término “arilo C5-7”, tal como se usa en la presente, hace referencia a un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo de un anillo aromático de un compuesto aromático, cuyo resto tiene de 5 a 7 átomos del anillo y el término “arilo C5-10”, tal como se usa en la presente, hace referencia a un resto monovalente que se obtiene al quitar un átomo de hidrógeno de un átomo de un anillo aromático de un compuesto aromático, cuyo resto tiene de 5 a 10 átomos del anillo. Preferentemente, cada anillo tiene de 5 a 7 átomos del anillo.
En este contexto, los prefijos (por ejemplo, C3-20, C5-7, C5-a, C5-10, etc.) indican la cantidad de átomos del anillo o el intervalo de números de átomos del anillo, ya sean átomos de carbono o heteroátomos. Por ejemplo, el término “arilo C5-a”, tal como se utiliza en la presente, hace referencia a un grupo arilo que tiene 5 o a átomos del anillo.
Todos los átomos del anillo pueden ser átomos de carbono, tal como en los “grupos carboarilo”.
Los ejemplos de grupos carboarilo incluyen, de modo no taxativo, aquellos derivados de benceno (es decir, fenilo) (Ca), naftaleno (C10), azuleno (C10), antraceno (C14), fenantreno (C14), naftaceno (C18) y pireno (C1a).
Los ejemplos de grupos arilo que comprenden anillo fusionados, de los cuales al menos uno es un anillo aromático, incluyen, de modo no taxativo, grupos derivados de indano (por ejemplo, 2,3-dihidro-1H-indeno) (C9), indeno (C9), isoindeno (C9), tetralina (1,2,3,4-tetrahidronaftaleno (C10), acenafteno (C12), fluoreno (C13), fenaleno (C13), acefenantreno (C15) y aceantreno (C1a).
De manera alternativa, los átomos del anillo pueden incluir uno o más heteroátomos, como en los “grupos heteroarilo”. Los ejemplos de grupos heteroarilo monocíclicos incluyen, de modo no taxativo, aquellos derivados de:
N1 : pirrol (azol) (C5), piridina (azina) (Ca);
O1 : furan (oxol) (C5);
S1 : tiofeno (tiol) (C5);
N1O1 : oxazol (C5), isoxazol (C5), isoxazina (Ca);
N2O1 : oxadiazol (furazan) (C5);
N3O1 : oxatriazol (C5);
N1S1 : tiazol (C5), isotiazol (C5);
N2: imidazol (1,3-diazol) (C5), pirazol (1,2-diazol) (C5), piridazina (1,2-diazina) (Ca), pirimidina (1,3-diazina) (Ca) (por ejemplo, citosina, timina, uracilo), pirazina (1,4-diazina) (Ca);
N3: triazol (C5), triazina (Ca); y,
N4: tetrazol (C5).
Los ejemplos de heteroarilo que comprende anillos fusionados incluyen, de modo no taxativo:
C9 (con 2 anillos fusionados) derivado de benzofurano (O1 ), isobenzofurano (O1), indol (N1 ), isoindol (N1), indolizina (N1 ), indolina (N1 ), isoindolina (N1 ), purina (N4) (por ejemplo, adenina, guanina), bencimidazol (N2), indazol (N2), benzoxazol (N1O1), benzisoxazol (N1O1), benzodioxol (O2), benzofurazano (N2O1 ), benzotriazol (N3), benzotiofurano (S1), benzotiazol (N1S1), benzotiadiazol (N2S);
C10 (con 2 anillos fusionados) derivado de cromeno (O1), isocromeno (O1), croman (O1 ), isocroman (O1), benzodioxano (O2), quinolina (N1), isoquinolina (N1), quinolizina (N1 ), benzoxazina (N1O1), benzodiazina (N2), piridopiridina (N2), quinoxalina (N2), quinazolina (N2), cinolina (N2), ftalazina (N2), naftiridina (N2), pteridina (N4); C11 (con 2 anillos fusionados) derivados de benzodiazepina (N2);
C13 (con 3 anillos fusionados) derivados de carbazol (N1 ), dibenzofurano (O1), dibenzotiofeno (S1 ), carbolina (N2), perimidina (N2), piridoindol (N2); y
C14 (con 3 anillos fusionados) derivados de acridina (N1 ), xanteno (O1), tioxanteno (S1 ), oxantreno (O2), fenoxatiin (O1S1 ), fenazina (N2), fenoxazina (N1O1 ), fenotiazina (N1S1), tiantreno (S2), fenantridina (N1), fenantrolina (N2),
fenazina (N2).
Los grupos anteriores, ya sea solos o como parte de otro sustituyente, pueden estar ellos mismos opcionalmente sustituidos con uno o más grupos seleccionados de ellos mismos y los sustituyentes adicionales indicados a continuación.
Halo: -F, -Cl, -Br, e -I.
Hidroxi: -OH.
Éter: -OR, donde R es un sustituyente éter, por ejemplo, un grupo alquilo C1-7 (también denominado grupo alquiloxi C1-7, descrito más adelante), un grupo heterociclilo C3-20(también denominado grupo heterocicliloxi C3-20), o un grupo arilo C5-20 (también denominado grupo ariloxi C5-20), preferentemente un grupo alquilo C1-7.
Alcoxi: -OR, donde R es un grupo alquilo, por ejemplo, un grupo alquilo C1-7. Los ejemplos de grupos alcoxi C1-7 incluyen, de modo no taxativo, -OMe (metoxi), -OEt (etoxi), -O(nPr) (n-propoxi), -O(iPr) (isopropoxi), -O(nBu) (nbutoxi), -O(sBu) (sec-butoxi), -O(iBu) (isobutoxi) y -O(tBu) (terc-butoxi).
Carboxi (ácido carboxílico): -C(=O)OH.
Éster (carboxilato, éster de ácido carboxílico, oxicarbonilo): -C(=O)OR, donde R es un sustituyente éster, por ejemplo, un grupo alquilo C1-7, un grupo heterociclilo C3-20 o un grupo arilo C5-20, preferentemente un grupo alquilo C1-7. Los ejemplos de grupos éster incluyen, de modo no taxativo, -C(=O)OCH3, -C(=O)OCH2CH3, -C(=O)OC(CH3)3 y -C(=O)OPh.
Amino: -NR1R2, donde R1 y R2 son independientemente sustituyentes de amino, por ejemplo, hidrógeno, un grupo alquilo C1-7 (al que también se hace referencia como alquilamino C ^ o alquilamino di-Cw), un grupo heterociclilo C3-20 , o un grupo arilo C5-20 , preferentemente H o un grupo alquilo C1-7, o, en el caso de un grupo amino “cíclico”, R1 y R2, junto con el átomo de nitrógeno al que se encuentran unidos, forman un anillo heterocíclico que tiene de 4 a 8 átomos del anillo. Los grupos amino pueden ser primarios (-NH2), secundarios (-NHR1) o terciarios (-NHR1R2), y en forma catiónica, pueden ser cuaternarios (-+NR1R2R3). Los ejemplos de grupos amino incluyen, de modo no taxativo, -NH2, -NHCH3, -NHC(CH3)2, -N(CH3)2, -N(CH2CH3)2 y -NHPh. Los ejemplos de grupos amino cíclicos incluyen, de modo no taxativo, aziridino, azetidino, pirrolidino, piperidino, piperazino, morfolino y tiomorfolino.
Amido (carbamoil, carbamilo, aminocarbonilo, carboxamida): -C(=O)NR1R2, donde R1 y R2 son independientemente sustituyentes amino, como se definió para los grupos amino. Los ejemplos de grupos amido incluyen, de modo no taxativo, -C(=O)NH2, -C(=O)NHCH3, -C(=O)N(CH3)2, -C(=O)NHCH2CH3 y -C(=O)N(CH2CH3)2, así como también grupos amido en los que R1 y R2, junto con el átomo de nitrógeno al que están unidos, forman una estructura heterocíclica como en, por ejemplo, piperidinocarbonilo, morfolinocarbonilo, tiomorfolinocarbonilo y piperazinocarbonilo.
Nitro: -NO2.
Azido: -N3.
Ciano (nitrilo, carbonitrilo): -CN.
Anticuerpo
En un aspecto, el anticuerpo es un anticuerpo que se une a AXL.
1H12
En algunas modalidades, el anticuerpo comprende un dominio VH que tiene una CDR3 de VH con la secuencia de aminoácidos del SEQ ID NO.7. En algunas modalidades, el dominio VH comprende además una CDR2 de VH con la secuencia de aminoácidos del SEQ ID NO.6 y/o una CDR1 de VH con la secuencia de aminoácidos del SEQ ID NO.5. En algunas modalidades, el anticuerpo comprende un dominio VH con una CDR1 de VH con la secuencia de aminoácidos del SEQ ID NO.5, una CDR2 de VH con la secuencia de aminoácidos del SEQ ID NO.6, y una CDR3 de VH con la secuencia de aminoácidos del SEQ ID NO.7. En modalidades preferidas, el anticuerpo comprende un dominio VH que tiene la secuencia de acuerdo con el SEQ ID NO. 1.
El anticuerpo puede comprender además un dominio de VL. En algunas modalidades, el anticuerpo comprende un dominio VL que tiene una CDR3 de VL con la secuencia de aminoácidos del SEQ ID NO.10. En algunas modalidades, el dominio VL comprende además una CDR2 de VL con la secuencia de aminoácidos del SEQ ID NO.9 y/o una CDR1 de VL con la secuencia de aminoácidos del SEQ ID NO.8. En algunas modalidades, el anticuerpo comprende un dominio VL con una CDR1 de VL con la secuencia de aminoácidos del SEQ ID NO.8, una CDR2 de VL con la secuencia
de aminoácidos del SEQ ID NO.9, y una CDR3 de VL con la secuencia de aminoácidos del SEQ ID NO.10. En modalidades preferidas, el anticuerpo comprende un dominio VL que tiene la secuencia de acuerdo con el SEQ ID NO. 2.
En modalidades preferidas, el anticuerpo comprende un dominio de VH y un dominio de VL. Preferentemente, el VH comprende la secuencia del SEQ ID NO.1 y el dominio de VL comprende la secuencia del SEQ ID NO.2.
Los dominios VH y VL pueden emparejarse de manera que formen un sitio de unión al antígeno del anticuerpo que se una a AXL.
En algunas modalidades, el anticuerpo es un anticuerpo intacto que comprende un dominio VH emparejado con un dominio VL, y los dominios VH y VL tienen las secuencias del SEQ ID NO.1 emparejadas con el SEQ ID NO.2.
En algunas modalidades, el anticuerpo comprende una cadena pesada que tiene la secuencia del SEQ ID NO. 3 emparejada con una cadena ligera que tiene la secuencia del SEQ ID NO.4. En algunas modalidades, el anticuerpo es un anticuerpo intacto que comprende dos cadenas pesadas que tienen la secuencia del SEQ ID NO.3, cada una de las cuales está emparejada con una cadena ligera que tiene la secuencia del SEQ ID NO.4.
En algunas modalidades, el anticuerpo comprende una cadena pesada que tiene la secuencia del SEQ ID NO. 24 emparejada con una cadena ligera que tiene la secuencia del SEQ ID NO.4. En algunas modalidades, el anticuerpo es un anticuerpo intacto que comprende dos cadenas pesadas que tienen la secuencia del SEQ ID NO.24, cada una de las cuales está emparejada con una cadena ligera que tiene la secuencia del SEQ ID NO.4.
En un aspecto, el anticuerpo es un anticuerpo tal como se describe en la presente que ha sido modificado (o modificado adicionalmente), tal como se describe más adelante. En algunas modalidades, el anticuerpo es una versión humanizada, desinmunizada o revestida de un anticuerpo descrito en la presente.
5F11
En algunas modalidades, el anticuerpo comprende un dominio VH que tiene una CDR3 de VH con la secuencia de aminoácidos del SEQ ID NO.15. En algunas modalidades, el dominio VH comprende además una CDR2 de VH con la secuencia de aminoácidos del SEQ ID NO.14 y/o una CDR1 de VH con la secuencia de aminoácidos del SEQ ID NO.13. En algunas modalidades, el anticuerpo comprende un dominio VH con una CDR1 de VH con la secuencia de aminoácidos del SEQ ID NO.13, una CDR2 de VH con la secuencia de aminoácidos del SEQ ID NO.14, y una CDR3 de VH con la secuencia de aminoácidos del SEQ ID NO.15.
En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de acuerdo con el SEQ ID NO: 11. En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de acuerdo con el SEQ ID NO: 19. En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de acuerdo con el SEQ ID NO: 20. En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de acuerdo con el SEQ ID NO: 21.
El anticuerpo puede comprender además un dominio de VL. En algunas modalidades, el anticuerpo comprende un dominio VL que tiene una CDR3 de VL con la secuencia de aminoácidos del SEQ ID NO.18. En algunas modalidades, el dominio VL comprende además una CDR2 de VL con la secuencia de aminoácidos del SEQ ID NO.17 y/o una CDR1 de VL con la secuencia de aminoácidos del SEQ ID NO.16. En algunas modalidades, el anticuerpo comprende un dominio VL con una CDR1 de VL con la secuencia de aminoácidos del SEQ ID NO.16, una CDR2 de VL con la secuencia de aminoácidos del SEQ ID NO.17, y una CDR3 de VL con la secuencia de aminoácidos del SEQ ID NO.18.
En algunas modalidades, el anticuerpo comprende un dominio VL que tiene la secuencia de acuerdo con el SEQ ID NO: 22.
En modalidades preferidas, el anticuerpo comprende un dominio de VH y un dominio de VL. En algunas modalidades, el VH comprende una CDR1 de VH con la secuencia de aminoácidos de SEQ ID NO.13, una CDR2 de VH con la secuencia de aminoácidos de SEQ ID NO.14, y una CDR3 de VH con la secuencia de aminoácidos de SEQ ID NO.15; y el dominio VL comprende una CDR1 de VL con la secuencia de aminoácidos de SEQ ID NO.16, una CDR2 de VL con la secuencia de aminoácidos de SEQ ID NO.17, y una CDR3 de VL con la secuencia de aminoácidos de SEQ ID NO.18.
En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de SEQ ID NO.19 y el dominio VL que tiene la secuencia de SEQ ID NO.22. En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de SEQ ID NO.20 y el dominio VL que tiene la secuencia de SEQ ID NO.22. En algunas modalidades, el anticuerpo comprende un dominio VH que tiene la secuencia de SEQ ID NO.21 y el dominio VL que tiene la secuencia de SEQ ID n O.22.
Los dominios VH y VL pueden emparejarse de manera que formen un sitio de unión al antígeno del anticuerpo que se
una a AXL.
En algunas modalidades, el anticuerpo es un anticuerpo intacto que comprende un dominio de VH emparejado con un dominio de VL.
En un aspecto, el anticuerpo es un anticuerpo tal como se describe en la presente que ha sido modificado (o modificado adicionalmente), tal como se describe más adelante. En algunas modalidades, el anticuerpo es una versión humanizada, desinmunizada o revestida de un anticuerpo descrito en la presente.
Terminología
El término “anticuerpo” en la presente se utiliza en su sentido más amplio y cubre específicamente los anticuerpos monoclonales, anticuerpos policlonales, dímeros, multímeros, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos), anticuerpos intactos y fragmentos de anticuerpos, siempre que exhiban la actividad biológica deseada, por ejemplo, la capacidad de unirse a AXL. Los anticuerpos pueden ser murinos, humanos, humanizados, quiméricos o derivados de otras especies. Un anticuerpo es una proteína generada por el sistema inmunitario capaz de reconocer y unirse a un antígeno específico. (Janeway, C., Travers, P., Walport, M., Shlomchik (2001) Immuno Biology, 5th Ed., Garland Publishing, Nueva York). Un antígeno diana tiene generalmente varios sitios de unión, también denominados epítopos, reconocidos por las CDR en múltiples anticuerpos. Cada anticuerpo que se une específicamente a un epítopo diferente tiene una estructura diferente. Por lo tanto, un antígeno puede tener más de un anticuerpo correspondiente. Un anticuerpo incluye una molécula de inmunoglobulina de longitud completa o una parte inmunológicamente activa de una inmunoglobulina de longitud completa, es decir, una molécula que contiene un sitio de unión al antígeno que se une de forma inmunoespecífica a un antígeno de una diana de interés o parte de esta, dichas dianas incluyen, de modo no taxativo, una o más células cancerosas que producen anticuerpos autoinmunitarios asociados a una enfermedad autoinmunitaria. La inmunoglobulina puede ser de cualquier tipo (por ejemplo, IgG, IgE, IgM, IgD e IgA), clase (por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA1 e IgA2) o subclase, o alotipo (por ejemplo, G1m1, G1m2, G1m3, no G1m1 [es decir, cualquier alotipo distinto a G1m1], G1m17, G2m23, G3m21, G3m28, G3m11, G3m5, G3m13, G3m14, G3m10, G3m15, G3m16, G3m6, G3m24, G3m26, G3m27, A2m1, A2m2, Km1, Km2 y Km3 humanos) de la molécula de inmunoglobulina. Las inmunoglobulinas pueden derivar de cualquier especie, lo que incluye origen humano, murino o de conejo.
Tal como se usa en la presente, “se une a AXL” hace referencia a que el anticuerpo se une a AXL con una afinidad más elevada que un compañero no específico, tal como albúmina de suero bovino (BSA, n.° de acceso de Genbank CAA76847, versión n.° CAA76847.1 GI:3336842, fecha de actualización del registro: 7 de enero de 2011 02:30 PM). En algunas modalidades, el anticuerpo se une a AXL con una constante de asociación (Ka) al menos 2, 3, 4, 5, 10, 20, 50, 100, 200, 500, 1000, 2000, 5000, 104, 105 o 106 veces más elevada que la constante de asociación del anticuerpo para BSA, cuando se mide en condiciones fisiológicas. Los anticuerpos de la invención se pueden unir a CD22 con una afinidad elevada. Por ejemplo, en algunas modalidades, el anticuerpo se puede unir a CD22 con una KD igual o menor de alrededor de 10-6 M, tal como 1 x 10-6, 10-7, 10-8, 10'9,10'1°, 10-11, 10-12, 10-13 o 10-14.
AXL es miembro de la familia TAM humana de receptores de tirosina cinasas. En algunas modalidades, el polipéptido AXL corresponde al n.° de acceso de Genbank AAH32229, versión n.° AAH32229.1 GI:21619004, fecha de actualización del registro: 6 de marzo de 2012 01:18 PM (SEQ ID NO.9). En una modalidad, el ácido nucleico que codifica el polipéptido AXL corresponde al n.° de acceso de Genbank M76125, versión n.° M76125.1 GI:292869, fecha de actualización del registro: 23 de junio de 2010 08:53 AM. En algunas modalidades, el polipéptido AXL tiene la secuencia del SEQ ID NO.23.
“Fragmentos de anticuerpo” comprende una parte de un anticuerpo de longitud completa, generalmente la región variable o de unión al antígeno de este. Los ejemplos de fragmentos de anticuerpo incluyen fragmentos Fab, Fab', F(ab')2 y scFv, diacuerpos, anticuerpos lineales, fragmentos producidos por una biblioteca de expresión de Fab, anticuerpos anti-idiotípicos (anti-Id), CDR (región determinante de complementariedad) y fragmentos de unión al epítopo de cualquiera de los anteriores que se unen de forma inmunoespecífica a los antígenos de células cancerosas, antígenos virales o antígenos microbianos, moléculas de anticuerpo de cadena simple, y anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpo.
La expresión “anticuerpo monoclonal”, tal como se usa en la presente, hace referencia a un anticuerpo obtenido de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que componen la población son idénticos excepto por posibles mutaciones de origen natural que pueden estar presentes en cantidades menores. Los anticuerpos monoclonales son altamente específicos y se dirigen a un único sitio antigénico. Además, a diferencia de las preparaciones de anticuerpos policlonales, que incluyen diferentes anticuerpos dirigidos contra diferentes determinantes (epítopos), cada anticuerpo monoclonal se dirige contra un único determinante en el antígeno. Además de su especificidad, los anticuerpos monoclonales tienen la ventaja de que se pueden sintetizar sin ser contaminados por otros anticuerpos. El modificador “monoclonal” indica el carácter del anticuerpo obtenido a partir de una población de anticuerpos considerablemente homogénea y no se debe interpretar que requiere la producción del anticuerpo a través de ningún método particular. Por ejemplo, los anticuerpos monoclonales para usar de acuerdo con la presente invención se pueden elaborar mediante el método de hibridoma, descrito por primera vez por Kohler
et ál (1975) Nature, 256:495, o se pueden elaborar mediante métodos de ADN recombinante (véase, US 4816567). Los anticuerpos monoclonales también pueden aislarse de bibliotecas de anticuerpos de fagos mediante el uso de las técnicas que se describen en Clackson et ál. (1991) Nature, 352:624-628; Marks et ál. (1991) J. Mol. Biol., 222:581-597 o a partir de ratones transgénicos que tienen un sistema de inmunoglobulina completamente humano (Lonberg (2008) Curr. Opinion 20(4):450-459).
Los anticuerpos monoclonales en la presente específicamente incluyen anticuerpos quiméricos en los que una parte de la cadena pesada y/o ligera es idéntica u homóloga a secuencias correspondientes en anticuerpos derivados de una especie particular o perteneciente a una clase o subclase de anticuerpo particular, mientras que el resto de la o las cadenas es idéntico u homólogo a las secuencias correspondientes en los anticuerpos derivados de otra especie o pertenecientes a otra clase o subclase de anticuerpo, así como también fragmentos de dichos anticuerpos, siempre y cuando exhiban la actividad biológica deseada (US 4816567 y Morrison et ál. (1984), Proc. Natl. Acad. Sci. u Sa , 81:6851-6855). Los anticuerpos quiméricos incluyen anticuerpos “primatizados” que comprenden secuencias de unión al antígeno de domino variable derivadas de un primate no humano (por ejemplo, mono del viejo mundo o simio) y secuencias de región constante humanas.
Un “anticuerpo intacto” de la presente, es uno que comprende dominios VL y VH, así como también un dominio constante de cadena ligera (CL) y dominios constantes de cadena pesada CH1, CH2 y CH3. Los dominios constantes pueden ser dominios constantes de una secuencia natural (por ejemplo, dominios constantes de una secuencia natural humana) o una variante de secuencia de aminoácidos de estos. El anticuerpo intacto puede tener una o más “funciones efectoras” que hacen referencia a aquellas actividades biológicas que se pueden atribuir a la región Fc (una región Fc de secuencia natural o una región Fc variante de secuencia de aminoácidos) de un anticuerpo. Los ejemplos de funciones efectoras de anticuerpo incluyen la unión a C1q, citotoxicidad dependiente de complemento, unión al receptor de Fc, citotoxicidad mediada por células dependiente del anticuerpo (ADCC), fagocitosis y regulación por disminución de los receptores de superficie celular, tales como receptor de linfocitos B y BCR.
En virtud de la secuencia de aminoácidos del dominio constante de sus cadenas pesadas, los anticuerpos intactos pueden dividirse en diferentes “clases”. Existen cinco clases principales de anticuerpos intactos: IgA, IgD, IgE, IgG, e IgM, y varios de estos pueden dividirse adicionalmente en “subclases” (isotipos), por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA e IgA2. Los dominios constantes de cadena pesada que corresponden a las diferentes clases de anticuerpos se llaman a, 8, £, y y M, respectivamente. Son conocidas las estructuras de las subunidades y las configuraciones tridimensionales de las diferentes clases de inmunoglobulinas.
Modificación de anticuerpos
Los anticuerpos descritos en la presente pueden estar modificados. Por ejemplo, para hacerlos menos inmunogénicos para un sujeto humano. Esto se puede lograr mediante cualquier cantidad de técnicas conocidas por el experto en la técnica. Algunas de estas técnicas se describen en más detalle más adelante.
Humanización
Las técnicas para reducir la inmunogenicidad in vivo de un anticuerpo o fragmento de anticuerpo no humano incluyen aquellas denominadas “de humanización”.
Un “anticuerpo humanizado” hace referencia a un polipéptido que comprende al menos una parte de una región variable modificada de un anticuerpo humano, en el que una parte de la región variable, preferentemente una parte sustancialmente menor que el dominio variable humano intacto, se ha sustituido con la secuencia correspondiente de una especie no humana, y en el que la región variable modificada se enlaza a al menos otra parte de otra proteína, preferentemente la región constante de un anticuerpo humano. La expresión “anticuerpos humanizados” incluye anticuerpos humanos, en los que uno o más residuos aminoácidos de región determinante de complementariedad (CDR) y/o uno o más residuos aminoácidos de región marco (FW o FR) se encuentran sustituidos con residuos aminoácidos de sitios análogos en anticuerpos de roedores u otros anticuerpos no humanos. La expresión “anticuerpo humanizado” también incluye una variante o un fragmento de secuencia de aminoácidos de inmunoglobulina de este, el que comprende una FR que tiene sustancialmente la secuencia de aminoácidos de una inmunoglobulina humana y una CDR que tiene sustancialmente la secuencia de aminoácidos de una inmunoglobulina no humana.
Las formas “humanizadas” de anticuerpos no humanos (por ejemplo, murinos) son anticuerpos quiméricos que contienen una secuencia mínima derivada de inmunoglobulina no humana. O, de otra manera, un anticuerpo humanizado es un anticuerpo humano que también contiene secuencias seleccionadas de anticuerpos no humanos (por ejemplo, murinos) en lugar de las secuencias humanas. Un anticuerpo humanizado puede incluir sustituciones conservadoras de aminoácidos o residuos no naturales de la misma especie o de otra distinta, las que no alteran significativamente su unión y/o actividad biológica. Dichos anticuerpos son anticuerpos quiméricos que contienen una secuencia mínima derivada de inmunoglobulinas no humanas.
Existe varias técnicas de humanización, las que incluyen “ injerto de CDR”, “selección guiada”, “desinmunización”, “revestimiento” (también conocido como “recubrimiento”), “anticuerpos compuestos”, “optimización de contenido de la
secuencia humana” y “trasposición de marco”.
Injertos en CDR
En esta técnica, los anticuerpos humanizados son inmunoglobulinas humanas (anticuerpo receptor) en las que se reemplazan residuos de una región determinante de complementariedad (CDR) del anticuerpo receptor con residuos de una CDR de una especie no humana (anticuerpo donante), tal como ratón, rata, camello, bovino, cabra o conejo, con las propiedades deseadas (de hecho, las CDR no humanas se injertan en el marco humano). En algunos casos, se reemplazan los residuos de región marco (FR) de la inmunoglobulina humana con residuos no humanos correspondientes (esto puede suceder cuando, por ejemplo, un residuo de FR particular tiene efecto significativo en la unión al antígeno).
Además, los anticuerpos humanizados pueden comprender residuos que no se encuentran ni en el anticuerpo receptor ni en las secuencias marco o CDR importadas. Estas modificaciones se realizan para refinar y maximizar adicionalmente el rendimiento del anticuerpo. Por lo tanto, en general, un anticuerpo humanizado comprenderá todos de al menos uno, y, en un aspecto, dos, dominios variables, en los que todos los bucles hipervariables corresponden a los de una inmunoglobulina no humana y todas o sustancialmente todas las regiones FR son las de una secuencia de inmunoglobulina humana. El anticuerpo humanizado también comprenderá opcionalmente al menos una parte de una región constante de inmunoglobulina (Fc) o la de una inmunoglobulina humana.
Selección guiada
El método consiste en la combinación del dominio Vh o Vl de un anticuerpo no humano dado específico para un epítopo particular con una biblioteca de Vh o Vl humanos y los dominios V humanos específicos se seleccionan en función del antígeno de interés. Luego, este VH humano seleccionado se combina con una biblioteca de VL para generar una combinación de VHxVL completamente humanos. El método se describe en Nature Biotechnology (N.Y.) 12, (1994) 899-903.
Anticuerpos compuestos
En este método, se combinan dos o más segmentos de secuencia de aminoácidos de un anticuerpo humano en la molécula del anticuerpo final. Se construyen mediante la combinación de múltiples segmentos de secuencia de VH y VL humanos en combinaciones que limitan o evitan los epítopos de linfocitos T humanos en las regiones V del anticuerpo compuesto final. En los casos que sea necesario, se limitan o se evitan los epítopos de linfocitos T mediante el intercambio de segmentos de región V que contribuyen o codifican un epítopo de linfocitos T con segmentos alternativos que evitan los epítopos de linfocitos T. Este método se describe en US 2008/0206239 A1.
Desinmunización
Este método implica remover epítopos de linfocitos T humanos (u otra especie secundaria) de las regiones V del anticuerpo terapéutico (u otra molécula). Se analizó la secuencia de región V de los anticuerpos terapéuticos para determinar la presencia de motivos de unión a MHC de clase II, por ejemplo, mediante su comparación con las bases de datos de motivos de unión a MHC (tal como la base de datos de “motivos” en www.wehi.edu.au). De manera alternativa, es posible identificar los motivos de unión a MHC de clase II mediante métodos con hilos informáticos, tal como los ideados por Altuvia et ál. (J. Mol. Biol. 249 244-250 (1995)); en estos métodos, se evalúan los péptidos superpuestos consecutivos de las secuencias de región V para determinar sus energías de unión a las proteínas de MHC de clase II. Luego, estos datos se pueden combinar con información sobre otras características de la secuencia que se relacionan con péptidos presentados de forma exitosa, tal como anfipaticidad, motivos Rothbard y sitios de escisión para la catepsina B y otras enzimas de procesamiento.
Una vez que se han identificado posibles epítopos de linfocitos T de especies secundarias (por ejemplo, humanos), estos se eliminan mediante la alteración de uno o más aminoácidos. Los aminoácidos modificados suelen encontrarse en el epítopo de linfocitos T en sí mismo, pero también se pueden encontrar adyacentes al epítopo en términos de la estructura primaria o secundaria de la proteína (y, por lo tanto, pueden no encontrarse adyacente en la estructura primaria). Más normalmente, la alteración es mediante sustitución pero, en algunas circunstancias, será más apropiada la adición o la eliminación de aminoácidos.
Todas las alteraciones se pueden lograr con tecnología de ADN recombinante, de manera que la molécula final se pueda preparar mediante la expresión de un hospedador recombinante a partir de métodos ya establecidos, tal como mutagénesis dirigida al sitio. Sin embargo, también es posible el uso de la química de proteínas o cualquier otro medio de alteración molecular.
Revestimiento
Este método implica:
(a) determinar la estructura conformacional de la región variable del anticuerpo (o fragmento de este) no humano (por ejemplo, roedor) al construir un modelo tridimensional de la región variable del anticuerpo no humano;
(b) generar alineamientos de secuencia con distribuciones de accesibilidad relativa a partir de estructuras cristalográficas de rayos x de una cantidad suficiente de cadenas pesada y ligera de región variable de anticuerpo no humano y humano para producir un conjunto de posiciones marco de cadena pesada y ligera, en las que las posiciones de alineamiento son idénticas en el 98 % de la cantidad suficiente de cadenas pesada y ligera de anticuerpo no humano,
(c) definir, para el anticuerpo no humano a ser humanizado, un conjunto de residuos aminoácidos expuestos en la superficie de cadena pesada y ligera con el conjunto de posiciones marco generadas en la etapa (b),
(d) identificar, a partir de secuencias de aminoácidos de anticuerpo humano, un conjunto de residuos aminoácidos expuestos en la superficie de cadena pesada y ligera, el que es más estrechamente idéntico al conjunto de residuos aminoácidos expuestos en la superficie que se defina en la etapa (c), en la que las cadenas pesada y ligera del anticuerpo humano se encuentran naturalmente emparejadas o no,
(e) sustituir, en la secuencia de aminoácidos del anticuerpo no humano a ser humanizado, el conjunto de residuos aminoácidos expuestos en la superficie de cadena pesada y ligera que se define en la etapa (c) con el conjunto de residuos aminoácidos expuestos en la superficie de cadena pesada y ligera que se identifica en la etapa (d), (f) construir un modelo tridimensional de la región variable del anticuerpo no humano que resulta de la sustitución especificada en la etapa (e),
(g) identificar, al comparar los modelos tridimensionales construidos en las etapas (a) y (f), cualquier residuo aminoácido de los conjuntos identificados en las etapas (c) o (d) que se encuentran a 5 Angstroms de cualquier átomo de cualquier residuo de las regiones determinantes de complementariedad del anticuerpo no humano a ser humanizado y
(h) cambiar cualesquiera residuos identificados en la etapa (g) del residuo aminoácido humano al no humano original, de forma que se define un conjunto humanizante de residuos aminoácidos expuestos en la superficie del anticuerpo no humano con la condición de que la etapa (a) no tenga por qué llevarse a cabo en primer lugar, sino que debe llevarse a cabo antes de la etapa (g).
Superhumanización
El método compara la secuencia no humana con el repertorio génico de la línea germinal humana funcional. Se seleccionan estos genes humanos que codifican las estructuras canónicas idénticas a las secuencias no humanas o estrechamente relacionadas con estas. Estos genes humanos seleccionados con homología más elevada en las CDR se eligen como donantes de FR. Por último, se injertan las CDR no humanas en estas FR humanas. Este método se describe en la patente WO 2005/079479 A2.
Optimización de contenido de la secuencia humana
Este método compara la secuencia no humana (por ejemplo, ratón) con el repertorio de genes de línea germinal humana y las diferencias se califican como contenido de la secuencia humana (HSC) que cuantifica una secuencia en el nivel de los posibles epítopos de MHC o linfocitos T. Luego, la secuencia diana se humaniza al maximizar su HSC en lugar de usar una medida de identidad global para generar múltiples variantes humanizadas diversas (descritas en Molecular Immunology, 44, (2007) 1986-1998).
Transposición de marco
Las CDR del anticuerpo no humano se fusionan en el marco con grupos de ADNc que comprenden todos los marcos de genes de línea germinal humana de cadena pesada y ligera conocidos. Luego, se seleccionan los anticuerpos humanizados, por ejemplo, mediante el cribado de la biblioteca de anticuerpos de expresión en fagos. Esto se describe en Methods 36, 43-60 (2005).
Modificación de anticuerpos con azida
El anticuerpo puede prepararse para su conjugación con el enlazador del fármaco a través de un proceso de tres etapas:
(1) La expresión del anticuerpo (Ab) con un núcleo N-glicano en un sistema de expresión adecuado (por ejemplo, una línea celular CHO). El núcleo N-glicano se conjuga normalmente a Asn-297 de la cadena pesada de acuerdo con el sistema de numeración de Kabat;
(2) cortar todas las isoformas de glicano (complejas, híbridas, con contenido alto de manosa) con una endoglicosidasa para dejar el núcleo GlcNAc; y
(3) transferencia enzimática al núcleo GlcNAc de un residuo de N-acetilgalactosa que presenta un grupo azida para la conjugación del enlazador del fármaco.
Se presenta un resumen del proceso anterior en van Geel, R., et ál., Bioconjugate Chemistry, 2015, 26, 2233-2242; DOI: 10.1021/acs.bioconjchem.5b00224. De manera alternativa, se puede usar un proceso en un solo recipiente, ver los ejemplos.
Modalidades
X
En algunas modalidades, X es un enlace simple.
En otras modalidades, X es -CH2-.
En modalidades adicionales, X es -C2H4-.
En algunas modalidades, n es 1 a 4.
En algunas de estas modalidades, n es 1.
En otras de estas modalidades, n es 2.
En otras de estas modalidades, n es 4.
R7
En una modalidad, R7 es metilo.
En otra modalidad, R7 es fenilo.
R2
Cuando existe un enlace doble presente entre C2' y C3', R2 se selecciona de:
(a) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3;
(b) un alquilo alifático C1-5 saturado;
(c) un cicloalquilo C3-6 saturado;
(d)

