ES2895150T3 - Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas - Google Patents
Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas Download PDFInfo
- Publication number
- ES2895150T3 ES2895150T3 ES19157147T ES19157147T ES2895150T3 ES 2895150 T3 ES2895150 T3 ES 2895150T3 ES 19157147 T ES19157147 T ES 19157147T ES 19157147 T ES19157147 T ES 19157147T ES 2895150 T3 ES2895150 T3 ES 2895150T3
- Authority
- ES
- Spain
- Prior art keywords
- formulation
- concentration
- vwf
- storage
- formulations
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39591—Stabilisation, fragmentation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/36—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against blood coagulation factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/22—Immunoglobulins specific features characterized by taxonomic origin from camelids, e.g. camel, llama or dromedary
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/569—Single domain, e.g. dAb, sdAb, VHH, VNAR or nanobody®
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/94—Stability, e.g. half-life, pH, temperature or enzyme-resistance
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Diabetes (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Dermatology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Una formulación que comprende un agente de unión a factor de von Willebrand (vWF) y un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0, en la que dicho agente de unión a vWF comprende al menos un dominio variable individual de inmunoglobulina que se une a SEQ ID NO: 20, en la que dicha formulación comprende sacarosa a una concentración del 7%; y/o dicho tampón preferiblemente tiene una osmolalidad en el intervalo de 290 ± 60 mOsm/kg, más preferiblemente en el intervalo de 290 ± 20 mOsm/kg; y/o en la que dicha formulación es preferiblemente adecuada para administración parenteral, tal como una o más seleccionadas entre inyección intravenosa, inyección subcutánea, inyección intramuscular o inyección intraperitoneal; y/o en la que dicha formulación es está preferiblemente en forma líquida, liofilizada, secada por pulverización, liofilizada reconstituida o congelada; y/o en la que dicha formulación tiene: (i) menos del 5% de especies de alto peso molecular (HMW) tras el almacenamiento durante al menos 12 meses a 5ºC; y/o (ii) menos del 5% de especies de bajo peso molecular (LMW) tras el almacenamiento durante al menos 12 meses a 5ºC; y/o en la que al menos el 80%, preferiblemente al menos el 90%, más preferiblemente al menos el 95% o incluso al menos el 99% del agente de unión a vWF retiene su actividad de unión tras el almacenamiento en comparación con la actividad de unión antes del almacenamiento, dicha actividad de unión tal como se mide mediante ELISA y/o Biacore; y/o en la que dicho agente de unión tiene una concentración en el intervalo de 0,1 a 80 mg/ml.
Description
DESCRIPCIÓN
Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas
1. Campo de la invención
La presente invención se refiere a formulaciones estables tal como se definen en las reivindicaciones, que comprenden un agente de unión a factor de von Willebrand (vWF) que comprende al menos un dominio variable individual de inmunoglobulina que se une a (SEQ ID NO: 20), tal como dominios variables individuales de inmunoglobulina según las SEQ ID NO: 1-19, específicamente la s Eq ID NO: 1, es decir el Nanobody ALX-0081. La invención proporciona formulaciones que son estables en almacenamiento durante periodos de tiempo prolongados y a lo largo de un amplio intervalo de temperaturas. Las formulaciones de la invención garantizan una alta estabilidad del polipéptido, permitiendo múltiples ciclos de congelación-descongelación sin deterioro químico o físico, y proporcionan estabilidad en relación a la tensión mecánica, tal como tensión de oscilación, cizalladura o agitación. Son adecuadas para preparaciones farmacéuticas y de diagnóstico y compatibles con diluyentes farmacéuticamente aceptables, tales como solución salina, disolución de Ringer o disolución de glucosa/dextrosa. La presente invención también se refiere a métodos de preparación, a métodos para el almacenamiento y a usos de las formulaciones. La invención se refiere además a formas unitarias de dosificación, a kits y a usos médicos de las formulaciones.
2. Antecedentes de la invención
Los dominios variables individuales de inmunoglobulina, tales como los dominios VHH de camélidos, los dominios VH camelizados o los dominios VHH humanizados, representan una clase de productos terapéuticos de anticuerpos en rápido crecimiento. Por ejemplo, se han descrito dominios variables individuales de inmunoglobulina contra vWF en los documentos WO2004/015425, WO2004/062551, WO2006/074947, WO2006/122825, WO2009/115614 y WO2011/067160.
Proteínas tales como dominios variables individuales de inmunoglobulina (ISVD) normalmente deben almacenarse y transportarse entre la fabricación inicial y su uso, por ejemplo, administración a un paciente. Los procesos de transporte, fabricación, almacenamiento y suministro pueden ejercer múltiples tensiones sobre el dominio variable individual de inmunoglobulina, tales como tensiones químicas y físicas. Durante el almacenamiento pueden producirse modificaciones químicas, tales como, por ejemplo, desamidación, racemización, hidrólisis, oxidación, isomerización, eliminación beta o intercambio de disulfuro. Las tensiones físicas pueden causar desnaturalización y desplegamiento, agregación, formación de material particulado, precipitación, opalescencia o adsorción.
Sigue habiendo la necesidad de proporcionar formulaciones para dominios variables individuales de inmunoglobulina, por ejemplo tal como se definen en el presente documento, que potencian la estabilidad, preservan el agente activo frente a la tensión química y/o mecánica y, por tanto, permiten cambios de temperatura y almacenamiento sin deterioro físico o químico significativo, permanecen estables durante períodos de tiempo prolongados y/o son respetuosos con el paciente, por ejemplo, en las que el agente activo es soluble a alta concentración.
3. Sumario de la invención
Se sabe que las tensiones mencionadas anteriormente pueden afectar a la integridad fisicoquímica de los productos terapéuticos de proteínas, por ejemplo, productos terapéuticos de anticuerpos. Por ejemplo, se han descrito la agregación, desamidación y oxidación como las causas más comunes de degradación de anticuerpos (Cleland et al., 1993, Crit. Rev. Ther. Drug Carrier Systems 10, 307-377). Al mismo tiempo, es fundamental que se proporcionen formulaciones que preserven la integridad física y química de los dominios variables individuales de inmunoglobulina. Se requiere integridad química y física para su uso como, por ejemplo, agente terapéutico, y normalmente también están asociadas con la actividad biológica. Aunque el conocimiento sobre la estabilidad de las proteínas está aumentando, la optimización de las condiciones de la formulación para suprimir o minimizar completamente estas múltiples tensiones y garantizar una vida útil de almacenamiento prolongada sigue siendo un gran desafío.
Poco se sabe acerca de formulaciones adecuadas de dominios variables individuales de inmunoglobulina. El documento WO2010/077422 describe una formulación de un Nanobody de unión a TNF que comprende un lioprotector, un tensioactivo y un tampón elegido de tampón histidina y tampón Tris-HCl a un pH de entre 5.0 y 7.5. El documento US 2012/0225072 A1 describe formulaciones estables de dominios variables individuales de inmunoglobulina, y en particular dominios variables individuales de inmunoglobulina dirigidos contra CXCR4.
Se han sometido a prueba agentes de unión de vWF específicos, y en particular dominios variables individuales de inmunoglobulina con alta afinidad por vWF, tales como ALX-0081 [INN: caplacizumab], como terapia adyuvante para
pacientes con síndrome coronario agudo (SCA) sometidos a intervención coronaria percutánea (ICP) y se desarrollan como tratamiento de púrpura trombocitopénica trombótica (PTT). Se han completado ensayos clínicos de fase I con éxito y actualmente están en marcha pruebas en ensayos de fase II. Hasta ahora, ALX-0081 se ha presentado como una formulación líquida basada en fosfato que contiene 5 mg/ml del principio farmacéutico activo (API) en D-PBS, glicina 200 mM y Tween-80 al 0,02% (v/v).
Aunque esta formulación demostró ser eficaz, puede mejorarse de varias maneras. En primer lugar, la concentración actual probablemente requeriría múltiples inyecciones subcutáneas (suponiendo que el volumen por inyección subcutánea se restringe a aproximadamente 1 ml), reduciendo así la comodidad y conveniencia para el paciente. En segundo lugar, la estabilidad en almacenamiento y la vida útil de almacenamiento de la formulación actual de ALX-0081 (a continuación en el presente documento, también denominada ALX-0081 contemporánea) pueden mejorarse a temperaturas elevadas. La estabilidad en la presente formulación a altas temperaturas está determinada principalmente por modificaciones químicas en el polipéptido. Las modificaciones químicas pueden estar vinculadas con una pérdida de potencia.
Aunque puede lograrse una vida útil de almacenamiento factible almacenando el producto a -20°C, sin embargo, esto no se considera una opción favorable para la mayoría de los propósitos prácticos.
El secado por congelación es una técnica comúnmente empleada para conservar proteínas que sirve para eliminar el agua de la preparación de proteínas de interés. El secado por congelación, o liofilización, es un proceso mediante el cual el material que va a secarse se congela en primer lugar y luego el hielo o disolvente congelado se elimina por sublimación en un entorno de vacío. Puede incluirse un excipiente en formulaciones preliofilizadas para potenciar la estabilidad durante el proceso de secado por congelación y/o para mejorar la estabilidad del producto liofilizado después del almacenamiento (Arakawa et al. Pharm Res. 8 (3): 285-291 (1991)). La presente invención se refiere a una formulación tal como se define en las reivindicaciones que comprende un agente de unión a factor de von Willebrand (vWF) y un tampón citrato, con un pH en el intervalo de 6,0 a 7,0. En particular, la presente invención se refiere a una formulación tal como se reivindica, en la que dicho agente de unión a vWF comprende al menos un dominio variable individual de inmunoglobulina que se une a SEQ ID NO: 20.
Dicho dominio variable individual de inmunoglobulina comprende o consiste esencialmente en, pero no se limita a, un dominio variable individual de inmunoglobulina que es una secuencia de dominio variable de cadena pesada, más específicamente un dominio variable individual de inmunoglobulina que es una secuencia de dominio variable de cadena pesada que se deriva de un anticuerpo de cuatro cadenas convencional o una secuencia de dominio variable de cadena pesada que se deriva de un anticuerpo de cadena pesada, o un Nanobody (incluyendo pero sin limitarse a una secuencia de VHH), preferiblemente un Nanobody.
Además, la presente invención se refiere a una formulación tal como se reivindica, en la que dicho agente de unión a vWF comprende al menos una de SEQ ID NO: 1-19. Además, la presente invención se refiere a una formulación tal como se reivindica, en la que dicho agente de unión a vWF es un polipéptido de cadena sencilla que comprende uno o más dominios variables individuales de inmunoglobulina, preferiblemente en la que dicho agente de unión a vWF es monovalente o multivalente, en la que dicho agente de unión a vWF es monoespecífico o multiespecifico y/o en la que uno o más dominios variables individuales de inmunoglobulina están injertados con CDR, humanizados, camelizados, desinmunizados y/o generados in vitro (por ejemplo seleccionados mediante presentación en fago). La presente invención también se refiere a una formulación tal como se reivindica, en la que dicho agente de unión a vWF comprende una secuencia de aminoácidos que es al menos el 90% idéntica a SEQ ID NO: 1. La presente invención se refiere también a una formulación tal como se reivindica, en la que dicho agente de unión a vWF tiene una concentración en el intervalo de 0,1 a 80 mg/ml, y/o en la que dicho tampón tiene una concentración en el intervalo de 5-200 mM.
Adicionalmente, la presente invención se refiere a una formulación tal como se reivindica, que comprende además un excipiente, preferiblemente dicho excipiente tiene una concentración en el intervalo de 10-500 mM, más preferiblemente, en la que dicho excipiente se selecciona de la lista que consiste en sacarosa, glicina, manitol, trehalosa y NaCl, incluso más preferiblemente, en la que dicha sacarosa tiene una concentración en el intervalo del 1-15%, preferiblemente el 2-12%, preferiblemente el 4-10%, por ejemplo el 4, el 5, el 6, el 7, el 8 o el 9% (p/v), lo más preferiblemente el 7%.
La presente invención también se refiere a una formulación tal como se reivindica, en la que el tampón es un tampón citrato, dicho tampón citrato tiene un pH de entre 6,0 y 7,0, preferiblemente 6,5.
Además, la presente invención se refiere a una formulación tal como se reivindica, que comprende además un detergente no iónico, tal como Tween-80, preferiblemente en una concentración de entre el 0,001 y el 0,5% (v/v), más preferiblemente el 0,01-0,02% (v/v).
Además, la presente invención se refiere a una formulación tal como se reivindica, en la que dicho tampón es un tampón citrato a pH 6,5 ±0,5, por ejemplo 6,2, 6,3, 6,4, 6,5, 6,6, 6,7 ó 6,8, más específicamente 6,5, y en la que dicha formulación comprende además sacarosa que tiene una concentración en el intervalo del 1-15%,
preferiblemente el 2-12%, preferiblemente el 4-10%, por ejemplo el 4, el 5, el 6, el 7, el 8 o el 9% (p/v), lo más preferiblemente el 7%, y preferiblemente comprende además un detergente no iónico tal como Tween-80, preferiblemente a una concentración del 0,01% (v/v).
Además, la presente invención se refiere a una formulación tal como se reivindica, en la que dicha formulación tiene una osmolalidad en el intervalo de 290 ± 60 mOsm/kg, más preferiblemente en el intervalo de 290 ± 20 mOsm/kg. La presente invención se refiere además a una formulación tal como se define en las reivindicaciones que comprende:
(a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) un excipiente elegido de sacarosa, glicina, manitol, trehalosa o NaCl a una concentración de aproximadamente el 1% a aproximadamente el 15% (p/v);
(c) Tween-80 a una concentración de aproximadamente el 0,001% al 0,5% (v/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0, en la que el agente de unión a vWF en la formulación retiene al menos aproximadamente el 80% de su estabilidad tras el almacenamiento durante al menos 12 meses a 5°C o incluso 24 meses a 5°C.
La invención también se refiere a una formulación tal como se reivindica que tiene menos del 5% de especies de alto peso molecular (HMW) tras el almacenamiento durante al menos 12 meses a 5°C o incluso 24 meses a 5°C; y/o menos del 5% de especies de bajo peso molecular (LMW) tras el almacenamiento durante al menos 12 meses a 5°C o incluso 24 meses a 5°C.
La invención se refiere además a una formulación tal como se reivindica en la que al menos el 80%, preferiblemente al menos el 90%, más preferiblemente al menos el 95% o incluso al menos el 99% del agente de unión a vWF retiene su actividad de unión tras el almacenamiento en comparación con la actividad de unión antes del almacenamiento, dicha actividad de unión tal como se mide mediante ELISA y/o Biacore.
Además, la invención se refiere a una formulación tal como se reivindica, en la que dicha formulación está en una forma líquida, liofilizada, secada por pulverización, liofilizada reconstituida o congelada, más específicamente la invención concierne a una formulación líquida o liofilizada reconstituida tal como se reivindica que comprende: (a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración de aproximadamente el 1% a aproximadamente el 15% (p/v);
(c) Tween-80 a una concentración de aproximadamente el 0,001%-0,5% (v/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0.
La formulación liofilizada puede reconstituirse entonces según sea necesario mezclando la forma liofilizada con un diluyente adecuado (por ejemplo agua) para volver a solubilizar los componentes de la formulación original hasta una concentración deseada.
La presente invención también se refiere a una formulación tal como se reivindica, en la que dicha formulación es una formulación de almacenamiento a granel tal como se reivindica que comprende:
(a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración de aproximadamente el 1% a aproximadamente el 15%;
(c) Tween-80 a una concentración de aproximadamente el 0,001%-0,5% (p/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0, en la que al menos 100 litros de la formulación se almacenan en condiciones por debajo de la congelación.
Además, la presente invención se refiere a una formulación tal como se reivindica, en la que dicha formulación es
adecuada para administración parenteral a un sujeto, por ejemplo, un sujeto humano (por ejemplo un paciente que tiene un trastorno relacionado con vWF). La formulación puede administrarse al sujeto mediante inyección (por ejemplo, intravenosa, subcutánea, intramuscular o intraperitoneal).
Además, la presente invención proporciona una formulación tal como se reivindica, para su uso en un método de tratamiento de un sujeto humano o animal, preferiblemente para su uso en el tratamiento de trastornos relacionados con vWF, tales como por ejemplo síndrome coronario agudo (SCA), accidente isquémico cerebral transitorio, angina de pecho estable o inestable, accidente cerebrovascular, infarto de miocardio o purpura trombocitopénica trombótica (PTT), lo más preferiblemente para su uso en el tratamiento de PTT o SCA. Además, la presente invención concierne a un método o procedimiento de preparación de la formulación tal como se reivindica. El método o procedimiento incluye expresar el agente de unión a vWF en un cultivo celular; purificar el agente de unión a vWF haciendo pasar el agente de unión a vWF a través de al menos una de una etapa de purificación cromatográfica y una etapa de ultrafiltración/diafiltración; ajustar la concentración del agente de unión a vWF a de aproximadamente 0,1 a 80 mg/ml en una formulación que contiene un lioprotector, un tensioactivo y un tampón tal como se reivindica, es decir, sacarosa a una concentración de 7%; Tween-80 a una concentración de aproximadamente el 0,001% a aproximadamente el 0,5% (p/v); y un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0; y opcionalmente que comprende una etapa de confeccionar la formulación en una forma unitaria de dosificación.
La invención también presenta un método o procedimiento para preparar una formulación reconstituida que contiene un agente de unión a vWF, por ejemplo, ALX-0081 tal como se reivindica. El método incluye: liofilizar una mezcla de un agente de unión a vWF, un lioprotector, un tensioactivo y un tampón, formando de ese modo una mezcla liofilizada; y reconstituir la mezcla liofilizada en un diluyente, preparando de ese modo una formulación que comprende (a) un agente de unión a vWF, por ejemplo, ALX-0081 a una concentración de aproximadamente 0,1 a aproximadamente 80 mg/ml; (b) sacarosa a una concentración de 7% (p/v); (c) Tween-80 a una concentración de aproximadamente el 0,001% a aproximadamente el 0,5% (v/v); y (d) un tampón citrato a una concentración de aproximadamente 5 a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0; y opcionalmente que comprende una etapa de confeccionar la formulación en una forma unitaria de dosificación.
La presente invención se refiere además a un método para estabilizar un agente de unión a vWF tal como se reivindica, preferiblemente un polipéptido que comprende al menos una de SEQ ID NO: 1-19 para el almacenamiento, que comprende preparar una formulación tal como se reivindica.
Además, la invención se refiere a un método para almacenar un agente de unión a vWF tal como se reivindica, preferiblemente un polipéptido que comprende al menos una de SEQ ID NO: 1-19, que comprende preparar una formulación tal como se reivindica.
También se proporcionan composiciones farmacéuticas o de diagnóstico que comprenden cualquiera de las formulaciones reivindicadas u obtenibles mediante los métodos reivindicados.
Además, la divulgación presenta un método de análisis de un producto o procedimiento, por ejemplo, un procedimiento de fabricación. El método incluye proporcionar una formulación de un agente de unión a vWF, por ejemplo, ALX-0081 tal como se describe en el presente documento, y evaluar un parámetro de la formulación, tal como color, claridad, viscosidad o una cantidad de una o más especies de HMW, LMW, tal como se describe en el presente documento. La evaluación puede incluir una evaluación de uno o más parámetros, tal como determinar si el parámetro cumple un criterio preseleccionado, por ejemplo, determinar si el criterio preseleccionado está presente, o está presente en un intervalo preseleccionado, analizando de ese modo el procedimiento. Por ejemplo, la evaluación del procedimiento incluye una medida de la estabilidad de la formulación de agente de unión a vWF. La estabilidad de la formulación de ALX-0081 puede medirse, por ejemplo, mediante la formación de agregados, que se somete a ensayo, por ejemplo, mediante cromatografía de líquidos de alta presión de exclusión molecular (SE-HPLC), mediante el color, la claridad o la viscosidad tal como se describe en el presente documento.
Además, el método puede comprender además comparar dos o más formulaciones de muestras en un método de monitorización o control de la variación lote a lote, comparar una preparación con un patrón de referencia, clasificar, seleccionar, aceptar o desechar, liberar o retener, procesar para dar un producto farmacológico, enviar, desplazar a una ubicación diferente, formular, etiquetar o envasar la formulación, basándose en la comparación. Además, el método puede comprender además proporcionar un registro que incluye datos referentes al parámetro evaluado de la formulación y opcionalmente incluye un identificador para un lote de la formulación; someter dicho registro a un tomador de decisiones; opcionalmente, recibir una comunicación de dicho tomador de decisiones; opcionalmente, decidir si se lanza o comercializa el lote de la formulación basándose en la comunicación del tomador de decisiones.
También se proporcionan kits o artículos de fabricación que comprenden un recipiente que contiene la formulación de la invención e instrucciones para su uso, por ejemplo, por un profesional sanitario. Los kits o artículos de fabricación pueden incluir un vial o una jeringa que contiene la formulación de la invención tal como se reivindica. Preferiblemente, el vial o la jeringa se compone de vidrio, plástico o un material polimérico elegido de un polímero o
copolímero de olefina cíclica. Además, la formulación también puede estar presente en un dispositivo inyectable (por ejemplo, una jeringa inyectable, por ejemplo una jeringa inyectable precargada).
La invención proporciona además formas de dosificación unitarias farmacéuticas que comprenden las formulaciones estables de la invención que son adecuadas para administración parenteral (por ejemplo, por vía intradérmica, por vía intramuscular, por vía intraperitoneal, por vía intravenosa y por vía subcutánea) de la formulación de la invención a un paciente humano.
Además, las formulaciones de la invención pueden usarse para el almacenamiento de un agente de unión a vWF, preferiblemente un polipéptido que comprende al menos una de SEQ ID NO: 1-19, tal como ALX-0081 tal como se describe en el presente documento, en la que dicho almacenamiento es de 1-36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses, preferiblemente al menos 12 meses, por ejemplo a una temperatura de entre -70°C y 40°C, tal como -70°C, -20°C, 5°C, 25°C o 40°C, preferiblemente una temperatura de entre -70°C y 25°C.
La presente divulgación también se refiere a un método de tratamiento o prevención de un trastorno relacionado con vWF, tal como por ejemplo síndrome coronario agudo (SCA), accidente isquémico cerebral transitorio, angina de pecho estable o inestable, accidente cerebrovascular, infarto de miocardio o purpura trombocitopénica trombótica (PTT); comprendiendo dicho método administrar a un sujeto una composición farmacéutica que comprende la formulación, reduciendo de ese modo uno o más síntomas asociados con dicho trastorno relacionado con vWF. En particular, dicho trastorno relacionado con vWF es PTT.
4. Breve descripción de las figuras
Figura 1. Diagrama de flujo que representa las diferentes etapas del programa de liofilización de 65 h convencional realizado para ALX-0081.
Figura 2A. Parte relevante de los cromatogramas de RP-HPLC de ALX-0081 después de 1 y 2 meses de almacenamiento a -70°C, 5°C y 25°C; mAU: unidad de miliabsorbancia.
Figura 2B. Acercamiento de la parte relevante de los cromatogramas de RP-HPLC de ALX-0081 después de 0, 4 y 8 semanas de incubación a 37°C. Se observa división del pico principal de RP-HPLC como resultado de una incubación prolongada a 37°C (0, 4, 8 w); mAU: unidad de miliabsorbancia.
Figura 3A. Superposición de perfiles de SE-HPLC del tampón citrato en blanco (bcb) y ALX-0081 en citrato 20 mM, pH 7,0 a 55,9 mg/ml antes (a) y después de 10 ciclos de congelación-descongelación (FT) a -20°C (c) y -70°C (b) ( l = 280 nm). Se observó un pico de citrato secundario para muestras prediluidas en tampón de ejecución; mAU: unidad de miliabsorbancia.
Figura 3B. Superposición de perfiles de SE-HPLC del tampón citrato en blanco (bcb) y ALX-0081 en citrato 20 mM, pH 7,0, a 55,9 mg/ml después de un almacenamiento de ± 1 semana a 4°C ( l = 280 nm). ALX-0081 se resolvió en un pico principal (97%) correspondiente a ALX-0081 intacto, sin modificar y pequeños picos previos que representan solo el 3% del área de superficie total. Se observó un pico de citrato minoritario para ALX-0081 prediluido en tampón de ejecución; mAU: unidad de miliabsorbancia.
Figura 4A. Intensidad de dispersión de muestras de ALX-0081 agitadas en citrato 50 mM, pH 6,0, citrato 50 mM, pH 6,0 Tween-80 al 0,01% (v/v) y citrato 50 mM, pH 6,0 Tween-80 al 0,02% (v/v) a 25°C. ‘+’ representan muestras en citrato 50 mM, pH 6,0 (y = 0,0044x 3,5962, R2= 0,9549); ‘o’ representan muestras en citrato 50 mM, pH 6,0 Tween-80 al 0,02% (v/v) (y = 0,0002x 1,0447, R2= 0,4673); ‘x’ representa muestras en citrato 50 mM, pH 6,0 Tween-80 al 0,01% (v/v) (y = 0,0004x 0,5125, R2= 0,6804); (eje x = tiempo en segundos; eje y = intensidad de dispersión).
Figura 4B. Intensidad de dispersión de muestras de ALX-0081 agitadas en citrato 50 mM, pH 6,5, citrato 50 mM, pH 6,5 Tween-80 al 0,01% (v/v) y citrato 50 mM, pH 6,5 Tween-80 al 0,02% (v/v) a 25°C. ‘+’ representan muestras en citrato 50 mM pH 6,5 (y = 0,0041x 4,7667, R2= 0,9431); ‘o’ representan muestras en citrato 50 mM pH 6,5 Tween-80 al 0,02% (v/v) (y = 0,0004x - 0,0208, R2= 0,9391); ‘x’ representan muestras en citrato 50 mM pH 6,5 Tween-80 al 0,01% (v/v) (y = 0,0001x - 1,8853, R2= 0,0376); (eje x = tiempo en segundos; eje y = intensidad de dispersión).
Figura 5. Superposición de perfiles de cIEF de ALX-0081 a 5 mg/ml en D-PBS glicina 200 mM Tween-80 al 0,01% después de 1 mes de almacenamiento a 40°C (a) y -70°C (b); ( l = 280 nm). AU: unidad de absorbancia; pxlpos: posición de píxel.
Figura 6. Fotografía de las formulaciones de ALX-0081 liofilizadas (forma 3 = citrato/sacarosa, pH 6,0; forma 7 = citrato/sacarosa, pH 6,5; forma 17 = D-PBS/glicina) antes (panel A) y después de la reconstitución con agua Milli-Q (panel B).
Figura 7. Fotografía de formulaciones liofilizadas de ALX-0081 basadas en citrato/sacarosa.
Figura 8. Fotografías de formulaciones líquidas de ALX-0081 a 28 mg/ml que contienen citrato 15, 20, 25, 30, 40 y 50 mM, pH 6,5 después de 4 días de almacenamiento a 25°C (panel A) o 5°C (panel B). Se incluye tampón citrato en blanco (50 mM) como referencia.
Figura 9. Fotografías de formulaciones de ALX-0081 líquidas a 20 mg/ml que contienen citrato 15 mM, pH 6,5 y diferentes cantidades de sacarosa y Tween-80 después de 4 días de almacenamiento a 25°C (panel A) o 5°C (panel B). Se incluye tampón citrato en blanco (50 mM) como referencia.
Figura 10. Predicción del porcentaje de piroglutamato en el producto farmacológico liofilizado de ALX-0081 en función del tiempo cuando se almacena a 5°C.
Figura 11. Predicción del porcentaje de piroglutamato en el producto farmacológico liofilizado de ALX-0081 en función del tiempo cuando se almacena a 25°C.
5. Descripción detallada de la invención
A menos que se indique otra cosa, todos los métodos, etapas, técnicas y manipulaciones que no se describen específicamente en detalle pueden realizarse y se han realizado de una manera conocida per se, tal como quedará claro para el experto en la técnica. Se hace referencia por ejemplo de nuevo a los manuales convencionales y la técnica anterior general mencionada en el presente documento y a las referencias adicionales mencionadas en la misma; así como a por ejemplo las siguientes revisiones: Presta, Adv. Drug Deliv. Rev. 2006, 58 (5-6): 640-56; Levin y Weiss, Mol. Biosyst. 2006, 2(1): 49-57; Irving et al., J. Immunol. Methods, 2001, 248(1-2), 31-45; Schmitz et al., Placenta, 2000, 21 supl. A, S106-12, Gonzales et al., Tumour Biol., 2005, 26(1), 31-43, que describen técnicas para la ingeniería de proteínas, tales como maduración por afinidad y otras técnicas para mejorar la especificidad y otras propiedades deseadas de proteínas tales como inmunoglobulinas.
Se ha encontrado ahora sorprendentemente que agentes de unión a vWF, y en particular ALX-0081 (SEQ ID NO: 1), pueden administrarse en regímenes de dosificación particulares en seres humanos. Se ha encontrado que los agentes de unión a vWF, y en particular ALX-0081, producen un efecto farmacodinámico, con un comienzo rápido de la acción inmediatamente al final de la dosificación y mantiene su eficacia durante hasta aproximadamente 12-24 h. Adicionalmente, se ha encontrado que los agentes de unión a vWF, y en particular ALX-0081 (SEQ ID NO: 1), se toleran bien y son seguros en voluntarios masculinos sanos. Estos resultados indican que los agentes de unión a vWF y en particular ALX-0081 (SEQ ID NO: 1) son adecuados para el tratamiento agudo en pacientes con angina de pecho estable que se someten a intervención coronaria percutánea programada (a continuación en el presente documento también “PCI”) y tratamiento en pacientes con purpura trombocitopénica trombótica (a continuación en el presente documento también “PTT”).
No obstante, las formulaciones actuales de los agentes de unión a vWF y en particular ALX-0081 (SEQ ID NO: 1) administradas a receptores humanos son propensas a mejora.
La invención de reformulación para los agentes de unión a vWF, y en particular ALX-0081, descrita en el presente documento produjo una nueva formulación basada en citrato/sacarosa con solubilidad aumentada (hasta 80 mg/ml) y estabilidad en almacenamiento líquido significativamente mejorada (por ejemplo se produce menos oxidación cuando se almacena la nueva formulación con respecto a la formulación original en su estado líquido). Además, en la forma liofilizada, esencialmente no pudo detectarse oxidación o isomerización de asp tras 12 meses de almacenamiento a 40°C o incluso 24 meses de almacenamiento a 40°C. Se observó todavía una formación residual de pequeñas cantidades de piroglutamato. La optimización adicional de la concentración de citrato y sacarosa dio como resultado una reducción del contenido en humedad del producto liofilizado, minimizando de ese modo la tasa de formación de piroglutamato residual.
Por consiguiente, la presente invención proporciona formulaciones liofilizadas y líquidas estables de agentes de unión anti-vWF (por ejemplo ALX-0081). También se divulgan usos de las mismas para tratar o prevenir trastornos relacionados con vWF.
5.1 Polipéptido(s) usado(s) en la invención
Los agentes de unión a vWF usados en la presente invención son proteínas o polipéptidos que se unen a factor de von Willebrand humano (vWF, SEQ ID NO: 20). Los agentes de unión a vWF son proteínas o polipéptidos que comprenden o consisten en al menos unas secuencias de inmunoglobulina, tales como un dominio variable individual de inmunoglobulina (ISVD) tal como se define en las reivindicaciones. Preferiblemente, los agentes de unión a vWF usados en la presente invención son proteínas o polipéptidos que comprenden o consisten en SEQ ID NO: 1-19, y lo más preferiblemente SEQ ID NO: 1. Los agentes de unión a vWF pueden usarse como terapia adyuvante para pacientes con SCA que se someten a PCI o como tratamiento de purpura trombocitopénica trombótica (PTT). Los términos “proteína”, “polipéptido” y “secuencia de aminoácidos” se usan de manera
intercambiable en el presente documento. Por tanto, una secuencia de aminoácidos usada en la invención es un agente de unión a vWF.
Por tanto, por ejemplo, los agentes de unión a vWF adecuados para su uso en la invención pueden incluir los compuestos en la tabla A-1, por ejemplo SEQ ID NO: 1-19, o un compuesto que tiene el 80% o más, más preferiblemente el 85% o más, lo más preferido el 90%, el 95%, el 96%, el 97%, el 98%, el 99% o más, de identidad de secuencia de aminoácidos con un compuesto de la tabla A-1 (véase la sección de definiciones para “identidad de secuencia”).
Preferiblemente, los agentes de unión a vWF para su uso en la invención son compuestos de tipo 12A02H1. Para los propósitos de la presente descripción, un compuesto de tipo 12A02H1 es un compuesto que comprende 12A02H1 (es decir, SEQ ID NO: 19) o un compuesto que tiene el 80% o más, más preferiblemente el 85% o más, lo más preferiblemente el 90%, el 95%, el 96%, el 97%, el 98%, el 99% o más, de identidad de secuencia de aminoácidos (tal como se define adicionalmente en el presente documento) con 12A02H1 (SEQ ID NO: 19). Un agente de unión a vWF particularmente preferido es ALX-0081 (SEQ ID NO: 1).
Todos los agentes de unión a vWF mencionados anteriormente se conocen bien de la bibliografía. Esto incluye su fabricación (véanse en particular por ejemplo el documento WO2006/122825 pero también WO2004/062551). Por ejemplo, ALX-0081 se prepara tal como se describe por ejemplo en el documento WO2006/122825 o WO2009/115614.
A menos que se indique otra cosa, el término “secuencia de inmunoglobulina”, ya se use en el presente documento para referirse a un anticuerpo de cadena pesada o a un anticuerpo de 4 cadenas convencional, se usa como término general que incluye tanto el anticuerpo de tamaño completo, las cadenas individuales del mismo, así como todas las partes, dominios o fragmentos del mismo (incluyendo pero sin limitarse a dominios de unión a antígeno o fragmentos tales como dominios Vhh o dominios Vh/Vl, respectivamente). Los términos molécula de unión a antígeno o proteína de unión a antígeno se usan de manera intercambiable con secuencia de inmunoglobulina, e incluyen dominios variables individuales de inmunoglobulina, tales como Nanobodies®.
Realizaciones de la invención se refieren a secuencias de inmunoglobulina que son dominios variables individuales de inmunoglobulina, tales como secuencias de dominio variable de cadena ligera (por ejemplo una secuencia de Vl), o secuencias de dominio variable de cadena pesada (por ejemplo una secuencia de Vh); más específicamente, secuencias de dominio variable de cadena pesada que se derivan de un anticuerpo de cuatro cadenas convencional o secuencias de dominio variable de cadena pesada que se derivan de un anticuerpo de cadena pesada (por ejemplo una secuencia de Vhh).
