ES2907992T3 - Agentes novedosos de obtención de imágenes para detectar disfunciones neurológicas - Google Patents
Agentes novedosos de obtención de imágenes para detectar disfunciones neurológicas Download PDFInfo
- Publication number
- ES2907992T3 ES2907992T3 ES09711194T ES09711194T ES2907992T3 ES 2907992 T3 ES2907992 T3 ES 2907992T3 ES 09711194 T ES09711194 T ES 09711194T ES 09711194 T ES09711194 T ES 09711194T ES 2907992 T3 ES2907992 T3 ES 2907992T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- compound
- mmol
- aryl
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000012216 imaging agent Substances 0.000 title description 9
- 230000009251 neurologic dysfunction Effects 0.000 title description 2
- 208000015015 neurological dysfunction Diseases 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 135
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 102
- -1 nitro, hydroxyl Chemical group 0.000 claims abstract description 49
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 32
- 239000001257 hydrogen Substances 0.000 claims abstract description 32
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 31
- 125000001072 heteroaryl group Chemical group 0.000 claims abstract description 31
- 125000006652 (C3-C12) cycloalkyl group Chemical group 0.000 claims abstract description 30
- 150000003839 salts Chemical class 0.000 claims abstract description 25
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 18
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 17
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 16
- 125000005914 C6-C14 aryloxy group Chemical group 0.000 claims abstract description 14
- 125000005553 heteroaryloxy group Chemical group 0.000 claims abstract description 14
- 125000001424 substituent group Chemical group 0.000 claims abstract description 14
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 12
- 125000000000 cycloalkoxy group Chemical group 0.000 claims abstract description 11
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 9
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 5
- 125000005114 heteroarylalkoxy group Chemical group 0.000 claims abstract description 4
- 125000001475 halogen functional group Chemical group 0.000 claims abstract 3
- 239000000203 mixture Substances 0.000 claims description 135
- 208000024827 Alzheimer disease Diseases 0.000 claims description 73
- 125000003118 aryl group Chemical group 0.000 claims description 57
- 230000027455 binding Effects 0.000 claims description 50
- 210000004556 brain Anatomy 0.000 claims description 41
- 238000000034 method Methods 0.000 claims description 39
- 210000005013 brain tissue Anatomy 0.000 claims description 21
- 238000003384 imaging method Methods 0.000 claims description 21
- 241000124008 Mammalia Species 0.000 claims description 19
- 229920000642 polymer Polymers 0.000 claims description 19
- 208000037259 Amyloid Plaque Diseases 0.000 claims description 10
- 238000002600 positron emission tomography Methods 0.000 claims description 9
- 230000008499 blood brain barrier function Effects 0.000 claims description 7
- 210000001218 blood-brain barrier Anatomy 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 6
- 238000002603 single-photon emission computed tomography Methods 0.000 claims description 6
- 201000010099 disease Diseases 0.000 claims description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 4
- 210000002682 neurofibrillary tangle Anatomy 0.000 claims description 4
- 231100000189 neurotoxic Toxicity 0.000 claims description 4
- 230000002887 neurotoxic effect Effects 0.000 claims description 4
- 238000011282 treatment Methods 0.000 claims description 4
- 238000009826 distribution Methods 0.000 claims description 3
- 230000000971 hippocampal effect Effects 0.000 claims description 3
- 208000019901 Anxiety disease Diseases 0.000 claims description 2
- 208000008589 Obesity Diseases 0.000 claims description 2
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims description 2
- 206010034912 Phobia Diseases 0.000 claims description 2
- 230000036506 anxiety Effects 0.000 claims description 2
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 206010015037 epilepsy Diseases 0.000 claims description 2
- 238000011503 in vivo imaging Methods 0.000 claims description 2
- 238000010253 intravenous injection Methods 0.000 claims description 2
- 238000012633 nuclear imaging Methods 0.000 claims description 2
- 235000020824 obesity Nutrition 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims description 2
- 208000019899 phobic disease Diseases 0.000 claims description 2
- 201000000980 schizophrenia Diseases 0.000 claims description 2
- 208000011580 syndromic disease Diseases 0.000 claims description 2
- 238000002591 computed tomography Methods 0.000 claims 1
- 125000005915 C6-C14 aryl group Chemical group 0.000 abstract 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 202
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 202
- 235000019439 ethyl acetate Nutrition 0.000 description 101
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 93
- 239000007787 solid Substances 0.000 description 88
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 87
- 238000006243 chemical reaction Methods 0.000 description 82
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 78
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 74
- 239000000047 product Substances 0.000 description 68
- 238000005160 1H NMR spectroscopy Methods 0.000 description 62
- 230000015572 biosynthetic process Effects 0.000 description 51
- 238000002360 preparation method Methods 0.000 description 47
- 239000000243 solution Substances 0.000 description 47
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 41
- 238000003786 synthesis reaction Methods 0.000 description 41
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 40
- 239000012043 crude product Substances 0.000 description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 35
- 238000004587 chromatography analysis Methods 0.000 description 33
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 32
- 125000000609 carbazolyl group Chemical class C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 30
- 238000000746 purification Methods 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 29
- 239000000377 silicon dioxide Substances 0.000 description 28
- 238000004128 high performance liquid chromatography Methods 0.000 description 27
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 26
- 239000012298 atmosphere Substances 0.000 description 26
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 25
- 238000003756 stirring Methods 0.000 description 25
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 23
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 23
- 239000002243 precursor Substances 0.000 description 23
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 22
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 21
- 238000002474 experimental method Methods 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 20
- IAVCEBMLYVGBLA-UHFFFAOYSA-N 2-[1-[6-[2-fluoroethyl(methyl)amino]naphthalen-2-yl]ethylidene]propanedinitrile Chemical compound C1=C(C(C)=C(C#N)C#N)C=CC2=CC(N(CCF)C)=CC=C21 IAVCEBMLYVGBLA-UHFFFAOYSA-N 0.000 description 19
- 238000010828 elution Methods 0.000 description 18
- 239000008223 sterile water Substances 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 17
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 16
- 239000000706 filtrate Substances 0.000 description 16
- KRHYYFGTRYWZRS-BJUDXGSMSA-M fluorine-18(1-) Chemical compound [18F-] KRHYYFGTRYWZRS-BJUDXGSMSA-M 0.000 description 16
- 239000000700 radioactive tracer Substances 0.000 description 16
- 239000012267 brine Substances 0.000 description 15
- 229910000024 caesium carbonate Inorganic materials 0.000 description 15
- 239000003446 ligand Substances 0.000 description 15
- 239000003921 oil Substances 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- 239000002904 solvent Substances 0.000 description 15
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 14
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 13
- 235000019341 magnesium sulphate Nutrition 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 239000000741 silica gel Substances 0.000 description 12
- 229910002027 silica gel Inorganic materials 0.000 description 12
- 210000004885 white matter Anatomy 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- 241000699670 Mus sp. Species 0.000 description 10
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 10
- 238000003682 fluorination reaction Methods 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 235000011181 potassium carbonates Nutrition 0.000 description 10
- 238000010186 staining Methods 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N benzo-alpha-pyrone Natural products C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 9
- 239000013020 final formulation Substances 0.000 description 9
- 238000005342 ion exchange Methods 0.000 description 9
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 9
- 238000011894 semi-preparative HPLC Methods 0.000 description 9
- 239000011734 sodium Substances 0.000 description 9
- MHUIIHOKCRBQES-UHFFFAOYSA-N 2-[2-(2-fluoroethoxy)ethoxy]ethyl 4-methylbenzenesulfonate Chemical compound CC1=CC=C(S(=O)(=O)OCCOCCOCCF)C=C1 MHUIIHOKCRBQES-UHFFFAOYSA-N 0.000 description 8
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 8
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000003153 chemical reaction reagent Substances 0.000 description 8
- ADEBPBSSDYVVLD-UHFFFAOYSA-N donepezil Chemical compound O=C1C=2C=C(OC)C(OC)=CC=2CC1CC(CC1)CCN1CC1=CC=CC=C1 ADEBPBSSDYVVLD-UHFFFAOYSA-N 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 8
- 229910052763 palladium Inorganic materials 0.000 description 8
- 238000012546 transfer Methods 0.000 description 8
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 description 7
- CKEKFQLHCAZGSP-UHFFFAOYSA-N 2-(trifluoromethyl)-3h-benzimidazol-5-amine Chemical compound NC1=CC=C2N=C(C(F)(F)F)NC2=C1 CKEKFQLHCAZGSP-UHFFFAOYSA-N 0.000 description 7
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 7
- 230000029936 alkylation Effects 0.000 description 7
- 238000005804 alkylation reaction Methods 0.000 description 7
- 229910052786 argon Inorganic materials 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 239000012467 final product Substances 0.000 description 7
- 229910052736 halogen Inorganic materials 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 235000013824 polyphenols Nutrition 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- JTLAIKFGRHDNQM-UHFFFAOYSA-N 1-bromo-2-fluoroethane Chemical compound FCCBr JTLAIKFGRHDNQM-UHFFFAOYSA-N 0.000 description 6
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 235000001671 coumarin Nutrition 0.000 description 6
- VRZVPALEJCLXPR-UHFFFAOYSA-N ethyl 4-methylbenzenesulfonate Chemical compound CCOS(=O)(=O)C1=CC=C(C)C=C1 VRZVPALEJCLXPR-UHFFFAOYSA-N 0.000 description 6
- 150000002367 halogens Chemical class 0.000 description 6
- 125000005842 heteroatom Chemical group 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- JADVWWSKYZXRGX-UHFFFAOYSA-M thioflavine T Chemical compound [Cl-].C1=CC(N(C)C)=CC=C1C1=[N+](C)C2=CC=C(C)C=C2S1 JADVWWSKYZXRGX-UHFFFAOYSA-M 0.000 description 6
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 6
- WGWUHHIPQPGNBS-UHFFFAOYSA-N 4-(2-nitro-4-phenylmethoxyphenyl)phenol Chemical compound C1=CC(O)=CC=C1C(C(=C1)[N+]([O-])=O)=CC=C1OCC1=CC=CC=C1 WGWUHHIPQPGNBS-UHFFFAOYSA-N 0.000 description 5
- UYDCNLOPKRFYGG-UHFFFAOYSA-N 7-phenylmethoxy-9h-carbazol-2-ol Chemical compound C=1C(O)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 UYDCNLOPKRFYGG-UHFFFAOYSA-N 0.000 description 5
- 238000006069 Suzuki reaction reaction Methods 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000000544 cholinesterase inhibitor Substances 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 238000007069 methylation reaction Methods 0.000 description 5
- 230000009871 nonspecific binding Effects 0.000 description 5
- 238000012216 screening Methods 0.000 description 5
- GNDRTCLMOHNBGG-UHFFFAOYSA-N tert-butyl n-(7-hydroxy-9h-carbazol-2-yl)carbamate Chemical compound OC1=CC=C2C3=CC=C(NC(=O)OC(C)(C)C)C=C3NC2=C1 GNDRTCLMOHNBGG-UHFFFAOYSA-N 0.000 description 5
- AIFRHYZBTHREPW-UHFFFAOYSA-N β-carboline Chemical compound N1=CC=C2C3=CC=CC=C3NC2=C1 AIFRHYZBTHREPW-UHFFFAOYSA-N 0.000 description 5
- LDCSOYKUKWPTHL-UHFFFAOYSA-N 2-(2-fluoroethoxy)-9h-carbazole Chemical compound C1=CC=C2C3=CC=C(OCCF)C=C3NC2=C1 LDCSOYKUKWPTHL-UHFFFAOYSA-N 0.000 description 4
- QBPAUIDFWFXLKB-UHFFFAOYSA-N 9h-carbazol-1-ol Chemical compound N1C2=CC=CC=C2C2=C1C(O)=CC=C2 QBPAUIDFWFXLKB-UHFFFAOYSA-N 0.000 description 4
- GWPGDZPXOZATKL-UHFFFAOYSA-N 9h-carbazol-2-ol Chemical compound C1=CC=C2C3=CC=C(O)C=C3NC2=C1 GWPGDZPXOZATKL-UHFFFAOYSA-N 0.000 description 4
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 4
- 101150041968 CDC13 gene Proteins 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 101100208721 Mus musculus Usp5 gene Proteins 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 229940039856 aricept Drugs 0.000 description 4
- 239000000090 biomarker Substances 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 229960000956 coumarin Drugs 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 210000004884 grey matter Anatomy 0.000 description 4
- 238000012744 immunostaining Methods 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- DNKXMKNQJFMUHE-UHFFFAOYSA-N n-(7-hydroxy-9h-carbazol-2-yl)formamide Chemical compound O=CNC1=CC=C2C3=CC=C(O)C=C3NC2=C1 DNKXMKNQJFMUHE-UHFFFAOYSA-N 0.000 description 4
- 239000012188 paraffin wax Substances 0.000 description 4
- 230000004224 protection Effects 0.000 description 4
- 230000002285 radioactive effect Effects 0.000 description 4
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 4
- 235000010378 sodium ascorbate Nutrition 0.000 description 4
- 229960005055 sodium ascorbate Drugs 0.000 description 4
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 4
- MGHMPUOTDNPBTM-UHFFFAOYSA-N tert-butyl n-[7-(4-nitrophenoxy)-9h-carbazol-2-yl]carbamate Chemical compound C=1C(NC(=O)OC(C)(C)C)=CC=C(C2=CC=3)C=1NC2=CC=3OC1=CC=C([N+]([O-])=O)C=C1 MGHMPUOTDNPBTM-UHFFFAOYSA-N 0.000 description 4
- 150000003852 triazoles Chemical class 0.000 description 4
- 239000001993 wax Substances 0.000 description 4
- WKGZJBVXZWCZQC-UHFFFAOYSA-N 1-(1-benzyltriazol-4-yl)-n,n-bis[(1-benzyltriazol-4-yl)methyl]methanamine Chemical compound C=1N(CC=2C=CC=CC=2)N=NC=1CN(CC=1N=NN(CC=2C=CC=CC=2)C=1)CC(N=N1)=CN1CC1=CC=CC=C1 WKGZJBVXZWCZQC-UHFFFAOYSA-N 0.000 description 3
- ZVHZPFSWZWSDEN-UHFFFAOYSA-N 1-bromo-1-fluoroethane Chemical compound CC(F)Br ZVHZPFSWZWSDEN-UHFFFAOYSA-N 0.000 description 3
- KVBJIVDDLMDAPB-UHFFFAOYSA-N 1-chloro-2-nitro-4-phenylmethoxybenzene Chemical compound C1=C(Cl)C([N+](=O)[O-])=CC(OCC=2C=CC=CC=2)=C1 KVBJIVDDLMDAPB-UHFFFAOYSA-N 0.000 description 3
- IBXNCJKFFQIKKY-UHFFFAOYSA-N 1-pentyne Chemical compound CCCC#C IBXNCJKFFQIKKY-UHFFFAOYSA-N 0.000 description 3
- LZIPBJBQQPZLOR-UHFFFAOYSA-N 2-(4-methylphenyl)sulfonyloxyethyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCCOS(=O)(=O)C1=CC=C(C)C=C1 LZIPBJBQQPZLOR-UHFFFAOYSA-N 0.000 description 3
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- OWVZHJLQWZMVEQ-UHFFFAOYSA-N 6-bromo-9h-carbazol-2-ol Chemical compound C1=C(Br)C=C2C3=CC=C(O)C=C3NC2=C1 OWVZHJLQWZMVEQ-UHFFFAOYSA-N 0.000 description 3
- OMDJLZIXYTWROG-UHFFFAOYSA-N 7-(2-fluoroethoxy)-9h-carbazol-2-ol Chemical compound FCCOC1=CC=C2C3=CC=C(O)C=C3NC2=C1 OMDJLZIXYTWROG-UHFFFAOYSA-N 0.000 description 3
- VQUIURGGROVPHM-UHFFFAOYSA-N 7-(2-fluoroethoxy)-n-methyl-9h-carbazol-2-amine Chemical compound FCCOC1=CC=C2C3=CC=C(NC)C=C3NC2=C1 VQUIURGGROVPHM-UHFFFAOYSA-N 0.000 description 3
- AOJTUFCIVLPRCW-UHFFFAOYSA-N 7-(2-fluoroethylamino)-9h-carbazol-2-ol Chemical compound FCCNC1=CC=C2C3=CC=C(O)C=C3NC2=C1 AOJTUFCIVLPRCW-UHFFFAOYSA-N 0.000 description 3
- PFXJNZRKHXNVLV-UHFFFAOYSA-N 7-[2-fluoroethyl(methyl)amino]-9h-carbazol-2-ol Chemical compound OC1=CC=C2C3=CC=C(N(CCF)C)C=C3NC2=C1 PFXJNZRKHXNVLV-UHFFFAOYSA-N 0.000 description 3
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 229930040373 Paraformaldehyde Natural products 0.000 description 3
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 3
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 3
- 229930013930 alkaloid Natural products 0.000 description 3
- 150000001540 azides Chemical class 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 150000001721 carbon Chemical group 0.000 description 3
- 208000010877 cognitive disease Diseases 0.000 description 3
- 230000003920 cognitive function Effects 0.000 description 3
- 229940125773 compound 10 Drugs 0.000 description 3
- 229910000365 copper sulfate Inorganic materials 0.000 description 3
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 3
- 150000004775 coumarins Chemical class 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 230000002349 favourable effect Effects 0.000 description 3
- 229930003944 flavone Natural products 0.000 description 3
- 235000011949 flavones Nutrition 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 230000026030 halogenation Effects 0.000 description 3
- 238000005658 halogenation reaction Methods 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 3
- INKLOBOQIFYNAT-UHFFFAOYSA-N n-methyl-6-phenylmethoxy-9h-carbazol-3-amine Chemical compound C1=C2C3=CC(NC)=CC=C3NC2=CC=C1OCC1=CC=CC=C1 INKLOBOQIFYNAT-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 229920002866 paraformaldehyde Polymers 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000001632 sodium acetate Substances 0.000 description 3
- 235000017281 sodium acetate Nutrition 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 102000013498 tau Proteins Human genes 0.000 description 3
- 108010026424 tau Proteins Proteins 0.000 description 3
- QSPZPPNAGGOCFE-UHFFFAOYSA-N tert-butyl n-(4-chloro-3-nitrophenyl)carbamate Chemical compound CC(C)(C)OC(=O)NC1=CC=C(Cl)C([N+]([O-])=O)=C1 QSPZPPNAGGOCFE-UHFFFAOYSA-N 0.000 description 3
- ZHEYWXGLPOUWEY-UHFFFAOYSA-N tert-butyl n-(7-phenylmethoxy-9h-carbazol-2-yl)carbamate Chemical compound C=1C(NC(=O)OC(C)(C)C)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 ZHEYWXGLPOUWEY-UHFFFAOYSA-N 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- XXCHNRKACRXBGR-UHFFFAOYSA-N (6-bromo-9h-carbazol-2-yl) acetate Chemical compound C1=C(Br)C=C2C3=CC=C(OC(=O)C)C=C3NC2=C1 XXCHNRKACRXBGR-UHFFFAOYSA-N 0.000 description 2
- KVFDJBFPVMQYFO-UHFFFAOYSA-N 1-[2-hydroxy-4-(methoxymethoxy)phenyl]ethanone Chemical compound COCOC1=CC=C(C(C)=O)C(O)=C1 KVFDJBFPVMQYFO-UHFFFAOYSA-N 0.000 description 2
- WMCUHRDQSHQNRW-UHFFFAOYSA-N 1-bromo-4-fluorobutane Chemical compound FCCCCBr WMCUHRDQSHQNRW-UHFFFAOYSA-N 0.000 description 2
- LBBJNGFCXDOYMQ-UHFFFAOYSA-N 1-methyl-2,9-dihydropyrido[3,4-b]indol-7-one Chemical compound C1=CC(=O)C=C2NC3=C(C)NC=CC3=C21 LBBJNGFCXDOYMQ-UHFFFAOYSA-N 0.000 description 2
- SULYEHHGGXARJS-UHFFFAOYSA-N 2',4'-dihydroxyacetophenone Chemical compound CC(=O)C1=CC=C(O)C=C1O SULYEHHGGXARJS-UHFFFAOYSA-N 0.000 description 2
- IBGBGRVKPALMCQ-UHFFFAOYSA-N 3,4-dihydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1O IBGBGRVKPALMCQ-UHFFFAOYSA-N 0.000 description 2
- ZFCWURBGTORKKO-UHFFFAOYSA-N 3,4-dihydroxychromen-2-one Chemical compound C1=CC=CC2=C1OC(=O)C(O)=C2O ZFCWURBGTORKKO-UHFFFAOYSA-N 0.000 description 2
- TUUQYQCDTCXIEH-UHFFFAOYSA-N 3-(methoxymethoxy)-4-phenylmethoxybenzaldehyde Chemical compound COCOC1=CC(C=O)=CC=C1OCC1=CC=CC=C1 TUUQYQCDTCXIEH-UHFFFAOYSA-N 0.000 description 2
- HUNGEAFDVLSPAM-UHFFFAOYSA-N 3-hydroxy-4-phenylmethoxybenzaldehyde Chemical compound OC1=CC(C=O)=CC=C1OCC1=CC=CC=C1 HUNGEAFDVLSPAM-UHFFFAOYSA-N 0.000 description 2
- QBGFOPPJYCVPPT-UHFFFAOYSA-N 3-methoxy-6-nitro-9h-carbazole Chemical compound C1=C([N+]([O-])=O)C=C2C3=CC(OC)=CC=C3NC2=C1 QBGFOPPJYCVPPT-UHFFFAOYSA-N 0.000 description 2
- FRLSFSKSFCJBGQ-UHFFFAOYSA-N 4-nitro-n-(4-phenylmethoxyphenyl)aniline Chemical compound C1=CC([N+](=O)[O-])=CC=C1NC(C=C1)=CC=C1OCC1=CC=CC=C1 FRLSFSKSFCJBGQ-UHFFFAOYSA-N 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- NLPZFGLHKNHASG-UHFFFAOYSA-N 6-phenylmethoxy-9h-carbazol-3-amine Chemical compound C1=C2C3=CC(N)=CC=C3NC2=CC=C1OCC1=CC=CC=C1 NLPZFGLHKNHASG-UHFFFAOYSA-N 0.000 description 2
- AQZUMUBUJIKXHQ-UHFFFAOYSA-N 7-phenylmethoxy-9h-carbazol-2-amine Chemical compound C=1C(N)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 AQZUMUBUJIKXHQ-UHFFFAOYSA-N 0.000 description 2
- RQTYNXBODFYTDX-UHFFFAOYSA-N 9-nitro-3-phenylmethoxycarbazole Chemical compound C=1C=C2N([N+](=O)[O-])C3=CC=CC=C3C2=CC=1OCC1=CC=CC=C1 RQTYNXBODFYTDX-UHFFFAOYSA-N 0.000 description 2
- RZPPWGSAHMLQHK-UHFFFAOYSA-N 9h-carbazol-2-yl acetate Chemical compound C1=CC=C2C3=CC=C(OC(=O)C)C=C3NC2=C1 RZPPWGSAHMLQHK-UHFFFAOYSA-N 0.000 description 2
- 102100033639 Acetylcholinesterase Human genes 0.000 description 2
- 108010022752 Acetylcholinesterase Proteins 0.000 description 2
- 229940100578 Acetylcholinesterase inhibitor Drugs 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- SATMZMMKDDTOSQ-UHFFFAOYSA-N Harmol Natural products C12=CC=C(O)C=C2NC2=C1C=CN=C2C SATMZMMKDDTOSQ-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 240000005523 Peganum harmala Species 0.000 description 2
- 235000005126 Peganum harmala Nutrition 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- OUAWMKXLNFVZDA-UHFFFAOYSA-N [3-(dimethylamino)-6-phenylmethoxycarbazol-9-yl]methanol Chemical compound C1=C2C3=CC(N(C)C)=CC=C3N(CO)C2=CC=C1OCC1=CC=CC=C1 OUAWMKXLNFVZDA-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 2
- 229960004373 acetylcholine Drugs 0.000 description 2
- 229940022698 acetylcholinesterase Drugs 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 238000000376 autoradiography Methods 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- OGHNVEJMJSYVRP-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=C1C1=CC=CC=C1N2 OGHNVEJMJSYVRP-UHFFFAOYSA-N 0.000 description 2
- 229960004195 carvedilol Drugs 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052804 chromium Inorganic materials 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- 230000006999 cognitive decline Effects 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 230000006735 deficit Effects 0.000 description 2
- 125000000950 dibromo group Chemical group Br* 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 150000002212 flavone derivatives Chemical class 0.000 description 2
- 150000002213 flavones Chemical class 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 150000002460 imidazoles Chemical class 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 210000005171 mammalian brain Anatomy 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- YKTYQEDBWQCKII-UHFFFAOYSA-N n-(4-methoxyphenyl)-4-nitroaniline Chemical compound C1=CC(OC)=CC=C1NC1=CC=C([N+]([O-])=O)C=C1 YKTYQEDBWQCKII-UHFFFAOYSA-N 0.000 description 2
- YEOXBUAQBMMZFX-UHFFFAOYSA-N n-(7-hydroxy-9h-carbazol-2-yl)-n-methylformamide Chemical compound OC1=CC=C2C3=CC=C(N(C=O)C)C=C3NC2=C1 YEOXBUAQBMMZFX-UHFFFAOYSA-N 0.000 description 2
- KHJKOGMFMOMJGN-UHFFFAOYSA-N n-(7-phenylmethoxy-9h-carbazol-2-yl)formamide Chemical compound C=1C(NC=O)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 KHJKOGMFMOMJGN-UHFFFAOYSA-N 0.000 description 2
- SLPNVYWMRHYMNQ-UHFFFAOYSA-N n-[4-(2-nitro-4-phenylmethoxyphenyl)phenyl]formamide Chemical compound C=1C=C(C=2C=CC(NC=O)=CC=2)C([N+](=O)[O-])=CC=1OCC1=CC=CC=C1 SLPNVYWMRHYMNQ-UHFFFAOYSA-N 0.000 description 2
- UPJWMCLDJBWFNP-UHFFFAOYSA-N n-[7-(4-fluorobutoxy)-9h-carbazol-2-yl]formamide Chemical compound O=CNC1=CC=C2C3=CC=C(OCCCCF)C=C3NC2=C1 UPJWMCLDJBWFNP-UHFFFAOYSA-N 0.000 description 2
- ZTZLNDWKVPMJJT-UHFFFAOYSA-N n-methyl-7-phenylmethoxy-9h-carbazol-2-amine Chemical compound C=1C(NC)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 ZTZLNDWKVPMJJT-UHFFFAOYSA-N 0.000 description 2
- WZPBJXOCNOKIQU-UHFFFAOYSA-N n-methyl-n-(7-phenylmethoxy-9h-carbazol-2-yl)formamide Chemical compound C=1C(N(C=O)C)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 WZPBJXOCNOKIQU-UHFFFAOYSA-N 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Substances [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical class C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 2
- 229930185107 quinolinone Natural products 0.000 description 2
- PUMYFTJOWAJIKF-UHFFFAOYSA-N ro5-4864 Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=C(Cl)C=C1 PUMYFTJOWAJIKF-UHFFFAOYSA-N 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- LSODCCFQUIMPIG-UHFFFAOYSA-N tert-butyl 2-[formyl(methyl)amino]-7-hydroxycarbazole-9-carboxylate Chemical compound OC1=CC=C2C3=CC=C(N(C=O)C)C=C3N(C(=O)OC(C)(C)C)C2=C1 LSODCCFQUIMPIG-UHFFFAOYSA-N 0.000 description 2
- WJGWOUVJOHLKSO-UHFFFAOYSA-N tert-butyl 2-[formyl(methyl)amino]-7-phenylmethoxycarbazole-9-carboxylate Chemical compound C=1C(N(C=O)C)=CC=C(C2=CC=3)C=1N(C(=O)OC(C)(C)C)C2=CC=3OCC1=CC=CC=C1 WJGWOUVJOHLKSO-UHFFFAOYSA-N 0.000 description 2
- QWGVSLOJOVUSQO-UHFFFAOYSA-N tert-butyl n-(2-oxo-1,9-dihydropyrido[2,3-b]indol-7-yl)carbamate Chemical compound OC1=CC=C2C3=CC=C(NC(=O)OC(C)(C)C)C=C3NC2=N1 QWGVSLOJOVUSQO-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- KQBDLOVXZHOAJI-UHFFFAOYSA-N (4-phenylmethoxyphenyl)azanium;chloride Chemical compound Cl.C1=CC(N)=CC=C1OCC1=CC=CC=C1 KQBDLOVXZHOAJI-UHFFFAOYSA-N 0.000 description 1
- VHNYOQKVZQVBLC-RTCGXNAVSA-N (4r,7e,9as)-7-[[3-methoxy-4-(4-methylimidazol-1-yl)phenyl]methylidene]-4-(3,4,5-trifluorophenyl)-1,3,4,8,9,9a-hexahydropyrido[2,1-c][1,4]oxazin-6-one Chemical compound C1([C@@H]2COC[C@@H]3CC\C(C(N32)=O)=C/C=2C=C(C(=CC=2)N2C=C(C)N=C2)OC)=CC(F)=C(F)C(F)=C1 VHNYOQKVZQVBLC-RTCGXNAVSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- BOKLPUXSDLSCNH-XYOKQWHBSA-N (e)-1-[2-hydroxy-4-(methoxymethoxy)phenyl]-3-[3-(methoxymethoxy)-4-phenylmethoxyphenyl]prop-2-en-1-one Chemical compound OC1=CC(OCOC)=CC=C1C(=O)\C=C\C(C=C1OCOC)=CC=C1OCC1=CC=CC=C1 BOKLPUXSDLSCNH-XYOKQWHBSA-N 0.000 description 1
- 125000003363 1,3,5-triazinyl group Chemical group N1=C(N=CN=C1)* 0.000 description 1
- JMXJCFJYPRPNOY-UHFFFAOYSA-N 1-[2-[2-[2-(2-fluoroethoxy)ethoxy]ethoxy]carbazol-9-yl]ethanone Chemical compound C1=C(OCCOCCOCCF)C=C2N(C(=O)C)C3=CC=CC=C3C2=C1 JMXJCFJYPRPNOY-UHFFFAOYSA-N 0.000 description 1
- HCMZFUYXZAURKZ-UHFFFAOYSA-N 1-azido-2-ethyl-9H-carbazole Chemical compound C1=CC=C2NC3=C(N=[N+]=[N-])C(CC)=CC=C3C2=C1 HCMZFUYXZAURKZ-UHFFFAOYSA-N 0.000 description 1
- ZDFBKZUDCQQKAC-UHFFFAOYSA-N 1-bromo-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(Br)C=C1 ZDFBKZUDCQQKAC-UHFFFAOYSA-N 0.000 description 1
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 1
- SCCCFNJTCDSLCY-UHFFFAOYSA-N 1-iodo-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(I)C=C1 SCCCFNJTCDSLCY-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- ONRILIKKLGEWHW-UHFFFAOYSA-N 2-(9h-carbazol-2-yloxy)ethyl 2-nitrobenzenesulfonate Chemical compound [O-][N+](=O)C1=CC=CC=C1S(=O)(=O)OCCOC1=CC=C2C3=CC=CC=C3NC2=C1 ONRILIKKLGEWHW-UHFFFAOYSA-N 0.000 description 1
- XNDCPFTULXRWQH-HNENSFHCSA-N 2-[(2e)-2-(1-azabicyclo[2.2.2]octan-3-ylidene)-2-fluoroethoxy]-9h-carbazole Chemical compound C/1N(CC2)CCC2C\1=C(/F)COC1=CC=C2C3=CC=CC=C3NC2=C1 XNDCPFTULXRWQH-HNENSFHCSA-N 0.000 description 1
- JWNAVCUBHGOIGR-UHFFFAOYSA-N 2-[(7-formamido-9h-carbazol-2-yl)oxy]ethyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCCOC1=CC=C2C3=CC=C(NC=O)C=C3NC2=C1 JWNAVCUBHGOIGR-UHFFFAOYSA-N 0.000 description 1
- BBFZZKXFLJFNQR-UHFFFAOYSA-N 2-[2-[2-[[7-[formyl(methyl)amino]-9h-carbazol-2-yl]oxy]ethoxy]ethoxy]ethyl 4-methylbenzenesulfonate Chemical compound C=1C(N(C=O)C)=CC=C(C2=CC=3)C=1NC2=CC=3OCCOCCOCCOS(=O)(=O)C1=CC=C(C)C=C1 BBFZZKXFLJFNQR-UHFFFAOYSA-N 0.000 description 1
- ZQAQXZBSGZUUNL-BJUDXGSMSA-N 2-[4-(methylamino)phenyl]-1,3-benzothiazol-6-ol Chemical compound C1=CC(N[11CH3])=CC=C1C1=NC2=CC=C(O)C=C2S1 ZQAQXZBSGZUUNL-BJUDXGSMSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- ZVZPFTCEXIGSHM-UHFFFAOYSA-N 2-fluoropropanoic acid Chemical compound CC(F)C(O)=O ZVZPFTCEXIGSHM-UHFFFAOYSA-N 0.000 description 1
- JXPDNDHCMMOJPC-UHFFFAOYSA-N 2-hydroxybutanedinitrile Chemical compound N#CC(O)CC#N JXPDNDHCMMOJPC-UHFFFAOYSA-N 0.000 description 1
- MXNLRVZITPPZHT-UHFFFAOYSA-N 2-phenylmethoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound O1C(C)(C)C(C)(C)OB1C(C=N1)=CC=C1OCC1=CC=CC=C1 MXNLRVZITPPZHT-UHFFFAOYSA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- PCYGLFXKCBFGPC-UHFFFAOYSA-N 3,4-Dihydroxy hydroxymethyl benzene Natural products OCC1=CC=C(O)C(O)=C1 PCYGLFXKCBFGPC-UHFFFAOYSA-N 0.000 description 1
- XHLOLFKZCUCROE-UHFFFAOYSA-N 3,6-dihydroxy-2-phenylchromen-4-one Chemical compound OC=1C(=O)C2=CC(O)=CC=C2OC=1C1=CC=CC=C1 XHLOLFKZCUCROE-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- BICZJRAGTCRORZ-UHFFFAOYSA-N 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(O)C=C1 BICZJRAGTCRORZ-UHFFFAOYSA-N 0.000 description 1
- FOHHWGVAOVDVLP-UHFFFAOYSA-N 4-chloro-3-nitroaniline Chemical compound NC1=CC=C(Cl)C([N+]([O-])=O)=C1 FOHHWGVAOVDVLP-UHFFFAOYSA-N 0.000 description 1
- JUIKCULGDIZNDI-UHFFFAOYSA-N 4-chloro-3-nitrophenol Chemical compound OC1=CC=C(Cl)C([N+]([O-])=O)=C1 JUIKCULGDIZNDI-UHFFFAOYSA-N 0.000 description 1
- JXRGUPLJCCDGKG-UHFFFAOYSA-N 4-nitrobenzenesulfonyl chloride Chemical compound [O-][N+](=O)C1=CC=C(S(Cl)(=O)=O)C=C1 JXRGUPLJCCDGKG-UHFFFAOYSA-N 0.000 description 1
- 125000001826 4H-pyranyl group Chemical group O1C(=CCC=C1)* 0.000 description 1
- YDEUKNRKEYICTH-UHFFFAOYSA-N 5-aminoquinolin-8-ol Chemical compound C1=CC=C2C(N)=CC=C(O)C2=N1 YDEUKNRKEYICTH-UHFFFAOYSA-N 0.000 description 1
- MXYUEMWMHJSSSJ-KWCOIAHCSA-N 5-fluoranylpent-1-yne Chemical compound [18F]CCCC#C MXYUEMWMHJSSSJ-KWCOIAHCSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- KIDHTYCXMSTLHB-UHFFFAOYSA-N 6-(methylamino)-9h-carbazol-3-ol Chemical compound C1=C(O)C=C2C3=CC(NC)=CC=C3NC2=C1 KIDHTYCXMSTLHB-UHFFFAOYSA-N 0.000 description 1
- HYMHLDYNCYFHHL-UHFFFAOYSA-N 6-methoxy-9h-carbazol-3-amine Chemical compound C1=C(N)C=C2C3=CC(OC)=CC=C3NC2=C1 HYMHLDYNCYFHHL-UHFFFAOYSA-N 0.000 description 1
- JRVIIPJSVKTPBK-UHFFFAOYSA-N 7,8-dihydroxy-4-phenyl-2h-chromen-2-one Chemical compound C=1C(=O)OC2=C(O)C(O)=CC=C2C=1C1=CC=CC=C1 JRVIIPJSVKTPBK-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- HVLNOWLSMIOOTG-UHFFFAOYSA-N 7-[2-[2-(2-fluoroethoxy)ethoxy]ethoxy]-9h-carbazol-2-amine Chemical compound FCCOCCOCCOC1=CC=C2C3=CC=C(N)C=C3NC2=C1 HVLNOWLSMIOOTG-UHFFFAOYSA-N 0.000 description 1
- XWWZUJSAORWKRB-UHFFFAOYSA-N 7-[2-[2-(2-fluoroethoxy)ethoxy]ethoxy]-n-methyl-9h-carbazol-2-amine Chemical compound FCCOCCOCCOC1=CC=C2C3=CC=C(NC)C=C3NC2=C1 XWWZUJSAORWKRB-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- ZIKXHZSORLSPCO-UHFFFAOYSA-N 9h-carbazol-3-ol Chemical class C1=CC=C2C3=CC(O)=CC=C3NC2=C1 ZIKXHZSORLSPCO-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 101710137189 Amyloid-beta A4 protein Proteins 0.000 description 1
- 101710151993 Amyloid-beta precursor protein Proteins 0.000 description 1
- 102100022704 Amyloid-beta precursor protein Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241001266506 Banisteriopsis caapi Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 108010053652 Butyrylcholinesterase Proteins 0.000 description 1
- OZKYVNKXVHASGF-UHFFFAOYSA-N CC1=CC=C(O)C(C=2ON=CC=2)=C1 Chemical compound CC1=CC=C(O)C(C=2ON=CC=2)=C1 OZKYVNKXVHASGF-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108010009685 Cholinergic Receptors Proteins 0.000 description 1
- 102100032404 Cholinesterase Human genes 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 108010044266 Dopamine Plasma Membrane Transport Proteins Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 241001248531 Euchloe <genus> Species 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- BXNJHAXVSOCGBA-UHFFFAOYSA-N Harmine Chemical class N1=CC=C2C3=CC=C(OC)C=C3NC2=C1C BXNJHAXVSOCGBA-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000009032 Imidazoline Receptors Human genes 0.000 description 1
- 108010049134 Imidazoline Receptors Proteins 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 102000010909 Monoamine Oxidase Human genes 0.000 description 1
- 108010062431 Monoamine oxidase Proteins 0.000 description 1
- 208000019022 Mood disease Diseases 0.000 description 1
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 102000004108 Neurotransmitter Receptors Human genes 0.000 description 1
- 108090000590 Neurotransmitter Receptors Proteins 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 238000012879 PET imaging Methods 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- XSVMFMHYUFZWBK-NSHDSACASA-N Rivastigmine Chemical compound CCN(C)C(=O)OC1=CC=CC([C@H](C)N(C)C)=C1 XSVMFMHYUFZWBK-NSHDSACASA-N 0.000 description 1
- 102100040119 SH3 domain-binding protein 5 Human genes 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 102100033928 Sodium-dependent dopamine transporter Human genes 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- GAMYVSCDDLXAQW-AOIWZFSPSA-N Thermopsosid Natural products O(C)c1c(O)ccc(C=2Oc3c(c(O)cc(O[C@H]4[C@H](O)[C@@H](O)[C@H](O)[C@H](CO)O4)c3)C(=O)C=2)c1 GAMYVSCDDLXAQW-AOIWZFSPSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 102000034337 acetylcholine receptors Human genes 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- 125000005354 acylalkyl group Chemical group 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004171 alkoxy aryl group Chemical group 0.000 description 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000005275 alkylenearyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 230000006933 amyloid-beta aggregation Effects 0.000 description 1
- DZHSAHHDTRWUTF-SIQRNXPUSA-N amyloid-beta polypeptide 42 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(O)=O)[C@@H](C)CC)C(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC(O)=O)C(C)C)C(C)C)C1=CC=CC=C1 DZHSAHHDTRWUTF-SIQRNXPUSA-N 0.000 description 1
- 229940082992 antihypertensives mao inhibitors Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 125000005334 azaindolyl group Chemical group N1N=C(C2=CC=CC=C12)* 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 150000005347 biaryls Chemical group 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 230000003925 brain function Effects 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- YRIBGSCJIMXMPJ-UHFFFAOYSA-N butyrylcholine Chemical compound CCCC(=O)OCC[N+](C)(C)C YRIBGSCJIMXMPJ-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- BQXQGZPYHWWCEB-UHFFFAOYSA-N carazolol Chemical compound N1C2=CC=CC=C2C2=C1C=CC=C2OCC(O)CNC(C)C BQXQGZPYHWWCEB-UHFFFAOYSA-N 0.000 description 1
- 229960004634 carazolol Drugs 0.000 description 1
- OKTJSMMVPCPJKN-BJUDXGSMSA-N carbon-11 Chemical compound [11C] OKTJSMMVPCPJKN-BJUDXGSMSA-N 0.000 description 1
- 125000005518 carboxamido group Chemical group 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- PUXBGTOOZJQSKH-UHFFFAOYSA-N carprofen Chemical compound C1=C(Cl)C=C2C3=CC=C(C(C(O)=O)C)C=C3NC2=C1 PUXBGTOOZJQSKH-UHFFFAOYSA-N 0.000 description 1
- 229960003184 carprofen Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000003710 cerebral cortex Anatomy 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 235000012721 chromium Nutrition 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- ZPUCINDJVBIVPJ-LJISPDSOSA-N cocaine Chemical class O([C@H]1C[C@@H]2CC[C@@H](N2C)[C@H]1C(=O)OC)C(=O)C1=CC=CC=C1 ZPUCINDJVBIVPJ-LJISPDSOSA-N 0.000 description 1
- 231100000870 cognitive problem Toxicity 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 1
- 230000001054 cortical effect Effects 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000009109 curative therapy Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- ZFTFAPZRGNKQPU-UHFFFAOYSA-N dicarbonic acid Chemical compound OC(=O)OC(O)=O ZFTFAPZRGNKQPU-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 229950010286 diolamine Drugs 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- XGGQGEAXLNJTTO-UHFFFAOYSA-N ethane 4-methylbenzenesulfonic acid Chemical compound C1(=CC=C(C=C1)S(=O)(=O)O)C.C1(=CC=C(C=C1)S(=O)(=O)O)C.CC XGGQGEAXLNJTTO-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 230000003400 hallucinatory effect Effects 0.000 description 1
- 230000004217 heart function Effects 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- RJTZUHVCZIGJMB-UHFFFAOYSA-N hydron;1h-indole;chloride Chemical compound Cl.C1=CC=C2NC=CC2=C1 RJTZUHVCZIGJMB-UHFFFAOYSA-N 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000006192 iodination reaction Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-BJUDXGSMSA-N iodomethane Chemical compound I[11CH3] INQOMBQAUSQDDS-BJUDXGSMSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000007787 long-term memory Effects 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000007257 malfunction Effects 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- LDDHMLJTFXJGPI-UHFFFAOYSA-N memantine hydrochloride Chemical compound Cl.C1C(C2)CC3(C)CC1(C)CC2(N)C3 LDDHMLJTFXJGPI-UHFFFAOYSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000006386 memory function Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- OIRDBPQYVWXNSJ-BJUDXGSMSA-N methyl trifluoromethanesulfonate Chemical compound [11CH3]OS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-BJUDXGSMSA-N 0.000 description 1
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 1
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 1
- 229960000907 methylthioninium chloride Drugs 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 235000019799 monosodium phosphate Nutrition 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- UMBVAWHYPCOPSB-UHFFFAOYSA-N n,n-dimethyl-7-phenylmethoxy-9h-carbazol-2-amine Chemical compound C=1C(N(C)C)=CC=C(C2=CC=3)C=1NC2=CC=3OCC1=CC=CC=C1 UMBVAWHYPCOPSB-UHFFFAOYSA-N 0.000 description 1
- CDYFHMUSLGPPMI-UHFFFAOYSA-N n-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]formamide Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(NC=O)C=C1 CDYFHMUSLGPPMI-UHFFFAOYSA-N 0.000 description 1
- ZUEHIFZFZABEJE-UHFFFAOYSA-N n-[7-(2-fluoroethoxy)-9h-carbazol-2-yl]formamide Chemical compound O=CNC1=CC=C2C3=CC=C(OCCF)C=C3NC2=C1 ZUEHIFZFZABEJE-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005487 naphthalate group Chemical group 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 238000002610 neuroimaging Methods 0.000 description 1
- 230000003961 neuronal insult Effects 0.000 description 1
- 230000004112 neuroprotection Effects 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- LEVJVKGPFAQPOI-UHFFFAOYSA-N phenylmethanone Chemical compound O=[C]C1=CC=CC=C1 LEVJVKGPFAQPOI-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000005499 phosphonyl group Chemical group 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 229940043131 pyroglutamate Drugs 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000000275 quality assurance Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 239000012217 radiopharmaceutical Substances 0.000 description 1
- 229940121896 radiopharmaceutical Drugs 0.000 description 1
- 230000002799 radiopharmaceutical effect Effects 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 239000003642 reactive oxygen metabolite Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 229960004136 rivastigmine Drugs 0.000 description 1
- 239000012146 running buffer Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 230000002000 scavenging effect Effects 0.000 description 1
- 150000003334 secondary amides Chemical class 0.000 description 1
- 210000002265 sensory receptor cell Anatomy 0.000 description 1
- 230000006403 short-term memory Effects 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000004059 squalene synthase inhibitor Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 229940114926 stearate Drugs 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000004697 synapse damage Effects 0.000 description 1
- 230000015883 synaptic transmission, dopaminergic Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- NWUCRXPGKIYXLM-UHFFFAOYSA-N tert-butyl 2-(dimethylamino)-7-hydroxycarbazole-9-carboxylate Chemical compound OC1=CC=C2C3=CC=C(N(C)C)C=C3N(C(=O)OC(C)(C)C)C2=C1 NWUCRXPGKIYXLM-UHFFFAOYSA-N 0.000 description 1
- GGDJJCLKIQLHGP-UHFFFAOYSA-N tert-butyl 2-(dimethylamino)-7-phenylmethoxycarbazole-9-carboxylate Chemical compound C=1C(N(C)C)=CC=C(C2=CC=3)C=1N(C(=O)OC(C)(C)C)C2=CC=3OCC1=CC=CC=C1 GGDJJCLKIQLHGP-UHFFFAOYSA-N 0.000 description 1
- JOIWBKKEHBOENG-UHFFFAOYSA-N tert-butyl carbazole-9-carboxylate Chemical compound C1=CC=C2N(C(=O)OC(C)(C)C)C3=CC=CC=C3C2=C1 JOIWBKKEHBOENG-UHFFFAOYSA-N 0.000 description 1
- OSCNJFAGWRCOCE-UHFFFAOYSA-N tert-butyl n-(2-phenylmethoxy-9h-pyrido[2,3-b]indol-7-yl)carbamate Chemical compound C=1C(NC(=O)OC(C)(C)C)=CC=C(C2=CC=3)C=1NC2=NC=3OCC1=CC=CC=C1 OSCNJFAGWRCOCE-UHFFFAOYSA-N 0.000 description 1
- KSIMSECYSHEOMF-UHFFFAOYSA-N tert-butyl n-[3-nitro-4-(6-phenylmethoxypyridin-3-yl)phenyl]carbamate Chemical compound [O-][N+](=O)C1=CC(NC(=O)OC(C)(C)C)=CC=C1C(C=N1)=CC=C1OCC1=CC=CC=C1 KSIMSECYSHEOMF-UHFFFAOYSA-N 0.000 description 1
- GDKMAZKXMNSDKA-UHFFFAOYSA-N tert-butyl n-[4-(2-nitro-4-phenylmethoxyphenyl)phenyl]carbamate Chemical compound C1=CC(NC(=O)OC(C)(C)C)=CC=C1C(C(=C1)[N+]([O-])=O)=CC=C1OCC1=CC=CC=C1 GDKMAZKXMNSDKA-UHFFFAOYSA-N 0.000 description 1
- UXWVQHXKKOGTSY-UHFFFAOYSA-N tert-butyl phenyl carbonate Chemical compound CC(C)(C)OC(=O)OC1=CC=CC=C1 UXWVQHXKKOGTSY-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- VHBFFQKBGNRLFZ-UHFFFAOYSA-N vitamin p Natural products O1C2=CC=CC=C2C(=O)C=C1C1=CC=CC=C1 VHBFFQKBGNRLFZ-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229940045860 white wax Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Anesthesiology (AREA)
- Child & Adolescent Psychology (AREA)
- Diabetes (AREA)
- Hospice & Palliative Care (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Indole Compounds (AREA)
- Quinoline Compounds (AREA)
- Pyrane Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un compuesto de la siguiente Fórmula I: **(Ver fórmula)** y sales farmacéuticamente aceptables de la misma; en la que A es N o CR1; B es N o CR2, J es N o CR3, K es N o CR4; L es N o CR5; M es N; P es N o CR7; y Q es N o CR8; siempre que no más de dos de A, B, J, K, L, M, P y Q puedan ser N; en la que X es un enlace; en el que R1, R2, R4, R5, R7, y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroarilalcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquil C1-5 NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)-, y arilo C6-10C(O)O- y R3 se selecciona del grupo que consiste en cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroaril alcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)-, y arilo C6-10C(O)O-; en la que al menos cuatro de R1, R2, R4, R5, R7, y R son hidrógenos; en la que R9 es hidrógeno; en la que cada Res independientemente H o alquilo C1-6 ; y en el que los sustituyentes en R1, R2, R3, R4, R5, R7 R8 y Restán opcionalmente sustituidos adicionalmente por sustituyentes seleccionados del grupo que consiste en amino, halo, cyano, nitro, hidroxilo, -SH, -S alquilo C1-6, -C(O)NH2, -C(S)NH2, halo alquilo C1-6, perhalo alquilo C1-6, alquilo C1-6, cicloalquilo C3-6, cicloalquilo C3- 12, arilo C6-14 y heteroarilo.
Description
DESCRIPCIÓN
Agentes novedosos de obtención de imágenes para detectar disfunciones neurológicas
SOLICITUDES RELACIONADAS:
La presente solicitud se basa en, y reivindica la prioridad de la solicitud provisional de EE.UU No. 60/066.101, presentada el 14 de febrero de 2008.
La enfermedad de Alzheimer (EA), una de las principales causas de demencia, se desarrolla en el uno por ciento de la población entre los 65 y los 69 años, y aumenta hasta el 40-50% en los mayores de 95 años. Los pacientes con AD presentan síntomas clínicos reveladores que incluyen el deterioro cognitivo y los déficits en la función de la memoria. En estos pacientes, la fuerte carga de placas seniles encontrada en la corteza cerebral, verificada por el examen histopatológico post mortem, confirma la presencia de EA. Las placas seniles maduras consisten en ovillos neurofibrilares (NFT) intracelulares derivados de filamentos de proteínas tau hiperfosforiladas, y péptidos p-amiloides extracelulares derivados del procesamiento enzimático de la proteína precursora amiloide. Curiosamente, a pesar del desarrollo y la presencia de placas seniles en personas mayores con una función cognitiva normal, la gravedad de la deposición de NFT y placas seniles supuestamente se correlaciona con una pérdida de la función cognitiva y el deterioro de los circuitos neuronales.
A pesar de que la enfermedad de Alzheimer es la cuarta causa de muerte en los Estados Unidos, la intervención farmacéutica aún no ha comercializado una terapia curativa. En cambio, los médicos prescriben actualmente inhibidores de la colinesterasa a los pacientes con problemas cognitivos. La rivastigmina, un tratamiento terapéutico tanto para los pacientes con AD como con la enfermedad de Parkinson, inhibe tanto la acetilcolinesterasa como la butirilcolinesterasa, impidiendo la descomposición de la acetil y la butirilcolina. La galantamina, un inhibidor natural de la acetilcolinesterasa, aumenta los receptores colinérgicos nicotínicos para liberar acetilcolina en el cerebro. Como último ejemplo, el inhibidor de la acetilcolinesterasa Aricept ralentiza la progresión de la AD en los pacientes al inhibir la acetilcolinesterasa y aumentar así la acetilcolina cortical. En un ensayo clínico reciente, la eficacia de Aricept ralentizó la progresión de la AD en los pacientes, pero los efectos terapéuticos desaparecieron al cabo de 36 meses. El efecto de tratar a los pacientes con AD con una combinación terapéutica de Aricept y memantina provocó un aumento de la función cognitiva en esos pacientes con AD en relación con los que sólo recibieron Aricept. A pesar de la utilidad de los inhibidores de la colinesterasa, la actual gama de terapias para la AD sólo puede retrasar la aparición de la enfermedad en aproximadamente dos o tres años, tras lo cual son terapéuticamente ineficaces para inhibir el deterioro cognitivo. Se ha informado de que retrasar la aparición de la AD en cinco años es suficiente para reducir el número de casos de AD a la mitad y, dadas las deficiencias actuales de los inhibidores de la colinesterasa, es necesario seguir investigando para alcanzar ese objetivo.
La obtención de imágenes neurológicas de la AD ha visto la aparición de trazadores de obtención de imágenes que parecen confirmar la presencia de la AD con base en la captación del trazador mediada por la placa y la fibrilla y, posteriormente, están siendo objeto de un amplio examen clínico. Muchos de estos trazadores contienen quimiotipos derivados de colorantes fluorescentes (Tabla 1). Por ejemplo, el aumento de la captación y la unión del derivado de la naftilina 18F-FDDNP en cerebros vivos se correlaciona bien con la presencia de AD en comparación con personas normales cognitivamente funcionales de edad similar. [Liu, J., et al., High-Yield, Automated Radiosynthesis of 2-(1 -{6-[(2-[18F]Fluoroetil)(metil)amino]-2-naphtil}etilidene)malonitrile ([18F]FDDNP) Ready for Animal or Human Administration. Molecular Imaging and Biology, 2007. 9: p. 6-16.] Un compuesto competidor, 11C-PIB, muestra una mayor captación en las regiones cerebrales frontotemporales e hipocampales de los pacientes con AD en comparación con los normales sanos.
Sin embargo, hay varias cuestiones que cuestionan la validez de la obtención de imágenes de las placas y ovillos seniles. En primer lugar, el conjunto actual de agentes de obtención de imágenes de la AD sólo puede confirmar la manifestación bien establecida de la AD y este diagnóstico en fase tardía ofrece poca defensa contra la progresión de la enfermedad más allá de los 36 meses. En segundo lugar, la detección de placas y ovillos seniles puede no estar relacionada con el desarrollo de las primeras fases de la EA. Datos recientes sugieren que el modelo de cascada amiloide [Hardy, J. y D. Selkoe, The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science, 2002. 297: p. 353-356] no describe con precisión los factores principales que conducen al deterioro cognitivo en pacientes con AD y que otros factores contribuyentes, tal como los oligómeros solubles neurotóxicos y los agregados, pueden desempeñar un papel contribuyente en la neurodegeneración. [Talaga, P., Inhibitors of beta-amyloid aggregation: still an issue of structure and function? Drug Discovery Today: Therapeutic Strategies, 2004. 1: p. 7-12]. Hasta la fecha, no se sabe que el FDDNP y el PIB se unan a los oligómeros y agregados solubles neurorotóxicos y, por lo tanto, no se espera que puedan diferenciar con precisión las fases tempranas de la AD de las avanzadas en los pacientes.
Como se resumió en un reciente grupo de discusión del 5 de diciembre de 2006 (Biochemical Pharmacology Discussion Group, copatrocinado por la sección de Nueva York de la American Chemical Society), los investigadores se están centrando ahora en procedimientos que se dirigen a los precursores de la AD bloqueando la producción de la proteína p-amiloide (BAP) o controlando la formación de la proteína tau mutante. Claramente, este esfuerzo de
investigación centrado tiene como objetivo controlar la formación de precursores de la AD que potencialmente conducen a la EA, y esta nueva estrategia podría retrasar la aparición de la AD de forma más eficaz que los tratamientos actuales. Paralelamente, las imágenes neurológicas deben reflejar la tendencia terapéutica mediante la identificación de precursores de la AD en un doble esfuerzo para complementar el desarrollo terapéutico de la AD y, además, identificar a los pacientes presintomáticos de riesgo de la EA.
Un número de procedimientos de diagnóstico médico, incluyendo PET y SPECT utilizan compuestos radiomarcados, son bien conocidos en la técnica. La PET y la SPECT son técnicas muy sensibles y requieren pequeñas cantidades de compuestos radiomarcados, llamados trazadores. Los compuestos marcados son transportados, acumulados y convertidos in vivo exactamente de la misma manera que el correspondiente compuesto no radiactivo. Los trazadores, o sondas, pueden ser radiomarcados con un radionúclido útil para la obtención de imágenes PET, tales como 11C, 13N, 15O, 18F, 64Cu y 124I, o con un radionúclido útil para la obtención de imágenes SPECT, tales como 99Tc, 77Br, 61Cu, 153Gd, 123I, 125I, 131I y 32P.
La PET crea imágenes con base en la distribución de los trazadores de obtención de imágenes moleculares que llevan isótopos emisores de positrones en el tejido del paciente. El procedimiento PET tiene el potencial de detectar un mal funcionamiento a nivel celular en los tejidos u órganos investigados. La PET se ha utilizado en oncología clínica, tal como para la obtención de imágenes de tumores y metástasis, y se ha empleado para el diagnóstico de ciertas enfermedades cerebrales, así como para la cartografía de la función cerebral y cardíaca. Del mismo modo, SPECT puede utilizarse para complementar cualquier estudio de gammagrafía, en el que una verdadera representación en 3D puede ser útil, por ejemplo, para obtener imágenes de tumores, infecciones (leucocitos), tiroides o huesos. El documento WO2007/014467 divulga procedimientos para el diagnóstico, el tratamiento y la monitorización de los trastornos del estado de ánimo.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN:
La presente invención se dirige a compuestos que tienen la Fórmula I estructural, donde los radicales tienen los significados indicados anteriormente.
Por "halógeno" o "halo" se entiende F, Cl, Br e I.
Por "alquilo" se entiende un radical hidrocarbonado monovalente saturado que tiene fracciones rectas o ramificadas. Algunos ejemplos de grupos alquilos son metilo, etilo, n-propilo, isopropilo y t-butilo.
Por "alquenilo" se entiende una molécula de alquilo que tiene al menos un doble enlace carbono-carbono, en el que el alquilo es como se ha definido anteriormente. Ejemplos de alquenilo son etenilo y el propenilo.
Por "alquinilo" se entienden las moléculas de alquilo que tienen al menos un triple enlace carbono-carbono, en el que el alquilo es como se ha definido anteriormente. Algunos ejemplos de grupos alquilos son etilo y 2-propinilo.
Por "alquileno" o "alquenilenilo" se entiende un radical hidrocarbonado saturado y divalente, es decir, generalmente presente como grupo puente o de enlace entre otros dos grupos, con fracciones rectas o ramificadas. Algunos ejemplos de grupos alquilenos son CH2-(metileno), -CH2CH2-(etileno), -CH2CH2CH2-(propileno), -CH(CH3)CH2-(isopropileno), etc.
Por "amino" se entiende una fracción de nitrógeno con dos sustituyentes adicionales donde un átomo de hidrógeno o de carbono está unido al nitrógeno. Por ejemplo, los grupos amino representativos incluyen -NH2, -NHCH3, -N(CH3)2, -NHC2-3-alkyl, -N(C2-3-alquil)2. A menos que se indique lo contrario, los compuestos de la invención que contienen fracciones amino pueden incluir derivados protegidos de los mismos. Los grupos protectores adecuados para las fracciones amino incluyen acetilo, tert-butoxicarbonilo y benzoxicarbonilo.
Por "arilo" se entiende un radical orgánico derivado de un hidrocarburo aromático por eliminación de un hidrógeno, tal como fenilo, naftilo, indenilo, indanilo y fluorenilo. "Arilo" abarca los grupos de anillos fusionados en los que al menos un anillo es aromático.
Por "cicloalquilo" se entienden las fracciones alquilo cíclicas saturadas no aromáticas formadas por uno o más anillos, en los que dichos anillos (si son más de uno) comparten al menos un átomo de carbono, siendo el alquilo como se ha definido anteriormente. Ejemplos de cicloalquilos son ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, biciclo-[3.1.0]-hexilo, biciclo-[2.2.1]-hept-1-ilo, norbornilo, espiro[4.5]decilo, espiro[4.4]nonilo, espiro[4.3]octilo, espiro[4.2]heptilo y adamantanilo.
Por "Haloalquilo C1-6" se entiende un grupo alquilo C1-6 que está sustituido con al menos un átomo de halógeno en un átomo de carbono del grupo alquilo. Ejemplos representativos de tales haloalquilos C1-6 incluyen F-CH2-, F-CH2CH2-, F-CH2CH2CH2-, CHF2-, CHF2CH2-, CHF2CH2CH2-, Br-CH2-, BPCH2CH2- BPCH2CH2CH2-, CHBr2-, CHBr2CH2-, yCHBr2CH2CH2-.
Por "heterocíclico" o "heterocicloalquilo" se entiende un grupo cíclico no aromático que consiste en uno o más anillos, en el que los anillos (si hay más de uno) comparten uno o dos átomos y cada anillo contiene hasta cuatro heteroátomos
(es decir, de cero a cuatro heteroátomos, siempre que al menos un anillo contenga al menos un heteroátomo). Los grupos heterocíclicos de esta invención también pueden incluir sistemas de anillos sustituidos con uno o más O, S(O)0-2 , y/o N-R10 como heteroátomos, en los que R10 es como se define en el presente documento, y en los que el subíndice
"0-2" de S(O)2 representa un número entero de 0, 1 o 2. Así, S(O)0-2 representa el grupo formado por S, S(=O) y S(O)2.
Ejemplos de grupos heterocíclicos no aromáticos son aziridinilo, azetidinilo, el pirrolidinilo, piperidinilo, azepinilo, piperazinilo, 1,2,3,6-tetrahidropiridinilo, oxiranilo, oxetanilo, tetrahidrofuranilo, tetrahidrotiranilo, tetrahidrotiopiranilo, morfolino, tiomorfolino, tioxanilo, pirrolinilo, indolinilo, 2H-piranilo, 4H-piranilo, dioxanilo, 1,3-dioxolanilo, pirazolinilo, dihidropiranilo, dihidrotienilo, dihidrofuranilo, pirazolidinilo, imidazolinilo, imidazolidinilo, 3-azabiciclo[3.1.0]hexanilo, 3-azabiciclo[4.1.0]heptanilo, quinolizinilo, quinuclidinilo, 1,4-dioxaspiro[4.5]decilo, 1,4-dioxaspiro[4.4]nonilo, 1,4-dioxaspiro[4.3]octilo y 1,4-dioxaspiro[4.2]heptilo.
Por "heteroarilo" se entiende un grupo aromático que contiene uno o más heteroátomos (O, S o N), preferentemente de uno a cuatro heteroátomos. Un heteroarilo puede ser un grupo monocíclico o policíclico. Ejemplos de grupos heteroarilo son piridinilo, piridazinilo, imidazolilo, pirimidinilo, pirazolilo, triazolilo, pirazinilo, quinolilo, isoquinolilo, 1,2,3,4-tetrahidroguinolilo tetrazolilo, furilo, tienilo, isoxazolilo, tiazolilo, oxazolilo, isotiazolilo, pirrolilo, indolilo, bencimidazolilo, benzofuranilo, indazolilo, indolizinilo, ftalazinilo, triazinilo, 1,3,5-triazinilo, isoindolilo, purinilo, oxadiazolilo, tiadiazolilo, furazanilo, benzofurazanilo, benzotiofenilo, benzootriazolilo, benzotiazolilo, benzoxazolilo, quinazolinilo, quinoxalinilo, naftiridinilo, dihidroquinolilo, tetrahidroquinolilo, dihidroisoquinolilo, tetrahidroisoquinolilo, benzofurilo, furopiridinilo, pirolopirimidinilo y azaindolilo. En ciertos aspectos de la presente solicitud, el heteroarilo es un 1H-1,2-3-triazol-1-il sustituido.
Tal como se utiliza en el presente documento, cuando un grupo divalente, tal como un enlazador por ejemplo, se representa mediante una estructura -A-B-, como se muestra a continuación, se pretende representar también un grupo que puede estar unido en ambas permutaciones posibles, como se observa en las dos estructuras siguientes.
S-A-B-s - también puede ser
Por ejemplo, cuando se proporciona un grupo divalente tal como el grupo "-N(R10)C(O)-", se pretende que el grupo incluya también el grupo divalente -N(R10)C(O)- y también el grupo divalente -C(O)N(R10)-.
Los sustituyentes o los grupos alquilo C1 -6 , cicloalquilo C3-6, cicloalquilo C3-12 alquilo C1 - 5 , arilo C6-14, ariloxi C6 -14 , arilo
Ca-10 alquilo C1-4 , heteroarilo, heteroariloxi, etc... de las variables R1, R2, R3, R4, R5, R7, R8, opcionalmente sustituidos por sustituyentes seleccionados del grupo que consiste en amino, halo, ciano, nitro, hidroxilo, -SH, alquilo -SC1-6, -C(O)NH2, -C(S)NH2, haloalquilo C1-6, perhaloalquilo C1-6, alquilo C1-6 , cicloalquilo C3-6, cicloalquilo C3 -12 , arilo Ca-14 y heteroarilo.
Por ejemplo, en cierto aspecto de la presente solicitud, el sustituyente heteroarilo es un 4-sustituido-1H-1,2-3-triazol-1-ilo.
El término "opcionalmente sustituido" o "sustituido" se refiere a los sustituyentes o grupos específicos en los que de uno a cuatro átomos de hidrógeno del grupo pueden ser sustituidos por uno a cuatro sustituyentes, por ejemplo seleccionados independientemente de los sustituyentes amino, halo, ciano, nitro, hidroxilo, -SH, alquilo -SC1-6 , -C(O)NH2, -C(S)NH2, haloalquilo C1-6, perhaloalquilo C1-6, alquilo C1 - 6 , cicloalquilo C3 - 6 , cicloalquilo C3 -12 , arilo Ca-14 y heteroarilo, o como se divulga específicamente en el presente documento. Además, los sustituyentes también pueden incluir alquilo, arilo, alquileno-arilo, hidroxi, alcoxi, ariloxi, perhaloalcoxi, heterociclilo, azido, amino, guanidino, amidino, halo, alquiltio, oxo, acilalquilo, ésteres carboxílicos, carboxilo, carboxamido, aciloxi, aminoalquilo, alquilaminoarilo, alquilaminoalquilo, alcoxiarilo, arilamino, fosfonilo, sulfonilo, carboxamidoarilo, hidroxialquilo, haloalquilo, alcoxialquilo y perhaloalquilo. Además, el término "opcionalmente sustituido" o "sustituido" en referencia a las variables R1, R2, R3,
R4, R5, R7, R8, R9 y R10, incluye los grupos sustituidos por uno a cuatro sustituyentes, como se ha identificado anteriormente, que comprenden además un emisor de positrones o de rayos gamma. Dichos emisores de positrones incluyen 11C, 13N, 15O, 18F, 123I, 124I, 125I, 131I y 77Br.
El término "compuesto radiomarcado", tal como se utiliza en el presente documento, se refiere a los compuestos que tienen un átomo o grupo que puede proporcionar un radiomarcado o que puede convertirse en un radiomarcado, tal como por ejemplo de un átomo no radiactivo a un radionúclido que es activo, tal como por ejemplo, 11C, 13N, 15O, 18F, 123I, 124I, 125I, 131I y 77Br. Además, a efectos de la presente solicitud, dicho "compuesto radiomarcado" también puede referirse a un átomo o un grupo, que comprende un nucleído no activo, como un halógeno, tal como 19F por ejemplo, en el que el compuesto puede utilizarse y administrarse en una cantidad terapéuticamente eficaz.
Los compuestos de la Fórmula I pueden tener centros ópticos y, por lo tanto, pueden presentarse en diferentes configuraciones enantioméricas y diastereoméricas. La presente invención incluye todos los enantiómeros,
diastereómeros y otros estereoisómeros de dichos compuestos de la Fórmula I, así como los compuestos racémicos y las mezclas racémicas y otras mezclas de estereoisómeros de los mismos. Las sales farmacéuticamente aceptables de los compuestos de Fórmula I incluyen las sales de adición de ácido y de base de los mismos. Las sales de adición de ácido adecuadas se forman a partir de ácidos que forman sales no tóxicas. Los ejemplos incluyen acetato, adipato, aspartato, benzoato, besilato, bicarbonato/carbonato, bisulfato/sulfato, borato, citrato, formiato, fumarato, gluconato, glucuronato, clorhidrato/cloruro, hidrobromuro/bromuro, hidroyoduro/yoduro, lactato, malato, maleato, malonato, mesilato, metilsulfato, naftilato, oxalato, palmitato, fosfato/fosfato de hidrógeno/fosfato de dihidrógeno, piroglutamato, salicilato, estearato, succinato, sulfonato, tartrato, tosilato y sales de trifluoroacetato. Las sales de base adecuadas se forman a partir de bases que forman sales no tóxicas. Algunos ejemplos son las sales de aluminio, arginina, benzatina, calcio, colina, dietilamina, diolamina, glicina, lisina, magnesio, potasio, sodio, trometamina y zinc. También pueden formarse hemisales de ácidos y bases, por ejemplo, sales de hemisulfato y hemicálcicas. Para una revisión de las sales adecuadas, véase Handbook of Pharmaceutical Salts: Properties, Selection, and Use de Stahl y Wermuth (Wiley-VCH, 2002). Las sales farmacéuticamente aceptables de los compuestos de Fórmula I pueden prepararse por uno o más de tres procedimientos: (i) haciendo reaccionar el compuesto de Fórmula I con el ácido o la base deseados; (ii) eliminando un grupo protector lábil al ácido o a la base de un precursor adecuado del compuesto de Fó'mula I; o (iii) convirtiendo una sal del compuesto de Fórmula I en otra sal mediante la reacción con un ácido o una base adecuados o por medio de una columna de intercambio iónico adecuada.
SUMARIO DE LA INVENCIÓN:
Los aspectos y las realizaciones de la invención se exponen en las reivindicaciones adjuntas.
En una realización, se proporciona un compuesto de la Fórmula I:

Fómi ula I
en la que:
A es N o CR1; B es N o CR2; J es N o CR3; K es N o CR4; L es N o CR5; M es N; P es N o CR7; y Q es N o CR8, siempre que no más de dos de A, B, J, K, L, M, P y Q puedan ser N;
X es un enlace;
R1, R2, R4, R5, R7y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxiC1-5, heteroarilalcoxiC2-5, ariloxi C6-14, aril C6-10 alcoxiC1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C^C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)- y arilo C6-10C(O)O-; y R3 se selecciona del grupo que consiste en cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroaril alcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C^C(O)O-, arilo C6-10C(O)-, y arilo C6-10C(O)O-;
en el que al menos cuatro de R1, R2, R4, R5, R7 y R8 son hidrógenos;
R9 es hidrógeno ; cada R10 es independientemente H o alquilo C1-6;
y en el que los sustituyentes en R1, R2, R3, R4, R5, R7 R8y R10 están opcionalmente sustituidos por sustituyentes seleccionados del grupo formado por amino, halo, ciano, nitro, hidroxilo, -SH, alquilo -SC1-6 , -C(O)NH2, -C(S)NH2, haloalquilo C1-6, perhaloalquilo C1-6, alquilo C1-6, cicloalquilo C3-6, cicloalquilo C3-12, arilo C6-14 y heteroarilo;
y sus sales farmacéuticamente aceptables.
En otra realización, se proporciona un compuesto de la Fórmula I que está radiomarcado, en el que el radiomarcado comprende un radionúclido seleccionado del grupo que consiste en 11C, 13N, 15O, 18F, 123I, 124I, 125I, 131I y 77Br, y sales farmacéuticamente aceptables del mismo.
En una variación del compuesto anterior:
A es N o CR1; B es N o CR2; J es N o CR3; K es N o CR4; L es N o CR5; M es N; P es N; y Q es N o CR8, siempre que no más de dos de A, B, J, K, L, M, P y Q puedan ser N;
X es un enlace;
R1, R2, R4, R5, y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en halo alquilo C1-6, perhalo alquilo C1-6, alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxiC1-5, heteroaril alcoxi C2-5, ariloxi C6-14, aril C6-10 alcoxiC1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, halo alquilo C1-5NR10C(O)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)-y arilo C6-10C(O)O-; y R3 se selecciona del grupo que consiste en cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, heteroaril alcoxi C2-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, haloalquilo C1-5NR10C(O)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)- y arilo C6-10C(O)O;
y sus sales farmacéuticamente aceptables.
En otra variación del compuesto anterior:
A es N o CR1; B es N o CR2; J es N o CR3; K es N o CR4; L es N o CR5; M es N; P es N o CR7; y Q es N o CR8, siempre que no más de dos de A, B, J, K, L, M, P y Q puedan ser N;
X es un enlace;
R1, R2, R4, R5, R7y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste haloalquilo C1-6, perhalo alquilo C1-6, alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroaril alcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo; and R3 is selected from the group consisting of cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroaril alcoxi C2-5, arilo C6-14, heteroarilo;
y sus sales farmacéuticamente aceptables.
En otra variación del compuesto anterior:
y Q es N o CR8, siempre que no más de dos de  A, B, J, K, L, M, P y Q puedan ser N;
A, B, J, K, L, M, P y Q puedan ser N;
X es un enlace;
R1, R2, R4, R5, R7 y R8 son cada uno independientemente hidrógeno o son cada uno independientemente haloalquilo C1-5 NR10C(O)-; y R3 es haloalquilo C1-5 NR10C(O)-;
y sus sales farmacéuticamente aceptables.
En una variación del compuesto anterior:
y Q es N o CR8, siempre que no más de dos de  A, B, J, K, L, M, P y Q puedan ser N;
A, B, J, K, L, M, P y Q puedan ser N;
X es un enlace;
R1, R2, R4, R5, R7y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, heteroaril alcoxi C2-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)- y arilo C6-10C(O)O-; and R3 is selected from the group consisting of cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, heteroaril alcoxi C2-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)- y arilo C6-10C(O)O-;
y sus sales farmacéuticamente aceptables.
En otra variación del compuesto anterior:
X es un enlace;
R1, R2, R4, R5, R7y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en haloalquilo C1-6, perhalo alquilo C1-6, alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, halo alquilo C1-5NR10C(O)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)-y arilo C6-10C(O)O-; y R3 se selecciona del grupo que consiste en cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, heteroaril alcoxi C2-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)- y arilo C6-10C(O)O-;
en el que al menos cuatro de R1, R2, R4, R5, R7 y R8 son hidrógenos;
R9 es hidrógeno; cada R10 es independientemente H o alquilo C1-6;
y sus sales farmacéuticamente aceptables.
En otra variación de lo anterior: R1, R2, R4, R5, R7 y R8 son cada uno independientemente hidrógeno o son cada uno independientemente seleccionados del grupo que consiste en, haloalquilo C1-6 y perhaloalquilo C1-6; y sales farmacéuticamente aceptables de los mismos.
En aún otra variación:
R1, R2, R4, R5, R7y R8 son cada uno independientemente hidrógeno o se seleccionan del grupo que consiste en F-alquil C1-6 , 4-(F-alquil C1-6)-1H-1,2,3-triazol-1-il-(alcoxi C2-5) - y F-alquil C1-5 NR10C(O)-; y R3 se selecciona del grupo que consiste en 4-(F-alquil C1-6)-1H-1,2,3-triazol-1-il-(alcoxi C2-5) y F-alquil C1-5 NR10C(O)-; y sales farmacéuticamente aceptables de los mismos.
En otra variación particular de lo anterior:
al menos cuatro de R1, R2, R4, R5, R7 y R8 son hidrógenos; y sales farmacéuticamente aceptables de los mismos.
En otra variación más de cada uno de los anteriores, el grupo amino se selecciona del grupo que consiste en NH2-, CH3NH-, (CH3)2N-, alquil C1-3-NH-, F- alquil C2-3-NH-, F-(alquil C2-3-O)1-4-alquil-NH-, (alquil C1-3-)2N-, alquil C1-6NH-, (alquil C1-6)2N-, cicloalquil C3-6NH-, (cicloalquil C3-6)2N-, cicloalquil C3-12 alquil C1-5NH-, aril C6-14NH-, aril C6-10 alquil C1.
4NH-, heteroaril NH-, ariloxi C6-14NH-, aril C6-10 alcoxiC1-4 NH-y heteroariloxi NH-.
En una variación particular de la Fórmula I, X es un enlace y R9 es hidrógeno.
En otra realización, se proporciona una composición farmacéutica para la obtención de imágenes in vivo de depósitos de amiloide, que comprende (a) un compuesto de la Fórmula I que está radiomarcado como se ha descrito anteriormente, y (b) un portador farmacéuticamente aceptable. En el presente documento se divulga un procedimiento para diagnosticar la enfermedad de Alzheimer o una predisposición a la misma en un mamífero, comprendiendo el procedimiento : a) administrar al mamífero una cantidad diagnóstica eficaz de cualquiera de los compuestos anteriores, en el que el compuesto pasa la barrera hematoencefálica y se une preferentemente a los oligómeros, polímeros y fibrillas solubles de la AD en un tejido cerebral; b) permitir que el compuesto se distribuya en el tejido cerebral; y c) obtener imágenes del tejido cerebral, en el que un aumento de la unión del compuesto al tejido cerebral en comparación con un nivel de control normal de unión indica que el mamífero padece o está en riesgo de desarrollar la Enfermedad de Alzheimer.
En otra realización, se proporciona el compuesto radiomarcado de la Fórmula I como se ha descrito anteriormente o la composición que comprende un compuesto de la Fórmula I que está radiomarcado como se ha descrito anteriormente para su uso en un procedimiento de diagnóstico de la enfermedad de Alzheimer o de una predisposición a la misma en un mamífero, comprendiendo el procedimiento a) administrar al mamífero una cantidad diagnóstica eficaz del compuesto radiomarcado de la Fórmula I como se ha descrito anteriormente o la composición que comprende un compuesto de la Fórmula I radiomarcado como se ha descrito anteriormente, en la que el compuesto pasa la barrera hematoencefálica y se une preferentemente a oligómeros, polímeros y fibrillas solubles de AD en un tejido cerebral; b) permitir que el compuesto se distribuya en el tejido cerebral; y c) obtener imágenes del tejido cerebral, en las que un aumento de la unión del compuesto al tejido cerebral en comparación con un nivel de control normal de unión indica que el mamífero sufre o corre el riesgo de desarrollar la enfermedad de Alzheimer.
En una variación del uso anterior, el compuesto radiomarcado se une preferentemente a las fibrillas. En otra variación del uso anterior, el tejido cerebral comprende una región frontotemporal o la región del hipocampo. En una variación particular del uso anterior, el aumento de la unión es al menos 10% mayor que dicho valor de control normal. En otra variación de cada uno de los usos anteriores, el compuesto se administra por inyección intravenosa.
En el presente documento se divulga un procedimiento para detectar la enfermedad de Alzheimer o una predisposición a la misma en un cerebro vivo de un mamífero, comprendiendo el procedimiento a) administrar al mamífero una cantidad diagnóstica eficaz de cualquiera de los compuestos anteriores que atraviesa la barrera hematoencefálica y se une preferentemente a los oligómeros, polímeros y fibrillas solubles de la AD en el cerebro, en el que el compuesto marcado de forma detectable es un compuesto de cualquiera de los anteriores; b) permitir que el compuesto se distribuya en el tejido cerebral; y c) obtener imágenes del tejido cerebral, en las que un aumento de la unión del compuesto al tejido cerebral en comparación con un nivel de control normal de unión indica que el mamífero padece o corre el riesgo de desarrollar la enfermedad de Alzheimer.
En el presente documento se divulga un procedimiento para detectar la enfermedad de Alzheimer o una predisposición a la misma en un cerebro vivo de un mamífero, comprendiendo el procedimiento a) administrar al mamífero una cantidad diagnóstica eficaz de cualquiera de los compuestos anteriores de cualquiera de los anteriores, en el que el compuesto pasa la barrera hematoencefálica y se une preferentemente a unos oligómeros, polímeros y fibrillas solubles de AD en el cerebro; b) permitir que el compuesto se distribuya en el tejido cerebral; y c) obtener imágenes del tejido cerebral, en el que un aumento de la unión del compuesto al tejido cerebral en comparación con un nivel de control normal de unión indica que el mamífero sufre o está en riesgo de desarrollar la Enfermedad de Alzheimer.
En otra realización, se proporciona el compuesto radiomarcado de la Fórmula I descrito anteriormente para su uso en un procedimiento de diagnóstico de la enfermedad de Alzheimer o de una predisposición a la misma en un mamífero, comprendiendo el procedimiento a) administrar al mamífero una cantidad diagnóstica eficaz del compuesto radiomarcado de la Fórmula I como se ha descrito anteriormente, en el que el compuesto pasa la barrera hematoencefálica y se une preferentemente a oligómeros, polímeros, fibrillas, tau hiperfosforilada, ovillos neurofibrilares, filamentos helicoidales emparejados y/u oligómeros solubles neurotóxicos de la AD en un cerebro, y en el que el compuesto radiomarcado es un compuesto como el divulgado en el presente documento; y (b) emplear una técnica de obtención de imágenes nuclear seleccionada del grupo que consiste en tomografía por emisión de positrones (PET) y tomografía computarizada por emisión de fotón único (SPECT) para monitorizar o visualizar una distribución del compuesto radiomarcado dentro del cerebro o dentro de una porción del mismo. En otra realización, se proporciona el compuesto de la Fórmula I y el compuesto radiomarcado de la Fórmula I como se ha descrito anteriormente para su uso en el tratamiento de una enfermedad o condición, en un mamífero que lo necesite, seleccionado del grupo que consiste en ansiedad, depresión, esquizofrenia, enfermedad de Alzheimer, enfermedad relacionada con el estrés, pánico, una fobia, trastorno obsesivo compulsivo, obesidad, síndrome de estrés postraumático o epilepsia. En el presente documento se divulga el uso anterior, en el que el compuesto se administra por vía rectal, tópica, oral, sublingual o parenteral. En el presente documento se divulga el uso anterior, en el que el compuesto se administra de aproximadamente 0,001 a aproximadamente 100 mg/kg de peso corporal del mamífero por día. En el presente documento se divulga el uso anterior, en el que el compuesto se administra de aproximadamente 0,1 a aproximadamente 50 mg/kg de peso corporal del mamífero por día. En el presente documento se divulga el compuesto radiomarcado de la Fórmula I como se ha descrito anteriormente o la composición que comprende un compuesto de la Fórmula I que está radiomarcado como se ha descrito anteriormente para su uso en los procedimientos de detección que detectan con precisión el inicio temprano de la AD antes de la sintomatología clínica, en el que el enfoque puede dirigirse a los precursores de la placa senil, en lugar de las propias placas y/o fibrillas. En consecuencia, una estrategia potencialmente más eficaz para detectar y posiblemente tratar la AD se basaría en la detección de biomarcadores como los oligómeros solubles neurotóxicos, que están relacionados con el daño sináptico y neuronal de la EA, en lugar de los biomarcadores de placas y fibrillas en fase tardía asociados con la AD totalmente avanzada.
T l 1: Tin fl r n n n i n im n iiv r l AD n i