donde cada uno de R21, R22 y R23 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R2 no es mayor de 5; (e)

donde uno de R25a y R25b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y
(f)

donde R24 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo.
Cuando R2 es un grupo arilo C5-10, puede ser un grupo arilo C5-7. Un grupo arilo C5-7 puede ser un grupo fenilo o un grupo heteroarilo C5-7, por ejemplo, furanilo, tiofenilo y piridilo. En algunas modalidades, R2 es preferentemente fenilo. En otras modalidades, R12 es preferentemente tiofenilo, por ejemplo, tiofen-2-ilo y tiofen-3-ilo.
Cuando R2 es un grupo arilo C5-10, puede ser un grupo arilo C8-10, por ejemplo, un grupo quiniolinilo o isoquinolinilo. El grupo quinolinilo o isoquinolinilo puede estar unido al núcleo de PBD a través de cualquier posición disponible del anillo. Por ejemplo, el quinolinilo puede ser quinolin-2-ilo, quinolin-3-ilo, quinolin-4ilo, quinolin-5-ilo, quinolin-6-ilo,
quinolin-7-ilo y quinolin-8-ilo. De estos, se pueden preferir quinolin-3-ilo y quinolin-6-ilo. El isoquinolinilo puede ser isoquinolin-1-ilo, isoquinolin-3- ilo, isoquinolin-4ilo, isoquinolin-5-ilo, isoquinolin-6-ilo, isoquinolin-7-ilo e isoquinolin-8-ilo. De estos, se pueden preferir isoquinolin-3-ilo e isoquinolin-6-ilo.
Cuando R2 es un grupo arilo C5-10, puede tener cualquier cantidad de grupos sustituyentes. Preferentemente, tiene de 1 a 3 grupos sustituyentes, con preferencia de 1 y 2, y se prefieren incluso más grupos con un único sustituyente. Los sustituyentes se pueden encontrar en cualquier posición.
En los casos en los que R2 es un grupo arilo C5-7, un sustituyente único se encuentra preferentemente en un átomo de anillo que no se encuentra adyacente al enlace al resto del compuesto, es decir, es preferentemente 3 respecto al enlace al resto del compuesto. Por consiguiente, en el caso en el que el grupo arilo C5-7 es fenilo, el sustituyente se encuentra preferentemente en posiciones meta o para y más preferentemente, en posición para.
En el caso en el que R2 es un grupo arilo C8-10, por ejemplo, quinolinilo o isoquinolinilo, puede presentar cualquier cantidad de sustituyentes en cualquier posición de los anillos de quinolina o isoquinolina. En algunas modalidades, presenta uno, dos o tres sustituyentes, los que se pueden encontrar ya sea en los anillos próximos como distales, o en ambos (si hubiera más de un sustituyente).
Los sustituyentes de R2, cuando R2 es un grupo arilo C5-10
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es halo, es preferentemente F o Cl, más preferentemente Cl.
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es éter, en algunas modalidades puede ser un grupo alcoxi, por ejemplo, un grupo alcoxi C1-7 (por ejemplo, metoxi, etoxi) o en algunas modalidades puede ser un grupo ariloxi C5-7 (por ejemplo, fenoxi, piridiloxi, furaniloxi). El grupo alcoxi se puede encontrar adicionalmente sustituido, por ejemplo, con un grupo amino (por ejemplo, dimetilamino).
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es alquilo C1-7, puede ser preferentemente un grupo alquilo C1-4 (por ejemplo, metilo, etilo, propilo, butilo).
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es heterociclilo C3-7, en algunas modalidades puede ser un grupo heterociclilo C6 que contiene nitrógeno, por ejemplo, morfolino, tiomorfolino, piperidinilo, piperazinilo. Dichos grupos se pueden enlazar al resto del resto de PBD a través del átomo de nitrógeno. Dichos grupos se pueden sustituir adicionalmente, por ejemplo, con grupos alquilo C1-4. Si el grupo heterociclilo C6 que contiene nitrógeno es piperazinilo, dicho sustituyente adicional se puede encontrar en el segundo átomo anular de nitrógeno.
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es bis-oxi-alquileno C1-3, preferentemente es bis-oxi-metileno o bis-oxi-etileno.
Si un sustituyente en R2 cuando R2 es un grupo arilo C5-10 es éster, preferentemente es éster metílico o éster etílico.
Los sustituyentes particularmente preferidos cuando R2 es un grupo arilo C5-10 incluyen metoxi, etoxi, flúor, cloro, ciano, bis-oxi-metileno, metil-piperazinilo, morfolino y metil-tiofenilo. Otros sustituyentes particularmente preferidos para R2 son dimetilaminopropiloxi y carboxi.
Los grupos R2 sustituidos particularmente preferidos cuando R2 es un grupo arilo C5-10 incluyen, de modo no taxativo, 4-metoxi-fenilo, 3-metoxifenilo, 4-etoxi-fenilo, 3-etoxi-fenilo, 4-fluoro-fenilo, 4-cloro-fenilo, 3,4-bisoximetilen-fenilo, 4-metiltiofenilo, 4-cianofenilo, 4-fenoxifenilo, quinolin-3-ilo y quinolin-6-ilo, isoquinolin-3-ilo e isoquinolin-6-ilo, 2-tienilo, 2-furanilo, metoxinaftilo y naftilo. Otro grupo R2 sustituido posible es 4-nitrofenilo. Los grupos R2 de interés particular incluyen 4-(4- metilpiperazin-1-il)fenilo y 3,4-bisoximetilen-fenilo.
Cuando R2 es alquilo alifático C1-5 saturado, puede ser metilo, etilo, propilo, butilo o pentilo. En algunas modalidades, puede ser metilo, etilo o propilo (n-pentilo o isopropilo). En algunas de estas modalidades, puede ser metilo. En otras modalidades, puede ser butilo o pentilo, los que pueden ser lineales o ramificados.
Cuando R2 es cicloalquilo C3-6 saturado, puede ser ciclopropilo, ciclobutilo, ciclopentilo o ciclohexilo. En algunas modalidades, puede ser ciclopropilo.
Cuando R2 es

cada uno de R21, R22 y R23 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R2 no es mayor de 5. En algunas modalidades, la cantidad total de átomos de carbono en el grupo R2 no es mayor de 4 o no mayor de 3.
En algunas modalidades, uno de R21, R22 y R23 es H, y los otros dos grupos se seleccionan de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo.
En otras modalidades, dos de R21, R22 y R23 son H, y el otro grupo se selecciona de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo.
En algunas modalidades, los grupos que no son H se seleccionan de metilo y etilo. En algunas de dichas modalidades, los grupos que no son H son metilo.
En algunas modalidades, R21 es H.
En algunas modalidades, R22 es H.
En algunas modalidades, R23 es H.
En algunas modalidades, R21 y R22 son H.
En algunas modalidades, R21 y R23 son H.
En algunas modalidades, R22 y R23 son H.
Un grupo R2 de interés particular es:

Cuando R2 es

uno de R25a y R25b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo. En algunas modalidades, el grupo que no es H es fenilo opcionalmente sustituido. Si el sustituyente opcional de fenilo es halo, preferentemente es flúor. En algunas modalidades, el grupo fenilo no se encuentra sustituido.
Cuando R2 es

R24 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo. Si el sustituyente opcional de fenilo es halo, preferentemente es flúor. En algunas modalidades, el grupo fenilo no se encuentra sustituido.
En algunas modalidades, R24 se selecciona de H, metilo, etilo, etenilo y etinilo. En algunas de dichas modalidades, R24 se selecciona de H y metilo.
Cuando existe un enlace simple entre C2 y C3,
R2 es

donde R26a y R26b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R26a y R26b es H, el otro se selecciona de nitrilo y un alquiléster C1-4.
En algunas modalidades, se prefiere que tanto R26a como R26b sean H.
En otras modalidades, se prefiere que R26a y R26b sean metilo.
En modalidades adicionales, se prefiere que uno de R26a y R26b sea H y el otro se selecciona de alquilo C1-4 saturado y alquenilo C2-3, con los grupos alquilo y alquenilo opcionalmente sustituidos. En dichas modalidades adicionales, se puede preferir adicionalmente que el grupo que no sea H se seleccione de metilo y etilo.
R12
Las preferencias anteriores para R2 aplican de igual manera a R12.
En una modalidad de la invención, DL es

Carga de fármaco
La carga de fármaco es la cantidad promedio de fármacos de PBD por anticuerpo, por ejemplo, anticuerpo.
La cantidad promedio de fármacos por anticuerpo en las preparaciones de ADC a partir de reacciones de conjugación puede caracterizarse mediante medios convencionales tales como UV, HPLC de fase inversa, HIC, espectroscopía de masas, ensayo de ELISA y electroforesis. También es posible determinar la distribución cuantitativa de ADC en términos de p. Se puede determinar mediante ELISA el valor promediado de p en una preparación particular de ADC (Hamblett et al (2004) Clin. Cancer Res. 10:7063-7070; Sanderson et al (2005) Clin. Cancer Res. 11:843-852). Sin embargo, no es posible discernir la distribución de valores de p (fármaco) mediante la unión de antígeno a anticuerpo y la limitación de detección de ELISA. Además, el ensayo de ELISA para detectar conjugados de fármaco y anticuerpo no determina dónde se unen los restos de fármaco al anticuerpo, como fragmentos de cadena pesada o cadena ligera, o los residuos aminoácidos particulares. En algunos casos, es posible lograr la separación, purificación y caracterización de ADC homogéneos con p como un valor determinado de ADC con otras cargas de fármaco a través de medios tales como HPLC de fase inversa o electroforesis. Dichas técnicas también se pueden aplicar a otros tipos de conjugados.
Para los conjugados de anticuerpos y fármacos de la presente, p está limitado por la cantidad de sitios de unión en el anticuerpo, es decir, la cantidad de grupos azida. Por ejemplo, el anticuerpo solo puede tener uno o dos grupos azida al cual el enlazador del fármaco puede estar unido.
Usualmente, durante una reacción de conjugación, se conjuga menos que el máximo teórico de restos de fármacos a un anticuerpo. Es posible controlar la carga (relación fármaco/anticuerpo) de un ADC en varias formas diferentes que incluyen: (i) limitar el exceso molar de intermedio de enlazador y fármaco (D-L) o reactivo enlazador respecto al anticuerpo y (ii) limitar la temperatura o tiempo de reacción de conjugación.
En los casos en los que más de un grupo nucleofílico o electrofílico del anticuerpo reacciona con un intermedio de enlazador y fármaco o un reactivo enlazador seguido de un reactivo de resto de fármaco, el producto resultante es una mezcla de compuestos de ADC y fármaco con una distribución de restos de fármacos unidos a un anticuerpo, por ejemplo, 1, 2, 3, etc. Los métodos de cromatografía líquida tales como de interacción hidrofóbica (HIC) y fase inversa polimérica (PLRP) pueden separar compuestos en la mezcla conforme al valor de carga de fármacos. Es posible aislar preparaciones de ADC con un valor de carga simple de fármaco (p). Sin embargo, dichos ADC de valor de carga simple también pueden ser mezclas heterogéneas debido a que los restos de fármacos pueden unirse en sitios diferentes del anticuerpo a través del enlazador.
Por lo tanto, las composiciones de conjugado de anticuerpo y fármaco de la invención incluyen mezclas de compuestos de conjugado de anticuerpo y fármaco en las que el anticuerpo tiene uno o más restos de fármacos de PBD y en las que los restos de fármacos se pueden unir al anticuerpo en varios residuos aminoácidos.
En una modalidad, la cantidad promedio de grupos pirrolobenzodiazepina diméricos por anticuerpo se encuentra en el intervalo de 1 a 8. En algunas modalidades el intervalo se selecciona de 1 a 4, 1 a 4, 2 a 4, y 1 a 3.
En algunas modalidades, hay uno o dos grupos pirrolobenzodiazepina diméricos por anticuerpo.
Incluye otras formas
A menos que se especifique lo contrario, lo anterior incluye las formas iónicas, de sal, de solvato y protegidas conocidas de dichos sustituyentes. Por ejemplo, una referencia a ácido carboxílico (-COOH) también incluye la forma aniónica (carboxilato) (-COO-), una sal o solvato de este, así como formas protegidas convencionales. De manera similar, una referencia a un grupo amino incluye la forma protonada (-N+HR1R2), una sal o solvato del grupo amino, por ejemplo, una sal de clorhidrato, así como formas protegidas convencionales de un grupo amino. De manera similar, una referencia a un grupo hidroxilo también incluye la forma aniónica (-O-), una sal o solvato de este, así como formas protegidas convencionales.
Sales
Puede ser conveniente o deseable preparar, purificar y/o manipular una sal correspondiente del compuesto activo, por ejemplo, una sal farmacéuticamente aceptable. Se describen ejemplos de sales farmacéuticamente aceptables en Berge, et ál., J. Pharm. Sci., 66, 1-19 (1977).
Por ejemplo, si el compuesto es aniónico o tiene un grupo funcional que puede ser aniónico (por ejemplo, -COOH puede ser -COO'), la sal se puede formar con un catión adecuado. Los ejemplos de cationes inorgánicos adecuados incluyen, de modo no taxativo, iones de metales alcalinos tales como Na+ y K+, cationes alcalinotérreos tales como Ca2+ y Mg2+, y otros cationes tales como Al3+. Los ejemplos de cationes orgánicos adecuados incluyen, de modo no taxativo, ion amonio (es decir, NH4+) e iones amonio sustituidos (por ejemplo, NH3R+, NH2R2+, NHR3+, NR4+). Los ejemplos de algunos iones de amonio sustituido adecuados son aquellos derivados de: etilamina, dietilamina, diciclohexilamina, trietilamina, butilamina, etilendiamina, etanolamina, dietanolamina, piperazina, bencilamina, fenilbencilamina, colina, meglumina y trometamina, así como aminoácidos, tales como lisina y arginina. Un ejemplo de un ion de amonio cuaternario común es N(CH3)4+.
Si el compuesto es catiónico, o tiene un grupo funcional que puede ser catiónico, (por ejemplo, -NH2 puede ser -NH3+), se puede formar una sal con un anión adecuado. Los ejemplos de aniones inorgánicos adecuados incluyen, de modo no taxativo, los derivados de los siguientes ácidos inorgánicos: clorhídrico, bromhídrico, yodhídrico, sulfúrico, sulfuroso, nítrico, nitroso, fosfórico y fosforoso.
Los ejemplos de aniones orgánicos adecuados incluyen, de modo no taxativo, los derivados de los siguientes ácidos orgánicos: 2-acetoxibenzoico, acético, ascórbico, aspártico, benzoico, alcanforsulfónico, cinámico, cítrico, edético, etandisulfónico, etansulfónico, fumárico, glucoheptónico, glucónico, glutámico, glicólico, hidroximaleico, hidroxinaftaleno carboxílico, isetiónico, láctico, lactobiónico, láurico, maleico, málico, metansulfónico, múcico, oleico, oxálico, palmítico, pamoico, pantoténico, fenilacético, fenilsulfónico, propiónico, pirúvico, salicílico, esteárico, succínico, sulfanílico, tartárico, toluensulfónico, trifluoroacético y valérico. Los ejemplos de aniones orgánicos poliméricos adecuados incluyen, de modo no taxativo, aquellos derivados de los siguientes ácidos poliméricos: ácido tánico, carboximetilcelulosa.
Solvatos
Puede ser conveniente o deseable preparar, purificar y/o manipular un solvato correspondiente del compuesto activo. En la presente, el término “solvato” se utiliza en el sentido convencional para hacer referencia a un complejo de soluto (por ejemplo, compuesto activo, sal de compuesto activo) y solvente. Si el solvente es agua, se puede hacer referencia convenientemente al solvato como un hidrato, por ejemplo, un monohidrato, un dihidrato, un trihidrato, etc.
La invención incluye compuestos en los que un solvente se agrega a través del enlace imina del resto de PBD, lo que se ilustra a continuación donde el solvente es agua o un alcohol (RAOH, en el que RA es alquilo C1-4):

Estas formas se pueden denominar formas carbinolamina y éter de carbinolamina de PBD (tal como se describió en la sección previa con referencia a R10). El equilibrio de dichos equilibrios depende de las condiciones en las que se encuentran los compuestos, así como de la naturaleza del resto en sí mismo.
Estos compuestos particulares se pueden aislar en forma sólida, por ejemplo, mediante liofilización.
Isómeros
Determinados compuestos de la invención pueden existir en un o más formas particulares geométricas, ópticas, enantioméricas, diasterioméricas, epiméricas, atrópicas, estereoisoméricas, tautoméricas, conformacionales o anoméricas, incluso, entre otras, formas cis y trans; formas E y Z; formas c, t y r; formas endo y exo; formas R, S y meso; formas D y L; formas d y l; formas (+) y (-); formas ceto, enol y enolato; sin y anti; formas sinclinales y anticlinales; formas a y p; formas axiales y ecuatoriales; formas de barco, silla, recodo, sobre y media silla; y combinaciones de estas, en lo sucesivo denominadas en forma colectiva “ isómeros” (o “formas isoméricas”).
El término “quiral” se refiere a moléculas que tienen la propiedad de no superponerse al compañero de imagen especular, mientras que el término “aquiral” hace referencia a moléculas que son suporponibles a su compañero de imagen especular.
El término “estereoisómeros” hace referencia a los compuestos que tienen idéntica constitución química, pero que difieren con respecto a la disposición de los átomos o de los grupos en el espacio.
“Diastereómero” hace referencia a un estereoisómero con dos o más centros quirales y cuyas moléculas no son imágenes especulares una de otra. Los diastereómeros difieren en sus propiedades físicas, p. ej., puntos de fusión, puntos de ebullición, propiedades espectrales y reactividades. Las mezclas de diasterómeros se pueden separar en procedimientos analíticos de alta resolución, tal como electroforesis y cromatografía.
“Enantiómeros” se refiere a dos estereoisómeros de un compuesto que son imágenes especulares no superponibles entre sí.
Las definiciones y convenciones estereoquímicas que se utilizan en la presente siguen en general a S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, Nueva York; y Eliel, E. y Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., Nueva York, 1994. Los compuestos de la presente invención pueden contener centros asimétricos o quirales y, por lo tanto, existen en diferentes formas estereoisoméricas. Se pretende que formen parte de la presente invención todas las formas estereoisoméricas de los compuestos de la invención, los que incluyen, de modo no taxativo, diastereómeros, enantiómeros y atropisómeros, así como mezclas de estos, tales como mezclas racémicas. Muchos compuestos orgánicos existen en formas ópticamente activas, es decir, tienen la capacidad de rotar el plano de luz polarizada en un plano. Al describir un compuesto ópticamente activo, los prefijos D y L o R y S, se utilizan para indicar la configuración absoluta de la molécula alrededor de su o sus centros quirales. Los prefijos d y l o (+) y (-) se emplean para indicar el signo de rotación de luz polarizada en un plano por parte del compuesto, donde (-) o l significa que el compuesto es levógiro. Un compuesto con el prefijo (+) o d es dextrógiro. Para una estructura química dada, estos estereoisómeros son idénticos excepto que son imágenes especulares entre sí. También puede hacerse referencia a un estereoisómero específico como un enantiómero y una mezcla de dichos isómeros generalmente se denomina mezcla enantiomérica. Se hace referencia a una mezcla de enantiómeros 50:50 como una mezcla racémica o un racemato, lo cual puede ocurrir en casos en los que no ha habido estereoselección o estereoespecificidad en una reacción o proceso químico. Las expresiones “mezcla racémica” y “racemato” hacen referencia a una mezcla equimolar de dos especies enantioméricas sin actividad óptica.
Se debe observar que, salvo por lo que se discute más adelante para formas tautoméricas, el término “ isómeros”, tal
como se utiliza en la presente, excluye específicamente isómeros estructurales (o constitucionales) (es decir, isómeros que difieren en la conexión entre átomos, en lugar de diferir meramente en la posición de los átomos en el espacio). Por ejemplo, una referencia al grupo metoxi, -OCH3, no se interpreta como una referencia a su isómero estructural, un grupo hidroximetilo, -CH2OH. De forma similar, una referencia al orto-clorofenil no se interpreta como una referencia a su isómero estructural, meta-clorofenil. Sin embargo, una referencia a una clase de estructuras puede incluir formas estructuralmente isoméricas que se encuentran dentro de esa clase (por ejemplo, alquilo C1-7 incluye n-propilo e isopropilo; butilo incluye n-, iso-, sec-y terc-butilo; metoxifenilo incluye orto-, meta-y para-metoxifenilo).
La exclusión anterior no pertenece a formas tautoméricas, por ejemplo, formas ceto, enol y enolato, como en, por ejemplo, los siguientes pares tautoméricos: ceto/enol (ilustrado abajo), imina/enamina, amida/iminoalcohol, amidina/amidina, nitroso/oxima, tiocetona/enetiol, N-nitroso/hidroxiazo y nitro/aci-nitro.