El término “dominio variable individual de inmunoglobulina” define moléculas en las que el sitio de unión a antígeno está presente en, y formado por, un dominio de inmunoglobulina individual o fragmentos adecuados del mismo. Esto establece dominios variables individuales de inmunoglobulina aparte de inmunoglobulinas “convencionales” o sus fragmentos, en los que dos dominios de inmunoglobulina, en particular dos dominios variables interaccionan para formar un sitio de unión a antígeno. Normalmente, en inmunoglobulinas convencionales, un dominio variable de cadena pesada (Vh) y un dominio variable de cadena ligera (Vl) interaccionan para formar un sitio de unión a antígeno. En este caso, las regiones determinantes de complementariedad (CDR) de tanto Vh como Vl contribuirán al sitio de unión a antígeno, es decir un total de 6 CDR estarán implicadas en la formación del sitio de unión a antígeno.
En cambio, el sitio de unión a antígeno de un dominio variable individual de inmunoglobulina se forma mediante un único dominio de Vh o Vl. Por tanto, el sitio de unión a antígeno de un dominio variable individual de inmunoglobulina se forma mediante no más de tres CDR, por ejemplo una, dos o tres CDR.
El término “dominio variable individual de inmunoglobulina” por tanto no comprende inmunoglobulinas convencionales o sus fragmentos que requieren interacción de al menos dos dominios variables para la formación de un sitio de unión a antígeno. Este es también el caso de realizaciones de la invención que “comprenden” o “contienen” un dominio variable individual de inmunoglobulina. En el contexto de la presente invención, tales realizaciones excluyen inmunoglobulinas convencionales o sus fragmentos. Por tanto, una composición que “comprende” o “contiene” un dominio variable individual de inmunoglobulina puede referirse a por ejemplo constructos que comprenden más de un dominio variable individual de inmunoglobulina.
Alternativamente, puede haber constituyentes adicionales distintos de los dominios variables individuales de inmunoglobulina, por ejemplo agentes auxiliares de diferentes clases, etiquetas proteicas, colorantes, tintes, etc. Sin embargo, estos términos sí comprenden fragmentos de inmunoglobulinas convencionales en los que el sitio de unión a antígeno se forma mediante un único dominio variable.
Según la invención, el polipéptido usado en la invención, más específicamente las secuencias de inmunoglobulina, pueden consistir en, o comprender uno o más de los siguientes: anticuerpos de dominio, o secuencias de aminoácidos que son adecuadas para su uso como anticuerpos de dominio, anticuerpos de un solo dominio, o
secuencias de aminoácidos que son adecuadas para su uso como anticuerpos de un solo dominio, “dAb”, o secuencias de aminoácidos que son adecuadas para su uso como dAb, o Nanobodies®, incluyendo pero sin limitarse a secuencias de VHH, tales como secuencias de VHH humanizadas o secuencias de VH camelizadas, y preferiblemente son Nanobodies®.
La presente invención abarca fragmentos de dominios variables individuales de inmunoglobulina. “Fragmentos adecuados” de dominios variables individuales de inmunoglobulina se refieren a polipéptidos que contienen menos aminoácidos que un dominio variable individual de inmunoglobulina nativo, pero que todavía muestra actividad de unión a antígeno (que entonces contendrá habitualmente al menos algunos de los residuos de aminoácido que forman al menos una de las CDR, tal como se describe adicionalmente en el presente documento). Tales dominios variables individuales de inmunoglobulina y fragmentos comprenden lo más preferiblemente un plegamiento de inmunoglobulina o son capaces de formar, en condiciones adecuadas, un plegamiento de inmunoglobulina. Más específicamente, dominios variables individuales de inmunoglobulina y sus fragmentos son tales que son capaces de unirse al antígeno diana. Como tal, el dominio variable individual de inmunoglobulina puede comprender por ejemplo una secuencia de dominio variable de cadena ligera (por ejemplo una secuencia de VL) o un fragmento adecuado de la misma; o una secuencia de dominio variable de cadena pesada (por ejemplo una secuencia de Vh o secuencia de Vhh) o un fragmento adecuado de la misma; siempre que sea capaz de formar una única unidad de unión a antígeno (es decir, una unidad de unión a antígeno funcional que consiste esencialmente en el dominio variable individual de inmunoglobulina, de manera que el único dominio de unión a antígeno no necesita interaccionar con otro dominio variable para formar una unidad de unión a antígeno funcional, tal como es por ejemplo el caso de dominios variables que están presentes en, por ejemplo, anticuerpos y fragmentos scFv convencionales que necesitan interaccionar con otro dominio variable, por ejemplo a través de una interacción VH/VL para formar un dominio de unión a antígeno funcional).
Las secuencias de inmunoglobulina usadas en la invención están preferiblemente en forma esencialmente aislada. Las secuencias de inmunoglobulina usadas en la invención forman parte de una proteína o polipéptido usado en la invención (tal como se define en las reivindicaciones), que comprende o consiste esencialmente en una o más secuencias de aminoácidos usadas en la invención y que pueden comprender además opcionalmente una o más secuencias de aminoácidos adicionales (todas opcionalmente unidas por medio de uno o más ligadores adecuados). Por ejemplo, y sin limitación, la una o más secuencias de aminoácidos usadas en la invención pueden usarse como una unidad de unión en una proteína o polipéptido de este tipo, que puede contener opcionalmente una o más secuencias de aminoácidos adicionales que pueden servir como unidad de unión, para proporcionar un polipéptido monovalente, multivalente o multiespecífico usado en la invención, respectivamente, todo tal como se describe en el presente documento. Una proteína o polipéptido de este tipo también puede estar en forma esencialmente aislada.
La invención se refiere a secuencias de inmunoglobulina de diferente origen, que comprende secuencias de inmunoglobulina de ratón, rata, conejo, asno, humano y camélido. La invención también incluye secuencias de inmunoglobulina completamente humanas, humanizadas o quiméricas. Por ejemplo, la invención comprende secuencias de inmunoglobulina de camélido y secuencias de inmunoglobulina de camélido humanizadas, o anticuerpos de dominio camelizados, por ejemplo dAb camelizado tal como describen Ward et al (véase por ejemplo el documento WO 94/04678 y Davies y Riechmann (1994 y 1996)). Además, la invención comprende secuencias de inmunoglobulina fusionadas, por ejemplo formando un constructo multivalente y/o multiespecífico (para polipéptidos multivalentes y multiespecíficos que contienen uno o más dominios VHH y su preparación, se hace referencia también a Conrath et al., J. Biol. Chem., vol. 276, 7346-7350, 2001, así como a por ejemplo los documentos WO96/34103 y WO99/23221), y secuencias de inmunoglobulina que comprenden etiquetas u otros restos funcionales, por ejemplo toxinas, marcadores, productos radioquímicos, etc., que pueden derivarse de las secuencias de inmunoglobulina usadas en la presente invención. También se han descrito dominios variables individuales de inmunoglobulina en tiburones (también denominados “IgNAR”, tal como se describe por ejemplo en el documento WO03/014161 o Streltsov, 2005).
En una realización particular, los dominios variables individuales de inmunoglobulina usados en la invención son Nanobodies®, en particular dominios VHH de camélido, dominios VHH humanizados o dominios VH camelizados. El experto en la técnica está bien familiarizado con la humanización de dominios Vhh y/o camelización de dominios Vh.
La secuencia de aminoácidos y la estructura de una secuencia de inmunoglobulina, en particular un Nanobody®, puede considerarse, sin embargo, sin limitarse a lo mismo, que está compuesta por cuatro regiones de entramado o “FR”, que se denominan en la técnica y en el presente documento “región de entramado 1” o “FR1”; “región de entramado 2” o “FR2”; “región de entramado 3” o “FR3”; y “región de entramado 4” o “FR4”, respectivamente; regiones de entramado que están interrumpidas por tres regiones determinantes de complementariedad o “CDR”, que se denominan en la técnica “región determinante de complementariedad 1” o “CDR1”; “región determinante de complementariedad 2” o “CDR2” ; y “región determinante de complementariedad 3” o “CDR3”, respectivamente.
El número total de residuos de aminoácido en un Nanobody® puede estar en la región de 110-120, es preferiblemente de 112-115 y es lo más preferiblemente de 113. Sin embargo, debe indicarse que partes, fragmentos, análogos o derivados (tal como se describe adicionalmente en el presente documento) de un Nanobody® no están particularmente limitados en cuanto a su longitud y/o tamaño, siempre que tales partes,
fragmentos, análogos o derivados cumplan los requisitos adicionales explicados resumidamente en el presente documento y también sean preferiblemente adecuados para los propósitos descritos en el presente documento.
Por tanto, generalmente, los dominios variables individuales de inmunoglobulina serán secuencias de aminoácidos que consisten en, o esencialmente consisten en 4 regiones de entramado (FR1 a FR4 respectivamente) y 3 regiones determinantes de complementariedad (CDR1 a CDR3 respectivamente). “Consiste esencialmente” en este contexto significa que pueden estar presentes elementos adicionales tales como por ejemplo etiquetas usadas para la purificación o el marcaje, pero tales elementos adicionales son pequeños en comparación con el dominio variable individual de inmunoglobulina per se, y no interfieren con la actividad de unión a antígeno del dominio variable individual de inmunoglobulina.
Tal como se usa en el presente documento, el término “secuencias de inmunoglobulina” o “dominios variables individuales de inmunoglobulina” se refiere a tanto las secuencias de ácido nucleico que codifican para el polipéptido, como al polipéptido per se. Cualquier significado más limitante resultará evidente a partir del contexto específico.
En particular, la secuencia de aminoácidos usada en la invención puede ser un Nanobody® o un fragmento adecuado del mismo. Para una descripción adicional de VHH y Nanobodies, se hace referencia al artículo de revisión de Muyldermans en Reviews in Molecular Biotechnology 74(2001), 277-302; así como a las siguientes solicitudes de patente, que se mencionan como técnica anterior general: WO94/04678, WO95/04079 y WO96/34103 de la Vrije Universiteit Brussel; WO94/25591, WO99/37681, WO00/40968, WO00/43507, WO00/65057, WO01/40310, WO01/44301, EP1134231 y WO02/48193 de Unilever; WO97/49805, WO01/21817, WO03/035694, WO03/054016 y WO03/055527 del Vlaams Instituut voor Biotechnologie (VIB); WO03/050531 de Algonomics N.V. y Ablynx N.V.; w O 01/90190 del National Research Council of Canada; WO03/025020 (= EP1433793) del Institute of Antibodies; así como WO04/041867, WO04/041862, WO04/041865, WO04/041863, WO04/062551, WO05/044858, WO06/40153, WO06/079372, WO06/122786, WO06/122787 y WO06/122825, de Ablynx N.V. y las solicitudes de patente publicadas adicionales de Ablynx N.V. Se hace referencia también a la técnica anterior adicional mencionada en estas aplicaciones, y en particular a la lista de referencias mencionadas en las páginas 41-43 de la solicitud internacional WO06/040153. Tal como se describe en estas referencias, los Nanobodies (en particular secuencias de VHH y Nanobodies parcialmente humanizados) pueden caracterizarse en particular por la presencia de uno o más "residuos distintivos" en una o más de las secuencias de entramado. Una descripción adicional de los Nanobodies, incluyendo la humanización y/o camelización de Nanobodies, así como otras modificaciones, partes o fragmentos, derivados o “fusiones de Nanobody”, constructos multivalentes (incluyendo algunos ejemplos no limitativos de secuencias de ligador) y diferentes modificaciones para aumentar la semivida de los Nanobodies y sus preparaciones pueden encontrarse por ejemplo en el documento WO07/104529.
Los dominios variables individuales de inmunoglobulina usados en la invención están preferiblemente en forma aislada o forma esencialmente aislada. Las secuencias de inmunoglobulina usadas en la invención pueden formar parte también de una proteína o polipéptido usado en la invención, que comprende o consiste esencialmente en uno o más dominios variables individuales de inmunoglobulina y que pueden comprender además opcionalmente una o más secuencias de aminoácidos adicionales (todas opcionalmente unidas por medio de uno o más ligadores adecuados). Por ejemplo, y sin limitación, el uno o más dominios variables individuales de inmunoglobulina pueden usarse como unidad de unión en una proteína o polipéptido de este tipo, que puede contener opcionalmente una o más secuencias de aminoácidos adicionales que pueden servir como unidad de unión, para proporcionar un polipéptido monovalente, multivalente o multiespecífico usado en la invención, respectivamente, todo tal como se describe en el presente documento. Una proteína o polipéptido de este tipo también puede estar en forma aislada o esencialmente aislada. Por tanto, según la invención, dominios variables individuales de inmunoglobulina comprenden constructos que comprenden dos o más unidades de unión a antígeno en forma de dominios individuales, tal como se explicó de manera resumida anteriormente. Por ejemplo, dos (o más) dominios variables individuales de inmunoglobulina con la misma especificidad de antígeno o diferente pueden unirse para formar por ejemplo un constructo bivalente, trivalente o multivalente. Combinando dominios variables individuales de inmunoglobulina de dos o más especificidades, pueden formarse constructos biespecíficos, triespecíficos, etc. Por ejemplo, un polipéptido usado en la invención puede comprender dos dominios variables individuales de inmunoglobulina dirigidos contra la diana A, y un dominio variable individual de inmunoglobulina contra la diana B, haciéndolo bivalente para A y monovalente para B. Tales constructos y modificaciones de los mismos, que el experto en la técnica puede prever fácilmente, se abarcan todos en la presente invención. En realizaciones particulares, la invención se refiere a constructos biparatópicos que comprenden al menos dos dominios variables individuales de inmunoglobulina dirigidos a diferentes epítopos dentro del mismo antígeno diana.
Todas estas moléculas se denominan también “polipéptido usado en la invención”, que es sinónimo con “secuencias de inmunoglobulina” o “dominios variables individuales de inmunoglobulina” usados en la invención.
Además, el término “secuencia” tal como se usa en el presente documento (por ejemplo en términos como “secuencia de inmunoglobulina”, “secuencia de anticuerpo”, “secuencia de dominio variable”, “secuencia de VHH” o “secuencia de proteína”), debe entenderse generalmente que incluye tanto las secuencias de aminoácidos relevantes así como secuencias de ácidos nucleicos o secuencias de nucleótidos que codifican para las mismas, a
menos que el contexto requiera una interpretación más limitada.
Según una realización no limitativa de la invención, las secuencias de inmunoglobulina, Nanobody® o polipéptido usados en la invención están glicosiladas. Según otra realización no limitativa de la invención, las secuencias de inmunoglobulina, Nanobody® o polipéptido usados en la invención no están glicosiladas.
5.2 "Unión" a un antígeno
La invención se refiere a secuencias de inmunoglobulina que pueden unirse a y/o tienen afinidad por un antígeno, que es el factor de von Willebrand. En el contexto de la presente invención, “que se une a y/o que tiene afinidad por” un determinado antígeno tiene el significado habitual en la técnica tal como se entiende por ejemplo en el contexto de anticuerpos y sus respectivos antígenos.
En realizaciones particulares de la invención, el término “se une a y/o que tiene afinidad por” significa que la secuencia de inmunoglobulina interacciona específicamente con un antígeno, y se usa de manera intercambiable con secuencias de inmunoglobulina “contra” dicho antígeno.
El término “especificidad” se refiere al número de diferente tipos de antígenos o determinantes antigénicos a los que puede unirse una secuencia de inmunoglobulina, molécula de unión a antígeno o proteína de unión a antígeno particular (tal como un dominio variable individual de inmunoglobulina, un Nanobody® o un polipéptido usados en la invención). La especificidad de una proteína de unión a antígeno puede determinarse basándose en la afinidad y/o avidez. La afinidad, representada por la constante de equilibrio para la disociación de un antígeno con una proteína de unión a antígeno (KD), es una medida de la fuerza de unión entre un determinante antigénico y un sitio de unión a antígeno en la proteína de unión a antígeno: cuando menor es el valor de la KD, más fuerte es la fuerza de unión entre un determinante antigénico y la molécula de unión a antígeno (alternativamente, la afinidad también puede expresarse como la constante de afinidad (KA), que es 1/KD). Tal como quedará claro para el experto en la técnica (por ejemplo basándose en la divulgación adicional en el presente documento), la afinidad puede determinarse de una manera conocida per se, dependiendo del antígeno de interés específico. La avidez es la medida de la fuerza de unión entre una molécula de unión a antígeno (tal como un dominio variable individual de inmunoglobulina, un Nanobody® o polipéptido usados en la invención) y el antígeno pertinente. La avidez se refiere a tanto la afinidad entre un determinante antigénico y su sitio de unión a antígeno en la molécula de unión a antígeno como al número de sitios de unión pertinentes presentes en la molécula de unión a antígeno.
Normalmente, las secuencias de inmunoglobulina usadas en la presente invención (tales como las secuencias de aminoácidos, dominios variables individuales de inmunoglobulina, Nanobodies® y/o polipéptidos usados en la invención) se unirán a su antígeno con una constante de disociación (KD) de 10-5 a 10-12 moles/litro o menos, y preferiblemente de 10-7 a 10-12 moles/litro o menos y más preferiblemente de 10-8 a 10-12 moles/litro (es decir con una constante de asociación (KA) de 105 a 1012 litros/moles o más, y preferiblemente de 107 a 1012 litros/moles o más y más preferiblemente de 108 a 1012 litros/moles), y/o se unen a su antígeno tal como se define en el presente documento con una velocidad kon de entre 102 M-1s-1 y aproximadamente 107 M-1s-1, preferiblemente entre 103 M-1s-1 y 107 M-1s-1, más preferiblemente entre 104 M-1s-1 y 107 M-1s-1, tal como entre 105 M-1s-1 y 107 M-1s-1 ; y/o se unen a su antígeno tal como se define en el presente documento con una velocidad koff de entre 1 s-1 (t1/2=0,69 s) y 10-6 s-1 (proporcionando un complejo casi irreversible con un t1/2 de múltiples días), preferiblemente entre 10-2 s-1 y 10-6 s-1, más preferiblemente entre 10-3 s-1 y 10-6 s-1, tal como entre 10-4 s-1 y 10-6 s-1.
Cualquier valor de KD mayor de 10-4 M (o cualquier valor de KA menor de 104 M-1) se considera generalmente que indica unión inespecífica.
Preferiblemente, una secuencia de inmunoglobulina monovalente usada en la invención se unirá al antígeno deseado con una afinidad menor de 500 nM, preferiblemente menor de 200 nM, más preferiblemente menor de 10 nM, tal como menor de 500 pM.
La unión específica de una proteína de unión a antígeno a un antígeno o determinante antigénico puede determinarse de cualquier manera adecuada conocida per se, incluyendo, por ejemplo, análisis de Scatchard y/o ensayos de unión competitiva, tales como radioinmunoensayos (RIA), inmunoensayos enzimáticos (EIA) y ensayos de competición de tipo sándwich, y las diferentes variantes de los mismos conocidos per se en la técnica; así como las otras técnicas mencionadas en el presente documento.
La constante de disociación (KD) puede ser la constante de disociación real o aparente, tal como quedará claro para el experto en la técnica. Los métodos para determinar la constante de disociación estarán claros para el experto en la técnica, y por ejemplo incluyen las técnicas mencionadas en el presente documento. En este sentido, también estará claro que puede no ser posible medir constantes de disociación de más de 10-4 moles/litro o 10-3 moles/litro (por ejemplo, de 10-2 moles/litro). Opcionalmente, tal como quedará claro para el experto, la constante de disociación (real o aparente) puede calcularse basándose en la constante de asociación (KA) (real o aparente), por medio de la relación [KD = 1/Ka ].
La afinidad indica la fuerza o estabilidad de una interacción molecular. La afinidad se proporciona comúnmente como mediante la KD, o constante de disociación, que tiene unidades de mol/litro (o M). La afinidad también puede expresarse como una constante de asociación, KA, que es igual a 1/KD y tiene unidades de (mol/litro)-1 (o M-1). En la presente memoria descriptiva, la estabilidad de la interacción entre dos moléculas (tal como una secuencia de aminoácidos, secuencia de inmunoglobulina, dominio variable individual de inmunoglobulina, Nanobody® o polipéptido usados en la invención y su diana prevista) se expresará principalmente en cuanto al valor de KD de su interacción; quedará claro para el experto en la técnica que en vista de la relación KA =1/KD, la especificación de la fuerza de interacción molecular mediante su valor de KD también puede usarse para calcular el valor de KA correspondiente. El valor de KD caracteriza la fuerza de una interacción molecular también en un sentido termodinámico ya que se refiere a la energía libre (DG) de unión mediante la relación bien conocida DG=RT.ln(KD) (de manera equivalente DG=-RT.ln(KA)), en donde R es igual a la constante de gases, T es igual a la temperatura absoluta y ln indica el logaritmo natural.
La KD para interacciones biológicas, tales como la unión de las secuencias de inmunoglobulina usadas en la invención a vWF tal como se define en el presente documento, que se considera significativa (por ejemplo específica) está normalmente en el intervalo de 10-10 M (0,1 nM) a 10-5 M (10000 nM). Cuanto más fuerte es una interacción, menor es su KD.
La KD también puede expresarse como la razón de la constante de velocidad de disociación de un complejo, indicada como koff, con respecto a la velocidad de su asociación, indicada kon (de modo que KD =koff/kon y KA = kon/koff). La velocidad de disociación koff tiene unidades de s-1 (en donde s es la notación de unidad del SI de segundo). La velocidad de asociación kon tiene unidades de M-1s-1.
Con respecto a las secuencias de inmunoglobulina usadas en la invención, la velocidad de asociación puede variar entre 102 M-1s-1 y aproximadamente 107 M-1s-1, que se aproxima a la constante de velocidad de asociación limitada por la difusión para interacciones bimoleculares. La velocidad de disociación se refiere a la semivida de una interacción molecular dada mediante la relación t1 /2=ln(2)/koff. La velocidad de disociación de secuencias de inmunoglobulina usadas en la invención puede variar entre 10-6 s-1 (complejo casi irreversible con un t1/2 de múltiples días) y 1 s-1 (t1/2=0,69 s).
La afinidad de una interacción molecular entre dos moléculas puede medirse por medio de diferentes técnicas conocidas per se, tales como la técnica de biosensor de resonancia de plasmón superficial (SPR) bien conocida (véase por ejemplo Ober et al., Intern. Immunology, 13, 1551-1559, 2001) en donde se inmoviliza una molécula sobre el chip biosensor y la otra molécula se hace pasar sobre la molécula inmovilizada en condiciones de flujo produciendo mediciones de kon, koff y por tanto valores de KD (o KA). Esto puede realizarse por ejemplo usando los instrumentos Biacore bien conocidos.
También quedará claro para el experto que la KD medida puede corresponder a la KD aparente si el proceso de medición influye algo en la afinidad de unión intrínseca de las moléculas implicadas, por ejemplo mediante artefactos relacionados con el recubrimiento sobre el biosensor de una molécula. Además, puede medirse una KD aparente si una molécula contiene más de un sitio de reconocimiento para la otra molécula. En tal situación, la afinidad medida puede verse afectada por la avidez de la interacción mediante las dos moléculas.
Otro enfoque que puede usarse para evaluar la afinidad es el procedimiento de ELISA (ensayo de inmunoabsorción ligado a enzimas) de 2 etapas de Friguet et al. (J. Immunol. Methods, 77, 305-19, 1985). Este método establece una medición en equilibrio de unión en fase de disolución y evita posibles artefactos referentes a la adsorción de una de las moléculas sobre un soporte tal como plástico.
Sin embargo, la medición precisa de KD puede ser bastante laboriosa y, como consecuencia, a menudo se determinan valores de KD aparente para evaluar la fuerza de unión de dos moléculas. Debe indicarse que siempre que todas las mediciones se realicen de un modo sistemático (por ejemplo manteniendo las condiciones de ensayo sin cambios), pueden usarse las mediciones de KD aparente como una aproximación de la KD verdadera y por tanto en el presente documento KD y KD aparente deben tratarse con igual importancia o relevancia.
Finalmente, debe indicarse que en muchas situaciones el científico experimentado puede considerar conveniente determinar la afinidad de unión en relación con alguna molécula de referencia. Por ejemplo, para evaluar la fuerza de unión entre las moléculas A y B, puede usarse por ejemplo una molécula de referencia C que se sabe que se une a B y que está adecuadamente marcada con un grupo fluoróforo o cromóforo u otro resto químico, tal como biotina para una detección fácil en un ELISA o FACS (clasificación celular activada por fluorescencia) u otro formato (el fluoróforo para la detección de fluorescencia, el cromóforo para la detección de absorción de luz, la biotina para la detección de ELISA mediada por estreptavidina). Normalmente, la molécula de referencia C se mantiene a una concentración fija y la concentración de A se varía para una concentración o cantidad dada de B. Como resultado, se obtiene un valor de CI50 correspondiente a la concentración de A al que la señal medida para C en ausencia de A es la mitad. Siempre que se conozca KD ref, la KD de la molécula de referencia, así como la concentración total cref de la molécula de referencia, puede obtenerse la KD aparente para la interacción A-B a partir de la siguiente fórmula: KD = CI50/(1+cref/ KD ref). Obsérvese que si cref << KD ref, KD = CI50. Siempre que la medición de la
CI50 se realice de un modo sistemático (por ejemplo manteniendo cref fija) para los agentes de unión que se comparan, la fuerza o estabilidad de una interacción molecular puede evaluarse mediante la CI50 y esta medición se considera equivalente a KD o a KD aparente a lo largo de todo este texto.
5.3 Antígeno diana
Los dominios variables individuales de inmunoglobulina usados en la presente invención se unen a y/o tienen afinidad por vWF. En el contexto de la presente invención, “vWF” significa vWF humano, es decir SEQ ID NO: 20 o entrada de GenBank: NP_000543. Sin embargo, esto no excluye que un agente de unión a vWF también pueda unirse a, por ejemplo, vWF de cinomólogo, babuino, cerdo, cobaya y/o ratón.
5.4 Realizaciones específicas de secuencias de inmunoglobulina
La presente invención se refiere a dominios variables individuales de inmunoglobulina descritos en, u obtenibles mediante, los métodos divulgados en los documentos WO2004/015425, WO2004/062551, WO2006/074947, WO2006/122825, WO2009/115614 o WO2011/067160, todos a nombre del presente solicitante.
La invención también se refiere a variantes optimizadas de estas secuencias de aminoácidos. Generalmente, una “variante optimizada” de una secuencia de aminoácidos usada en la invención es una variante que comprende una o más sustituciones beneficiosas tales como unas sustituciones que aumentan i) el grado de “humanización”, ii) la estabilidad química y/o iii) el nivel de expresión; mientras que la potencia (medida por ejemplo mediante el ensayo de potencia descrito en la parte experimental del documento WO2006/122825 sigue siendo comparable (es decir, dentro de una desviación del 10%) con el 12A02 de tipo natural (tal como se define en el documento WO2006/122825) o comparable al 12A02H1 variante (SEQ ID NO: 19), también tal como se define en el documento WO2006/122825. Preferiblemente, en comparación con la secuencia de tipo natural de 12A02, una secuencia de aminoácidos usada en la invención contiene al menos una sustitución de este tipo, y preferiblemente al menos dos sustituciones de esta tipo, y preferiblemente al menos tres sustituciones humanizantes y preferiblemente al menos 10 sustituciones humanizantes de este tipo.
En un aspecto particular, las secuencias de aminoácidos usadas en la invención contienen un total de entre 1 y 15, preferiblemente entre 2 y 14, tal como entre 9 y 13, por ejemplo 10, 11 ó 12 sustituciones de aminoácidos en comparación con la secuencia de tipo natural 12A02. Tal como se mencionó, estas diferencias comprenden preferiblemente al menos una y preferiblemente al menos dos, tal como tres, cuatro o cinco o diez sustituciones humanizantes, y pueden comprender opcionalmente una o más sustituciones adicionales (tal como una cualquiera de, o cualquier combinación adecuada de dos cualesquiera o más de, las sustituciones adicionales (a) a (c) tal como se menciona en el presente documento). De nuevo, basándose la divulgación en el presente documento y opcionalmente tras un grado limitado de ensayo y error, el experto en la técnica podrá seleccionar (una combinación adecuada de) una o más de tales sustituciones humanizantes y/o adicionales.
La presente invención se refiere a secuencias de polipéptido tal como se definen en las reivindicaciones que son altamente similares a cualquiera de los ejemplos específicos proporcionados en el presente documento, o cualquiera de los ejemplos específicos definidos por la referencia anterior.
Altamente similar significa una identidad de aminoácidos de al menos el 90%, por ejemplo el 95, el 97, el 98 o el 99%. Las secuencias de polipéptido altamente similares tendrán la misma función que la secuencia de la que se derivan, es decir se unirán a vWF, más específicamente se unirán a e inhibirán la interacción entre vWF y las plaquetas.
En una realización particular, la invención se refiere a secuencias altamente similares a una cualquiera de SEQ ID NO: 1 -19, en particular SEQ ID NO: 1. Sin embargo, para cada secuencia variante tiene que evaluarse la estabilidad en la formulación tal como se define en el presente documento, de manera que la invención en particular se refiere a variantes o secuencias altamente similares que son estables en las formulaciones definidas en el presente documento.
Se conocen ampliamente métodos para generar secuencias de polipéptido usadas en la invención e incluyen por ejemplo síntesis o expresión recombinante. El experto está bien familiarizado con la tecnología de expresión adecuada, por ejemplo células huésped y vectores recombinantes adecuados, por ejemplo células huésped de levadura o bacterianas. El experto está también bien familiarizado con protocolos y técnicas de purificación adecuadas.
5.5 Formulaciones de la invención
La presente invención proporciona formulaciones de polipéptidos dirigidos contra vWF tal como se reivindican, que comprenden al menos un dominio variable individual de inmunoglobulina que se une a SEQ ID NO: 20, que son estables, y preferiblemente adecuadas para usos farmacéuticos, que comprenden la preparación de medicamentos.
Una formulación de un agente de unión a vWF, por ejemplo, un ISVD, incluye un ISVD, un compuesto que puede servir como crioprotector y/o lioprotector, y un tampón. El pH de la formulación es pH 6,0 - 7,0. En algunas realizaciones, una formulación se almacena como un líquido. En otras realizaciones, una formulación se prepara como un líquido y luego se seca, por ejemplo, mediante liofilización o secado por pulverización, antes del almacenamiento. Puede usarse una formulación secada (es decir, el liofilizado) como un compuesto seco, por ejemplo, como un aerosol o polvo, o reconstituirse hasta su concentración original u otra, por ejemplo, usando agua, un tampón, u otro líquido apropiado (diluyente).
El proceso de purificación del agente de unión a vWF está diseñado para permitir la transferencia del agente de unión a vWF a una formulación adecuada para almacenamiento a largo plazo, por ejemplo como un líquido congelado y/o posteriormente para el secado por congelación (por ejemplo, usando una formulación de citrato/sacarosa). La formulación se liofiliza con la proteína, por ejemplo agente de unión a vWF a una concentración específica. La formulación liofilizada puede reconstituirse entonces según sea necesario con un diluyente adecuado (por ejemplo, agua) para resolubilizar los componentes originales de la formulación hasta una concentración deseada, generalmente la misma concentración o superior en comparación con la concentración antes de la liofilización. La formulación liofilizada puede reconstituirse para producir una formulación que tiene una concentración que difiere de la concentración original (es decir, antes de la liofilización), dependiendo de la cantidad de diluyente añadida al liofilizado en relación con el volumen de líquido que se secó por congelación originalmente. Pueden identificarse formulaciones adecuadas sometiendo a ensayo uno o más parámetros de la integridad del agente de unión a vWF. Los parámetros sometidos a ensayo son generalmente el porcentaje de especies de alto peso molecular (HMW) o el porcentaje de especies de bajo peso molecular (LMW) mediante HPLC de exclusión molecular (SE-HPLC).
Por consiguiente, la presente invención proporciona formulaciones tal como se reivindican caracterizadas por un grado adecuado de pureza y a concentraciones adecuadas según se requiera por ejemplo para propósitos farmacéuticos. Las formulaciones proporcionan los polipéptidos que comprenden al menos un dominio variable individual de inmunoglobulina tal como se reivindica en una forma estable a lo largo de un gran intervalo de concentraciones, y un gran intervalo de condiciones de almacenamiento, por ejemplo temperaturas, incluyendo condiciones con tensión tales como temperaturas elevadas (por ejemplo 25°C o superiores), liofilización, agitación u otras formas de tensión física.
La formulación comprende un portador acuoso. El portador acuoso es un tampón citrato.
La invención, sin embargo, también abarca productos obtenibles mediante procesamiento adicional de una formulación líquida, tal como un producto congelado, liofilizado o secado por pulverización. Tras la reconstitución, estos productos sólidos pueden convertirse en formulaciones líquidas tal como se describe en el presente documento (pero sin limitarse a lo mismo). En su sentido más amplio, por tanto, el término “formulación” abarca tanto formulaciones líquidas como sólidas. Sin embargo, se entiende que las formulaciones sólidas pueden derivarse de las formulaciones líquidas (por ejemplo mediante congelación, secado por congelación o secado por pulverización), y por tanto tienen diversas características que se definen mediante las características especificadas para formulaciones líquidas en el presente documento. La invención no excluye reconstitución que conduce a una composición que se desvía de la composición original antes de por ejemplo el secado por congelación o pulverización.