(continuación)

Se desarrolló un ensayo utilizando un instrumento Biacore que introdujo ligandos de cribado sobre proteínas objetivo inmovilizadas en la superficie de oro y midió las tasas resultantes de asociación y disociación con el fin de cribado de diversos compuestos que se unen a oligómeros, polímeros y fibrillas solubles de EA. En la Figura 1, la porción izquierda de la curva representa la unión de los ligandos a un sustrato específico. La porción derecha de la curva representa la disociación del ligando del sustrato. Los ligandos que se asociaron rápidamente y se disociaron lentamente, en relación con un ligando de control, se consideraron éxitos.
Figura 1: Resultados del ensayo de unión Biacore
Se examinaron varios cientos de compuestos de manera similar y se identificaron varias clases comunes de compuestos como éxitos que caen en siete quimiotipos comunes: flavonas, cumarinas, carbazoles, quinolinonas, cromoonas, imidazoles y triazoles.
Figura 2: Andamios representativos de compuestos que se unen a oligómeros, polímeros y/o fibrillas.
A partir de la biblioteca de cribado, se identificaron 38 compuestos como aglutinantes de los oligómeros/polímeros solubles de Ap42 (Tabla 2). Entre estos 38 compuestos, 23 compuestos se unieron a las fibrillas sintéticas de Ap42. Varios compuestos pertenecientes a las arquitecturas de la flavona y la cumarina se unieron muy fuertemente a los oligómeros, polímeros y fibrillas. Se comprobó que los cromoos, carbazoles y diaril triazoles se unen a los oligómeros y polímeros preferentemente a las fibrillas. Varios de estos compuestos pueden prepararse como análogos radiomarcados para su uso en la detección de biomarcadores en pacientes con EA.
Tabla 2: Compuestos identificados como aglutinantes de oligómeros, polímeros y/o fibrillas. Un signo "+" representa un acierto y el aumento de los signos "+" se relaciona con el aumento de la afinidad de unión. El signo "-" representa que no hay vinculación. Estos compuestos son sólo de referencia y no forman parte de la invención.
NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol
APi- Fibrillas de

#42: 3,6-Dihidroxiflavona

NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol
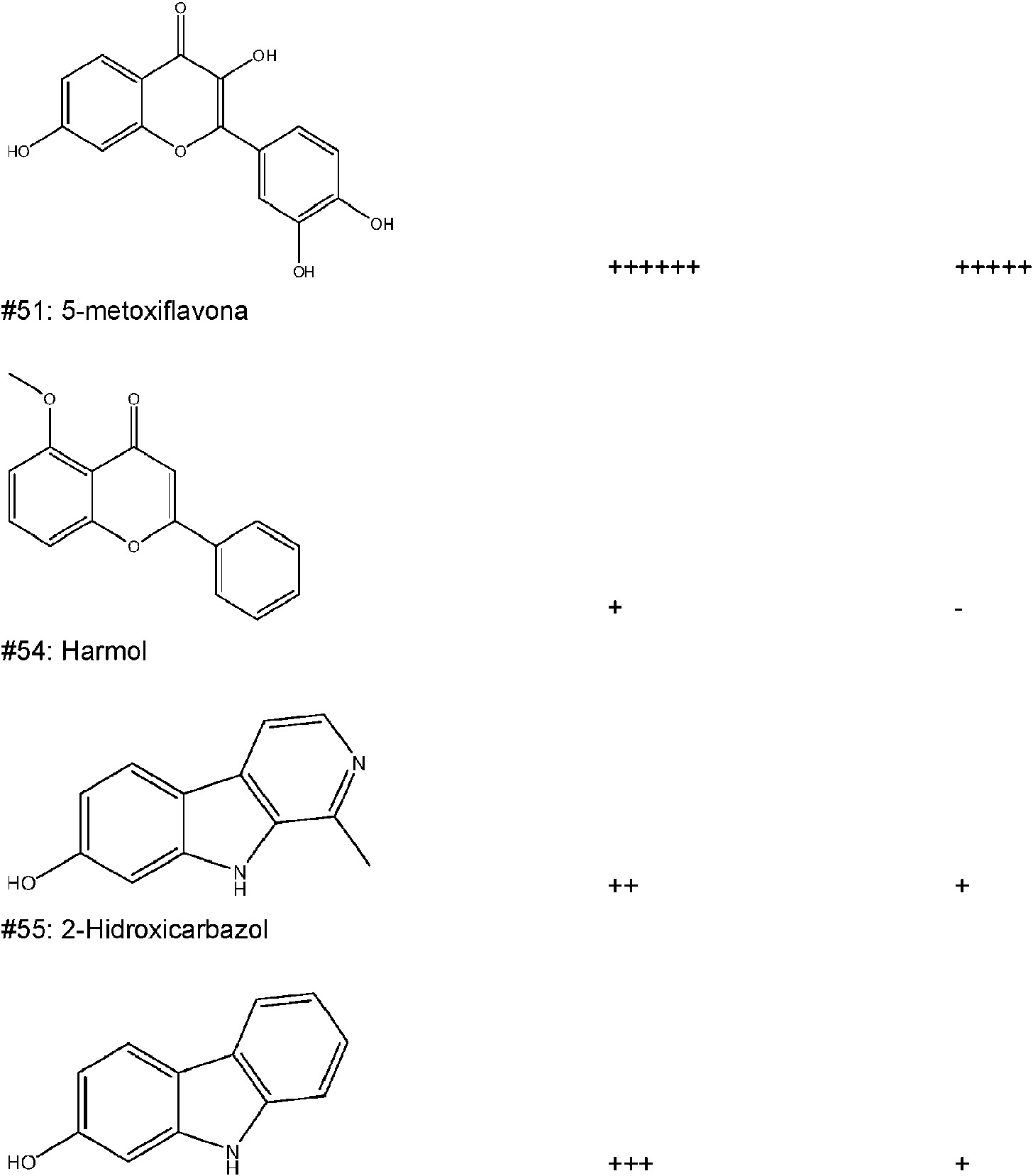
#73: 7,8-Dihidroxi-4-fenilcumarina

#84: 1 -Cloro-4-hidroxisoquinolina
NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol

#89: 2-(5-Isoxazolil)-4-metilfenol

1-metil-6,7-dihidroxi-1,2,3,4-tetrahidroisoquinol




NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol

#200: 6-Etil-3-(4-fluorofenil)-7-hidroxi-2-metil-4H-c

#201: 7,8-Dimetoxi-2-metil-3-fenil-4H-cromo-4-ona

#202: 7-Hidroxi-2,8-dimetil-3-(4-fenil-4H-1,2,4-triazo
+
na-4(1H)-ona


#205: 2-(3,4-Dimetoxifenil)-3,6-dimetoxi-4H-cromo-4


#207: 3,6-Dihidroxi-2-(3-hidroxifeml)-4H-cromo-4-ona


#216: 2-(Furan-2-yl)-3-hidroxi-6,8-dimetil-4H-cromo-4

NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol #217: 3-hidroxi-6,8-dimetil-2-(tiofen-2-il)-4H-cromo
+++

NIVELES DE UNIÓN AL #25: 5-Amino-2-(trifluorometil)benzimidazol

imidazol
La Tabla 3 proporciona ejemplos de agentes de obtención de imágenes derivados de los andamiajes golpeados. Los fluoruros se muestran en las estructuras como equivalentes al fluoruro 18F y los grupos metílicos son equivalentes a los grupos metílicos de carbono 11C.
Tabla 3: Ejemplos de análogos radiomarcados útiles para la detección de biomarcadores de la AD in vivo. Estos m n l r f r n i n f rm n r l inv ni n.

(continuación)

(continuación)
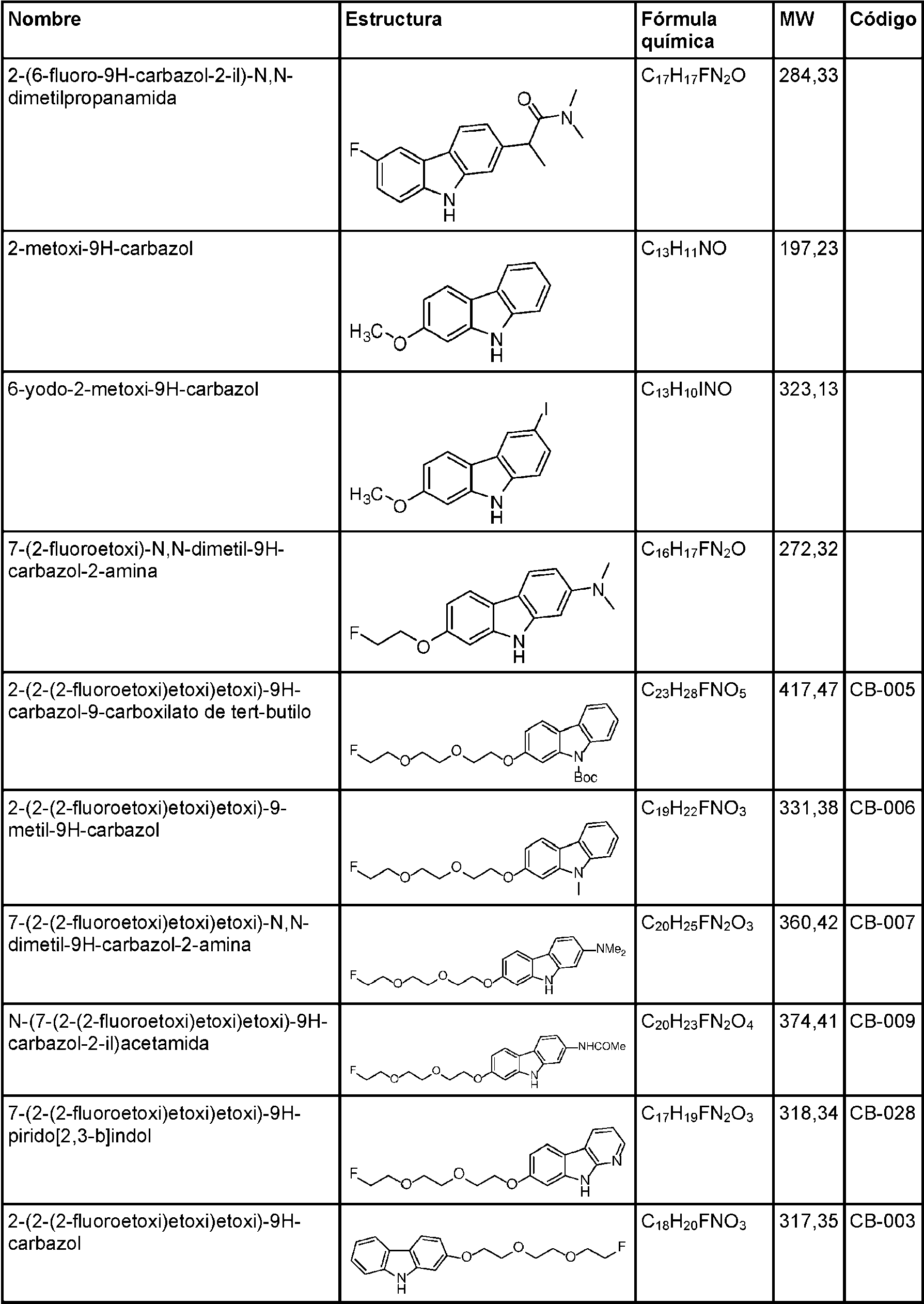
(continuación)

(continuación)
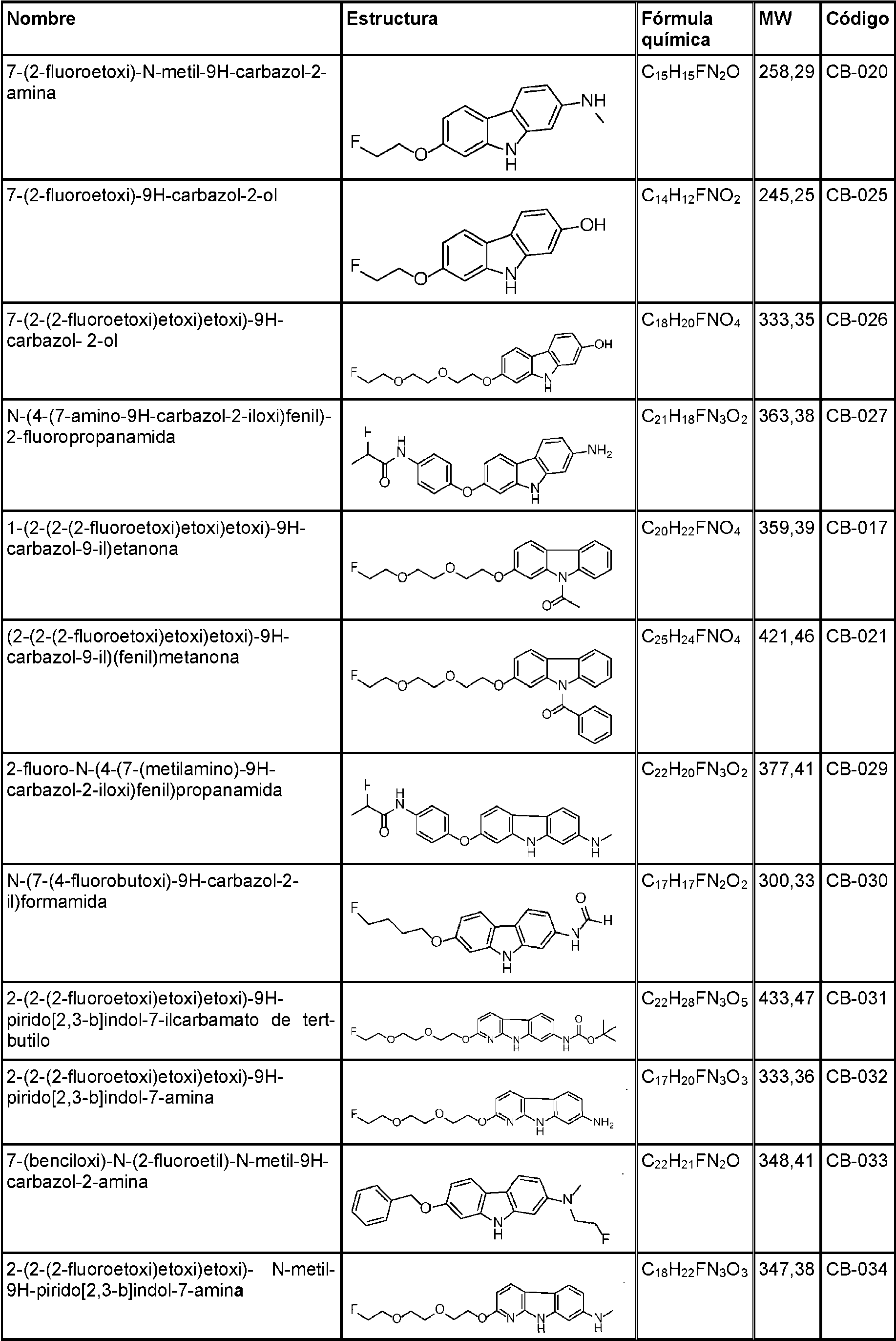
(continuación)

(continuación)

(continuación)

Síntesis de ligandos y sus precursores de etiquetado:
Halogenación y radiohalogenación:
Como se divulga en el presente documento, para un número de diferentes ligandos AD, tales como flavonas, cumarinas, carbazoles, quinolinonas, cromoonas, imidazoles trisustituidos y sus derivados como se divulga en el presente documento, el átomo radiomarcado, tal como un átomo de halógeno, por ejemplo, puede ser fácilmente introducido en el ligando utilizando un número de diferentes procedimientos bien conocidos la técnica. En consecuencia, los compuestos radiomarcados de la Fórmula I de la presente solicitud pueden prepararse utilizando procedimientos estándar conocidos en la técnica para preparar dichos compuestos radiomarcados que tienen un sustituyente particular, en los que el compuesto puede incorporarse con un radionúclido particular seleccionado del grupo que consiste en 11C, 13N, 15O, 18F, 1231,1241,1251,131I y 77Br.
En un ejemplo particular, el halógeno puede ser introducido por un procedimiento que utiliza un procedimiento de intercambio de estaño por halógeno. Por ejemplo, un halógeno no radiactivo, tal como el yodo, puede ser sustituido por un compuesto de organoestaño a través de un metal, tal como una composición de paladio, para formar el precursor de estaño radiomarcado, como se representa a continuación. Este precursor se somete entonces a la halogenación radiactiva mediante el desplazamiento con la fuente Na125I , por ejemplo, para obtener el ligando
radiactivo.

Alternativamente, el halógeno radioetiquetado puede introducirse fácilmente mediante halogenación directa. Por ejemplo, para un ligando que comprende un anillo aromático como parte del andamio, o un sustituyente aromático de un ligando, el anillo aromático puede ser yodado directamente utilizando un procedimiento de radoiodinación bien establecido. Uno de estos ejemplos se representa a continuación utilizando un ligando de carbazol

Para los compuestos marcados con 11C, el compuesto marcado puede prepararse mediante la alquilación o metilación de un grupo hidroxilo, como por ejemplo con [11C]CH3I para proporcionar el correspondiente derivado metoxi marcado con C-11. Por ejemplo, dicho proceso está representado por la reacción del derivado de la flavona que se muestra a continuación.

Otros procedimientos de preparación de ligandos radiomarcados son bien conocidos en la técnica. Ejemplos de tales procedimientos se divulgan en, por ejemplo: 1)Jewett, D.M. (1992) A Simple Synthesis of [11C]Metil Triflate Appl. Radiat. Isot. 43, 1383-1385; 2) Crouzel, C. Langstrom, B., Pike, V.W., y Coenen, H.H. (1987) Recommendations for a practical production of [11C]methyl iodide Appl. Radiat. Isot. Int. J. Appl. Instrum. Part A 38, 601-603; Dannals, R.F., Ravert, H.T.; 3) Wilson, A.A. (1990) Radiochemistry of Tracers for Neurotransmitter Receptor Studies. In: Quantitative Imaging: Neuroreceptors, Neurotransmitters, and Enzymes. (Editado por Frost, J.J. Wagner Jr., H.N. pp. 19-35, Raven Press, Nueva York; 4) Jewett, D.M., Manger, T.J., y Watkins, G.L. (1991) Captive Solvent Methods for Fast Simple Carbon-11 Radioalkylations. In: New Trends in Radiopharmaceutical Synthesis, Quality Assurance and Regulatory Control (Editado por Emran, A.M.) pp. 387-391. Plenum Press, Nueva York; 5) Marazano, C., Maziere, M., Berger, G., y Comar, D. (1977) Synthesis of metil iodide-11C and formaldehyde-11C Appl. Radiat. Isot. 28, 49-52; 6) Watkins, G., Jewett, D., Mulholland, G., Kitbourn, M., y Toorongian, S. (1988) A Captive Solvent Method for Rapid N-[11C]Metilation of Secondary Amides: Application to the Benzodiazepine, 4'-Chlorodiazepam (RO5-4864) Appl. Radiat. Isot. 39, 441 444y 7) Wilson, A. A., DaSilva, J.N., y Houle, S. (1996) In vivo evaluation of [11C] and [15F]-labelled cocaine analogues as potential dopamine transporter ligands for positron emission tomography Nucl. Med. Biol. 23, 141-146.
Síntesis de AD-CB-WZ01013

Al hidroxicarbazol (73 mg, 0,4 mmol) en 1 m de NMP se añadió CS2CO3(130 mg, 0,4 mmol) y bromofluoroetano (51 mg, 0,4 mmol). La mezcla se agitó ata durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 1 M (30 ml) y agua (2x40 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (4% EtOAc en hexano al 25 %) para obtener el producto deseado (36 mg) como un sólido blanquecino.
1H RMN (400 MHz, CDCk/acetona-da) 89,98 (s, 1 H), 7,95 (t, J = 8,8 Hz, 2 H), 7,40 (d, J = 8,0 Hz, 1 H), 7,28 (t, J = 8 Hz, 1 H), 7,13 (t, J = 8,0 Hz, 1 H), 7.00 (d, J = 2 Hz, 1 H), 6,83 (dd, J = 8,8, 2,0 Hz, 1 H), 4,85 (t, J = 4 Hz, 1 H), 4,73 (t, J = 4 Hz, 1 H), 4,35 (t, J = 4 Hz, 1 H), 4,28 (t, J = 4 Hz, 1 H); MS(ESI) m/z 230 (M+H+).
Síntesis de AD-C-WZ01011

Al hidroxicarbazol (183 mg, 1 mmol) en 4 ml de NMP se añadió Cs2CO3 (326 mg, 1 mmol) y etilendi-tosilato (370 mg, 1 mmol). La mezcla se agitó ata durante 15 h y se diluyó con Et2O (80 ml). Se lavó con HCl 1 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (50% DCM en hexano a 100% DCM) para obtener el producto deseado (75 mg) como un sólido blanquecino.
1H RMN (400 MHz, acetona-da) 810,21 (s, 1 H), 8,00 (d, J = 8,0 Hz, 1 H), 7,95 (d, J = 8,4 Hz, 1 H), 7,84 (d, J = 8,4 Hz, 2 H), 7,45 (m, 3 H), 7,30 (t, J = 8,0 Hz, 1 H), 7.13 (t, J = 8,0 Hz, 1 H), ); 6,98 (s, 1 H), 6,73 (d, J = 8,4 Hz, 1 H), 4,44 (t, J = 4,0 Hz, 2 H), 4,30 (t, J = 4,0 Hz, 2 H), 2,42 (s, 3 H); MS(ESI) m/z 382 (M+H+), 404 (M+Na+).
Síntesis de AD-CB-001P-WZ-01019 marcado con 18F ([18F]2-(2-Fluoro-etoxi)-9H-carbazol)
k 18f Kryptof
eCN.'DMSQ  Dmin
Dmin
Semi-prep HPLC


El [18F]Fluoruro (600 - 900 mCi) como solución enriquecida en H218O fue entregado al módulo de síntesis. El [18F]fluoruro se atrapó en una columna de intercambio iónico y se eludió en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añadió el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evaporó hasta sequedad. Se añadió al reactor el
precursor del éster 2-(9H-carbazol-2-iloxi)-etilo del ácido tolueno-4-sulfónico (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) y luego se calentó la reacción de fluoración a 115 °C durante 10 min. La mezcla de reacción cruda se purificó por HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H20 (+0,05 % TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA); Caudal: 5 ml/min).
El pico correspondiente al [18F]2-(2-fluoro-etoxi)-9H-carbazol se recogió y se diluyó simultáneamente con agua estéril (10 ml). La mezcla resultante se pasó por un C-18 Sep-Pak para que el producto quedara atrapado y el acetonitrilo residual se lavó con más agua (10 ml). el [18F]2-(2-Fluoro-etoxi)-9H-carbazol se eludió entonces en el vial del producto con etanol de grado USP (0,5 ml) y se diluyó con agua estéril (9,5 ml) para proporcionar una formulación final (19 - 34 mCi en 10 ml) adecuada para la inyección (rendimiento corregido por decaimiento del 7,5 %, pureza radioquímica del 100 %).
La pureza se determinó por HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC para el correspondiente estándar de referencia no marcado.
Figura 3: Análisis por UV y HPLC gamma de AD-CB-001P-WZ-01019
Síntesis de AD-CB-002P-WZ01031.