Las expresiones “tautómero” o “forma tautomérica” hacen referencia a isómeros estructurales de distintas energías que son interconvertibles a través de una barrera de baja energía. Por ejemplo, los tautómeros por transferencia de protones (también conocidos como tautómeros prototrópicos) incluyen interconversiones a través de la migración de un protón, tales como isomerizaciones cetoenólicas e imina-enamina. Los tautómeros de valencia incluyen interconversiones mediante la reorganización de algunos de los electrones de unión.
Cabe destacar que el término “ isómero” incluye específicamente compuestos con una o más sustituciones isotópicas. Por ejemplo, H puede encontrarse en cualquier forma isotópica, lo cual incluye 1H, 2H (D) y 3H (T); C puede encontrarse en cualquier forma isotópica, lo cual incluye 12C, 13C y 14C; O puede encontrarse en cualquier forma isotópica, lo cual incluye 16O y 18O; y similares.
Los ejemplos de isótopos que se pueden incorporar en los compuestos de la invención incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, fluoro y cloro, como, entre otros, 2H (deuterio, D), 3H (tritio), 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36Cl y 125I. Se incorporan varios compuestos marcados de forma isotópica de la presente invención, por ejemplo, aquellos que incorporan isótopos radioactivos tales como 3H, 13C y 14C. Esos compuestos con etiquetas isotópicas pueden ser útiles en estudios metabólicos, estudios cinéticos de reacción, técnicas de detección o imagenología, tales como tomografía por emisión de positrones (PET, por su sigla en inglés) o tomografía computarizada de emisión monofotónica (SPECT, por su sigla en inglés), incluidos ensayos de distribución de sustrato o fármaco en los tejidos o en el tratamiento radiactivo de pacientes. Los compuestos terapéuticos sustituidos o etiquetados con deuterio de la invención pueden tener propiedades de DMPK (farmacocinética y metabolismo del fármaco) mejoradas, con respecto a la distribución, el metabolismo y la excreción (ADME). La sustitución con isótopos más pesados, tales como deuterio, puede proporcionar determinadas ventajas terapéuticas como resultado de una mayor estabilidad metabólica, por ejemplo, una mayor semivida in vivo o menos requisitos de dosificación. Un compuesto marcado con 18F puede ser útil para estudios PET o SPECT. Los compuestos con etiquetas isotópicas de la presente invención y sus profármacos pueden prepararse por lo general mediante la realización de los procedimientos descritos en los esquemas o en los ejemplos y las preparaciones que se describirán más adelante mediante la sustitución de un reactivo con etiqueta isotópica fácilmente disponible por un reactivo sin etiqueta isotópica. Además, la sustitución con isótopos más pesados, particularmente con deuterio (es decir, 2H o D), puede generar determinadas ventajas terapéuticas debido a mayor estabilidad metabólica, por ejemplo, mayor semivida in vivo, menores requisitos de dosis o mejor índice terapéutico. Se entiende que, en este contexto, el deuterio se considera un sustituyente. Un factor de enriquecimiento isotópico puede definir la concentración de un isótopo más pesado de ese tipo, específicamente el deuterio. En los compuestos de la presente invención, se pretende que cualquier átomo no designado específicamente como un isótopo particular represente cualquier isótopo estable de ese átomo.
A menos que se especifique lo contrario, una referencia a un compuesto particular incluye todas las formas isoméricas, las que incluyen (de forma total o parcial) mezclas racémicas u otras mezclas de estas. Se conocen en la técnica métodos para preparar (por ejemplo, síntesis asimétrica) y separar (por ejemplo, medios cromatográficos y cristalización fraccionada) dichas formas isoméricas o se obtienen fácilmente mediante la adaptación de métodos indicados en la presente, o métodos conocidos, en una forma conocida.
Actividad biológica
Ensayos de proliferación celular in vitro
Generalmente, la actividad citotóxica o citostática de un conjugado de anticuerpo-fármaco (ADC) se mide mediante: exposición de células de mamífero que tienen proteínas de receptor en el anticuerpo del ADC en un medio de cultivo celular; cultivo de las células durante un período de alrededor de 6 horas a alrededor de 5 días; y medición de la viabilidad celular. Se utilizan ensayos con base celular in vitro para medir la viabilidad (proliferación), la citotoxicidad y la inducción de apoptosis (activación de caspasa) de un ADC de la invención.
Se puede medir la potencia de los conjugados de anticuerpo y fármaco in vitro mediante un ensayo de proliferación celular. El ensayo de viabilidad celular por luminiscencia CellTiter-Glo® es un método de ensayo homogéneo comercialmente disponible (Promega Corp., Madison, WI) con base en la expresión recombinante de luciferasa de Coleóptera (patentes estadounidenses n.° 5583024, 5674713 y 5700670). Este ensayo de proliferación celular determina la cantidad de células viables en el cultivo con base en la cuantificación del ATP presente, un indicador de células metabólicamente activas (Crouch et ál. (1993) J. Immunol. Meth. 160:81-88; US 6602677). El ensayo CellTiter-Glo® se lleva a cabo en 96 pocillos, por lo que es posible someterlo a análisis de alto rendimiento (HTS) automático (Cree et ál. (1995) AntiCancer Drugs 6:398-404). El procedimiento de ensayo homogéneo implica la adición del único reactivo (reactivo CellTiter-Glo®) directamente a las células cultivadas en medio complementado con suero. No son necesarias etapas de lavado celular, extracción de medio y múltiples transferencias con pipeta. El sistema detecta hasta 15 células/pocillo en un formato de 384 pocillos en 10 minutos después de agregar el reactivo y mezclar. Las células se pueden tratar continuamente con ADC, o se pueden tratar y separar de ADC. Generalmente, las células tratadas brevemente, es decir, 3 horas, mostraron los mismos efectos de potencia que las células tratadas continuamente.
El formato homogéneo de “adición, mezcla y medida” produce lisis celular y generación de una señal luminiscente proporcional a la cantidad de ATP presente. La cantidad de ATP es directamente proporcional a la cantidad de células presentes en el cultivo. El ensayo CellTiter-Glo® genera una señal luminiscente tipo brillo producida por la reacción de la luciferasa y que tiene una semivida generalmente mayor de cinco horas, conforme al tipo de célula y el medio utilizado. Las células utilizadas se reflejan en unidades relativas de luminiscencia (RLU). El sustrato, Beetle Luciferina, se descarboxila de manera oxidativa mediante la luciferasa de mariposa recombinante con una conversión concomitante de ATP a AMP y la generación de fotones.
También se puede medir la potencia de los conjugados de anticuerpo y fármaco in vitro mediante un ensayo de citotoxicidad. Las células adherentes cultivadas se lavan con PBS, se desvinculan con tripsina, se diluyen en medio completo que contiene FCS al 10 %, se centrifugan, se resuspenden en medio nuevo y se usa un hemocitómetro para su conteo. El recuento de los cultivos de suspensión se realiza directamente. Las suspensiones celulares monodispersas adecuadas para recuento pueden requerir agitación de la suspensión mediante aspiración repetida para romper las aglomeraciones de células.
La suspensión celular se diluye hasta la densidad de siembra deseada y se coloca en placas negras de 96 pocillos (100 pl por pocillo). Las placas de líneas de células adherentes se incuban durante la noche para permitir la adherencia. Es posible utilizar los cultivos de células en suspensión el día de la siembra.
Se produce una solución madre (1 ml) de ADC (20 pg/ml) en el medio de cultivo celular adecuado. Se realizan diluciones seriales de 10 veces de ADC madre en tubos de centrífuga de 15 ml mediante la transferencia en serie de 100 pl a 900 pl de medio de cultivo celular.
Se producen cuatro pocillos idénticos de cada dilución de ADC (100pl) en placas negras de 96 pocillos, previamente en placas con suspensión celular (100pl), lo que produce un volumen final de 200 pl. Se coloca medio de cultivo de control (100 pl) en los pocillos de control.
Si el tiempo de duplicación de la línea celular es mayor de 30 horas, la incubación con ADC se extiende 5 días, de lo contrario se hace una incubación de cuatro días.
Al finalizar el período de incubación, se evalúa la viabilidad celular con el ensayo de azul Alamar. Se coloca AlamarBlue (Invitrogen) en toda la placa (20 pl por pocillo) y se incubó durante 4 horas. Se midió la fluorescencia de azul Alamar a una excitación de 570 nm y una emisión de 585 nm en el lector de placas ultrarrápido Varioskan. Se calcula el porcentaje de supervivencia celular a partir de la fluorescencia promedio en los pocillos tratados con ADC en comparación con la fluorescencia promedio en los pocillos de control.
Uso
Es posible utilizar los conjugados de la invención para proporcionar un compuesto de PBD en una ubicación diana.
Preferentemente, la ubicación diana es una población de células proliferativas. El anticuerpo es un anticuerpo para un antígeno presente en una población de células proliferativas.
En una modalidad, el antígeno está ausente o presente a un nivel reducido en una población celular no proliferativa en comparación con la cantidad de antígeno presente en la población de células proliferativas, por ejemplo, una
población de células tumorales.
El enlazador se puede escindir en la ubicación diana de forma de liberar un compuesto RelA. Por lo tanto, el conjugado se puede usar para proporcionar de manera selectiva un compuesto RelA a la ubicación diana.
Una enzima presente en la ubicación diana puede escindir el enlazador.
La ubicación diana se puede encontrar in vitro, in vivo o ex vivo.
Los compuestos de conjugado de anticuerpo y fármaco (ADC) de la invención incluyen aquellos útiles en actividad contra el cáncer. En particular, los compuestos incluyen un anticuerpo conjugado, es decir, unido de forma covalente mediante un enlazador, a un resto de fármaco de PBD, es decir, una toxina. Cuando el fármaco no se conjuga con un anticuerpo, el fármaco de PBD tiene un efecto citotóxico. Por lo tanto, la actividad biológica del resto de fármaco de PBD se modula mediante la conjugación a un anticuerpo. Los conjugados de anticuerpo y fármaco (ADC) de la invención suministran selectivamente una dosis eficaz de un agente citotóxico al tejido tumoral, de forma que se logra mayor selectividad, es decir, una dosis eficaz más baja.
Por lo tanto, en un aspecto, la presente invención proporciona un compuesto conjugado como se describe en la presente para usar en la terapia.
En un aspecto adicional también se proporciona un compuesto conjugado como se describe en la presente para usar en el tratamiento de una enfermedad proliferativa. También se desvela en el presente documento el uso de un compuesto conjugado en la fabricación de un medicamento para tratar una enfermedad proliferativa.
Un experto en la técnica puede determinar fácilmente si un conjugado candidato trata o no una afección proliferativa para cualquier tipo de célula particular. Por ejemplo, en los ejemplos que se encuentran más adelante se describen ensayos que se pueden utilizar de forma conveniente para evaluar la actividad que ofrece un compuesto particular.
La expresión “enfermedad proliferativa” hace referencia a una proliferación celular no deseada o descontrolada de células anormales o excesivas que es indeseable, tal como, crecimiento hiperplásico o neoplásico, ya sea in vitro o in vivo.
Ejemplos de afecciones proliferativas incluyen, de modo no taxativo, la proliferación celular benigna, pre-maligna y maligna que incluye, de modo no taxativo, neoplasmas y tumores (por ejemplo, histiocitoma, glioma, astrocitoma, osteoma), cánceres (por ejemplo, cáncer de pulmón, cáncer pulmonar microcítico, cáncer gastrointestinal, cáncer intestinal, cáncer de colon, carcinoma de mama, carcinoma ovárico, cáncer esofágico, cáncer de próstata, cáncer testicular, cáncer de hígado, cáncer de riñón, cáncer de vejiga, cáncer de páncreas, cáncer cerebral, sarcoma, osteosarcoma, sarcoma de Kaposi, melanoma), linfomas, leucemias, psoriasis, enfermedades óseas, trastornos fibroproliferativos (por ejemplo, de tejidos conectivos) y ateroesclerosis. Los cánceres de interés particular incluyen, de modo no taxativo, leucemias y cánceres de ovario.
Es posible tratar cualquier tipo de célula, lo que incluye, de modo no taxativo, células de pulmón, gastrointestinales (que incluyen, por ejemplo, intestino, colon), de mama (mamarias), de ovario, de próstata, de hígado (hepáticas), de riñón (renales), de vejiga, de páncreas, cerebrales y de piel.
Los trastornos de interés particular incluyen, de modo no taxativo, cánceres, que incluyen cánceres metastásicos y células cancerosas metastásicas, tales como células tumorales en circulación, las que se pueden encontrar en circulación en fluidos corporales, tal como sangre o linfa. Los cánceres de interés particular incluyen cáncer de mama, pulmón, gástrico, cabeza y cuello, colorrectal, renal, pancreático, uterino, hepático, vejiga, endometrio y próstata, así como también linfomas (por ejemplo, linfoma no Hodgkin, NHL) y leucemia (en particular, leucemia mieloide aguda, AML).
Otros trastornos de interés incluyen cualquier afección en la que Axl esté sobreexpresado, o donde el antagonismo de Axl proporcionará un beneficio clínico. Estos incluyen trastornos inmunitarios, trastornos cardiovasculares, trombosis, diabetes, trastornos del punto de control inmunitario, trastornos fibróticos (fibrosis) o enfermedades proliferativas, tales como cáncer, particularmente, cáncer metastásico. Además, se sabe que Axl cumple una función en muchos cánceres de origen epitelial.
Los trastornos fibróticos de interés incluyen estrabismo, escleroderma, queloide, fibrosis sistémica nefrogénica, fibrosis pulmonar, fibrosis pulmonar idiopática (IPF), fibrosis cística (CF), esclerosis sistémica, fibrosis cardíaca, esteatohepatitis no alcóholica (NASH), otros tipos de fibrosis hepática, cirrosis biliar primaria, fibrosis renal, cáncer y aterosclerosis. En dichas enfermedades, el desarrollo de fibrosis crónico en el tejido lleva a alteraciones marcadas en la arquitectura de los órganos afectados y posteriormente causa la función deficiente de los órganos. Como consecuencia de este proceso de desgaste sostenido de los órganos, muchas enfermedades que implican fibrosis suelen ser afecciones progresivas y tienen un pronóstico a largo plazo poco favorable (ver Rockey, D.C., Bell, P.D. y Hill, J.A. (2015), N. Engl. Med., tomo 372, págs. 1138-1149).
Se contempla la posibilidad de utilizar los conjugados de anticuerpo y fármaco (ADC) de la presente invención para tratar diversas enfermedades o trastornos, por ejemplo, aquellos caracterizados por la sobreexpresión de un antígeno tumoral. Ejemplos de afecciones o trastornos hiperproliferativos incluyen tumores malignos o benignos, leucemia, neoplasias malignas linfoides y hematológicas. Otros incluyen trastornos neuronales, gliales, astrocitales, hipotalámicos, glandulares, macrofáicos, epitealiales, estromales, blastocélicos, inflamatorios, angiogénicos e inmunológicos, lo que incluye trastornos autoinmunes.
En general, la enfermedad o trastorno a tratar es una enfermedad hiperproliferativa, tal como cáncer. Ejemplos de cánceres a tratar de la presente incluyen, de modo no taxativo, carcinoma, linfoma, blastoma, sarcoma y leucemia o neoplasias malignas linfoides. Los ejemplos más particulares de dichos cánceres incluyen cáncer de célula escamosa (por ejemplo, cáncer de célula escamosa epitelial), cáncer de pulmón incluyendo cáncer microcítico de pulmón, cáncer no microcítico de pulmón, adenocarcinoma de pulmón y carcinoma escamoso de pulmón, cáncer del peritoneo, cáncer hepatocelular, cáncer gástrico o de estómago incluyendo cáncer gastrointestinal, cáncer pancreático, glioblastoma, cáncer del cuello uterino, cáncer de ovario, cáncer de hígado, cáncer de vejiga, hepatoma, cáncer de mama, cáncer de colon, cáncer rectal, cáncer colorrectal, carcinoma endometrial o uterino, carcinoma de glándula salival, cáncer renal o de riñón, cáncer de próstata, cáncer de vulva, cáncer de tiroides, carcinoma hepático, carcinoma anal, carcinoma de pene, así como también cáncer de cabeza y cuello.
Las enfermedades autoinmunitarias para las que es posible utilizar los compuestos de ADC como parte del tratamiento incluyen trastornos reumatológicos (tales como, por ejemplo, artritis reumatoide, síndrome de Sjogren, escleroderma, lupus, tal como lupus sistémico eritematoso (SLE) y nefritis lúpica, polimiositis/dermatomiositis, crioglobulinemia, síndrome de anticuerpos antifosfolípidos y artritis psoriásica), osteoartritis, trastornos gastrointestinales y hepáticos autoinmunitarios (tales como, por ejemplo, enfermedades intestinales inflamatorias (por ejemplo, colitis ulcerativa y enfermedad de Crohn), anemia perniciosa y gastritis autoinmunitaria, hepatitis autoinmunitaria, cirrosis biliar primaria, colangitis esclerosante primaria y enfermedad celíaca), vasculitis (tal como, por ejemplo, vasculitis asociada a ANCA, lo que incluye vasculitis de Churg-Strauss, granulomatosis de Wegener y poliarteritis), trastornos neurológicos autoinmunitarios (tales como, por ejemplo, esclerosis múltiple, síndrome de opsoclono mioclono, miastenia gravis, neuromielitis óptica, enfermedad de Parkinson, enfermedad de Alzheimer y polineuropatías autoinmunitarias), trastornos renales (tales como, por ejemplo, glomerulonefritis, síndrome de Goodpasture y enfermedad de Berger), trastornos dermatológicos autoinmunitarios (tales como, por ejemplo, psoriasis, urticaria, erupciones, pénfigo vulgar, pénfigo buloso y lupus cutáneo eritematoso), trastornos hematológicos (tales como, por ejemplo, púrpura trombocitopénica, púrpura trombocitopénica trombótica, púrpura postransfusional y anemia hemolítica autoinmunitaria), ateroesclerosis, uveitis, enfermedades de audición autoinmunitarios (tales como, por ejemplo, enfermedades del oído interno y pérdida auditiva), enfermedad de Behcet, síndrome de Raynaud, trasplante de órganos y trastornos endócrinos autoinmunitarios (tales como, por ejemplo, trastornos autoinmunitarios relativos a la diabetes, tales como diabetes mellitus tipo 1, enfermedad de Addison y enfermedades tiroideas autoinmunitarias (por ejemplo, tiroiditis y enfermedad de Graves)). Las enfermedades más preferidas de estas incluyen, por ejemplo, artritis reumatoide, colitis ulcerativa, vasculitis asociada a ANCA, lupus, esclerosis múltiple, síndrome de Sjogren, enfermedad de Graves, IDDM, anemia perniciosa, tiroiditis y glomerulonefritis.
Métodos de tratamiento
Es posible utilizar los conjugados de la presente invención en un método de tratamiento. También se desvela en la presente un método de tratamiento que comprende la administración de una cantidad terapéuticamente eficaz de un compuesto de conjugado de la invención a un sujeto que requiere tratamiento. La expresión “cantidad terapéuticamente eficaz” es una cantidad suficiente para lograr beneficios en un paciente. Dicho beneficio puede ser al menos la mejora de al menos un síntoma. La cantidad real que se administra y la tasa y el transcurso de tiempo de la administración dependerán de la naturaleza y la gravedad de lo que se trata. La prescripción del tratamiento, por ejemplo, decisiones relativas a la dosis, es parte de las responsabilidades de los médicos y otros profesionales de la salud.
Un compuesto de la invención se puede administrar por sí solo o en combinación con otros tratamientos, ya sea de forma simultánea o secuencial, de acuerdo con la afección que se trata. Ejemplos de tratamientos y terapias incluyen, de modo no taxativo, quimioterapia (la administración de agentes activos que incluyen, por ejemplo, fármacos tales como agentes quimioterapéuticos), cirugía y radioterapia.
Un “agente quimioterapéutico” es un compuesto químico útil para el tratamiento del cáncer, independientemente del mecanismo de acción. Las clases de agentes quimioterapéuticos incluyen, de modo no taxativo: agentes alquilantes, antimetabolitos, alcaloides antimitóticos de origen vegetal, antibióticos citotóxicos/antitumorales, inhibidores de la topoisomerasa, anticuerpos, fotosensibilizantes e inhibidores de cinasa. Los agentes quimioterapéuticos incluyen compuestos usados en la “terapia dirigida” y en la quimioterapia convencional.
Los ejemplos de agentes quimioterapéuticos incluyen: erlotinib (TARCEVA®, Genentech/OSI Pharm.), docetaxel (TAx Ot ERE®, Sanofi-Aventis), 5-FU (fluorouracilo, 5-fluorouracilo, CAS N.° 51-21-8), gemcitabina (GEMZAR®, Lilly), PD-0325901 (CAS N.° 391210-10-9, Pfizer), cisplatino (cis-diamina, dicloroplatino(II), CAS N.° 15663-27-1),
carboplatino (CAS N.° 41575-94-4), paclitaxel (TAXOL®, Bristol-Myers Squibb Oncology, Princeton, N.J.), trastuzumab (HERCEPTlN®, Genentech), temozolomida (4-metil-5-oxo- 2,3,4,6,8-pentazabiciclo [4.3.0] nona-2,7,9-trien-9-carboxamida, CAS N.° 85622-93-1, TEMODAR®, TEMODAL®, Schering Plough), tamoxifeno ((Z)-2-[4-(1,2-difenilbut-1-enil)fenoxi]-N,A/-dimetil-etanamina, NOLVADEX®, ISTUBAL®, VALODEX®) y doxorrubicina (ADRIAMYCIN®), Akti-1/2, HPPD y rapamicina.
Más ejemplo de agentes quimioterapéuticas incluyen: oxaliplatino (ELOXATIN®, Sanofi), bortezomib (VELCADE®, Millennium Pharm.), sutent (SUNITINIB®, SU11248, Pfizer), letrozol (FEMARA®, Novartis), mesilato de imatinib (GLEEVEC®, Novartis), XL-518 (inhibidor de Mek, Exelixis, WO 2007/044515), ARRY-886 (inhibidor de Mek, AZD6244, Array BioPharma, Astra Zeneca), SF-1126 (inhibidor de PI3K, Semafore Pharmaceuticals), BEZ-235 (inhibidor de PI3K, Novartis), XL-147 (inhibidor de PI3K, Exelixis), PTK787/ZK 222584 (Novartis), fulvestrant (FASLODEX®, AstraZeneca), leucovorina (ácido folínico), rapamicina (sirolimus, RAPAMUNE®, Wyeth), lapatinib (TYKERB®, GSK572016, Glaxo Smith Kline), lonafarnib (SARASAR™, SCH 66336, Schering Plough), sorafenib (NEXAVAR®, BAY43-9006, Bayer Labs), gefitinib (IRESSA®, AstraZeneca), irinotecán (CAMPTOSAR®, CPT-11, Pfizer), tipifarnib (ZARNESTrA™, Johnson & Johnson), ABRAXANE™ (sin Cremophor), formulaciones de nanopartículas de paclitaxel modificadas genéticamente con albúmina (American Pharmaceutical Partners, Schaumberg, Il), vandetanib (rINN, ZD6474, ZACTIMA®, AstraZeneca), cloranmbucilo, AG1478, AG1571 (SU 5271; Sugen), temsirolimus (TORISEL®, Wyeth), pazopanib (GlaxoSmithKline), canfosfamida (TELCYTA®, Telik), tiotepa y ciclosfosfamida (CYTOXAN®, NEOSAr ®); alquil sulfonatos tales como busulfán, improsulfán y piposulfán; aziridines tales como benzodopa, carbocuona, meturedopa y uredopa; etileniminas y metilamelaminas que incluyen altretamina, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida y trimetilomelamina; acetogeninas (especialmente bulatacina y bulatacinona); una camptotecina (que incluyen el análogo sintético topotecán); briostatina; calistatina; CC-1065 (que incluye sus análogos sintéticos de adozelesina, carzelesina y bizelesina); criptoficinas (particularmente criptoficina 1 y criptoficina 8); dolastatina; duocarmicina (que incluye los análogos sintéticos, KW-2189 y CB1-TM1); eleuterobina; pancratistatina; una sarcodictiina; spongistatina; mostazas de nitrógeno tal como clorambucilo, clornafazina, clorofosfamida, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalán, novembiquina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo; nitrosoureas tal como carmustina, clorozotocina, fotemustina, lomustina, nimustina y ranimnustina; antibióticos tal como antibióticos de enediina (por ejemplo, calicheamicina, calicheamicina gamma1I, calicheamicina omegaI1 (Angew Chem. Intl. Ed. Engl. (1994) 33:183-186); dinemicina, dinemicina A; bisfosfonatos, tales como clodronato; una esperamicina; así como también cromóforo de neocarcinostatina y cromóforos de antibióticos de enedina de cromoproteínas relacionados), aclacinomisinas, actinomicina, autramicina, azaserina, bleomicinas, cactinomicina, carabicina, carminomicina, carcinofilina, cromomicinas, dactinomicina, daunorrubicina, detorrubicina, 6-diazo-5-oxo-L-norleucina, morfolinodoxorrubicina, cianomorfolino-doxorrubicina, 2-pirrolino-doxorrubicina y desoxidoxorrubicina), epirrubicina, esorrubicina, idarrubicina, nemorrubicina, marcelomicina, mitomicinas tales como mitomicina C, ácido micofenólico, nogalamicina, olivomicinas, peplomicina, porfiromicina, puromicina, quelamicina, rodorrubicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorrubicina; antimetabolitos tales como metotrexato y 5-fluorouracilo (5-FU); análogos de ácido fólico tales como denopterina, metotrexato, pteropterina, trimetrexato; análogos de purina tales como fludarabina, 6-mercaptopurina, tiamiprina, tioguanina; análogos de pirimidina tales como ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina; andrógenos tales como calusterona, propionato de dromostanolona, epitiostanol, mepitiostano, testolactona; agentes antisuprarrenales tales como aminoglutetimida, mitotano, trilostano; rellenador de ácido fólico tal como ácido frolínico; aceglatona; glicósido de aldofosfamida; ácido aminolevulínico; eniluracilo; amsacrina; bestrabucilo; bisantreno; edatraxato; defofamina; demecolcina; diazicuona; elfornitina; acetato de eliptinio; una epotilona; etoglúcido; nitrato de galio; hidroxiurea; lentinan; lonidainina; maitansinoides tales como maitansina y ansamitocinas; mitoguazona; mitoxantrona; mopidanmol; nitraerina; pentostatina; fenamet; pirarrubicina; losoxantrona; ácido podofilínico; 2-etilhidrazida; procarbazina; complejo polisacárido PSK® (JHS Natural Products, Eugene, OR); razoxana; rizoxina; sizofiran; espirogermanio; ácido tenuazónico; triazicuona; 2,2',2”-triclorotrietilamina; tricotecenas (especialmente, toxina T-2, verracurina A, roridina A y anguidina); uretano; vindesina; dacarbazina; mannomustina; mitobronitol; mitolactol; pipobromano; gacitosina; arabinósido (Ara-C); ciclofosfamida; tiotepa; 6-tioguanina; mercaptopurina; metotrexato; análogos de platino como cisplatino y carboplatino; vinblastina; etopósido (VP-16); ifosfamida; mitoxantrona; vincristina; vinorelbina (NAVELBINE®); novantrona; tenipósido; edatrexato; daunomicina; aminopterina; capecitabina (XELODA®, Roche); ibandronato; CPT-11; inhibidor de la topoisomerasa RFS 2000; difluorometilornitina (DMFO); retinoides tal como ácido retinoico; y sales, ácidos y derivados farmacéuticamente aceptables de cualquiera de los anteriores.
La definición de “agente quimioterapéutico” incluye también: (i) agentes antihormonales que actúan para regular o inhibir la acción hormonal sobre tumores tales como antiestrógenos y moduladores de receptores de estrógeno selectivos (SERM), lo que incluye, por ejemplo, tamoxifeno (lo que incluye NOLVADEX®; citrato de tamoxifeno), raloxifeno, droloxifeno, 4-hidroxitamoxifeno, trioxifeno, keoxifeno, LY117018, onapristona y FARESTON® (citrato de toremifina); (ii) inhibidores de aromatasa que inhiben la enzima aromatasa, lo que regula la producción de estrógeno en las glándulas suprarrenales, tales como, por ejemplo, 4(5)-imidazol, aminoglutetimida, MEGASE® (acetato de megestrol), AROMASIN® (exemestano; Pfizer), formestano, fadrozol, RIVISOR® (vorozol), FEMARA® (letrozol; Novartis) y ARIMIDEX® (anastrozol; AstraZeneca); (iii) antiandrógenos tales como flutamida, nilutamida, bicalutamida, leuprolida y goserelina, así como troxacitabina (un análogo de nucleósido de 1,3-dioxolano de citosina); (iv) inhibidores de proteína cinasa, tal como inhibidores de MEK (WO 2007/044515); (v) inhibidores de lípido cinasa; (vi) oligonucleótidos antisentido, particularmente los que inhiben la expresión de genes en las vías de señalización
implicadas en la proliferación celular anormal, por ejemplo, PKC-alfa, Raf y H-Ras, tal como oblimersen (GENASENSE®, Genta Inc.); (vii) ribozimas tales como inhibidores de expresión de VEGF (por ejemplo, ANGIOZYME®) e inhibidores de expresión de HER2; (viii) vacunas tales como vacunas de terapia génica, por ejemplo, ALLOVECTIN®, LEUVECTIN® y VAXID®; PROLEUKIN®, rIL-2; un inhibidor de topoisomerasa 1 tal como LURTOTECAN®; ABARELIX® rmRH; (ix) agentes antiangiogénicos como bevacizumab (AVASTIN®, Genentech) y derivados, ácidos y sales farmacéuticamente aceptables de cualquiera de los anteriores.
También se incluyen en la definición de “agente quimioterapéutico” anticuerpos terapéuticos tales como alemtuzumab (Campath), bevacizumab (AVASTIN®, Genentech); cetuximab (ERBITUX®, Imclone); panitumumab (VECTIBIX®, Amgen), rituximab (RIt Ux AN®, Genentech/Biogen Idec), ofatumumab (ARz Er RA®, GSK), pertuzumab (PERJETA™, OMNITARG™, 2C4, Genentech), trastuzumab (HERCEPTIN®, Genentech), tositumomab (Bexxar, Corixia) y el conjugado de anticuerpo y fármaco, ozogamicina de gemtuzumab (MYLOTARG®, Wyeth).
Los anticuerpos monoclonales humanizados con potencial terapéutico como agentes quimioterapéuticos en combinación con los conjugados de la invención incluyen: alemtuzumab, apolizumab, aselizumab, atlizumab, bapineuzumab, bevacizumab, bivatuzumab mertansina, cantuzumab mertansina, cedelizumab, certolizumab pegol, cidfusituzumab, cidtuzumab, daclizumab, eculizumab, efalizumab, epratuzumab, erlizumab, felvizumab, fontolizumab, gemtuzumab ozogamicina, inotuzumab ozogamicina, ipilimumab, labetuzumab, lintuzumab, matuzumab, mepolizumab, motavizumab, motovizumab, natalizumab, nimotuzumab, nolovizumab, numavizumab, ocrelizumab, omalizumab, palivizumab, pascolizumab, pecfusituzumab, pectuzumab, pertuzumab, pexelizumab, ralivizumab, ranibizumab, reslivizumab, reslizumab, resyvizumab, rovelizumab, ruplizumab, sibrotuzumab, siplizumab, sontuzumab, tacatuzumab tetraxetano, tadocizumab, talizumab, tefibazumab, tocilizumab, toralizumab, trastuzumab, tucotuzumab celmoleucina, tucusituzumab, umavizumab, urtoxazumab y visilizumab.
Las composiciones farmacéuticas de acuerdo con la presente invención y para utilizar de acuerdo con la presente invención pueden comprender, además del ingrediente activo, es decir, un compuesto conjugado, un excipiente farmacéuticamente aceptable, vehículo, solución amortiguadora, estabilizante u otros materiales conocidos para los expertos en la técnica. Tales materiales deben ser no tóxicos y no deben interferir con la eficacia del ingrediente activo. La naturaleza exacta del vehículo u otro material dependerá de la vía de administración, la que puede ser oral o mediante inyección, por ejemplo, cutánea, subcutánea o intravenosa.
Las composiciones farmacéuticas para administración oral se pueden encontrar en forma de comprimido, cápsula, en polvo o líquida. Un comprimido puede comprender un vehículo sólido o un adyuvante. En general, las composiciones farmacéuticas líquidas comprenden un vehículo líquido, tal como agua, petróleo, aceites animales o vegetales, aceite mineral o aceite sintético. Se pueden incluir solución salina fisiológica, dextrosa u otra solución de sacárido o glicoles, tal como etilenglicol, propilenglicol o polietilenglicol. Una cápsula puede comprender un vehículo sólido, tal como gelatina.
Para la inyección intravenosa, cutánea o subcutánea o la inyección en el sitio de aflicción, el ingrediente activo se encontrará en forma de una solución acuosa parenteralmente aceptable libre de pirógenos y con pH, isotonicidad y estabilidad adecuados. Los expertos en la técnica pertinente son plenamente capaces de preparar soluciones adecuadas, por ejemplo, con vehículos isotónicos tales como inyección de cloruro de sodio, inyección de Ringer, inyección de Ringer lactato. De ser necesario, se pueden incluir conservantes, estabilizantes, amortiguadores, antioxidantes y/u otros aditivos.
Formulaciones
Aunque es posible utilizar el compuesto conjugado (por ejemplo, administrarlo) por sí solo, a menudo se prefiere su presentación como una composición o formulación.
En una modalidad, la composición es una composición farmacéutica (por ejemplo, formulación, preparación, medicamento) que comprende un compuesto de conjugado, tal como se describió en la presente, y un vehículo, diluyente o excipiente farmacéuticamente aceptable.
En una modalidad, la composición es una composición farmacéutica que comprende al menos un compuesto de conjugado, tal como se describió en la presente, junto con uno o más ingredientes farmacéuticamente aceptables diferentes conocidos por los expertos en la técnica y que incluyen, de modo no taxativo, vehículos, diluyentes, excipientes, adyuvantes, rellenos, amortiguadores, conservantes, antioxidantes, lubricantes, estabilizadores, solubilizantes, tensioactivos (por ejemplo, agentes humectantes), agentes de enmascaramiento, agentes colorantes, agentes saborizantes y agentes endulzantes farmacéuticamente aceptables.
En una modalidad, la composición también comprende otros agentes activos, por ejemplo, otros agentes profilácticos o terapéuticos.
Es posible encontrar vehículos, diluyentes, excipientes adecuados, etc., en textos farmacéuticos estándar. Véase, por ejemplo, Handbook of Pharmaceutical Additives, 2.a edición (eds. M. Ash y I. Ash), 2001 (Synapse Information Resources, Inc., Endicott, Nueva York, EUA), Remington's Pharmaceutical Sciences, 20.a edición, pub. Lippincott,
Williams & Wilkins, 2000; y Handbook of Pharmaceutical Excipients, 2.a edición, 1994.
También se desvelan en la presente a métodos de hacer una composición farmacéutica que comprende mezclar al menos un conjugado radioetiquetado [11C] o compuesto tipo conjugado, como se define en la presente, junto con uno o más ingredientes farmacéuticamente aceptables bien conocidos para los expertos en la técnica, por ejemplo, vehículos, diluyentes, excipientes, etc. Si se formula como unidades individuales (por ejemplo, comprimidos, etc.), cada unidad contiene una cantidad predeterminada (dosis) del compuesto activo.
Tal como se usa en la presente, la expresión “farmacéuticamente aceptable” hace referencia a compuestos, ingredientes, materiales, composiciones, formas de dosificación, etc. que, conforme al criterio médico bien fundado, son adecuadas para utilizar en contacto con los tejidos del sujeto en cuestión (por ejemplo, un ser humano) sin irritación, respuesta alérgica, toxicidad excesiva u otro problema o complicación, adecuado para una relación entre riesgo y beneficio razonable. Además, cada vehículo, diluyente, excipiente, etc. debe ser “aceptable” en el sentido de ser compatible con los otros ingredientes de la formulación.
Las formulaciones se pueden preparar mediante cualesquiera métodos conocidos en la técnica farmacéutica. Dichos métodos incluyen la etapa de asociación del compuesto activo con un vehículo que constituye uno o más ingredientes adicionales. En general, las formulaciones se preparan al asociar de forma uniforme e íntima el compuesto activo con vehículos (por ejemplo, vehículos líquidos, vehículo sólido finamente dividido, etc.) y, posteriormente, darle forma al producto, si fuera necesario.
Es posible preparar la formulación de forma que sea de liberación rápida o lenta, liberación inmediata, demorada, programada o continua, o una combinación de estas.
Las formulaciones adecuadas para administración parenteral (por ejemplo, mediante inyección) incluyen líquidos acuosos o no acuosos, isotónicos, libres de pirógenos, estériles (por ejemplo, soluciones, suspensiones), en los que el ingrediente activo se disuelve, suspende o se provee de otra forma (por ejemplo, en un liposoma u otro microparticulado). Dichos líquidos pueden contener también otros ingredientes farmacéuticamente aceptables, tales como antioxidantes, soluciones amortiguadoras, conservantes, estabilizadores, agentes bacteriostáticos, agentes de suspensión, agentes espesantes y solutos que tornan la formulación isotónica con la sangre (u otro fluido corporal relevante) del receptor deseado. Ejemplos de excipientes incluyen, por ejemplo, agua, alcoholes, polioles, glicerol, aceites vegetales y similares. Ejemplos de vehículos isotónicos adecuados a utilizar en dichas formulaciones incluyen inyección de cloruro de sodio, solución de Ringer o inyección de solución de Ringer con lactato. Usualmente, la concentración del ingrediente activo en el líquido es de entre alrededor de 1 ng/ml y alrededor de 10 ng/ml, por ejemplo, entre alrededor de 10 ng/ml y alrededor de 1 pg/ml. La formulación se puede presentar en recipientes sellados con dosis unitarias o dosis múltiples, por ejemplo, ampollas y viales, y se puede almacenar en condiciones de liofilización que solo requieren la adición del vehículo líquido estéril, por ejemplo, agua para inyección, inmediatamente antes de su uso. Es posible preparar soluciones y suspensiones para inyección improvisadas a partir de polvos estériles, gránulos y comprimidos.
Dosificación
Un experto en la técnica observará que las dosis adecuadas del compuesto de conjugado, y composiciones que comprenden el compuesto de conjugado, pueden variar entre pacientes. En general, la determinación de la dosis óptima involucrará el equilibrio del nivel de beneficio terapéutico contra cualquier riesgo o efectos secundarios perjudiciales. El nivel de dosis seleccionado dependerá de varios factores que incluyen, de modo no taxativo, la actividad del compuesto particular, la vía de administración, el momento de administración, la velocidad de excreción del compuesto, la duración del tratamiento, otros fármacos, compuestos y/o materiales utilizados en combinación, la gravedad de la afección y la especie, sexo, edad, peso, afección, estado de salud general e historial médico previo del paciente. En última instancia, la cantidad de compuesto y la vía de administración quedarán a discreción del médico, veterinario o profesional de salud, aunque, en general, la dosis se seleccionará para lograr concentraciones locales en el sitio de acción que logren el efecto deseado sin provocar efectos secundarios perjudiciales o dañinos considerables.
La administración se puede llevar a cabo en una dosis, de forma continua o intermitente (por ejemplo, en dosis divididas en intervalos adecuados) durante el trascurso del tratamiento. Los expertos en la técnica conocen métodos para determinar los medios y las dosis a administrar más eficaces, los que variarán conforme a la formulación utilizada en la terapia, el objetivo de la terapia, las células diana que se tratan y el sujeto que se trata. Son posibles administraciones únicas o múltiples con el nivel de dosis y el patrón seleccionados por el médico, veterinario o profesional de la salud tratante.
En general, una dosis adecuada del compuesto activo se encuentra en el rango de alrededor de 100 ng a alrededor de 25 mg (más usualmente, alrededor de 1 pg a alrededor de 10 mg) por kilogramo de peso corporal del sujeto por día. En los casos en los que el compuesto activo es una sal, un éster, una amida, un profármaco o similares, la cantidad que se administra se calcula con base en el compuesto original, por lo que el peso real a utilizar se aumenta de forma proporcional.
En una modalidad, el compuesto activo se administra a un paciente humano de acuerdo con el siguiente régimen de dosificación: alrededor de 100 mg, 3 veces al día.
En una modalidad, el compuesto activo se administra a un paciente humano de acuerdo con el siguiente régimen de dosificación: alrededor de 150 mg, 2 veces al día.
En una modalidad, el compuesto activo se administra a un paciente humano de acuerdo con el siguiente régimen de dosificación: alrededor de 200 mg, 2 veces al día.
Sin embargo, en una modalidad, el compuesto conjugado se administra a un paciente humano de acuerdo con el siguiente régimen de dosificación: alrededor de 50 mg o alrededor de 75 mg, 3 o 4 veces al día.
En una modalidad, el compuesto conjugado se administra a un paciente humano de acuerdo con el siguiente régimen de dosificación: alrededor de 100 mg o alrededor de 125 mg, 2 veces al día.
Las cantidades de dosificación anteriormente descritas se pueden aplicar al conjugado (lo que incluye el resto de PBD y el enlazador al anticuerpo) o a la cantidad eficaz del compuesto de PBD provisto, por ejemplo, la cantidad de compuesto que se puede liberar luego de la escisión del enlazador.
La dosis adecuada de un ADC de la invención para la prevención o el tratamiento de una enfermedad dependerá del tipo de enfermedad a ser tratada, tal como se definió anteriormente, la gravedad y la evolución de la enfermedad, el hecho de que la molécula se administre con fines preventivos o terapéuticos, los tratamientos previos, el historial clínico del paciente y la respuesta al anticuerpo, así como del criterio del médico tratante. Se administra la molécula en forma adecuada al paciente en un momento o en una serie de tratamientos. De acuerdo con el tipo y la gravedad de la enfermedad, una dosis de alrededor de 1 |ig/kg a 15 mg/kg (por ejemplo, 0,1-20 mg/kg) de molécula es una dosis inicial posible a administrar al paciente, ya sea, por ejemplo, mediante una o más administraciones independientes o mediante infusión continua. Una dosificación diaria típica puede variar de alrededor de 1 |ig/kg a 100 mg/kg o más, dependiendo de los factores antes mencionados. Un ejemplo de dosis de ADC a administrar a un paciente se encuentra en el intervalo de alrededor de 0,1 a alrededor de 10 mg/kg de peso del paciente. En el caso de administraciones repetidas durante varios días o más, de acuerdo con la afección, el tratamiento se mantiene hasta que se produce una supresión deseada de síntomas de la enfermedad. Un ejemplo de régimen de dosificación comprende la administración de una dosis de carga inicial de alrededor de 4 mg/kg, seguida de dosis adicionales cada semana, cada dos semanas o cada tres semanas de un ADC. Pueden resultar útiles otros regímenes de dosificación. Es posible controlar la evolución de esta terapia mediante ensayos y técnicas convencionales.
Tratamiento
Tal como se utiliza en la presente en el contexto del tratamiento de una afección, el término “tratamiento” hace referencia, en general, al tratamiento y la terapia, ya sea de un ser humano o un animal (por ejemplo, en aplicaciones veterinarias), en los que se logra cierto efecto terapéutico deseado, por ejemplo, la inhibición de la evolución de la afección y los que incluyen una reducción de la velocidad de evolución, una detención de la velocidad de evolución, la regresión de la afección, la mejora de la afección y la cura de la afección. También se incluye el tratamiento como una medida profiláctica (es decir, profilaxis, prevención).
Tal como se utiliza en la presente, la expresión “cantidad terapéuticamente eficaz” hace referencia a dicha cantidad de un compuesto activo o material, composición o forma de dosificación que comprende un compuesto activo que produce de forma eficaz cierto efecto terapéutico proporcional a una relación entre riesgo y beneficio razonable cuando se administra de acuerdo con un régimen de tratamiento deseado.
De manera similar, tal como se utiliza en la presente, la expresión “cantidad profilácticamente eficaz” hace referencia a dicha cantidad de un compuesto activo o material, composición o forma de dosificación que comprende un compuesto activo que produce de forma eficaz cierto efecto profiláctico proporcional a una relación entre riesgo y beneficio razonable cuando se administra de acuerdo con un régimen de tratamiento deseado.
Preparación de conjugados de fármaco
Los conjugados de fármacos y anticuerpos de la presente invención se pueden preparar conjugando el siguiente enlazador de fármacos:
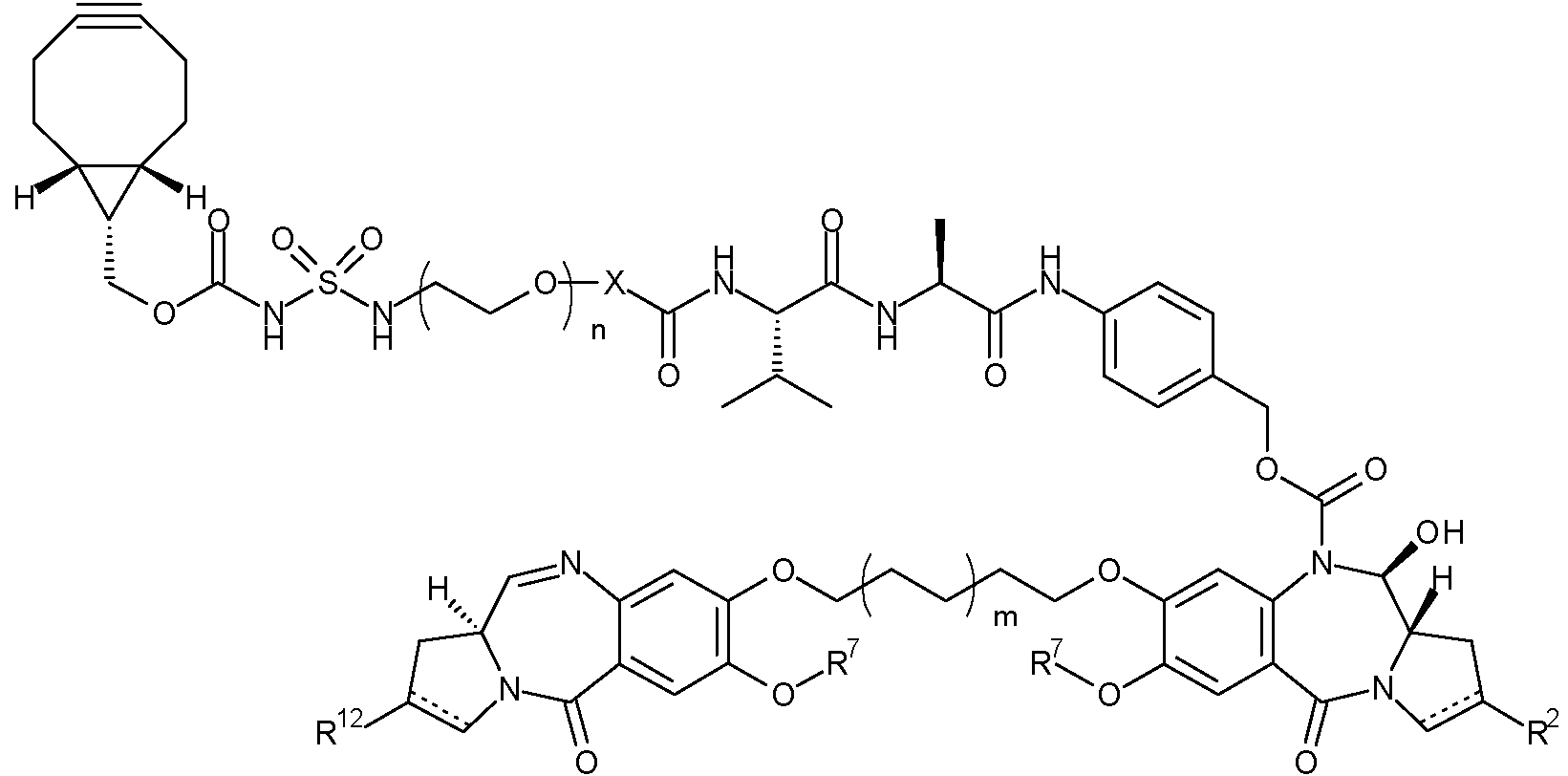
con el anticuerpo que contiene azida con los métodos que se describen en, por ejemplo, van Geel, R., et ál., Bioconjugate Chemistry, 2015, 26, 2233-2242; DOI: 10.1021/acs.bioconjchem.5b00224. Los métodos adecuados incluyen, de modo no taxativo, conjugación libre de cobre en, por ejemplo, condiciones acuosas con un cosolvente opcional seleccionado de DMF, DMSO y DMA.
El enlazador del fármaco se puede sintetizar de acuerdo con los ejemplos, con modificaciones adecuadas, por ejemplo, con referencia a WO 2016/053107, para la síntesis del enlazador y los siguientes documentos para el dímero de PBD, por ejemplo: WO 2011/130598, WO2013/055987, WO2014/057074.
El sujeto/paciente
El sujeto o paciente puede ser un animal, mamífero, un mamífero placentario, un marsupial (por ejemplo, un canguro, un wombat), un monotrema (por ejemplo, un ornitorrinco), un roedor (por ejemplo, un cobayo, un hámster, una rata, un ratón), murino (por ejemplo, un ratón), un lagomorfo (por ejemplo, un conejo), ave (por ejemplo, un pájaro), canino (por ejemplo, un perro), felino (por ejemplo, un gato), equino (por ejemplo, un caballo), porcino (por ejemplo, un cerdo), ovino (por ejemplo, una oveja), bovino (por ejemplo, una vaca), un primate, un simio (por ejemplo, un mono o un simio), un mono (por ejemplo, un mono tití, un babuino), un simio (por ejemplo, un gorila, un chimpancé, un orangután, un gibón) o un ser humano.
Además, el sujeto o paciente puede estar en cualquiera de sus formas de desarrollo, por ejemplo, un feto. En una modalidad preferida, el sujeto o paciente es un ser humano.
Ejemplos
Síntesis del intermedio 3