Las formulaciones de la invención comprenden al menos un agente de unión a vWF que comprende al menos un dominio variable individual de inmunoglobulina tal como se define en las reivindicaciones. En realizaciones particulares, la formulación comprende uno o más polipéptidos seleccionados de SEQ ID NO: 1-19, preferiblemente SEQ ID NO: 1. Los polipéptidos pueden tener además una semivida prolongada por ejemplo incorporando un dominio de unión o péptido de unión a albúmina sérica, que puede ser cualquier dominio de unión o péptido de unión a albúmina sérica adecuado capaz de aumentar la semivida del constructo (en comparación con el mismo constructo sin el dominio de unión o péptido de unión a albúmina sérica), y pueden ser en particular péptidos de unión a albúmina sérica tal como se describen en el documento WO2008/068280 por el solicitante (y en particular los documentos WO2009/127691 y WO2011/095545, ambos del solicitante), o un dominio variable individual de inmunoglobulina de unión a albúmina sérica (tal como un Nanobody de unión a albúmina sérica; por ejemplo Alb-1 o una versión humanizada de Alb-1 tal como Alb-8, para lo que se hace referencia por ejemplo al documento WO06/122787). Los medios alternativos para prolongar la semivida que también abarca la presente divulgación incluyen por ejemplo pegilación (PEG) tal como se conoce ampliamente en la técnica, incluyendo pegilación al azar o específica de sitio, preferiblemente pegilación específica de sitio. Puede usarse PEG con un peso molecular por encima de 5000, por ejemplo entre 10.000 y 200.000, preferiblemente en el intervalo entre 20.000 y 100.000. En cualquier aspecto de extensión de la semivida, se prevé que la actividad del polipéptido tal como se define en el presente documento no se vea comprometida, por ejemplo retiene al menos el 75%, el 80%, el 85%, el 90% o el 95% de la actividad del mismo polipéptido sin extensión de la semivida. La actividad puede referirse a por ejemplo la unión al antígeno diana, y/o la potencia en un bioensayo. El experto en la técnica podrá determinar también que la tecnología de extensión de la semivida elegida es adecuada porque no aumenta o incluso disminuye la inmunogenicidad.
5.5.1 Tampón
La formulación de la invención comprende un tampón citrato tal como se define en las reivindicaciones. En una realización particular, el tampón citrato se prepara usando ácido cítrico monohidratado y citrato de trisodio deshidratado, por ejemplo ácido cítrico monohidratado 0,2154 g/l y citrato de trisodio deshidratado 5,5805 g/l. Tal como se determina midiendo las temperaturas de fusión en un ejemplo no limitativo, estos tampones potencian la estabilidad de los agentes de unión a vWF, en comparación con otros tampones sometidos a prueba.
La formulación según la invención comprende un tampón citrato a una concentración en el intervalo de 5-200 mM, por ejemplo 5, 7,5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190 ó 200 mM, preferiblemente 5-100 mM, más preferiblemente 7,5-80 mM, incluso más preferiblemente 10-50, por ejemplo 10, 15, 20, 25 ó 30 mM, y lo más preferiblemente 20 mM, en la que cada valor se entiende que abarca opcionalmente un intervalo de ±5 mM. La formulación según la divulgación puede comprender un tampón fosfato a una concentración en el intervalo de 5-200 mM, por ejemplo 5, 7,5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190 ó 200 mM, preferiblemente 5-80 mM, más preferiblemente 7,5-60 mM, incluso más preferiblemente 10-40, por ejemplo 10, 15, 20, 25 ó 30 mM, y lo más preferiblemente 10 mM, en la que cada valor se entiende que abarca opcionalmente un intervalo de ±5 mM. Se entenderá que una concentración inferior del tampón tiene un efecto sobre la osmolalidad final, y correspondientemente sobre los solutos adicionales que pueden haberse añadido.
El pH de la formulación de la invención está en el intervalo 6,0 a 7,0, en el que cada valor se entiende que abarca un intervalo de ±0,2. Pueden seleccionarse ejemplos específicos de valores de pH preferidos para formulaciones de la invención de la lista no limitativa que comprende pH de 6,0, 6,2, 6,5, 6,7, 7,0, preferiblemente 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8 ó 6,9, por ejemplo 6,5, en el que cada valor se entiende que abarca opcionalmente un intervalo de ±0,2. Inesperadamente, los tampones citrato y fosfato tienen intervalos de pH solapantes con por ejemplo tampones histidina y Tris-HCI, y favorecen aún la estabilidad.
El pH más ventajoso dependerá del tampón comprendido en la formulación. Por tanto, la divulgación se refiere particularmente a una formulación que comprende un tampón fosfato, que tiene preferiblemente un pH en el intervalo de 6,5 a 7,5, preferiblemente 6,9, 7,0, 7,1, por ejemplo 7,1.
Se mostró que una formulación que comprende un tampón citrato era extraordinariamente adecuada para el almacenamiento y uso. Sin embargo, en contraposición al saber convencional, las formulaciones líquidas que comprenden un tampón citrato eran lo más estables a un pH de aproximadamente 6,0, mientras que las formulaciones liofilizadas que comprende un tampón citrato eran lo más estables a un pH de aproximadamente 6,5. Por tanto, la presente invención se refiere a una formulación que comprende un tampón citrato tal como se reivindica, que tiene un pH de aproximadamente 6,0 a 7,0, más preferiblemente 6,1, 6,2, 6,3, 6,4, 6,5, 6,6, 6,7, 6,8 ó 6,9, por ejemplo 6,5, en el que cada valor se entiende que abarca opcionalmente un intervalo de ±0,2.
5.5.2 Concentración
Las formulaciones de la invención comprenden los agentes de unión a vWF tal como se definen en las reivindicaciones, en particular los polipéptidos que comprenden al menos un dominio variable individual de inmunoglobulina tal como se reivindica a una concentración que es adecuada para propósitos clínicos, que incluye concentraciones usadas en disoluciones madre para la dilución antes de su uso en paciente. Aparte de una estabilización mejorada, las formulaciones de la invención permiten mayores concentraciones de los agentes de unión a vWF, por ejemplo ISVD o polipéptidos. En particular, las formulaciones de la invención permanecían físicamente estables, es decir la ausencia de turbidez y/o formación de partículas pequeñas, tal como se confirmó mediante inspección visual, microscopía, SE-HPLC y DLS. El almacenamiento a temperaturas elevadas durante tiempos prolongados y ciclos de congelación-descongelación repetidos aparentemente no afectó a la estabilidad física de los agentes de unión a vWF en estas formulaciones.
Las concentraciones típicas del agente activo, es decir agentes de unión a vWF o los polipéptidos usados en la invención, en las formulaciones de la invención comprenden los ejemplos no limitativos de concentraciones en el intervalo de 0,1 a 80 mg/ml, preferiblemente 1-70 mg/ml, 5-60 mg/ml, 7,5-50 mg/ml o 10-40 mg/ml, tal como 5, 7,5, 10, 12,5, 15, 17,5, 20, 25, 30, 35, 40, 45, 50 ó 60 mg/ml, preferiblemente 12,5 mg/ml o 10 mg/ml, en las que cada valor se entiende que abarca opcionalmente un intervalo del ±20% (por ejemplo un valor de 10 abarca opcionalmente un intervalo de 8 a 12 mg/ml).
5.5.3 Excipientes
Las formulaciones según la invención pueden comprender también opcionalmente uno o más excipientes. El término “excipiente” tal como se usa en el presente documento se refiere a una sustancia inerte que se usa comúnmente como diluyente, vehículo, conservante, lioprotector, aglutinante o agente estabilizante para compuestos que
confieren una propiedad física beneficiosa a una formulación. El experto en la técnica está familiarizado con excipientes adecuados para propósitos farmacéuticos, que pueden tener funciones particulares en la formulación, tales como lioprotección, estabilización, conservación, etc. El experto en la técnica conoce bien estabilizadores y conservantes comúnmente usados (véase por ejemplo el documento WO2010/077422). Los portadores farmacéuticamente aceptables que pueden usarse en estas composiciones incluyen, pero no se limitan a, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, proteínas séricas, tales como albúmina sérica humana, sustancias tampón tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, hidrogenofosfato de disodio, hidrogenofosfato de potasio, cloruro de sodio, sales de zinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros de bloque de polietileno-polioxipropileno, polietilenglicol y lanolina. En realizaciones ventajosas, el excipiente puede ser uno o más, seleccionados de la lista que consiste en NaCl, trehalosa, sacarosa, manitol o glicina.
Al apreciar la invención, el experto en la técnica puede determinar fácilmente concentraciones adecuadas de los excipientes que van a añadirse a las formulaciones. En realizaciones a modo de ejemplo, el NaCl tiene una concentración en el intervalo de 10-500 mM, tal como 25, 30, 40, 50, 60, 70, 100, 150, 250 ó 500 mM, preferiblemente 50-150 mM, por ejemplo 75 ó 140 mM, en el que cada valor se entiende que abarca opcionalmente un intervalo de ±5 mM; y/o el manitol tiene una concentración del 1-10%, preferiblemente el 2-4%, por ejemplo el 2, el 3 o el 4% (p/p), en el que cada valor se entiende que abarca opcionalmente un intervalo de ±0,5%; y/o la sacarosa tiene una concentración del 1-15%, preferiblemente el 2-12% o el 4-10%, por ejemplo el 4, el 5, el 6, el 7, el 8 o el 9% (p/p), y lo más preferiblemente el 7%, en el que cada valor se entiende que abarca opcionalmente un intervalo del ±0,5%; y/o la glicina tiene una concentración en el intervalo de 10-500 mM, tal como 25, 30, 40, 50, 60, 70, 75, 100, 150, 250 ó 500 mM, preferiblemente 50-400 mM, 75-300 mM, 100-250 mM, por ejemplo 140 ó 200 mM, en el que cada valor se entiende que abarca opcionalmente un intervalo de ±5 mM; y/o la trehalosa tiene una concentración en el intervalo de 10-500 mM, tal como 25, 30, 40, 50, 60, 70, 75, 100, 150, 250 ó 500 mM, preferiblemente 100-300 mM, 150-280 mM, por ejemplo 160 mM ó 260 mM, en el que cada valor se entiende que abarca opcionalmente un intervalo de ±5 mM.
En una realización preferida, las formulaciones según cualquier aspecto de la invención son isotónicas en relación con la sangre humana. Las disoluciones isotónicas presentan la misma presión osmótica que el plasma sanguíneo y, de ese modo, pueden infundirse por vía intravenosa en un sujeto sin cambiar la presión osmótica del plasma sanguíneo del sujeto. La tonicidad puede expresarse en cuanto a osmolalidad, que puede ser una osmolalidad teórica, o preferiblemente una osmolalidad determinada experimentalmente. La osmolalidad estará normalmente en el intervalo de 290 ± 60 mOsm/kg, preferiblemente 290 ± 20 mOsm/kg.
Por tanto, en la selección de excipientes (si los hay) el experto en la técnica considerará la concentración de tampón y las concentraciones del uno o más excipientes y preferiblemente llegará a una formulación con una osmolalidad en los intervalos especificados anteriormente. El experto en la técnica está familiarizado con cálculos para estimar la osmolalidad (véase por ejemplo el documento WO2010/077422). Si se requiere, el experto en la técnica puede incluir además un compuesto para ajustar la osmolalidad de la formulación. Los compuestos a modo de ejemplo, incluyen pero no se limitan a los excipientes mencionados anteriormente y/o uno o más de sorbitol, metionina, dextrosa, inositol, arginina o clorhidrato de arginina.
Se ha mostrado que una formulación que comprende sacarosa era particularmente adecuada para mantener la estabilidad física, durante por ejemplo el almacenamiento y la congelación-descongelación, de los polipéptidos. Por consiguiente, la presente invención se refiere a formulaciones que comprenden aproximadamente el 5-9%, más preferiblemente el 6-8% e incluso más preferiblemente el 7% de sacarosa, en las que cada valor se entiende que abarca opcionalmente un intervalo de ±0,5%.
Las formulaciones de la invención pueden comprender también compuestos que son específicamente útiles para proteger al polipéptido usado en la invención durante el secado por congelación. Tales compuestos se conocen también como lioprotectores, y los conoce bien el experto en la técnica. Los ejemplos específicos incluyen, pero no se limitan a azúcares como sacarosa, sorbitol o trehalosa; aminoácidos tales como glutamato, en particular glutamato de monosodio o histidina; betaína, sulfato de magnesio, alcoholes de azúcar, propilenglicol, polietilenglicoles y combinaciones de los mismos. Al apreciar la invención, la cantidad requerida de un compuesto de este tipo que va a añadirse puede determinarla el experto en la técnica considerando la estabilidad de la formulación en forma líquida y cuando se somete a liofilización. Las formulaciones que son particularmente adecuadas para secado por congelación pueden comprender además agentes de carga. El experto en la técnica conoce ampliamente agentes adecuados. Se ha mostrado que una formulación que comprende sacarosa no sólo era particularmente adecuada para mantener la estabilidad física durante, por ejemplo, el almacenamiento y la congelación-descongelación de los agentes de unión a vWF, sino también como lioprotector.
5.5.4 Detergente
En una realización adicional de la invención, la formulación según cualquier aspecto de la invención puede comprender además un detergente o tensioactivo. Los detergentes o tensioactivos adecuados para su uso con la invención incluyen, pero no se limitan a, ésteres de ácidos grasos de polioxietileno-sorbitano, por ejemplo polisorbato-20, -40, -60, -65, -80 u -85. Los nombres comerciales comunes para polisorbatos incluyen Alkest, Canarcel y Tween. El experto en la técnica conoce ejemplos no limitativos adicionales de detergentes, tales como los enumerados por ejemplo en el documento WO2010/077422. En una realización preferida, el detergente es un detergente no iónico. Más específicamente, el detergente es polisorbato-80, también designado Tween-80 a continuación en el presente documento. El experto en la técnica puede determinar fácilmente una concentración adecuada de detergente para una formulación de la invención. Normalmente, la concentración será tan baja como sea posible, mientras que se mantienen los efectos beneficiosos de los detergentes, por ejemplo un efecto estabilizante en condiciones de tensión de cizalladura, por ejemplo agitación, que reduce la agregación de los agentes de unión a vWF formulados. En realizaciones a modo de ejemplo, no limitativas, la concentración del detergente puede estar en el intervalo del 0,001 al 0,5%, por ejemplo el 0,001%, el 0,002%, el 0,003%, el 0,004%, el 0,005%, el 0,01%, el 0,015%, el 0,02%, el 0,025%, el 0,03%, el 0,035%, el 0,04%, el 0,045%, el 0,05%, el 0,1%, el 0,2%, el 0,3%, el 0,4% o el 0,5%, preferiblemente en una concentración de entre el 0,01 y el 0,05%, más preferiblemente entre el 0,01 y el 0,02%, por ejemplo el 0,01% (v/v).
5.5.5 Combinaciones
Las diversas realizaciones descritas anteriormente en las secciones 5.5.1 a 5.5.4 pueden combinarse sin limitaciones en formulaciones de la invención. Por ejemplo, se pretende incluir intervalos de valores usando una combinación de cualquiera de los valores mencionados anteriormente como límites superior y/o inferior. Sin embargo, los ejemplos no limitativos preferibles de formulaciones incluyen formulaciones en las que el tampón es un tampón citrato a aproximadamente pH 6,5, preferiblemente a una concentración de 20 mM, y la formulación comprende además sacarosa, preferiblemente a una concentración de aproximadamente el 7% (p/v), y opcionalmente comprende además un detergente no iónico tal como Tween-80, preferiblemente a una concentración del 0,01% (v/v).
5.6 Procesamiento adicional
Tal como se explicó de manera resumida, cualquiera de las formulaciones anteriores puede procesarse adicionalmente por ejemplo mediante liofilización, secado por pulverización o congelación, por ejemplo congelación a granel. El producto procesado resultante tiene características derivadas de la formulación líquida de partida, tal como se definió anteriormente. Cuando es necesario, pueden incluirse agentes adicionales para el procesamiento adicional, tal como, por ejemplo, lioprotectores, etc.
5.6.1 Congelación
En algunos casos, las formulaciones que contienen agentes de unión a vWF se congelan para su almacenamiento. Por consiguiente, es deseable que la formulación sea relativamente estable en tales condiciones, tales como ciclos de congelación-descongelación (FT). Un método de determinación de la idoneidad de una formulación es someter una formulación de muestra a al menos dos, por ejemplo, tres, cuatro, cinco, ocho, diez, o más ciclos de congelación (a, por ejemplo -20°C o -70°C) y descongelación (por ejemplo mediante descongelación rápida en baño de agua a 25°C o descongelación lenta a de 2°C a 8°C), determinar la recuperación de masa del producto original y la presencia y/o cantidad de especies de LMW y/o especies de HMW que se acumulan tras los ciclos de FT y compararla con la cantidad de especies de LMW o especies de HMW presentes en la muestra antes del procedimiento de FT, por ejemplo mediante SE-HPLC. Un aumento en las especies de LMW o HMW indica una disminución de la estabilidad.
5.6.2 Liofilización
Las formulaciones pueden almacenarse tras la liofilización. Por tanto, someter a prueba una formulación para determinar la estabilidad del componente polipeptídico de la formulación tras la liofilización es útil para determinar la idoneidad de una formulación. El método es similar al descrito anteriormente para la congelación, excepto porque la formulación de muestra se liofiliza en lugar de congelarse, se reconstituye hasta su volumen original y se somete a prueba para determinar la presencia de especies de LMW y/o especies de HMW. La formulación de muestra liofilizada se compara con una formulación de muestra que no estaba liofilizada. Un aumento en las especies de LMW o HMW en la muestra liofilizada en comparación con la muestra correspondiente indica una disminución de la estabilidad de la muestra liofilizada. En general, un protocolo de liofilización incluye cargar una muestra en un liofilizador o secador por congelación, un periodo de enfriamiento previo, congelar, inicio del vacío, elevación hasta la temperatura de secado primario, secado primario, elevación hasta la temperatura de secado secundaria, secado secundario y tapado de la muestra. Aunque el procedimiento de secado por congelación se conoce bien en la técnica, diversos factores determinan las características de secado por congelación de una muestra, incluyendo: la temperatura de transición vítrea (Tg’) y la temperatura de colapso (Tc). Los parámetros adicionales que pueden seleccionarse para un protocolo de liofilización incluyen vacío (por ejemplo, en micrómetros) y temperatura del condensador.
Las velocidades de elevación adecuadas para la temperatura están entre aproximadamente 0,12C/min y 22C/min, por ejemplo entre 0,1°C/min y 1,0°C/min, entre 0,1°C/min y 0,5°C/min, entre 0,2°C/min y 0,5°C/min, entre 0,1°C/min, 0,2°C/min, 0,3°C/min, 0,4°C/min, 0,5°C/min, 0,6°C/min, 0,7°C/min, 0,82C/min, 0,9°C/min y 1,0°C/min. Temperaturas de anaquel adecuadas durante la congelación para un ciclo de liofilización son generalmente de desde aproximadamente -55°C hasta -5°C, de -25°C a -5°C, de -20°C a -5°C, de -15°C a -5°C, de -10 C a -5°C, -10°C, -112C, -12°C, -13°C, -14°C, -15°C, -16°C, -172C, -182C, -192C, -202C, -212C, -222C, -232C, -242C o -252C. Las temperaturas de anaquel pueden ser diferentes para el secado primario y secado secundario, por ejemplo, el secado primario puede realizarse a una temperatura inferior al secado secundario. En un ejemplo no limitativo, el secado primario puede ejecutarse a 0°C o alternativamente a 5°C y el secado secundario a 25°C. En algunos casos, se usa un protocolo de apareamiento durante la congelación y antes del inicio del vacío. En tales casos, el tiempo de apareamiento debe seleccionarse y la temperatura está generalmente por encima de la temperatura de transición vitrea de la composición. En general, el tiempo de apareamiento es de aproximadamente 2 a 20 horas, de aproximadamente 3 a 19 horas, de aproximadamente 2 a 10 horas, de aproximadamente 3 a 5 horas, de aproximadamente 3 a 4 horas, de aproximadamente 2 horas, aproximadamente 3 horas, aproximadamente 5 horas, aproximadamente 8 horas, aproximadamente 10 horas, aproximadamente 12 horas, aproximadamente 15 horas o aproximadamente 19 horas.
La temperatura para el apareamiento es generalmente de desde aproximadamente -35°C hasta aproximadamente -5°C, por ejemplo desde aproximadamente -25°C hasta aproximadamente -8°C, de aproximadamente -20°C a aproximadamente -10°C, de aproximadamente -25°C, aproximadamente -20°C, aproximadamente -15°C, aproximadamente 0°C o aproximadamente -5°C. En algunos casos, la temperatura de apareamiento es generalmente de desde -35°C hasta 5°C, por ejemplo desde -25°C hasta -8°C, de -20°C a -10°C, -25°C, -20°C, -15°C, 02C, 5°C.
La estabilidad de las formulaciones descritas en el presente documento puede someterse a prueba usando una variedad de parámetros de liofilización incluyendo: las temperaturas de anaquel del secado primario de desde -25°C hasta 30°C, y duraciones del secado secundario de 2 horas a 33 horas a de 0° a 30°C. La temperatura para el secado secundario debe ser tan alta como sea posible, sin provocar degradación del principio farmacéutico activo.
Un excipiente que va a usarse en una formulación de esta invención debe satisfacer preferiblemente uno o más de los siguientes parámetros: ser farmacológicamente inerte; ser compatible con los requisitos de procesamiento; tolerarse bien por el paciente; no ser dañina para el material activo; proporcionar un producto soluble, absorbible; proporcionar un producto estable en anaquel; y proporcionar un producto comercialmente aceptable.
En una realización, la formulación de la presente invención se prepara mediante secado por congelación, por ejemplo tal como se explica resumidamente en la figura 1 o la tabla 14.
Se demostró que la liofilización de formulaciones basadas en citrato/sacarosa mejora drásticamente la estabilidad de los agentes de unión a vWF. En particular, las formulaciones basadas en citrato/sacarosa impiden esencialmente modificaciones químicas que se producen en la forma líquida, pero con la excepción de pequeñas cantidades de formación de piroglutamato. Inesperadamente, la disminución de la concentración de citrato y al mismo tiempo el aumento de la concentración de sacarosa mejoraron la estabilidad química, por ejemplo disminuyeron la formación de piroglutamato. Se demostró que el agente de unión a vWF es robusto tras la liofilización hasta temperaturas de producto extremas. De hecho, el perfil de estabilidad era idéntico para material que se había preparado usando una variedad de ciclos de secado por congelación.
En general, un ciclo de liofilización puede ejecutarse desde 10 horas hasta 100 horas, por ejemplo, de 20 horas a 80 horas, de 30 horas a 70 horas, de 40 horas a 60 horas, de 45 horas a 50 horas, de 50 horas a 66 horas.
En un ejemplo no limitativo, una formulación de citrato 20 mM, sacarosa al 7%, Tween-80 al 0,01%, pH 6,5, a una concentración de proteína de 12,5 mg/ml de agente de unión a vWF se formuló a granel y se liofilizó.
Ejemplos no limitativos del intervalo de temperatura para el almacenamiento de una formulación de la invención son de aproximadamente -20°C a aproximadamente 50°C, por ejemplo, de aproximadamente -15°C a aproximadamente 40°C, de aproximadamente -15°C a aproximadamente 30°C, de aproximadamente -15°C a aproximadamente 20°C, de aproximadamente 5°C a aproximadamente 25°C, de aproximadamente 5°C a aproximadamente 20°C, de aproximadamente 5°C de aproximadamente 15°C, de aproximadamente 2°C a aproximadamente 12°C, de aproximadamente 2°C a aproximadamente 10°C, de aproximadamente 2°C a aproximadamente 8°C, de aproximadamente 2°C a aproximadamente 6°C o de aproximadamente 2°C, 3°C, 4°C, 5°C, 6°C, 7°C, 8°C, 10°C, 15°C, 25°C, 30°C o 40°C. Pese a las temperaturas de almacenamiento, en determinados casos, las muestras son estables bajo cambios de temperatura que pueden producirse transitoriamente durante las condiciones de almacenamiento y transporte que pueden anticiparse para tales composiciones.
Se ha establecido que trabajando con las formulaciones de la invención tal como se definen en las reivindicaciones,
es posible obtener un polvo secado resultante que presenta un tamaño de partícula adecuado para la retención confortable y una disolución rápida del material activo. La formulación secada según la invención comprende partículas que permanecen estables y uniformes a lo largo de todo el procesamiento, acabado final, almacenamiento y distribución. La formulación es estable en anaquel y fluye libremente, no presenta problemas cuando se dispensa en su recipiente final y es sencilla de administrar por el paciente.
5.6.3 Secado por pulverización
En algunos casos, una formulación se seca por pulverización y luego se almacena. El secado por pulverización se realiza usando métodos conocidos en la técnica, y pueden modificarse para usar secado por pulverización líquido o congelado (por ejemplo, usando métodos tales como los de Niro Inc. (Madison, WI), Upperton Particle Technologies (Nottingham, Inglaterra), o las publicaciones de patente estadounidense n.os 2003/0072718 y 2003/0082276), o Buchi (Brinkman Instruments Inc., Westbury, NY).
5.6.4 Diluyente
Las formulaciones liofilizadas descritas en el presente documento pueden reconstituirse según sea necesario mezclando la forma liofilizada con un diluyente adecuado para resolubilizar los componentes originales de la formulación hasta una concentración deseada. El término “diluyente” tal como se usa en el presente documento se refiere a un disolvente farmacéuticamente aceptable (seguro y no tóxico para su administración a un humano) para alterar o lograr una concentración apropiada tal como se describe en el presente documento. Los diluyentes a modo de ejemplo incluyen, pero no se limitan a, agua estéril (por ejemplo WFI, agua Milli-Q), solución salina, glucosa, dextrosa, Ringer y disoluciones de tampón acuoso.
5.7 Composiciones farmacéuticas
Las formulaciones de la presente invención son preferiblemente adecuadas para su uso en métodos de terapia del cuerpo humano o animal. Por tanto, la invención se refiere a composiciones farmacéuticas o de diagnóstico que comprenden una formulación del polipéptido según cualquier aspecto de la invención u obtenible mediante cualquier método o procedimiento de la invención.
Las formulaciones de la invención son preferiblemente formulaciones farmacéuticas. En particular, las formulaciones son adecuadas para administración parenteral a un humano, por ejemplo administración subcutánea, intravenosa, intramuscular, intradérmica o intraperitoneal, preferiblemente administración intravenosa o subcutánea. La administración abarca cualquier modo de administración de una formulación líquida, en particular inyección. Otras formas de administración sistémica, por ejemplo por medio de dispositivos implantables, bombas de microinfusión (opcionalmente implantables) y/o formulaciones de liberación sostenida (implantables), por ejemplo depósitos, geles, formulaciones de polímeros biodegradables también están dentro del alcance de la presente invención. Las composiciones farmacéuticas son estériles y estables durante la fabricación y el almacenamiento, ya que no se desean en un entorno clínico derivados/productos de degradación de los agentes de unión a vWF. La composición también será de alta pureza, por ejemplo excluirá la presencia de productos bacterianos tales como LPS. Las formulaciones pueden esterilizarse por cualquier medio, por ejemplo filtración estéril, irradiación y combinaciones de las mismas, etc. Preferiblemente, las composiciones farmacéuticas están adaptadas a administración parenteral (especialmente intravenosa, intraarterial o transdérmica). Se considera que la administración intravenosa es de particular importancia. Preferiblemente el agente de unión a vWF está en forma de una forma parenteral, lo más preferiblemente formas intravenosa y subcutánea.
Para que sea adecuada como formulación farmacéutica, la formulación de la invención normalmente comprenderá el polipéptido usado en la invención (es decir, el agente activo) en una razón adecuada con respecto al volumen. Por ejemplo, para inyección subcutánea la concentración de agente activo puede ser superior, con el fin de permitir que se administre la dosis farmacéutica necesaria en un volumen más pequeño, en comparación con una formulación para inyección intrevenosa. Sin embargo, en algunas realizaciones la concentración de agente activo será idéntica para inyección subcutánea o intravenosa, y puede estar en los intervalos a modo de ejemplo definidos en el presente documento.
En algunas realizaciones, las formulaciones de la invención pueden comprender agentes adicionales, por ejemplo agentes activos adicionales, excipientes, estabilizadores, conservantes tales como agentes antimicrobianos, etc. Las formulaciones de la invención se encuentran preferiblemente en una dosis que se aplica a un paciente que lo necesita. No obstante, el modo de administración particular y la dosificación puede seleccionarlos el médico encargado teniendo en cuenta los detalles del paciente, especialmente la edad, el peso, el estilo de vida, el nivel de actividad y el estado médico general según sea apropiado. Más específicamente, ALX-0081 se administra por vía intravenosa o por vía subcutánea en un intervalo de dosis de 24 h. Incluso más preferiblemente, ALX-0081 se administra por vía intravenosa o por vía subcutánea en un intervalo de dosis de 24 h tras la consideración de la actividad de agregación, por ejemplo medida mediante RIPA, agregación plaquetaria inducida por ristocetina -(Favaloro EJ. Clin Haematol 2001; 14: 299-319) y/o ensayo de aglutinación plaquetaria con cofactor de ristocetina (Howard MA, Firkin BG. Ristocetin - a new tool in the investigation of platelet aggregation. Thrombosis et Diathesis Haemorrhagica 1971; 26: 362-9). Por ejemplo, no se administra una dosis adicional si se estima que la actividad de agregación permanece por debajo del 10% medida mediante RIPA o permanece por debajo del 20% medida mediante RICO durante las siguientes 6 horas (inhibición clínicamente relevante).
Sin embargo, en general la dosificación de los agentes de unión a vWF puede depender de diversos factores, tales como eficacia y duración de acción del principio activo, especie de sangre caliente y/o sexo, edad, peso y condición individual del animal de sangre caliente.
Normalmente la dosificación es tal que una única dosis de un agente de unión a vWF, se estima por ejemplo basándose en resultados in vitro, o por ejemplo basándose en resultados de un estudio de escalada de la dosis para someter a prueba la toxicidad subcrónica en monos cynomolgus. Basándose en un conjunto de datos preclínicos de este tipo, puede determinarse una dosis inicial y de escalada posterior para un agente de unión a vWF. Por ejemplo una dosis puede ser de desde 0,5 - 50 mg, especialmente 1-30 mg, y se administra a un animal de sangre caliente que pesa aproximadamente 75 (+/-30) kg (pero puede ser diferente también a esta norma). Si se desea, esta dosis puede tomarse también en varias dosis parciales, opcionalmente iguales, (“mg” significa mg de fármaco por mamífero, incluyendo humano, que va a tratarse).
La dosis mencionada anteriormente, o bien administrada como una única dosis (que es una realización) o bien en varias dosis parciales, puede repetirse, tal como se mencionó anteriormente por ejemplo una vez cada seis horas, una vez cada 12 horas o una vez al día. En otras palabras, las composiciones farmacéuticas pueden administrarse en regímenes que oscilan entre terapia cada 6 horas continua hasta terapia de dosificación de intervalo más largo.
Preferiblemente, los agentes de unión a vWF se administran en dosis que son del mismo orden de magnitud que las usadas en el tratamiento adjunto en pacientes que necesitan PCI tal como se sugiere en el presente documento para ALX-0081. Por ejemplo, para los agentes de unión a vWF que contienen 12A02H1 preferidos, por ejemplo ALX-0081 y variantes funcionales de los mismos, pueden usarse dosis de agentes de unión a vWF en el intervalo de desde aproximadamente 0,5 hasta aproximadamente 40 mg, preferiblemente desde aproximadamente 1 hasta aproximadamente 35 mg o desde aproximadamente 2 hasta aproximadamente 30 mg, incluso más preferiblemente desde aproximadamente 3 hasta aproximadamente 25 mg o desde aproximadamente 4 hasta aproximadamente 20 mg, o desde aproximadamente 5 hasta aproximadamente 17,5 mg, o incluso desde aproximadamente 6 hasta aproximadamente 16 mg, o desde aproximadamente 7,5 hasta aproximadamente 15 mg, o incluso desde aproximadamente 10 hasta aproximadamente 14 mg, de manera más preferible aproximadamente 10, aproximadamente 12,5 o aproximadamente 13,8 mg, para el tratamiento agudo en pacientes humanos.
Las formulaciones en forma unitaria de dosis individual contienen preferiblemente desde aproximadamente 0,5 hasta aproximadamente 40 mg, preferiblemente desde aproximadamente 1 hasta aproximadamente 35 mg o desde aproximadamente 2 hasta aproximadamente 30 mg, incluso más preferiblemente desde aproximadamente 3 hasta aproximadamente 25 mg o desde aproximadamente 4 hasta aproximadamente 20 mg, o desde aproximadamente 5 hasta aproximadamente 17,5 mg, o incluso desde aproximadamente 6 hasta aproximadamente 16 mg, o desde aproximadamente 7,5 hasta aproximadamente 15 mg, o incluso desde aproximadamente 10 hasta aproximadamente 14 mg, de manera más preferible aproximadamente 10, aproximadamente 12,5 o aproximadamente 13,8 mg y las formulaciones que no están en forma unitaria de dosis individual contienen preferiblemente desde aproximadamente 0,5 hasta aproximadamente 40 mg, preferiblemente desde aproximadamente 1 hasta aproximadamente 35 mg, o desde aproximadamente 2 hasta aproximadamente 30 mg, incluso más preferiblemente desde aproximadamente 3 hasta aproximadamente 25 mg o desde aproximadamente 4 hasta aproximadamente 20 mg, o desde aproximadamente 5 hasta aproximadamente 17,5 mg, o incluso desde aproximadamente 6 hasta aproximadamente 16 mg, o desde aproximadamente 7,5 hasta aproximadamente 15 mg, o incluso desde aproximadamente 10 hasta aproximadamente 14 mg, de manera más preferible aproximadamente 10, aproximadamente 12,5 o aproximadamente 13,8 mg del principio activo.
Preparaciones farmacéuticas para administración parenteral son, por ejemplo, aquellas en formas unitarias de dosificación, tales como ampollas. Se preparan de manera conocida per se, por ejemplo por medio de procesos de liofilización, disolución o mezclado convencionales.
Formulaciones parenterales son especialmente fluidos inyectables que son eficaces de diversas maneras, tales como en el sitio de PCI, por vía intraarterial, por vía intramuscular, por vía intraperitoneal, por vía intranasal, por vía intradérmica, por vía subcutánea o preferiblemente por vía intravenosa. Tales fluidos son preferiblemente disoluciones o suspensiones acuosas isotónicas que pueden prepararse antes de su uso, por ejemplo a partir de preparaciones liofilizadas o concentrado que contienen el principio activo solo o junto con un portador farmacéuticamente aceptable. Las preparaciones farmacéuticas pueden esterilizarse y/o contener adyuvantes, por ejemplo conservantes, estabilizadores, agentes humectantes y/o emulsionantes, solubilizantes, sales para regular la presión osmótica y/o tampones.
Las formulaciones adecuadas para aplicación transdérmica incluyen una cantidad eficaz del principio activo con portador. Los portadores ventajosos incluyen disolventes farmacológicamente aceptables absorbibles para ayudar al
paso a través de la piel del huésped. De manera característica, los dispositivos transdérmicos están en forma de un vendaje que comprende un elemento de soporte, un depósito que contiene el compuesto opcionalmente con portadores, opcionalmente una barrera de control de la velocidad para administrar el principio activo a la piel del huésped a una velocidad controlada y predeterminada a lo largo de un periodo de tiempo prolongado, y medios para sujetar el dispositivo a la piel.
La siguiente tabla proporciona algunos ejemplos no limitativos de formulaciones basadas en tampón fosfato y citrato de la presente divulgación. Todas las formulaciones pueden ajustarse a una osmolalidad de 290 ± 60 mOsm/kg añadiendo un excipiente adecuado, si se desea. Las formulaciones pueden comprender uno cualquiera o más de los polipéptidos usados en la presente invención, por ejemplo SEQ ID NO: 1 -19, particularmente SEQ ID NO: 1.