Al hidroxicarbazol (92 mg, 0,5 mmol) en 2 ml de NMP se añadió Cs2CO3 (163 mg, 0,5 mmol) y etiltosilato de azida (121 mg, 0,5 mmol). La mezcla se agitó ata durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 0,5 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (80 % DCM en hexano a 100 % DCM) para obtener el producto deseado (76 mg) como sólido blanco.
1H RMN (400 MHz, CDCh/acetona-da) 59,98 (s, 1 H), 7,95 (m, 2 H), 7,41 (d, J = 8,4 Hz, 1 H), 7,29 (t, J = 8,0 Hz, 1 H), 7,14 (t, J = 8.0 Hz, 1 H); 7,01 (s, 1 H), 6,84 (d, J = 8,4 Hz, 1 H), 4,28 (t, J = 4,8 Hz, 2 H), 3,67 (t, J = 4,8 Hz, 2 H); MS(ESI) m/z 253 (M+H+).
Síntesis de AD-CB-002S-WZ01033

Al azido carbazol (32 mg, 0,127 mmol) en 0,5 ml de DMF se añadió Cul (7,6 mg, 0,04 mmol), DIPEA (16,4 mg, 0,127 mmol) y fluoropentina (16,4 mg, 0,19 mmol). La mezcla de reacción se agitó enérgicamente durante 1 h y se diluyó con EtOAc (30 ml). Se lavó con agua (50 ml), HCl 0,5 M (30 ml), agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se preabsorbió en sílice (3 g) y se cargó en una columna de sílice de 4 g y se eludiócon EtOAc al 30 % en hexano al 50 % para obtener el compuesto deseado (20 mg).
1H RMN (400 MHz, CDCls/CDsOD) 57,95 (d, J = 7,6 Hz, 1 H), 7,91 (d, J = 8,4 Hz, 1 H), 7,76 (s, 1 H), 7,40 (d, J = 8,0 Hz, 1 H), 7,31 (t, J = 7,6 Hz, 1 H), 7,14 (t, J = 7,6 Hz, 1 H), 6.94 (d, J = 2,4 Hz, 1 H), 6,78 (dd, J = 8,8, 2,4 Hz, 1 H), 4,83-4,78 (m, 2 H), 4,53-4,48 (m, 3 H), 4,40 (t, J = 6,0 Hz, 1 H), 2,85 (t, J = 7,6 Hz, 2 H), 2,10-1,99 (m, 2 H); MS(ESI) m/z 339 (M+H+).
Síntesis de AD-CB-002S-WZ01033 marcado con 18F:
Preparación de [18F] 5-Fluoro-pent-l-ina
i) K 18F, Kryptofix-2.2.2

El [18F] fluoruro (600 - 900 mCi) como solución enriquecida en H218O se entrega al módulo de síntesis. El [18F]fluoruro se atrapa en una columna de intercambio iónico y se elude en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añade el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evapora hasta sequedad.
Se añade al reactor el éster de pent-4-inilo del ácido tolueno-4-sulfónico (20 mg en 0,8 ml de MeCN) y se calienta la reacción de fluoración a 110 °C durante 5 min. Tras la fluoración, la mezcla cruda de reacción se purifica por destilación y se obtiene [18F] 5-fluoro-pent-1-ino como solución en acetonitrilo (atrapada a -78 °C debido a la volatilidad del producto).
Preparación del triazol:
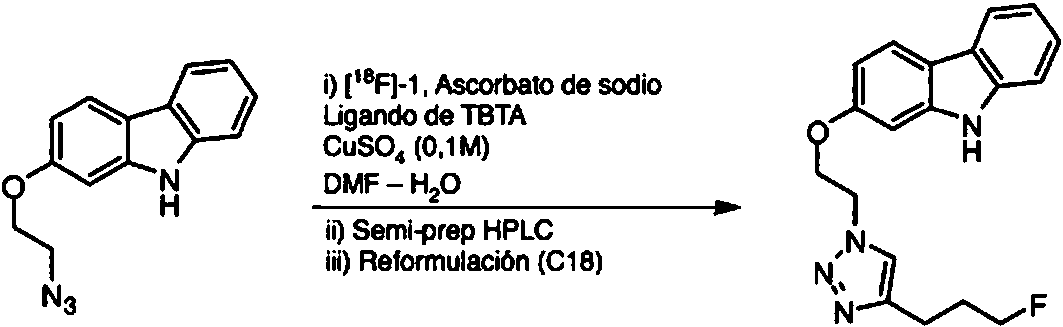
Se añade una mezcla de precursor de azida (5 mg), ascorbato de sodio (40 mg), fr/s-(benciltriazolilmetil) amina (TBTA, 25 mg) y solución acuosa de sulfato de cobre (0,1 M, 0,25 ml) en DMF (0,4 ml) y agua (0,1 ml) a la solución de pentina enfriada descrita anteriormente. La mezcla de reacción se calienta hastata y se agita durante 30 minutos. Transcurrido este tiempo, la reacción se purifica por HPLC semipreparativa. Se recoge el pico correspondiente al producto y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak y el acetonitrilo residual se lava con agua adicional (10 ml). El producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final adecuada para la inyección.
La pureza se determina por HPLC analítica equipada con un detector de radiactividad y la identidad se confirma por comparación con los datos de HPLC para el correspondiente estándar de referencia no etiquetado.
Síntesis de CB-003 marcado con 18F

El [18F] fluoruro (600 - 900 mCi) como solución enriquecida en H218O se entrega al módulo de síntesis. El [18F]fluoruro se atrapa en una columna de intercambio iónico y se elude en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añade el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evapora hasta sequedad. Se añade el precursor (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) al reactor y se calienta la reacción de fluoración a 115 °C durante 10 min. La mezcla se enfrió a 55 °C y la mayor parte del acetonitrilo se evaporó al vacío y con una corriente de argón como antes. Al producto bruto protegido con Boc se le añadió ácido clorhídrico acuoso (1,0 M, 1,0 ml), y la mezcla se calentó a 105 °C durante
3 minutos. Tras enfriar a 35 °C, se añadió acetato de sodio acuoso (2,0 M, 0,5 ml) con agitación. A continuación, la mezcla de reacción cruda se purifica mediante HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H2O (+0,05% TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA); Caudal: 5 ml/min; tiempo = 25 min). Se recoge el pico correspondiente al producto final y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak para que el producto quede atrapado y el acetonitrilo residual se lava con más agua (10 ml). A continuación, el producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final apta para la inyección (31 % de rendimiento de decadencia no corregido, 100% de pureza radioquímica). La pureza se determinó mediante HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC del correspondiente estándar de referencia no marcado.
Síntesis de CB-004 marcado con 18F

El [18F] fluoruro (600 - 900 mCi) como solución enriquecida en H218O se entrega al módulo de síntesis. El [18F]fluoruro se atrapa en una columna de intercambio iónico y se elude en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añade el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evapora hasta sequedad. Se añade el precursor (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) al reactor y se calienta la reacción de fluoración a 115 °C durante 10 min. La mezcla se enfrió a 55 °C y la mayor parte del acetonitrilo se evaporó al vacío y con una corriente de argón como antes. Al producto bruto protegido con Boc se le añadió ácido clorhídrico acuoso (1,0 M, 1,0 ml), y la mezcla se calentó a 105 °C durante 3 minutos. Tras enfriar a 35 °C, se añadió acetato de sodio acuoso (2,0 M, 0,5 ml) con agitación. A continuación, la mezcla de reacción cruda se purifica mediante HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H20 (+0,05 % TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA); Caudal: 5 ml/min; tiempo = 25 min). Se recoge el pico correspondiente al producto final y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak para que el producto quede atrapado y el acetonitrilo residual se lava con más agua (10 ml). A continuación, el producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final apta para la inyección (3% de rendimiento de decadencia no corregido, 100% de pureza radioquímica). La pureza se determinó mediante HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC del correspondiente estándar de referencia no marcado.
Síntesis de CB-007 marcado con 18F

El [18F] fluoruro (600 - 900 mCi) como solución enriquecida en H218O se entrega al módulo de síntesis. El [18F]fluoruro se atrapa en una columna de intercambio iónico y se elude en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añade el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evapora hasta sequedad. Se añade el precursor (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) al reactor y se calienta la reacción de fluoración a 115 °C durante 10 min. La mezcla se enfrió a 55 °C y la mayor parte del acetonitrilo se evaporó al vacío y con una corriente de argón como antes. Al producto bruto protegido con Boc se le añadió ácido clorhídrico acuoso (1,0 M, 1,0 ml), y la mezcla se calentó a 105 °C durante 3 minutos. Tras enfriar a 35 °C, se añadió acetato de sodio acuoso (2,0 M, 0,5 ml) con agitación. A continuación, la mezcla de reacción cruda se purifica mediante HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H20 (+0,05 % TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA);
Caudal: 5 ml/min; tiempo = 25 min). Se recoge el pico correspondiente al producto final y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak para que el producto quede atrapado y el acetonitrilo residual se lava con más agua (10 ml). A continuación, el producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final apta para la inyección (1,2% de rendimiento de decadencia no corregido, 100% de pureza radioquímica). La pureza se determinó mediante HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC del correspondiente estándar de referencia no marcado.
Síntesis de CB-012 marcado con 18F

El [18F]Fluoruro (600 - 900 mCi) como solución enriquecida en H218O fue entregado al módulo de síntesis. El [18F]fluoruro se atrapó en una columna de intercambio iónico y se eludió en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añadió el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evaporó hasta sequedad. Se añadió al reactor el precursor del éster 2-(9H-carbazol-2-iloxi)-etilo del ácido tolueno-4-sulfónico (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) y luego se calentó la reacción de fluoración a 115 °C durante 10 min. La mezcla de reacción cruda se purificó por HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H20 (+0,05 % TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA); Caudal: 5 ml/min). Se recogió el pico correspondiente al producto y se diluyó simultáneamente con agua estéril (10 ml). La mezcla resultante se pasó por un C-18 Sep-Pak para que el producto quedara atrapado y el acetonitrilo residual se lavó con más agua (10 ml). El [18F]2-(2-Fluoro-etoxi)-9H-carbazol se eludió entonces en el vial del producto con etanol de grado USP (0,5 ml) y se diluyó con agua estéril (9,5 ml) para obtener una formulación final (19 - 34 mCi en 10 ml) apta para la inyección (2 % de rendimiento sin corregir, 100 % de pureza radioquímica). La pureza se determinó mediante HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC del correspondiente estándar de referencia no marcado.
Ensayos de Derivados de Carbazol:
A partir del ensayo Biacore, dos derivados de carbazol mostraron afinidades de unión prometedoras a oligómeros/polímeros y fibrillas (Tabla 4). La beta-carbolina Harmol, un miembro de los alcaloides harmala, es el metabolito urinario de la harmina. Los alcaloides de la harmala son inhibidores de la MAO y se encuentran comúnmente en la ruda siria, Peganum harmala, y en la enredadera sudamericana Banisteriopsis caapi, ambas supuestamente con fuertes efectos alucinógenos. Los beta-carbolenos tienen un efecto variado sobre el sistema nervioso central que incluye la unión a los receptores 5-HT2, 5-HT1a, glutamato NMDA e imidazolina; la inhibición de la enzima MAO-A y la interferencia en la transmisión dopaminérgica. Y aunque se cree que las beta-carbolinas son citotóxicas, también mantienen propiedades neuroprotectoras ofreciendo supuestamente neuroprotección contra la dopamina y el glutamato y, además, eliminando las especies reactivas del oxígeno. Un informe reciente demostró que los alcaloides beta-carbolina inducen una facilitación de la memoria a corto y largo plazo en tareas de reconocimiento de objetos en ratones, aunque no está claro el medio por el que los alcaloides ejercen su efecto. Moura, D.J., et al., Effects of b-carboline alkaloids in the object recognition task in mice. Life Sciences, 2006, 79: p. 2099-2104.
El segundo carbazol activo descubierto en el ensayo es el 2-hidroxicarbazol. recientemente se ha demostrado que el 2-hidroxicarbazol libera iones de Ca2+ del músculo esquelético y cardíaco a través de una vía farmacológica distinta. El andamio genérico del carbazol existe en varias terapias, incluyendo el antiinflamatorio no esteroideo carprofeno, el carazolol (un betabloqueante) y el YM-53601 (un inhibidor de la escualeno sintasa). Trabajos recientes han demostrado que los derivados del carbazol pueden actuar como moduladores de la y-secretasa. [Narlawar, R., et al., N-Substituted carbazolyloxyacetic acids modulate Alzheimer associated g-secretas. Bioorganic & Medicinal Chemistry Letters, 2007, 17: p. 176-182] En otro proyecto relacionado con la EA, Howlett descubrió que los carbazoles altamente elaborados, tal como carvedilol, inhiben la formación de fibrillas, aunque no se determinaron las afinidades de unión a las mismas. [Howlett, D.R., et al., Common Structural Features Determine the Effectiveness of Carvedilol, Daunomycin and Rotiletracycline as Inhibitors of Alzheimer b-Amyloid Fibril Formation. Biochemical Journal, 1999, 343: p. 419-423] Curiosamente, un artículo que pretende determinar la viabilidad del uso de carbazoles como inhibidores de fibrillas con base en la permeabilidad celular sugiere que es poco probable que los carbazoles crucen la barrera hematoencefálica, ya que son sustratos de la PGP, lo que impide su uso como terapéutica para la inhibición de fibrillas.
[Saengkhae, C., et al., Ability of Carbazole Salts, Inhibitors of Alzheimer b-Amyloid Fibril Formation, to Cross Cellular Membranes. European Journal of Pharmacology, 2007, 559: p. 124-131]
Utilizando una modalidad de obtención de imágenes apropiada, el patrón de biodistribución de un trazador se vuelve instantáneamente visible y accesible. Por ejemplo, mediante el uso de trazadores marcados con 18F se puede cuantificar fácilmente la captación de un trazador en el cerebro y su eliminación mediante la tomografía por emisión de positrones (PET). Los trazadores con alta captación y lenta eliminación en cerebros normales generan una baja relación señal a ruido. Los trazadores con alta captación y rápido lavado en cerebros normales tienen una alta relación señal/ruido y se consideran ideales. los carbazoles marcados con 18F poseen propiedades ideales para la obtención de imágenes cerebrales. Por ejemplo, se preparó un carbazol marcado con 18Fy se administró a una rata blanca normal Sprague-Dawley (Figura 6). En pocos minutos, el trazador entró en el cerebro y se eliminó durante varios minutos. El carbazol no radiactivo también compite con éxito tanto con la tioflavina T como con el FDDNP en secciones de tejido cerebral, lo que sugiere que el trazador se une a sitios de unión similares (Figuras 4 y 5).
Tabla 4: Resultados del ensayo Biacore con base en carbazoles. Un signo "+" representa un acierto y el aumento de los signos "+" se relaciona con el aumento de la afinidad de unión. El signo "-" representa que no hay vinculación.
Unión a oligómeros/polímeros (Ap1- Unión a fibrillas (Ap1-42) 42)
#54: Harmol
+

En la Tabla 5 se muestra una lista de ejemplos de agentes de obtención de imágenes con base en el carbazol. Muchos de los compuestos están marcados con 18F o 11C. Estos compuestos son sólo de referencia y no forman parte de la invención.
T l : E m l n n i n im n n n r z l

(continuación)

(continuación)

Protocolo detallado del ensayo Biacore:
Agregados solubles de p-Amiloide (Ap42) (oligómeros/polímeros solubles). La biotina-LC-Ap42 se mezcló con AP42 en una relación de 3:2. Tras disolverla en NH4OH al 1% y dH2O, la mezcla (concentración de 40 uM) se incubó en tampón PBS 1X (pH 7,4) a ta durante 6 horas para formar oligómeros/polímeros solubles. El monómero libre de Ap42 en la muestra se eliminó utilizando un tubo de filtro centrífugo Microcon con un corte de 10 KDa de MW. Los oligómeros/polímeros Biotin-LC-Ap42 se inmovilizaron en el chip SA mediante la captura de estreptavidina-biotina.
Agregados insolubles (fibrillas) de p-Amiloide (Ap42). Las fibrillas se prepararon de acuerdo con los procedimientos publicados anteriormente (Agdeppa e D et al. 2001). Brevemente, se disolvieron 0,5 mg de Ap42 (Biotin-LC-Ap42:Ap42 = 1:1) en 1 ml de PBS, pH 7,4, y se mezclaron con una barra de agitación magnética durante 3 d a 37 °C, dando lugar a una solución visiblemente turbia. Las pellas de fibrillas se recogieron por centrifugación. Las fibrillas Biotin-LC-Ap42 se inmovilizaron en el chip SA mediante la captura de estreptavidina-biotina.
Cribado de compuestos de unión a amiloide con Biacore (Análisis de Resonancia de Plasmón de Superficie). Los oligómeros/solubles de Ap42 o las fibrillas se inmovilizaron en la celda de flujo 2 (Fc2) o en la celda de flujo 3 (Fc3) del chip sensor, mientras que Fc1 sirvió de control. Los compuestos de cribado a una concentración de 10 uM fueron volados a través de Fcl, Fc2 y Fc3 durante 2 minutos a un flujo de 30 ul/minuto. A continuación, las células de flujo se lavaron con tampón de funcionamiento (1X PBS) durante 2 minutos y se regeneraron con 50 mM de NaOH durante 30 segundos. La interacción en tiempo real entre el compuesto de cribado y los agregados amiloides inmovilizados en la superficie del chip se registró en el sensorgrama.
Inmunotinción de secciones cerebrales con tioflavina T . Las muestras cerebrales de donantes con enfermedad de Alzheimer fueron infiltradas en cera de parafina después de la fijación. Los bloques de parafina con muestras cerebrales incrustadas se montaron en un microtomo y se seccionaron. A continuación, las secciones se desparafinaron e hidrataron, y se incubaron con o sin AD-CB-001S-WZ01013. La tinción se realizó con 1 uM de tioflavina T. Las imágenes se obtuvieron con un microscopio de fluorescencia.
Figura 4: Inmunotinción de secciones de cerebro con tioflavina T, tioflavina T con trazador y sin tioflavina T.
Inmunotinción de secciones cerebrales con FDDNP. Las muestras de cerebro de donantes con enfermedad de Alzheimer se infiltraron en cera de parafina tras su fijación. Los bloques de parafina con muestras cerebrales incrustadas se montaron en un microtomo y se seccionaron. A continuación, las secciones se desparafinaron e hidrataron, y se incubaron con o sin AD-CB-001S-WZ01013. La tinción se realizó con 1 uM de FDDNP. Las imágenes se obtuvieron con un microscopio de fluorescencia.
Figura 5: Inmunotinción de secciones cerebrales con FDDNP, FDDNP con trazador y sin FDDNP.
Resultados de la obtención de imágenes de AD-CB-001
Una rata blanca Sprague-Dawley fue inyectada por la vena de la cola con -850 uCi AD-CB-001, formulada en 10 % EtOH: agua. Se realizó una exploración dinámica durante 30 minutos en un escáner R4 microPET. Los datos se reconstruyeron utilizando un encuadre de 1 minuto. En pocos minutos, el trazador entró en el cerebro de la rata y se eliminó rápidamente.
Figura 6: Cortes coronales de un cerebro de rata blanca utilizando un encuadre de 1 minuto. Después de 2 minutos, la concentración del trazador alcanza un nivel máximo en el cerebro y se elimina por completo después de 7 minutos.
Preparación de derivados de la cumarina:

A la cumarina (120 mg, 0,5 mmol) en 2 ml de NMP se añadió CS2CO3 (163 mg, 0,5 mmol) y etilendi-tosilato (185 mg, 0,5 mmol). La mezcla se agitó a ta durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 1 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (d Cm 100 % y luego 0,3 % de MeOH en DCM) para obtener el producto deseado (51 mg) como un aceite claro.
1H RMN(400 MHz, CDCk) 8 7,82 (d, J = 8,4 Hz, 2 H), 7,52 (m, 3 H), 7,43 (m, 2 H), 7,37 (m, 3 H), 6,75 (s, 1 H), 671 (d, J = 9,2 Hz, 1 H), 6,23 (s, 1 H), 4,41 (t, J = 4,4 Hz, 2 H), 4,22 (t, J = 4,4 Hz, 2 H), 2,46 (s, 3 H); MS(ESI) m/z 437 (M+H+).
Síntesis de AD-C-003S-WZ01041

A la cumarina (238 mg, 1 mmol) en 4 ml de NMP se añadió Cs2CO3 (326 mg, 1 mmol) y bromofluoroetano (152 mg, 1,2 mmol). La mezcla se agitó a ta durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 1 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (DCM 80 % en hexano al 100 %) para obtener el producto deseado (160 mg) como sólido blanco.
1H RMN (400 MHz, ace tona^) 8 7,60-7,56 (m, 5 H), 7,43 (d, J = 8 , 8 Hz, 1 H), 7,03 (d, J = 2,4 Hz, 1 H), 6,96 (dd, J = 8 ,8 , 2.4 Hz, 1 H), 6,18 (s, 1 H), 4,91-4,89 (m, 1 H), 4,79-4,77 (m, 1 H), 4,49-4,47 (m, 1 H), 4,42-4,40 (m, 1 H); MS(ESI) m/z 285 (M+H+).
Síntesis de AD-C-003S-WZ01041 marcado con 18F

El [18F] fluoruro (600 - 900 mCi) como solución enriquecida en H218O se entrega al módulo de síntesis. El [18F]fluoruro se atrapa en una columna de intercambio iónico y se elude en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añade el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evapora hasta sequedad. El precursor (4 mg en 0,9 ml de MeCN / 0,1 ml de DMSO) se añade al reactor y la reacción de fluoración se calienta a 115 °C durante 10 min. La mezcla de reacción cruda se purifica por HPLC semipreparativa (Columna: Phenomenex Luna C-18, 250 mm x 10 mm; Gradiente de la fase móvil 95:5 H20 (+0,05 % TFA) : MeCN (+0,05 % TFA) a 100 % MeCN (+0,05 % TFA); Caudal: 5 ml/min; tiempo = 25 min).
Se recoge el pico correspondiente al producto y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak para que el producto quede atrapado y el acetonitrilo residual se lava con más agua (10 ml). A continuación, el producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final adecuada para la inyección.
La pureza se determina por HPLC analítica equipada con un detector de radiactividad y la identidad se confirma por comparación con los datos de HPLC para el correspondiente estándar de referencia no etiquetado.
Síntesis de AD-C-002P-WZ01029

A la dihidroxicumarina (254 mg, 1 mmol) en 4 ml de NMP se añadió Cs2CO3 (326 mg, 1 mmol) y tosilato de etilo (241 mg, 1 mmol). La mezcla se agitó ata durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 1 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (DCM en hexano del 80 % al 100 %) para obtener el producto monoalquilado deseado (72 mg) como un sólido blanquecino.
1H RMN (400 MHz, CDCfe) 87,53-7,50 (m, 3 H), 7,45-7,43 (m, 2 H), 7,00 (d, J = 8,8 Hz, 1 H), 6,82 (d, J = 8.8 Hz, 1 H), 6,26 (s, 1 H), 5,92 (s, 1 H), 4,31 (t, J = 5,0 Hz, 2 H), 3,72 (t, J = 5,0 Hz, 2 H); MS(ESI) m/z 324 (M+H+).
Síntesis de AD-C-002S-WZ01035

A la azidoetil cumarina (42 mg, 0,13 mmol) en 1 ml de metanol se añadió CuSO4 (21 mg, 0,13 mmol), ascorbato de sodio (28 mg, 0,13 mmol) y fluoropentina (16,3 mg, 0,19 mmol). La mezcla de reacción se agitó enérgicamente durante 1 h y se diluyó con EtOAc (30 ml). Se lavó con agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (eludido con EtOAc al 5 % en hexano al 60 %) para obtener el compuesto deseado (42 mg).
1H RMN (400 MHz, CDCfe/CDaOD) 87,95 (s, 1H), 7,54-7,52 (m, 3 H), 7,48-7,44 (m, 2 H), 6,94 (d, J = 8,8 Hz, 1 H), 6,86 (d, J = 8,8 Hz, 1 H), 4,86 (t, J = 4,8 Hz, 2 H), 4,55-4,51 (m, 3 H), 4,41 (t, J = 6,0 Hz, 1 H), 2,83 (t, J = 7,2 Hz, 2 H), 2,14- 2,02 (m, 2 H); MS(ESI) m/z 410 (M+H+).
n es s e - - - - marca o con
Preparación de [18F] 5-Fluoro-pent-1-ina
i) K ,8F, Kryptofix-2.2.2

El [18F]Fluoruro (600 - 900 mCi) como solución enriquecida en H218O fue entregado al módulo de síntesis. El [18F]Fluoruro se atrapó en una columna de intercambio iónico y se eludió en el recipiente de reacción utilizando carbonato potásico acuoso (3,0 mg en 0,4 ml de H2O). Se añadió el reactivo de transferencia de fase Kryptofix-2.2.2 (20,0 mg en 1,0 ml de MeCN) y el azeótropo agua-acetonitrilo se evaporó hasta sequedad. Se añadió al reactor el éster pent-4-quinilo del ácido tolueno (20 mg en 0,8 ml de MeCN) y luego se calentó la reacción de fluoración a 110 °C durante 5 min. Tras la fluoración, la mezcla de reacción cruda se purificó por destilación para obtener[18F]55-fluoropent-1-ino como solución en acetonitrilo (atrapada a -78 °C debido a la volatilidad del producto).
Preparación del triazol:

Se añadió una mezcla de precursor de azida (5 mg), ascorbato de sodio (40 mg), fr/s-(benciltriazolilmetil) amina (TBTA, 25 mg) y solución acuosa de sulfato de cobre (0,1 M, 0,25 ml) en DMF (0,4 ml) y agua (0,1 ml) a la solución de pentina enfriada descrita anteriormente. La mezcla de reacción se calentó hastata y se agitó durante 30 minutos. Transcurrido este tiempo, la reacción se purificó por HPLC semipreparativa. Se recogió el pico correspondiente al producto y se diluyó simultáneamente con agua estéril (10 ml). La mezcla resultante se pasó por un C-18 Sep-Pak para que el producto quedara atrapado y el acetonitrilo residual se lavó con más agua (10 ml). A continuación, el producto se eludió en el vial del producto con etanol de grado USP (0,5 ml) y se diluyó con agua estéril (9,5 ml) para obtener una formulación final (19 mCi en 10 ml) apta para la inyección (rendimiento corregido por decaimiento del 10 %, pureza radioquímica del 100%).
La pureza se determinó por HPLC analítica equipada con un detector de radiactividad y la identidad se confirmó por comparación con los datos de HPLC para el correspondiente estándar de referencia no marcado.
Síntesis de AD-CB-002P-WZ01031

Al hidroxicarbazol (92 mg, 0,5 mmol) en 2 ml de NMP se añadió Cs2CO (163 mg, 0,5 mmol) y tosilato de etilo (121 mg, 0,5 mmol). La mezcla se agitó a ta durante 15 h y se diluyó con Et2O (50 ml). Se lavó con HCl 0,5 M (50 ml) y agua (2x50 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (80 % DCM en hexano a 100 % DCM) para obtener el producto deseado (76 mg) como sólido blanco.
1H RMN (400 MHz, CDCl3/acetona-da) ó 9,98 (s, 1 H), 7,95 (m, 2 H), 7,41 (d, J = 8,4 Hz, 1 H), 7,29 (t, J = 8,0 Hz, 1 H), 7,14 (t, J = 8.0 Hz, 1 H); 7,01 (s, 1 H), 6,84 (d, J = 8,4 Hz, 1 H), 4,28 (t, J = 4,8 Hz, 2 H), 3,67 (t, J = 4,8 Hz, 2 H)
MS(ESI) m/z 253 (M+H+).
Síntesis de AD-CB-002S-WZ01033

Al etilazido carbazol (32 mg, 0,127 mmol) en 0,5 ml de DMF se añadió CuI (7,6 mg, 0,04 mmol), DIPEA (16,4 mg, 0,127 mmol) y fluoropentina (16,4 mg, 0,19 mmol). La mezcla de reacción se agitó enérgicamente durante 1 h y se diluyó con EtÜAc (30 ml). Se lavó con agua (50 ml), HCl 0,5 M (30 ml), agua (2x50 ml), se secó sobre MgSÜ4 y se concentró. El producto crudo se preabsorbió en sílice (3 g) y se cargó en una columna de sílice de 4 g y se eludió con EtOAc al 30 % en hexano al 50 % para obtener el compuesto deseado (20 mg).
1H RMN (400 MHz, CDCb/CDsOD) 57,95 (d, J = 7,6 Hz, 1 H), 7,91 (d, J = 8,4 Hz, 1 H), 7,76 (s, 1 H), 7,40 (d, J = 8,0 Hz, 1 H), 7,31 (t, J = 7,6 Hz, 1 H), 7,14 (t, J = 7,6 Hz, 1 H); 6.94 (d, J = 2,4 Hz, 1 H), 6,78 (dd, J = 8,8, 2,4 Hz, 1 H), 4,83-4,78 (m, 2 H), 4,53-4,48 (m, 3 H), 4,40 (t, J = 6,0 Hz, 1 H), 2,85 (t, J = 7,6 Hz, 2 H), 2,10-1,99 (m, 2 H); MS(ESI) m/z 339 (M+H+).
Síntesis de AD-CB-002S-WZ01033 marcado con 18F
Preparación del triazol

Se añade una mezcla de precursor de azida (5 mg), ascorbato de sodio (40 mg), f/7s-(benciltriazolilmetil) amina (TBTA, 25 mg) y solución acuosa de sulfato de cobre (0,1 M, 0,25 ml) en DMF (0,4 ml) y agua (0,1 ml) a la solución de pentina enfriada descrita anteriormente. La mezcla de reacción se calienta hasta ta y se agita durante 30 min. Transcurrido este tiempo, la reacción se purifica por HPLC semipreparativa. Se recoge el pico correspondiente al producto y se diluye simultáneamente con agua estéril (10 ml). La mezcla resultante se pasa por un C-18 Sep-Pak y el acetonitrilo residual se lava con agua adicional (10 ml). El producto se elude en el vial del producto con etanol de grado USP (0,5 ml) y se diluye con agua estéril (9,5 ml) proporcionando una formulación final adecuada para la inyección.
La pureza se determina por HPLC analítica equipada con un detector de radiactividad y la identidad se confirma por comparación con los datos de HPLC para el correspondiente estándar de referencia no etiquetado.
Síntesis de AD-C-WZ01011