3
Se enfrió una solución de alcohol de BCN (0,384 g, 2,55 mmol) en MeCN (25 ml) en una atmósfera de N2 a 0 °C y se agregó isocianato de clorosulfonilo (CSI) gota a gota (0,255 ml, 415 mg, 2,93 mmol, 1,15 equiv.). Después de agitar durante 15 minutos, se agregó Et3N por goteo (1,42 ml, 1,03 g, 10,2 mmol, 4 equiv.) y la agitación continuó durante 10 minutos adicionales. Después, se agregó una solución de ácido 2-(2-(2-aminoetoxi)etoxi)acético (1,0 g, 6,1 mmol, 2,4 equiv.) en H2O (5 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 2 h. Después de este tiempo, se agregó CHCh (50 ml) y H2O (100 ml) y las capas se separaron. A la capa acuosa en un embudo de separación se agregó CH2Ch (100 ml) y el pH se ajustó a 4 con HCl 1 N, antes de la separación de las capas. La capa de agua se extrajo dos veces con C ^ C h (2 * 100 ml), las capas orgánicas se combinaron y se secaron (Na2SO4), se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna de matraz en sílice, se eluyó con CH2Cl2 a 20% MeOH en CH2Ch. Rendimiento 0,42 g (1,0 mmol, 39%) de 3 como una cera pegajosa incolora.
Síntesis del enlazador del fármaco


El compuesto 1 se puede sintetizar como se describe en la WO2014/057074 - ver el compuesto 22.
(a) Se pesa tetraquistrifenilfosfina de paladio (Pd(PPh3)4, 4,8 mg, 4,15 |jmol) y se coloca en una atmósfera inerte. Se desgasificó una solución de pirrolidina (5,0 jl, 4,3 mg, 60 jm ol) en DCM (1 ml) mediante burbujeo de N2 a través de la solución. Se desgasificó una solución de 1 (27 mg, 24 jm ol) en DCM (6 ml) mediante burbujeo de N2 a través de la solución. Si bien se sigue burbujeando N2 en la solución, se agrega la solución desgasificada de pirrolidina. Se disuelve el Pd(PPh3)4 pesado en DCM (1 ml) y se agrega 0,9 ml de esta solución. Después de 50 min de burbujeo de N2, se agrega DCM (25 ml) y la mezcla se lava con NH4O (25 ml) saturado acuoso. Después de la separación, la capa acuosa se extrae con DCM (2 * 25 ml). Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron. El residuo se purificó mediante RP-HPLC (30-90% MeCN (ácido fórmico al 0,1%) en H2O (ácido fórmico al 0,1%). Las fracciones combinadas se pasan a través de columnas SPE (HCO3- ) y se concentran. Después de la adición de MeCN (50 ml), la mezcla se vuelve a concentrar. El residuo resultante 2 se usó en la siguiente etapa.
La conversión de la reacción se puede controlar a través del análisis LCMS. Columna: Columna XBridge BEH C18 Intelligent Speed (IS), 130Á, 3,5 |jm (4,6 mm x 20 mm). Fase móvil A: Agua (ácido fórmico al 0,1%), Fase móvil B (ácido fórmico al 0,1%). Detección con PDA y ESI+. Las muestras se pueden preparar diluyendo la mezcla de reacción con MeCN.
(b) A una solución del residuo anterior 2 en CHCh (5 ml) se agrega una solución de 3 (15 mg, 36 jmol, mw 418 g/mol) en CHCh (0,8 ml). La mezcla resultante se agrega a EDC.HCl (4,7 mg, 25 jm ol) sólido, se agregó CHCh (5 ml) y luego la mezcla se agitó durante 30 minutos. Se agregó DCM (30 ml) y la mezcla resultante se lavó con agua (30 ml). Después de la separación, la fase acuosa se extrae con DCM (30 ml). Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron. El residuo se purificó mediante RP-HPLC (30-90% MeCN (sin ácido) en H2O (ácido fórmico al 0,01%). Los tubos de recolección de HPLC se llenaron con (NH4)HCO3 acuoso al 5 % antes de la recolección. Las fracciones de HPLC combinadas se extrajeron con DCM (3 * 20 ml). Las capas orgánicas combinadas se secaron (Na2SO4) y se concentraron. El producto 4 se obtiene como un aceite levemente amarillo/blanco (21 mg, 16 jmol, mw 1323 g/mol, 67% en dos etapas).
La conversión de la reacción se puede controlar a través del análisis LCMS. Columna: Columna XBridge BEH C18 Intelligent Speed (IS), 130Á, 3,5 jm (4,6 mm x 20 mm). Fase móvil A: Agua (ácido fórmico al 0,1%), Fase móvil B (ácido fórmico al 0,1%). Detección con PDA y ESI+.
Modificación de anticuerpos
Condiciones de reacción
Las condiciones de reacción para el remodelado de glicano de un solo recipiente son:
15 mg/ml de anticuerpo (~0,1 mM)
0. 15 mg/ml de EndoSH (1% p/p) de Streptococcus pyogenes
1,13 mg/ml de His-TnGalNAcT (7,5% p/p) enzima Galactosa-N-acetil Transferasa (GalNAcT)
2.5 mM de 6 -N3GalNAc-UDP (25 eq. en comparación con IgG)
10mM de MnCh
25 mM de TrisHCl pH 8,0
150 mM de NaCl
Incubar 16 horas a 30°C
Esto se llevó a cabo en AXL y B12.
Procedimiento
Este ejemplo se realiza a escala de 25 mg, que puede modificarse según sea necesario. Los componentes individuales se agregan en orden y se mezclan:
106.5 |jL 25 mM de Tris pH 8,0, 150 mM de NaCl (para obtener un volumen final de 1667 |jL)
1 ml 25 mg/ml de anticuerpo en 25 mM de Tris pH 8,0, 150 mM de NaCl
71.4 j L 3,5 mg/ml de EndoSH en 25 mM de Tris pH 8,0
389 j L 4,82 mg/ml de His-TnGalNAcT en 25 mM de Tris pH 8,0
16,7 j L de MnCl21M en MQ
83.4 j L de 6 -N3GalNAc-UDP 0,1 M en MQ
Esta se mezcló durante aproximadamente 16 horas a 30 °C. Es posible evaluar si se completó el residuo de galactosa modificado al someter una muestra a análisis MS. Después de la purificación de afinidad de la proteína A, se puede reducir una pequeña muestra del producto con DTT y posteriormente se puede someter a análisis de MS. Un espectro de masas típico de una reacción de transferencia exitosa muestra la formación de un producto principal de (90 % de la cadena pesada total), que resulta de una transferencia de galactosa modificada al Ab sustituido por GlcNAc(Fuc) de núcleo, y un producto menor (±10 % de la cadena pesada total), que resulta de una transferencia de galactosa modificada al Ab sustituido (sin Fucosa) GlcNAc de núcleo.
Procedimiento de purificación
Amortiguadores
Amortiguador de unión/lavado (TBS pH 7,5):
20 mM de TrisHCl pH 7,5
NaCl 150 mM
Amortiguador de lavado para retiro de endotoxina (TBS pH 7,5 Triton-X100):
20 mM de TrisHCl pH 7,5
NaCl 150 mM
Triton X-100 al 0,2%
Solución amortiguadora de elución:
Glicina 0,1 M, pH 2,7
Amortiguador CIP:
NaOH 0,5 M
Procedimiento
1. Lavar la columna MabSelectSure de 5 ml (5 ml/min) con los siguientes amortiguadores para limpiar la columna antes de aplicar la muestra:
Lavar la columna con al menos 5 volúmenes de columna (VC) TBS pH 7,5
Lavar columna con 15 VC de NaOH 0,5 M
Lavar columna con 5 VC de TBS pH 7,5
Lavar columna con 5 VC de Glicina pH 2,7
Lavar columna con TBS pH 7,5 hasta obtener un pH natural
2. Retirar precipitación de la mezcla de reacción mediante centrifugación (5 min a 4000g) o mediante filtración (filtro de 0,22 o 0,45 |jm)
3. Cargar muestra a 2 ml/min y realizar las siguientes etapas con 5 ml/min:
Lavar con al menos 20 VC de TBS = Triton X-100 al 0,2%
Lavar con al menos 20 VC de TBS
Eluir con Glicina 0,1 M, pH 2,7
4. Neutralizar inmediatamente las fracciones al agregar 1/5 volumen de Tric-HCl 1 M pH 8,0 y mezclar
5. Dializar muestra contra 3 x >50 volúmenes de PBS pH 7,4 a 4°C (3 x >1 hora)
6. Concentrar muestra usando dispositivos de filtro de centrifugadora hasta ~20 mg/ml
Conjugación de 4 al anticuerpo modificado para producir ConjA y ConjB
Condiciones de reacción
15 mg/ml de anticuerpo modificado con azido (IgG 0,1 M)
4 0,5 mM (5 eq. en comparación con IgG = 2,5 eq por azida)
DMF al 10% o propilenglicol al 25%
PBS pH7,4
Procedimiento
1. Agregar 9 volúmenes de 16,67 mg/ml de anticuerpo modificado con azido en PBS pH7
2. Agregar 1 vol de 5 mM de 4 en DMF y mezclar inmediatamente.
3. Incubar durante la noche.
4. Medir conversión mediante RP-HPLC y MS.