Las concentraciones de tampón en esta tabla se entiende que abarcan opcionalmente ±5 mM. Los valores de pH se entiende que abarcan opcionalmente ±0,2. Cada uno de los tampones anteriores puede combinarse con uno o más excipientes seleccionados de por ejemplo NaCl a una concentración de por ejemplo 25, 30, 40, 50, 60, 70, 100, 150, 250 ó 500 mM; manitol a una concentración de por ejemplo el 2, el 3 o el 4% (p/v); glicina a una concentración de por ejemplo 25, 30, 40, 50, 60, 70, 100, 150, 250 ó 500 mM; trehalosa a una concentración de por ejemplo 25, 30, 40, 50, 60, 70, 100, 150, 250 ó 500 mM y sacarosa a una concentración de por ejemplo el 4, el 5, el 6, el 7, el 8 o el 9% (p/v), y/o un tensioactivo, por ejemplo Tween-80 a una concentración del 0,001%, el 0,002%, el 0,003%, el 0,004%, el 0,005%, el 0,01%, el 0,015%, el 0,02%, el 0,025%, el 0,03%, el 0,035%, el 0,04%, el 0,045%, el 0,05%, el 0,1%, el 0,2%, el 0,3%, el 0,4% o el 0,5% (v/v).
5.8 Efectos de la invención
La invención proporciona formulaciones estables de los agentes de unión a vWF tal como se reivindican, por ejemplo los dominios variables individuales de inmunoglobulina tal como se definen en el presente documento, por ejemplo SEQ ID NO: 1 -19, en particular SEQ ID NO: 1. “Estable” significa generalmente que los dominios variables individuales de inmunoglobulina no experimentan cambios físicos o químicos significativos tras el almacenamiento durante periodos de tiempo prolongados, por ejemplo de 1 mes a 36 meses, incluso si se exponen a una o más tensiones físicas o químicas tales como temperaturas elevadas (iguales o superiores a 25°C), o tensiones físicas tales como agitación o remoción. Más en particular, “estable” significa que tras el almacenamiento durante periodos prolongados (tal como se define) en condiciones (tal como se define) hay sólo una formación limitada (tal como se define) de uno o más productos de degradación, por ejemplo derivados de bajo peso molecular (LMW) (fragmentos) de los polipéptidos usados en la invención; y/o derivados químicos o modificaciones tales como por ejemplo variantes de piroglutamato; y/o derivados de alto peso molecular (HMW) (oligómeros o polímeros) formados por ejemplo mediante agregación.
El experto en la técnica está muy familiarizado con técnicas para evaluar el tamaño de proteínas, por ejemplo cromatografía de exclusión molecular-HPLC o para evaluar la formación de derivados químicos, por ejemplo HPLC
de fase inversa. El experto está familiarizado también con aparatos y herramientas de software comúnmente usados para realizar tales análisis. Por ejemplo, el experto en la técnica conoce software comúnmente usado para analizar ejecuciones cromatográficas por ejemplo en cuanto a área de pico relativa. Los ejemplos incluyen (pero no se limitan a) el sistema de HPLC Agilent 1200 equipado con el software ChemStation (Agilent Technologies, Palo Alto, EE.UU., Rev B) o el sistema de HPLC Dionex Ultimate 3000 equipado con el software Chromeleon (Dionex Corporation, Sunnyvale, CA, EE.UU., V6.8).
Las técnicas generales que pueden usarse para evaluar la estabilidad de una proteína, por ejemplo un dominio variable individual de inmunoglobulina, incluyen dispersión de luz estática, filtración de flujo tangencial, espectroscopía infrarroja con transformada de Fourier, dicroismo circular, desplegamiento de proteínas inducido por urea, fluorescencia de triptófano intrínseca y/o unión de proteína a ácido 1-anilin-8-naftalenosulfónico. Además, la formulación de la invención muestra poca o ninguna pérdida de potencia/actividad biológica en el transcurso del almacenamiento y/o bajo la influencia de una o más tensiones tal como se define en el presente documento. La actividad biológica y/o potencia puede determinarse tal como se describe por ejemplo en el documento WO2006/122825.
5.8.1 Estabilidad térmica
Las formulaciones de la presente invención se caracterizan por proporcionar una alta estabilidad térmica de los agentes de unión a vWF tal como se define en las reivindicaciones. La estabilidad térmica puede evaluarse por ejemplo determinando la temperatura de fusión (Tm). Se conocen técnicas adecuadas para determinar la temperatura de fusión e incluyen por ejemplo un ensayo de desplazamiento térmico (TSA) por ejemplo tal como se describe en el presente documento. Más específicamente, las formulaciones de la presente invención conducen a un aumento de Tm para los dominios variables individuales de inmunoglobulina tal como se determina mediante TSA en comparación con otras formulaciones. Este efecto se ejemplifica en la tabla 1 de la sección experimental.
Tal como puede determinarse a partir de la sección experimental, una alta estabilidad térmica, es decir alta Tm puede tomarse como una indicación de la estabilidad en almacenamiento.
Según la presente invención, las formulaciones de la invención tienen una influencia positiva sobre la Tm a lo largo de un amplio intervalo de valores de pH, por ejemplo entre 6,0 y 7,0 para tampón citrato, y de 6,5 a 7,5 para tampón fosfato. El efecto más ventajoso sobre la Tm puede observarse para tampón citrato a pH 6-7, y en particular para pH 6,5±0,2 y tampón fosfato a pH de 6,5 a 7,5, en particular pH 7,1±0,2.
La adición de excipientes puede tener un efecto positivo o negativo adicional sobre la Tm (tabla 1). Por ejemplo, la trehalosa puede aumentar la Tm (en el contexto de un tampón particular) por ejemplo entre 150 mM y 300 mM. Además el manitol o la sacarosa tenían un claro efecto positivo sobre la Tm. Estos excipientes pueden encontrar uso en realizaciones particulares de la invención, por ejemplo formulaciones en las que son ventajosos un agente de carga o lioprotectores. Estas realizaciones a modo de ejemplo no excluyen el uso de agentes de carga o lioprotectores conocidos adicionales, o bien solos o bien en combinación con manitol o sacarosa.
Tal como se evidencia mediante la sección experimental de esta descripción, la Tm tal como se determina mediante TSA sirve como indicador valioso de la estabilidad de los agentes de unión a vWF usados en la invención.
5.8.2 Estabilidad con respecto a la tensión mecánica
Las formulaciones de la invención se caracterizan por una alta estabilidad con respecto a la tensión mecánica, tal como tensión por agitación, remoción o cizalladura. Un posible ensayo para evaluar la estabilidad bajo tensión mecánica es monitorizar la señal de dispersión a 500 nm en un espectrofluorómetro o por medio de espectrofotometría UV, por ejemplo a 340 nm. Un aumento en la dispersión o absorción UV refleja la formación de agregados. Cuando se forman agregados (HMW), el aumento a lo largo del tiempo sigue una curva lineal para la que puede determinarse una pendiente (intensidad de dipsersión/tiempo o unidades de absorbancia/s). Preferiblemente, las formulaciones de la presente invención se caracterizan por una pendiente de menos de 0,0006, por ejemplo menos de 0,0005, por ejemplo entre 0 y 0,0004 (véanse las figuras 4A y B).
Las formulaciones que comprenden tampones citrato se prefieren particularmente y tienen un efecto positivo sobre la recuperación de proteína tras por ejemplo agitación tal como se definió anteriormente. Por ejemplo, la recuperación de masa es de al menos el 90%, el 95%, el 98% o el 100%. La recuperación de proteína se determina en comparación con el contenido en proteína total antes de someter a tensión la muestra, por ejemplo mediante agitación. Las formulaciones que comprenden tampones fosfato dan como resultado una recuperación de al menos el 75%, el 80%, el 85% o incluso más tras la agitación tal como se definió anteriormente.
A una concentración a modo de ejemplo, no limitativa de 5 mg/ml, las formulaciones de la invención sólo forman agregados reversibles en respuesta a la agitación en ausencia de Tween. Por tanto, las formulaciones de la invención impiden la formación de agregados irreversibles bajo tensión mecánica. Por consiguiente, en una realización adicional de la invención, las formulaciones de la invención pueden comprender un detergente no iónico
tal como se definió anteriormente, por ejemplo Tween-80, por ejemplo a una concentración tal como se definió anteriormente, por ejemplo entre el 0,01% y el 0,02% (v/v). La adición del detergente puede mejorar además la estabilidad física de la formulación. Por ejemplo, a una concentración a modo de ejemplo no limitativa de 5 mg/ml, la adición del detergente puede impedir la formación de agregados (reversibles e irreversibles) tal como se determina por ejemplo monitorizando la señal de dispersión a 500 nm en un espectrofluorómetro o mediante espectrofotometría UV (340 nm) (figuras 4A y B).
La estabilidad física de las formulaciones de la presente invención también puede demostrarse mediante SE-HPLC. Diferentes formulaciones no limitativas de dominios variables individuales de inmunoglobulina de la presente invención pueden soportar tensión mecánica, por ejemplo tensión por agitación, sin formar oligómeros (HMW) o productos de degradación (LMW). Las formulaciones de la invención permanecen estables sin degradación u oligomerización, tal como se determina por ejemplo tras 1,5 horas de agitación mediante análisis de SE-HPLC. No se detecta oligomerización ni degradación (por ejemplo tal como se determina mediante el perfil de RP-HPLC (sólo degradación) o SE-HPLC) en ninguna de las formulaciones. Por tanto, según una realización preferida de la invención, las formulaciones comprenden un tampón citrato y muestran una recuperación de al menos el 70%, el 75%, el 80%, el 85%, el 90%, el 95%, el 98% o incluso aproximadamente el 100%, por ejemplo en condiciones descritas anteriormente, en las que la recuperación se determina por ejemplo mediante RP-HPLC o SE-HPLC en comparación con una muestra no sometida a tensión. Ventajosamente, el excipiente en el contexto de un tampón citrato puede ser sacarosa, y la recuperación tal como se definió anteriormente es de al menos el 80%, el 85%, el 90%, el 95%, el 98% o incluso aproximadamente el 100%.
5.8.3 Pruebas de estabilidad de formulaciones líquidas
5.8.3.1 Estabilidad en almacenamiento
Las formulaciones líquidas de la invención proporcionan buena estabilidad cuando se almacenan, por ejemplo a una temperatura de -70°C, -20°C, 5°C, 25°C o 40°C, por ejemplo durante 1 -36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses. El resultado más ventajoso puede obtenerse con formulaciones basadas en tampón citrato tal como se ejemplifica en la tabla 5.
El experto en la técnica reconocerá además que el almacenamiento a 25°C, y más en particular 40°C representan condiciones de almacenamiento con tensión. Se espera que tales condiciones aumenten y aceleren cualquier signo de inestabilidad, por ejemplo inestabilidad química o física. Por tanto, un almacenamiento relativamente corto a por ejemplo 25 o 40°C proporciona una buena indicación de la estabilidad en almacenamiento a largo plazo en condiciones más suaves (por ejemplo 5°C o congelado).
5.8.32 Estabilidad en almacenamiento en cuanto a recuperación de proteína
Por ejemplo, las formulaciones de la presente invención proporcionan una recuperación de proteína de al menos el 95%, por ejemplo al menos el 96, el 97, el 98, el 99 o incluso aproximadamente el 100% tras el almacenamiento a una temperatura de entre -70°C y 40°C. La recuperación de proteína puede determinarse mediante cualquier medio conocido para cuantificar proteínas, por ejemplo mediante RP-HPLC o SE-HPLC, tal como se ejemplifica en la tabla 5 en comparación con una muestra de referencia mantenida a -70°C. Estos resultados pueden observarse por ejemplo tras el almacenamiento a la temperatura indicada de 1 mes, 1,5 meses, 3 meses, 6 meses, 9 meses, 12 meses, 18 meses, 24 meses, 30 meses o incluso a los 36 meses.
5.8.3.3 Estabilidad en almacenamiento en cuanto a derivados químicos/productos de degradación
Además, las formulaciones de la presente invención minimizan la producción de derivados químicos, por ejemplo variantes de piroglutamato, de menos del 5,0% de tamaño de pico tal como se determina por ejemplo mediante RP-HPLC (véase la tabla 5). En este tipo de análisis, el área de un pico dado se compara con el área total del cromatograma, y se asigna un área relativa a cada pico. El experto en la técnica conoce medios de análisis adecuados, por ejemplo software adecuado, para analizar los cromatogramas (los ejemplos específicos, no limitativos incluyen el sistema de HPLC Agilent 1200 equipado con el software ChemStation (Agilent Technologies, Palo Alto, EE.Uu ., Rev B) o el sistema de HPLC Dionex Ultimate 3000 equipado con el software Chromeleon (Dionex Corporation, Sunnyvale, CA, EE.UU., V6.8). Por tanto, preferiblemente, la variante de piroglutamato contribuye a un área de pico de menos del 5%, preferiblemente menos del 4,6%, por ejemplo el 4,5, el 4,3, el 4,2, el 4,0 o incluso menos del 3,8% tal como se determina mediante RP-HPLC tras el almacenamiento a temperaturas de entre -70°C y 40°C, por ejemplo 40°C, por ejemplo tras el almacenamiento durante una duración definida anteriormente, por ejemplo 1 mes.
Las formulaciones de la presente invención también minimizan la oxidación, tal como la formación de productos oxidados (tal como se determina por ejemplo mediante RP-HPLC) a lo largo de un periodo de almacenamiento tal como se definió anteriormente, por ejemplo 1 mes a una temperatura de entre -70°C y 40°C (véase la tabla 5). Por tanto, las formulaciones de la presente invención dan como resultado variantes de oxidación con un área de pico de
menos del 3%, preferiblemente menos del 2,7%, preferiblemente menos del 2,5%, por ejemplo menos del 2,3%, el 2,2%, por ejemplo el 2,0, o incluso menos tal como el 1,7% o el 1,5% tras el almacenamiento a temperaturas de entre -702C y 40°C, por ejemplo 40°C, por ejemplo tras el almacenamiento durante una duración tal como se definió anteriormente, por ejemplo 1 mes (tal como se determina por ejemplo mediante RP-HPLC).
5.8.3.4 Estabilidad en almacenamiento en cuanto a oligomerización
Las formulaciones de la invención también proporcionan estabilidad en almacenamiento, de manera que no se forma material oligomérico soluble aparente (tal como se define por ejemplo mediante SE-HPLC) a temperaturas de almacenamiento de entre -70°C y 40°C, tras duraciones de almacenamiento definidas anteriormente, por ejemplo 1 mes; o se forma menos del 1%, preferiblemente menos del 0,5%, por ejemplo el 0,3% de material oligomérico soluble (tal como se define por ejemplo mediante SE-HPLC) a temperaturas de almacenamiento de entre -70°C y 40°C, por ejemplo 40°C, tras duraciones de almacenamiento definidas anteriormente, por ejemplo 1 mes.
La presente invención tiene también el efecto de proporcionar un índice de agregación tal como se determina mediante valores de absorbancia [(100xA340)/(A280-A340)] que permanecen por debajo de 0,15, preferiblemente por debajo de 0,1 tras el almacenamiento a -70°C o 40°C durante un almacenamiento de una duración definida anteriormente, por ejemplo 1 mes.
5.8.3.5 Estabilidad en almacenamiento tal como se refleja en la recuperación del producto principal
Las formulaciones de la invención tienen el efecto de que el área de pico de producto principal, tal como se determina por ejemplo mediante RP-HPLC (véase la tabla 5) es de aproximadamente el 90% tras el almacenamiento entre -70°C y 40°C tras una duración de almacenamiento indicada anteriormente, por ejemplo 1 mes; o el pico de producto principal, tal como se determina por ejemplo mediante RP-HPLC (véase la tabla 5) es de al menos el 85%, 0 más, tal como el 86%, el 87% o el 88%. Más preferiblemente, el pico principal es el 90%, el 92% o el 95%, por ejemplo al menos el 97%, más preferiblemente el 100% tras el almacenamiento entre -70°C y 40°C, por ejemplo 40°C tras una duración de almacenamiento indicada anteriormente, por ejemplo 1 mes; o el pico de producto principal, tal como se determina por ejemplo mediante SE-HPLC es al menos el 85%, al menos el 90%, preferiblemente al menos el 95%, por ejemplo al menos el 98% o incluso aproximadamente el 100% tras el almacenamiento entre -70°C y 40°C, por ejemplo 40°C, tras una duración de almacenamiento indicada anteriormente, por ejemplo 1 mes.
Las formulaciones según la presente invención tienen también el efecto de que el área de pico principal tal como se determina mediante RP-HPLC tras el almacenamiento por ejemplo a una concentración de hasta 20 mg/ml a entre -70°C y 25°C durante entre 1 y 3 meses permanece sin cambios en comparación con la formulación antes del almacenamiento, y representa al menos el 90%, más preferiblemente al menos el 95% de los picos totales, en las que la muestra de referencia tiene un pico principal de por ejemplo el 95%. Tras el almacenamiento a 40°C durante 1 mes la formulación de la presente invención retiene el pico principal tal como se determina mediante RP-HPLC de al menos el 80%, el 85% o el 90%; tras el almacenamiento durante 2 meses de al menos el 80% o el 85%, y tras el almacenamiento durante 3 meses de al menos el 75% o el 80%.
Además, tal como se determina mediante clEF, la formulación de la presente invención tiene el efecto de proporcionar recuperación del producto principal tras el almacenamiento a una concentración de por ejemplo hasta 20 mg/ml durante entre 1-3 meses a una temperatura de entre -70°C y 40°C que es comparable a la muestra de referencia (formulación sin almacenamiento, el pico principal es de al menos el 98%), por ejemplo el pico principal es al menos el 85%, o más, tal como el 86%, el 87% o el 88%. Más preferiblemente, el pico principal es el 90%, el 92% o el 95%, por ejemplo al menos el 97%, más preferiblemente el 100% tras el almacenamiento entre -70°C y 40°C.
5.8.3.6 Estabilidad en condiciones de congelación-descongelación
Además de proporcionar estabilidad de las formulaciones en condiciones de almacenamiento que permanecen contantes a lo largo del tiempo (por ejemplo almacenamiento a 5°C), o incluyen un único ciclo de FT (por ejemplo almacenamiento a -20°C o -70°C), un efecto adicional de la invención es la estabilidad en condiciones de ciclos de FT repetidos. Cada transición entre estado líquido y congelado y viceversa impone condiciones particularmente estresantes sobre los dominios variables individuales de inmunoglobulina.
Las formulaciones de la invención tienen también el efecto de proporcionar buena estabilidad en condiciones de FT. Por ejemplo las formulaciones de la invención pueden someterse a por ejemplo 10 ciclos de FT entre -70°C y temperatura ambiente (por ejemplo 25°C), o -20°C y temperatura ambiente. Los dominios variables individuales de inmunoglobulina comprendidos en las formulaciones soportarán estas condiciones sin deterioro significativo, tal como se determina por ejemplo mediante RP-HPLC o SE-HPLC. Se evaluó el efecto de ciclos de FT repetitivos sobre diferentes realizaciones no limitativas de formulaciones de la invención, y revelan que en todos los casos se ha conservado la integridad química y física de los agentes de unión a vWF, por ejemplo dominios variables individuales de inmunoglobulina. La recuperación global estaba en el intervalo entre el 95 y el 100%, preferiblemente al menos el 95, el 98 o el 99%. La proporción relativa de los diferentes picos permaneció sin cambios en
comparación con un control sometido a sólo un ciclo de FT.
Más específicamente, a una concentración de entre 5 mg/ml y 20 mg/ml, 10 ciclos de FT dieron como resultado una recuperación (tal como se determinó basándose en por ejemplo el área de pico total, es decir AU de polipéptido, tal como se determina mediante o bien RP-HPLC o bien s E-HPLC que es de al menos el 90%, el 95%, el 98% o el 100%; en la que en una realización particular el perfil de RP-HPLC o SE-HPLC estaba sin cambios en comparación con una muestra de referencia (1 ciclo de FT).
5.8.3.7 Estabilidad en cuanto a potencia
El experto en la técnica conoce diversos modos para determinar la potencia de agentes de unión a vWF, en particular dominios variables individuales de inmunoglobulina, más específicamente polipéptidos según una cualquiera de SEQ ID NO: 1-19, por ejemplo SEQ ID NO: 1 (véase, por ejemplo, la sección experimental del documento WO2006/122825, por ejemplo los ejemplos 3-6, 18 y 19, o la sección experimental del documento WO2009/115614).
En una realización, la potencia del polipéptido usado en la presente invención puede determinarse mediante la unión a su antígeno mediante un ensayo convencional, por ejemplo ELISA, Biacore, RIA, FACS, etc.
La potencia de los agentes de unión a vWF siguió siendo aceptable en las formulaciones de la invención tal como se sometió a prueba en condiciones con tensión, es decir 4 semanas de almacenamiento a 40°C.
5.8.3.8 Estabilidad en cuanto a compatibilidad
Las formulaciones de la presente invención también son compatibles con una gama de diferentes diluyentes. Por ejemplo, las formulaciones pueden mezclarse/diluirse con tales diluyentes, sin afectar a la estabilidad física y química de los dominios variables individuales de inmunoglobulina.
Por tanto, las formulaciones de la presente invención también proporcionan estabilidad a lo largo de un amplio intervalo de concentraciones, tal como se define en el presente documento.
5.8.3.9 Sumario de efectos estabilizantes
Las formulaciones de la presente invención tienen el efecto de mantener la integridad química y física de los polipéptidos usados en la presente invención incluso tras un almacenamiento prolongado, por ejemplo durante duraciones definidas anteriormente, a temperaturas de entre -70°C y 25°C.
El almacenamiento de dominios variables individuales de inmunoglobulina tal como se definen en el presente documento, en particular ALX-0081 a -70°C durante 1 mes no afectó a sus características fisicoquímicas para cualquiera de las formulaciones de la invención, en particular los ejemplos no limitativos de tampones sometidos a prueba en la sección experimental. El almacenamiento no tuvo un efecto significativo sobre los perfiles de RP-HPLC, SE-HPLC o clEF.
5.8.4 Pruebas de estabilidad de formulaciones liofilizadas
Además, la invención proporciona formulaciones estables de los agentes de unión a vWF tal como se definen en las reivindicaciones, por ejemplo SEQ ID NO: 1-19, preferiblemente que comprenden SEQ ID NO: 1, que son particularmente útiles para la liofilización. Las formulaciones de la invención dieron como resultado una solubilidad mejorada y estabilidad en almacenamiento mejorada tras la liofilización.
5.8.4.1 Estabilidad en almacenamiento
Las formulaciones de la invención pueden proporcionar una buena estabilidad tras la liofilización cuando se almacenan, por ejemplo a una temperatura de -70°C, -20°C, 5°C, 25°C o 40°C, por ejemplo durante 1 -36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses. Los resultados más ventajosos pueden obtenerse con formulaciones basadas en tampón citrato, por ejemplo las formulaciones 3 y 7 tal como se ejemplifica en la sección experimental (por ejemplo buena formación de la torta y sin signos visuales de deterioro, figura 6). El experto en la técnica puede reconocer que en la discusión a continuación los valores preferidos reflejan composiciones de tampón citrato, por ejemplo tal como se ejemplifican en la tabla 8.
El experto también reconocerá que el almacenamiento a 25°C, y más en particular 40°C representan condiciones de almacenamiento con tensión. Se espera que tales condiciones aumenten y aceleren cualquier signo de inestabilidad, por ejemplo inestabilidad química o física. Por tanto, un almacenamiento relativamente corto a por ejemplo 25°C o 40°C proporciona una buena indicación de la estabilidad en almacenamiento prolongada en condiciones más suaves (por ejemplo 5°C o congelado).
5.8.4.2 Estabilidad en almacenamiento en cuanto a recuperación de proteína
Por ejemplo, las formulaciones de la presente invención proporcionan una recuperación de proteína tras la liofilización de al menos el 95%, por ejemplo al menos el 96, el 97, el 98, el 99 o incluso aproximadamente el 100% tras el almacenamiento a una temperatura de entre -702C y 402C. La recuperación de proteína puede determinarse mediante cualquier medio conocido para cuantificar proteínas, por ejemplo mediante el contenido, RP-HPLC o SE-HPLC. Estos resultados pueden observarse por ejemplo tras el almacenamiento a la temperatura indicada de 1-36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses.
5.8.4.3 Estabilidad en almacenamiento en cuanto a derivados químicos/productos de degradación
Además, las formulaciones de la presente invención pueden prevenir y minimizar la producción de derivados químicos tras la liofilización, tal como se confirma mediante por ejemplo mediante SE-HPLC.
5.8.4.4 Estabilidad en almacenamiento en cuanto a oligomerización
Las formulaciones de la invención pueden proporcionar también estabilidad en almacenamiento tras la liofilización, de manera que no se forma material oligomérico soluble aparente (tal como se define por ejemplo mediante SE-HPLC) a temperaturas de almacenamiento de entre -70°C y 40°C, tras duraciones de almacenamiento definidas anteriormente, por ejemplo 1 mes; o se forma menos del 1%, preferiblemente menos del 0,5%, por ejemplo el 0,3% de material oligomérico soluble (tal como se define por ejemplo mediante SE-HPLC) a temperaturas de almacenamiento de entre -70°C y 40°C, por ejemplo 40°C, tras duraciones de almacenamiento definidas anteriormente, por ejemplo 1 -36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses.
5.8.4.5 Estabilidad en almacenamiento tal como se refleja en la recuperación de producto principal
Las formulaciones de la invención también pueden tener el efecto de que tras la liofilización el pico de producto principal, tal como se determina por ejemplo mediante SE-HPLC (véase la tabla 18 y las tablas 27-29) es de aproximadamente el 100% tras el almacenamiento entre -70°C y 40°C tras una duración de almacenamiento tal como se indicó anteriormente, por ejemplo 1, 3, 6, 9, 12, 18 ó 24 meses; o el pico de producto principal, tal como se determina por ejemplo mediante SE-HPLC (véase la tabla 18 y las tablas 27-29) es de al menos el 85%, o más, tal como el 86%, el 87% o el 88%. Más preferiblemente, el pico principal es el 90%, el 92% o el 95%, por ejemplo al menos el 97%, más preferiblemente el 100% tras el almacenamiento entre -70°C y 40°C, por ejemplo 25°C tras una duración de almacenamiento tal como se indicó anteriormente, por ejemplo 1, 3, 6, 9, 12, 18 ó 24 meses; o el pico de producto principal, tal como se determina por ejemplo mediante SE-HPLC es de al menos el 85%, al menos el 90%, preferiblemente al menos el 95%, por ejemplo al menos el 98% o incluso aproximadamente el 100% tras el almacenamiento entre -70°C y 40°C, por ejemplo 40°C, tras una duración de almacenamiento tal como se indicó anteriormente, por ejemplo 1,3, 6, 9, 12, 18 ó 24 meses.
Las formulaciones según la presente invención tienen también el efecto de que tras la liofilización el pico principal tal como se determina mediante RP-HPLC tras el almacenamiento por ejemplo a una concentración de 12,5 mg/ml a entre -70°C y 40°C durante entre 1 y 12 meses permanece sin cambios en comparación con la formulación antes del almacenamiento, y representa al menos el 90%, más preferiblemente al menos el 93% de los picos totales, en el que la muestra de referencia tiene un pico principal de por ejemplo el 93% (véase la tabla 15). Tras el almacenamiento tras la liofilización a 40°C durante hasta 12 meses, la formulación de la presente invención retiene el pico principal tal como se determina mediante RP-HPLC de al menos el 91%, el 92% o el 93%.
Además, tal como se determina mediante clEF (véanse las tablas 27-29), las formulaciones de la presente invención tienen el efecto de proporcionar tras la liofilización la recuperación del producto principal tras el almacenamiento a una concentración de por ejemplo 12,7 mg/ml durante entre 1-24 meses a una temperatura de entre -70°C y 40°C que es comparable con la muestra de referencia (formulación sin almacenamiento, el pico principal es al menos el 96%), por ejemplo el pico principal es al menos el 85%, o más, tal como el 86%, el 87% o el 88%. Más preferiblemente, el pico principal es el 90%, el 92%, el 93%, el 94%, el 95% o el 96%, por ejemplo al menos el 97%, más preferiblemente el 100% tras el almacenamiento entre -70°C y 40°C.
5.8.4.6 Estabilidad en condiciones de congelación-descongelación
Las formulaciones de la invención tienen también el efecto de proporcionar buena estabilidad tras la liofilización en condiciones de FT. Por ejemplo las formulaciones de la invención pueden someterse a por ejemplo 5 ciclos de FT entre -20°C y temperatura ambiente (por ejemplo 25°C). Los dominios variables individuales de inmunoglobulina comprendidos en las formulaciones soportarán estas condiciones sin deterioro significativo, tal como se determina por ejemplo mediante RP-HPLC o SE-HPLC. En todos los casos, se ha conservado la integridad física y química de los agentes de unión a vWF, por ejemplo dominios variables individuales de inmunoglobulina. La recuperación global estaba en el intervalo entre el 95 y el 100%, preferiblemente al menos el 95, el 98 o el 99% en comparación con una muestra de control líquida almacenada a -70°C.
Más específicamente, a una concentración de 16 mg/ml, 5 ciclos de FT dieron como resultado una recuperación (tal como se determina basándose en por ejemplo el área total, es decir AU) de polipéptido, tal como se determina mediante o bien RP-HPLC o bien SE-HPLC que es de al menos el 90%, el 95%, el 98%, el 99% o el 100%; en el que en una realización particular el perfil de RP-HPLC o SE-HPLC permanecía sin cambios en comparación con una muestra de referencia (muestra de control líquida almacenada a -70°C) (véase la tabla 12).
5.8.4.7 Estabilidad en cuanto a potencia
El experto en la técnica conoce diversos modos para determinar la potencia de agentes de unión a vWF, en particular dominios variables individuales de inmunoglobulina, más específicamente polipéptidos según una cualquiera de SEQ ID NO: 1-19, por ejemplo SEQ ID NO: 1 (véase, por ejemplo, la sección experimental del documento WO2006/122825, por ejemplo los ejemplos 3-6, 18 y 19, o la sección experimental del documento WO2009/115614). La potencia de los agentes de unión a vWF tras la liofilización no se vio afectada tras ciclos de FT repetidos en las formulaciones. En particular, la potencia de agentes de unión a vWF permaneció estable en las formulaciones de la invención tal como se sometió a prueba en condiciones con tensión, es decir hasta 12 meses de almacenamiento a 40°C (tabla 23) e incluso hasta 24 meses de almacenamiento a 40°C (tabla 29). En una realización, la potencia del polipéptido usado en la presente invención tras la liofilización puede determinarse mediante unión a su antígeno mediante un ensayo convencional, por ejemplo ELISA, Biacore, RIA, FACS, etc. Más específicamente, en las formulaciones de la presente invención al menos el 80%, preferiblemente al menos el 90%, más preferiblemente al menos el 95% o incluso al menos el 99% del agente de unión a vWF retiene su actividad de unión tras el almacenamiento en las condiciones de tensión anteriores en comparación con la actividad de unión antes del almacenamiento.
En un aspecto adicional, las formulaciones de la presente invención casi no presentan pérdida de actividad biológica cuando se compara la formulación líquida de ALX-0081 con la formulación liofilizada, tal como se evalúa mediante diversos ensayos inmunológicos incluyendo, pero sin limitarse a ensayo de Biacore, ensayo de inmunoabsorción ligado a enzimas (ELISA), ensayo de actividad de cofactor inducida por ristocetina (RICO) y/o ensayo basado en Gyrolab (véanse la sección 7.13 y la tabla 24).
5.8.4.8 Sumario de efectos estabilizantes
Las formulaciones de la presente invención tienen el efecto tras la liofilización de mantener la integridad física y química de los polipéptidos usados en la presente invención, en particular ALX-0081, es decir incluso tras almacenamiento prolongado, por ejemplo durante duraciones definidas anteriormente, a temperaturas de entre -70°C y 40°C, el perfil de pureza/impureza del producto permanece esencialmente sin cambios. Por ejemplo, el almacenamiento prolongado tras la liofilización no tuvo un efecto significativo sobre los perfiles de RP-HPLC, SE-HPLC o clEF tal como queda respaldado por la sección experimental.
5.9 Métodos de la invención
Los agentes de unión a vWF usados en la invención pueden producirse mediante cualquier método usado comúnmente. Los ejemplos típicos incluyen la expresión recombinante en sistemas huésped adecuados, por ejemplo bacterias o levadura. Los agentes de unión a vWF se someterán a un régimen de purificación adecuado antes de formularse según la presente invención.