A la dihidroxicumarina (100 mg, 0,4 mmol) en 2,5 ml de NMP se añadió CS2CO3 (130 mg, 0,4 mmol) y bromofluoroetano (46 mg, 0,36 mmol). La mezcla se agitó ata durante 18 h y se diluyó con Et2O (50 ml). Se lavó con HCl 1 M (50 ml) y agua (2x50 ml) y se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (MeOH en DCM de 0% a 1%) para obtener el producto monoalquilado deseado (25 mg) como sólido blanco.
1H RMN (400 MHz, CDCl3/CD3OD) 87,55-7,48 (m, 5 H), 6,96 (q, J = 7,6 Hz, 2 H), 6,19 (s, 1 H), 4,86 (m, 1 H), 4,75 (m, 1 H), 4,43 (m, 1 H), 4,37 (m, 1 H); MS(ESI) m/z 301 (M+H+).
Procedimiento general para la protección del carbazol N-Boc:
En un matraz de fondo redondo equipado con una barra de agitación magnética, un septo de goma y una entrada de argón que contenía THF (40 vol) se colocó carbazol (1,0 equiv). A esta solución se añadió NaH (60 % de dispersión en aceite, 3 equiv) a 0 °C y la reacción se dejó agitar a 0 °C durante 30 min. A esta reacción se añadió (Boc)2O (1,2 equiv) a 0 °C y la reacción se dejó agitar durante 1 h. Una vez completada la reacción por LCMS, se vertió en agua (25 vol) y se extrajo en EtOAc (3 * 20 vol). Los extractos orgánicos combinados se lavaron con agua (2 * 25 vol), se secaron (Na2SO4) y se concentraron in vacuo. El residuo se purificó sobre gel de sílice utilizando Hexanos: EtOAc como eluyente para obtener el producto final.
Procedimiento general para la N-metilación de carbazoles:
En un matraz de fondo redondo equipado con una barra de agitación magnética, un septo de goma y una entrada de argón que contenía THF (50 vol) se colocó carbazol (1,0 equiv). A esta solución se añadió NaH (60 % de dispersión en aceite, 3 equiv) a 0 °C y la reacción se dejó agitar a 0 °C durante 30 min. A esta reacción se añadió MeOTf (1,0 equiv) a 0 °C y la reacción se dejó agitar durante 1 h. Una vez completada la reacción por LCMS, se vertió en agua (25 vol) y se extrajo en EtOAc (3 * 20 vol). Los extractos orgánicos combinados se lavaron con agua (2 * 25 vol), se secaron (Na2SO4) y se concentraron in vacuo. El residuo se purificó sobre gel de sílice utilizando Hexanos: EtOAc como eluyente para obtener el producto final.
Procedimiento experimental general para la alquilación fenólica:
En un matraz de fondo redondo equipado con una barra de agitación magnética que contenía DMF (20 vol) se colocó fenol (1 equiv). A esta solución se añadió el agente alquilante (1,0 equiv), Cs2CO3 (1,2 equiv) y se dejó que la reacción se agitara a 60 °C durante 16 h. A continuación, la reacción se vertió en agua (25 vol) y se extrajo en EtOAc (3 * 20 vol). Los extractos orgánicos combinados se lavaron con agua (2 * 25 vol), se secaron (Na?SO4) y se concentraron in vacuo. El residuo se purificó sobre gel de sílice utilizando Hexanos: EtOAc como eluyente para obtener el producto final.
Procedimiento experimental general para la reacción de acoplamiento de Suzuki:
En un matraz de fondo redondo equipado con una barra de agitación magnética y con una entrada de argón que contenía tolueno:H2O (1:1, 40 vol) se colocó el compuesto de cloro (1 equiv). A esta solución se añadió ácido borónico (1,5 equiv), Pd(PPh3)4 (0,02 equiv), K2CO3 y se dejó agitar la reacción a 110 °C durante 16 h. A continuación, se vertió en agua (25 vol) y se extrajo en EtOAc (3 * 20 vol). Los extractos orgánicos combinados se lavaron con agua (2 * 25 vol), se secaron (Na2SO4) y se concentraron in vacuo. El residuo se purificó sobre gel de sílice utilizando Hexanos: EtOAc como eluyente para obtener el producto final.
Procedimiento experimental general para la formación de carbazoles utilizando P(OEt)3:
En un matraz de fondo redondo equipado con una barra de agitación magnética que contenía P(OEt¡3 (25 vol) se colocó biarilo (1 equiv). La reacción se dejó agitar a 150 °C durante 16 h. Una vez completada la reacción, el P(OEt)3
se eliminó in vacuo. El residuo se purificó sobre gel de sílice utilizando Hexanos: EtOAc como eluyente para obtener el compuesto final.
Síntesis del precursor del CBI-nosilato:

En un matraz de fondo redondo de 50 ml equipado con una barra de agitación magnética que contenía DCM (10 ml) se colocó 1,2-etanodiol (0,25 g, 4,0 mmol). A esta solución se añadió cloruro de nosilo (1,9 g, 8,5 mmol) y Et3N (0,90 g, 8,9 mmol) a 0 °C y se dejó que la reacción se agitara a temperatura ambiente durante 16 h. Una vez completada la reacción, el sólido blanco se filtró, se lavó con DCM (100 ml) y se secó in vacuo para obtener DHK-4-14 (1,3 g, 75 %) como sólido incoloro. MS: [M+Na]+: 455,0
Preparación del 2-(9H-carbazol-2-iloxi)etil 2-nitrobencenosulfonato (DHK-4-15):
En un matraz de fondo redondo de 25 ml equipado con una barra de agitación magnética que contenía DMF (5 vol) se colocó carbazol (0,2 g, 1,1 mmol). A esta solución se añadió el DHK-4-14 (0,52 g, 1,2 mmol), Cs2CO3 (0,43 g, 1,3 mmol) y se dejó que la reacción se agitara a temperatura ambiente durante 16 h. A continuación, la reacción se vertió en agua (25 ml) y se extrajo con EtOAc (4 * 50 ml). Los extractos orgánicos combinados se secaron (Na2SO4) y se concentraron in vacuo. El residuo crudo se purificó por cromatografía flash utilizando Hexanos: EtOAc (50:50) en un sistema de purificación Combiflash para obtener DHK-4-15 como un sólido blanco (0,28 g, 62 %). MS: [M+Nâ : 435,0 Síntesis de CB-5:

Preparación de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol-9-carboxilato de tert-butilo : CB-5: DHK-4-27 Se siguió el procedimiento experimental general para la protección N-Boc del carbazol. La reacción se realizó en una escala de 0,03 g. El producto se eludió en una mezcla de 30-35 % de EtOAc: Hexanos en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,03 g (74 %) de CB-5 como un aceite incoloro. MS: [M+H]+: 418,0
Síntesis de CB-6: DHK-4-28

Preparación de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9-metil-9H-carbazol: CB-6
Se siguió el procedimiento experimental general para la N-metilación de carbazoles. La reacción se realizó en una escala de 0,05 g. El producto se eludió en una mezcla de 40-45 % de EtOAc: Hexanos en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,04 g (78 %) de CB-6 como un sólido blanco. MS: [M+H]+:
332,1.
Síntesis del precursor CB-3 protegido por N-Boc:

Preparación del 2-(2-(2-(tosiloxi)etoxi)etoxi)etoxi)-9H-carbazol-9-carboxilato de tert-butilo: DHK-4-32
Se siguió el procedimiento experimental general para la protección N-Boc del carbazol. La reacción se realizó en una escala de 0,07 g. El producto se eludió en una mezcla de 40 % EtOAc: Hexanos en una elución de gradiente en un sistema de purificación Combiflash. Se aislaron 0,07 g (82 %) de DHK-4-32 como sólido blanco. MS: [M+Na]+: 592.
Síntesis del precursor N-metil CB-3:

Preparación de 2-(2-(9-metil-9H-carbazol-2-iloxi)etoxi)etoxi)etil 4-metilbencenosulfonato: DHK-4-30
Se siguió el procedimiento experimental general para la N-metilación de carbazoles. La reacción se realizó en una escala de 0,075 g. El producto se eludió en una mezcla de 40 % EtOAc:Hexanos en una elución de gradiente en un sistema de purificación Combiflash. Se aislaron 0,07 g (91 % )de DHK-4-30 como un sólido blanco. MS: [M+H]+: 484,2 Síntesis de CB-7 Std:

Preparación de 1-cloro-4-(2-(2-fluoroetoxi)etoxi)etoxi)-2-mtrobenceno: DHK-4-51
Se siguió el procedimiento experimental general para la alquilación fenólica. La reacción se realizó en una escala de 0,25 g. El producto se eludió en una mezcla de 20-30 % de EtOAc:Hexanos en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,44 g (99 %) de DHK-4-51 como aceite amarillo. MS: [M+H]+: 308,0.
Preparación de 4'-(2-(2-fluoroetoxi)etoxi)etoxi)-N,N-dimetil-2'-nitrobifenil-4-amina: DHK-4-26
Se siguió el procedimiento experimental general para la reacción de acoplamiento de Suzuki. La reacción se realizó en una escala de 0,11 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 50-60 % en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,06 g (43 %) de DHK-4-26 como aceite amarillo. MS:
[M+H]+: 393,1
Preparación de 7-(2-(2-fluoroetoxi)etoxi)ethoxy)-N,N-dimetil-9H-carbazol-2-amina: DHK-4-29: CB-7
Se siguió el procedimiento experimental general para la formación de carbazoles utilizando P(OEt)3. La reacción se realizó en una escala de 0,06 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 70-80% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,03 g (49 %) de DHK-4-29 CB-7 como sólido blanco. MS: [M+H]+: 361,1.
Síntesis de CB-9 Std:

Preparación de 1-cloro-4-(2-(2-fluoroetoxi)etoxi)etoxi)-2-nitrobenceno: DHK-4-51
Se siguió el procedimiento experimental general para la alquilación fenólica. La reacción se realizó en una escala de 0,25 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 20-30% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,44 g (99 %) de DHK-4-51 como aceite amarillo. MS: [M+H]+: 308,0.
Preparación de N-(4'-(2-(2-(2-fluoroetoxi)etoxi)etoxi)-2'-nitrobifenM-4-N)acetamida: DHK-4-31
Se siguió el procedimiento experimental general para la reacción de acoplamiento de Suzuki. La reacción se realizó en una escala de 0,11 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 80-90% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,14 g (100 %) de DHK-4-31 como aceite amarillo. MS: [M+H]+: 407,0.
Preparación de N-(7-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol-2-il)acetamida: DHK-4-33: CB-9
Se siguió el procedimiento experimental general para la formación de carbazoles utilizando P(OEt)3. La reacción se realizó en una escala de 0,15 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 90% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,03 g (49 %) de CB-9 como sólido blanco. MS:
[M+H]+: 375,1.
Síntesis de CB-28 Std:

Se siguió el procedimiento experimental general para la alquilación fenólica. La reacción se realizó en una escala de 0,25 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 20-30% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,44 g (99 %) de DHK-4-51 como aceite amarillo. MS: [M+H]+: 308,0.
Preparación de 3-(4-(2-(2-fluoroetoxi)etoxi)etoxi)-2-nitrofenil)piridina: DHK-4-56
Se siguió el procedimiento experimental general para la reacción de acoplamiento de Suzuki. La reacción se realizó en una escala de 0,095 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 40-50% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,01 g (9%) de DHK-4-56 como aceite amarillo. MS:
[M+H]+: 351,1.
Preparación de 7-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-pirido[2,3-b]indol DHK-4-58: CB-28
Se siguió el procedimiento experimental general para la formación de carbazoles utilizando P(OEt)3. La reacción se realizó en una escala de 0,01 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 50 % en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,002 g (22 %) de CB-28 como sólido blanco. MS:
[M+H]+: 319
Síntesis del precursor CB-7

DHK-4-71
Preparación de 4-(benciloxi)-1-cloro-2-nitrobenceno: DHK-4-63
Se siguió el procedimiento experimental general para la alquilación fenólica. La reacción se realizó en una escala de 1 g. Se utilizó K2CO3 como base y acetona como disolvente. El tiempo de reacción fue de 4 h. El producto se eludió en una mezcla de EtOAc:Hexanos al 20-30 % en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 1,45 g (95 %) de DHK-4-63 como sólido cristalino blanco. MS: [M+H]+: 264,0
Preparación de 34'-(benciloxi)-N,N-dimetil-2'-nitrobifenil-4-amina: DHK-4-66
Se siguió el procedimiento experimental general para la reacción de acoplamiento de Suzuki. La reacción se realizó en una escala de 0,47 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 20-30% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,21 g (34 %) de DHK-4-66 como sólido naranja. MS:
[M+H]+: 349,1
Preparación de la 7-(benciloxi)-N,N-dimetil-9H-carbazol-2-amina DHK-4-68:
Se siguió el procedimiento experimental general para la formación de carbazoles utilizando P(OEt)3. La reacción se realizó en una escala de 0,21 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 20-30% en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,13 g ( 6 8 %) de DHK-4-68 como sólido blanco. MS:
[M+H]+: 317,1
Preparación de 2-(benciloxi)-7-(dimetilamino)-9H-carbazol-9-carboxilato de tert-butilo: DHK-4-69
Se siguió el procedimiento experimental general para la protección N-Boc del carbazol. La reacción se realizó en una escala de 0,13 g. La reacción se llevó a cabo a temperatura ambiente durante 16 h. El producto se eludió en una mezcla de EtOAc:Hexanos al 10 % en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,12 g (70 %) de DHK-4-69 como sólido blanco. MS: [M+H]+: 417,2
Preparación del 2-(dimetilamino)-7-hidroxi-9H-carbazol-9-carboxilato de tert-butilo: DHK-4-71 En un matraz de fondo redondo de 50 ml equipado con una barra de agitación magnética que contenía EtOAc (50 ml) se colocó DHK-4-69 (0,11 g, 0,19 mmol). A esta solución se añadió Pd/C (10 %, 20 mg) y la reacción se dejó agitar bajo H2 (1 atm) a ta durante 16 h. Una vez completada la reacción, la mezcla de reacción se filtró a través de celita y los volátiles se eliminaron in vacuo para obtener DHK-4-71 (0,09 g, 100 %) como sólido blanco.
Preparación de 2-(dimetilamino)-7-(2-(2-(tosiloxi)etoxi)etoxi)etoxi)-9H-carbazol-9-carboxilato de tert-butilo: DHK-4-72: Precursor del CB-7:
Se siguió el procedimiento experimental general para la alquilación fenólica. La reacción se realizó en una escala de 0,09 g. El producto se eludió en una mezcla de EtOAc:Hexanos al 45 % en una elución en gradiente en un sistema de purificación Combiflash. Se aislaron 0,07 g (41 %) del precursor CB-7 como sólido blanco. MS: [M+H]+: 613,2. Síntesis de AD-CB-003S-WZ0129

Al 2,2'-(etano-1,2-diilbis(oxi))bis(etano-2,1 -diil) bis(4-metilbencenosulfonato) (8,7 g, 19 mmol) se añadió TBAF (22,8 ml, solución 1,0 M de t Hf , 22,8 mmol). La mezcla se calentó a reflujo durante 1 h bajo atmósfera de Ar y se enfrió a ta y se concentró a presión reducida. El material crudo se purificó con cromatografía de sílice (5 % a 40 % de THF en hexano) para obtener 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato como un aceite claro (2,5 g, 43 % ).1H RMN(400 MHz, CDCls) 57,80 (d, J = 8,4 Hz, 2 H), 7,34 (d, J = 8,4 Hz, 2 H), 4,61 (m, 1 H), 4,49 (m, 1 H), 4,16 (m, 2 H), 3,75 (m, 1 H), 3,71-3,67 (m, 3 H), 3,62 (m, 4 H); MS(ESI) m/z 307 (M+H+).
Al 2-hidroxicarbazol (45 mg, 0,25 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (82 mg, 0,27 mmol) en 0,5 ml de NMP se añadió Cs2CO33 (82 mg, 0,25 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con Et2O (50 ml). Se lavó con agua (3x 50 ml) y se secó sobre MgSO4. El disolvente se eliminó a presión reducida y el producto bruto se purificó con cromatografía de sílice (5 % a 50 % de EtOAc en hexano) para obtener el producto deseado como sólido blanco (37 mg, 47 %). 1H RMN (400 MHz, CDCls) 58,02 (s, 1H), 7,96 (d, J = 7,2 Hz, 1H), 7,91 (d, J = 8,4 Hz, 1H), 7,38-7,33 (m, 2 H), 7,20 (m, 1 H), 6,91 (d, J = 2,4 Hz, 1 H), 6.86 (dd, J = 8,8, 2,4 Hz, 1 H), 4,63 (m, 1 H), 4,51 (m, 1 H), 4,21 (m, 2 H), 3,90 (m, 2 H), 3,80-3,76 (m, 3 H), 3,74-3,71 (m, 3 H); MS(ESI) m/z 318 (M+H+).
Síntesis de AD-CB-003P-WZ0141

Al 2-hidroxicarbazol (183 mg, 1 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (687 mg, 1,5 mmol) en 5 ml de NMP se añadió Cs2CO3(326 mg, 1 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con Et2O (100 ml). Se lavó con agua (3x 100 ml) y se secó sobre MgSO4. El disolvente se eliminó a presión reducida y el producto bruto se purificó con cromatografía de sílice (5 % a 60 % de EtOAc en hexano) para obtener el producto deseado como sólido blanco (165mg, 35 %). 1H RMN (400 MHz, CDCh) 58,21 (s, 1H), 7,95 (d, J = 7,2 Hz, 1H), 7,91 (d, J = 8,4 Hz, 1H), 7,77-7,75 (m, 2 H), 7,37-7,30 (m, 2 H), 7,28 (s, 1 H), 7,25 (m, 1 H), (td, J = 7,6, 1,2 Hz, 1 H), 6,92 (d, J = 2,4 Hz, 1 H), 6,83 (dd, J = 8,8, 2,4 Hz, 1 H), 4,15 (m, 4 H), 3,84 (m, 2 H), 3,69-3,65 (m, 4 H), 3,62-3,59 (m, 2 H), 2,38 (s, 3 H); MS(ESI) m/z 470 (M+H+), 492 (M+Na+).
AD-CB-004S-WZ01165

Al 4-cloro-3-nitrofenol (1,74 g, 10 mmol) y al bromuro de bencilo (2,05 g, 12 mmol) en 25 ml de acetona se añadió K2CO3 (2,76 g, 20 mmol). La mezcla se calentó a 60 °C durante 4 h bajo atmósfera de Ar y se enfrió ta. Se filtró y el sólido se lavó con éter (80 ml) y el filtrado combinado se concentró y se cromatografió (EtOAc en hexano, gradiente del 3% al 30%) para obtener 4-(benciloxi)-1-cloro-2-nitrobenceno como un sólido amarillo claro (2,5 g, 95 %).1H RMN (400 MHz, CDCk) 8 7,46 (d, J = 2,8 Hz, 1H), 7,42-7,34 (m, 5 H), 7,11 (dd, J = 8 ,8 , 2,8 Hz, 1 H), 5,08 (s, 2 H); MS(ESI) m/z 264 (M+H+).
Al 4-(benciloxi)-1-cloro-2-nitrobenceno (526 mg, 2 mmol) y al 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenilcarbamato de tert-butilo(670 mg, 2.1 mmol) en 12 ml de dioxano se añadieron 4 ml de una solución de Na2CO3 (aq) 1 M y Tetrakis(trifenilfosfina)paladio (69 mg, 0,06 mmol). La suspensión se calentó a reflujo durante 15 horas bajo atmósfera de Ar y se enfrió a ta. Se añadió EtOAc (100 ml) y se lavó con salmuera (80 ml), agua (80 ml) y se secó sobre MgSO4. Tras eliminar el disolvente, el residuo se cromatografió (hexano/EtOAc) para obtener 4-(benciloxi)-2'-nitrobifenil-4-ilcarbamato de tert-butilo como un sólido amarillo (740 mg, 8 8 %). 1H RMN(400 MHz, CDCk) 8 7,44-7,34 (m, 8 H), (d, J = 8,4 Hz, 1 H), 7,20-7,16 (m, 3 H), 6,50 (s, 1 H), 5,12 (s, 2 H), 1,51 (s, 9 H); MS(ESI) m/z 443 (M+Na+).
Una suspensión de 4'-(benciloxi)-2'-nitrobifenil-4-ilcarbamato de tert-butilo (740 mg, 1,67 mmol) en 2 ml de fosfito de tritilo se calentó a 145 °C durante 15 h bajo atmósfera de Ar y se enfrió ata. Se añadió 10 ml de hexano y se dejó reposar durante 10 min. El sólido se recogió por filtración y se lavó con éter/hexano (v:v 1/1, 10 ml) y se secó bajo alto vacío para obtener 7-(benciloxi)-9H-carbazol-2-ilcarbamato de tert-butilo como un sólido blanquecino (480 mg, 74 %). 1H RMN (400 MHz, CDQ3) 8 7,89 (s, 1H), 7,83-7,78 (m, 3 H), 7,46 (d, J = 7,2 Hz, 2 H), 7,38 (m, 2 H), 7,32 (d, J = 7,2 Hz, 1 H), 6 , 94 (d, J = 2,0 Hz, 1 H), 6 , 8 8 (dd, J = 8 ,8 , 2,4 Hz, 1 H), 6,83 (dd, J = 8,4, 2,0 Hz, 1 H), 6,60 (s, 1 H), 5,15 (s, 2 H), 1,52 (s, 9 H); MS(ESI) m/z 389 (M+H+).
Al 7-(benciloxi)-9H-carbazol-2-ilcarbamato de tert-butilo (220 mg, 0,56 mmol) en 50 ml de MeOH se añadió paladio sobre carbón activado (80 mg). La mezcla se agitó ata bajo atmósfera de H2 durante 3h. El sólido se filtró y el filtrado se concentró para obtener 7-hidroxi-9H-carbazol-2-ilcarbamato de tert-butilo como sólido marrón (165 mg, 100 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 619 (2M+Na+).
Al 7-hidroxi-9H-carbazol-2-ilcarbamato de tert-butilo (165 mg, 0,55 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (202 mg, 0,66 mmol) en 2 ml de NMP se añadió Cs2CO3 (179 mg, 0,55 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con EtOAc (50 ml). Se lavó con agua (3x50 ml) y se secó sobre MgSO4. Tras eliminar el disolvente, el residuo se cromatografió (hexano/EtOAc) para obtener 7-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol-2-ilcarbamato de tert-butilo como sólido blanco (130 mg, 55 %). 1H RMN (400 MHz, CDCl3) 8 7,94 (s, 1H), 7,83-7,79 (m, 3 H), 6,91 (d, J = 2,0 Hz, 1 H), 6 , 8 6 (dd, J = 8,4, 2,0 Hz, 1 H), 6,83 (dd, J =
8,8, 2.4 Hz, 1 H), 6,63 (s, 1 H), 4,64 (m, 1 H), 4,51 (m, 1 H), 4,21 (m, 2 H), 3,91 (m, 2 H), 3,81-3,71 (m, 6 H), 1,55 (s, 9 H); MS(ESI) m/z 433 (M+H+).
Al 7-(2-(2-(fluoroetoxi)etoxi)etoxi)etoxi)-9H-carbazol-2-ilcarbamato de tert-butilo (130 mg, 0,3 mmol) se añadieron 10 ml de una solución de HCl 4 M en dioxano. La mezcla se agitó ata durante 5 h y se concentró a presión reducida. El residuo se lavó con éter (15 ml) y se suspendió en EtOAc (50 ml). A esta suspensión se añadieron 10 ll de NaHCO3 (sat.) y la mezcla se agitó durante 5 min. La capa orgánica se secó sobre MgSO4 y se concentró para obtener 7-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol-2-amina como un sólido marrón (95 mg, 95 %). MS(ESI) m/z 333 (M+H+).
Una mezcla de 7-(2-(2-(fluoroetoxi)etoxi)etoxi)-9H-carbazol-2-amina (95 mg, 0,28 mmol), paraformaldehído (43 mg, 1,43 mmol) y NaOMe (492 mg, solución de MeOH al 25 %, 2,3 mmol) en 8 ml de MeOH se calentó a reflujo durante 1,5 h bajo atmósfera de Ar y se enfrió ata. A esta mezcla se añadió NaBH4 (54 mg, 1,43 mmol) y la mezcla se calentó a reflujo durante 2 h. Tras enfriar a ta, la mezcla se apagó en hielo. Se extrajo con éter (3x30 ml) y la fase orgánica combinada se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 7-(2-(2-(2-fluoroetoxi)etoxi)etoxi)-N-metil-9H-carbazol-2-amina (AD-CB-003P-WZ0141) como un sólido marrón claro (55 mg, 56 %). 1H RMN (400 MHz, CDCh) 87,76 (s, 1 H), 7,77 (t, J = 8,8 Hz, 2 H), 6,78 (dd, J = 8,0, 2,0 Hz, 1 H), 6,77 (s, 1 H), 6,53 (dd, J = 8,4, 2,0 Hz, 1 H), 6,46 (s, 1 H), 4,62 (9m, 1 H), 4,50 (m, 1 H), 4,13 (t, J = 5,2 Hz, 2 H), 3,85 (t, J = 5,2 Hz, 2 H), 3,83 (s, 1 H), 3,79-3,67 (m, 6 H), 2,87 (s, 3 H); MS(ESI) m/z 347 (M+H+).
AD-CB-004Pa-WZ01179

Al 7-(benciloxi)-9H-carbazol-2-ilcarbamato de tert-butilo (200 mg, 0,51 mmol) se añadieron 10 ml de una solución de HCl 4 M en dioxano. La mezcla se agitó ata durante 4 h y se concentró a presión reducida. El residuo se lavó con éter (15 ml) y se suspendió en EtOAc (50 ml). A esta suspensión se añadieron 10 ml de NaHCO3 (sat.) y la mezcla se agitó durante 5 min. La capa orgánica se secó sobre MgSO4 y se concentró para obtener la 7-(benciloxi)-9H-carbazol-2-amina como un sólido marrón (150 mg, 100 %). 1H RMN (400 MHz, DMSO-d6) 811,33 (s, 1 H), 7,99 (d, J = 8,4 Hz, 1 H), 7,92 (d, J = 8,8 Hz, 1 H), 7,42 (d, J = 6,8 Hz, 2 H), 7,34-7,21 (m, 3 H), 7,27-7,23 (m, 1 H), 7,00-6,97 (m, 2 H), 6,81 (dd, J = 8,8, 2m4 Hz, 1 H), 5,12 (s, 2 H); MS(ESI) m/z 289 (M+H+).
Una mezcla de 7-(benciloxi)-9H-carbazol-2-amina (150 mg, 0,52 mmol), paraformaldehído (78 mg, 2,6 mmol) y NaOMe (900 mg, solución de MeOH al 25 %, 4,16 mmol) en 15 ml de MeOH se calentó a reflujo durante 2 h bajo atmósfera de Ar y se enfrió a ta. A esta mezcla se añadió NaBH4 (98 mg, 2,6 mmol) y la mezcla se calentó a reflujo durante 2 h. Tras enfriar a ta, la mezcla se apagó en hielo (30 g). Se extrajo con EtOAc (3x50 ml) y la fase orgánica combinada se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 7-(benciloxi)-N-metil-9H-carbazol-2-amina como un sólido marrón claro (130 mg, 82 %). 1H RMN (400 MHz, acetona-d6) 8 9,78 (s, 1 H), 7,72 (d, J = 8,4 Hz, 1 H), 7,66 (d, J = 8,8 Hz, 1 H), 7,49 (d, J = 7,2 Hz, 2 H), 7,37 (m, 2 H), 7,32-7,28 (m, 1 H), 6,98 (d, J = 2.4 Hz, 1 H), 6,78 (dd, J = 8,4, 2,4 Hz, 1 H), 6,56 (d, J = 2,0 Hz, 1 H), 6,49 (dd, J = 8,4, 2,4 Hz, 1 H), 5,13 (s, 2 H), 4,96 (s, 1 H), 2,82 (s, 3 H); MS(ESI) m/z 303 (M+H+).
A la 7-(benciloxi)-N-metil-9H-carbazol-2-amina (120 mg, 0,4 mmol), el ácido fórmico (55 mg, 1,2 mmol) y DMAP (5 mg, 0,04 mmol) en 3 ml de piridina se añadió porciones de EDC (230 mg, 1,2 mmol). La mezcla se agitó ata durante 3 h bajo atmósfera de Ar y se concentró a presión reducida. El residuo se diluyó con EtOAc (50 ml) y se lavó con agua (2x50 ml), HCl 0,5 M (2x50 ml) y salmuera (50 ml), y se secó sobre MgSO4. Tras eliminar el disolvente, el producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener N-(7-(benciloxi)-9H-carbazol-2-il)-N-metilformamida
como sólido blanco (110 mg, 83 %). 1H RMN (400 MHz, acetona-d6) 5 10,34 (s, 1 H), 8,49 (s, 1 H), 8,02 (d, J = 8,4 Hz, 1 H), 7,98 (d, J = 8,8 Hz, 1 H), 7,51 (d, J = 7,2 Hz, 2 H), 7,39 (m, 2 H), 7.34-7,28 (m, 1 H), 7,13 (d, J = 2,4 Hz, 1 H), 7,08 (dd, J = 8,4, 2,4 Hz, 1 H), 6,91 (dd, J = 8,4, 2,4 Hz, 1 H), 5,19 (s, 2 H), 3,31 (s, 3 H); MS(ESI) m/z 331 (M+H+).
A la N-(7-(benciloxi)-9H-carbazol-2-il)-N-metilformamida (110 mg, 0,33 mmol) en 50 ml de MeOH se añadió paladio sobre carbón activado (50 mg). La mezcla se agitó a ta bajo atmósfera de H2 durante 15h. El sólido se filtró y el filtrado se concentró para obtener N-(7-hidroxi-9H-carbazol-2-il)-N-metilformamida como sólido marrón (75 mg, 94 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 241 (M+H+).
A la N-(7-hidroxi-9H-carbazol-2-il)-N-metilformamida (45 mg, 0,187 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (172 mg, 0,38 mmol) en 0,5 ml de NMP se añadió Cs2cO 3 (65 mg, 0,2 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con EtOAc (50 ml). Se lavó con agua (2x 50 ml), HCl 0,5 M (50 ml) y salmuera (50 ml), y se secó sobre MgSO4. El disolvente se eliminó bajo presión reducida y el producto bruto se purificó con cromatografía de sílice (hexano/EtOAc) para obtener 2-(2-(2-(7-(N-metilformamido)-9H-carbazol-2-iloxi)etoxi)etoxi)etil 4-metilbencenosulfonato (AD-CB-004Pa-WZ01179) como un aceite marrón claro (48 mg, 48 %). 1H RMN (400 MHz, CDCls) 58,52 (s, 1H), 8,45 (s, 1 H), 7,95 (d, J = 8,4 Hz, 1 H), 7,90 (d, J = 8,8 Hz, 1 H), 7.80-7,77 (m, 2 H), 7,30 (d, J = 8,0 Hz, 2 H), 7,17 (d, J = 2,4 Hz, 1 H), 7,02 (d, J = 2,0 Hz, 1 H), 7.01 (dd, J = 8,0, 2,0 Hz, 1 H), 6,89 (dd, J = 8,8, 2,4 Hz, 1 H), 4,23 (m, 1 H), 4,17 (m, 2 H), 3.88 (m, 2 H), 3,72-3,68 (m, 4 H), 3,66-3,61 (m, 2 H), 3,39 (s, 3 H), 2,41 (s, 3 H); MS(ESI) m/z 527 (M+H+).
AD-CB-004Pb-WZ01191

A la N-(7-(benciloxi)-9H-carbazol-2-il)-N-metilformamida (140 mg, 0,42 mmol) en 5 ml de THF seco a 0 °C bajo atmósfera de Ar se añadió NaH (50 mg, 60 % en aceite, 1,26 mmol) en 4 porciones. A continuación, la mezcla se agitó a ta durante 20 min. y se añadió carbonato de tert-butilfenilo (244 mg, 1,26 mmol) con una jeringa. La reacción se dejó agitar a ta durante 3 h y se apagó en hielo (30 g). La mezcla se extrajo con EtOAc (2x40 ml) y la fase orgánica combinada se secó sobre MgSO4. Tras la eliminación del disolvente, el residuo se cromatografió para obtener 2-(benciloxi)-7-(N-metilformamido)-9H-carbazol-9-carboxilato de tert-butilo como sólido blanco (120 mg, 66%). 1H RMN(400 MHz, CDCls) 58,56 (s, 1 H), 8,15 (s, 1 H), 7,98 (s, 1 H), 7,86 (d, J = 8,4 Hz, 1 H), 7,83 (d, J = 8,4 Hz, 1 H), 7,50-7,49 (m, 2 H), 7,43-7,39 (m, 2 H), 7,37-7,32 (m, 1 H), 7,13 (dd, J = 8,4, 2,0 Hz, 1 H), 7,05 (dd, J = 8,8, 2,4 Hz, 1 H), 5,18 (s, 2 H), 3,41 (s, 3 H), 1,75 (s, 9 H); MS(ESI) m/z 431 (M+H+).
Al 2-(benciloxi)-7-(N-metilformamido)-9H-carbazol-9-carboxilato de tert-butilo (120 mg, 0,28 mmol) en 50 ml de MeOH se añadió paladio sobre carbón activado (50 mg). La mezcla se agitó a ta bajo atmósfera de H2 durante 3h. El sólido se filtró y el filtrado se concentró para obtener 2-hidroxi-7-(N-metilformamido)-9H-carbazol-9-carboxilato de tert-butilo como sólido marrón (95 mg, 100 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 341 (M+H+).
Al 2-hidroxi-7-(N-metilformamido)-9H-carbazol-9-carboxilato de tert-butilo (65 mg, 0,19 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (174 mg, 0,38 mmol) en 0,5 ml de NMP se añadió Cs2CO3 (68 mg, 0,21 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con EtOAc (80 ml). Se lavó con agua (3x 50 ml) y se secó sobre MgSO4. El disolvente se eliminó bajo presión reducida y el producto bruto se purificó con cromatografía de sílice (hexano/EtOAc) para obtener 2-(N-metilformamido)-7-(2-(2-(tosiloxi)etoxi)etoxi)etoxi)-9H-carbazol-9-carboxilato de tert-butilo (AD-cB-004Pb-WZ01191) como aceite claro (75 mg, 63 %). 1H RMN (400 MHz, CDCh) 58,56 (s, 1 H), 8,14 (s, 1 H), 7,89 (s, 1 H), 7,87 (d, J = 8,0 Hz, 1 H), 7,83 (d, J = 8,8 Hz, 1 H), 7,79 (m, 2 H), 7,32 (d, J = 8,4 Hz, 2 H), 7,15 (dd, J = 8,4, 2.0 Hz, 1 H), 7,00 (dd, J = 8,8, 2,4 Hz, 1 H), 4,23 (m, 1 H), 3,89 (m, 2 H), 3,88 (m, 2 H), 3,73-3,68 (m, 4 H), 3,66-3,63 (m, 2 H), 3,41 (s, 3 H), 2,42 (s, 3 H), 1,76 (s, 9 H); MS(ESI) m/z 527 (M+H+).
AD-CB-010S-WZ01183

A la 4-(bencNoxi)-1-doro-2-nitrobenceno (394 mg, 1,5 mmol) N-(4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)formamida (370 mg, 1,5 mmol) en 6 ml de dioxano se añadieron 3 ml de una solución de Na2CO3 (aq) 1 M y Tetrakis(trifenilfosfina)paladio (52 mg, 0,045 mmol). La suspensión se calentó a reflujo durante 15 horas bajo atmósfera de Ar y se enfrió ata. Se añadió EtOAc (80 ml) y se lavó con salmuera (50 ml), agua (2x80 ml) y se secó sobre MgSO4. Tras eliminar el disolvente, el residuo se cromatografió (hexano/EtOAc) para obtener N-(4'-(bencioloxi)-2'-nitrobifenil-4-il)formamida como sólido amarillo (395 mg, 75 %). MS(ESI) m/z 349 (M+Hf).
Una suspensión de N-(4'-(benciloxi)-2'-nitrobifenil-4-il)formamida (350 mg, 1 mmol) en 2 ml de fosfito de tritilo se calentó a 145 C durante 15 h bajo atmósfera de Ar y se enfrió a ta. Se añadió 10 ml de hexano y se dejó reposar durante 10 min. El sólido se recogió por filtración y se lavó con éter/hexano (v:v 1/1, 10 ml) y se secó bajo alto vacío para obtener N-(7-(benciloxi)-9H-carbazol-2-il)formamida como un sólido marrón claro (280 mg, 88 %). Ms (ESI) m/z 317 (M+H+).
A la N-(7-(benciloxi)-9H-carbazol-2-il)formamida (250 mg, 0,79 mmol) en 50 ml de MeOH se añadió paladio sobre carbón activado (60 mg). La mezcla se agitó a ta bajo atmósfera de H2 durante 15h. La mezcla se concentró bajo presión reducida y se secó a alto vacío para obtener N-(7-hidroxi-9H-carbazol-2-il)formamida mezclada con el catalizador como un sólido negro (240 mg). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 227 (M+H+).
A la N-(7-hidroxi-9H-carbazol-2-il)formamida (30 mg) y 2-(2-(2-fluoroetoxi)etoxi)etil) 4-metilbencenosulfonato de etilo (48 mg, 0,156 mmol) en 0,3 ml de NMP se añadió Cs2CO3 (42 mg, 0,13 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con EtOAc (30 ml). Se lavó con agua (3x30 ml) y se secó sobre MgSO4. Tras eliminar el disolvente, el residuo se cromatografió (hexano/EtOAc) hasta obtener N-(7-(2-fluoroetoxi)-9H-carbazol-2-il)formamida (AD-CB-010S-WZ01183)como sólido blanco (17 mg, 36 %). Para el rotómero mayor: 1H RMN (400 MHz, acetona-d6) 510,10 (s, 1 H), 9,28 (s, 1 H), 8,39 (d, J = 1,6 Hz, 1 H), 8,11 (d, J = 2,0 Hz, 1 H), 7,91 (s, 1 H), 7,87 (d, J = 8,4, Hz, 2 H), 7,17 (dd, J = 8,4, 2.0 Hz, 1 H), 7,01 (d, J = 2,0 Hz, 1 H), 6,80 (dd, J = 8,4, 2,0 Hz, 1 H), 4,58 (m, 1 H), 4,46 (m, 1 H), 4,21 (m, 2 H), 3,88 (m, 2 H), 3,77 (m, 1 H), 3,73-3,66 (m, 5 H); MS(ESI) m/z 361 (M+H+).
AD-CB-012S-WZ01185