Purificación de ADC
Preparación de muestras
Se deberían cumplir los siguientes requisitos antes de cargarse en la columna:
Solvente orgánico <5%
Volumen total de la muestra <3% del VC (<720 jL para Superdex 200 10/300 GL, y <10 ml para Superdex 200 HiLoad 26/600)
Sin precipitantes
Es posible cumplir con los siguientes requisitos usando el siguiente procedimiento:
1. Diluir la muestra con PBS pH7,4 hasta una concentración final de solvente orgánico de <5%
2. Si el volumen supera el 3% del VC, se concentró la muestra usando filtros centrífugos Amicon Ultra (MWCO 10 kDa)
3. La posible precipitación se retira mediante centrifugación (10 min a 13000 rpm en una centrífuga de mesa) Purificación
La purificación se llevó a cabo usando una columna Superdex 200 10/300 GL (VC = 23 ml, GE healthcare) en un Akta Purifier-10. Las siguientes etapas de lavado se realizan con una velocidad de flujo de 0,5 ml/min:
Lavar la columna con 1 VC de agua
Lavar columna con 1 VC de NaOH 0,5 M
Equilibrar columna con PBS pH 7,4 (Sigma, D8537) hasta obtener pH neutro.
Se inyecta la muestra con PBS 0,5 ml/min pH7,4 y se recogen fracciones de 1 ml (ejecución total = 1,5VC). Las fracciones de monómero se agrupan y someten a diálisis a 4°C contra 3x1L de amortiguador de formulación (30 mM de histidina, 200 mM de sorbitol, 0,02% (p/v) tween-20, pH 6,0). Las muestras se esterilizan mediante filtración usando un filtro de 0,22jm, se congelan rápidamente usando nitrógeno líquido y se almacenan a -80°C.
El análisis de espectro de masas de la muestra digerida con fabricator mostró un producto principal (masa observada 25691 Da, aproximadamente 90% de fragmento Fc/2 total), que corresponde al fragmento Fc/2 conjugado. El análisis de RP-HPLC de la muestra reducida indicó un promedio DAR de 1,98.
Citotoxicidad in vitro
Se obtuvieron células H1299 a partir de ATCC (número ATCC CRL-5803). El medio H1299 fue medio Eagle modificado de Dulbecco (DMEM) complementado con Gibco FBS al 10%. Se desarrollaron las células a 37 °C, CO2 al 5% en una incubadora humidificada. Las suspensiones celulares se dispensaron en placas de fondo redondo de 96 pocillos (104 células por pocillo). Se preparó un conjunto de diluciones 8 x 10 veces de ADC madre en medio de cultivo celular. Se dispensó cada dilución de ADC (50 |jl por pocillo) en 4 pocillos replicados de la placa de 96 pocillos que contenía una suspensión celular. Los pocillos de control se prepararon al agregar el mismo volumen del medio de cultivo solamente. Después de la incubación durante 96 horas, se midió la viabilidad celular mediante ensayo de 3-(4,5-dimetiltiazol-2-il)-5-(3-carboximetoxi-fenil)-2-(4-sulfofenil)-2H-tetrazolio (MTS) (Promega, número de catálogo G5421) según las instrucciones del fabricante. La absorbancia se midió a 490 nm. Se calculó la supervivencia celular (%) a partir de la absorbancia promedio en los 4 pocillos tratados con ADC en comparación con la absorbancia promedio en los 4 pocillos de control (100 %). Se generaron curvas de respuesta a la dosis a partir de los datos promedio de 3 experimentos replicados y se determinaron los valores de EC50 ajustando los datos a una curva de respuesta a la dosis sigmoidal con pendiente variable usando Prism (GraphPad, San Diego, CA). Las barras de error indican desviación estándar (SD, por sus siglas en inglés).
Se descubrió que la EC50 de ConjA era de 0,0554 |jg/ml.
Estudio de unión al antígeno
Se recubrieron placas Maxisorp a 4°C durante la noche con antígeno Axl humano (50 ng/pocillo; lote en PBS. Los sitios no reactivos se bloquearon con amortiguador SuperBlock (durante la noche a 4°C o temperatura ambiente). Se preparó un conjunto de diluciones 8 x 3 veces o 5 veces de ADC madre en amortiguador madre/PBS/Tween20. Se dispensó cada dilución de ADC (60 jl/pocillo) en 4 pocillos replicados de la placa recubierta. Los pocillos de control se prepararon al agregar el mismo volumen del amortiguador de muestra/PBS/Tween20. Se usó conjugado de peroxidasa de rábano picante (HRP)-IgG kappa antihumana como anticuerpo secundario (1:5000, 1 hora a temperatura ambiente). Se detectó HRP con una solución de sustrato Ultra TMB-ELISA de 1 etapa (75 jl/pocillo; 5 minutos a temperatura ambiente). Se detuvo la reacción de sustrato con HCl 0,6 M (75 jl/pocillo). Se midió la densidad óptica a 450 nm en Envision usando programa de 450 nm de Peroxidasa. Se generaron las curvas de unión al antígeno a partir de los datos promedio de 3 experimentos replicados usando Prism (GraphPad, San Diego, CA). La Figura 1 muestra los resultados obtenidos, donde Aes ConjA. Las barras de error indican el error estándar del promedio (SEM). ConjA se unió con gran afinidad al dominio extracelular de AXL recubierto en placas.
Estudio de eficacia in vivo - MDA-MB-231
Se implantaron por vía subcutánea 5 x 106 células tumorales MDA-MB-231 a ratones lampiños atímicos hembra. Se inició la dosificación de ADC o del artículo de prueba cuando los volúmenes tumorales alcanzaron 88-172 mm3. Se administró ConjA por vía intravenosa (i.v.) mediante inyección en la vena de la cola una vez con un nivel de dosis de 1 mg/kg. El volumen de dosificación fue 10 ml/kg de peso corporal y aumentó hasta el peso corporal de cada animal individual. Los animales fueron sometidos a eutanasia si su volumen tumoral alcanzó el volumen final de 1500 mm3 o al final del estudio, lo que ocurriera primero. Se controlaron el peso de los animales, los síntomas de efectos secundarios adversos relacionados con el tratamiento y los síntomas clínicos durante el período de estudio. Para el cálculo del volumen tumoral promedio del grupo, se aplicó la siguiente regla: cuando un animal salió del estudio debido al tamaño del tumor, se incluyó el volumen tumoral final registrado para el animal con los datos usados para calcular el volumen promedio en momentos posteriores. Los valores de volumen tumoral y peso corporal no se usaron para calcular volúmenes tumorales promedio/peso corporal de un grupo cuando menos del 50% de los animales en un grupo permaneció en el estudio. Se usó Prism (GraphPad, San Diego, CA) para presentaciones gráficas y análisis estadísticos. La Figura 2 muestra los resultados obtenidos, donde Aes ConjA, y O es el vehículo solo. Las barras de error indican SEM.
Una única dosis de 1 mg/kg de ConjA inhibió mucho el crecimiento tumoral donde 10/10 de los ratones estaban libres de tumor 60 días después de la dosificación.
Estudio de toxicología en ratas
Método
Se evaluó ConjA en un estudio de tolerabilidad de rata con dosis intravenosa única. Las ratas Sprague Dawley macho (n=3 /grupo) recibieron una dosis a 3 y 6 mg/kg para ConjA el día 1, con necropsia el día 21 después de la dosis. Se controlaron con frecuencia los pesos corporales y el consumo de alimento con la muestra en vivo para la patología
clínica (sangre los días 8 y 21) y muestras repetidas para la farmacocinética. En la necropsia, se realizaron observaciones macroscópicas con órganos seleccionados que se pesaron y se retuvieron para una posible histopatología.
ConjA se toleró clínicamente a 3 y 6 mg/kg. Se redujo el aumento de peso corporal en 11 y 21% en los grupos de 3 y 6 mg/kg, respectivamente, lo que concuerda con la reducción del consumo de alimento. Se redujeron varios parámetros de hematología el día 8 , principalmente en el grupo de dosis de 6 mg/kg (reticulocitos (-76%), hemoglobina (-29%) glóbulos blancos (-66 %) y plaquetas (-37%)), con evidencia de recuperación al día 21. Al momento de la necropsia, se observó una reducción del peso del timo en todos los animales. Por lo tanto, la dosis máxima tolerada (MTD) para ConjA fue 6 mg/kg.
Estudio de eficacia in vivo - SN12C
Los ratones inmunodeficientes combinados hembra (Fox Chase SCID®, CB17/Icr-Prkdcscid/IcrIcoCrl, Charles River) tenían diez semanas de edad con un intervalo de peso corporal (BW) de 18,0 a 21,6 g el Día 1 del estudio. El día del implante de tumor, cada ratón de prueba recibió 5 x 106 células SN12C (0,1 ml de suspensión celular en Matrigel® Matrix 50 % (Corning®) en solución salina tamponada con fosfato) implantadas subcutáneamente en el flanco derecho. SN12C es un modelo de xenoinjerto derivado de carcinoma de células renales humanas con nivel elevado de expresión de AXL (~88,000 copias por célula).
El crecimiento tumoral se controló a medida que el tamaño promedio alcanzaba el intervalo diana de 100 a 150 mm3. Se midieron los tumores en dos dimensiones usando calibres, y el volumen se calculó usando la fórmula:
Volumen tumoral (mm3) = w2 x l/2
donde w = ancho y l = longitud, en mm, del tumor. El peso del tumor se puede estimar con la suposición de que 1mg es equivalente a 1 mm3 de volumen de tumor.
Veinticinco días después del implante tumoral, designado como el día 1 del estudio, los animales se clasificaron en cinco grupos (n=8 ) con volúmenes tumorales individuales de 108 a 172 mm3 y volúmenes tumorales con un promedio grupal de 120 a 123 mm3. Todos los tratamientos se administraron i.v. en la vena de la cola lateral en una única inyección el día 1 del estudio. El volumen de dosificación fue 0,2 ml por 20 gramos de peso corporal (10 ml/kg), y se ajustó al peso corporal de cada animal individual. Los tumores se midieron usando calibres dos veces por semana, y todos los animales se sometieron a eutanasia cuando su tumor alcanzó el volumen de destino de 2000 mm3 o al final del estudio, lo que ocurriera primero. El estudio terminó el Día 60. Con una dosis de 1 mg/kg, ConjA dio como resultado 7/8 pacientes con respuesta completa (CR) y 6/8 pacientes con supervivencia sin tumor (TFS) al final del estudio el día 60.
La Figura 3 muestra los resultados obtenidos, donde:
O es el vehículo solo;
O es ConjB dosificado a 1 mg/kg;
□ es ConjA dosificado a 0,3 mg/kg;
A es ConjA dosificado a 0,6 mg/kg;
V es ConjA dosificado a 1 mg/kg.
Las barras de error indican SEM.
Estudio de eficacia in vivo - Karpas299 (negativo a AXL)
Los ratones inmunodeficientes combinados hembra (Fox Chase SCID®, CB17/Icr-Prkdcscid/IcrIcoCrl, Charles River) tenían nueve semanas de edad con un intervalo de peso corporal (BW) de 17,0 a 22,5 g el Día 1 del estudio. El día del implante de tumor, cada ratón de prueba recibió 1 x 107 células Karpas-299 (0,1 ml de suspensión celular en PBS) implantadas por vía subcutánea en el flanco derecho.
El crecimiento tumoral se controló a medida que el tamaño promedio alcanzaba el intervalo diana de 100 a 150 mm3. Se midieron los tumores en dos dimensiones usando calibres, y el volumen se calculó usando la fórmula:
Volumen tumoral (mm3) = w2 x l/2
donde w = ancho y l = longitud, en mm, del tumor. El peso del tumor se puede calcular con la suposición de que 1 mg es equivalente a 1 mm3 de volumen tumoral.
Diez días después del implante tumoral, designado como el día 1 del estudio, los animales se clasificaron en cuatro grupos con volúmenes tumorales individuales de 108 a 126 mm3 y volúmenes tumorales con un promedio grupal de 113 a 117 mm3. Todos los tratamientos se administraron i.v. en la vena de la cola lateral en una única inyección el día 1 del estudio. El volumen de dosificación fue 0,2 ml por 20 gramos de peso corporal (10 ml/kg), y se ajustó al peso
corporal de cada animal individual. Los tumores se midieron usando calibres dos veces por semana, y todos los animales se sometieron a eutanasia cuando su tumor alcanzó el volumen de destino de 2000 mm3 o al final del estudio, lo que ocurriera primero. El estudio finalizó el Día 29.
La Figura 4 muestra los resultados obtenidos, donde:
O es el vehículo solo;
□ es ConjA dosificado a 1 mg/kg.
Las barras de error indican SEM.
Estudio de eficacia in vivo - Modelo de xenoinjerto derivado de paciente (PDX) con cáncer pancreático PAXF 1657
Los ratones nu/nu hembra (NU-Foxn1nu) de Charles River tenían al menos 8 semanas de edad con un intervalo de peso corporal (BW) de 22,0 a 30,0 g el Día 0. El día del implante, se obtuvieron fragmentos del tumor de xenoinjertos en ratones lampiños. Después de la eliminación de los ratones donantes, los tumores se cortaron en fragmentos (3-4 mm de longitud de borde) y se colocaron en PBS que contenía 10% de penicilina/estreptomicina. Los animales receptores fueron anestesiados mediante inhalación de isoflurano y recibieron implantes tumorales unilaterales o bilaterales por vía subcutánea en el costado.
Se controlaron los animales e implantes tumorales dado que sus volúmenes de implantes alcanzaron el intervalo diana de 50 a 250 mm3 en una cantidad suficiente de animales. Se midieron los tumores en dos dimensiones usando calibres, y el volumen se calculó usando la fórmula:
Volumen tumoral (mm3) = w2 x l/2
donde w = ancho y l = longitud, en mm, del tumor.
El día de la aleatorización se designó como el día 0 del experimento. El día 1 del experimento, se clasificaron ratones nu/nu hembra que tienen xenoinjertos PAXF 1657 subcutáneos (volúmenes tumorales promedio grupales 109,0-110,1 mm3) en grupos (n = 8 por grupo) y se inició la dosificación. El volumen de dosificación fue 0,1 ml cada 20 gramos de peso corporal (5 ml/kg), y se ajustó al peso corporal de cada animal individual. Todos los tratamientos se administraron por vía intravenosa (i.v.) en una única inyección el día 1 (qd x 1). Los tumores se midieron usando calibres dos veces por semana, y todos los animales se sometieron a eutanasia cuando su tumor alcanzó el volumen de destino de 2000 mm3 o al final del estudio, lo que ocurriera primero. El estudio finalizó el Día 42. Cada dosis única de ConjA (0,3, 0,6 y 1 mg/kg) dio como resultado la erradicación completa de los tumores al final del estudio.
La Figura 5 muestra los resultados obtenidos, donde:
O es el vehículo solo;
O es ConjB dosificado a 1 mg/kg;
□ es ConjA dosificado a 0,3 mg/kg;
A es ConjA dosificado a 0,6 mg/kg;
V es ConjA dosificado a 1 mg/kg.
Las barras de error indican SEM. La línea punteada vertical indica el inicio de la dosificación (día 1).
Estudio de eficacia in vivo - Modelo de xenoinjerto derivado de paciente (PDX) con cáncer esofágico ES0195
Los ratones lampiños Balb/c hembra de Beijing AniKeeper Bio-Technology Co. Ltd. tenían 5-6 semanas de edad con un intervalo de peso corporal de 20,2-24,6 g al inicio del estudio. Los fragmentos tumorales (2-3mm de diámetro, en medio RPMI1640 helado sin suero) se inocularon por vía subcutánea en el flanco derecho de 24 ratones lampiños Balb/c hembra.
Todos los animales se asignaron aleatoriamente a los 3 grupos de estudio distintos. Se realizó aleatorización usando el método de múltiples tareas en el software Study Log el día 0. El volumen tumoral promedio (mm3) para cada grupo SD en la aleatorización fue el siguiente, ~141mm3 ± 47mm3.
Todos los tratamientos se administraron por vía intravenosa (i.v.) en una única inyección el día 1 (qd x 1). Se midieron los pesos corporales dos veces a la semana durante la duración del estudio. Los volúmenes tumorales se midieron dos veces a la semana. El estudio terminó en el día 51 después del inicio de la dosificación.
La Figura 6 muestra los resultados obtenidos, donde:
O es el vehículo solo;
□ es ConjA dosificado a 1 mg/kg;
A es ConjB dosificado a 1 mg/kg;
Las barras de error indican SEM. La línea punteada vertical indica el inicio de la dosificación (día 1).
Estudio de actividad de espectador in vitro
La citotoxicidad in vitro de ConjA y un control de isotipo ADC, ConjB, se comparó en líneas celulares SN12C y Karpas-299 - Karpas299 fueron negativas para AXL. SN12C son positivas para AXL. Las células SN12C adherentes se tripsinizaron, volvieron a suspender en medio de cultivo 1 ml y se mezclaron suavemente antes del conteo para determinar la densidad celular. Las células Karpas299 en suspensión se contabilizaron sin un pretratamiento. Se determinó la densidad celular en duplicado mediante ensayo de exclusión de azul de tripán usando un contabilizador de células automático LUNA-II™. Se diluyó la suspensión de células SN12C hasta 1 x 104 células/ml en medio de cultivo celular específico y se dispensaron 100 pl/pocillo en microplacas de fondo redondo de 96 pocillos blancas estériles y se incubaron durante la noche para permitir que las células se adhieran. Se sedimentaron las células Karpas299 el mismo día que la aplicación de ADC.
Las diluciones en serie de las ADC esterilizadas por filtro se realizaron en una relación 1:10 y se repitieron para producir ocho diluciones en serie, usando una concentración de ADC inicial de 20 pg/ml y diluyendo en medio de cultivo celular específico en placas de polipropileno de 96 pocillos estériles. Se dispensaron diluciones de ADC (que incluyen la solución madre) en 2 pocillos replicados, 100 pl/pocillo, de la placa de fondo redondo de 96 pocillos blanca etiquetada, que contiene una suspensión de células sembradas de 100 pl. Para los pocillos de control de medio, se dispensaron 100 pl de medio de cultivo celular en 2 pocillos replicados, y para los pocillos de control de línea celular, se dispensaron 100 pl de medio de cultivo en 100 pl de suspensión celular, dispensados previamente, en 2 pocillos replicados. Todas las placas se incubaron durante 5 días en un incubador gasificado con CO2 (5%) a 37°C. Los ensayos se llevaron a cabo usando las mismas densidades de siembra de células y los mismos tiempos de incubación para ambas líneas celulares; 1 x 103 células/pocillo incubadas durante 5 días cada una (estas condiciones se habían optimizado previamente para este estudio).
Después de un período de incubación de 5 días, las placas se centrifugaron a 600 G (20°C) durante 5 min, antes de transferir cuidadosamente 100 pl/pocillo en placas de fondo redondo de 96 pocillos blancas recientemente preparadas, que contienen 100 pl de células Karpas-299 sembradas (previamente dispensadas). Todas las placas se incubaron durante 5 días en un incubador gasificado con CO2 (5%) a 37°C. Después del período de incubación, las placas se centrifugaron a 600 G (20°C) durante 5 min, antes de retirar cuidadosamente y desechar 100 pl/pocillo, y se midió la viabilidad celular en el medio restante en los pocillos usando el ensayo CellTiter-Glo®. Las placas se leyeron en Envision usando el protocolo de luminiscencia y se analizaron los datos usando el software Graphpad Prism.

SECUENCIAS
SEQ ID NO.1 MH 1H12, CDR subravadal
QVQLVESGGGVVQPGRSLRLSCAASGFTFSSYGMSWVRQAPGKGLEWVATISSGGSYTYYPDSVKGRFTISRD
NSKNTLYLQMNSLRAEDTAVYYCARHPIYYTYDDTMDYWGQGTTVTVSS
SEQ ID NO.2 ÍVL 1H12, CDR subrayada! EIVLTQSPGTLSLSPGERATLSCSASSSVSSGNFHWYQQKPGLAPRLLIYRTSNLASGIPARFSGSGSGTDFTLTIS SLEPEDFAVYYCQQWSGYPWTFGGGTKLEIK
SEQ ID NO.3 TCadena pesada de 1H12!
QVQLVESGGGVVQPGRSLRLSCAASGFTFSSYGMSWVRQAPGKGLEWVATISSGGSYTYYPDSVKGRFTISRD
NSKNTLYLQMNSLRAEDTAVYYCARHPIYYTYDDTMDYWGQGTTVTVSSASTKGPSVFPLAPSSKSTSGGTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEP KSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPR
EEQYN*STYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLT
CLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQK
SLSLSPGK
N* indica Asn297
SEQ ID NO.4 TCadena ligera de 1H12! EIVLTQSPGTLSLSPGERATLSCSASSSVSSGNFHWYQQKPGLAPRLLIYRTSNLASGIPARFSGSGSGTDFTLTIS
SLEPEDFAVYYCQQWSGYPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKV
DNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
SEQ ID NO.5 TCDR1 VH 1H121
SYGMS SEQ ID NO.6 TCDR2 VH 1H121
TISSGGSYTYYPDSVKG SEQ ID NO.7 TCDR3 VH 1H121
HPIYYTYDDTMDY SEQ ID NO.8 TCDR1 VL 1H121
SASSSVSSGNFH SEQ ID NO.9 TCDR2 VL 1H121
RTSNLAS SEQ ID NO.10 TCDR3 VL 1H121
QQWSGYPWT SEQ ID NO.11 TVH 5F11 murino, CDR subrayada1
EVKLLESGGGLVQPGGSLKLSCAASGFDFSRYWMSWVRQAPGKGLEWIGEINPDSSTINYTPSLKDKFIISRDNA KNTLYLQMSKVRSEDTALYYCASPYYYGPFAYWGQGTLVTVSS SEQ ID NO.12 TVL 5F11 murino, CDR subrayada1
DIVLTQSPASLAVSLGQRAIISCKASQSVSFAGTSLMHWYQQKPGQQPKLLIYRASNLEAGFPTRFSGSGSRTDFT LNIHPVEEEDAATYYCQQSREYPRTFGGGTKLEVK SEQ ID NO.13 TCDR1 VH 5F111
RYWMS SEQ ID NO.14 TCDR2 VH 5F111
EINPDSSTINYTPSLKD SEQ ID NO.15 TCDR3 VH 5F111
PYYYGPFAY SEQ ID NO.16 TCDR1 VL 5F111
KASQSVSFAGTSLMH SEQ ID NO.17 TCDR2 VL 5F111
RASNLEA SEQ ID NO.18 TCDR3 VL 5F111
QQSREYPRT SEQ ID NO.19 TRHA 5F111
QVQLVESGGGVVQPGRSLRLSCAASGFTFSRYWMSWVRQAPGKGLEWVAEINPDSSTINYTPSLKDRFAISRDN SKNTLYLQMNSLRAEDTAVYYCASPYYYGPFAYWGQGTLVTVS SEQ ID NO.20 TRHB 5F111
EVQLVESGGGLVQPGGSLRLSCAASGFTFSRYWMSWVRQAPGKGLEWVAEINPDSSTINYTPSLKDRFTISRDN AKNSLYLQMNSLRAEDTAVYYCASPYYYGPFAYWGQGTLVTVS SEQ ID NO.21 TRHC 5F111
EVQLLESGGGLVQPGGSLRLSCAASGFTFSRYWMSWVRQAPGKGLEWVSEINPDSSTINYTPSLKDRFTISRDN SKNTLYLQMNSLRAEDTAVYYCASPYYYGPFAYWGQGTLVTVS SEQ ID NO.22 TRKA 5F111
EIVLTQSPLSLPVTPGEPASISCKASQSVSFAGTSLMHWYLQKPGQSPQLLIYRASNLEAGVPDRFSGSGSGTDFT LKISRVEAEDVGVYYCQQSREYPRTFGQGTKVEIK SEQ ID NO.23 TAxl humano1
MAWRCPRMGRVPLAWCLALCGWACMAPRGTQAEESPFVGNPGNITGARGLTGTLRCQLQVQGEPPEVHWLR DGQILELADSTQTQVPLGEDEQDDWIVVSQLRITSLQLSDTGQYQCLVFLGHQTFVSQPGYVGLEGLPYFLEEPE DRTVAANTPFNLSCQAQGPPEPVDLLWLQDAVPLATAPGHGPQRSLHVPGLNKTSSFSCEAHNAKGVTTSRTAT ITVLPQQPRNLHLVSRQPTELEVAWTPGLSGIYPLTHCTLQAVLSDDGMGIQAGEPDPPEEPLTSQASVPPHQLRL
Claims (26)
1. Un conjugado de la fórmula (I):
Ab - (DL)p (I)
en la que:
Ab es un anticuerpo que se une a AXL;
DL es