La presente invención abarca métodos de producción de las formulaciones tal como se definen en las reivindicaciones.
Las etapas de purificación y formulación pueden coincidir, por ejemplo cuando los agentes de unión a vWF usados en la invención se eluyen de una columna usando un tampón usado en la presente invención. Alternativamente, las formulaciones de la invención pueden prepararse intercambiando un tampón por cualquier medio adecuado, por ejemplo medios ampliamente usados en la técnica tales como diálisis, ultrafiltración, etc.
En algunas realizaciones el método de producción de una formulación de la invención también puede referirse a la reconstitución de una formulación liofilizada o secada por pulverización, por ejemplo mediante la adición de agua o un tampón adecuado (que puede comprender opcionalmente excipientes adicionales).
Los métodos para preparar una formulación según la presente invención pueden abarcar etapas adicionales, tales como llenado de viales adecuados para uso clínico, tales como recipientes sellados y/o confeccionando una forma unitaria de dosificación. Los métodos pueden comprender también etapas adicionales tales como secado por pulverización, liofilización o congelación, por ejemplo congelación a granel. La invención también abarca los recipientes, formas unitarias de dosificación u otros productos obtenibles mediante cualquiera de los métodos tal como se definen en las reivindicaciones.
Las formulaciones de la presente invención pueden usarse para almacenar los agentes de unión a vWF, por ejemplo ISVD tal como se define en el presente documento. Por tanto, la invención abarca un método de almacenamiento de
un agente de unión a vWF tal como se define en las reivindicaciones, caracterizado por el uso de una formulación tal como se define en el presente documento. Más específicamente, la divulgación abarca métodos para estabilizar un agente de unión a vWF tal como se define en las reivindicaciones para almacenamiento, que comprende por ejemplo la preparación de una formulación tal como se describe en el presente documento. El almacenamiento puede ser de 1-36 meses, tal como 1, 1,5, 3, 6, 9, 12, 18, 24, 30 ó 36 meses, por ejemplo al menos 12 o incluso 24 meses, opcionalmente a una temperatura de entre -70°C y 40°C, tal como -70°C, -20°C, 5°C, 25°C o 40°C, preferiblemente una temperatura de entre -70°C y 25°C, más preferiblemente a una temperatura de entre -20°C y 5°C. Por tanto, el almacenamiento puede abarcar congelación, secado por congelación (liofilización) y/o secado por pulverización. Los métodos de almacenamiento pueden comprender además la evaluación de la integridad física y química de los agentes de unión a vWF tal como se define en el presente documento.
La presente divulgación también se refiere a métodos para analizar formulaciones que comprenden al menos uno de los agentes de unión a vWF tal como se definen en el presente documento. Las formulaciones pueden analizarse para detectar cualquier signo de inestabilidad química o física de los agentes de unión a vWF tal como se definen en el presente documento. Por ejemplo, las formulaciones pueden evaluarse para detectar la presencia de productos de degradación, por ejemplo derivados de bajo peso molecular tales como fragmentos proteolíticos; y/o para detectar derivados químicos, por ejemplo variantes de piroglutamato; y/o para detectar derivados de alto peso molecular tales como agregados, aglomerados, etc. La formulación también puede evaluarse para detectar el contenido en proteína total y/o la potencia. Cada uno de los diversos métodos de ensayo a los que se hace referencia en el presente documento puede usarse en el método de análisis de la presente divulgación.
Por tanto, la presente divulgación también se refiere a un método para monitorizar y/o evaluar la calidad y/o estabilidad de una formulación, por ejemplo durante uno o más de fabricación, almacenamiento y uso. La divulgación también se refiere a un método de control de calidad de una formulación, por ejemplo para evaluar que la formulación cumple las especificaciones del producto tal como se describe adicionalmente en el presente documento. La divulgación en cualquiera de estos aspectos comprende uno o más seleccionados de la comparación con una o más muestras de referencia, el análisis de la variación lote a lote y la monitorización en curso de un proceso de producción.
La presente invención se refiere a cualquier producto que esté asociado con las formulaciones de la presente invención, por ejemplo porque las comprende, o porque es necesario para su producción o confección, sin ninguna limitación.
Por ejemplo, la presente invención se refiere a un artículo de fabricación, por ejemplo un recipiente sellado que comprende una o más de las formulaciones según la presente invención. La invención también se refiere a una forma de dosificación unitaria farmacéutica, por ejemplo una forma de dosificación adecuada para administración parenteral a un paciente, preferiblemente un paciente humano, que comprende una o más de la formulación según cualquier realización descrita en el presente documento. La forma unitaria de dosificación puede estar por ejemplo en el formato de una jeringa precargada, una ampolla, cartucho o un vial. La jeringa, ampolla, cartucho o vial puede fabricarse de cualquier material adecuado, tal como vidrio o plástico y puede incluir materiales de caucho, tales como tapones de caucho para los viales y émbolos de caucho y sellos de caucho para jeringas y cartuchos. La invención también se refiere a un kit que comprende una o más de las formulaciones según la presente invención. El kit puede comprender además instrucciones para su uso y/o un prospecto clínico.
En cualquier realización de los productos tal como se definen en el presente documento, la invención también abarca la presencia de material de envasado, instrucciones para su uso y/o prospectos clínicos, por ejemplo según se requiera por los aspectos regulatorios.
5.10 Definiciones
5.10.1 Identidad
Para los propósitos de comparar dos o más secuencias de aminoácidos, el porcentaje de "identidad de secuencia" entre una primera secuencia de aminoácidos y una segunda secuencia de aminoácidos (también denominada en el presente documento "identidad de aminoácidos") puede calcularse dividiendo [el número de residuos de aminoácido en la primera secuencia de aminoácidos que son idénticos a los residuos de aminoácido en las posiciones correspondientes en la segunda secuencia de aminoácidos] entre [el número total de residuos de aminoácido en la primera secuencia de aminoácidos] y multiplicando por [700%], en el que cada deleción, inserción, sustitución o adición de un residuo de aminoácido en la segunda secuencia de aminoácidos, en comparación con la primera secuencia de aminoácidos, se considera como una diferencia en un único residuo de aminoácido (posición), es decir como una “diferencia de aminoácido” tal como se define en el presente documento.
Alternativamente, el grado de identidad de secuencia entre dos o más secuencias de aminoácidos puede calcularse usando un algoritmo informático conocido para la alineación de secuencias tal como NCBI Blast v2.0 usando parámetros convencionales.
Algunas otras técnicas, algoritmos informáticos y parámetros para determinar el grado de identidad de secuencia se describen por ejemplo en los documentos WO04/037999, EP0967284, EP1085089, WO00/55318, WO00/78972, WO98/49185 y GB2357768-A.
Habitualmente, para el propósito de determinar el porcentaje de “identidad de secuencia” entre dos secuencias de aminoácidos según el método de cálculo explicado de manera resumida anteriormente en el presente documento, la secuencia de aminoácidos con el mayor número de residuos de aminoácido se tomará como la “primera” secuencia de aminoácidos, y la otra secuencia de aminoácidos se tomará como la “segunda” secuencia de aminoácidos.
Además, al determinar el grado de identidad de secuencia entre dos secuencias de aminoácidos, el experto puede tener en cuenta las denominadas sustituciones de aminoácidos “conservativas”, que pueden describirse generalmente como sustituciones de aminoácido en las que un residuo de aminoácido se reemplaza por otro residuo de aminoácido de estructura química similar y que tiene poca o esencialmente ninguna influencia sobre la función, actividad u otras propiedades biológicas del polipéptido. Tales sustituciones de aminoácidos conservativas se conocen bien en la técnica, por ejemplo a partir de los documentos WO04/037999, GB2357768-A, WO98/49185, WO00/46383 y WO01/09300; y los tipos y/o combinaciones (preferidos) de tales sustituciones pueden seleccionarse basándose en las enseñanzas pertinentes del documento WO04/037999 así como WO98/49185 y de las referencias adicionales citadas en los mismos. Tales sustituciones conservativas son preferiblemente sustituciones en las que un aminoácido dentro de los siguientes grupos (a) -(e) se sustituye por otro residuo de aminoácido dentro del mismo grupo: (a) residuos alifáticos pequeños, apolares o ligeramente polares: Ala, Ser, Thr, Pro y Gly; (b) residuos polares, cargados negativametne y sus amidas (no cargadas): Asp, Asn, Glu y Gln; (c) residuos polares, cargados positivamente: His, Arg y Lys; (d) residuos alifáticos grandes, apolares: Met, Leu, Ile, Val y Cys; y (e) residuos aromáticos: Phe, Tyr y Trp. Sustituciones conservativas particularmente preferidas son las siguientes: Ala a Gly o a Ser; Arg a Lys; Asn a Gln o a His; Asp a Glu; Cys a Ser; Gln a Asn; Glu a Asp; Gly a Ala o a Pro; His a Asn o a Gln; Ile a Leu o a Val; Leu a Ile o a Val; Lys a Arg, a Gln o a Glu; Met a Leu, a Tyr o a Ile; Phe a Met, a Leu o a Tyr; Ser a Thr; Thr a Ser; Trp a Tyr; Tyr a Trp; y/o Phe a Val, a Ile o a Leu. Cualquier sustitución de aminoácido aplicada a los polipéptidos descritos en el presente documento puede basarse también en el análisis de las frecuencias de variaciones de aminoácidos entre proteínas homólogas de diferentes especies desarrollado por Schulz et al., Principles of Protein Structure, Springer-Verlag, 1978, en los análisis de potenciales de formación de estructuras desarrollados por Chou y Fasman, Biochemistry 13: 211, 1974 y Adv. Enzymol., 47: 45-149, 1978, y en el análisis de patrones de hidrofobicidad en proteínas desarrollado por Eisenberg et al., Proc. Natl. Acad. Sci. USA 81: 140-144, 1984; Kyte & Doolittle; J Molec. Biol. 157: 105-132, 1981 y Goldman et al., Ann. Rev. Biophys. Chem. 15: 321-353, 1986. Se facilita información sobre la estructura primaria, secundaria y terciaria de Nanobodies® en la descripción en el presente documento y en la técnica anterior general mencionada anteriormente. Además, para este propósito, la estructura cristalina de un dominio Vhh de una llama se facilita por ejemplo por Desmyter et al., Nature Structural Biology, vol. 3, 9, 803 (1996); Spinelli et al., Nature Structural Biology (1996); 3, 752-757; y Decanniere et al., Structure, vol. 7, 4, 361 (1999). Puede encontrarse información adicional sobre algunos de los residuos de aminoácido que en dominios VH convencionales forman la superficie de contacto VH/VL y posibles sustituciones camelizantes en estas posiciones en la técnica anterior mencionada anteriormente.
6. Abreviaturas
API Principio farmacéutico activo
clEF Isoelectroenfoque capilar
DLS Dispersión dinámica de la luz
DOE Diseño de experimentos
DP Producto farmacológico
DS Sustancia farmacológica
FT Congelación-descongelación
HMW Alto peso molecular
LMW Bajo peso molecular
MALS Dispersión de luz multiángulo
HR Humedad relativa
RPC Cromatografía de fase inversa
RP-HPLC Cromatografía de líquidos de alto rendimiento de fase inversa
SE-HPLC Cromatografía de líquidos de alto rendimiento de exclusión molecular
SOP Procedimiento de funcionamiento convencional
Tm Temperatura de fusión (°C)
TSA Ensayo de desplazamiento térmico
vWF Factor de von Willebrand
WFI Agua para inyección
La invención se describirá adicionalmente ahora por medio de los siguientes aspectos, ejemplos y figuras no limitativos preferidos:
7. EJEMPLOS
Se diseñó un conjunto de experimentos con el fin de obtener un tampón de formulación mejorado, destinado a satisfacer una amplia gama de objetivos diferentes y aparentemente incongruentes. En particular, se proporcionan formulaciones a modo de ejemplo en el presente documento que son capaces de mantener la estabilidad, actividad biológica, pureza y calidad de ALX-0081 y esto a lo largo de un periodo de tiempo prolongado, estable a diversas tensiones tales como congelación, liofilización, calor y/o reconstitución.
Se ha presentado DS de ALX-0081 contemporánea como una formulación líquida que contiene 5 mg/ml del principio farmacéutico activo (API) en un tampón basado en fosfato (D-PBS) que contiene glicina 200 mM y Tween-80 al 0,02% (v/v), pH 7,1 (DS). Aunque esta formulación se ha aplicado durante ensayos clínicos iniciales, puede mejorarse de varias maneras. En primer lugar, la concentración relativamente baja probablemente necesitaría múltiples inyecciones subcutáneas (suponiendo que el volumen por inyección subcutánea se restringe a aproximadamente 1 ml), reduciendo así la comodidad de uso para el paciente. En segundo lugar, la estabilidad en almacenamiento a 3-8°C o temperatura ambiente de la formulación actual de ALX-0081 es limitada. La vida útil de almacenamiento limitada en la presente formulación se determina principalmente mediante modificación química (véase la sección 7.2). Pueden vincularse modificaciones químicas con pérdida de potencia. Aunque puede lograse una vida útil de almacenamiento factible almacenando el producto a -20°C, sin embargo esto no se considera una opción favorable para la mayoría de los propósitos prácticos.
7.1 Métodos
Se analizaron muestras esencialmente según procedimientos de funcionamiento convencionales para evaluar el contenido, la potencia y la pureza, precipitación, concentración, degradación, agregación y potencia. Además, todas las muestras se inspeccionaron visualmente para detectar turbidez o la presencia de agregados proteicos o precipitación. El contenido en humedad residual de muestras liofilizadas específicas se determinó por medio de valoración de Karl-Fischer.
Se usaron tres programas de liofilización diferentes en el presente estudio para secar por congelación ALX-0081, una ejecución de 65 h convencional (figura 1), una ejecución de 37 h acortada y un ciclo de liofilización más largo de 66 h optimizado para reducir el contenido en humedad residual tal como se describe en la tabla 14.
En resumen, al final de procedimiento de liofilización de 37 h acortado, la temperatura de anaquel era de 20°C y se llevó a -50°C en 2 horas. A continuación, se creó un vacío de 0,04 mbar en 1 hora. Tras alcanzar el vacío de 0,04 mbar, se mantuvo la temperatura de anaquel a -50°C durante 4 horas. Tras estas 4 horas, se aumentó gradualmente la temperatura hasta 0°C en 15 horas (es decir, etapa de secado primario, que elimina agua congelada). La temperatura de anaquel de 0°C se mantuvo durante 7 horas mientras se mantenía un vacío de 0,04 mbar. Tras 7 horas, se elevó la temperatura hasta 25°C en 3 horas y posteriormente se mantuvo a 25°C durante 5 horas (es decir, etapa de secado secundario, que elimina agua no congelada). Se cerraron los viales bajo un vacío de ±0,400 mbar tras lo cual se restableció la presión normal.
Se aplicó el mismo método para la ejecución convencional de 65 h y sólo difiere en la segunda etapa de secado que se prolongó con 28 h a 25°C a vacío dando como resultado un tiempo de ciclo total de aproximadamente 65 h. En la figura 1 se muestra una visión general esquemática de las diferentes etapas en la ejecución de liofilización de 65 h convencional. Durante el proceso de liofilización, se monitorizó la temperatura de producto de tres viales en posiciones estratégicas. Finalmente, se modificó la ejecución de 65 h convencional durante la ejecución según la lectura de las sondas de temperatura dando como resultado ciclos de liofilización prolongados tal como se describe en la tabla 14.
7.2 Estabilidad química de la formulación de ALX-0081 contemporánea
RP-HPLC es uno de los métodos más informativos para evaluar la estabilidad química de una sustancia farmacéutica (DS).
RP-HPLC resolvió la DS ALX-0081 en varias especies diferentes. Además del pico principal, pudieron discernirse picos anteriores (sustancia que eluye antes del material intacto sin modificar) y varios picos posteriores. En los lotes producidos hasta ahora, los picos previos y pico posterior 1 representaron de manera consistente aproximadamente el 2% y el 3,6% de la DS respectivamente, mientras que los otros picos posteriores representaron menos del 1% del DS.
Sin embargo, durante el almacenamiento en condiciones aceleradas (+5°C) o con tensión (+25°C y 372C/+402C), la abundancia relativa de ciertas variantes relacionadas con el producto aumentó con el tiempo y la temperatura, tal como se muestra en la figura 2A. Además, el pico principal de RP-HPLC parece dividirse en varias especies diferentes durante la incubación prolongada, especialmente a temperaturas elevadas (> 25°C) (figura 2B). Los datos indican que algunas especies moleculares nuevas que eluyen antes se generan durante el almacenamiento. Las modificaciones más importantes que estaban presentes en DS ALX-0081 en el momento de la fabricación o que surgieron durante el almacenamiento son las siguientes: (i) pico anterior 1 (oxidación); (ii) pico posterior 1 (variante de nor-leu); (iii) pico posterior 2 (formación de piroglutamato) y (iv) división del pico principal (isomerización). Las modificaciones (i), (ii) y (iii) no afectaron significativamente a la potencia (datos no mostrados). En cambio, se demostró que la isomerización de los residuos de ácido aspártico en las posiciones 105 y 236 de SEQ ID NO: 1, que se encuentran en la región CDR3, es el mecanismo molecular predominante subyacente a una posible pérdida de potencia de ALX-0081 (véase (iv) anteriormente).
Algunas de las variantes relacionadas con el producto de ALX-0081, presentes en el momento de la fabricación o que surgen durante el almacenamiento, también podrían detectarse por clEF. Este fue el caso de la modificación de piroglutamato que apareció como un pico posterior (véase (iii) anteriormente). Además, de manera similar a lo que se observó en el análisis de RP-HPLC, los eventos de isomerización en la posición 105 en ambos dominios 12A02H1 dieron como resultado un ensanchamiento del pico principal y finalmente la división del pico principal de clEF (véase (iv) anteriormente).
7.3 Examen de tampones y excipientes.
Con el fin de desarrollar adicionalmente la formulación de los agentes de unión a vWF, se diseñó un conjunto complejo de experimentos que elaboran diversos parámetros que se influyen todos entre sí, incluyendo (i) diferentes tampones, (ii) a diferentes concentraciones, (iii) cada tampón a distintos pH; y (iv) cada uno combinado con diferentes excipientes.
Los sistemas de tampón deben tener una capacidad de tamponamiento tan baja como sea posible, para no perturbar significativamente el sistema de tamponamiento del cuerpo cuando se inyecta. Además, el tipo de tampón y la concentración en la actividad del principio farmacéutico activo (API) deben evaluarse con mucho cuidado.
En general, niveles aumentados de estabilidad de proteínas se han atribuido a altas temperaturas de fusión. Por consiguiente, se monitorizaron las propiedades térmicas de ALX-0081 en presencia de diversas composiciones. En particular, se realizó un experimento con TSA en 192 formulaciones isotónicas diferentes, para lo cual los resultados se incorporaron a un diseño de experimentos (DOE) para evaluar el efecto del tampón, la concentración, la fuerza iónica, el pH y los excipientes sobre la estabilidad térmica de ALX 0081. La lectura fue la temperatura de fusión (Tm) de ALX-0081, que es indicativa de la estabilidad térmica de la proteína en las diversas composiciones sometidas a prueba.
Brevemente, el ensayo de desplazamiento térmico (TSA) empleado sigue los cambios de señal de un colorante de fluorescencia, como Sypro Orange, mientras que la proteína experimenta un desplegamiento térmico. Cuando se agrega Sypro Orange a una disolución de proteína correctamente plegada, no puede unirse a ninguna superficie de la proteína y su señal de fluorescencia se extingue. Cuando la temperatura aumenta, la proteína experimenta un desplegamiento térmico y expone su región central hidrófoba. Sypro Orange se une entonces a las regiones hidrófobas y no se extingue, lo que da como resultado el aumento de la señal de fluorescencia. El ensayo se realizó en disoluciones que contenían diferentes formulaciones que van a analizarse, ALX-0081 a 0,2 mg/ml y 10x Sypro Orange. El programa consistió en los siguientes pasos: calentar hasta 37°C a una velocidad de rampa de 4,4°C/s y mantener durante 10 s; calentar hasta 90°C a una velocidad de rampa continua de 0,02°C/s (20 adquisiciones por °C); y enfriar hasta 37°C a una velocidad de rampa de 2,2°C/s y mantener durante 10 s.
El siguiente conjunto de tampones con concentraciones variables (10-200 mM), valores de pH y excipientes se exploraron en el presente documento:
- citrato pH 6,0-6,5-7,0
- histidina pH 5,5-6,0-6,5
- fosfato pH 6,5-7,0-7,5
- Tris-HCl pH 7,4-7,7-8,0
- NaCl Intervalo de concentración de 0-140 mM
- glicina Intervalo de concentración de 0-270 mM
- manitol Intervalo de concentración de 0-270 mM
- sacarosa Intervalo de concentración de 0-270 mM
- trehalosa Intervalo de concentración de 0-270 mM
Las temperaturas de fusión (Tm) obtenidas se importaron en el programa Design Expert para el análisis del experimento de examen factorial para predecir 50 formulaciones que dieron como resultado la mayor estabilidad térmica (véase la tabla 1).
Los valores más altos de Tm se predijeron para fosfato (pH 7,0-7,5) y citrato (pH 6,2-7,0) que contiene trehalosa, sacarosa, manitol o glicina. De forma totalmente inesperada, los resultados del estudio sugieren que los tampones basados en Tris-HCI (pH 7,8-8,0) e histidina-HCI (pH 6,5) regresan a temperaturas de fusión significativamente más bajas, aunque previamente se eligieron como el sistema de tampón de elección para controlar el pH de la disolución de dominios variables individuales de inmunoglobulina tal como se describe en el documento WO2010/077422.
Por consiguiente, se llegó a la conclusión de que las formulaciones de fosfato y citrato que contenían trehalosa, sacarosa, glicina o manitol, tienen un rendimiento especialmente bueno en la estabilización de los agentes de unión a vWF, por ejemplo ALX-0081.
7.4 Pruebas de solubilidad
Con el fin de evaluar si la solubilidad de ALX-0081 podría mejorarse aún más, se realizó un examen inicial en varias formulaciones. Se sometió ALX-0081 a intercambio de tampón a la formulación de interés (excluyendo Tween-80) y se concentró adicionalmente en una celda de agitación (por ejemplo, tipo Amicon) equipada con un filtro de corte de 5 kDa. Tan pronto como se produjo precipitación o turbidez visible, la muestra se filtró y se midió la concentración de proteína. La tabla 2 muestra un resumen de los resultados obtenidos.
La concentración en tampones basados en fosfato e histidina dio como resultado turbidez de la muestra y formación de precipitación a concentraciones de proteína relativamente bajas (< 10 mg/ml). En cambio, ALX-0081 permaneció físicamente estable en tampón de citrato, incluso después de alcanzar una concentración de ~56 mg/ml. Además de la inspección visual, se confirmó la ausencia de materia particulada o especies de HMW mediante microscopía de fluorescencia (tinción con Nile Red, mediante SE-HPLC y DLS). Además, someter la disolución de ~56 mg/ml a o bien 10 ciclos de FT a -20°C o -70°C, o bien almacenamiento a 4°C durante aproximadamente 1 semana no pareció afectar a la estabilidad física de la molécula tal como se evidencia mediante análisis de SE-HPLC (véanse las figuras 3A y 3B respectivamente).
La comparativamente alta solubilidad de ALX-0081 en el tampón citrato se corroboró mediante un ensayo de precipitación con PEG (datos no mostrados).
7.5 Tween-80
Para determinar si el tensioactivo no iónico polisorbato, también denominado Tween, (monolaurato de polioxietileno (N) sorbitano; en el que N = 20, 40, 60, 65, 80 u 85) se requiere en la formulación de ALX-0081, se realizaron varios experimentos de tensión por agitación en tampón citrato 50 mM a pH 6,0 y 6,5. Se evaluó el efecto de las diferentes concentraciones de Tween-80 (sin Tween-80 frente al 0,01% frente al 0,02% (v/v)) sobre la estabilidad física de ALX-0081 a 5 mg/ml monitorizando la señal de dispersión a 500 nm en un espectrofluorómetro.
Tween-80 evitó un aumento en la señal de dispersión en ambos tampones, lo que demuestra su efecto protector (figuras 4A y 4B). No se observaron diferencias significativas entre muestras que contienen el 0,01% o el 0,02% de Tween-80 (v/v). Además, los perfiles de SE-HPLC de las muestras antes y después de la agitación no mostraron diferencias: se logró una recuperación del 95-100% y no pudo detectarse oligomerización o degradación.
Basándose en estos resultados, se decidió incluir Tween-80 al 0,01% (v/v) en la formulación de agentes de unión a vWF, por ejemplo ALX-0081.
7.6 Tween
Con el fin de determinar si los otros miembros en la gama de polisorbato, que difieren en la longitud de la cadena de polioxietileno y el resto éster de ácido graso, por ejemplo Tween-20, Tween-40, Tween-60, Tween-65 y Tween-85 se requieren en la formulación de agentes de unión anti-vWF, se realizan varios experimentos de tensión por agitación en tampón de citrato 50 mM a pH 6,0 y 6,5, esencialmente como se describe en la sección 7.5 anterior. El efecto de las diferentes concentraciones de los diversos miembros de Tween (sin Tween frente al 0,01% frente al 0,02% (v/v)) sobre la estabilidad física del agente de unión a vWF se evalúa a 5 mg/ml monitorizando la señal de dispersión a 500 nm en un espectrofluorómetro.
Tween-20, Tween-40, Tween-60, Tween-65 y Tween-85 dan sustancialmente el mismo resultado beneficioso que Tween-80.
7.7 Pruebas de estabilidad de formulaciones líquidas.
Se realizó un estudio más completo para evaluar la estabilidad de ALX-0081 en diferentes formulaciones isotónicas basadas en citrato a una concentración de 20 mg/ml. La tabla 3 ofrece una visión general de las diferentes formulaciones que se sometieron a prueba.
El objetivo principal fue evaluar el efecto del pH (6,0-6,5-7,0) y el tipo de excipiente (NaCl, manitol, sacarosa o glicina) sobre la estabilidad del producto líquido. Para propósitos de comparación directa y control, el estudio también incluyó ALX-0081 contemporáneo formulado a 5 mg/ml en D-PBS y glicina (idéntico a la formulación actual, excepto por la concentración más baja de Tween-80), así como las diferentes disoluciones isotónicas de ALX-0081 basadas en citrato mencionadas anteriormente pero formuladas a una concentración de 5 mg/ml en lugar de 20 mg/ml. Conjuntamente, esto dio como resultado 17 formulaciones líquidas diferentes (formulación n.° 1-17) que se sometieron a estudios de estabilidad profundos. Para excluir el efecto de las diferencias en la concentración de Tween, todas las formulaciones contenían un 0,01% de Tween-80 (v/v).
7.7.1 Estabilidad frente a la congelación y descongelación
Se evaluó el efecto de los ciclos repetitivos de FT sobre la estabilidad de ALX-0081 como una formulación líquida. Se sometieron alícuotas de las diferentes formulaciones (0,5 ml/tubo) a hasta 10 ciclos de FT a -70°C o -20 °C. Un ciclo incluía congelación durante ± 20 min seguido de descongelación durante 5 min en un baño de agua a 25°C. Después de este tratamiento, todas las formulaciones permanecieron visiblemente transparentes. Los análisis de RP-HPLC mostraron una buena recuperación (el 95-100%) y no pudo detectarse ninguna diferencia significativa en el perfil, lo que sugiere que la calidad de los agentes de unión a vWF, por ejemplo ALX-0081 no se ve afectada por la congelación-descongelación repetida en las 17 formulaciones líquidas diferentes sometidas a prueba.
7.7.2 Estabilidad en almacenamiento
También se evaluó la estabilidad de las 17 formulaciones diferentes almacenando alícuotas (0,5 ml/tubo) en condiciones de tensión, es decir, 40°C; se incluyó la condición de almacenamiento a largo plazo a -70°C como referencia. Los análisis se centraron en RP-HPLC porque este método se conoce generalmente como un método particularmente informativo para revelar modificaciones químicas que se producen durante el almacenamiento (véase la tabla 4). Esta sección ofrece una descripción general de los datos obtenidos después de 1 mes de almacenamiento; los resultados confirman los hallazgos en los puntos de tiempo anteriores, es decir, después de 1 semana y 2 semanas.
(a) RP-HPLC
Tal como se indicó anteriormente en la sección 7.2 anterior, el análisis de RP-HPLC resolvió la DS ALX-0081 contemporánea (formulación de D-PBS/glicina) en algunas variantes relacionadas con el producto e impurezas. Brevemente, en condiciones de tensión (por ejemplo, 40°C), la pureza (% del pico principal) disminuyó concomitantemente con un aumento en algunos de los picos anteriores/posteriores existentes, así como la formación de otros adicionales.
Los datos de RP-HPLC obtenidos en el presente estudio se resumen en la tabla 5.
En general, los resultados obtenidos indicaron que tuvieron lugar esencialmente las mismas modificaciones en los diferentes tampones citrato observados en el presente tampón de formulación (es decir, D-PBS/glicina), aunque pudieron observarse algunas diferencias en el área de pico relativa. En particular, el aumento en el área de pico anterior (oxidación) fue más lento en las formulaciones de citrato (especialmente a pH 6,0) en comparación con la formulación de D-PBS/glicina. Con respecto a esta acumulación de pico anterior, la glicina parece ser la menos favorable entre los diferentes excipientes. El perfil de los diferentes picos posteriores después del almacenamiento durante 1 mes a 40°C fue comparable para todas las formulaciones, aunque el segundo pico posterior (es decir, la variante de piroglutamato) parecía ser más pronunciada a pH 7,0 que a pH 6,0-6,5. El alcance de la ampliación/división del pico principal, el resultado de la isomerización de asp, es difícil de cuantificar debido a la mala resolución; el porcentaje de área del pico del hombro no pudo estimarse con precisión y, por lo tanto, se
incluyó en el área de superficie relativa notificada en la tabla 5 para el pico principal. No obstante, los cromatogramas de RP-HPLC correspondientes (datos no mostrados) permitieron realizar una evaluación cualitativa; estos datos sugieren que el alcance de la isomerización es bastante similar en las diversas formulaciones.
(b) clEF
De manera similar a RP-HPLC, el método de clEF permite la detección de ciertas variantes de productos que se producen durante el almacenamiento en condiciones de tensión (véase la sección 7.2 para detalles). Esto se ejemplifica en la figura 5, que compara los electroferogramas de ALX-0081 contemporáneo después del almacenamiento durante un mes a -70°C y 40°C.
Los datos de clEF obtenidos en el presente estudio (datos no mostrados) confirman básicamente las conclusiones alcanzadas por el análisis de RP-HPLC, es decir, tiene lugar el mismo tipo de modificaciones aproximadamente en la misma medida en los diferentes tampones citrato observados en el presente tampón de formulación, representado en el presente documento por la formulación 17 tal como se muestra en la figura 5 (es decir, D-PBS/glicina). Sin embargo, pudieron observarse algunas diferencias en el área de pico relativa. En particular, el pico posterior (es decir, la variante de piroglutamato) parecía ser más pronunciado a un pH de 7,0 que a un pH de 6,0 a 6,5, lo que está de acuerdo con los hallazgos de RP-HPLC tal como se resumieron anteriormente en la tabla 5.
(c) SE-HPLC
Se realizó análisis de SE-HPLC para examinar la estabilidad física de ALX-0081, es decir, para detectar especies de HMW y/o productos de degradación que podrían haberse formado durante el almacenamiento en condiciones de tensión. Para todas las formulaciones que se sometieron a prueba en este caso, la prueba de tensión no pareció tener un efecto significativo sobre los cromatogramas de SE-HPLC.
(d) Conclusión
En la tabla 5 se muestra un resumen de los hallazgos más importantes con respecto a la estabilidad en almacenamiento de las diferentes formulaciones líquidas de ALX-0081. Sólo se enumeraron los datos más informativos basados en el análisis de RP-HPLC. Estos datos sugieren una mayor estabilidad química en citrato 50 mM a pH 6,0-6,5. Con la excepción de la glicina, el tipo de excipiente no tuvo un efecto significativo sobre la estabilidad. Con respecto a la estabilidad física, no pudieron observarse diferencias entre las diferentes formulaciones. Esto último se evidenció por la recuperación de ±100% observada para todas las muestras en los diversos análisis de HPLC, así como por los cromatogramas de SE-HPLC que demuestran la ausencia de agregación/degradación.