El compuesto AD-CB-012S-WZ01185 se preparó utilizando el mismo procedimiento para la preparación de AD-CB-010S-WZ01183. Para el rotómero mayor: 1H RMN (400 MHz, acetona-d6) 510,08 (s, 1 H), 9,19 (s, 1 H), 8,26(d, J = 1,6 Hz, 1 H), 8,00 (d, J = 2,0 Hz, 1 H), 7,84-7,77 (m, 3 H), 7,07 (dd, J = 8,4, 2.0 Hz, 1 H), 6,93 (d, J = 2,0 Hz, 1 H), 6,69 (dd, J = 8,4, 2,0 Hz, 1 H), 4,73 (m, 1 H), 4,61 (m, 1 H), 4,24 (m, 1 H), 4,17 (m, 1 H); MS(ESI) m/z 273 (M+Hf).
AD-CB-024S-WZ02033

El compuesto AD-CB-024S-WZ02033se preparó utilizando el mismo procedimiento para la preparación de AD-CB-010S-WZ01183. Para el rotómero mayor: 1H RMN (400 MHz, acetona-d6) 510,19 (s, 1 H), 9,31 (s, 1 H), 8,38(d, J = 1,6 Hz, 1 H), 8,11 (d, J = 2,0 Hz, 1 H), 7,88 (d, J = 8,2 Hz, 2 H), 7,19 (dd, J = 8,4, 2,0 Hz, 1 H), 7,03 (d, J = 2,0 Hz, 1 H), 6,79 (dd, J = 8,4, 2,0 Hz, 1 H), 4,62 (m, 1 H), 4,50 (m, 1 H), 4,20 (m, 2 H), 3,88 (m, 2 H), 3,83 (m, 1 H), 3,75 (m, 1 H); MS(ESI) m/z 317 (M+H+).
AD-CB-013S-WZ-02001

Una mezcla de acetato de paladio (37 mg, 0,165 mmol) y BINAP (154 mg, 0,248 mmol) en 5 ml de dioxano se agitó durante 10 min bajo atmósfera de Ar. A esta mezcla se añadió 1-bromo-4-nitrobenceno (1,11 g, 5,5 mmol), 4-metoxianilina (745 mg, 6,07 mmol), CsCO3 (2,5 g, 7,73 mmol) y 10 ml de dioxano. La mezcla se calentó a reflujo durante 15 h, se enfrió y se diluyó con éter (80 ml). El sólido se eliminó por filtración y el filtrado se concentró. El residuo se cromatografió (hexano/EtOAc) para obtener 4-metoxi-N-(4-nitrofenil)anilina como un sólido amarillo (786 mg, 58 %). MS(ESI) m/z 245 (M+H+).
A la 4-metoxi-N-(4-nitrofenil)anilina (785 mg, 3,2 mmol) en 5 ml de AcOH se añadió Pd(OAc)2 (1,43 g, 6,4 mmol). La mezcla se calentó a 100 °C durante 15 h bajo atmósfera de aire, se enfrió a ta y se concentró a presión reducida. El residuo se tomó en EtOAc (100 ml) y se lavó con NaHCO3 (2x100 ml) y agua (100 ml). Tras eliminar el disolvente, el crudo se purificó con cromatografía (hexano/EtOAc) para obtener 3-metoxi-6-nitro-9H-carbazol como un sólido naranja (495 mg, 64 %). 1H RMN (400 MHz, acetona-d6) 510,90 (s, 1 H), 9,09 (d, J = 2,4 Hz, 1 H), 8,27 (dd, J = 9,2, 2,4 Hz, 1 H), 7,96 (d, J = 2,4 Hz, 1 H), 7,62 (d, J = 9,2 Hz, 1 H), 7,53 (d, J = 8,8 Hz, 1 H), 7,14 (dd, J = 8,8, 2,8 Hz, 1 H), 3,92 (s, 3 H); MS(ESI) m/z 243 (M+H+).
Al 3-metoxi-6-nitro-9H-carbazol (100 mg, 0,41 mmol) en 40 ml de MeOH se añadió paladio sobre carbón activado (50 mg). La mezcla se agitó a ta bajo atmósfera de H2 durante 5 h. El sólido se filtró y el filtrado se concentró para obtener 6-metoxi-9H-carbazol-3-amina como sólido marrón (80 mg, 92 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 213 (M+H+).
A la 6-metoxi-9H-carbazol-3-amina (16 mg, 0,075 mmol) y al 1-bromo-2-fluoroetano (48 mg, 0,375 mmol) en 0,3 ml de NMP se añadió Cs2CO3 (30 mg, 0,09 mmol). La mezcla se agitó a ta durante 72 h bajo atmósfera de Ar y se diluyó con EtOAc (30 ml). Se lavó con agua (3x30 ml) y se secó sobre MgSO4. Tras eliminar el disolvente, el residuo se purificó mediante HPLC en fase inversa (tampón A: 0,05 % TFA acuoso; tampón B: 0,05,% TFA en MeCN) para obtener una cera de color marrón claro (5 mg, 26 %). 1H RMN (400 MHz, acetona-d6) 57,75 (s, 1 H), 7,67 (s, 1 H), 7,52 (d, J = 2,4 Hz, 1 H), 7,35 (t, J = 9,6 Hz, 2 H), 7,14 (d, J = 8,0 Hz, 1 H), 7,00 (dd, J = 8,8, 2,4 Hz, 1 H), 4,81 (t, J = 5,2 Hz, 1 H), 4,69 (t, J = 4,8 Hz, 1 H), 3,89 (s, 3 H); MS(ESI) m/z 259 (M+H+).

A la 7,8-dihidroxi-4-fenil-2H-cromo-2-ona (500 mg, 2 mmol) y al 3,4-dihidro-2H-pirano en 4 ml de THF y 4 ml de DCM se añadió paratoluenosulfonato de piridinio (PPTS, 8 mg). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con EtOAc (50 ml). Se lavó con NaHCO3 (sat. 30 ml) y agua (50 ml) y se secó sobre MgSO4 y se concentró a presión reducida. El residuo se cromatografió (hexano/EtOAc) para obtener 8-hidroxi-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-2-ona como sólido hueco (180 mg, 26 %). m S(ESI) m/z 339 (M+H+).
A la 8-hidroxi-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-2-ona (40 mg, 0,12 mmol) y al 1-bromo-2-fluoroetano (22 mg, 0,17 mmol) en 0,4 ml de NMP se añadió Cs2CO3 (46 mg, 0,14 mmol). La mezcla se agitó a ta durante 5 h bajo atmósfera de Ar y se diluyó con éter (40 ml). Se lavó con agua (3x30 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 8-(2-fluoroetoxi)-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-2-ona como sólido blanco (34 mg, 73 %). MS(ESI) m/z 385 (M+H+).
A la 8-(2-fluoroetoxi)-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-2-ona se añadieron 1,5 ml de una solución de HCl 4 M en dioxano. La mezcla se agitó a ta durante 30 min bajo atmósfera de Ar y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 8-(2-fluoroetoxi)-7-hidroxi-4-fenil-2H-cromo-2-ona (AD-C-004S-WZ01055) como sólido blanco (24 mg, 90 %). 1H RMN (400 MHz, CDC13) 58,96 (s, 1 H), 7,57-7,54 (m, 3 H), 7,52-7,49 (m, 2 H), 7,08 (dd, J = 8,8, 1,2 Hz, 1 H), 6,87 (dd, J = 8,8, 1.2 Hz, 1 H), 6,11 (dd, J = 1,2 Hz, 1 H), 4,86 (m, 1 H), 4,74 (m, 1 H), 4,50 (m, 1 H), 4,43 (m, 1 H); MS(ESI) m/z 301 (M+H+), 323 (M+ H+).
AD-C-004P-WZ01051

A la 8-hidroxi-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-2-ona (115 mg, 0,34 mmol) y al bis(4-metilbencenosulfonato) de etano-1,2-diilo (188 mg, 0,51 mmol) en 1 ml de NMP se añadió Cs2cO 3 (133 mg, 0,41 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con éter (50 ml). Se lavó con agua (3x50 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 2-(2-oxo-4-fenil-7-(tetrahidro-2H-pirano-2-iloxi)-2H-cromo-8-iloxi)etil 4-metilbencenosulfonato como una cera blanca (97 mg, 53 %). 1H RMN (400 MHz, CDCl3) 57,83 (d, J = 8,4 Hz, 1 H), 7,53-7,50 (m, 3 H), 7,43-7,41 (m, 2 h), 7,36 (d, J = 8,0 Hz, 1 H), 7,16 (d, J = 8,8 Hz, 1 H), 7,06 (d, J = 9,2 Hz, 1 H), 622 (s, 1 H), 5,56 (t, J = 2,8 Hz, 1 H), 4,41 (m, 4 H), 3,86 (td, J = 10,8, 2,8 Hz, 1 H), 3,61 (m, 1 H), 2,08-1,86 (m, 3 H), 1,76-1,63 (m, 3 H); MS(ESI) m/z 537 (M+H+).
AD-F-001S-WZ01067

A la 1-(2,4-dihidroxifenil)etanona (1,52 g, 10 mmol) en 12 ml de acetona se añadió K2CO3 (1,38 g, 10 mmol) seguido de la adición lenta de cloro(metoxi)metano (885 mg, 11 mmol) con agitación bajo atmósfera de Ar ata. La mezcla de reacción se agitó a ta durante 4 h y se filtró. El sólido se lavó con EtOAc (80 ml) y el filtrado combinado se lavó con NaH2PO4 (sat. 50 ml) y agua (80 ml) y se secó sobre MgSO4 y se concentró. El producto crudo se purificó con cromatografía de sílice (hexano/EtOAc) para obtener 1-(2-hidroxi-4-(metoximetoxi)fenil)etanona como un sólido blanquecino (1,2 g, 61%). MS(ESI) m/z 197 (M+H+).
Una mezcla de 3,4-dihidroxibenzaldehído (1,38 g, 10 mmol), bromuro de bencilo (1,71 g, 10 mmol) y K2CO3 (1,24 g, 9 mmol) en 20 ml de acetona se agitó a ta durante 15 h bajo atmósfera de Ar. El sólido se filtró y el filtrado se diluyó con EtOAc (100 ml) y se lavó con NaH2PO4 (sat. 100 ml), y se secó sobre MgSO4 y se concentró. El residuo se
cromatografió (hexano/EtOAc) para obtener 4-(benciloxi)-3-hidroxibenzaldehído como un sólido blanco (1,15 g, 50 %). 1H RMN(400 MHz, CDC13) 89,84 (s, 1 H), 7,46 (d, J = 2,0 Hz, 1 H), 7,44-7,40 (m, 6 H), 7,04 (d, J = 8,0 Hz, 1 H), 5,80 (s, 1 H), 5,21 (s, 2 H); MS(ESI) m/z 229 (M+H+).
Una mezcla de 4-(benciloxi)-3-hidroxibenzaldehído (912 mg, 4 mmol), cloro(metoxi)metano (480 mg, 6 mmol) y DIPEA (1.03 g, 8 mmol) en 20 ml de DCM se agitó a ta bajo atmósfera de Ar durante 15 h. Se diluyó con éter (100 ml) y se lavó con HCl 0,5 M (2x50 ml) y agua (2x80 ml), se secó sobre MgSo4 y se concentró. El producto bruto se purificó con cromatografía de sílice (hexano/EtOAc) para obtener 4-(benciloxi)-3-(metoximetoxi)benzaldehído como un aceite claro (960 mg, 88%). MS(ESI) m/z 273 (M+H+).
A la 1-(2-hidroxi-4-(metoximetoxi)fenil)etanona (618 mg, 3,15 mmol) y al 4-(benciloxi)-3-(metoximetoxi)benzaldehído (816 mg, 3 mmol) en un matraz de fondo redondo de 25 ml bajo atmósfera de Ar se añadieron 2,5 ml de una solución de KOH en EtOH al 5 % recién hecha. La mezcla se agitó enérgicamente a ta hasta que se solidificó y se mantuvo a ta durante 6 días. Se tomó en éter (100 ml) y se añadió HCl 0,5 M a pH=5, y se agitó durante 3 min. La mezcla se lavó con agua (2*100 ml) y se secó sobre MgSo4 y se concentró. El residuo se cromatografió (hexano/EtOAc) para obtener (E)-3-(4-(benciloxi)-3-(metoximetoxi)fenil)-1-(2-hidroxi-4-(metoximetoxi)fenil)prop-2-en-1-ona como un aceite claro (780 mg, 58 %). MS(ESI) m/z 451 (M+H+).
A la (E)-3-(4-(benicloxi)-3-(metoximetoxi)fenil)-1-(2-hidroxi-4-(metoximetoxi)fenil)prop-2-en-1-ona (450 mg, 1 mmol) en 1,5 ml de MeOH se añadieron 4 ml de una solución de NaOH al 15 %, seguida de peróxido de hidrógeno (113 mg, solución al 30%). La mezcla se agitó a ta durante 2 h y se añadieron 226 mg adicionales de peróxido de hidrógeno. La reacción se agitó durante 15 y se apagó en NaH2PO4 (sat. 50 ml). Se extrajo con EtOAc (3x50 ml) y la fase orgánica combinada se secó sobre MgSO4 y se concentró. El residuo se cromatografió (hexano/EtOAc) para obtener 2-(4-(benciloxi)-3-(metoxietoxi)fenil)-3-hidroxi-7-(metoximetoxi)-4H-cromo-4-ona como un sólido blanquecino (55 mg, 12 %). MS(ESI) m/z 465 (M+H+).
A la 2-(4-(benciloxi)-3-(metoximetoxi)fenil)-3-hidroxi-7-(metoximetoxi)-4H-cromo-4-ona (55 mg, 0,12 mmol) en 2 ml de DCM se añadió DIPEA (31 mg, 0,24 mmol), seguido de la adición lenta de cloro(metoxi)metano (15 mg, 0,18 mmol). La mezcla de reacción se agitó a ta bajo atmósfera de Ar durante 3 h y se diluyó con éter (30 ml). Se lavó con agua (3x30 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía de sílice (hexano/EtOAc) para obtener 2-(4-(benciloxi)-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona como sólido blanco (55 mg, 90 %). 1H RMN (400 MHz, CDC13) 88,14 (d, J = 8,8 hz, 1 H), 7,91 (d, J = 2,0 Hz, 1 H), 7,71 (dd, J = 8,4 Hz, 2,0 Hz, 1 H), 7,46-7,44 (m, 2 H), 7,39 (m, 2 H), 7,34-7,31 (m, 1 H), 7,12 (d, J = 2.4 Hz, 1 H), 7,05 (dd, J = 8,8, 2,4 Hz, 1 H), 7,03 (d, J = 8,4 Hz, 1 H), 5,30 (s, 2 H), 5,28 (s, 2 H), 5.24 (s, 2 H), 5,21 (s, 2 H), 3,55 (s, 3 H), 3,51 (s, 3 H), 3,18 (s, 3 H); MS(ESI) m/z 509 (M+H+).
A la 2-(4-(benciloxi)-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (55 mg, 0,108 mmol) en 10 ml de MeOH se añadió paladio sobre carbón activado (20 mg). La mezcla se agitó a ta bajo atmósfera de H2 durante 2h. El sólido se filtró y el filtrado se concentró para obtener 2-(4-hidroxi-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona como sólido amarillo (45 mg, 99 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 419 (M+H+).
A la 2-(4-hidroxi-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (20 mg, 0,048 mmol) y al 1-bromo-2-fluoroetano (18 mg, 0,14 mmol) en 0,3 ml de NMP se añadió Cs2CO3 (39 mg, 0,12 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con éter (30 ml). Se lavó con NaH2PO4 (sat. 30 ml) y agua (2*30 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) para obtener 2-(4-(2-fluoroetoxi)-3-(metoximeetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona como sólido blanco (19 mg, 85 %). 1H RMN (400 MHz, CDCla) 88,15 (d, J = 8,8 Hz, 1 H), 7,91 (d, J = 2,0 Hz, 1 H), 7,76 (dd, J = 8,4 Hz, 2,0 Hz, 1 H), 7,13 (d, J = 2,4 Hz, 1 H), 7,05 (dd, J = 8,8, 2,4 Hz, 1 H), 7.03 (d, J = 8,4 Hz, 1 H), 5,29 (s, 4 H), 5,22 (s, 2 H), 4,89 (m, 1 H), 4,78 (m, 1 H), 4,40 (m, 1 H), 4,32 (m, 1 H), 3,56 (s, 3 H), 3,51 (s, 3 H) 3,20 (s, 3 H); MS(ESI) m/z 465 (M+H+).
A la 2-(4-(2-fluoroetoxi)-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (19 mg, 0,041 mmol) se añadió 1 ml de solución de HCl 4 M en dioxano. La mezcla se agitó a ta durante 3 h. Los volátiles se eliminaron a presión reducida y el residuo se lavó con éter (2*1 ml) y se secó a alto vacío para obtener 2-(4-(2-fluoroetoxi)-3-hidroxifenil)-3,7-dihidroxi-4H-cromo-4-ona (AD-F-001S-WZ01067) como un sólido amarillo (11 mg, 81 %). 1H RMN (400 MHz, DMSO-d6) 810,73 (s, 1 H), 9,39 (s, 1 H), 9,17 (s, 1 H), 7,89 (d, J = 9,6 Hz, 1 H), 7,69 (d, J = 2,0 Hz, 1 H), 7,59 (dd, J = 8,4 Hz, 2.0 Hz, 1 H), 7,06 (d, J = 8,8 Hz, 1 H), 6,87 (m, 2 H), 4,80 (m, 1 H), 4,68 (m, 1 H), 4,30 (m, 1 H), 4,23 (m, 1 H); MS(ESI) m/z 333 (M+H+), 355 (M+Na+).
AD-C-001P-WZ01079

A la 2-(4-hidroxi-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (19 mg, 0,045 mmol) y al 2-bromoetanol (44 mg, 0,36 mmol) en 0,2 ml de NMP se añadió Cs2CO3 (29 mg, 0,09 mmol). La mezcla se agitó a ta durante 15 h bajo atmósfera de Ar y se diluyó con éter (30 ml). Se lavó con NaH2PO4 (sat. 30 ml) y agua (2x30 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó con cromatografía (hexano/EtOAc) hasta obtener 2-(4-(2-hidroxietoxi)-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (18 mg, 86 %). m S(ESI) m/z 463 (M+H+).
A la 2-(4-(2-hidroxietoxi)-3-(metoximetoxi)fenil)-3,7-bis(metoximetoxi)-4H-cromo-4-ona (18 mg, 0,039 mmol) y DIPEA (15 mg, 0,11 mmol) se añadió cloruro de 4-metilbenceno-1-sulfonilo (11 mg, 0,058 mmol). La mezcla se agitó ata durante 15 h bajo atmósfera de Ar y se diluyó con éter (30 ml). Se lavó con HCl 0,5 M (2x30 ml) y agua (50 ml), se secó sobre MgSO4 y se concentró. El producto bruto se cromatografió para obtener 2-(4-(3,7-bis(metoximetoxi)-4-oxo-4H-cromo-2-il)-2-(metoximetoxi)fenoxi)etil 4-metilbencenosulfonato(AD-C-001P-WZ01079) como una cera amarilla (20 mg, 83 %). 1H RMN(400 MHz, CDC13) 58,15 (dd, J = 8,8, 2,0 Hz, 1 H), 7,89 (d, J = 2,0 Hz, 1 H), 7,76 (dd, J = 8,4 Hz, 2,0 Hz, 2 H), 7,73 (dd, J = 8.4, 2,0 Hz, 1 H), 7,36 (dd, J = 6,8 Hz, 2 H), 7,27 (d, J = 2,0 Hz, 1 H), 7,14 (d, J = 2,4 Hz, 1 H), 7,05 (dd, J = 8,8, 2,0 Hz, 1 H), 6,94 (dd, J = 8,8, 2,0 Hz, 1 H), 5,29 (d, J = 1,6 Hz, 2 H), 5,22 (d, J = 2,0 Hz, 4 H), 4,42 (t, J = 3,0 Hz, 2 H), 4,32 (t, J = 3,0 Hz, 2 H), 3,53 (d, J = 2,4 Hz, 3 H), 3,52 (d, J = 2,0 Hz, 3 H), 3,20 (d, J = 2,4 Hz, 3 H), 2,46 (s, 3 H); MS(ESI) m/z 617 (M+H+)

-((4-fluorobutil)(metil)amino)-9H-carbazol-2-ol (CB-14)

A un matraz de fondo redondo que contenía el compuesto 6 (21 mg, 0,073 mmol) en DMF (1 ml), se añadió carbonato de cesio (28,5 mg, 0,087 mmol) y 1-bromo-4-fluorobutano (56,4 mg, 0,364 mmol). La reacción se agitó a ta durante 30 min. La reacción se trabajó con EtOAc (15 ml x 3) y agua (10 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron y se concentraron in vacuo. El residuo se disolvió en MeOH (10 ml). A la mezcla de reacción se añadió Pd/C (22 mg). La mezcla se agitó a ta durante la noche bajo hidrógeno (1 atm). La reacción se filtró a través de un tapón de celita, se concentró in vacuo y se purificó por HPLC para obtener CB-14 (11 mg, 0,029 mmol, rendimiento del 40,3 %). 1H-RMN (400 MHz, CD3OD) 5: 8,74 (d, J = 8,4 Hz, 1H), 7,85 (d, J = 8,4 Hz, 1H), 7,53 (s, 1H), 7,25 (d, J = 8,0 Hz, 1H), 6,85 (s, 1H), 6,73 (d, J = 8.4 Hz, 1H), 4,44 (m, 1H), 4,32 (m, 1H), 3,70 (m, 2H), 3,35 (s, 3H), 1,74-1,67 (m, 4H); LRMS para C19H19F4N2O2+H+, calc'd: 384,1, encontrado: 287,2 (M+H+-TFA).
7-((2-fluoroetil)(metil)amino)-9H-carbazol-2-ol (CB-15)

A un matraz de fondo redondo que contenía el compuesto 6 (37 mg, 0,122 mmol) en DMF (0,5 ml), se añadió carbonato de cesio (47,8 mg, 0,147 mmol) y 1-bromo-2-fluoroetano (78 mg, 0,612 mmol). La reacción se agitó a ta durante 30 min. La reacción se trabajó con EtOAc (15 ml x 3 ) y agua (10 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron y se concentraron in vacuo. El residuo se disolvió en MeOH (10 ml). A la mezcla de reacción se añadió Pd/C (22 mg). La mezcla se agitó a ta durante la noche bajo hidrógeno (1 atm). La reacción se filtró a través de un tapón de celita, se concentró in vacuo y se purificó por HPLC para obtener CB-15 (5 mg, 0,019 mmol, rendimiento del 7,3 %). 1H-RMN (400 MHz, CD3CN) 5: 7,96 (d, J = 8,0 Hz, 1H), 7,87 (d, J = 8,4 Hz, 1H), 7,31 (d, J = 2,0 Hz, 1H), 7,05 (dd, J = 8,0 Hz, 2,0 Hz, 1H), 6,90 (d, J = 2,0 Hz, 1H), 6,76 (dd, J = 8,0 Hz, 2,0 Hz, 1H), 4,86 (m, 1H), 4,74 (m, 1H), 4,60-4,52 (m, 2H), 3,28 (br, 1H), 3,03 (s, 3H); LRMS para C17H15F4N2O2+H+, calc'd: 356,1, encontrado: 259,2 (M+H+-TFA).
7-(2-fluoroetilamino)-9H-carbazol-2-ol (CB-16)

A un matraz de fondo redondo que contenía el compuesto 5 (21 mg, 0,073 mmol) en DMF (1 ml), se añadió carbonato de cesio (28,5 mg, 0,087 mmol) y 1-bromo-2-fluoroetano (46 mg, 0,36 mmol). La reacción se agitó a ta durante 72 horas. La reacción se trabajó con EtOAc (15 ml x 3 ) y agua (10 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron y se concentraron in vacuo. El residuo se disolvió en MeOH (10 ml). A la mezcla de reacción se añadió Pd/C (20 mg). La mezcla se agitó a ta durante la noche bajo hidrógeno (1 atm). La reacción se filtró a través de un tapón de celita, se concentró in vacuo y se purificó por HPLC para obtener CB-16 (5 mg, 0,015 mmol, rendimiento del 20 %). 1H-RMN (400 MHz, CD3CN) 5: 9,00 (br, 1H), 7,77-7,73 (m, 2H), 6,82 (s, 1H), 6,81 (s, 1H), 6,72-6,65 (m, 2H), 4,71 (m, 1H), 4,60 (m, 1H), 3,60-3,50 (m, 2H); LRMS para C16H13F4N2O2+H+, calc'd: 342,3, encontrado: 245,1 (M+H+-TFA).
7-((2-(2-fluoroetoxi)etoxi)etil)(metil)amino)-9H-carbazol-2-ol (CB-19)

A un matraz de fondo redondo que contenía el compuesto 6 (41 mg, 0,14 mmol) en DMF (0,5 ml), se añadieron carbonato de cesio (53 mg, 0,16 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (125 mg, 0,407 mmol). La reacción se agitó a ta durante 4 semanas. La reacción se trabajó con EtOAc (15 ml x 3 ) y agua (10 ml). Las capas orgánicas se lavaron con salmuera (10 ml), se secaron y se concentraron in vacuo. El residuo se disolvió en MeOH
(10 ml). A la mezcla de reacción se añadió Pd/C (20 mg). La mezcla se agitó a ta durante la noche bajo atmósfera de hidrógeno (1 atm). La reacción se filtró a través de un tapón de celita, se concentró in vacuo y se purificó por HPLC para obtener CB-19 (7 mg, 0,020 mmol, 14 % de rendimiento.1H-RMN(400 MHz, CD3CN) 5: 9,43 (br, 1H), 8,07 ((d, J = 8,4 Hz, 1H), 7,91 (d, J = 8,4 Hz, 1H), 7,58 (d, J = 2,4 Hz, 1H), 7,24 (dd, J = 8,0 Hz, 2,0 Hz, 1H), 6,93 (d, J = 2,0 Hz, 1H), 6,77 (dd, J = 8,0 Hz, 2,0 Hz, 1H), 4,59 (m, 1H), 4,52 (m, 1H), 3,74-3,50 (m, 10H), 3,28 (s, 3H); LRMS para C21H2sF4 N2O4+Na+, calc'd: 444,2, encontrado: 347,2 (M+H+-TFA).
7-(2-fluoroetoxi)-N-metil-9H-carbazol-2-amina (CB-20)

A un matraz de fondo redondo que contenía el compuesto 6 (90 mg, 0,29 mmol) en MeOH (10 ml), se añadió Pd/C (20 mg). La reacción se purgó con hidrógeno y se agitó a ta durante 2 h bajo atmósfera de hidrógeno (1 atm). La reacción se filtró a través de un tapón de celita y se concentró in vacuo para obtener un sólido oscuro (60 mg, 0,28 mmol, rendimiento del 95 %). A un matraz de fondo redondo que contenía el sólido oscuro anterior (15 mg, 0,071 mmol) en DMF (0,5 ml), se añadió carbonato de cesio (21 mg, 0,65 mmol) y 2-bromo-1-fluoroetano (8,1 mg, 0,065 mmol). La reacción se agitó a ta durante la noche. La reacción se concentró in vacuo mediante coevacuación de MecN. El residuo se purificó por HPLC para obtener CB-20 (7,0 mg, 0,027 mmol, 38 % de rendimiento). 1H RMN (400 MHz, CD3CN) 5: 9,52 (br, 1H), 7,91-7,86 (m, 2H), 7,13 (s, 1H), 7,02 (s, 1H), 6 , 8 8 (d, J = 7,6 Hz, 1H), 6,82 (dd, J = 7,6 Hz, J = 2,4 Hz 1H), 4,85 (m, 1H), 4,72 (m, 1H), 4,34-4,25 (m, 2H), 2,96 (s, 3H); LRMS para C17H15N2O2+H+, calc'd: 356,1, encontrado: 259,1 (M+H+-TFA)

4'-(benciloxi)-2'-nitrobifenil-4-ol (Compuesto 7)

Un matraz de fondo redondo cargado con el compuesto 2 (1,96 g, 7,44 mmol), éster de pinacol del ácido 4 hidroxifenilborónico (1,56 g, 7,09 mmol), terakis(trifenilfosfina) paladio (0,410 g, 0,354 mmol), se purgó con argón. A la mezcla se añadió DME (10 ml) y carbonato potásico (1,96 g, 14,2 mmol) en agua (2 ml). La mezcla se calentó durante 60 horas. La reacción se diluyó con HCl (1N, 10 ml) y salmuera (40 ml), y se extrajo con EtOAc (50 ml * 3). La capa orgánica combinada se lavó con salmuera (50 ml), se secó (MgSo4) y se concentró in vacuo. El residuo se purificó en una columna de gel de sílice (EtOAc : Hexanos = 1 : 4) para obtener el Compuesto 7 como un sólido amarillo (2 g, 6,22 mmol, 88 % de rendimiento). 1H-RMN (400 MHz, CDQs) 5: 7,45-7,33 (m, 7H), 7,37-7,15 (m, 3H), 6,88-6,85 (m, 2H), 5,14 (s, 2H), 5,03 (s, 1H); LRMS para C19H15NO4+H+, calc'd: 322,1, encontrado: 322,1 (M+H+).
7-(benciloxi)-9H-carbazol-2-ol (Compuesto 8)

En un vial resistente a la presión, se añadió el compuesto 7 (2,00 g, 6,22 mmol y fosfito de tritilo (6,53 ml, 37,3 mmol). La mezcla se calentó a 160 °C durante la noche. La mezcla de reacción se concentró in vacuo. El residuo se suspendió en cloroformo ( 2 0 ml), se formó un precipitado sólido que se filtró y se lavó con éter ( 10 ml x 2 ) para obtener el Compuesto 8 (900 mg, 3,11 mmol, rendimiento del 50,0 %). 1H-RMN (400 MHz, DMSO) 5: 10,81 (br, 1H), 9,25 (br, 1H), 7,80 (d, J = 8,4 Hz, 1H), 7,72 (d, J = 8,4 Hz , 1H), 7,50-7,33 (m, 5H), 6,95 (s, 1H), 6-78-6,76 (m, 2H), 6,56 (dd, J = 8,4, 2,0 Hz 1H), 5,16 (s, 2H); LRMS para C19H15NO2+H+, calc'd: 290,1, encontrado: 290,1 (M+H+).
7-(2-fluoroetoxi)-9H-carbazol-2-ol (CB-25)

A un matraz de fondo redondo que contenía el Compuesto 8 (50 mg, 0,17 mmol) en DMF (1 ml), se añadió carbonato de cesio (62 mg, 0,19 mmol) y 1-bromo-2-fluoroetano (33 mg, 0,26 mmol). La reacción se agitó a ta durante 15 h y luego se diluyó con agua (15 ml). El precipitado blanco (50 mg) se recogió por filtración y se secó in vacuo. El sólido se disolvió en MeOH (10 ml). A la reacción se añadió Pd/C (30 mg) y ácido acético (5 gotas). La mezcla se agitó bajo atmósfera de hidrógeno (1 atm) durante 2 0 h y luego se filtró a través de un tapón de celita, concentrándose in vacuo. El residuo se purificó por HPLC para obtener CB-25 (18 mg, 0,053 mmol, 31 % de rendimiento). 1H RMN (400 MHz, CD3CN) 5: 8,99 (br, 1H), 7,72 (d, J = 8,4 Hz, 1H), 7,67 (d, J = 8,4 Hz, 1H), 6 , 8 8 (d, J = 2,0 Hz, 1H), 6,76 (d, J = 2,0 Hz, 1H), 6,67 (dd, J = 8.0 Hz, 2,0 Hz, 1H), 6,58 (dd, J = 8,0 Hz, 2,0 Hz, 1H), 4,75-4,74 (m, 1H), 4,63-4,61 (m, 1H), 4,23-4,13 (m, 2H); LRMS para C16H12F4NO3+H+, calc'd: 343,1, encontrado: 246,0 (M+H+-TFA).
7-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol-2-ol (CB-26)

A un matraz de fondo redondo que contenía el Compuesto 8 (50 mg, 0,17 mmol) en DMF (1 ml), se añadió carbonato de cesio (56 mg, 0,17 mmol) y 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (53 mg, 0,17 mmol). La reacción se agitó a ta durante 15 h y luego se diluyó con agua (15 ml). El precipitado blanco (72 mg) se recogió por filtración y se secó in vacuo. El sólido se disolvió en MeOH (10 ml). A la reacción se añadió Pd/C (20 mg) y ácido acético (5 gotas). La mezcla se agitó bajo atmósfera de hidrógeno (1 atm) durante 20 h y, a continuación, se filtró a través de un tapón de celita y se concentró in vacuo. El residuo se purificó por HPLC para obtener CB-26 (20 mg, 0,046 mmol, 27 % de rendimiento). 1H RMN (400 MHz, CD3CN) 5: 9,03 (br, 1H), 7,81-7,75 (m, 2H), 6,96 (d, J = 2,4 Hz, 1H), 6,84 (d, J = 2,4 Hz, 1H), 6,76 (dd, J = 7,6 Hz, 2,0 Hz, 1H), 6,67(dd, J = 7,6 Hz, 2,0 Hz, 1H), 4,59-4,57 (m, 1H), 4,47-4,45 (m, 1H), 4,17-4,15 (m, 2H), 3,83-3,63 (m, 8 H);LRMS para C20H20NO5+H+, calc'd: 431,1, encontrado: 334,1 (M+H+-TFA)
Esquema sintético de CB 27:

CB-27
7-hidroxi-9H-carbazol-2-ilcarbamato de tert-butilo (Compuesto 9)

A un matraz de fondo redondo que contenía el compuesto 4 (1,0 g, 2,6 mmol) en MeOH (150 ml), se añadió paladio sobre carbón (400 mg). El matraz se purgó con gas hidrógeno y se agitó bajo atmósfera de hidrógeno durante la noche. La mezcla de reacción se filtró a través de un tapón de celita y se concentró para obtener el Compuesto 9 como un sólido gris (700 mg, 2,34 mmol, rendimiento del 90 %). 1H RMN (400 MHz, (CÜ3)2CO) 5: 9,99 (br, iH), 8,41 (br, 1H), 8,24 (s, 1H), 7,86 (s, 1H), 7,81-7,78 (m, 2H), 7,18 (dd, J = 8,4 Hz, 2,0 Hz, 1H), 6,90 (d, J = 2,0 Hz, 1H), 6,70 (dd, J = 8,4 Hz, 2,0 Hz, 1H), 1,51 (s, 9H).
7-(4-nitrofenoxi)-9H-carbazol-2-ilcarbamato de tert-butilo (Compuesto 10)

A un matraz de fondo redondo que contenía el compuesto 9 (80 mg, 0,268 mmol) en DMF (2 ml) se añadió carbonato potásico (74,1 mg, 0,536 mmol) y 4-fluoro-nitrobenceno (41,6 mg, 0,295 mmol). La mezcla de reacción se calentó durante 20 min a 140 °C. Tras enfriar hasta ta, la mezcla se diluyó con agua (20 ml) y se extrajo con EtOAc (20 ml x 3). Las capas orgánicas se secaron y se concentraron. El residuo se purificó en una columna de gel de sílice (EtOAc : Hexanos = 3: 7) para obtener el Compuesto 10 como un sólido amarillo (50 mg, 0,12 mmol, 44 % de rendimiento). 1H RMN (400 MHz, CDCb) 5: 8,22 (d, J = 9,2 Hz, 2H), 8,10 (br, 1H), 8,00-7,90 (m, 3H), 7,12 (s, 1H), 7,06-6,90 (m, 4H), 6,70 (br, 1H), 1,56 (s, 9H).;LRMS para C23H21N3O5+H+, calc'd: 420,2, encontrado: 420,2 (M+H+).
7-(4-nitrofenoxi)-9H-carbazol-2-ilcarbamato de tert-butilo (CB-27)
A un matraz de fondo redondo que contenía el compuesto 10 (35 mg, 0,083 mmol) en MeOH (5 ml), se añadió paladio sobre carbón (10 mg). El matraz se purgó con gas hidrógeno y se agitó bajo atmósfera de hidrógeno durante la noche. La mezcla de reacción se filtró a través de un tapón de gel de sílice y se concentró para obtener el intermedio de amina (23 mg). A un vial que contenía ácido 2-fluoropropanoico (10,87 mg, 0,118 mmol) en DCM (1 ml), se añadió EDC (22,64 mg, 0,118 mmol) y DMAP (1 mg ). La mezcla se agitó a ta durante 5 min. La amina intermedia anterior se disolvió en DCM (1 ml) y se añadió al vial de reacción gota a gota. La mezcla de reacción se agitó a ta durante 3 horas. A continuación, la mezcla de reacción se lavó con agua (3 ml) y se concentró. El residuo se redisolvió en HCl (4,0 M en dioxano, 5 ml) y se agitó durante la noche. La mezcla se concentró y se purificó por HPLC para obtener CB-27 (12 mg, 0,026 mmol, 31 % de rendimiento). 1H RMN (400 MHz, CD3CN) 5: 9,42 (br, 1H), 8,69 (br, 1H), 7,92 (d, J = 8,4 Hz, 1H), 7,87 (d, J = 8,4 Hz, 1H), 7,60 (m, 2H), 7,04-7,01 (m, 4H), 6,86 (d, J = 8,0 Hz, 1H), 6,84 (d, J = 8,0 Hz, 1H), 5,11 (dt, J = 49,2, 6,8 Hz, 1H), 1,58 (dd, J = 24,8, 6,8 Hz, 3H); LRMS para C23H18F4 N3O3+H+, calc'd: 460,1, encontrado: 364,1 (M+H+-TFA).
Sección experimental para la preparación de derivados de carbazol

4- (benciloxi)-N-(4-nitrofenil)anilina 1: A un matraz secado al horno se cargó Pd(OAc)2 (81 mg, 0,36 mmol) y (S)-(-)-BINAP (336 mg, 0,54 mmol), seguido de tolueno (10 ml). La mezcla se agitó bajo Ar a temperatura ambiente durante 5 min. A esta mezcla se añadió 4-nitroyodobenceno (3,0 g, 12 mmol), clorhidrato de 4-benciloxianilina (3,39 g, 14,4 mmol), Cs2CO3 (9,8 g, 30 mmol) y tolueno (40 ml). La mezcla resultante se calentó bajo Ar a 100 °C durante 16 horas, y luego se enfrió a temperatura ambiente y se vertió en H2O (100 ml). Las capas fueron separadas. La capa acuosa se extrajo con EtOAc (3 x 20 ml). Las capas orgánicas combinadas se lavaron con salmuera (2 x 20 ml), se secaron (MgSO4) y se filtraron. El filtrado se concentró. El residuo se purificó mediante cromatografía en columna (gel de sílice, 5- 40 % EtOAc/hexano) para dar el producto deseado como un sólido naranja (1,2 g, 31 %). 1H RMN (CDCl3, 400 MHz) 5: 8,09 (d, J = 9,2 Hz, 2H), 7,30-7,49 (m, 5H), 7,15 (d, J = 9,2 Hz, 2H), 7,01 (d, J = 9,2 Hz, 2H), 6,77 (d, J = 8,8 Hz, 2H), 6,10 (br s, 1H), 5,09 (s, 2H). MS: m/z = 321 (M+H+)+.
3-(benciloxi)-9-nitro-9H-carbazol 2: Una mezcla de 4-(benciloxi)-N-(4-nitrofenil)anilina 1 (0,5 g, 1,56 mmol) y Pd(OAc)2 (0,8 g, 3,56 mmol) en ácido acético (20 ml) se sometió a reflujo y se monitorizó por TLC. Tras refluir durante 2 horas, TLC mostró que no había material de partida. Se concentró al vacío para eliminar el ácido acético. El residuo se diluyó con EtOAc (30 ml), se lavó con H2O (20 ml), sat. Solución de NaHCO3 (2 x 20 ml), salmuera (20 ml), y después se secó (MgSo4) y se filtró. El filtrado se concentró al vacío. El residuo se purificó mediante cromatografía en columna (gel de sílice, 5-40 % EtOAc/hexano) para dar el producto deseado 2 como un sólido amarillo oscuro (100 mg, 20%). 1H RMN(acetona-d6, 400 MHz) 5: 10,92 (br s, 1H), 9,08 (d, J = 2,0 Hz, 1H), 8,28 (dd, J = 8,8, 2,4 Hz, 1H), 8,07 (d, J = 2,4 Hz, 1H), 7,63 (d, J = 9,2 Hz, 1H), 7.55 (d, J = 8,8 Hz, 3H), 7,40 (t, J = 7,2 Hz, 2H), 7,33 (t, J = 7,2 Hz, 1H), 7,24 (dd, J = 8,8, 2,4 Hz, 1H), 5,26 (s, 2H). MS: m/z = 321 (M+H+)+.
3-Amino-6-(benciloxi)-9H-carbazol 3: A una suspensión de 3-(benciloxi)-9-nitro-9H-carbazol 2 (100 mg, 0,31 mmol) y Cu(OAc)2 (57 mg, 0,31 mmol) en EtOH (20 ml) se añadió NaBH4 (240 mg, 6,3 mmol). La mezcla resultante se agitó a temperatura ambiente durante 3 horas y luego se concentró al vacío. El residuo se disolvió en H2O (30 ml), se extrajo con EtOAc (2 x 30 ml). Las capas orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío para dar un sólido (90 mg). Se utilizó directamente en el siguiente paso sin ninguna otra purificación. 1H RMN(acetonad6, 400 MHz) 5: 9,67 (br s, 1H), 7,57 (d, J = 2,4 Hz, 1H), 7,52 (d, J = 6,8 Hz, 2H), 7,39 (t, J = 6,8 Hz, 2H), 7,26-7,33
(m, 3H), 7,19 (d, J = 8,8 Hz, 1H), 7,03 (dd, J = 8,8, 2,4 Hz, 1H), 6,81 (dd, J = 8,8, 2,4 Hz, 1H), 5,17 (s, 2H), 4,24 (br s, 2H). MS: m/z = 289 (M+H+)+.
6-(benciloxi)-N-metil-9H-carbazol-3-amina 4 : A una suspensión de 3-amino-6-(benciloxi)-9H-carbazol 3 (90 mg, 0,31 mmol) y paraformaldehído (47 mg, 1,57 mmol) en MeOH (20 ml) se añadió una solución de NaOMe en MeOH (0,32 ml, 1,56 mmol). La mezcla resultante se calentó a 80 °C durante 1 h y luego se añadió NaBH4 (59 mg, 1,55 mmol). La mezcla resultante se calentó a 80 °C durante 2 h y luego se enfrió a temperatura ambiente. A esta solución se añadió NaOH (1 N, 30 ml). La mezcla se extrajo entonces con CH2Cl2 (3 * 20 ml). Las capas orgánicas combinadas se secaron (MgSO4) y se filtraron. El filtrado se concentró al vacío para dar un sólido marrón (93 mg, 100 %). Se utilizó directamente en el siguiente paso sin ninguna otra purificación. 1H RMN(acetona-d6, 400 m Hz) 5: 9,68 (br s, 1H), 7,67 (d, J = 2,4 Hz, 1H), 7,53 (d, J = 7,6 Hz, 2H), 7,20-7,42 (m, 6H), 7,03 (dd, J = 8,8, 2,4 Hz, 1H), 6,79 (dd, J = 8,4, 2,4 Hz, 1H), 5,17 (s, 2H), 2,85 (s, 3H). MS: m/z = 303 (M+H+)+.
6-(Metilamino)-9H-carbazol-3-ol 5 : Una mezcla de 6-(benciloxi)-N-metil-9H-carbazol-3-amina 4 (93 mg, 0,31 mmol), Pd/C (10 mg) y ácido acético (10 gotas) en MeOH (10 ml) se hidrogenó a temperatura ambiente durante 1,5 h. Se pasó por una almohadilla corta de Celita. El filtrado se concentró al vacío para dar el producto deseado 5 (66 mg). Se utilizó directamente en el siguiente paso sin ninguna otra purificación. MS: m/z = 213 (M+H+)+.
[3-(Benciloxi)-6-(dimetilamino)-9H-carbazol-9-yl]metanol 7 : A una solución de 6-(benciloxi)-N-metil-9H-carbazol-3-amina 4 (110 mg, 0,38 mmol) y una solución acuosa de formaldehído (37%, 1,0 ml) en acetonitrilo (30 ml) se añadió NaB(OAc)3 (323 mg, 1,52 mmol). La mezcla resultante se agitó a temperatura ambiente durante 6 h y luego se concentró. El residuo se disolvió en H2O (30 ml), se extrajo con CH2Cl2 (2 * 30 ml). Las capas orgánicas combinadas se secaron (MgSO4) y se filtraron. El filtrado se concentró al vacío para dar el producto deseado (0,12 g). Se utilizó directamente en el siguiente paso sin ninguna otra purificación. MS: m/z = 321 (M+H+)+.
6-(Dimetilamino)-9-(metoximetil)-9H-carbazol-3-01 8: Una mezcla de [3-(benciloxi)-6-(dimetilamino)-9H-carbazol-9-il]metanol 7 (120 mg,), Pd/C (100 mg) y ácido acético (cantidad cat.) en MeOH (15 ml) se hidrogenó a temperatura ambiente durante 4 h. Se filtró a través de una almohadilla corta de Celita. El filtrado se concentró al vacío para dar el producto deseado (94 mg, 100 %). 1H RMN(acetona-d6, 400 MHz) 5: 7,38-7,50 (m, 4H), 7,05 (dd, J = 8,8, 2,4 Hz, 1H), 6,97 (dd, J = 8,4, 2,4 Hz, 1H), 5,62 (s, 2H), 3,20 (s, 3H), 2,94 (s, 6H). MS: m/z = 271 (M+H+)+.

Procedimientos generales para la preparación de derivados de carbazol O-alquilados: A una solución de derivados de carbazol-3-ol (1 eq.) y Cs2CO3 (1,5 eq.) en DMF (10 ml) se añadió una solución de 2-(2-(2-fluoroetoxi)etoxi)etoxi)etil-4-metilbencenosulfonato (1,2 eq.) en DMF (1,0 ml). La mezcla resultante se agitó a temperatura ambiente durante una noche y luego se concentró al vacío. El residuo se purificó mediante cromatografía en columna (gel de sílice, 5-50 % EtOAc/hexano) para obtener los productos deseados.
6-(2-(2-(2-Fluoroetoxi)etoxi)etoxi)-N-mef//-9H-carbazo/-3-am/ne 6: (3 mg, 5 %). 1H RMN(acetona-d6, 400 MHz) 5: 7,59 (d, J = 2,4 Hz, 1H), 7,28-7,33 (m, 2H), 7,26 (d, J = 8,4 Hz, 1H), 6,97 (dd, J = 8,8, 2,4 Hz, 1H), 6.85 (dd, J = 8,8, 2,0 Hz, 1H), 4,51 (dt, J = 48, 4,0 Hz, 2H), 4,19 (t, J = 4,4 Hz, 2H), 3,61-3,88 (m, 8H), 3,87 (s, 3H). MS: m/z = 347 (M+H+)+.
6-(2-(2-Fluoroetoxi)etoxi)etoxi)-9-(metoximetil)-N,N-dimetil-9H-carbazol-3-amina 9: (50 mg, 36 %). 1H RMN(acetona-d6, 400 MHz) 5: 7,68 (d, J = 2,4 Hz, 1H), 7,46-7,52 (m, 3H), 7,04-7,08 (m, 2H), 5,66 (s, 2H), 4,52 (dt, J = 48,4, 4,4 Hz, 2H), 4,21 (t, J = 4,8 Hz, 2H), 3,63-3,87 (m, 8H). MS: m/z = 405 (M+H+)+.

Procedimientos generales para la preparación de derivados de carbazol acilados: A una solución de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-carbazol (1,0 eq.) en DMF (3,0 ml) se añadió NaH (en exceso). Tras agitar a temperatura ambiente durante 5 minutos, se añadió un haluro de acilo (en exceso). La mezcla resultante se agitó a temperatura
ambiente durante una noche y luego se concentró al vacío. El residuo se purificó mediante cromatografía en columna (gel de sílice, 0-40 % EtOAc/hexano) para dar el producto deseado

1-(2-(2-(2-(2-Fluoroetoxi)etoxi)etoxi)-9H-carbazol-9-yl)etanona: (4 mg, 36 %). 1H RMN (CDCls, 400 MHz) 5: 8,21 (d, J = 8,0 Hz, 1H), 7,99-8,25 (m, 2H), 7,94 (d, J = 2,4 Hz, 1H), 7,36-7,46 (m, 2H), 7,06 (dd, J = 8,4, 2,4 Hz, 1H), 4,52 (dt, J = 48, 4,4 Hz, 2H), 4,27 (t, J = 4,4 Hz, 2H), 3,89 (t, J = 8,8 Hz, 2H), 3,64-3,78 (m, 6H), 2,91 (s, 3H). MS: m/z= 360 (M+H+)+.

1-(2-(2-(2-Fluoroetoxi)etoxi)etoxi)-9H-carbazol-9-il)fenilmetanona: (51 mg, 78 %). 1H RMN (CDCl3, 400 MHz) 5: 7,84-7,92 (m, 2H), 7,62-7,74 (m, 3H), 7,53 (t, J = 8,0 Hz, 2H), 7,27-7,33 (m, 2H), 7,17-7,23 (m, 1H), 6.99 (dd, J = 8,4, 2,4 Hz, 1H), 4,57 (dt, J = 47,6, 4,4 Hz, 2H), 4,06 (t, J = 4,8 Hz, 2H), 3,70-3,87 (m, 8H). MS: m/z = 321 (M+H+)+.
Preparación de 2-(7-formamido-9H-carbazol-2-iloxi)etil 4-metilbencenosulfonato: AD-CB-012P-WZ02039

El Compuesto 2-(7-formamido-9H-carbazol-2-iloxi)etil 4-metilbencenosulfonato(AD-CB-012P-WZ02039) se preparó utilizando el mismo procedimiento para la preparación de AD-CB-012S-WZ01185) a partir de N-(7-hidroxi-9H-carbazol-2-il)formamida (100 mg) y etano-1,2-diil bis(4-metilbencenosulfonato) (325 mg). (sólido blanco, 22 mg, 12 %). Para el rotómero mayor: 1H RMN (400 MHz, acetona-d6) 510,19 (s, 1 H), 9,31 (s, 1 H), 8,38(d, J = 1,6 Hz, 1 H), 8,11 (d, J = 2,0 Hz, 1 H), 7,90-7,81 (m, 4 H), 7,45 (d, J = 8,4 Hz, 2 H), 7.19 (dd, J = 8,4, 2,0 Hz, 1 H), 6,95 (d, J = 2,0 Hz, 1 H), 6,69 (dd, J = 8,4, 2,0 Hz, 1 H), 4,43-4,41 (m, 2 H), 4,29-4,27 (m, 2 H); MS(ESI) m/z 425 (M+H+).
Preparación de N-(7-(4-fluorobutoxi)-9H-carbazol-2-il)formamida: AD-CB-30S-WZ02055

El compuesto N-(7-(4-fluorobutoxi)-9H-carbazol-2-il)formamida(AD-CB-30S-WZ02055) se preparó utilizando el mismo procedimiento para la preparación de AD-CB-012S-WZ01185) a partir de N-(7-hidroxi-9H-carbazol-2-il)formamida (20 mg) y 1-bromo-4-fluorobutano (27 mg). (sólido blanco, 11 mg, 42 %). 1H Rm N (400 MHz, acetona-d6) 510,18 (s, 1 H), 9,31 (s, 1 H), 8,39 (d, J = 2,0 Hz, 1 H), 8,11 (d, J = 2,0 Hz, 1 H), 7,95 (d, J = 1,6 Hz, 2 H), 7,88 (d, J = 2,0 Hz, 1 H), 720 (dd, J = 8,4, 2,0 Hz, 1 H), 7,03 (d, J = 2,4 Hz, 1 H), 6,79 (dd, J = 8,4, 2,4 Hz, 1 H), 4,61 (m, 1 H), 4,49 (m, 1 H), 4,11 (m, 2 H), 1,97-1,88 (m, 4 H); MS(ESI) m/z 301 (M+H+).
Preparación del clorhidrato de 2-(2-(2-fluoroetoxi)etoxi)-9H-pirido[2,3-b]indol-7-amina:

Preparación de WZ02045:
A la 4-cloro-3-nitroanilina (2,5 g, 14,5 mmol) en 40 ml de DCM se añadió TEA (2,9 g, 29 mmol), DMAP (177 mg, 1,45 mmol) y dicarbonato de d-tert-butilo (4,7 g, 21,7 mmol). La mezcla se agitó ata durante 24 h y se concentró. El residuo se diluyó con Et2O (100 ml), se lavó con salmuera (100 ml), agua (100 ml), HCl 0,5 M (2x100 ml) y salmuera (100 ml), se secó sobre MgSO4 y se concentró. El producto crudo se purificó por cromatografía de sílice (EtOAc/hexano) para obtener el 4-cloro-3-nitrofenilcarbamato de tert-butilo (WZ02045) como un sólido amarillo (1,5 g, 38%). MS(ESI) m/z 295 (M+Na+).
Preparación de WZ02049:
Una mezcla de 4-cloro-3-nitrofenilcarbamato de tert-butilo (818 mg, 3 mmol), 2-(benciloxi)-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridina (933 mg, 3 mmol), tetrakis(trifenilfosfina)paladio (104 mg, 0,09 mmol), 10 ml de dioxano y 6 ml de Na2CO3 1 M se calentó a reflujo durante 15 h. Se diluyó con 50 ml de Et2O y se lavó con salmuera (2x50 ml) y se secó sobre MgSO4 y se concentró. El producto bruto se purificó mediante cromatografía de sílice (EtOAc/hexano) para obtener 4-(6-(benciloxi)piridin-3-il)-3-nitrofenilcarbamato de tert-butilo (WZ02049) como una cera amarilla (1,2 g, 95 %). MS(ESI) m/z 444 (M+Na+).
Preparación de WZ02057:
Una suspensión del compuesto anterior (800 mg, 1,9 mmol) en 2 ml de fosfito de tritilo se calentó a 148 °C durante 15 h. Después de enfriar, se concentró a presión reducida para eliminar los volátiles. El producto bruto se purificó mediante cromatografía de sílice (EtOAc/hexano) para obtener 2-(benciloxi)-9H-pirido[2,3-b]indol-7-ilcarbamato de tert-butilo (WZ02057) como un sólido blanquecino (400 mg, 54 %). Ms (ESI) m/z 390 (M+HT).
Preparación de WZ02061:
Al compuesto anterior (220 mg, 0,56 mmol) disuelto en 80 ml de MeOH se añadió paladio sobre carbón activado (80 mg). La mezcla se agitó ata bajo atmósfera de H2 durante 15 h. El sólido se filtró y el filtrado se concentró para obtener 2-hidroxi-9H-pirido[2,3-b]indol-7-ilcarbamato de tert-butilo (WZ02061) como un sólido blanco (105 mg, 100 %). Este material se utilizó directamente para la siguiente reacción sin purificación. MS(ESI) m/z 300 (M+H+).
Preparación de WZ02063:
Al compuesto anterior (50 mg, 0,167 mmol) en 1 nl de NMP se añadió 2-(2-(2-fluoroetoxi)etoxi)etil 4-metilbencenosulfonato (76 mg, 0,25 mmol), y Cs2CÜ3 (65 mg, 0,2 mmol). La mezcla se agitó a ta durante 15 h y se diluyó con Et2O (40 ml), se lavó con agua (3x30 ml), se secó sobre MgSO4 y se concentró. El producto bruto se purificó mediante cromatografía de sílice (EtOAc/hexano) para obtener 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-pirido[2,3-b]indol-7-ilcarbamato de tert-butilo (WZ02063) como una cera clara (45 mg, 62 %). MS(ESl) m/z 434 (M+H+).
Preparación de AD-CB-032S-WZ02067:
El compuesto anterior (45 mg, 0,1 mmol) se trató con 2 ml de una solución de HCl 4 M en dioxano a ta durante 5 h y se concentró a presión reducida. El residuo se lavó con éter (5 ml) y se secó a alto vacío para obtener clorhidrato de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-pirido[2,3-b]indol-7-amina (Ad -c B-032S-WZ02067) como un sólido amarillo claro (23 mg, 62 %). 1H RMN (400 MHz, metanol-d4) 58,42 (d, J = 8,4 Hz, 1 H), 8,12 (d, J = 8,4 Hz, 1 H), 7,53 (d, J = 2,0Hz, 1 H), 7,21 (dd, J = 8,4, 2,0 Hz, 1 H), 6.77 (d, J = 8,4 Hz, 1 H), 4,58-4,54 (m 3 H), 4,43 (m, 1 H), 3,91 (m, 2 H), 3,76 3,72 (m, 3 H), 3,70-3,66 (m, 3 H); MS(ESI) m/z 334 (M+H+).
Preparación de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-N-metil-9H-pirido[2,3-b]indol-7-amina: AD-CB-034S-WZ02069 El compuesto AD-CB-034S-WZ02069 se preparó utilizando el mismo procedimiento para la preparación de AD-CB-004S a partir de clorhidrato de 2-(2-(2-fluoroetoxi)etoxi)etoxi)-9H-pirido[2,3-b]indol-7-amina (AD-CB-032S-WZ02067, 20 mg) (10 mg, 53 %). 1H RMN (400 MHz, metanol-d4) 58,06 (d, J = 8,0 Hz, 1 H), 7,66 (d, J = 8,4 Hz, 1 H), 6,65 (d, J = 2,0 Hz, 1 H), 6,58 (dd, J = 8,4, 2,0 Hz, 1 H), 6.53 (d, J = 8,0 Hz, 1 H), 4,58 (m 1 H), 4,53-4,45 (m, 3 H), 3,88 (m, 2 H), 3,76 (m, 1 H), 3,73-3,67 (m, 5 H), 3,03 (s, 3 H); MS(ESI) m/z 348 (M+H+).
Preparación del 6-bromo-9H-carbazol-2-ol: W138

Preparación de WZ02013:
A 9H-carbazol-2-ol (915 mg, 5 mmol) en 10 ml de DMF y 20 ml de DCM se añadió TEA (1,0 g, 10 mmol), seguido de cloruro de acetilo (589 mg, 7,5 mmol) a 0 °C. La mezcla de reacción se agitó a ta durante 1 h y se vertió en hielo (50 g). La mezcla se extrajo con EtOAc (2x60 ml) y la fase orgánica combinada se secó sobre MgSO4 y se concentró. El producto crudo se purificó por cromatografía de sílice para obtener acetato de 9H-carbazol-2-il (WZ02013) como un sólido blanquecino (800 mg, 71 %). MS(ESI) m/z 348 (M+H+).
Preparación de WZ02025:
A una solución de acetato de 9H-carbazol-2-ilo (500 mg, 2,2 mmol) en DCM (40 ml) se añadió una solución de NBS en 25 ml de DCM gota a gota a ta. La mezcla de reacción se agitó en la oscuridad durante 5 h. Se lavó con agua (3x50 ml), se secó sobre MgSO4 y se concentró. El producto bruto se purificó mediante cromatografía de sílice (EtOAc/hexano) para obtener acetato de 6-bromo-9H-carbazol-2-ilo (WZ02025) como un sólido blanquecino (250 mg, que contenía 17 % de producto dibromado). MS(ESI) m/z 305 (M+H+).
Preparación de W138:
Una suspensión de acetato de 6-bromo-9H-carbazol-2-ilo (200 mg, 0,65 mmol) en 30 ml de MeOH y 4 ml de LiOH acuoso 1,0 M se agitó durante 5 h. Se neutralizó con HCl 1 M y se concentró. El producto bruto se purificó mediante cromatografía de sílice (EtOAc/hexano) para obtener 6-bromo-9H-carbazol-2-ol(W138) como un sólido blanquecino (125 mg, que contenía 15 % de producto dibromado). 1H RMN (400 MHz, acetona-d6) 58,58 (s, 1 H), 8,10 (d, J = 2,0 Hz, 1 H), 7,92 (d, J = 8,8 Hz, 1 H), 7,42 (dd, J = 8,4, 2,0 Hz, 1 H), 7,35 (s, 1 H), 7,13 (d, J = 8,4 Hz, 1 H), 6,92 (d, J = 2,0 Hz, 1 H), 6,76 (dd, J = 8,8, 2,0 Hz, 1 H); MS(ESI) m/z 263 (M+H+).
Figura 7: Ensayo de competencia ex vivo con tinción autorradiográfica de amiloide (corte de cerebro de paciente con EA).
Tinción de autorradiografía de amiloide (ex vivo) del cerebro de un paciente con AD con [18F]-CB-001 muestra una buena unión del amiloide y poca/ninguna unión de la materia blanca
Figura 8: Los estudios de competencia de [18F]-CB001 en rebanadas del cerebro de la AD demuestran la unión reversible de la placa y la competencia con PiB, y poca/ninguna unión de la materia blanca.
La serie de carbazoles de los agentes de obtención de imágenes de la AD muestran cualidades sorprendentes cuando se comparan con los resultados previamente establecidos por otros. Los datos del estado de la técnica sugieren que los compuestos con valores LogP más altos tienen afinidades amiloides más altas, pero estos mismos compuestos también pueden sufrir una unión no específica alta, es decir, un lavado cerebral deficiente (J. Molecular Neuroscience 2003,20, 255-260). Para los estudios divulgados en esta solicitud, se utilizaron los valores cLogP en lugar de los valores LogP.
Se realizó un estudio para examinar las relaciones de unión entre la materia gris y la materia blanca para 4 trazadores diferentes: CB-001, CB-003, FDDNP y F-PiB. En este estudio no se examinó un agente de obtención de imágenes conocido que contiene carbazol, el 18F-fluorocarazolol, debido a su valor cLogP relativamente bajo (2,77) en comparación con el FDDNP y el PiB, y a su captación específica competitiva en los beta-adrenoceptores. Además, no hay datos del estado de la técnica que sugieran que el 18F-fluorocarazolol se une a las placas de la EA. Después de incubar los cortes de cerebro humano de pacientes con AD con un trazador determinado durante 30 minutos, los cortes se lavaron con varias soluciones de EtOH: agua en un intento de optimizar las proporciones de materia gris y blanca. Los resultados fueron sorprendentes e inesperados a la vista de los trabajos anteriores realizados por otros investigadores. El CB-001 tiene un PCLc ligeramente más alto que el FDDNP (3,8 frente a 3,4) y se esperaría que tuviera un peor lavado que el FDDNP con base en estos valores. Sin embargo, a pesar de la diferencia en los valores de cLogP, el CB-001 tiene una menor propensión de unión no específica y muestra una relación mucho mejor entre la materia gris y la materia blanca en comparación con el FDDNP (véase la sección anterior, "lavado original"). Más concretamente, la unión a la materia blanca de FDDNP es varios tonos más oscura que la unión a la materia blanca de CB-001, lo que indica una baja unión no específica de CB-001. Por el contrario, el F-PiB, que tiene un valor de cLogP de 3,99, también muestra unos proporciones de unión razonables, similares a los del CB-001, aunque mostrando una señal global muy débil. Los datos de lavado sugieren que los carbazoles son un objetivo viable y novedoso para la obtención de imágenes de objetivos relacionados con la AD debido a sus propiedades únicas de unión y lavado.
Para ampliar estos resultados, se preparó y probó el CB-003, un trazador con un valor cLogP similar al FDDNP. Utilizando unas condiciones de lavado mucho más suaves que las duras, el CB-003 mostró unas excelentes proporciones de unión entre la materia gris y la blanca que son muy superiores a los resultados obtenidos con el FDDNP, el PiB y el CB-001. Estos resultados favorables y únicos sugieren que el CB-003 tendría un lavado cerebral más favorable en sistemas vivos, lo que llevaría a una captación más específica y a una menor unión no específica, lo que supondría una clara ventaja sobre el FDDNP y la imagen PiB.
Resumen de los resultados del lavado:

(continuación)
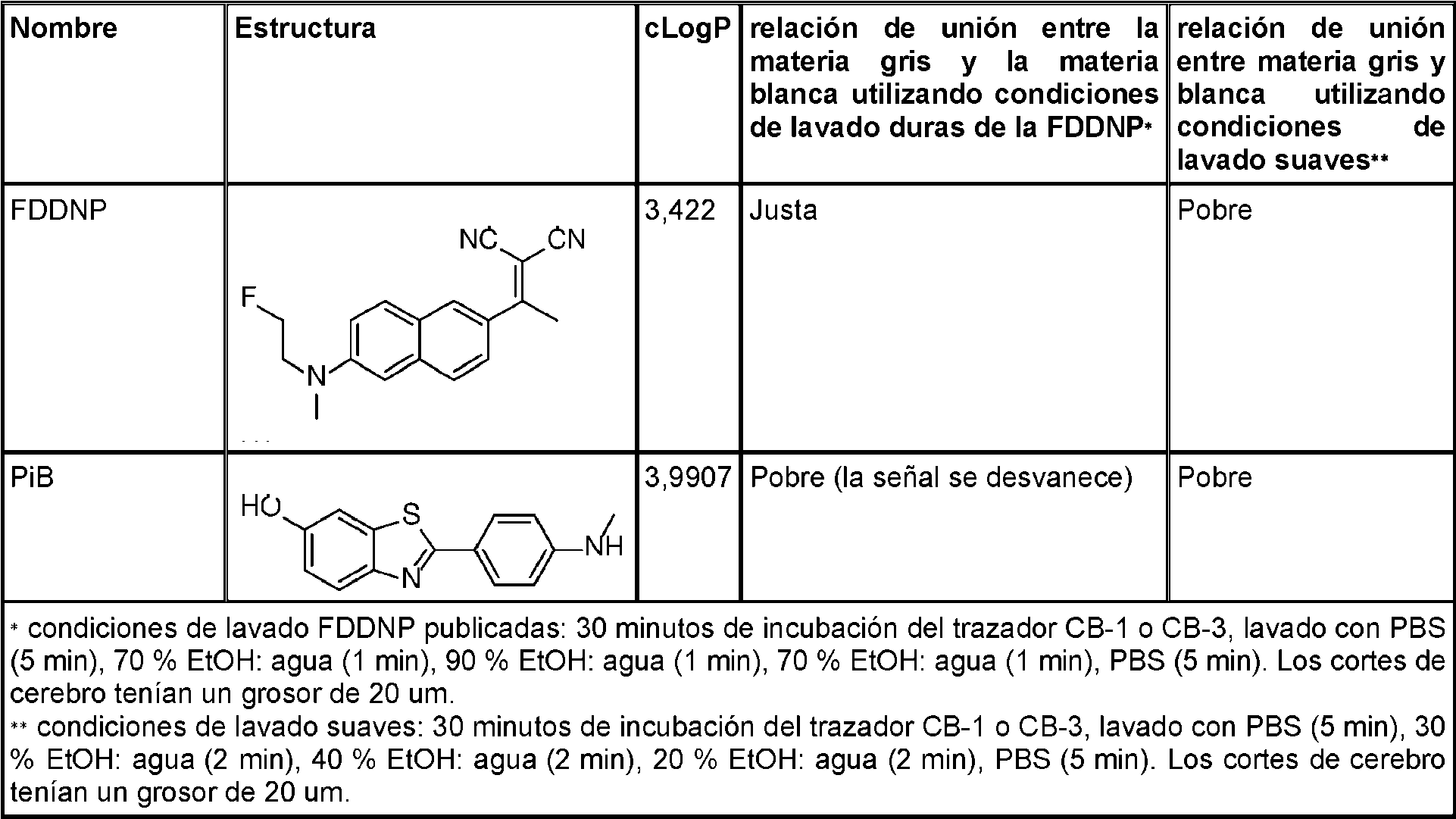
Los resultados demuestran que 1) PiB bloquea la tinción con [18F]-CB001 con concentraciones crecientes, sugiriendo que los dos compuestos compiten por los mismos bolsillos de unión del amiloide; 2) PiB parece bloquear la unión del trazador con la misma fuerza que el CB001 frío, sugiriendo que ambos tienen afinidades de unión similares; 3) FDDNP es mucho menos capaz de bloquear la tinción con [18F]-CB001, debido a su menor afinidad de unión al amiloide. Figura 9: El protocolo óptimo de tinción y lavado que indica el trazador específico.
Estos datos sugieren el siguiente orden de unión (no específica) de la materia blanca:
FDDNP > CB001 > [18F]-PiB > CB003
Figura 10: el [18F]-CB003 distingue claramente entre los cerebros con Alzheimer y los normales.
Figura 11A: Bloqueo dependiente de la concentración de la unión tisular de [18F]-PiB con PiB y CB003.
Figura 11B: Determinación del IC50 con [18F]-PiB mediante ensayo de competencia ex vivo utilizando tinción de autorradiografía.