en la que:
X se selecciona del grupo que comprende: un enlace simple, -CH2- y -C2H4-;
n es de 1 a 8;
m es 0 o 1;
R7 es metilo o fenilo;
cuando existe un enlace doble entre C2 y C3, R2 se selecciona del grupo que consiste en:
(ia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3; (ib) un alquilo alifático C1-5 saturado;
(ic) un cicloalquilo C3-6 saturado;
(id)

en la que cada uno de R21, R22y R23 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R2 no es mayor de 5;
(ie)

en la que uno de R25a y R25b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y
(if)
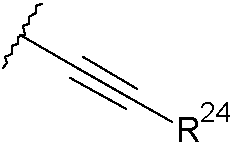
donde R24 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo;
cuando existe un enlace simple entre C2 y C3, R2 es

donde R26a y R26b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R26a y R26b es H, el otro se selecciona de nitrilo y un alquiléster C1-4;
cuando existe un enlace doble entre C2' y C3', R12 se selecciona del grupo que consiste en:
(iia) un grupo arilo C5-10 opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que comprende: halo, nitro, ciano, éter, carboxi, éster, alquilo C1-7, heterociclilo C3-7 y bis-oxi-alquileno C1-3;
(iib) un alquilo alifático C1-5 saturado;
(iic) un cicloalquilo C3-6 saturado;
(iid)

en la que cada uno de R31, R32y R33 se selecciona de forma independiente de H, alquilo C1-3 saturado, alquenilo C2-3, alquinilo C2-3 y ciclopropilo, donde la cantidad total de átomos de carbono en el grupo R12 no es mayor de 5;
(iie)
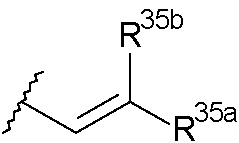
en la que uno de R35a y R35b es H y el otro se selecciona de: fenilo, dicho fenilo se encuentra opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo; y
(iif)

donde R34 se selecciona de: H; alquilo C1-3 saturado; alquenilo C2-3; alquinilo C2-3; ciclopropilo; fenilo, dicho fenilo opcionalmente sustituido con un grupo seleccionado de halo, metilo, metoxi; piridilo y tiofenilo;
cuando existe un enlace simple entre C2' y C3', R12 es

donde R36a y R36b se seleccionan independientemente de H, F, alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo están opcionalmente sustituidos por un grupo que se selecciona de alquilamido C1-4 y alquiléster C1-4; o, cuando uno de R36a y R36b es H, el otro se selecciona de nitrilo y un alquiléster C1-4;
en donde en (ia) y (iia), alquilo C1-7 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un átomo de carbono de un compuesto de hidrocarburo que tiene de 1 a 7 átomos de carbono, que puede ser alifático o alicíclico, y que puede ser saturado o insaturado; y
en (if) y (iif), alquilo C1-4 pertenece a un resto monovalente obtenido eliminando un átomo de hidrógeno de un compuesto de hidrocarburo que tiene de 1 a 4 átomos de carbono, que puede ser alifático o alicíclico, y que puede ser saturado o insaturado;
y p es de 1 a 8.
2. El conjugado de acuerdo con la reivindicación 1, en donde X es -CH2-.
3. El conjugado de acuerdo con las reivindicaciones 1 o 2, en donde n es 1 a 4.
4. El conjugado de acuerdo con la reivindicación 3, en donde n es 2.
5. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 4, en donde existe un enlace doble entre C2 y C3, y R2 es un grupo alquilo alifático saturado C5-7.
6. Un compuesto de acuerdo con la reivindicación 5, en donde R2 es metilo, etilo o propilo.
7. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 4, en donde existe un enlace doble entre C2 y C3, y R2 es:
(a) fenilo, que lleva de uno a tres grupos sustituyentes, en donde los sustituyentes pueden seleccionarse de metoxi, etoxi, flúor, cloro, ciano, bis-oxi-metileno, metil-piperazinilo, morfolino y metiltiofenilo; o
(b) ciclopropilo; o
(c) un grupo de fórmula:

donde la cantidad total de átomos de carbono en el grupo R2 no es mayor de 3; o
(d) el grupo:

o
(e) un grupo de fórmula:

en la que R24 se selecciona de H y metilo.
8. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 4, en donde existe un enlace simple entre C2 y C3, y R2 es

y
(a) R26a y R26b son ambos H; o
(b) R26a y R26b son ambos metilo; o
(c) uno de R26a y R26b es H, y el otro se selecciona de alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo se encuentran opcionalmente sustituidos.
9. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8, en donde existe un enlace doble entre C2' y C3', y R12 es un grupo alquilo alifático saturado C1-5.
10. Un compuesto de acuerdo con la reivindicación 9, en donde R12 es metilo, etilo o propilo.
11. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8, en donde existe un enlace doble entre C2' y C3', y R12 es:
a) fenilo, que lleva de uno a tres grupos sustituyentes, en donde los sustituyentes pueden seleccionarse de metoxi, etoxi, flúor, cloro, ciano, bis-oxi-metileno, metil-piperazinilo, morfolino y metiltiofenilo; o
(b) ciclopropilo; o
(c) un grupo de fórmula:

en donde la cantidad total de átomos de carbono en el grupo R12 no es mayor de 3; o
(d) el grupo:

o
(e) un grupo de fórmula:

en la que R34 se selecciona de H y metilo.
12. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 8, en donde hay un enlace simple entre C2' y C3', R12 es