Basándose en los resultados anteriores, se decidió explorar adicionalmente el potencial de las formulaciones de citrato/sacarosa a un pH de 6,0-6,5.
7.8 Pruebas de estabilidad de formulaciones liofilizadas.
Se evaluó el efecto de la liofilización comparando la estabilidad en almacenamiento de ALX-0081 en formulaciones líquidas y liofilizadas de citrato/sacarosa (API 20 mg/ml a pH 6,0-6,5). En la tabla 6 se proporciona una visión general de las formulaciones sometidas a prueba. La formulación basada en D-PBS/glicina de la técnica anterior (API 5 mg/ml) se incluyó para comparación. ALX-0081 líquido (es decir, antes de la liofilización) y liofilizado se mantuvieron congelados (-70°C para las muestras líquidas y -20°C para las formulaciones liofilizadas), así como a 5°C, 25°C y 40°C y las muestras se analizaron después de 2 semanas y 1,5 meses de almacenamiento.
El panel A de la figura 6 muestra una imagen de los viales después del proceso de liofilización usando la ejecución de 65 h convencional tal como se muestra en la figura 1. La liofilización de las formulaciones que contienen citrato/sacarosa dio como resultado una buena formación de torta, mientras que las muestras formuladas en D-PBS/glicina no produjeron una torta decente. Todas las muestras pudieron resolubilizarse fácilmente con agua Milli-Q y las soluciones eran transparentes e incoloras (figura 6 , panel B).
7.8.1 Evaluación del producto antes y después de la liofilización.
El análisis de RP-HPLC y SE-HPLC no reveló diferencias significativas en cuanto a las características fisicoquímicas entre el producto líquido de partida (mantenido a < -70°C) y el producto después de la liofilización y reconstitución para cualquiera de las formulaciones sometidas a prueba. Además, se demostró la recuperación completa de la muestra para todas las formulaciones (tabla 7).
7.8.2 Evaluación del producto liofilizado después de 1,5 meses de almacenamiento.
(a) Inspección visual y contenido
La torta de las muestras liofilizadas no mostró signos visuales de deterioro después de 1,5 meses de almacenamiento a -20°C, 5°C, 25°C o 40°C.
Las muestras eran transparentes e incoloras después de la reconstitución con agua Milli-Q. Además, el almacenamiento no tuvo un efecto significativo sobre el contenido, medido después de la reconstitución (tabla 8). (b) RP-HPLC
Se compararon los perfiles de las 3 formulaciones liofilizadas diferentes (n.° 3, 7 y 17) después de 1,5 meses de almacenamiento a -20°C, 5°C, 25°C y 40°C respectivamente. Una comparación en las condiciones más extenuantes (+40°C) revela mejor el impacto de la liofilización sobre la estabilidad química. Los resultados correspondientes se resumen en la tabla 8. Tal como puede observarse, el almacenamiento en forma congelada no parece afectar a ALX-0081 en ninguna de las formulaciones sometidas a prueba en el presente estudio.
Globalmente, la conclusión predominante a partir de los datos obtenidos es que la liofilización de una formulación basada en citrato/sacarosa previene esencialmente las modificaciones químicas que se producen en forma líquida, con la excepción de algunas cantidades menores de modificación de piroglutamato. En estas formulaciones liofilizadas, no hubo un aumento en el porcentaje de área de los picos previos ni un signo de mayor ensanchamiento/división del pico. En cambio, la liofilización de la formulación basada en D-PBS/glicina no dio como resultado una mejora significativa de la estabilidad química. La formación de piroglutamato en la formulación liofilizada de citrato/sacarosa parece ser ligeramente más pronunciada a pH 6,0 que a pH 6,5. Esto se fundamenta en los datos a 25°C, temperatura a la que la tasa de formación de piroglutamato es menor, pero muestra la misma dependencia del pH. Tal como se esperaba, el almacenamiento durante hasta 1,5 meses a -20°C o 5°C no provocó ningún deterioro detectable de ALX-0081 liofilizado (datos no mostrados).
Sorprendentemente, a 40°C se obtuvo una estabilidad mejorada en las formulaciones basadas en citrato/sacarosa en comparación con la formulación basada en D-PBS/glicina, mostrando esta última una susceptibilidad significativamente mayor a las modificaciones químicas.
Para las formulaciones líquidas, el almacenamiento durante hasta 1,5 meses a -70°C, 5°C y 25°C no tuvo un efecto significativo sobre ALX-0081 (datos no mostrados). El deterioro observado después de 1,5 meses de almacenamiento a 40°C, está aproximadamente de acuerdo con observaciones anteriores (véase la sección 7.7.2). (c) cIEF
Los resultados obtenidos por el análisis de clEF están de acuerdo con los de RP-HPLC. Lo más notablemente, la liofilización de una formulación basada en citrato/sacarosa no puede evitar completamente la modificación de piroglutamato. De hecho, el almacenamiento del producto liofilizado a 40°C durante 1,5 meses dio como resultado un aumento en el pico posterior. Nuevamente, se observó una formación más rápida de piroglutamato en citrato/sacarosa a pH 6,0 que a pH 6,5.
(d) SE-HPLC/MALS/DLS
El almacenamiento durante hasta 1,5 meses a -70°C/-20°C, 5°C y 25°C no tuvo efecto sobre los perfiles de SE-HPLC de formulaciones líquidas o liofilizadas de ALX-0081 (datos no mostrados). Sin embargo, a 40°C pudo observarse un ensanchamiento del pico y la formación de picos de hombro en todas las formulaciones líquidas. El análisis MALS mostró que estos picos de hombro corresponden a ALX-0081 monomérico (datos no mostrados). Los datos apuntan a un cambio conformacional en una subpoblación de ALX-0081 como resultado del almacenamiento con tensión. Sorprendentemente, el perfil SE-HPLC de formulaciones de citrato/sacarosa liofilizadas no se vio afectado por la prueba de tensión a 40°C, lo que indica que estas formulaciones liofilizadas también mejoran la estabilidad física de ALX-0081. Sin embargo, este no fue el caso de la formulación liofilizada de D-PBS/glicina; someter a tensión esta formulación a 40°C no solo dio como resultado un pico de hombro sino, aparentemente, también algunas especies de peso molecular más alto, visibles como un amplio pico previo (tabla 8). El análisis de DLS no detectó ninguna especie oligomérica grande en ninguna de las formulaciones (datos no mostrados).
(e) Conclusión
En la tabla 8 se muestra un resumen de los hallazgos más importantes con respecto a la estabilidad en almacenamiento de las formulaciones de ALX-0081 liofilizadas sometidas a prueba. Globalmente, solo se observaron diferencias limitadas en la estabilidad entre las formulaciones de citrato/sacarosa, aunque inesperadamente ALX-0081 pareció ser menos propenso a la formación de piroglutamato a pH 6,5 que a pH 6,0. Por tanto, el trabajo de reformulación adicional para ALX-0081 se centró en formulaciones basadas en citrato/sacarosa a pH 6,5.
7.9 Optimización adicional de la formulación basada en citrato/sacarosa.
Los datos recopilados hasta ahora muestran que una formulación basada en citrato/sacarosa mejora la solubilidad y que la liofilización de esta formulación mejora drásticamente la estabilidad en almacenamiento de ALX-0081. Sin embargo, el almacenamiento de ALX-0081 liofilizado a temperaturas más altas, aunque limitado, todavía da como resultado la formación de piroglutamato. Es razonable suponer que esta modificación puede limitar la vida útil de almacenamiento del producto liofilizado (incluso cuando se almacena a 52C). Queda por aclarar por qué la liofilización no pudo evitar esta modificación.
Se planteó la hipótesis de que el agua que queda en el producto liofilizado desempeña un papel clave.
Si esta hipótesis fuera cierta, entonces el agua restante puede minimizarse optimizando los parámetros físicos de la liofilización, tales como el tiempo de secado, las temperaturas, el vacío, etc., tal como se enumeró anteriormente, pero al mismo tiempo, los otros parámetros del agente de unión a vWF deben permanecer constantes. Otro enfoque es modificar la formulación, pero de nuevo al mismo tiempo, los otros parámetros del agente de unión a vWF deben permanecer constantes. Además, puede usarse el ajuste de los parámetros físicos de la liofilización en combinación con la modificación de la formulación.
7.9.1 Optimización de los parámetros de liofilización.
La optimización de los parámetros físicos de la liofilización, incluyendo (i) tiempos de secado, (ii) temperaturas de las diferentes etapas, (iii), vacío y una combinación de (i)-(iii) no fue satisfactoria, es decir, un efecto inadecuado o ninguno adecuado sobre el contenido de humedad residual o que afecte a los parámetros de los agentes de unión a vWF.
7.9.2 Optimización de la formulación para la liofilización.
El efecto del contenido de humedad sobre la estabilidad química del producto liofilizado se investigó ajustando las concentraciones del tampón citrato y el excipiente de sacarosa. Además, se investigó un tiempo de secado secundario durante el programa de liofilización.
7.10 Efecto del contenido en humedad sobre la estabilidad del producto liofilizado.
Tres formulaciones isotónicas diferentes de ALX-0081 con concentraciones variables de citrato y sacarosa (las tres a pH 6,5) se sometieron a dos programas de liofilización diferentes: la ejecución de 65 h convencional por un lado y una ejecución de 37 horas acortada por otro lado. En la tabla 9 se presenta una visión general de las formulaciones sometidas a prueba. La figura 7 muestra los viales obtenidos después de la liofilización. La liofilización dio como resultado una buena formación de torta para todas las formulaciones.
Se analizaron las muestras liofilizadas de ALX-0081 después de 2 y 4 semanas de almacenamiento tanto a -20°C como a 40°C. En el presente experimento, se decidió realizar una prueba exhaustiva para probar aún más la utilidad de las formulaciones.
En primer lugar, durante el almacenamiento a 40°C durante hasta 4 semanas, la torta de las muestras liofilizadas se mantuvo intacta y la reconstitución produjo disoluciones transparentes. El ciclo de liofilización no parece tener un efecto significativo sobre el contenido (medido espectrofométricamente a 277 nm) ni la osmolalidad. De acuerdo con experimentos anteriores, el almacenamiento de 4 semanas a 40°C no tuvo un efecto sobre la estabilidad física de ALX-0081, basándose en los análisis SE-HPLC, MALS y DLS (datos no mostrados). Además, se encontró que la potencia de ALX-0081 determinada por el ensayo basado en Biacore no se vio afectada por el proceso de liofilización y el almacenamiento posterior (datos no mostrados). Sin embargo, los análisis de RP-HPLC demostraron que el almacenamiento dio como resultado nuevamente la formación de cantidades, aunque menores, de la variante de piroglutamato. Esto fue un poco más pronunciado para la formulación que contiene la mayor concentración de citrato y la menor concentración de sacarosa (tabla 10). Además, para cada formulación liofilizada, el contenido total de humedad se determinó por medio de valoración de Karl Fisher. En la tabla 10 se muestra un resumen de estos datos junto con la cantidad de piroglutamato detectado en las muestras sometidas a tensión correspondientes. A partir de los datos obtenidos para cada programa de liofilización por separado se desprende que un mayor contenido de humedad da como resultado una mayor susceptibilidad a la formación de piroglutamato. Esto sugiere que el agua residual presente en el producto liofilizado promueve modificaciones químicas.
En conclusión, los resultados indican que reducir el contenido de humedad de los agentes de unión a vWF liofilizados, por ejemplo ALX-0081, es beneficioso para su estabilidad química.
7.11 Efecto de la reducción de la fuerza del tampón y el aumento del contenido de sacarosa
Los datos obtenidos en la sección anterior muestran que reducir la concentración de citrato al tiempo que se aumenta la concentración de sacarosa (manteniendo de ese modo una disolución isotónica) es beneficioso para la estabilidad del producto liofilizado. Al mismo tiempo, se obtuvo evidencia de que ALX-0081 requería una concentración suficientemente alta de citrato para obtener una solubilidad mejorada. Por tanto, se decidió evaluar el
efecto de las concentraciones de citrato y sacarosa sobre el aspecto de la disolución durante el almacenamiento a 5°C y 25°C, y volver a evaluar la estabilidad frente a la congelación y descongelación en presencia de concentraciones más bajas de citrato.
7.11.1 Evaluación del impacto de la concentración de citrato/sacarosa
En un primer experimento, se almacenaron 12 formulaciones diferentes de ALX-0081 a 5°C y 25°C durante hasta 4 días. Las muestras se inspeccionaron regularmente para detectar turbidez o presencia de precipitados. Las imágenes tomadas de las muestras después de 4 días de almacenamiento se muestran en las figuras 8 y 9. En la tabla 11 se presenta una visión general de las diferentes formulaciones y los resultados correspondientes. Después de 4 días de almacenamiento a 25°C, todas las muestras permanecieron transparentes e incoloras (figura 8 , panel A) . En cambio, a 5°C, la mayoría de las formulaciones de citrato sin excipientes se volvieron turbias (figura 8 , panel B) . Claramente, el grado de turbidez es inversamente proporcional a la concentración de citrato, permaneciendo la formulación de citrato 50 mM transparente. Además, la recuperación de la muestra para la muestra que contenía citrato 15 mM fue del 68% (basándose en A277 después de 20 h de almacenamiento), mientras que otras recuperaciones variaron desde el 90 hasta el 100% (datos no mostrados). La adición de sacarosa a la formulación de citrato 15 mM evitó la turbidez de la muestra, aunque a la concentración más baja de sacarosa (es decir, 5%), se detectó una pequeña turbidez a 5°C (figura 9, panel B).
Las observaciones confirman la importancia de una concentración suficientemente alta de citrato en el mantenimiento de ALX-0081 soluble, particularmente a baja temperatura. Sin embargo, el aumento de la concentración de citrato dio lugar a un mayor contenido de humedad. Inesperadamente, la reducción de la concentración de citrato puede compensarse mediante la adición de sacarosa. No se observó ningún efecto de Tween-80 sobre la solubilidad.
7.11.2 Evaluación de la estabilidad frente a FT
Un experimento de seguimiento se centró en la estabilidad frente a FT de varias formulaciones basadas en citrato/sacarosa. Nueve formulaciones diferentes de ALX-0081 se sometieron a 5 ciclos de FT consecutivos a -20°C. En la tabla 12 se muestra una visión general de las formulaciones sometidas a prueba y los resultados correspondientes. Todas las muestras permanecieron transparentes y los ciclos FT no afectaron a la estabilidad física de los agentes de unión a vWF, por ejemplo ALX-0081, basándose en el análisis del contenido y los datos de SE-HPLC.
7.11.3 Optimización de la concentración de sacarosa y citrato en vista de la isotonicidad.
Basándose en los resultados de almacenamiento y FT mencionados anteriormente, se seleccionó la concentración óptima de tampón de citrato como 20 mM. Se realizó un experimento final sobre tres formulaciones que diferían en la concentración de sacarosa. El objetivo de este experimento fue establecer la concentración óptima de sacarosa para lograr una fórmula isotónica y confirmar la estabilidad frente a FT de ALX-0081 a 20 mg/ml. Además de a 5 ciclos de FT consecutivos, cada formulación también se sometió a 1 ciclo de FT seguido de 24 horas de almacenamiento a 25°C y un ciclo de FT adicional para imitar las etapas de manipulación durante la fabricación. En la tabla 13 se facilita un resumen de los resultados. Todas las formulaciones sometidas a prueba eran transparentes y las diversas manipulaciones no tuvieron un efecto sobre el contenido/recuperación u osmolalidad. Basándose en los valores de osmolalidad, parece que una concentración del 7% de sacarosa es óptima para obtener una disolución isotónica.
7.12 Estudio de estabilidad de formulaciones liofilizadas de ALX-0081 almacenadas a diversas temperaturas durante hasta 12 meses
Se liofilizó ALX-0081 formulado a 12,5 mg/ml en tampón de citrato 20 mM pH 6,5, el 7% de sacarosa (p/v) y el 0,01% de Tween-80 (v/v) según las condiciones establecidas en la tabla 14. Las muestras se almacenaron posteriormente a -202C (±5°C), 5°C (±3°C), 25°C (±2°C/60 ± 5% de HR) y 40°C (±2°C/75 ± 5% de HR).
La estabilidad de las formulaciones liofilizadas se evaluó en diferentes puntos de tiempo, es decir, inicial, 1 mes, 3 meses, 6 meses, 9 meses y 12 meses, y se evaluó la pureza, aspecto, propiedades fisicoquímicas y potencia. Se proporcionan datos detallados de la caracterización de la muestra en las tablas 15 a 23. La pureza de las muestras se evaluó mediante RP-HPLC en la que se determinó el porcentaje del área del pico medio así como el porcentaje de áreas de pico anterior y pico posterior. La concentración de proteína se determinó por absorbancia UV. Además, las muestras liofilizadas se inspeccionaron visualmente, se reconstituyeron y la formulación reconstituida se inspeccionó visualmente. Se midió el pH de las muestras después de la reconstitución y se determinó el contenido de humedad del polvo liofilizado mediante valoración coulométrica (Karl Fischer). Se realizaron mediciones de recuento de materia particulada para contar partículas de > 10 pm y > 25 pm. Las muestras se caracterizaron adicionalmente para la función biológica utilizando un ensayo basado en Biacore. La potencia se expresó como porcentaje de potencia relativa del material de referencia.
Los datos de estabilidad obtenidos muestran que las características del producto ALX-0081 liofilizado no se ven afectadas significativamente por un almacenamiento de 12 meses a o bien -20°C o bien 5°C. Se encontró que los datos recopilados a lo largo de todo el estudio de estabilidad a esas temperaturas eran comparables a los generados en el tiempo cero.
Se observaron varios cambios menores en las muestras almacenadas a 25°C o 40°C, que pueden atribuirse a las condiciones de almacenamiento acelerado o con tensión. Las principales observaciones fueron:
° A 25°C y a 40°C, se observó un aumento en el pico posterior 2 en RP-HPLC durante los 12 meses de almacenamiento, lo que corresponde a la formación de la variante de piroglutamato desde el 0,7% hasta el 1,1% o hasta el 2,4% respectivamente.
° A 40°C, se observó un aumento en el contenido de humedad desde el 0,7% hasta el 2,1% (p/p) después de 12 meses de almacenamiento. Esto podría atribuirse posiblemente a la toma de humedad del entorno de almacenamiento (es decir, el 75% de HR) por el tapón, con la posterior difusión gradual al producto.
Los resultados obtenidos en condiciones de tensión sugieren una correlación entre el contenido de humedad y la estabilidad química del producto; esto se corresponde con los datos notificados anteriormente en la Sección 7.10. Por tanto, estos datos indican la importancia de controlar el contenido de humedad del producto DP durante el almacenamiento. Considerando que el almacenamiento a 40°C puede considerarse predictivo para la estabilidad a largo plazo a 25°C, los datos de estabilidad a 12 meses incluidos en el presente documento proporcionan una buena indicación de la estabilidad en almacenamiento a largo plazo a temperatura ambiente (tal como 18, 24, 30 o 36 meses) e incluso estabilidades prolongadas cuando se almacenan en condiciones más suaves (por ejemplo, 5°C o congelado).
7.13 Estudio de comparabilidad in vitro sobre la actividad biológica de la formulación de producto farmacéutico liofilizado y líquido del Nanobody anti-vWF caplacizumab (ALX-0081)
7.13.1 Objetivo
Se usaron varios ensayos para evaluar la comparabilidad in vitro de DP ALX-0081 contemporáneo [formulación líquida que contiene 5 mg/ml del principio farmacéutico activo (API) en un tampón basado en fosfato (D-PBS) que contiene glicina 200 mM y Tween-80 al 0,02% (v/v), pH 7,1] y la formulación liofilizada de DP ALX-0081 tal como se presentó anteriormente [formulada a 12,5 mg/ml en tampón de citrato 20 mM, pH 6,5, sacarosa al 7% (p/v) y Tween-80 al 0 ,01% (v/v)] con respecto a la actividad biológica y la unión a la diana:
a) Ensayo de potencia basado en Biacore
b) Ensayo de potencia basado en ELISA
c) Ensayo de biomarcadores farmacodinámicos de actividad de cofactor inducida por ristocetina (RICO) d) Determinación de afinidad basada en Gyrolab
Estos ensayos permitieron una comparación lado a lado del producto de fármaco líquido y liofilizado de ALX-0081 (caplacizumab). Se usaron criterios de comparabilidad predefinidos para evaluar la comparabilidad para cada ensayo, y se enumeran en la tabla 24.
7.13.2 Métodos
a) El ensayo de Biacore se basa en la tecnología de resonancia de plasmón superficial (SPR) y mide la unión ávida de ALX-0081 al dominio A1 de vWF humano inmovilizado en un chip sensor. El ensayo se ha seleccionado para pruebas de potencia en el momento de la liberación y en la estabilidad.
b) El ensayo de potencia basado en ELISA es un método ortogonal para las pruebas de potencia de ALX-0081 que se ha desarrollado para una caracterización adicional de la capacidad de neutralización de la diana de caplacizumab. Este ensayo mide la inhibición de la unión inducida por ristocetina del factor de Von Willebrand (vWF) a la plaqueta unida por caplacizumab.
c) El ensayo RICO se utiliza como marcador farmacodinámico para la actividad farmacológica de caplacizumab. El ensayo mide la velocidad y el grado en que plaquetas liofilizadas humanas forman agregados después de la adición del antibiótico ristocetina, que imita la activación de vWF inducida por cizallamiento.
d) El ensayo basado en Gyrolab analiza las interacciones cinéticas de caplacizumab con su diana multimérica
vWF y determina la constante de afinidad de caplacizumab a vWF multimérico humano. En resumen, la determinación de la afinidad en la plataforma Gyroloab se estableció de la siguiente manera: se utilizaron Gyrolab Bioaffy 1000 CD. Como herramienta de captura, se aplicaron 3000 nM de vWF purificado biotinilado en el laboratorio (HaemateP purificado usando cromatografía de exclusión molecular) sobre las columnas que se empaquetaron previamente con cuentas recubiertas con estreptavidina. Se utilizó D-PBS esterilizado por filtración que contenía Tween-20 al 0 ,01% para la dilución de la herramienta de captura. Se preincubó una serie de dilución 1/3 de vWF HaemateP purificado durante 24 horas a TA (+20°C) en una placa de 96 pocillos en un rotor a 600 rpm con una concentración fija de caplacizumab (5 pM) en tampón AD1 (tampón de diluyente de ensayo para la curva de respuesta a la dosis). Después de 24 horas, se centrifugó la placa durante 1 minuto a 200 g. 70 ml de la mezcla de preincubación, que contenía las moléculas de caplacizumab libres, se llevaron a una placa de PCR de pocillos profundos. Entonces, esta mezcla se hizo fluir sobre la columna para que el caplacizumab libre pudiera unirse a vWF biotinilado inmovilizado en la columna. El sistema Gyrolab transfirió automáticamente la mezcla por triplicado a los CD. Se detectó caplacizumab libre con anticuerpo monoclonal anti-caplacizumab marcado con AlexaFluor647 50 nM diluido en tampón Rexxip F (tampón de detección disponible comercialmente). Se realizaron tres experimentos independientes para determinar la KD final. Los fluorocromos se excitaron mediante el láser rojo, por lo que se obtuvieron señales fluorescentes y se amplificaron mediante un tubo foto multiplicador (PMT). El nivel de amplificación de este ensayo fue del 1% de PMT. Se usó un modelo de análisis de ligando desconocido para la determinación de KD de caplacizumab. El análisis se realizó con el software de ajuste XL de la estación de trabajo Gyrolab.
7.13.3 Resultados
a) La potencia relativa de las muestras de prueba de ALX-0081 líquidas y liofilizadas se midió en un ensayo de potencia Biacore, en relación con el material de referencia ALX-0081 usado en el ensayo de potencia, también designado patrón de referencia maestro 2 (MRS-2). La potencia relativa fue del 102,8% y el 102,9%, respectivamente, lo que indica una comparabilidad total con respecto a la potencia biológica determinada a través de Biacore (véase la tabla 24).
b) La potencia relativa de las muestras de prueba de ALX-0081 líquidas y liofilizadas se determinó en el ensayo de potencia basado en ELISA, en relación con MRS-2. Los valores de potencia relativa fueron del 99,4% y el 109,5%, respectivamente, y por tanto, dentro de los criterios de comparabilidad (véase la tabla 24). Por tanto, estos resultados indican que ambas formulaciones son comparables con respecto a la potencia determinada a través de ELISA.
c) La actividad RICO de las muestras de prueba de ALX-0081 líquidas y liofilizadas se midió en una comparación lado a lado, y se determinó la concentración para bloquear completamente la actividad RICO (< 20%). La concentración para bloquear completamente la actividad RICO (< 20%) fue < 0,4 mg/ml para ambas formulaciones. Estos resultados están dentro de los criterios de comparabilidad (véase la tabla 24) e indican la comparabilidad completa con respecto a la actividad farmacodinámica de ambas formulaciones.
d) La constante de afinidad (valor de Kd) de las muestras de prueba de ALX-0081 líquidas y liofilizadas también se determinó en una comparación lado a lado en el ensayo basado en Gyrolab. Los valores de Kd fueron de 6,84 pM y 4,46 pM, respectivamente, con intervalos de confianza solapantes. Por tanto, estos resultados indican una comparabilidad total de ambas formulaciones con respecto a la afinidad por el vWF diana multimérico (véase la tabla 24).
7.13.4 Conclusión
El objetivo de este estudio fue evaluar la comparabilidad in vitro del producto farmacéutico líquido y liofilizado de ALX-0081 (caplacizumab) por medio de cuatro ensayos, capaces de evaluar la actividad biológica y unión a diana in vitro:
a) Ensayo de potencia basado en Biacore
b) Ensayo de potencia basado en ELISA
c) Ensayo de biomarcadores farmacodinámicos de actividad de cofactor inducida por ristocetina (RICO) d) Determinación de afinidad basada en Gyrolab
Todos los ensayos in vitro cumplieron con los criterios de aceptación predefinidos y mostraron que ambas formulaciones de ALX-0081 son comparables en cuanto a actividad biológica y unión a la diana (véase la tabla 24). Las formulaciones líquidas y liofilizadas de DP ALX-0081 sometidas a prueba mostraron:
• una potencia relativa similar determinada por ensayo Biacore y ELISA
• una actividad farmacodinámica comparable in vitro (neutralización de la diana) a través del ensayo RICO • una afinidad por la diana comparable a través del ensayo Gyrolab.
7.14 Pruebas de estabilidad aceleradas y a largo plazo de formulaciones de ALX-0081 líquidas y liofilizadas.
Como complemento al ejemplo 7.12, se realizaron experimentos de estabilidad independientes usando un lote diferente de ALX-0081 de la misma formulación [tampón de citrato 20 mM, pH 6,5, sacarosa al 7% (p/v) y Tween-80 al 0 ,01 % (v/v)].
La estabilidad tanto de la formulación liofilizada como de la líquida se sometió a prueba a diferentes temperaturas:
• La formulación líquida de ALX-0081 13,8 mg/ml en tampón citrato 20 mM, pH 6,5, sacarosa al 7% (p/v) y Tween-80 al 0,01% (v/v) se almacenó a temperaturas < -60°C y 5°C (± 3°C) y se sometió a prueba la estabilidad a diferentes puntos de tiempo, es decir, inicial, 9 meses, 12 meses, 18 meses y 24 meses. • La formulación liofilizada de ALX-0081 12,7 mg/ml en tampón citrato 20 mM, pH 6,5, sacarosa al 7% (p/v) y Tween-80 (v/v) al 0,01% se almacenó a 52C (±3°C ), 25°C (±2°C/60 ± 5% de HR) y 40°C (±2°C/75 ± 5% de HR). De manera similar a la formulación líquida, la estabilidad de la formulación liofilizada se determinó a los 0, 9, 12, 18 y 24 meses.
En cada punto de tiempo, se monitorizó la estabilidad química y física de las muestras usando varias técnicas analíticas, incluyendo clEF, RP-HPLC, SE-HPLC, aspecto visual, pH y absorción de UV. El contenido de humedad del polvo liofilizado se determinó mediante valoración coulométrica. La potencia relativa de las muestras líquidas y liofilizadas se midió en Biacore en relación con un patrón de referencia de ALX-0081 interno.
Los datos detallados de la caracterización de las muestras para las formulaciones líquidas y liofilizadas se proporcionan en las tablas 25 a 26 y en las tablas 27 a 29, respectivamente. Las muestras que cumplían con los criterios establecidos en la columna 2 de cada una de las tablas mencionadas anteriormente se consideraron que estaban dentro de las especificaciones del producto.
Los datos obtenidos demuestran que la formulación de la invención es altamente estable durante al menos 24 meses. Las características fisicoquímicas, así como la actividad biológica de ALX-0081 liofilizado, no se vieron afectadas significativamente por el almacenamiento de 24 meses a 5°C o 25°C. Cuando se hizo hincapié en ALX-0081 durante 24 meses a 40°C, se observó un aumento en el pico posterior 2, que corresponde a la formación de la variante de piroglutamato desde el 1,1% en el material de partida hasta el 2,8%, el 3,2%, el 4,2% y el 6 ,2% después de 9, 12, 18 y 24 meses, respectivamente. El almacenamiento de formulaciones líquidas de ALX-0081 durante al menos 24 meses a temperaturas < -60°C o a 5°C, no afectó significativamente a su estabilidad fisicoquímica: los valores de contenido fueron estables, las muestras se mantuvieron transparentes y los perfiles de cIEF, RP-HPLC y SE-HPLC del material inicial fueron comparables con los de las muestras de estabilidad.
Los cambios notificados para muestras liofilizadas almacenadas a 40°C pueden atribuirse a las condiciones de almacenamiento con tensión y proporcionan una buena indicación de la estabilidad en almacenamiento a largo plazo en condiciones más suaves.
Predicción de la estabilidad a largo plazo
Las especificaciones actuales del producto farmacéutico establecen que el porcentaje permitido de piroglutamato es < 4%. Basándose en esta especificación y los datos de estabilidad actuales, se empleó la ecuación de Arrhenius para predecir la vida útil de almacenamiento del producto farmacéutico liofilizado a 5°C y 25°C. La ecuación de Arrhenius es una fórmula precisa que describe la dependencia de la temperatura de las velocidades de reacción que se usa comúnmente en la industria farmacéutica. Tal como se muestra en las figuras 10 y 11, se espera que el producto farmacéutico liofilizado permanezca dentro de las especificaciones durante al menos 500 meses cuando se almacena a 5°C y durante al menos 60 meses cuando se almacena a 25°C.
7.15 Conclusión general
La invención de la reformulación para los agentes de unión a vWF, y especialmente ALX-0081 descrito en el presente documento, brindó una nueva formulación basada en citrato/sacarosa con una solubilidad mejorada (hasta 80 mg/ml) y una estabilidad en almacenamiento líquido significativamente mejorada (por ejemplo, menos oxidación en comparación con su formulación original). Además, en la forma liofilizada, esencialmente no pudo detectarse oxidación o isomerización de asp después de 12 o incluso 24 meses de almacenamiento a 40°C. La optimización adicional de la concentración de citrato y sacarosa dio como resultado una reducción del contenido de humedad del producto liofilizado, minimizando de ese modo la velocidad de formación de piroglutamato. Se demostró que cada
característica fisicoquímica del agente de unión a vWF estaba influenciada de manera diferente por los diferentes constituyentes, tanto físicos como químicos, de la formulación, tales como la elección del tampón, el pH, la concentración, el excipiente, etc. En este documento se proporcionan diversas formulaciones optimizadas para remediar o prevenir diferentes tensiones químicas y/o físicas.
Se diseñó un tampón de formulación que cumplía con los criterios más críticos: citrato 20 mM pH 6,5 sacarosa al 7,0% (p/v) Tween-80 al 0,01% (v/v). Usando esta formulación, se demostró que ALX-0081 es estable durante al menos 12 o incluso 24 meses a -20°C, 5°C, 25°C y 40°C. Estos datos apuntan claramente a una vida útil de almacenamiento considerablemente más larga a 5°C que la formulación líquida actual. Además, los inventores han demostrado ampliamente que la formulación contemporánea de ALX-0081 que se ha usado en estudios clínicos hasta la fecha es comparable a la formulación de ALX-0081 liofilizada recién optimizada presentada en el presente documento en cuanto a actividad biológica y unión a la diana in vitro.
Tabla A-1: Ejemplos de agentes de unión a vWF