Para demostrar aún más la eficacia del empleo de estos trazadores relacionados con el CB como agentes de obtención de imágenes de la EA, se utilizó el CB-003 para diferenciar claramente entre un cerebro sano y un cerebro con EA. Más concretamente, al utilizar el protocolo de lavado suave, los depósitos de amiloide eran claramente visibles en la materia gris con poca captación de materia blanca. Los resultados fueron corroborados tanto por la IHC de anticuerpos como por la tinción de amiloide T, confirmando la especificidad de la captación. Estos sorprendentes resultados
demuestran que este trazador posee la cualidad única de un rápido lavado de la materia blanca y una alta y significativa captación en la materia gris que es específica para las placas de la EA.
Los carbazoles compiten directamente contra el 18F-PiB por los mismos sitios de unión en los cerebros humanos de la EA. Este sorprendente resultado no podría haberse predicho dadas sus diferentes estructuras y la falta de CB-003 de un grupo fenólico OH y NH-Me terminal, que se consideran esenciales para la unión a las placas de EA. A pesar de que el CB-003 carece de estos dos grupos funcionales, sigue compitiendo con el 18F-PiB por los sitios de unión en los cerebros humanos de la EA. Debido a la simplicidad de su estructura, los rendimientos de marcaje de CB-001 y CB-003 son excepcionalmente altos y mejores que los rendimientos de marcaje de 18F-PiB.
Ensayo de Resonancia Plasmónica Superficial (SPR)
Se desarrolló un ensayo utilizando un instrumento Biacore que introdujo los ligandos sobre proteínas objetivo inmovilizadas en la superficie de oro y midió las tasas resultantes de asociación y disociación para probar varios diversos que se unen a oligómeros, polímeros y fibrillas solubles de EA.
[Figuras 12 a 17]
La serie de carbazoles también demostró una capacidad única y sorprendente de unirse favorable y preferentemente a agregados insolubles (9 nM) sobre agregados solubles (262 nM). PiB también se une bien a los agregados insolubles (16 nM) pero también se une esencialmente igual de bien a los agregados solubles (48 nM). Para las aplicaciones de obtención de imágenes en las que es favorable distinguir entre la unión de un trazador a agregados insolubles frente a los solubles, el CB-003 proporciona una mayor relación de unión de 29:1, mientras que el PiB sólo proporciona una relación de 3:1. Por lo tanto, CB-003 puede proporcionar una información de unión más selectiva en relación con PiB. Los resultados indican que 1) para la unión de agregados solubles, PIB > CB3 > CB4; y 2) para la unión de agregados insolubles, PIB = CB3 > CB4.
Figura 18: La obtención de imágenes de microPET con [18F]-CB-001 en ratones App y WT demuestran una muy buena captación cerebral.
Figura 19: La obtención de imágenes de MicroPET con [18F]-CB-003 en ratones WT y App.
[Figuras 20 y 21]
Los resultados demuestran que 1) los ratones WT y App muestran diferencias estadísticamente significativas en la retención del trazador en el cerebro; 2) los ratones App muestran una relación cerebro / músculo hasta 25 % mayor en comparación con los ratones WT. Los carbazoles muestran tanto una sorprendente alta captación en los cerebros de los ratones (tanto WT como APP) como un lavado lo suficientemente lento como para poder distinguir los ratones WT de los APP. Sin estar atados a ninguna teoría propuesta en el presente documento, se especula que la razón detrás de estos resultados puede ser que el CB-003 posee una tasa de lavado más rápida que el 18F-PiB, lo cual es consistente con los datos de tinción: el 18F-PiB requiere unas condiciones de lavado más duras para dar una relación razonable de materia gris y blanca. El rápido lavado del CB-003 es presumiblemente un factor importante para su baja unión no específica, aunque el lavado es lo suficientemente lento como para distinguir el WT del APP. Esto sugiere que los carbazoles muestran una combinación única de excelentes propiedades de lavado y retención en los cerebros humanos de la AD que no son evidentes a partir de los datos de la técnica anterior. El CB-003, al ser un compuesto neutro, también poseería potencialmente mayores valores de captación frente a los agentes de obtención de imágenes con base en zwitteriónica, como tal el azul de metileno. La comparación de [18F]PiB y [18F]-CB003 en el cerebro de ratones WT y App indica que el aclaramiento cerebral de [18F]-CB-003 es mucho más rápido que con [18F]PiB.
Figura 22:
[18F]PiB.
Figura 23:
[18F]-CB003.
Claims (10)
1. Un compuesto de la siguiente Fórmula I:

F ó rm u la I
y sales farmacéuticamente aceptables de la misma;
en la que A es N o CR1; B es N o CR2, J es N o CR3, K es N o CR4; L es N o CR5; M es N; P es N o CR7; y Q es N o CR8; siempre que no más de dos de A, B, J, K, L, M, P y Q puedan ser N;
en la que X es un enlace;
en el que R1, R2, R4, R5, R67, y R8 son cada uno independientemente hidrógeno o se seleccionan cada uno independientemente del grupo que consiste en alquilo C1-6, cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroarilalcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxiC1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquil C1-5 NR10C(O)-, (alquil C1-6)2NC(O)CH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C1-5C(O)O-, arilo C6-10C(O)-, y arilo C6-10c (o )O- y R3 se selecciona del grupo que consiste en cicloalquilo C3-6, cicloalquil C3-12 alquilo C1-5, heteroaril alcoxi C2-5, arilo C6-14, aril C6-10 alquilo C1-4, heteroarilo, cicloalcoxi C3-6, cicloalquil C3-12 alcoxi C1-5, ariloxi C6-14, aril C6-10 alcoxi C1-4, heteroariloxi, alquilo C1-5NR10C(O)-, (alquil C1-6)2NC(O)cH(alquilo C1-5)-, alquilo C1-5NR10C(O)O-, alquilo C1-5C(O)-, alquilo C |-5C(O)O-, arilo C6-10C(O)-, y arilo C6-10C(O)O-;
en la que al menos cuatro de R1, R2, R4, R5, R7, y R son hidrógenos;
en la que R9 es hidrógeno;
en la que cada R10 es independientemente H o alquilo C1 - 6 ; y
en el que los sustituyentes en R1, R2, R3, R4, R5, R7 R8 y R10 están opcionalmente sustituidos adicionalmente por sustituyentes seleccionados del grupo que consiste en amino, halo, cyano, nitro, hidroxilo, -SH, -S alquilo C1-6, -C(O)NH2 , -C(S)NH2 , halo alquilo C1-6, perhalo alquilo C1.6, alquilo C1.6, cicloalquilo C3-6, cicloalquilo C3-12, arilo C6-14 y heteroarilo.
2. El compuesto de la reivindicación 1 que está radiomarcado, en el que el radiomarcado comprende un radionúclido seleccionado del grupo que consiste en 11C, 13N, 15O, 18F, 123I, 124I, 125I, 131I y 77Br, y sales farmacéuticamente aceptables de los mismos.
3. El compuesto de la reivindicación 1 o 2 para su uso en el tratamiento de una enfermedad o condición en un mamífero que lo necesite, seleccionado del grupo que consiste en ansiedad, depresión, esquizofrenia, enfermedad de Alzheimer, enfermedad relacionada con el estrés, pánico, una fobia, trastorno obsesivo compulsivo, obesidad, síndrome de estrés postraumático o epilepsia.
4. Una composición farmacéutica para la obtención de imágenes in vivo de los depósitos de amiloide, que comprende:
un compuesto de la reivindicación 2; y
un portador farmacéuticamente aceptable.
5. El compuesto o composición de la reivindicación 2 o 4 para su uso en un procedimiento de diagnóstico de la enfermedad de Alzheimer o de una predisposición a la misma en un mamífero, comprendiendo el procedimiento:
a. administrar al mamífero una cantidad diagnóstica eficaz del compuesto radiomarcado o de la composición de la reivindicación 2 o 4, en la que el compuesto pasa la barrera hematoencefálica y se une preferentemente a los oligómeros, polímeros y fibrillas solubles de la AD en un tejido cerebral;
b. permitir que el compuesto se distribuya en el tejido cerebral; y
c. obtener imágenes del tejido cerebral, en las que un aumento de la unión del compuesto al tejido cerebral en comparación con un nivel de control normal de unión indica que el mamífero padece o corre el riesgo de padecer la enfermedad de Alzheimer.
6. El compuesto o composición de la reivindicación 2 o 4 para su uso de acuerdo con la reivindicación 5, en el que el compuesto se une preferentemente a las fibrillas.
7. El compuesto o composición de la reivindicación 2 o 4 para su uso de acuerdo con la reivindicación 5, en el que el tejido cerebral comprende una región frontotemporal o la región del hipocampo.
8. El compuesto o composición de la reivindicación 2 o 4 para su uso de acuerdo con la reivindicación 5, en el que el aumento de la unión es al menos 10 % mayor que dicho valor de control normal.
9. El compuesto o composición de la reivindicación 2 o 4 para su uso de acuerdo con cualquiera de las reivindicaciones 5 a 8, en el que el compuesto se administra por inyección intravenosa.
10. El compuesto de la reivindicación 2 para su uso en un procedimiento de diagnóstico de la enfermedad de Alzheimer o de una predisposición a la misma en un mamífero, comprendiendo el procedimiento:
a. administrar al mamífero una cantidad diagnósticamente eficaz de un compuesto de la reivindicación 2, en el que el compuesto pasa la barrera hematoencefálica y se une preferentemente a oligómeros, polímeros, fibrillas, tau hiperfosforilada, ovillos neurofibrilares, filamentos helicoidales pareados y/u oligómeros solubles neurotóxicos de la AD en un cerebro;
b. emplear una técnica de obtención de imágenes nuclear seleccionada del grupo que consiste en la tomografía de emisión de positrones (PET) y la tomografía computarizada de emisión única (SPECT) para monitorizar o visualizar una distribución del compuesto radiomarcado dentro del cerebro o dentro de una porción del mismo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US6610108P | 2008-02-14 | 2008-02-14 | |
| PCT/US2009/000961 WO2009102498A1 (en) | 2008-02-14 | 2009-02-17 | Novel imaging agents for detecting neurological dysfunction |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2907992T3 true ES2907992T3 (es) | 2022-04-27 |
| ES2907992T5 ES2907992T5 (en) | 2025-03-19 |
Family
ID=40627498
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES09711194T Active ES2907992T5 (en) | 2008-02-14 | 2009-02-17 | Novel imaging agents for detecting neurological dysfunction |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8318132B2 (es) |
| EP (2) | EP2247558B2 (es) |
| JP (1) | JP2011512354A (es) |
| KR (1) | KR20100135235A (es) |
| CA (1) | CA2715390A1 (es) |
| CY (1) | CY1125258T1 (es) |
| DK (1) | DK2247558T4 (es) |
| ES (1) | ES2907992T5 (es) |
| FI (1) | FI2247558T4 (es) |
| HR (1) | HRP20220401T4 (es) |
| HU (1) | HUE058352T2 (es) |
| LT (1) | LT2247558T (es) |
| PL (1) | PL2247558T5 (es) |
| PT (1) | PT2247558T (es) |
| SI (1) | SI2247558T2 (es) |
| WO (1) | WO2009102498A1 (es) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2247558B2 (en) | 2008-02-14 | 2024-07-03 | Eli Lilly and Company | Novel imaging agents for detecting neurological dysfunction |
| US8932557B2 (en) | 2008-02-14 | 2015-01-13 | Eli Lilly And Company | Imaging agents for detecting neurological dysfunction |
| JP2011530495A (ja) * | 2008-08-08 | 2011-12-22 | ノイロサーチ アクティーゼルスカブ | ニコチン性アセチルコリン受容体のモジュレーターとして有用な新規のジフェニル1,2,3−トリアゾール誘導体 |
| JP5771525B2 (ja) | 2008-09-18 | 2015-09-02 | セダーズ−シナイ メディカル センター | アルツハイマー病を検出するための光学的方法 |
| US8691187B2 (en) * | 2009-03-23 | 2014-04-08 | Eli Lilly And Company | Imaging agents for detecting neurological disorders |
| GB201016038D0 (en) | 2010-09-24 | 2010-11-10 | Ge Healthcare Ltd | In vivo imaging method |
| RU2627694C2 (ru) * | 2010-10-29 | 2017-08-10 | Клино Лтд. | Производные хинолина, визуализирующий белок тау |
| WO2013020184A1 (en) * | 2011-08-11 | 2013-02-14 | Neuprotect Pty Ltd | Flavonoid compounds, and methods of use thereof |
| US20130315825A1 (en) * | 2012-05-03 | 2013-11-28 | Washington University | Tricyclic heteroaromatic compounds as alpha-synuclein ligands |
| CN104781234A (zh) * | 2012-05-22 | 2015-07-15 | 伊莱利利公司 | 用于检测神经功能障碍的基于咔啉和咔唑的成像剂 |
| DK2767532T3 (en) | 2012-12-21 | 2016-09-12 | Nat Inst For Quantum And Radiological Science And Tech | New connection for reproduction of tau protein heavy in the brain. |
| US20160115162A1 (en) | 2013-05-31 | 2016-04-28 | The General Hospital Corporation | Radiosynthesis of Tau Radiopharmaceuticals |
| AU2014302550A1 (en) | 2013-06-25 | 2016-02-11 | Bristol-Myers Squibb Company | Carbazole carboxamide compounds useful as kinase inhibitors |
| TWI648272B (zh) | 2013-06-25 | 2019-01-21 | 美商必治妥美雅史谷比公司 | 經取代之四氫咔唑及咔唑甲醯胺化合物 |
| CN105358558B (zh) * | 2013-10-08 | 2017-09-26 | 豪夫迈·罗氏有限公司 | 作为tau‑pet‑配体的二氮杂咔唑衍生物 |
| TWI667233B (zh) | 2013-12-19 | 2019-08-01 | 德商拜耳製藥公司 | 新穎吲唑羧醯胺,其製備方法、含彼等之醫藥製劑及其製造醫藥之用途 |
| WO2015110263A1 (en) * | 2014-01-21 | 2015-07-30 | Ac Immune Sa | Carbazole and carboline compounds for use in the diagnosis, treatment, alleviation or prevention of disorders associated with amyloid or amyolid-like proteins |
| HUE053939T2 (hu) | 2014-05-13 | 2021-08-30 | Hoffmann La Roche | Deuterált heterociklikus vegyületek és képalkotó kontrasztanyagként való használatuk |
| CN107074856A (zh) * | 2014-09-05 | 2017-08-18 | 阿略斯泰罗斯医疗公司 | CaMKII抑制剂及其用途 |
| KR20170052626A (ko) | 2014-09-05 | 2017-05-12 | 심바이오믹스 세러퓨틱스 엘엘씨 | 세균질증 치료에 사용하기 위한 세크니다졸 |
| MX2017005060A (es) | 2014-10-24 | 2017-07-05 | Bristol Myers Squibb Co | Compuestos atropisomeros triciclicos. |
| PE20190710A1 (es) | 2014-10-24 | 2019-05-17 | Bristol Myers Squibb Co | Compuestos de indol carboxamida utiles como inhibidores de cinasas |
| RS59707B1 (sr) | 2014-10-24 | 2020-01-31 | Bristol Myers Squibb Co | Derivati karbazola |
| EA036518B1 (ru) * | 2015-02-02 | 2020-11-18 | Юсб Байофарма Спрл | Производные 9h-пирролодипиридина |
| CN105524034B (zh) * | 2015-03-27 | 2018-08-17 | 北京大学 | 香豆素衍生物的制备、药理作用及治疗脑缺血的应用 |
| CN105566269B (zh) * | 2015-03-27 | 2018-07-10 | 北京大学 | 香豆素衍生物的制备、药理作用及治疗瘙痒的应用 |
| US11253501B2 (en) | 2015-06-01 | 2022-02-22 | Lupin Inc. | Secnidazole formulations and use in treating bacterial vaginosis |
| US20180264146A1 (en) | 2015-07-15 | 2018-09-20 | Ac Immune S.A. | Novel imaging compounds |
| EP3118202A1 (en) | 2015-07-15 | 2017-01-18 | AC Immune S.A. | Dihydropyridopyrrole derivatives as tau-pet-ligands |
| CA3003884C (en) | 2015-11-13 | 2020-04-21 | Eli Lilly And Company | Azetidine derivatives for tau imaging |
| EA035621B1 (ru) * | 2016-02-03 | 2020-07-16 | Янссен Фармацевтика Нв | Лиганды для pet-визуализации тау-белков |
| WO2018015546A1 (en) | 2016-07-22 | 2018-01-25 | Ac Immune S.A. | Compounds for imaging tau protein aggregates |
| KR102472003B1 (ko) | 2016-07-22 | 2022-11-29 | 에이씨 이뮨 에스에이 | 타우 단백질 응집체를 이미징하기 위한 화합물 |
| WO2018024643A1 (en) | 2016-08-02 | 2018-02-08 | Ucb Biopharma Sprl | 9h-pyrrolo-dipyridine derivatives |
| WO2018024642A1 (en) | 2016-08-02 | 2018-02-08 | Ucb Biopharma Sprl | 9h-pyrrolo-dipyridine derivatives |
| MX375720B (es) | 2016-09-09 | 2025-03-04 | Hoffmann La Roche | Proceso para preparacion de 2-(6-nitropiridin-3-il)-9h-dipirido[2,3-beta;3',4'-d]pirrol. |
| TW201906818A (zh) * | 2017-05-31 | 2019-02-16 | 美商511製藥公司 | 新穎氘取代之正子發射斷層掃描(pet)顯影劑及其藥理應用 |
| US20210041447A1 (en) | 2018-01-24 | 2021-02-11 | Ac Immune Sa | Azacarboline compounds for the detection of tau aggregates |
| EP3743423A1 (en) | 2018-01-24 | 2020-12-02 | AC Immune SA | Gamma-carboline compounds for the detection of tau aggregates |
| WO2019168170A1 (ja) * | 2018-03-02 | 2019-09-06 | 国立大学法人 長崎大学 | クロモン誘導体及びアミロイド関連疾患診断用組成物 |
| TW201946624A (zh) | 2018-05-09 | 2019-12-16 | 新旭生技股份有限公司 | 雜芳基化合物及其用途 |
| US12352719B2 (en) | 2019-09-20 | 2025-07-08 | KYCERA AVX Components Corporation | Somatic cell-based electrical biosensor |
| CN114728071B (zh) | 2019-11-13 | 2024-12-06 | 新旭生技股份有限公司 | 用于降解tau蛋白聚集体的化合物及其用途 |
| US12280037B2 (en) | 2020-09-22 | 2025-04-22 | Evofem Biosciences, Inc. | Method and pharmaceutical composition for treating or preventing trichomoniasis and uses thereof |
| WO2023034346A1 (en) * | 2021-08-30 | 2023-03-09 | Pretzel Therapeutics, Inc. | Chromen-2-one modulators of polrmt |
| WO2023034339A1 (en) * | 2021-08-30 | 2023-03-09 | Pretzel Therapeutics, Inc. | Hydroxy and alkoxy coumarins as modulators of polrmt |
| US11938127B2 (en) | 2021-09-28 | 2024-03-26 | Wayne State University | Methods and compositions relating to steroid hormone receptor-dependent proliferative disorders |
| PT4426434T (pt) | 2021-11-02 | 2025-11-24 | Flare Therapeutics Inc | Agonistas inversos de pparg e suas utilizações |
| EP4511361A1 (en) * | 2022-04-21 | 2025-02-26 | Virginia Commonwealth University | Nlrp3 inflammasome inhibitors and compositions and uses thereof |
| WO2023205463A1 (en) | 2022-04-22 | 2023-10-26 | Vertex Pharmaceuticals Incorporated | Heteroaryl compounds for the treatment of pain |
| KR20250137711A (ko) | 2023-02-02 | 2025-09-18 | 일라이 릴리 앤드 캄파니 | 타우 영상화를 위한 신규 화합물 |
| WO2025155625A1 (en) | 2024-01-16 | 2025-07-24 | Eli Lilly And Company | Novel compounds for tau imaging |
Family Cites Families (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5741476A (en) * | 1992-12-21 | 1998-04-21 | Mallinckrodt Medical, Inc. | Polyhydric phenol compounds |
| CN1048731C (zh) * | 1993-09-17 | 2000-01-26 | 史密丝克莱恩比彻姆公司 | 编码细胞因子抑制性抗炎药物结合蛋白质的多核苷酸 |
| US6417178B1 (en) | 1994-07-19 | 2002-07-09 | University Of Pittsburgh | Amyloid binding nitrogen-linked compounds for the antemortem diagnosis of alzheimer's disease, in vivo imaging and prevention of amyloid deposits |
| BR9611047A (pt) * | 1995-10-17 | 2000-03-08 | Searle & Co | Processo de deteção de ciclo oxigenase-2 |
| JPH09165378A (ja) * | 1995-12-15 | 1997-06-24 | Seitai Kinou Kenkyusho:Kk | N1−[18f]フルオロベンジル複素環化合物およびそれらの製造法 |
| ZA989365B (en) | 1997-10-15 | 1999-04-15 | Boehringer Mannheim Pharm Corp | Preparation for treating alzheimer's disease |
| JP2001048786A (ja) | 1999-08-05 | 2001-02-20 | Yamanouchi Pharmaceut Co Ltd | 三環式ヘテロアリール誘導体 |
| US20020115717A1 (en) | 2000-07-25 | 2002-08-22 | Francine Gervais | Amyloid targeting imaging agents and uses thereof |
| CA2444214A1 (en) | 2001-04-23 | 2002-10-31 | The Trustees Of The University Of Pennsylvania | Amyloid plaque aggregation inhibitors and diagnostic imaging agents |
| CN1299777C (zh) * | 2001-08-27 | 2007-02-14 | 宾夕法尼亚州大学理事会 | 茋衍生物及其用于结合和成像淀粉样蛋白斑的用途 |
| EA200400135A1 (ru) | 2001-08-31 | 2004-08-26 | Ньюрочем ( Интернэшнл) Лимитед | Применение производных амидинов для лечения амилоидоза |
| JP2005515173A (ja) | 2001-10-31 | 2005-05-26 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | ピリミド[4,5−b]インドール誘導体 |
| EP1572129A4 (en) * | 2002-06-07 | 2007-03-28 | Univ North Carolina | AMIDINE DERIVATIVES FOR THE TREATMENT OF AMYLOIDOSE |
| JP2006513996A (ja) * | 2002-10-24 | 2006-04-27 | メルク エンド カムパニー インコーポレーテッド | 代謝調節型グルタミン酸受容体結合用トレーサーとしてのアルキン誘導体 |
| WO2004043496A1 (de) | 2002-11-11 | 2004-05-27 | Austria Wirtschaftsservice Gesellschaft M.B.H. | Fluor-18-markierte fluorchinolone |
| GB0229686D0 (en) | 2002-12-20 | 2003-01-29 | Amersham Plc | Solid-phase fluorination of benzothiazoles |
| WO2004058764A1 (en) | 2002-12-27 | 2004-07-15 | Bayer Healthcare Ag | 4-phenyl-pyrimido [4,5-b] indole derivatives |
| EP1644325A2 (en) | 2003-06-23 | 2006-04-12 | Neurochem (International) Limited | Methods and compositions for treating amyloid-related diseases |
| CA2500358A1 (en) | 2003-08-13 | 2005-02-24 | Bf Research Institute, Inc. | Probes for diseases in which amyloid accumulates, agents for staining amyloid, drugs for treatment and prophylaxis of diseases with accumulated amyloid, and probes for diagnosis of neurofibrillary tangles and agents for staining neurofibrillary tangles |
| JP2006100537A (ja) | 2004-09-29 | 2006-04-13 | Konica Minolta Holdings Inc | 有機エレクトロルミネッセンス素子、照明装置及び表示装置 |
| US20060110787A1 (en) * | 2004-11-19 | 2006-05-25 | The Board Trustees Of The University Of Arkansas | Use of fluorine-18-labeled fluoroquinolone antibiotics for diagnosing and monitoring bacterial infection |
| JP4000353B2 (ja) * | 2004-11-26 | 2007-10-31 | 国立大学法人 長崎大学 | アミロイド関連疾患診断用組成物 |
| JP2007084526A (ja) * | 2004-11-26 | 2007-04-05 | Nagasaki Univ | アミロイド関連疾患診断用組成物 |
| EP2213652B1 (en) * | 2004-12-17 | 2014-10-22 | The Trustees of The University of Pennsylvania | Stilbene derivatives and their use for binding and imaging amyloid plaques |
| FR2885905A1 (fr) | 2005-05-23 | 2006-11-24 | Trophos Sa | Nouveaux composes chimiques et leurs utilisations comme medicament |
| JP2008546804A (ja) * | 2005-06-24 | 2008-12-25 | ザ・トラスティーズ・オブ・ザ・ユニバーシティ・オブ・ペンシルバニア | 造影剤として用いるためのリガンドの放射性標識ペグ化 |
| CA2616397A1 (en) | 2005-08-03 | 2007-02-08 | Centre For Addiction And Mental Health | Diagnosis and treatment of mood disorders |
| GB0523506D0 (en) * | 2005-11-18 | 2005-12-28 | Hammersmith Imanet Ltd | Novel in vivo imaging compounds |
| JP5249772B2 (ja) * | 2005-11-22 | 2013-07-31 | メルク・シャープ・アンド・ドーム・コーポレーション | キナーゼの阻害剤として有用な三環式化合物 |
| WO2007063946A1 (ja) | 2005-11-30 | 2007-06-07 | Fujifilm Ri Pharma Co., Ltd. | アミロイドの凝集及び/又は沈着に起因する疾患の診断薬及び治療薬 |
| AR059356A1 (es) | 2006-02-14 | 2008-03-26 | Astrazeneca Ab | Nuevos radioligandos |
| JP2007223952A (ja) | 2006-02-23 | 2007-09-06 | Tohoku Univ | アミロイドβ蛋白が蓄積する疾患の画像診断プローブ |
| WO2008118122A2 (en) * | 2006-05-08 | 2008-10-02 | Molecular Neuroimaging, Llc | Compounds and amyloid probes thereof for therapeutic and imaging uses |
| US7678819B2 (en) | 2006-12-07 | 2010-03-16 | The Trustees Of The University Of Pennsylvania | Acetylene derivatives and their use for binding and imaging amyloid plaques |
| CA2673038C (en) | 2006-12-22 | 2015-12-15 | Incyte Corporation | Substituted tricyclic heteroaryl compounds as janus kinase inhibitors |
| EP1944281A1 (en) | 2007-01-09 | 2008-07-16 | Technische Universität München | 18F-labeled compounds, method for the preparation and use thereof |
| BRPI0702640A (pt) | 2007-01-10 | 2008-08-26 | Comissao Nac Energia Nuclear | processo de radiomarcação de flavonóides e sua aplicação em diagnóstico in vivo de disfunções cerebrais relacionadas aos sìtios receptores benzodiazepìnicos |
| EP2144916A4 (en) | 2007-04-10 | 2012-01-04 | Univ Pennsylvania | PHEN-NAPHTHALENE AND PHEN-CHINOLINE DERIVATIVES AND THEIR USE FOR THE BINDING AND PICTURE OF AMYLOIDER PLAQUE |
| US20110158907A1 (en) | 2007-04-19 | 2011-06-30 | The Trustees Of The Univeristy Of Pennsylvania | Diphenyl-heteroaryl derivatives and their use for binding and imaging amyloid plaques |
| WO2008132454A1 (en) * | 2007-04-26 | 2008-11-06 | Ge Healthcare Limited | Carbolines and their use as imaging agents |
| US8942164B2 (en) | 2007-10-22 | 2015-01-27 | Texas Instruments Incorporated | Differential CQI for OFDMA systems |
| EP2172444A4 (en) | 2007-07-04 | 2010-12-22 | Univ Tohoku | PET PROBE WITH FLUORO-SUBSTITUTED ALKOXY GROUP AND HYDROXYGRUPPE |
| WO2009045535A2 (en) | 2007-10-04 | 2009-04-09 | Sloan-Kettering Institute For Cancer Research | Fluorine-18 derivative of dasatinib and uses thereof |
| EP2247558B2 (en) | 2008-02-14 | 2024-07-03 | Eli Lilly and Company | Novel imaging agents for detecting neurological dysfunction |
| JP2011529086A (ja) | 2008-07-24 | 2011-12-01 | シーメンス メディカル ソリューションズ ユーエスエー インコーポレイテッド | Ad病変を同定するために有用な造影剤 |
| JP2012051804A (ja) | 2008-12-26 | 2012-03-15 | Kyoto Univ | Eg5阻害剤 |
| EP2218464A1 (en) | 2009-02-11 | 2010-08-18 | Technische Universität München | Compounds for non-invasive measurement of aggregates of amyloid peptides |
| CN105358558B (zh) | 2013-10-08 | 2017-09-26 | 豪夫迈·罗氏有限公司 | 作为tau‑pet‑配体的二氮杂咔唑衍生物 |
| EA036518B1 (ru) | 2015-02-02 | 2020-11-18 | Юсб Байофарма Спрл | Производные 9h-пирролодипиридина |
-
2009
- 2009-02-17 EP EP09711194.2A patent/EP2247558B2/en active Active
- 2009-02-17 US US12/372,717 patent/US8318132B2/en active Active
- 2009-02-17 KR KR1020107020589A patent/KR20100135235A/ko not_active Ceased
- 2009-02-17 FI FIEP09711194.2T patent/FI2247558T4/fi active
- 2009-02-17 SI SI200932157T patent/SI2247558T2/sl unknown
- 2009-02-17 DK DK09711194.2T patent/DK2247558T4/da active
- 2009-02-17 HR HRP20220401TT patent/HRP20220401T4/hr unknown
- 2009-02-17 US US12/867,502 patent/US20110046378A1/en not_active Abandoned
- 2009-02-17 LT LTEPPCT/US2009/000961T patent/LT2247558T/lt unknown
- 2009-02-17 ES ES09711194T patent/ES2907992T5/es active Active
- 2009-02-17 PT PT97111942T patent/PT2247558T/pt unknown
- 2009-02-17 CA CA2715390A patent/CA2715390A1/en not_active Abandoned
- 2009-02-17 JP JP2010546795A patent/JP2011512354A/ja active Pending
- 2009-02-17 HU HUE09711194A patent/HUE058352T2/hu unknown
- 2009-02-17 WO PCT/US2009/000961 patent/WO2009102498A1/en not_active Ceased
- 2009-02-17 EP EP20130157487 patent/EP2599763A1/en not_active Withdrawn
- 2009-02-17 PL PL09711194.2T patent/PL2247558T5/pl unknown
-
2022
- 2022-03-30 CY CY20221100245T patent/CY1125258T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| KR20100135235A (ko) | 2010-12-24 |
| HRP20220401T3 (hr) | 2022-05-27 |
| US20110046378A1 (en) | 2011-02-24 |
| US20110091382A1 (en) | 2011-04-21 |
| SI2247558T1 (sl) | 2022-07-29 |
| DK2247558T4 (da) | 2024-07-29 |
| EP2247558B2 (en) | 2024-07-03 |
| CA2715390A1 (en) | 2009-08-20 |
| PT2247558T (pt) | 2022-03-21 |
| EP2247558B1 (en) | 2022-02-02 |
| EP2247558A1 (en) | 2010-11-10 |
| LT2247558T (lt) | 2022-04-11 |
| JP2011512354A (ja) | 2011-04-21 |
| ES2907992T5 (en) | 2025-03-19 |
| US8318132B2 (en) | 2012-11-27 |
| HRP20220401T4 (hr) | 2024-09-27 |
| PL2247558T3 (pl) | 2022-05-02 |
| WO2009102498A1 (en) | 2009-08-20 |
| DK2247558T3 (da) | 2022-02-21 |
| HUE058352T2 (hu) | 2022-07-28 |
| PL2247558T5 (pl) | 2024-10-14 |
| FI2247558T4 (fi) | 2024-09-19 |
| SI2247558T2 (sl) | 2024-10-30 |
| EP2599763A1 (en) | 2013-06-05 |
| CY1125258T1 (el) | 2024-02-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2907992T3 (es) | Agentes novedosos de obtención de imágenes para detectar disfunciones neurológicas | |
| KR101609504B1 (ko) | 신경계 기능장애를 검출하기 위한 카르볼린계 및 카르바졸계 영상화제 | |
| US8932557B2 (en) | Imaging agents for detecting neurological dysfunction | |
| ES2578228T3 (es) | Agentes de formación de imágenes para detectar trastornos neurológicos | |
| EP2411057B1 (en) | Imaging agents for detecting neurological disorders | |
| CN110678465A (zh) | 新型氘取代的正电子发射断层扫描(pet)显像剂及其药理学应用 | |
| EA017713B1 (ru) | [f-18]-меченая l-глутаминовая кислота, [f-18]-меченый l-глутамин, их производные и их применение, а также способ их получения | |
| JP2002525325A (ja) | 放射標識化ニューロキニン−1受容体拮抗薬 | |
| JP2026053514A (ja) | ハンチンチンタンパク質をイメージングするための複素環式化合物及びイメージング剤 | |
| CN102574859A (zh) | 作为体内显像剂的[18f]-标记氟马西尼类似物 | |
| EP2512525B1 (en) | Novel aryloxyanilide imaging agents | |
| TWI917391B (zh) | 用於亨丁頓(huntingtin)蛋白造影之雜環化合物及造影劑 | |
| KR20210126146A (ko) | Pet 방사선추적자로서의 [18f]-라벨링된 벤조티아졸 유도체 | |
| ES2354151T3 (es) | Antagonistas del receptor de neuroquinina-1 radiomarcados. | |
| JP6273251B2 (ja) | 芳香族アミノ酸誘導体およびそれを用いるpetプローブ | |
| KR102150377B1 (ko) | 플루오린-18이 치환된 피라졸 유도체 방사성의약품 제조방법 | |
| WO2025188917A1 (en) | Construction and use of reagents for conjugation to bioligands for imaging and radiopharmaceutical applications | |
| CN117586270A (zh) | 放射性2,7,9a-三氮杂苯并[cd]天青-6(7H)酮衍生物及其制备方法和应用 | |
| ITMC20080089A1 (it) | Sintesi chimica di i-124[6-oh-bta-1-3'-i] iodine-124[2-(3'-iodo-4'-methylaminophenyl)-6-hydroxy-benzothialzole] per indagini pet e radioterapia. |