y
(a) R36a y R36b son ambos H; o
(b) R36a y R36b son ambos metilo; o
(c) uno de R36a y R36b es H, y el otro se selecciona de alquilo C1-4 saturado, alquenilo C2-3, dichos grupos alquilo y alquenilo se encuentran opcionalmente sustituidos.
13. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 12, en donde el anticuerpo comprende un dominio VH que tiene una CDR3 de VH con la secuencia de aminoácidos del SEQ ID NO.7, una CDR2 de VH con la secuencia de aminoácidos del SEQ ID NO.6 y una CDR1 de VH con la secuencia de aminoácidos del SEQ ID NO.5.
14. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 13, en donde el anticuerpo comprende un dominio VH que tiene la secuencia del SEQ ID NO.1.
15. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 14, en donde el anticuerpo comprende un dominio VL que comprende una CDR3 de VL con la secuencia de aminoácidos del SEQ ID NO.10, una CDR2 de VL con la secuencia de aminoácidos del SEQ ID NO.9 y una CDR1 de VL con la secuencia de aminoácidos del SEQ ID NO.8.
16. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 15, en donde el anticuerpo comprende un dominio VH que tiene la secuencia del SEQ ID NO. 2.
17. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 16, en donde el anticuerpo comprende una cadena pesada que tiene la secuencia del SEQ ID NO. 3 o delel SEQ ID NO. 24.
18. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 17, en donde el anticuerpo comprende una cadena pesada que tiene la secuencia del SEQ ID NO. 3 emparejada con una cadena ligera que tiene la secuencia del SEQ ID NO. 4.
19. Una composición que comprende una mezcla de los compuestos de conjugados de anticuerpos y fármacos como se definen en cualquiera de las reivindicaciones 1 a 18, en donde la carga de fármaco promedio por anticuerpo en la mezcla de compuestos de conjugados de anticuerpos y fármacos es de 1 a 4.
20. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 18, para su uso en terapia.
21. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 18, para su uso en el tratamiento de una enfermedad proliferativa en un sujeto.
22. El conjugado para su uso de acuerdo con la reivindicación 21, en donde la enfermedad es cáncer.
23. El conjugado para su uso de acuerdo con cualquiera de las reivindicaciones 21 o 22, la enfermedad o el cáncer están caracterizados por la presencia de un neoplasma que comprende células AXL+ve y AXL-ve.
24. El conjugado de acuerdo con cualquiera de las reivindicaciones 1 a 18, para su uso en un método para tratar una enfermedad proliferativa, comprendiendo dicho método:
(i) identificar la presencia en el sujeto de un neoplasma que comprende células AXL+ve y células AXL-ve;
(ii) administrarle al sujeto el conjugado de anticuerpo y fármaco.
25. Una composición farmacéutica que comprende el conjugado de cualquiera de las reivindicaciones 1 a 18 y un diluyente, un vehículo o un excipiente farmacéuticamente aceptables.
26. La composición farmacéutica de la reivindicación 25 que comprende además una cantidad terapéuticamente eficaz de un agente quimioterapéutico.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1702029.8A GB201702029D0 (en) | 2017-02-08 | 2017-02-08 | Pyrrolobenzodiazepine-antibody conjugates |
| GBGB1702031.4A GB201702031D0 (en) | 2017-02-08 | 2017-02-08 | Pyrrolobenzodiazepine-antibody conjugates |
| GBGB1719906.8A GB201719906D0 (en) | 2017-11-30 | 2017-11-30 | Pyrrolobenzodiazepine-antibody conjugates |
| PCT/EP2018/053163 WO2018146189A1 (en) | 2017-02-08 | 2018-02-08 | Pyrrolobenzodiazepine-antibody conjugates |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2871001T3 true ES2871001T3 (es) | 2021-10-28 |
Family
ID=61198845
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18704956T Active ES2871001T3 (es) | 2017-02-08 | 2018-02-08 | Conjugados de pirrolobenzodiazepinas y anticuerpos |
Country Status (23)
| Country | Link |
|---|---|
| US (2) | US11160872B2 (es) |
| EP (1) | EP3544636B1 (es) |
| JP (1) | JP6671555B2 (es) |
| KR (2) | KR102181375B1 (es) |
| CN (1) | CN110267686B (es) |
| AU (1) | AU2018217926B2 (es) |
| BR (1) | BR112019016373B1 (es) |
| CA (1) | CA3047683C (es) |
| CY (1) | CY1124402T1 (es) |
| DK (1) | DK3544636T3 (es) |
| ES (1) | ES2871001T3 (es) |
| HR (1) | HRP20210979T1 (es) |
| HU (1) | HUE054689T2 (es) |
| LT (1) | LT3544636T (es) |
| MX (1) | MX375569B (es) |
| NZ (1) | NZ754810A (es) |
| PL (1) | PL3544636T3 (es) |
| PT (1) | PT3544636T (es) |
| RS (1) | RS61795B1 (es) |
| SI (1) | SI3544636T1 (es) |
| SM (1) | SMT202100543T1 (es) |
| UA (1) | UA125198C2 (es) |
| WO (1) | WO2018146189A1 (es) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201506411D0 (en) | 2015-04-15 | 2015-05-27 | Bergenbio As | Humanized anti-axl antibodies |
| GB201702031D0 (en) * | 2017-02-08 | 2017-03-22 | Medlmmune Ltd | Pyrrolobenzodiazepine-antibody conjugates |
| WO2018193102A1 (en) * | 2017-04-20 | 2018-10-25 | Adc Therapeutics Sa | Combination therapy with an anti-axl antibody-drug conjugate |
| WO2020127573A1 (en) | 2018-12-19 | 2020-06-25 | Adc Therapeutics Sa | Pyrrolobenzodiazepine resistance |
| GB201820725D0 (en) | 2018-12-19 | 2019-01-30 | Adc Therapeutics Sarl | Pyrrolobenzodiazepine resistance |
| GB201908128D0 (en) | 2019-06-07 | 2019-07-24 | Adc Therapeutics Sa | Pyrrolobenzodiazepine-antibody conjugates |
| KR20210086557A (ko) | 2019-12-31 | 2021-07-08 | 주식회사 레고켐 바이오사이언스 | 피롤로벤조디아제핀 유도체 및 이의 리간드-링커 접합체 |
| EP4172141A1 (en) | 2020-06-26 | 2023-05-03 | Synaffix B.V. | Methods for the preparation of linker payload constructs |
| GB202011993D0 (en) | 2020-07-31 | 2020-09-16 | Adc Therapeutics Sa | ANTI-IL 13Ra2 antibodies |
| NL2026400B1 (en) | 2020-09-02 | 2022-05-04 | Synaffix Bv | Methods for the preparation of bioconjugates |
| US20230372528A1 (en) | 2020-10-16 | 2023-11-23 | University Of Georgia Research Foundation, Inc. | Glycoconjugates |
| WO2022081895A1 (en) | 2020-10-16 | 2022-04-21 | University Of Georgia Research Foundation, Inc. | Glycoconjugates |
| CN113189089B (zh) * | 2021-03-19 | 2022-05-17 | 四川轻化工大学 | 一种臭氧检测试剂及其制备装置和制备方法 |
| EP4230222A1 (en) | 2022-02-17 | 2023-08-23 | Oxsonics Limited | Combination therapy with an anti-axl antibody-pbd conjugate and nanocups |
| EP4279090A1 (en) | 2022-05-20 | 2023-11-22 | ADC Therapeutics SA | Composition comprising a pyrrolobenzodiazepine-based antibody drug conjugate |
| EP4548932A1 (en) | 2022-06-30 | 2025-05-07 | Toray Industries, Inc. | Pharmaceutical composition for treating and/or preventing cancer |
| EP4389152A1 (en) * | 2022-12-23 | 2024-06-26 | Synaffix B.V. | Conjugates of pbd prodrugs |
Family Cites Families (422)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3361742A (en) | 1964-12-07 | 1968-01-02 | Hoffmann La Roche | 5-oxo-1h-pyrrolo-[2, 1-c][1, 4]-benzodiazepin-2-crylamides |
| US3523941A (en) | 1967-03-06 | 1970-08-11 | Hoffmann La Roche | Benzodiazepine compounds and process for their preparation |
| US3524849A (en) | 1967-10-27 | 1970-08-18 | Hoffmann La Roche | Process for the preparation of pyrrolo-benzodiazepine acrylamides and intermediates useful therein |
| JPS4843755B1 (es) | 1969-06-26 | 1973-12-20 | ||
| IL33558A (en) | 1968-12-30 | 1973-10-25 | Fujisawa Pharmaceutical Co | Antibiotic pyrrolo-benzodiazepine compound,its derivatives and processes for their production |
| DE1965304A1 (de) | 1968-12-30 | 1970-07-23 | Fujisawa Pharmaceutical Co | Benzdiazepinon-Verbindungen und Verfahren zu ihrer Herstellung |
| JPS5382792U (es) | 1976-12-10 | 1978-07-08 | ||
| JPS6053033B2 (ja) | 1976-12-28 | 1985-11-22 | 財団法人微生物化学研究会 | 新制癌抗生物質マゼスラマイシン及びその製造方法 |
| JPS585916B2 (ja) | 1977-12-27 | 1983-02-02 | 株式会社ミドリ十字 | 新規ベンゾジアゼピン系化合物 |
| JPS5615289A (en) | 1979-07-17 | 1981-02-14 | Green Cross Corp:The | Novel benzodiazepinnbased compound 3 |
| JPS57131791A (en) | 1980-12-31 | 1982-08-14 | Fujisawa Pharmaceut Co Ltd | Benzodiazepine derivative and its preparation |
| CA1184175A (en) | 1981-02-27 | 1985-03-19 | Walter Hunkeler | Imidazodiazepines |
| CA1185602A (en) | 1981-02-27 | 1985-04-16 | Emilio Kyburz | Imidazodiazepines |
| CA1173441A (en) | 1981-02-27 | 1984-08-28 | Hoffmann-La Roche Limited | Imidazodiazepines |
| JPS58180487A (ja) | 1982-04-16 | 1983-10-21 | Kyowa Hakko Kogyo Co Ltd | 抗生物質dc−81およびその製造法 |
| US4427588A (en) | 1982-11-08 | 1984-01-24 | Bristol-Myers Company | Process for conversion of oxotomaymycin to tomaymycin |
| US4427587A (en) | 1982-11-10 | 1984-01-24 | Bristol-Myers Company | Total synthesis of antitumor antibiotics BBM-2040A and BBM-2040B |
| JPS59152329A (ja) | 1983-02-17 | 1984-08-31 | Green Cross Corp:The | 局所障害抑制剤 |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| FR2586683B1 (fr) | 1985-08-29 | 1988-07-01 | Centre Nat Rech Scient | Nouveaux derives de neothramycine, leur procede de preparation et leur application en tant que medicaments |
| US5583024A (en) | 1985-12-02 | 1996-12-10 | The Regents Of The University Of California | Recombinant expression of Coleoptera luciferase |
| JP2660201B2 (ja) | 1988-08-05 | 1997-10-08 | 塩野義製薬株式会社 | 新規ピロロ[1,4]ベンゾジアゼピン誘導体および老人性痴呆薬 |
| AU6355090A (en) | 1989-08-23 | 1991-04-03 | Scripps Clinic And Research Foundation | Compositions and methods for detection and treatment of epstein-barr virus infection and immune disorders |
| JPH053790A (ja) | 1990-04-19 | 1993-01-14 | Fujisawa Pharmaceut Co Ltd | デヒドロペプチダーゼ−i |
| US5256643A (en) | 1990-05-29 | 1993-10-26 | The Government Of The United States | Human cripto protein |
| AU9016591A (en) | 1990-10-25 | 1992-05-26 | Tanox Biosystems, Inc. | Glycoproteins associated with membrane-bound immunoglobulins as antibody targets on B cells |
| ATE194385T1 (de) | 1991-03-29 | 2000-07-15 | Genentech Inc | Menschliche pf4a rezeptoren und ihre verwendung |
| US5543503A (en) | 1991-03-29 | 1996-08-06 | Genentech Inc. | Antibodies to human IL-8 type A receptor |
| US5440021A (en) | 1991-03-29 | 1995-08-08 | Chuntharapai; Anan | Antibodies to human IL-8 type B receptor |
| FR2676058B1 (fr) | 1991-04-30 | 1994-02-25 | Hoechst Lab | Prodrogues glycosylees, leur procede de preparation et leur utilisation dans le traitement des cancers. |
| FR2676230B1 (fr) | 1991-05-07 | 1993-08-27 | Centre Nat Rech Scient | Nouveaux derives de pyrrolo [1,4]-benzodiazepines, leur procede de preparation et medicaments les contenant. |
| JP3050424B2 (ja) | 1991-07-12 | 2000-06-12 | 塩野義製薬株式会社 | ヒトエンドセリンリセプター |
| US5264557A (en) | 1991-08-23 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Polypeptide of a human cripto-related gene, CR-3 |
| US5362852A (en) | 1991-09-27 | 1994-11-08 | Pfizer Inc. | Modified peptide derivatives conjugated at 2-hydroxyethylamine moieties |
| US6153408A (en) | 1991-11-15 | 2000-11-28 | Institut Pasteur And Institut National De La Sante Et De La Recherche Medicale | Altered major histocompatibility complex (MHC) determinant and methods of using the determinant |
| US6011146A (en) | 1991-11-15 | 2000-01-04 | Institut Pasteur | Altered major histocompatibility complex (MHC) determinant and methods of using the determinant |
| GB9205051D0 (en) | 1992-03-09 | 1992-04-22 | Cancer Res Campaign Tech | Pyrrolobenzodiazepine derivatives,their preparation,and compositions containing them |
| FR2696176B1 (fr) | 1992-09-28 | 1994-11-10 | Synthelabo | Dérivés de pipéridine, leur préparation et leur application en thérapeutique. |
| IL107366A (en) | 1992-10-23 | 2003-03-12 | Chugai Pharmaceutical Co Ltd | Genes coding for megakaryocyte potentiator |
| US5644033A (en) | 1992-12-22 | 1997-07-01 | Health Research, Inc. | Monoclonal antibodies that define a unique antigen of human B cell antigen receptor complex and methods of using same for diagnosis and treatment |
| US5869445A (en) | 1993-03-17 | 1999-02-09 | University Of Washington | Methods for eliciting or enhancing reactivity to HER-2/neu protein |
| US5801005A (en) | 1993-03-17 | 1998-09-01 | University Of Washington | Immune reactivity to HER-2/neu protein for diagnosis of malignancies in which the HER-2/neu oncogene is associated |
| US6214345B1 (en) | 1993-05-14 | 2001-04-10 | Bristol-Myers Squibb Co. | Lysosomal enzyme-cleavable antitumor drug conjugates |
| GB9316162D0 (en) | 1993-08-04 | 1993-09-22 | Zeneca Ltd | Fungicides |
| US5773223A (en) | 1993-09-02 | 1998-06-30 | Chiron Corporation | Endothelin B1, (ETB1) receptor polypeptide and its encoding nucleic acid methods, and uses thereof |
| EP0647450A1 (en) | 1993-09-09 | 1995-04-12 | BEHRINGWERKE Aktiengesellschaft | Improved prodrugs for enzyme mediated activation |
| US5750370A (en) | 1995-06-06 | 1998-05-12 | Human Genome Sciences, Inc. | Nucleic acid encoding human endothlein-bombesin receptor and method of producing the receptor |
| JPH08336393A (ja) | 1995-04-13 | 1996-12-24 | Mitsubishi Chem Corp | 光学活性なγ−置換−β−ヒドロキシ酪酸エステルの製造法 |
| US5707829A (en) | 1995-08-11 | 1998-01-13 | Genetics Institute, Inc. | DNA sequences and secreted proteins encoded thereby |
| US20020193567A1 (en) | 1995-08-11 | 2002-12-19 | Genetics Institute, Inc. | Secreted proteins and polynucleotides encoding them |
| JP3646191B2 (ja) | 1996-03-19 | 2005-05-11 | 大塚製薬株式会社 | ヒト遺伝子 |
| US6218519B1 (en) | 1996-04-12 | 2001-04-17 | Pro-Neuron, Inc. | Compounds and methods for the selective treatment of cancer and bacterial infections |
| EP0910636A1 (en) | 1996-05-17 | 1999-04-28 | Schering Corporation | Human b-cell antigens, related reagents |
| WO1998013059A1 (en) | 1996-09-27 | 1998-04-02 | Bristol-Myers Squibb Company | Hydrolyzable prodrugs for delivery of anticancer drugs to metastatic cells |
| US6759509B1 (en) | 1996-11-05 | 2004-07-06 | Bristol-Myers Squibb Company | Branched peptide linkers |
| US5945511A (en) | 1997-02-20 | 1999-08-31 | Zymogenetics, Inc. | Class II cytokine receptor |
| US20030185830A1 (en) | 1997-02-25 | 2003-10-02 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of prostate cancer |
| US7033827B2 (en) | 1997-02-25 | 2006-04-25 | Corixa Corporation | Prostate-specific polynucleotide compositions |
| US6541212B2 (en) | 1997-03-10 | 2003-04-01 | The Regents Of The University Of California | Methods for detecting prostate stem cell antigen protein |
| EP0981541B1 (en) | 1997-03-10 | 2004-06-02 | The Regents Of The University Of California | Psca: prostate stem cell antigen |
| US6261791B1 (en) | 1997-03-10 | 2001-07-17 | The Regents Of The University Of California | Method for diagnosing cancer using specific PSCA antibodies |
| US6555339B1 (en) | 1997-04-14 | 2003-04-29 | Arena Pharmaceuticals, Inc. | Non-endogenous, constitutively activated human protein-coupled receptors |
| US6319688B1 (en) | 1997-04-28 | 2001-11-20 | Smithkline Beecham Corporation | Polynucleotide encoding human sodium dependent phosphate transporter (IPT-1) |
| WO1998051805A1 (en) | 1997-05-15 | 1998-11-19 | Abbott Laboratories | Reagents and methods useful for detecting diseases of the prostate |
| WO1998051824A1 (en) | 1997-05-15 | 1998-11-19 | Abbott Laboratories | Reagents and methods useful for detecting disease of the urinary tract |
| US6602677B1 (en) | 1997-09-19 | 2003-08-05 | Promega Corporation | Thermostable luciferases and methods of production |
| US20030060612A1 (en) | 1997-10-28 | 2003-03-27 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of tumor |
| US20020034749A1 (en) | 1997-11-18 | 2002-03-21 | Billing-Medel Patricia A. | Reagents and methods useful for detecting diseases of the breast |
| US6110695A (en) | 1997-12-02 | 2000-08-29 | The Regents Of The University Of California | Modulating the interaction of the chemokine, B Lymphocyte Hemoattractant, and its Receptor, BLR1 |
| ES2313779T3 (es) | 1998-03-13 | 2009-03-01 | The Burnham Institute | Moleculas que migran a diversos organos o tejidos seleccionados. |
| US6534482B1 (en) | 1998-05-13 | 2003-03-18 | Epimmune, Inc. | Expression vectors for stimulating an immune response and methods of using the same |
| US20020187472A1 (en) | 2001-03-09 | 2002-12-12 | Preeti Lal | Steap-related protein |
| US20030064397A1 (en) | 1998-05-22 | 2003-04-03 | Incyte Genomics, Inc. | Transmembrane protein differentially expressed in prostate and lung tumors |
| EA003398B1 (ru) | 1998-05-22 | 2003-04-24 | Дайити Фармасьютикал Ко., Лтд. | Лекарственный комплекс c полимерным носителем |
| EP1093600B1 (en) | 1998-07-08 | 2004-09-15 | E Ink Corporation | Methods for achieving improved color in microencapsulated electrophoretic devices |
| GB9818732D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Collection of compounds |
| WO2000012130A1 (en) | 1998-08-27 | 2000-03-09 | Smithkline Beecham Corporation | Rp105 agonists and antagonists |
| GB9818731D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Compounds |
| DE69925133T2 (de) | 1998-08-27 | 2006-01-19 | Spirogen Ltd., Ryde | Pyrrolobenzodiazepine |
| GB9818730D0 (en) | 1998-08-27 | 1998-10-21 | Univ Portsmouth | Collections of compounds |
| JP4689781B2 (ja) | 1998-09-03 | 2011-05-25 | 独立行政法人科学技術振興機構 | アミノ酸輸送蛋白及びその遺伝子 |
| AU5963699A (en) | 1998-10-02 | 2000-04-26 | Mcmaster University | Spliced form of (erb)b-2/neu oncogene |
| US20020119158A1 (en) | 1998-12-17 | 2002-08-29 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US6962980B2 (en) | 1999-09-24 | 2005-11-08 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US6858710B2 (en) | 1998-12-17 | 2005-02-22 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US6468546B1 (en) | 1998-12-17 | 2002-10-22 | Corixa Corporation | Compositions and methods for therapy and diagnosis of ovarian cancer |
| US20030091580A1 (en) | 2001-06-18 | 2003-05-15 | Mitcham Jennifer L. | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US20030009013A1 (en) | 1998-12-30 | 2003-01-09 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| DE69939553D1 (de) | 1998-12-30 | 2008-10-23 | Beth Israel Hospital | Charakterisierung der proteinfamilie von soc/crac kalziumkanälen |
| CZ20012587A3 (cs) | 1999-01-29 | 2002-05-15 | Corixa Corporation | Izolovaný protein, nukleová kyselina, virový vektor, farmaceutický prostředek, izolovaná populace T buněk, způsob posílení imunitní odpovědi, způsob odstranění nádorových buněk, způsob stimulace a/nebo namnoľení T buněk a způsob přípravy fúzního proteinu |
| GB9905124D0 (en) | 1999-03-05 | 1999-04-28 | Smithkline Beecham Biolog | Novel compounds |
| AU3395900A (en) | 1999-03-12 | 2000-10-04 | Human Genome Sciences, Inc. | Human lung cancer associated gene sequences and polypeptides |
| US7304126B2 (en) | 1999-05-11 | 2007-12-04 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| US6268488B1 (en) | 1999-05-25 | 2001-07-31 | Barbas, Iii Carlos F. | Prodrug activation using catalytic antibodies |
| AU4952600A (en) | 1999-06-03 | 2000-12-28 | Takeda Chemical Industries Ltd. | Screening method with the use of cd100 |
| BR0012196A (pt) | 1999-06-25 | 2002-03-19 | Genentech Inc | Método de tratamento de um tumor em mamìferos através do uso de conjugados de maitansinóide e anticorpo receptor anti-erbb e artigo industrializado |
| US6302318B1 (en) | 1999-06-29 | 2001-10-16 | General Electric Company | Method of providing wear-resistant coatings, and related articles |
| US7589172B2 (en) | 1999-07-20 | 2009-09-15 | Genentech, Inc. | PRO256 polypeptides |
| US7297770B2 (en) | 1999-08-10 | 2007-11-20 | Genentech, Inc. | PRO6496 polypeptides |
| US7294696B2 (en) | 1999-08-17 | 2007-11-13 | Genentech Inc. | PRO7168 polypeptides |
| US6909006B1 (en) | 1999-08-27 | 2005-06-21 | Spirogen Limited | Cyclopropylindole derivatives |
| CA2380355A1 (en) | 1999-09-01 | 2001-03-08 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| US20030129192A1 (en) | 1999-09-10 | 2003-07-10 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US20030206918A1 (en) | 1999-09-10 | 2003-11-06 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| US20030232056A1 (en) | 1999-09-10 | 2003-12-18 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of ovarian cancer |
| ES2309012T3 (es) | 1999-10-29 | 2008-12-16 | Genentech, Inc. | Composiciones del anticuerpo anti-psca y a sus procedimientos contra celulas cancerigenas que expresen psca. |
| AU2048901A (en) | 1999-11-29 | 2001-06-04 | Trustees Of Columbia University In The City Of New York, The | Isolation of five novel genes coding for new Fc receptors-type melanoma involved in the pathogenesis of lymphoma/melanoma |
| EP1248800A2 (en) | 1999-11-30 | 2002-10-16 | Corixa Corporation | Compositions and methods for therapy and diagnosis of breast cancer |
| EP1239866A4 (en) | 1999-12-10 | 2005-02-09 | Epimmune Inc | INDUCTION OF HER2 / NEU CELLULAR IMMUNE RESPONSES USING PEPTIDE AND NUCLEIC ACID-CONTAINING COMPOSITIONS |
| EP1244708B8 (en) | 1999-12-23 | 2007-03-07 | ZymoGenetics, Inc. | Method for treating inflammation |
| NZ502058A (en) | 1999-12-23 | 2003-11-28 | Ovita Ltd | Isolated mutated nucleic acid molecule for regulation of ovulation rate |
| AU783473B2 (en) | 1999-12-23 | 2005-10-27 | Zymogenetics Inc. | Soluble interleukin-20 receptor |
| US6610286B2 (en) | 1999-12-23 | 2003-08-26 | Zymogenetics, Inc. | Method for treating inflammation using soluble receptors to interleukin-20 |
| EP1240337B1 (en) | 1999-12-24 | 2006-08-23 | Genentech, Inc. | Methods and compositions for prolonging elimination half-times of bioactive compounds |
| US20040001827A1 (en) | 2002-06-28 | 2004-01-01 | Dennis Mark S. | Serum albumin binding peptides for tumor targeting |
| US7294695B2 (en) | 2000-01-20 | 2007-11-13 | Genentech, Inc. | PRO10268 polypeptides |
| CA2398102A1 (en) | 2000-01-21 | 2001-07-26 | Corixa Corporation | Compounds and methods for prevention and treatment of her-2/neu associated malignancies |
| US20030224379A1 (en) | 2000-01-21 | 2003-12-04 | Tang Y. Tom | Novel nucleic acids and polypeptides |
| AU2001243142A1 (en) | 2000-02-03 | 2001-08-14 | Hyseq, Inc. | Novel nucleic acids and polypeptides |
| US20030219806A1 (en) | 2000-02-22 | 2003-11-27 | Millennium Pharmaceuticals, Inc. | Novel 18607, 15603, 69318, 12303, 48000, 52920, 5433, 38554, 57301, 58324, 55063, 52991, 59914, 59921 and 33751 molecules and uses therefor |
| US20020142377A1 (en) | 2000-02-22 | 2002-10-03 | Glucksmann Maria Alexandra | 18607, a novel human calcium channel |
| US20040052793A1 (en) | 2001-02-22 | 2004-03-18 | Carter Paul J. | Caspase activivated prodrugs therapy |
| US20040002068A1 (en) | 2000-03-01 | 2004-01-01 | Corixa Corporation | Compositions and methods for the detection, diagnosis and therapy of hematological malignancies |
| US20040005561A1 (en) | 2000-03-01 | 2004-01-08 | Corixa Corporation | Compositions and methods for the detection, diagnosis and therapy of hematological malignancies |
| EP1261743A2 (en) | 2000-03-07 | 2002-12-04 | Hyseq, Inc. | Novel nucleic acids and polypeptides |
| AU2001249411B2 (en) | 2000-03-24 | 2007-02-15 | Fahri Saatcioglu | Novel prostate-specific or testis-specific nucleic acid molecules, polypeptides,and diagnostic and therapeutic methods |
| WO2004043361A2 (en) | 2002-11-08 | 2004-05-27 | Genentech, Inc. | Compositions and methods for the treatment of natural killer cell related diseases |
| AU2001250412A1 (en) | 2000-03-31 | 2001-10-08 | Ipf Pharmaceuticals Gmbh | Diagnostic and medicament for analysing the cell surface proteome of tumour and inflammatory cells and for treating tumorous and inflammatory diseases, preferably using specific chemokine receptor analysis and the chemokine receptor-ligand interaction |
| US7279294B2 (en) | 2000-04-03 | 2007-10-09 | The United States Of America As Represented By The Secretary, Dept. Of Health And Human Services, Nih | Tumor markers in ovarian cancer |
| CN1420929A (zh) | 2000-04-07 | 2003-05-28 | 阿瑞那制药公司 | 非内源性组成型活化的已知g蛋白偶联受体 |
| US20030119115A1 (en) | 2000-05-17 | 2003-06-26 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| WO2001088133A2 (en) | 2000-05-18 | 2001-11-22 | Lexicon Genetics Incorporated | Human semaphorin homologs and polynucleotides encoding the same |
| AU2001274888A1 (en) | 2000-05-19 | 2001-12-03 | Human Genome Sciences, Inc. | Nucleic acids, proteins, and antibodies |
| AU2001275437A1 (en) | 2000-06-09 | 2001-12-17 | Idec Pharmaceuticals Corporation | Gene targets and ligands that bind thereto for treatment and diagnosis of ovarian carcinomas |
| WO2001098351A2 (en) | 2000-06-16 | 2001-12-27 | Incyte Genomics, Inc. | G-protein coupled receptors |
| EP1383892A2 (en) | 2000-06-30 | 2004-01-28 | Incyte Genomics, Inc. | Human extracellular matrix and cell adhesion polypeptides |
| EP1301524A1 (en) | 2000-06-30 | 2003-04-16 | Human Genome Sciences, Inc. | B7-like polynucleotides, polypeptides, and antibodies |
| WO2002002624A2 (en) | 2000-06-30 | 2002-01-10 | Amgen, Inc. | B7-like molecules and uses thereof |
| WO2002006339A2 (en) | 2000-07-03 | 2002-01-24 | Curagen Corporation | Proteins and nucleic acids encoding same |
| US20040044179A1 (en) | 2000-07-25 | 2004-03-04 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| US6891030B2 (en) | 2000-07-27 | 2005-05-10 | Mayo Foundation For Medical Education And Research | T-cell immunoregulatory molecule |
| DE60134178D1 (de) | 2000-07-28 | 2008-07-03 | Ulrich Wissenbach | Trp8 krebsmarker |
| US7229623B1 (en) | 2000-08-03 | 2007-06-12 | Corixa Corporation | Her-2/neu fusion proteins |
| EP1366153A2 (en) | 2000-08-14 | 2003-12-03 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of her-2/neu-associated malignancies |
| WO2002013847A2 (en) | 2000-08-14 | 2002-02-21 | Corixa Corporation | Methods for diagnosis and therapy of hematological and virus-associated malignancies |
| CA2420140A1 (en) | 2000-08-24 | 2002-02-28 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of tumor |
| GB0020953D0 (en) | 2000-08-24 | 2000-10-11 | Smithkline Beecham Biolog | Vaccine |
| EP1346040A2 (en) | 2000-09-11 | 2003-09-24 | Nuvelo, Inc. | Novel nucleic acids and polypeptides |
| US6613567B1 (en) | 2000-09-15 | 2003-09-02 | Isis Pharmaceuticals, Inc. | Antisense inhibition of Her-2 expression |
| US7582293B2 (en) | 2000-09-15 | 2009-09-01 | Genentech, Inc. | Anti-PRO6308 antibodies |
| ATE333888T1 (de) | 2000-09-15 | 2006-08-15 | Zymogenetics Inc | Verwendung eines polypeptids, welches die extrazelluläre domäne von il-20ra und il-20rb enthält, zur behandlung von entzündungen |
| UA83458C2 (uk) | 2000-09-18 | 2008-07-25 | Байоджен Айдек Ма Інк. | Виділений поліпептид baff-r (рецептор фактора активації в-клітин сімейства tnf) |
| NZ525380A (en) | 2000-09-18 | 2008-06-30 | Biogen Idec Inc | Non-fucosylated forms of Cripto and their use as tumor blocking agents |
| WO2002030894A2 (en) | 2000-09-19 | 2002-04-18 | Taiho Pharmaceutical Co., Ltd. | Compositions and methods of the use thereof achiral analogues of cc-1065 and the duocarmycins |
| EP1474528A4 (en) | 2000-10-13 | 2006-06-14 | Protein Design Labs Inc | METHODS OF DIAGNOSIS OF PROSTATE CANCER, COMPOSITIONS AND METHODS OF EXAMINING PROSTATE CANCER MODULATORS |
| ES2329012T3 (es) | 2000-11-07 | 2009-11-20 | Zymogenetics, Inc. | Receptor del factor de necrosis tumoral humano. |
| US20020150573A1 (en) | 2000-11-10 | 2002-10-17 | The Rockefeller University | Anti-Igalpha-Igbeta antibody for lymphoma therapy |
| US20040018194A1 (en) | 2000-11-28 | 2004-01-29 | Francisco Joseph A. | Recombinant anti-CD30 antibodies and uses thereof |
| WO2002061087A2 (en) | 2000-12-19 | 2002-08-08 | Lifespan Biosciences, Inc. | Antigenic peptides, such as for g protein-coupled receptors (gpcrs), antibodies thereto, and systems for identifying such antigenic peptides |
| WO2002054940A2 (en) | 2001-01-12 | 2002-07-18 | University Of Medicine & Dentistry Of New Jersey | Bone morphogenetic protein-2 in the treatment and diagnosis of cancer |
| US20030119126A1 (en) | 2001-01-16 | 2003-06-26 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| US20030119125A1 (en) | 2001-01-16 | 2003-06-26 | Genentech, Inc. | Secreted and transmembrane polypeptides and nucleic acids encoding the same |
| US7754208B2 (en) | 2001-01-17 | 2010-07-13 | Trubion Pharmaceuticals, Inc. | Binding domain-immunoglobulin fusion proteins |
| JP2005503760A (ja) | 2001-01-24 | 2005-02-10 | プロテイン デザイン ラブス, インコーポレイテッド | 乳癌の診断方法、組成物および乳癌のモジュレーターのスクリーニング方法 |
| WO2002060317A2 (en) | 2001-01-30 | 2002-08-08 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of pancreatic cancer |
| WO2002064780A1 (en) | 2001-02-12 | 2002-08-22 | Bionomics Limited | Dna sequences for human tumour suppressor genes |
| AU2002258518A1 (en) | 2001-03-14 | 2002-09-24 | Millennium Pharmaceuticals, Inc. | Nucleic acid molecules and proteins for the identification, assessment, prevention, and therapy of ovarian cancer |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| US20040236091A1 (en) | 2001-03-28 | 2004-11-25 | Chicz Roman M. | Translational profiling |
| US6362331B1 (en) | 2001-03-30 | 2002-03-26 | Council Of Scientific And Industrial Research | Process for the preparation of antitumor agents |
| WO2003008537A2 (en) | 2001-04-06 | 2003-01-30 | Mannkind Corporation | Epitope sequences |
| US6820011B2 (en) | 2001-04-11 | 2004-11-16 | The Regents Of The University Of Colorado | Three-dimensional structure of complement receptor type 2 and uses thereof |
| CA2443617C (en) | 2001-04-17 | 2013-12-10 | The Board Of Trustees Of The University Of Arkansas | Repeat sequences of the ca125 gene and their use for diagnostic and therapeutic interventions |
| AU2002309583A1 (en) | 2001-04-18 | 2002-11-05 | Protein Desing Labs, Inc. | Methods of diagnosis of lung cancer, compositions and methods of screening for modulators of lung cancer |
| PL207087B1 (pl) | 2001-04-26 | 2010-10-29 | Biogen | Przeciwciało, które wiąże się swoiście z białkiem Cripto, zawierająca je kompozycja farmaceutyczna i zastosowanie przeciwciała |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| JP2005504513A (ja) | 2001-05-09 | 2005-02-17 | コリクサ コーポレイション | 前立腺癌の治療及び診断のための組成物及び方法 |
| US20030078399A1 (en) | 2001-05-11 | 2003-04-24 | Sloan-Kettering Institute For Cancer Research | Nucleic acid sequence encoding ovarian antigen, CA125, and uses thereof |
| BRPI0209933B8 (pt) | 2001-05-24 | 2021-05-25 | Zymogenetics Inc | proteína de fusão, e, molécula de ácido nucleico |
| US7157558B2 (en) | 2001-06-01 | 2007-01-02 | Genentech, Inc. | Polypeptide encoded by a polynucleotide overexpresses in tumors |
| JP2005518185A (ja) | 2001-06-04 | 2005-06-23 | キュラジェン コーポレイション | 新規タンパク質およびそれをコード化する核酸 |
| AU2002314901A1 (en) | 2001-06-04 | 2002-12-16 | Eos Biotechnology, Inc. | Methods of diagnosis and treatment of androgen-dependent prostate cancer, prostate cancer undergoing androgen-withdrawal, and androgen-independent prostate cancer |
| EP1402053A4 (en) | 2001-06-05 | 2005-05-11 | Exelixis Inc | CHDS AS MODULATORS OF THE P53 PATH AND METHOD OF USE |
| DE60237917D1 (de) | 2001-06-05 | 2010-11-18 | Exelixis Inc | Gfats als modifikatoren des p53-wegs und verwendungsverfahren |
| US7235358B2 (en) | 2001-06-08 | 2007-06-26 | Expression Diagnostics, Inc. | Methods and compositions for diagnosing and monitoring transplant rejection |
| WO2002101075A2 (en) | 2001-06-13 | 2002-12-19 | Millennium Pharmaceuticals, Inc. | Novel genes, compositions, kits, and methods for identification, assessment, prevention, and therapy of cervical cancer |
| WO2002102235A2 (en) | 2001-06-18 | 2002-12-27 | Eos Biotechnology Inc. | Methods of diagnosis of ovarian cancer, compositions and methods of screening for modulators of ovarian cancer |
| US7189507B2 (en) | 2001-06-18 | 2007-03-13 | Pdl Biopharma, Inc. | Methods of diagnosis of ovarian cancer, compositions and methods of screening for modulators of ovarian cancer |
| WO2003004989A2 (en) | 2001-06-21 | 2003-01-16 | Millennium Pharmaceuticals, Inc. | Compositions, kits, and methods for identification, assessment, prevention, and therapy of breast cancer |
| US20030108958A1 (en) | 2001-06-28 | 2003-06-12 | Rene De Waal Malefyt | Biological activity of AK155 |
| WO2003004529A2 (en) | 2001-07-02 | 2003-01-16 | Licentia Ltd. | Ephrin-tie receptor materials and methods |
| US20040076955A1 (en) | 2001-07-03 | 2004-04-22 | Eos Biotechnology, Inc. | Methods of diagnosis of bladder cancer, compositions and methods of screening for modulators of bladder cancer |