Tabla A-2

Tabla 1. Visión general de 50 combinaciones de tampón/excipiente diferentes predichas mediante el programa Design Expert para proporcionar las temperaturas de fusión más altas para ALX-0081. Las combinaciones de tampón/excipiente se clasifican según el valor de Tm. Diferentes tipos de tampón se muestran en diferentes tonos de gris.



Tabla 2. Pruebas de solubilidad de ALX-0081 en diferentes tampones de formulación.

N.D. = no determinado
Tabla 3. Visión general de formulaciones de ALX-0081 líquidas evaluadas en un ensayo de estabilidad en FT y almacenamiento, junto con los valores de pH y osmolalidad medidos.

Tabla 4. Visión general del estudio de estabilidad en almacenamiento de diferentes formulaciones de ALX-0081. Se indican puntos de tiempo, temperaturas de almacenamiento y métodos.

Tabla 5. Datos de estabilidad en almacenamiento para las diferentes formulaciones de ALX-0081 líquidas. Se muestran las áreas de superficie relativa de los picos de RP-HPLC más relevantes tras 1 mes de almacenamiento a 40°C. Piro = piroglutamato, principal = pico principal (incluyendo el pico de hombro, cuando está presente), oxidación = picos previos conjuntamente. La codificación por colores indica la pureza relativa de la muestra: la pureza más alta en blanco, pureza intermedia en gris y la pureza más baja en negro. La recuperación fue de ±100% para todas las muestras.

Tabla 6. Visión general de formulaciones de ALX-0081 liofilizadas/líquidas evaluadas en un ensayo de estabilidad en almacenamiento.

Tabla 7. Recuperación de ALX-0081 en diferentes formulaciones tras la liofilización y reconstitución basándose en las áreas totales notificadas por RP-HPLC y SE-HPLC.

Tabla 8. Visión general de datos de estabilidad en almacenamiento de las diferentes formulaciones de ALX-0081 (1,5 meses de almacenamiento a -20°C, 5°C, 25°C y 40°C). El código de color representa una evaluación cualitativa de la estabilidad de la muestra, que oscila entre blanco (la estabilidad más alta) y tonos de gris a negro (la estabilidad más baja).
N.T. = no sometido a prueba. (* en comparación con muestra de control líquida mantenida a -70°C)

Tabla 9. Visión general de formulaciones de ALX-0081 evaluadas en un ensayo de estabilidad en almacenamiento.

Tabla 10. Contenido de humedad de muestras de ALX-0081 liofilizadas y cantidades relativas de piroglutamato detectadas en RP-HPLC tras 4 semanas de almacenamiento a 40°C.

Tabla 11. Resultados de la inspección visual de la formulación de ALX-0081 durante el almacenamiento a 5°C y 25°C. “ ” = transparente, “+/-” = ligeramente turbio, “-” = turbio “h” = hora, “d” = días.

Tabla 12. Resultados de la inspección visual, el contenido y análisis de SE-HPLC de formulaciones de ALX-0081 tras 5 ciclos consecutivos de FT a -20°C (*en comparación con muestra de control líquida mantenida a < -70°C).

Tabla 13. Resultados de las mediciones de inspección visual, recuperación y osmolalidad de formulaciones de ALX-0081 tras 5 ciclos consecutivos de FT a -20°C o tras 1 ciclo de FT 24 h de almacenamiento 1 ciclo de FT (*en comparación con muestra de control líquida mantenida a < -70°C).

Tabla 14. Parámetros de liofilización.
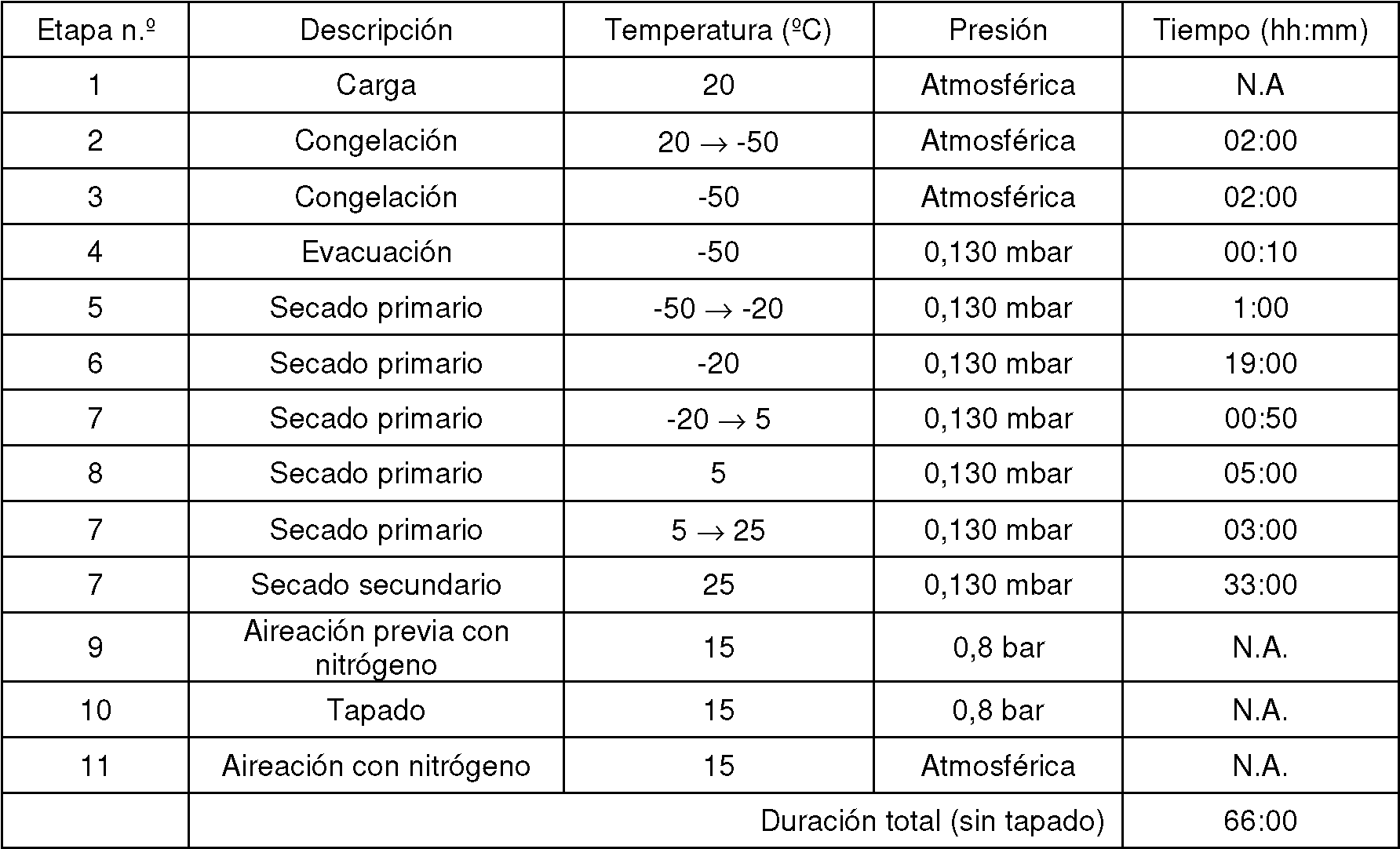
Tabla 15. Pico principal de análisis de RP-HPLC (pureza) de la formulación de ALX-0081 [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].



Tabla 17. Resultados de concentración de proteína mediante UV para la formulación de ALX-0081 [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Tabla 18. Análisis de SE-HPLC de la formulación de ALX-0081 [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Tabla 19. Resultados de pruebas físicas sobre ALX-0081 liofilizado almacenado a -202C [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].
Tabla 20. Resultados de pruebas físicas sobre ALX-0081 liofilizado almacenado a 52C [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Tabla 21. Resultados de pruebas físicas sobre ALX-0081 liofilizado almacenado a 252C/60% de HR [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Nota: *Valor promedio de 2 en lugar de 3 mediciones independientes
Tabla 22. Resultados de pruebas físicas sobre ALX-0081 liofilizado almacenado a 402C/75% de HR [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Nota: *Valor promedio de 2 en lugar de 3 mediciones independientes
Tabla 23. Resultados de potencia de la formulación de ALX-0081 [API 12,5 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Tabla 24. Resultados de comparabilidad in vitro de caplacizumab.


Tabla26ResudadosdeestabilidadenALX-0061liquidoalmacenadoa+5“C±3"C[API13.8mg/ml.Twoen-80al0.01%(vAr)ysacarosaal7%(pTv)enlampóncitrato20mMa pH6.5]

Tabla 27. Resultados de estabilidad en ALX-0081 liofilizado almacenado a 5°C ± 3°C [API 12,7 mg/ml, Tween-80 al 0,01 % (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH6,5]

Tabla 28. Resultados de estabilidad en ALX-0081 liofilizado almacenado a 25°C (±2®C/60 ± 5% de HR) [API 12,7 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5].

Tabla 29. Resultados de estabilidad en ALX-0081 liofilizado almacenado a 40*C (±2°C/75 ± 5% de HR) [API 12,7 mg/ml, Tween-80 al 0,01% (v/v) y sacarosa al 7% (p/v) en tampón citrato 20 mM a pH 6,5],
Claims (15)
1. Una formulación que comprende un agente de unión a factor de von Willebrand (vWF) y un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0, en la que dicho agente de unión a vWF comprende al menos un dominio variable individual de inmunoglobulina que se une a SEQ ID NO: 20, en la que dicha formulación comprende sacarosa a una concentración del 7%; y/o
dicho tampón preferiblemente tiene una osmolalidad en el intervalo de 290 ± 60 mOsm/kg, más preferiblemente en el intervalo de 290 ± 20 mOsm/kg; y/o
en la que dicha formulación es preferiblemente adecuada para administración parenteral, tal como una o más seleccionadas entre inyección intravenosa, inyección subcutánea, inyección intramuscular o inyección intraperitoneal; y/o
en la que dicha formulación es está preferiblemente en forma líquida, liofilizada, secada por pulverización, liofilizada reconstituida o congelada; y/o
en la que dicha formulación tiene:
(i) menos del 5% de especies de alto peso molecular (HMW) tras el almacenamiento durante al menos 12 meses a 5°C; y/o
(ii) menos del 5% de especies de bajo peso molecular (LMW) tras el almacenamiento durante al menos 12 meses a 5°C; y/o
en la que al menos el 80%, preferiblemente al menos el 90%, más preferiblemente al menos el 95% o incluso al menos el 99% del agente de unión a vWF retiene su actividad de unión tras el almacenamiento en comparación con la actividad de unión antes del almacenamiento, dicha actividad de unión tal como se mide mediante ELISA y/o Biacore; y/o en la que dicho agente de unión tiene una concentración en el intervalo de 0,1 a 80 mg/ml.
2. La formulación según la reivindicación 1, en la dicho agente de unión a vWF comprende:
- un dominio variable individual de inmunoglobulina que comprende una secuencia de dominio variable de cadena pesada que se deriva de un anticuerpo de cuatro cadenas convencional; y/o
- un dominio variable individual de inmunoglobulina que comprende una secuencia de dominio variable de cadena pesada que se deriva de un anticuerpo de cadena pesada o un Nanobody (incluyendo pero sin limitarse a una secuencia de VHH); y/o
- al menos una de las SEQ ID NO: 1-19; y/o
- una secuencia de aminoácidos que es al menos el 90% idéntica a SEQ ID NO: 1.
3. La formulación según cualquiera de las reivindicaciones 1-2, en la que dicho agente de unión a vWF es un polipéptido de cadena individual que comprende uno o más dominios variables individuales de inmunoglobulina, preferiblemente
- dicho agente de unión a vWF es monovalente o multivalente; y/o
- dicho agente de unión a vWF es monoespecífico o multiespecífico; y/o
- en la que uno o más dominios variables individuales de inmunoglobulina están injertados con CDR, humanizados, camelizados, desinmunizados y/o se seleccionan mediante presentación en fago.
4. La formulación según cualquiera de las reivindicaciones 1 a 3, que comprende además
- un detergente no iónico, tal como Tween-80, preferiblemente en una concentración de entre el 0,001 y el 0,5% (v/v), más preferiblemente el 0,01-0,02% (v/v).
5. Una formulación según cualquiera de las reivindicaciones 1 a 4 que comprende un agente de unión a vWF y un tampón citrato, en la que dicho tampón tiene un pH de 6,5 ±0,5, por ejemplo 6,2, 6,3, 6,4, 6,5, 6,6, 6,7 ó 6,8, más específicamente 6,5, y preferiblemente que comprende además un detergente no iónico, tal como Tween-80, preferiblemente en una concentración del 0,01% (v/v).
6. Una formulación según cualquiera de las reivindicaciones 1 a 5, que comprende:
(a) un agente de unión a vWF a una concentración de desde 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración del 7% (p/v); y
(c) Tween-80 a una concentración de aproximadamente el 0,001% a 0,5% (v/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0,
en la que el agente de unión a vWF en la formulación retiene al menos aproximadamente el 80% de su estabilidad tras el almacenamiento durante al menos 12 meses a 52C.
7. La formulación según cualquiera de las reivindicaciones 1-6, que es una formulación líquida o liofilizada reconstituida que comprende:
(a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración del 7% (p/v);
(c) Tween-80 a una concentración de aproximadamente el 0,001%-0,5% (v/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0.
8. La formulación según cualquiera de las reivindicaciones 1-7, que es una formulación de almacenamiento a granel que comprende:
(a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración del 7%;
(c) Tween-80 a una concentración de aproximadamente el 0,001%-0,5% (p/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0,
en la que al menos 100 litros de la formulación se almacenan en condiciones por debajo de la congelación.
9. Un método o procedimiento de preparación de una formulación según una cualquiera de las reivindicaciones 1 a 8, comprendiendo dicho método o procedimiento las etapas de:
- expresar el agente de unión a vWF en un cultivo celular;
- purificar el agente de unión a vWF haciendo pasar el agente de unión a vWF a través de al menos una de una etapa de purificación cromatográfica y una etapa de ultrafiltración/diafiltración;
- ajustar la concentración del agente de unión a vWF a de aproximadamente 0,1 a 80 mg/ml, en una formulación que contiene:
(i) sacarosa a una concentración del 7%;
(ii) Tween-80 a una concentración de aproximadamente el 0,001%-0,5% (p/v); y
(iii) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0.
10. Un método de preparación de una formulación reconstituida según una cualquiera de las reivindicaciones 1 a 8, comprendiendo dicho método las etapas de: (i) liofilizar una mezcla de un agente de unión a vWF, un lioprotector, un tensioactivo y un tampón, formando de ese modo una mezcla liofilizada; y (ii) reconstituir la mezcla liofilizada en un diluyente, preparando de ese modo la formulación, en el que la formulación reconstituida comprende
(a) un agente de unión a vWF a una concentración de desde aproximadamente 0,1 mg/ml hasta aproximadamente 80 mg/ml;
(b) sacarosa a una concentración del 7% (p/v);
(c) Tween-80 a una concentración de aproximadamente el 0,001-0,5% (v/v); y
(d) un tampón citrato a una concentración de aproximadamente 5 mM a aproximadamente 200 mM, de manera que el pH de la formulación es de aproximadamente 6,0 a 7,0.
11. Un método para estabilizar o almacenar un agente de unión a vWF, preferiblemente un polipéptido que comprende al menos una de SEQ ID NO: 1-19 para el almacenamiento, que comprende preparar una formulación según cualquiera de las reivindicaciones 1 a 8.
12. Un kit o artículo de fabricación que comprende un recipiente que contiene la formulación según cualquiera de las reivindicaciones 1 a 8, e instrucciones para su uso; preferiblemente la formulación está presente en un vial o una jeringa inyectable, tal como una jeringa inyectable precargada.
13. Una forma de dosificación unitaria farmacéutica para administración parenteral a un paciente, preferiblemente un paciente humano, que comprende una formulación según cualquiera de las reivindicaciones 1 a 8.
14. Una composición farmacéutica o de diagnóstico que comprende una formulación que comprende un agente de unión a vWF según cualquiera de las reivindicaciones 1 a 8 u obtenible mediante el método según las reivindicaciones 9 a 11.
15. La formulación según cualquiera de las reivindicaciones 1 a 8 para su uso en un método de tratamiento o prevención de un trastorno relacionado con vWF en un sujeto humano o animal, que comprende administrar a dicho sujeto una composición farmacéutica que comprende la formulación de cualquiera de las reivindicaciones 1-8, reduciendo de ese modo uno o más síntomas asociados con el trastorno relacionado con vWF, preferiblemente PTT.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361824523P | 2013-05-17 | 2013-05-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2895150T3 true ES2895150T3 (es) | 2022-02-17 |
Family
ID=49304267
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14727433T Active ES2731922T3 (es) | 2013-05-17 | 2014-05-16 | Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas |
| ES19157147T Active ES2895150T3 (es) | 2013-05-17 | 2014-05-16 | Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14727433T Active ES2731922T3 (es) | 2013-05-17 | 2014-05-16 | Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas |
Country Status (25)
| Country | Link |
|---|---|
| US (2) | US10035862B2 (es) |
| EP (2) | EP3511018B1 (es) |
| JP (2) | JP6433986B2 (es) |
| KR (1) | KR102166399B1 (es) |
| CN (3) | CN110063935B (es) |
| AU (3) | AU2014267222B2 (es) |
| BR (1) | BR112015028411B1 (es) |
| CA (1) | CA2912670A1 (es) |
| CY (1) | CY1122090T1 (es) |
| DK (1) | DK2996720T3 (es) |
| ES (2) | ES2731922T3 (es) |
| HR (1) | HRP20190954T1 (es) |
| HU (1) | HUE044392T2 (es) |
| LT (1) | LT2996720T (es) |
| MX (2) | MX387527B (es) |
| NL (1) | NL1040254C2 (es) |
| PH (1) | PH12015502591B1 (es) |
| PL (2) | PL3511018T3 (es) |
| PT (2) | PT2996720T (es) |
| RS (1) | RS58857B1 (es) |
| RU (1) | RU2671977C2 (es) |
| SI (1) | SI2996720T1 (es) |
| SM (1) | SMT201900359T1 (es) |
| TR (1) | TR201908663T4 (es) |
| WO (1) | WO2014184352A1 (es) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL1040254C2 (en) * | 2013-05-17 | 2014-11-24 | Ablynx Nv | Stable formulations of immunoglobulin single variable domains and uses thereof. |
| AU2015366284B2 (en) | 2014-12-19 | 2021-07-22 | Ablynx N.V. | Cysteine linked nanobody dimers |
| CN111035754A (zh) * | 2016-03-23 | 2020-04-21 | 兆科药业(合肥)有限公司 | 抗血小板溶栓素在制备治疗血栓性血小板减少性紫癜的药物中的应用 |
| GB201612043D0 (en) | 2016-07-11 | 2016-08-24 | Crescendo Biologics Ltd | Composition for treatment of disorders |
| TWI784982B (zh) * | 2016-11-02 | 2022-12-01 | 開曼群島商北極星藥業集團股份有限公司 | 聚乙二醇化精胺酸脫亞胺酶之調配物 |
| EP3749696A1 (en) | 2018-02-06 | 2020-12-16 | Ablynx N.V. | Methods of treating initial episode of ttp with immunoglobulin single variable domains |
| CN112805238A (zh) | 2018-07-03 | 2021-05-14 | 芬内克制药股份有限公司 | 无水硫代硫酸钠和其调配物 |
| JP7653352B2 (ja) * | 2018-09-05 | 2025-03-28 | エクスプレステック、リミテッド、ライアビリティ、カンパニー | 免疫グロブリンa製剤 |
| SG11202103670XA (en) * | 2018-10-10 | 2021-05-28 | Astellas Pharma Inc | Pharmaceutical composition containing tagged site-antihuman antibody fab fragment complex |
| CN110564841A (zh) * | 2019-09-19 | 2019-12-13 | 广东省中医院(广州中医药大学第二附属医院、广州中医药大学第二临床医学院、广东省中医药科学院) | 脑缺血相关基因作为缺血性卒中行为学特征分析的生物标记的应用 |
| CN110624105B (zh) * | 2019-09-24 | 2021-06-11 | 苏州大学 | 血管性血友病因子的结构敏感多肽抗原的序列 |
| JP7502861B2 (ja) * | 2019-12-27 | 2024-06-19 | 旭化成株式会社 | 標的物質と両親媒性高分子との複合体の形成反応の反応温度の評価方法 |
| CN113908254B (zh) * | 2021-10-19 | 2024-05-28 | 山西锦波生物医药股份有限公司 | 一种干粉吸入剂及其制备方法和用途 |
| CN115850493B (zh) * | 2022-11-08 | 2024-03-12 | 江苏耀海生物制药有限公司 | 一种二价纳米抗体Cablivi的分离纯化方法 |
| WO2025240928A1 (en) * | 2024-05-16 | 2025-11-20 | Vega Therapeutics, Inc. | Treatment of von willebrand disease |
Family Cites Families (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1621554T4 (da) | 1992-08-21 | 2012-12-17 | Univ Bruxelles | Immunoglobuliner blottet for lette kæder |
| ES2162863T3 (es) | 1993-04-29 | 2002-01-16 | Unilever Nv | Produccion de anticuerpos o fragmentos (funcionalizados) de los mismos derivados de inmunoglobulinas de cadena pesada de camelidae. |
| FR2708622B1 (fr) | 1993-08-02 | 1997-04-18 | Raymond Hamers | Vecteur recombinant contenant une séquence d'un gène de lipoprotéine de structure pour l'expression de séquences de nucléotides. |
| MX9605717A (es) | 1994-05-18 | 1998-05-31 | Inhale Therapeutic Syst | Metodos y composiciones para la formulacion de polvo seco de interferones. |
| EP0739981A1 (en) | 1995-04-25 | 1996-10-30 | Vrije Universiteit Brussel | Variable fragments of immunoglobulins - use for therapeutic or veterinary purposes |
| ES2294799T3 (es) | 1996-06-27 | 2008-04-01 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw. | Moleculas de anticuerpos que interactuan especificamente con el sitio activo o hendidura de una molecula diana. |
| US6329516B1 (en) | 1997-04-28 | 2001-12-11 | Fmc Corporation | Lepidopteran GABA-gated chloride channels |
| DK1027439T3 (da) | 1997-10-27 | 2010-05-10 | Bac Ip Bv | Multivalente antigenbindende proteiner |
| AU3596599A (en) | 1998-01-26 | 1999-08-09 | Unilever Plc | Method for producing antibody fragments |
| EP0967284A1 (en) | 1998-05-28 | 1999-12-29 | Pfizer Limited | Phosphodiesterases |
| EP1141711A1 (en) | 1999-01-05 | 2001-10-10 | Unilever Plc | Binding of antibody fragments to solid supports |
| DK1144616T4 (da) | 1999-01-19 | 2009-03-30 | Unilever Nv | Fremgangsmåde til fremstilling af antistoffragmenter |
| CA2361678A1 (en) | 1999-02-05 | 2000-08-10 | Rijksuniversiteit Leiden | Method of modulating metabolite biosynthesis in recombinant cells |
| DE60035163T2 (de) | 1999-03-15 | 2008-02-21 | University Of British Columbia, Vancouver | Abc1 polypeptide und verfahren und reagenzien zur modulation des cholesterolgehalts |
| CA2370351A1 (en) | 1999-04-22 | 2000-11-02 | Unilever Plc | Inhibition of viral infection using monovalent antigen-binding proteins |
| CN100513570C (zh) | 1999-06-18 | 2009-07-15 | Cv治疗公司 | 结合框运输蛋白abc1的调节作用 |
| IL147920A0 (en) | 1999-08-02 | 2002-08-14 | Keygene Nv | Method for generating cgmmv resistant plants, genetic constructs, and the obtained cgmmv-resistant plants |
| GB9922124D0 (en) | 1999-09-17 | 1999-11-17 | Pfizer Ltd | Phosphodiesterase enzymes |
| US6479280B1 (en) | 1999-09-24 | 2002-11-12 | Vlaams Interuniversitair Institutuut Voor Biotechnologie Vzw | Recombinant phages capable of entering host cells via specific interaction with an artificial receptor |
| DE19955408A1 (de) | 1999-11-18 | 2001-05-23 | Bayer Ag | GABA-B-Rezeptoren |
| WO2001040310A2 (en) | 1999-11-29 | 2001-06-07 | Unilever Plc | Immobilisation of proteins using a polypeptide segment |
| ES2331051T3 (es) | 1999-11-29 | 2009-12-21 | Bac Ip B.V. | Inmovilizacion de moleculas de union de antigenos de un dominio. |
| ES2324280T3 (es) | 2000-03-14 | 2009-08-04 | Unilever N.V. | Dominios variables de la cadena pesada de anticuerpos frente a lipasas dieteticas humanas y sus usos. |
| US20030190598A1 (en) | 2000-05-26 | 2003-10-09 | Jasmid Tanha | Single-domain antigen-binding antibody fragments derived from llama antibodies |
| ATE513854T1 (de) | 2000-12-13 | 2011-07-15 | Bac Ip B V | Proteinraster aus variablen domänen der schweren immunoglobulinkette von kamelen |
| PT1419179E (pt) | 2001-08-10 | 2010-03-16 | Univ Aberdeen | Domínios de ligação de antigenes |
| JP4213586B2 (ja) | 2001-09-13 | 2009-01-21 | 株式会社抗体研究所 | ラクダ抗体ライブラリーの作製方法 |
| US6723359B2 (en) | 2001-10-18 | 2004-04-20 | Firmenich Sa | Spray-dried compositions and method for their preparation |
| JP2005289809A (ja) | 2001-10-24 | 2005-10-20 | Vlaams Interuniversitair Inst Voor Biotechnologie Vzw (Vib Vzw) | 突然変異重鎖抗体 |
| EP1456410A2 (en) | 2001-12-11 | 2004-09-15 | AlgoNomics N.V. | Method for displaying loops from immunoglobulin domains in different contexts |
| EP1456237A2 (en) | 2001-12-21 | 2004-09-15 | Vlaams Interuniversitair Instituut voor Biotechnologie vzw. | Method for cloning of variable domain sequences |
| CA2471645A1 (en) | 2002-01-03 | 2003-07-10 | Vlaams Interuniversitair Instituut Voor Biotechnologie Vzw | Immunoconjugates useful for treatment of tumours |
| GB0202633D0 (en) * | 2002-02-05 | 2002-03-20 | Delta Biotechnology Ltd | Stabilization of protein preparations |
| AU2003251238A1 (en) | 2002-08-07 | 2004-02-25 | Umc Utrecht Holding B.V. | Modulation of platelet adhesion based on the surface exposed beta-switch loop of platelet glycoprotein ib-alpha |
| BRPI0315666B8 (pt) | 2002-10-23 | 2021-05-25 | Ludwig Inst For Cancer Res Ltd | dna de a34 e a33 do tipo 3, proteínas, seus anticorpos e métodos de tratamento usando os mesmos |
| WO2004041863A2 (en) | 2002-11-08 | 2004-05-21 | Ablynx N.V. | Single domain antibodies directed against interferon- gamma and uses therefor |
| EP1558645B1 (en) | 2002-11-08 | 2011-07-27 | Ablynx N.V. | Stabilized single domain antibodies in pharmaceutical composition adapted for inhalation |
| JP2006517789A (ja) | 2003-01-10 | 2006-08-03 | アブリンクス エン.ヴェー. | 治療用ポリペプチド、その相同物、その断片、および血小板媒介凝集の調節での使用 |
| ATE485307T1 (de) | 2003-11-07 | 2010-11-15 | Ablynx Nv | Camelidae schwere ketten antikörper vhhs gegen epidermalen wachstumfaktor rezeptor (egfr) und ihre verwendung |
| WO2006040153A2 (en) | 2004-10-13 | 2006-04-20 | Ablynx N.V. | Single domain camelide anti -amyloid beta antibodies and polypeptides comprising the same for the treatment and diagnosis of degenarative neural diseases such as alzheimer's disease |
| WO2006074947A2 (en) | 2005-01-14 | 2006-07-20 | Ablynx N.V. | METHODS AND ASSAYS FOR DISTINGUISHING BETWEEN DIFFERENT FORMS OF DISEASES AND DISORDERS CHARACTERIZED BY THROMBOCYTOPENIA AND/OR BY SPONTANEOUS INTERACTION BETWEEN VON WILLEBRAND FACTOR (vWF) AND PLATELETS |
| AU2005325801A1 (en) | 2005-01-31 | 2006-08-03 | Ablynx N.V. | Method for generating variable domain sequences of heavy chain antibodies |
| US8188223B2 (en) | 2005-05-18 | 2012-05-29 | Ablynx N.V. | Serum albumin binding proteins |
| ES2694247T3 (es) * | 2005-05-20 | 2018-12-19 | Ablynx N.V. | NanobodiesTM mejorados para el tratamiento de trastornos mediados por agregación |
| CA2611726C (en) * | 2005-06-10 | 2017-07-11 | Chugai Seiyaku Kabushiki Kaisha | Pharmaceutical compositions containing sc(fv)2 |
| US20110059101A9 (en) | 2006-03-13 | 2011-03-10 | Ablynx N.V. | Amino Acid Sequences Directed Against Il-6 And Polypetides Comprising The Same For The Treatment Of Diseases And Disorders Associated With Il-6 Mediated Signalling |
| US20080267949A1 (en) | 2006-12-05 | 2008-10-30 | Ablynx N.V. | Peptides capable of binding to serum proteins |
| EP2136839A4 (en) * | 2007-03-22 | 2010-04-07 | Imclone Llc | STABLE ANTIBODY FORMULATIONS |
| SG187410A1 (en) * | 2007-12-28 | 2013-02-28 | Baxter Int | Recombinant vwf formulations |
| CA2711984A1 (en) * | 2008-01-15 | 2009-07-23 | Abbott Gmbh & Co. Kg | Powdered protein compositions and methods of making same |
| EP2252303A2 (en) * | 2008-03-21 | 2010-11-24 | Ablynx NV | Von willebrand factor specific binders and methods of use therefor |
| WO2009127691A1 (en) | 2008-04-17 | 2009-10-22 | Ablynx N.V. | Peptides capable of binding to serum proteins and compounds, constructs and polypeptides comprising the same |
| EP2349314B1 (en) * | 2008-10-21 | 2013-02-27 | Baxter International Inc. | Lyophilized recombinant vwf formulations |
| CA2738243C (en) * | 2008-10-29 | 2020-09-29 | Wyeth Llc | Formulations of single domain antigen binding molecules |
| EP2196476A1 (en) * | 2008-12-10 | 2010-06-16 | Novartis Ag | Antibody formulation |
| US20120321640A1 (en) | 2009-12-01 | 2012-12-20 | Ablynx N.V. | Von willebrand factor specific binding agents and uses thereof |
| EP2531523A1 (en) | 2010-02-05 | 2012-12-12 | Ablynx N.V. | Peptides capable of binding to serum albumin and compounds, constructs and polypeptides comprising the same |
| DK2533761T3 (da) * | 2010-02-11 | 2019-06-24 | Ablynx Nv | Fremgangsmåder og sammensætninger til fremstilling af aerosoler |
| US20120225072A1 (en) * | 2011-03-02 | 2012-09-06 | Ablynx N.V. | Stable formulations of immunoglobulin single variable domains and uses thereof |
| NL1040254C2 (en) * | 2013-05-17 | 2014-11-24 | Ablynx Nv | Stable formulations of immunoglobulin single variable domains and uses thereof. |
-
2013
- 2013-06-14 NL NL1040254A patent/NL1040254C2/en active
-
2014
- 2014-05-16 HR HRP20190954TT patent/HRP20190954T1/hr unknown
- 2014-05-16 US US14/891,441 patent/US10035862B2/en active Active
- 2014-05-16 MX MX2019005269A patent/MX387527B/es unknown
- 2014-05-16 ES ES14727433T patent/ES2731922T3/es active Active
- 2014-05-16 SI SI201431229T patent/SI2996720T1/sl unknown
- 2014-05-16 PT PT14727433T patent/PT2996720T/pt unknown
- 2014-05-16 MX MX2015015722A patent/MX364664B/es active IP Right Grant
- 2014-05-16 RU RU2015154214A patent/RU2671977C2/ru active
- 2014-05-16 RS RS20190694A patent/RS58857B1/sr unknown
- 2014-05-16 ES ES19157147T patent/ES2895150T3/es active Active
- 2014-05-16 PT PT191571470T patent/PT3511018T/pt unknown
- 2014-05-16 BR BR112015028411-6A patent/BR112015028411B1/pt active IP Right Grant
- 2014-05-16 AU AU2014267222A patent/AU2014267222B2/en active Active
- 2014-05-16 JP JP2016513387A patent/JP6433986B2/ja active Active
- 2014-05-16 PL PL19157147T patent/PL3511018T3/pl unknown
- 2014-05-16 CN CN201811423557.0A patent/CN110063935B/zh active Active
- 2014-05-16 CA CA2912670A patent/CA2912670A1/en not_active Abandoned
- 2014-05-16 LT LTEP14727433.6T patent/LT2996720T/lt unknown
- 2014-05-16 HU HUE14727433 patent/HUE044392T2/hu unknown
- 2014-05-16 DK DK14727433.6T patent/DK2996720T3/da active
- 2014-05-16 WO PCT/EP2014/060107 patent/WO2014184352A1/en not_active Ceased
- 2014-05-16 KR KR1020157035523A patent/KR102166399B1/ko active Active
- 2014-05-16 EP EP19157147.0A patent/EP3511018B1/en active Active
- 2014-05-16 EP EP14727433.6A patent/EP2996720B1/en active Active
- 2014-05-16 SM SM20190359T patent/SMT201900359T1/it unknown
- 2014-05-16 CN CN201811422890.XA patent/CN110037983A/zh active Pending
- 2014-05-16 CN CN201480034630.9A patent/CN105339003B/zh active Active
- 2014-05-16 PL PL14727433T patent/PL2996720T3/pl unknown
- 2014-05-16 TR TR2019/08663T patent/TR201908663T4/tr unknown
-
2015
- 2015-11-16 PH PH12015502591A patent/PH12015502591B1/en unknown
-
2018
- 2018-06-14 US US16/008,103 patent/US20190010249A1/en active Pending
- 2018-11-07 JP JP2018209611A patent/JP6952019B2/ja active Active
-
2019
- 2019-02-25 AU AU2019201307A patent/AU2019201307B2/en active Active
- 2019-06-26 CY CY20191100669T patent/CY1122090T1/el unknown
-
2021
- 2021-01-05 AU AU2021200035A patent/AU2021200035A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2895150T3 (es) | Formulaciones estables de dominios variables individuales de inmunoglobulina y usos de las mismas | |
| US7951368B2 (en) | Compositions of specific binding agents to hepatocyte growth factor | |
| ES2893861T3 (es) | Formulaciones de anticuerpo estables, acuosas | |
| ES2831323T3 (es) | Formulaciones estables de polipéptidos y usos de las mismas | |
| JP7657545B2 (ja) | 室温で安定な凍結乾燥タンパク質 | |
| TW201414496A (zh) | 抗體及蛋白質配方 | |
| ES2915473T3 (es) | Dominios variables únicos de inmunoglobulina para su uso en métodos de tratamiento de TTP | |
| JPWO2008029908A1 (ja) | 抗体を含有する安定な凍結乾燥医薬製剤 | |
| RU2773172C2 (ru) | Стабильные композиции иммуноглобулинового одиночного вариабельного домена и их применение | |
| US20120225072A1 (en) | Stable formulations of immunoglobulin single variable domains and uses thereof | |
| HK1218258B (zh) | 免疫球蛋白单可变结构域的稳定制剂及其应用 | |
| WO2026025028A1 (en) | Methods of making ultra-high concentrated protein formulations using lyophilization | |
| CA3173706A1 (en) | Pharmaceutical formulation |