| WO2003003984A2 (en) | 2001-07-05 | 2003-01-16 | Curagen Corporation | Novel proteins and nucleic acids encoding same |
| WO2003055439A2 (en) | 2001-07-18 | 2003-07-10 | The Regents Of The University Of California | Her2/neu target antigen and use of same to stimulate an immune response |
| WO2003009814A2 (en) | 2001-07-25 | 2003-02-06 | Millennium Pharmaceuticals, Inc. | Novel genes, compositions, kits, and methods for identification, assessment, prevention, and therapy of prostate cancer |
| BR0211614A (pt) | 2001-08-03 | 2006-10-31 | Genentech Inc | polipeptìdeo tacis e br3 e empregos dos mesmos |
| AU2002324700A1 (en) | 2001-08-14 | 2003-03-03 | Bayer Ag | Nucleic acid and amino acid sequences involved in pain |
| US20030092013A1 (en) | 2001-08-16 | 2003-05-15 | Vitivity, Inc. | Diagnosis and treatment of vascular disease |
| WO2003018621A2 (en) | 2001-08-23 | 2003-03-06 | Oxford Biomedica (Uk) Limited | Genes |
| US6902930B2 (en) | 2001-08-29 | 2005-06-07 | Vanderbilt University | Human Mob-5 (IL-24) receptors and uses thereof |
| US20030124579A1 (en) | 2001-09-05 | 2003-07-03 | Eos Biotechnology, Inc. | Methods of diagnosis of ovarian cancer, compositions and methods of screening for modulators of ovarian cancer |
| EP2287186B1 (en) | 2001-09-06 | 2014-12-31 | Agensys, Inc. | Nucleic acid and corresponding protein entitled STEAP-1 useful in treatment and detection of cancer |
| AU2002330039A1 (en) | 2001-09-17 | 2003-04-01 | Eos Biotechnology, Inc. | Methods of diagnosis of cancer compositions and methods of screening for modulators of cancer |
| US20050004017A1 (en) | 2001-09-18 | 2005-01-06 | Yuval Reiss | Methods and compositions for treating hcap associated diseases |
| DE60238143D1 (de) | 2001-09-18 | 2010-12-09 | Genentech Inc | Zusammensetzungen und verfahren für die diagnose von tumoren |
| CA2460621A1 (en) | 2001-09-19 | 2003-03-27 | Nuvelo, Inc. | Novel nucleic acids and polypeptides |
| US7091186B2 (en) | 2001-09-24 | 2006-08-15 | Seattle Genetics, Inc. | p-Amidobenzylethers in drug delivery agents |
| WO2003026577A2 (en) | 2001-09-24 | 2003-04-03 | Seattle Genetics, Inc. | P-amidobenzylethers in drug delivery agents |
| US20030077644A1 (en) | 2001-09-28 | 2003-04-24 | Bing Yang | Diagnosis and treatment of diseases caused by mutations in CD72 |
| AU2002362454A1 (en) | 2001-10-03 | 2003-04-14 | Origene Technologies, Inc. | Regulated breast cancer genes |
| US20050130117A1 (en) | 2001-10-03 | 2005-06-16 | Davis Cong L. | Modulators of lymphocyte activation and migration |
| US20050123925A1 (en) | 2002-11-15 | 2005-06-09 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of tumor |
| WO2003034984A2 (en) | 2001-10-19 | 2003-05-01 | Genentech, Inc. | Compositions and methods for the diagnosis and treatment of inflammatory bowel disorders |
| CA2465268A1 (en) | 2001-10-24 | 2003-05-01 | National Jewish Medical And Research Center | Three-dimensional structures of tall-1 and its cognate receptors and modified proteins and methods related thereto |
| KR100996226B1 (ko) | 2001-10-31 | 2010-11-24 | 유니버시티 오브 노스 텍사스 헬스 사이언스 센터 | 뼈 형태발생 단백질(bmp), bmp 수용체 및 bmp결합 단백질 및 녹내장 진단 및 치료에서 그의 용도 |
| US20030232350A1 (en) | 2001-11-13 | 2003-12-18 | Eos Biotechnology, Inc. | Methods of diagnosis of cancer, compositions and methods of screening for modulators of cancer |
| WO2003042661A2 (en) | 2001-11-13 | 2003-05-22 | Protein Design Labs, Inc. | Methods of diagnosis of cancer, compositions and methods of screening for modulators of cancer |
| CA2467242A1 (en) | 2001-11-20 | 2003-05-30 | Seattle Genetics, Inc. | Treatment of immunological disorders using anti-cd30 antibodies |
| AU2002339691A1 (en) | 2001-11-29 | 2003-06-10 | Genset | Agonists and antagonists of prolixin for the treatment of metabolic disorders |
| AU2002349784A1 (en) | 2001-12-03 | 2003-06-17 | Asahi Kasei Pharma Corporation | Nf-kappab activating genes |
| AU2002359568B2 (en) | 2001-12-03 | 2008-02-21 | Alexion Pharmaceuticals, Inc. | Hybrid antibodies |
| AU2002366951A1 (en) | 2001-12-10 | 2003-07-09 | Nuvelo,Inc. | Novel nucleic acids and polypeptides |
| JP4360915B2 (ja) | 2002-01-07 | 2009-11-11 | ユーロスクリーン・ソシエテ・アノニム | Gタンパク質共役受容体gpr43のリガンドおよびその使用 |
| US20030134790A1 (en) | 2002-01-11 | 2003-07-17 | University Of Medicine And Dentistry Of New Jersey | Bone Morphogenetic Protein-2 And Bone Morphogenetic Protein-4 In The Treatment And Diagnosis Of Cancer |
| EP1467263A4 (en) | 2002-01-16 | 2009-12-16 | Japan Science & Tech Agency | HOLOGRAPHY REPRODUCTION DEVICE FOR MOVABLE IMAGES AND HOLOGRAPHY REPRODUCTION DEVICE FOR MOBILE COLOR PICTURES |
| US7452675B2 (en) | 2002-01-25 | 2008-11-18 | The Queen's Medical Center | Methods of screening for TRPM4b modulators |
| WO2003072736A2 (en) | 2002-02-21 | 2003-09-04 | Duke University | Reagents and treatment methods for autoimmune diseases |
| EP1575480A4 (en) | 2002-02-22 | 2008-08-06 | Genentech Inc | COMPOSITIONS AND METHODS OF TREATING IMMUNE-INFLAMMATORY DISEASES |
| AU2003213606A1 (en) | 2002-03-01 | 2003-10-13 | Exelixis, Inc. | MP53s AS MODIFIERS OF THE p53 PATHWAY AND METHODS OF USE |
| US20050287147A1 (en) | 2002-05-15 | 2005-12-29 | Reinhard Ebner | Cancer-linked gene as target for chemotherapy |
| US6660856B2 (en) | 2002-03-08 | 2003-12-09 | Kaohsiung Medical University | Synthesis of pyrrolo[2,1-c][1,4]benzodiazepine analogues |
| EP2258712A3 (en) | 2002-03-15 | 2011-05-04 | Multicell Immunotherapeutics, Inc. | Compositions and Methods to Initiate or Enhance Antibody and Major-histocompatibility Class I or Class II-restricted T Cell Responses by Using Immunomodulatory, Non-coding RNA Motifs |
| WO2004000997A2 (en) | 2002-03-19 | 2003-12-31 | Curagen Corporation | Therapeutic polypeptides, nucleic acids encoding same, and methods of use |
| WO2003081210A2 (en) | 2002-03-21 | 2003-10-02 | Sunesis Pharmaceuticals, Inc. | Identification of kinase inhibitors |
| JP2005520566A (ja) | 2002-03-22 | 2005-07-14 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | Cripto特異的抗体 |
| US7193069B2 (en) | 2002-03-22 | 2007-03-20 | Research Association For Biotechnology | Full-length cDNA |
| JP2005521429A (ja) | 2002-03-25 | 2005-07-21 | ユーエービー リサーチ ファウンデーション | Fcレセプターホモログ、試薬およびこれらの使用 |
| AU2003222103A1 (en) | 2002-03-28 | 2003-10-13 | Idec Pharmaceuticals Corporation | Novel gene targets and ligands that bind thereto for treatment and diagnosis of colon carcinomas |
| US20030194704A1 (en) | 2002-04-03 | 2003-10-16 | Penn Sharron Gaynor | Human genome-derived single exon nucleic acid probes useful for gene expression analysis two |
| EP1572916A4 (en) | 2002-04-05 | 2007-05-30 | Agensys Inc | NUCLEIC ACID AND CORRESPONDING PROTEIN WITH THE NAME 98P4B6, SUITABLE FOR THE TREATMENT AND DETECTION OF CANCER |
| US20040101874A1 (en) | 2002-04-12 | 2004-05-27 | Mitokor Inc. | Targets for therapeutic intervention identified in the mitochondrial proteome |
| JP2005536190A (ja) | 2002-04-16 | 2005-12-02 | ジェネンテック・インコーポレーテッド | 腫瘍の診断と治療のための組成物と方法 |
| WO2003089904A2 (en) | 2002-04-17 | 2003-10-30 | Baylor College Of Medicine | Aib1 as a prognostic marker and predictor of resistance to encocrine therapy |
| AU2003228869A1 (en) | 2002-05-03 | 2003-11-17 | Incyte Corporation | Transporters and ion channels |
| WO2003101388A2 (en) | 2002-05-30 | 2003-12-11 | Bristol-Myers Squibb Company | Human solute carrier family 7 member 11 (hslc7a11) |
| WO2003101283A2 (en) | 2002-06-04 | 2003-12-11 | Incyte Corporation | Diagnostics markers for lung cancer |
| AU2003239969A1 (en) | 2002-06-04 | 2003-12-19 | Avalon Pharmaceuticals, Inc. | Cancer-linked gene as target for chemotherapy |
| EP1513934B1 (en) | 2002-06-06 | 2011-03-02 | Oncotherapy Science, Inc. | Genes and polypeptides relating to human colon cancers |
| AU2003242633A1 (en) | 2002-06-06 | 2003-12-22 | Molecular Engines Laboratories | Dudulin genes, non-human animal model: uses in human hematological disease |
| CA2488629A1 (en) | 2002-06-07 | 2003-12-18 | Avalon Pharmaceuticals, Inc | Cancer-linked gene as target for chemotherapy |
| AU2003245441A1 (en) | 2002-06-12 | 2003-12-31 | Avalon Pharmaceuticals, Inc. | Cancer-linked gene as target for chemotherapy |
| US20040249130A1 (en) | 2002-06-18 | 2004-12-09 | Martin Stanton | Aptamer-toxin molecules and methods for using same |
| US20040022727A1 (en) | 2002-06-18 | 2004-02-05 | Martin Stanton | Aptamer-toxin molecules and methods for using same |
| EP1539218A4 (en) | 2002-06-20 | 2007-08-22 | Univ California | COMPOSITIONS AND METHODS FOR MODULATING LYMPHOCYTE ACTIVITY |
| EP2365004B1 (en) | 2002-06-21 | 2016-01-06 | Johns Hopkins University School of Medicine | Membrane associated tumor endothelium markers |
| DE10229834A1 (de) | 2002-07-03 | 2004-01-29 | Zinser Textilmaschinen Gmbh | Streckwerk für Spinnmaschinen mit nachgeordneter Verdichtungsvorrichtung |
| WO2004009622A2 (en) | 2002-07-19 | 2004-01-29 | Cellzome Ag | Protein complexes of cellular networks underlying the development of cancer and other diseases |
| WO2004011611A2 (en) | 2002-07-25 | 2004-02-05 | Genentech, Inc. | Taci antibodies and uses thereof |
| US20050180972A1 (en) | 2002-07-31 | 2005-08-18 | Wahl Alan F. | Anti-CD20 antibody-drug conjugates for the treatment of cancer and immune disorders |
| DK1545613T3 (da) | 2002-07-31 | 2011-11-14 | Seattle Genetics Inc | Auristatinkonjugater og deres anvendelse til behandling af cancer, en autoimmun sygdom eller en infektiøs sygdom |
| WO2004015426A1 (en) | 2002-08-06 | 2004-02-19 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with human cxc chemokine receptor 5(cxcr5) |
| JP2004121218A (ja) | 2002-08-06 | 2004-04-22 | Jenokkusu Soyaku Kenkyusho:Kk | 気管支喘息または慢性閉塞性肺疾患の検査方法 |
| MXPA05001933A (es) | 2002-08-19 | 2005-04-28 | Genentech Inc | Composiciones y metodos para el diagnostico y tratamiento de tumores. |
| AU2003278725A1 (en) | 2002-08-27 | 2004-03-19 | Bristol-Myers Squibb Company | Polynucleotide predictor set for identifying protein tyrosine kinase modulators |
| WO2004020595A2 (en) | 2002-08-29 | 2004-03-11 | Five Prime Therapeutics, Inc. | Novel human polypeptides encoded by polynucleotides |
| AU2003256038A1 (en) | 2002-08-30 | 2004-03-19 | Ramot At Tel Aviv University Ltd. | Self-immolative dendrimers releasing many active moieties upon a single activating event |
| AU2002951346A0 (en) | 2002-09-05 | 2002-09-26 | Garvan Institute Of Medical Research | Diagnosis of ovarian cancer |
| AU2003270311A1 (en) | 2002-09-06 | 2004-03-29 | Mannkind Corporation | Epitope sequences |
| EP1581648A2 (en) | 2002-09-09 | 2005-10-05 | Nura, Inc. | G protein coupled receptors and uses thereof |
| JP2004113151A (ja) | 2002-09-27 | 2004-04-15 | Sankyo Co Ltd | 癌遺伝子及びその用途 |
| CA2500978A1 (en) | 2002-10-03 | 2004-04-15 | Mcgill University | Antibodies and cyclic peptides which bind cea (carcinoembryonic antigen) and their use as cancer therapeutics |
| AU2003288918A1 (en) | 2002-10-04 | 2004-05-04 | Van Andel Research Institute | Molecular sub-classification of kidney tumors and the discovery of new diagnostic markers |
| US20040138269A1 (en) | 2002-10-11 | 2004-07-15 | Sugen, Inc. | Substituted pyrroles as kinase inhibitors |
| EP1578940A4 (en) | 2002-11-13 | 2007-12-12 | Genentech Inc | METHOD AND COMPOSITIONS FOR DYSPLASED DIAGNOSIS |
| GB0226593D0 (en) | 2002-11-14 | 2002-12-24 | Consultants Ltd | Compounds |
| WO2004043493A1 (en) | 2002-11-14 | 2004-05-27 | Syntarga B.V. | Prodrugs built as multiple self-elimination-release spacers |
| AU2003298650B2 (en) | 2002-11-15 | 2010-03-11 | Musc Foundation For Research Development | Complement receptor 2 targeted complement modulators |
| WO2004045553A2 (en) | 2002-11-15 | 2004-06-03 | The Board Of Trustees Of The University Of Arkansas | Ca125 gene and its use for diagnostic and therapeutic interventions |
| WO2004046342A2 (en) | 2002-11-20 | 2004-06-03 | Biogen Idec Inc. | Novel gene targets and ligands that bind thereto for treatment and diagnosis of carcinomas |
| EP2410332A1 (en) | 2002-11-21 | 2012-01-25 | The University Of Utah | Method for identifying purinergic modulators of the olfactory system |
| US20040253606A1 (en) | 2002-11-26 | 2004-12-16 | Protein Design Labs, Inc. | Methods of detecting soft tissue sarcoma, compositions and methods of screening for soft tissue sarcoma modulators |
| EP1581641A4 (en) | 2002-12-06 | 2006-11-15 | Diadexus Inc | COMPOSITIONS, SPREADING VARIATIONS AND METHODS RELATED TO OVARAL SPECIFIC GENES AND PROTEINS |
| JP2004198419A (ja) | 2002-12-13 | 2004-07-15 | Bayer Healthcare Llc | Timp1を用いた検出方法 |
| WO2004058171A2 (en) | 2002-12-20 | 2004-07-15 | Protein Design Labs, Inc. | Antibodies against gpr64 and uses thereof |
| AU2003299819A1 (en) | 2002-12-23 | 2004-07-22 | Human Genome Sciences, Inc. | Neutrokine-alpha conjugate, neutrokine-alpha complex, and uses thereof |
| CA2512536A1 (en) | 2003-01-08 | 2004-07-29 | Bristol-Myers Squibb Company | Biomarkers and methods for determining sensitivity to epidermal growth factor receptor modulators |
| WO2004063355A2 (en) | 2003-01-10 | 2004-07-29 | Protein Design Labs, Inc. | Novel methods of diagnosis of metastatic cancer, compositions and methods of screening for modulators of matastatic cancer |
| US20050227301A1 (en) | 2003-01-10 | 2005-10-13 | Polgen | Cell cycle progression proteins |
| WO2004065577A2 (en) | 2003-01-14 | 2004-08-05 | Bristol-Myers Squibb Company | Polynucleotides and polypeptides associated with the nf-kb pathway |
| EP1583821A4 (en) | 2003-01-15 | 2007-07-18 | Millennium Pharm Inc | METHODS AND COMPOSITIONS FOR TREATING UROLOGICAL DISORDERS USING GENES 44390, 54181, 211, 5687, 884, 1405, 636, 4421, 5410, 30905, 2045, 16405, 18560, 2047, 33751, 52872, 14063, 20739, 32544, 43239, 44373, 51164, 53010, 16852, 1587, 2207, 22245, 2387, 52908, 69112, 14990, 18547, 115, 579 |
| AU2004213432A1 (en) | 2003-02-14 | 2004-09-02 | Sagres Discovery, Inc. | Novel therapeutic targets in cancer |
| US20030224411A1 (en) | 2003-03-13 | 2003-12-04 | Stanton Lawrence W. | Genes that are up- or down-regulated during differentiation of human embryonic stem cells |
| ATE421967T1 (de) | 2003-03-31 | 2009-02-15 | Council Scient Ind Res | Nichtvernetzende pyrroloä2,1-cüä1, 4übenzodiazepine als potentielle antitumor- agentien und ihre herstellung |
| GB0321295D0 (en) | 2003-09-11 | 2003-10-15 | Spirogen Ltd | Synthesis of protected pyrrolobenzodiazepines |
| GB0416511D0 (en) | 2003-10-22 | 2004-08-25 | Spirogen Ltd | Pyrrolobenzodiazepines |
| WO2005040170A2 (en) | 2003-10-22 | 2005-05-06 | Government Of The United States Of America, Represented By The Secretary, Department Of Health And Human Services | Pyrrolobenzodiazepine derivatives, compositions comprising the same and methods related thereto |
| EP2478912B1 (en) | 2003-11-06 | 2016-08-31 | Seattle Genetics, Inc. | Auristatin conjugates with anti-HER2 or anti-CD22 antibodies and their use in therapy |
| EP1720908A2 (en) | 2004-02-17 | 2006-11-15 | Absalus, Inc. | Super-humanized antibodies against respiratory syncytial virus |
| CA2556752C (en) | 2004-02-23 | 2016-02-02 | Genentech, Inc. | Heterocyclic self-immolative linkers and conjugates |
| WO2005085251A1 (en) | 2004-03-01 | 2005-09-15 | Spirogen Limited | 11-hydroxy-5h-pyrrolo[2,1-c][1,4]benzodiazepin-5-one derivatives as key intermediates for the preparation of c2 substituted pyrrolobenzodiazepines |
| GB0404577D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0404574D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Amino acids |
| GB0404578D0 (en) | 2004-03-01 | 2004-04-07 | Spirogen Ltd | Pyrrolobenzodiazepines |
| DE102004010943A1 (de) | 2004-03-03 | 2005-09-29 | Degussa Ag | Verfahren zur Herstellung von N-geschützten 4-Ketprolinderivaten |
| WO2005085260A1 (en) | 2004-03-09 | 2005-09-15 | Spirogen Limited | Pyrrolobenzodiazepines |
| FR2869231B1 (fr) | 2004-04-27 | 2008-03-14 | Sod Conseils Rech Applic | Composition therapeutique contenant au moins un derive de la pyrrolobenzodiazepine et la fludarabine |
| EP1750614B1 (en) | 2004-05-11 | 2017-09-27 | The General Hospital Corporation | Methods for making oxidation resistant polymeric material |
| GB0410725D0 (en) | 2004-05-13 | 2004-06-16 | Spirogen Ltd | Pyrrolobenzodiazepine therapeutic agents |
| DK1791565T3 (en) | 2004-09-23 | 2016-08-01 | Genentech Inc | Cysteingensplejsede antibodies and conjugates |
| JP2008521828A (ja) | 2004-11-29 | 2008-06-26 | シアトル ジェネティックス, インコーポレイテッド | 操作された抗体およびイムノコンジュゲート |
| US7849909B2 (en) | 2004-12-24 | 2010-12-14 | Showa Denko K.K. | Production method of thermoelectric semiconductor alloy, thermoelectric conversion module and thermoelectric power generating device |
| AU2006210724A1 (en) | 2005-02-03 | 2006-08-10 | Antitope Limited | Human antibodies and proteins |
| JP5122441B2 (ja) | 2005-04-19 | 2013-01-16 | シアトル ジェネティックス, インコーポレイテッド | ヒト化抗cd70結合剤およびその使用 |
| US7612062B2 (en) | 2005-04-21 | 2009-11-03 | Spirogen Limited | Pyrrolobenzodiazepines |
| GB0508084D0 (en) | 2005-04-21 | 2005-06-01 | Spirogen Ltd | Pyrrolobenzodiazepines |
| US20070154906A1 (en) | 2005-10-05 | 2007-07-05 | Spirogen Ltd. | Methods to identify therapeutic candidates |
| US8637664B2 (en) | 2005-10-05 | 2014-01-28 | Spirogen Sarl | Alkyl 4- [4- (5-oxo-2,3,5, 11a-tetrahydo-5H-pyrrolo [2, 1-c] [1,4] benzodiazepine-8-yloxy)-butyrylamino]-1H-pyrrole-2-carboxylate derivatives and related compounds for the treatment of a proliferative disease |
| SI1934174T1 (sl) | 2005-10-07 | 2011-08-31 | Exelixis Inc | Inhibitorji MEK in postopki za njihovo uporabo |
| RS52060B (sr) | 2006-01-25 | 2012-04-30 | Sanofi | Citotoksični agensi koji sadrže nove derivate tomaimicina |
| MX2009000709A (es) | 2006-07-18 | 2009-02-04 | Sanofi Aventis | Anticuerpo antagonista contra epha2 para el tratamiento de cancer. |
| PL2383297T3 (pl) | 2006-08-14 | 2013-06-28 | Xencor Inc | Zoptymalizowane przeciwciała ukierunkowane na CD19 |
| US20080112961A1 (en) | 2006-10-09 | 2008-05-15 | Macrogenics, Inc. | Identification and Engineering of Antibodies with Variant Fc Regions and Methods of Using Same |
| EP1914242A1 (en) | 2006-10-19 | 2008-04-23 | Sanofi-Aventis | Novel anti-CD38 antibodies for the treatment of cancer |
| WO2008050140A2 (en) | 2006-10-27 | 2008-05-02 | Spirogen Limited | Compounds for treatment of parasitic infection |
| PT2099823E (pt) | 2006-12-01 | 2014-12-22 | Seattle Genetics Inc | Agentes de ligação ao alvo variantes e suas utilizações |
| CL2008001334A1 (es) | 2007-05-08 | 2008-09-22 | Genentech Inc | Anticuerpo anti-muc16 disenado con cisteina; conjugado que lo comprende; metodo de produccion; formulacion farmaceutica que lo comprende; y su uso para tratar el cancer. |
| SI2019104T1 (sl) | 2007-07-19 | 2013-12-31 | Sanofi | Citotoksična sredstva, ki obsegajo nove tomaimicinske derivate, in njihova terapevtska uporaba |
| NZ584514A (en) | 2007-10-19 | 2012-07-27 | Genentech Inc | Cysteine engineered anti-tenb2 antibodies and antibody drug conjugates |
| GB0722088D0 (en) | 2007-11-09 | 2007-12-19 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0722087D0 (en) | 2007-11-09 | 2007-12-19 | Spirogen Ltd | Polyamides |
| DK2265283T3 (da) | 2008-03-18 | 2014-10-20 | Seattle Genetics Inc | Auristatin-lægemiddel-linker-konjugater |
| EP2109244A1 (de) | 2008-04-09 | 2009-10-14 | Siemens Aktiengesellschaft | Verfahren zur sicherheitsgerichteten Übertragung Sicherheitsschaltgerät und Kontrolleinheit |
| NZ610239A (en) | 2008-04-30 | 2014-11-28 | Immunogen Inc | Cross-linkers and their uses |
| CA2727278A1 (en) | 2008-06-16 | 2010-01-21 | Immunogen, Inc. | Novel synergistic effects |
| GB0813432D0 (en) | 2008-07-22 | 2008-08-27 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0819097D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| GB0819095D0 (en) | 2008-10-17 | 2008-11-26 | Spirogen Ltd | Pyrrolobenzodiazepines |
| RU2683325C2 (ru) | 2009-02-05 | 2019-03-28 | Иммьюноджен, Инк. | Новые производные бензодиазепина |
| CN102421800A (zh) | 2009-02-23 | 2012-04-18 | 格兰马克药品股份有限公司 | 结合cd19的人源化抗体及其用途 |
| JP5382792B2 (ja) | 2009-08-14 | 2014-01-08 | 独立行政法人理化学研究所 | 光2次非線形薄膜における1次及び2次光感受率異方性同時測定方法、当該方法を実行する装置及び当該方法をコンピュータに実行させるプログラム |
| FR2949469A1 (fr) | 2009-08-25 | 2011-03-04 | Sanofi Aventis | Derives anticancereux, leur preparation et leur application en therapeutique |
| WO2011028811A2 (en) | 2009-09-01 | 2011-03-10 | Abbott Laboratories | Dual variable domain immunoglobulins and uses thereof |
| US20110070227A1 (en) | 2009-09-18 | 2011-03-24 | Anna-Marie Novotney-Barry | Treatment of Autoimmune and Inflammatory Diseases |
| JP2013505944A (ja) | 2009-09-24 | 2013-02-21 | シアトル ジェネティックス, インコーポレイテッド | Dr5リガンド薬物結合体 |
| US9040526B2 (en) | 2010-02-09 | 2015-05-26 | Bristol-Myers Squibb Company | Benzylpyrrolidinone derivatives as modulators of chemokine receptor activity |
| KR20130009760A (ko) | 2010-02-10 | 2013-01-23 | 이뮤노젠 아이엔씨 | Cd20 항체 및 이의 용도 |
| US9175086B2 (en) | 2010-02-10 | 2015-11-03 | Immunogen, Inc. | CD20 antibodies and uses thereof |
| JP5875083B2 (ja) | 2010-04-15 | 2016-03-02 | メディミューン リミテッド | 増殖性疾患治療用ピロロベンゾジアゼピン |
| WO2011130615A2 (en) | 2010-04-15 | 2011-10-20 | Dr. Reddy's Laboratories Ltd. | Preparation of lacosamide |
| GB201006340D0 (en) | 2010-04-15 | 2010-06-02 | Spirogen Ltd | Synthesis method and intermediates |
| CA2795349C (en) | 2010-04-15 | 2016-11-29 | Seattle Genetics, Inc. | Targeted pyrrolobenzodiazepine conjugates |
| SI2528625T1 (sl) | 2010-04-15 | 2013-11-29 | Spirogen Sarl | Pirolobenzodiazepini in njihovi konjugati |
| PT3056203T (pt) | 2010-04-21 | 2018-02-15 | Syntarga Bv | Conjugados de análogos de cc-1065 e ligantes bifuncionais |
| FR2963007B1 (fr) | 2010-07-26 | 2013-04-05 | Sanofi Aventis | Derives anticancereux, leur preparation et leur application therapeutique |
| WO2012064733A2 (en) | 2010-11-09 | 2012-05-18 | Medimmune, Llc | Antibody scaffold for homogenous conjugation |
| JP5826863B2 (ja) | 2011-02-15 | 2015-12-02 | イミュノジェン・インコーポレーテッド | 細胞傷害性ベンゾジアゼピン誘導体 |
| US9135118B2 (en) | 2011-03-07 | 2015-09-15 | Aptare, Inc. | System to catalog and search point-in-time instances of a file system |
| SG11201400770SA (en) | 2011-09-20 | 2014-04-28 | Spirogen Sarl | Pyrrolobenzodiazepines as unsymmetrical dimeric pbd compounds for inclusion in targeted conjugates |
| BR112014009050B1 (pt) | 2011-10-14 | 2022-06-21 | Medimmune Limited | Conjugado anticorpo-fármaco de pirrolbenzodiazepinas, composição farmacêutica que compreende o mesmo, bem como compostos de pirrolbenzodiazepinas |
| CN103997893B (zh) | 2011-10-14 | 2019-04-12 | 西雅图基因公司 | 吡咯并苯并二氮杂卓和靶向结合物 |
| BR112014009070B1 (pt) | 2011-10-14 | 2021-11-23 | Medimmune Limited | Método de síntese e intermediários úteis na preparação de pirrolobenzo-diazepinas |
| EP2751111B1 (en) | 2011-10-14 | 2017-04-26 | MedImmune Limited | Asymmetrical bis-(5H-Pyrrolo[2,1-c][1,4]benzodiazepin-5-one) derivatives for the treatment of proliferative or autoimmune diseases |
| CA2850373C (en) | 2011-10-14 | 2019-07-16 | Seattle Genetics, Inc. | Pyrrolobenzodiazepines and targeted conjugates |
| AU2012322932B2 (en) | 2011-10-14 | 2016-11-10 | Medimmune Limited | Pyrrolobenzodiazepines |
| JP2013194140A (ja) | 2012-03-19 | 2013-09-30 | Fuji Xerox Co Ltd | トナー用ポリエステル樹脂、静電荷像現像用トナー、静電荷像現像剤、トナーカートリッジ、プロセスカートリッジ、画像形成装置及び画像形成方法 |
| AU2013255612B2 (en) | 2012-04-30 | 2017-06-22 | Medimmune Limited | Pyrrolobenzodiazepines |
| ES2623042T3 (es) | 2012-04-30 | 2017-07-10 | Medimmune Limited | Pirrolobenzodiacepinas |
| US9062577B2 (en) | 2012-05-14 | 2015-06-23 | Southwest Research Institute | Diesel engine operation for fast transient response and low emissions |
| WO2013177481A1 (en) | 2012-05-25 | 2013-11-28 | Immunogen, Inc. | Benzodiazepines and conjugates thereof |
| AR091703A1 (es) | 2012-07-09 | 2015-02-25 | Genentech Inc | Anticuerpos e inmunoconjugados que comprenden anticuerpos anti-cd22 |
| IN2014DN10652A (es) | 2012-07-09 | 2015-09-11 | Genentech Inc | |
| JP2015528818A (ja) | 2012-08-02 | 2015-10-01 | ジェネンテック, インコーポレイテッド | 抗etbr抗体およびイムノコンジュゲート |
| HK1211208A1 (zh) | 2012-08-22 | 2016-05-20 | Immunogen, Inc. | 細胞毒性苯並二氮呯衍生物 |
| HRP20182129T1 (hr) | 2012-10-12 | 2019-02-08 | Adc Therapeutics Sa | Konjugati protutijelo - pirolobenzodiazepin |
| RS57694B1 (sr) | 2012-10-12 | 2018-11-30 | Adc Therapeutics Sa | Pirolobenzodiazepin - anti-psma konjugati antitela |
| MX338711B (es) | 2012-10-12 | 2016-04-28 | Medimmune Ltd | Pirrolobenzodiazepinas y conjugados de las mismas. |
| HRP20190366T1 (hr) | 2012-10-12 | 2019-04-19 | Medimmune Limited | Pirolobenzodiazepini i njihovi konjugati |
| JP6392765B2 (ja) | 2012-10-12 | 2018-09-19 | エイディーシー・セラピューティクス・エス・アーAdc Therapeutics Sa | ピロロベンゾジアゼピン−抗体結合体 |
| EP2906297B1 (en) | 2012-10-12 | 2017-12-06 | ADC Therapeutics SA | Pyrrolobenzodiazepine-antibody conjugates |
| EP2906296B1 (en) | 2012-10-12 | 2018-03-21 | ADC Therapeutics SA | Pyrrolobenzodiazepine-antibody conjugates |
| EP2906249B1 (en) | 2012-10-12 | 2018-06-27 | MedImmune Limited | Synthesis and intermediates of pyrrolobenzodiazepine derivatives for conjugation |
| SI2906252T1 (sl) * | 2012-10-12 | 2017-10-30 | Adc Therapeutics Sa | Pirolobenzodiazepin-anti-HER2 protitelesni konjugati |
| WO2014057118A1 (en) | 2012-10-12 | 2014-04-17 | Adc Therapeutics Sarl | Pyrrolobenzodiazepine-anti-cd22 antibody conjugates |
| ES2680153T3 (es) | 2012-10-12 | 2018-09-04 | Adc Therapeutics Sa | Conjugados de anticuerpos anti-PSMA-pirrolobenzodiazepinas |
| BR102013027577A2 (pt) | 2012-10-25 | 2016-03-29 | Memorialsloan Kettering Cancer Ct | método de detecção de um nível aumentado de axl ou gas6 e método de identificação de um paciente com câncer resistente a inibidor de egfr |
| CA2891280C (en) | 2012-11-24 | 2018-03-20 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
| CN105189507A (zh) | 2012-12-21 | 2015-12-23 | 斯皮罗根有限公司 | 吡咯并苯并二氮杂卓及其结合物 |
| CN110627797A (zh) | 2012-12-21 | 2019-12-31 | 麦迪穆有限责任公司 | 用于治疗增殖性和自身免疫疾病的非对称吡咯并苯并二氮杂卓二聚物 |
| ES2731681T3 (es) | 2013-02-22 | 2019-11-18 | Abbvie Stemcentrx Llc | Conjugados de anticuerpo anti-DLL3 y PBD y usos de los mismos |
| TWI680766B (zh) | 2013-03-13 | 2020-01-01 | 英商梅迪繆思有限公司 | 吡咯并苯并二氮呯及其共軛物 |
| AU2014229529B2 (en) | 2013-03-13 | 2018-02-15 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| MX362970B (es) | 2013-03-13 | 2019-02-28 | Medimmune Ltd | Pirrolobenzodiazepinas y conjugados de las mismas. |
| US20160106861A1 (en) | 2013-04-26 | 2016-04-21 | Spirogen Sarl | Axl antibody-drug conjugate and its use for the treatment of cancer |
| WO2015016253A1 (ja) | 2013-07-30 | 2015-02-05 | 東レ株式会社 | 分離膜エレメント |
| KR20160047567A (ko) | 2013-08-28 | 2016-05-02 | 스템센트알엑스 인코포레이티드 | 조작된 항-dll3 접합체 및 사용 방법 |
| NL2011583C2 (en) | 2013-10-10 | 2015-04-13 | Wwinn B V | Module, system and method for detecting acoustical failure of a sound source. |
| GB201318069D0 (en) | 2013-10-11 | 2013-11-27 | Maersk Olie & Gas | Seismic data processing method and apparatus |
| DE102013220528B4 (de) | 2013-10-11 | 2015-05-07 | Continental Automotive Gmbh | Einspritzventil und Verfahren zum Betreiben eines Einspritzventils |
| WO2015052534A1 (en) | 2013-10-11 | 2015-04-16 | Spirogen Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| GB201317982D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| EP3054983B1 (en) | 2013-10-11 | 2019-03-20 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| GB201317981D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| WO2015052535A1 (en) | 2013-10-11 | 2015-04-16 | Spirogen Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| AU2014333776B2 (en) | 2013-10-11 | 2021-01-28 | Cellectis | Methods and kits for detecting nucleic acid sequences of interest using DNA-binding protein domain |
| US20160256561A1 (en) | 2013-10-11 | 2016-09-08 | Medimmune Limited | Pyrrolobenzodiazepine-antibody conjugates |
| KR102337414B1 (ko) | 2013-12-16 | 2021-12-10 | 제넨테크, 인크. | 펩타이드 모방체 화합물 및 이의 항체-약물 컨쥬게이트 |
| WO2015112822A1 (en) | 2014-01-24 | 2015-07-30 | Kolltan Pharmaceuticals, Inc. | Antibody-drug conjugates targeting kit receptor and uses thereof |
| GB201406767D0 (en) | 2014-04-15 | 2014-05-28 | Cancer Rec Tech Ltd | Humanized anti-Tn-MUC1 antibodies anf their conjugates |
| GB201410826D0 (en) * | 2014-06-18 | 2014-07-30 | Bergenbio As | Anti-axl antibodies |
| EP3193940A1 (en) | 2014-09-10 | 2017-07-26 | Medimmune Limited | Pyrrolobenzodiazepines and conjugates thereof |
| MA40579A (fr) | 2014-09-12 | 2016-03-17 | Genentech Inc | Anticorps anti-cll-1 et immunoconjugués |
| GB201416112D0 (en) | 2014-09-12 | 2014-10-29 | Medimmune Ltd | Pyrrolobenzodiazepines and conjugates thereof |
| CN107073136A (zh) | 2014-09-17 | 2017-08-18 | 健泰科生物技术公司 | 吡咯并苯并二氮杂卓及其抗体二硫化物偶联物 |
| CN107106701B (zh) * | 2014-10-03 | 2020-11-06 | 西纳福克斯股份有限公司 | 磺酰胺接头、其缀合物及制备方法 |
| GB201506399D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Howard Philip W | Site-specific antibody-drug conjugates |
| GB201506388D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Cancer Res Technology Ltd And Howard Philip W | Site-specific antibody-drug conjugates |
| GB201506393D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Howard Philip W | Site-specific antibody-drug conjugates |
| GB201506402D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Howard Philip W | Site-specific antibody-drug conjugates |
| GB201506409D0 (en) | 2015-04-15 | 2015-05-27 | Williams David G And Berkel Patricius H C Van And Howard Philip W | Humanized anti-axl antibodies and their conjugates |
| GB201506407D0 (en) | 2015-04-15 | 2015-05-27 | Berkel Patricius H C Van And Howard Philip W | Site-specific antibody-drug conjugates |
| TW201709932A (zh) | 2015-06-12 | 2017-03-16 | 西雅圖遺傳學公司 | Cd123抗體及其共軛物 |
| EP3413920A1 (en) * | 2016-02-08 | 2018-12-19 | Synaffix B.V. | Bioconjugates containing sulfamide linkers for use in treatment |
-
2018
- 2018-02-08 RS RS20210560A patent/RS61795B1/sr unknown
- 2018-02-08 EP EP18704956.4A patent/EP3544636B1/en active Active
- 2018-02-08 PL PL18704956T patent/PL3544636T3/pl unknown
- 2018-02-08 SI SI201830277T patent/SI3544636T1/sl unknown
- 2018-02-08 JP JP2019542659A patent/JP6671555B2/ja not_active Expired - Fee Related
- 2018-02-08 MX MX2019009428A patent/MX375569B/es active IP Right Grant
- 2018-02-08 KR KR1020197023785A patent/KR102181375B1/ko active Active
- 2018-02-08 CN CN201880010619.7A patent/CN110267686B/zh not_active Expired - Fee Related
- 2018-02-08 BR BR112019016373-5A patent/BR112019016373B1/pt not_active IP Right Cessation
- 2018-02-08 KR KR1020207007470A patent/KR20200032243A/ko not_active Withdrawn
- 2018-02-08 LT LTEP18704956.4T patent/LT3544636T/lt unknown
- 2018-02-08 NZ NZ754810A patent/NZ754810A/en not_active IP Right Cessation
- 2018-02-08 SM SM20210543T patent/SMT202100543T1/it unknown
- 2018-02-08 ES ES18704956T patent/ES2871001T3/es active Active
- 2018-02-08 HR HRP20210979TT patent/HRP20210979T1/hr unknown
- 2018-02-08 UA UAA202004635A patent/UA125198C2/uk unknown
- 2018-02-08 HU HUE18704956A patent/HUE054689T2/hu unknown
- 2018-02-08 US US16/484,328 patent/US11160872B2/en not_active Expired - Fee Related
- 2018-02-08 AU AU2018217926A patent/AU2018217926B2/en active Active
- 2018-02-08 DK DK18704956.4T patent/DK3544636T3/da active
- 2018-02-08 WO PCT/EP2018/053163 patent/WO2018146189A1/en not_active Ceased
- 2018-02-08 CA CA3047683A patent/CA3047683C/en active Active
- 2018-02-08 PT PT187049564T patent/PT3544636T/pt unknown
-
2021
- 2021-06-10 CY CY20211100518T patent/CY1124402T1/el unknown
- 2021-09-24 US US17/484,467 patent/US11813335B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2871001T3 (es) | Conjugados de pirrolobenzodiazepinas y anticuerpos | |
| ES2890934T3 (es) | Conjugados de pirrolobenzodiazepinas y anticuerpos | |
| AU2015352545B2 (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| HK40056993A (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| EA040749B1 (ru) | Конъюгаты пирролобензодиазепин-антитело | |
| HK40012498A (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| HK40012498B (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| HK40012528A (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| HK40012528B (en) | Pyrrolobenzodiazepine-antibody conjugates | |
| EA039826B1 (ru) | Конъюгаты пирролобензодиазепин-антитело |