ES2944545T3 - Moduladores de piruvato quinasa y uso de los mismos - Google Patents
Moduladores de piruvato quinasa y uso de los mismos Download PDFInfo
- Publication number
- ES2944545T3 ES2944545T3 ES18766400T ES18766400T ES2944545T3 ES 2944545 T3 ES2944545 T3 ES 2944545T3 ES 18766400 T ES18766400 T ES 18766400T ES 18766400 T ES18766400 T ES 18766400T ES 2944545 T3 ES2944545 T3 ES 2944545T3
- Authority
- ES
- Spain
- Prior art keywords
- optionally substituted
- alkyl
- methyl
- compound
- protecting group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102000013009 Pyruvate Kinase Human genes 0.000 title description 20
- 108020005115 Pyruvate Kinase Proteins 0.000 title description 20
- 150000001875 compounds Chemical class 0.000 claims abstract description 280
- 238000000034 method Methods 0.000 claims abstract description 166
- 229910052739 hydrogen Inorganic materials 0.000 claims description 112
- 239000001257 hydrogen Substances 0.000 claims description 111
- -1 substituted Chemical class 0.000 claims description 103
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 95
- 150000003839 salts Chemical class 0.000 claims description 89
- 125000000623 heterocyclic group Chemical group 0.000 claims description 83
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 82
- 125000001072 heteroaryl group Chemical group 0.000 claims description 77
- 206010028980 Neoplasm Diseases 0.000 claims description 75
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 72
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 56
- 229910052736 halogen Inorganic materials 0.000 claims description 55
- 125000003118 aryl group Chemical group 0.000 claims description 50
- 150000002367 halogens Chemical group 0.000 claims description 49
- 125000000217 alkyl group Chemical group 0.000 claims description 47
- 201000010099 disease Diseases 0.000 claims description 42
- 201000011510 cancer Diseases 0.000 claims description 41
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 41
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 38
- 125000004432 carbon atom Chemical group C* 0.000 claims description 37
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 37
- 206010012601 diabetes mellitus Diseases 0.000 claims description 37
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 35
- 125000003107 substituted aryl group Chemical group 0.000 claims description 30
- 125000005017 substituted alkenyl group Chemical group 0.000 claims description 26
- 125000004426 substituted alkynyl group Chemical group 0.000 claims description 26
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 25
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 24
- 229910052799 carbon Inorganic materials 0.000 claims description 24
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 24
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 19
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 19
- 229910052705 radium Inorganic materials 0.000 claims description 16
- 229910052701 rubidium Inorganic materials 0.000 claims description 16
- 208000029078 coronary artery disease Diseases 0.000 claims description 15
- 125000002950 monocyclic group Chemical group 0.000 claims description 15
- 208000008589 Obesity Diseases 0.000 claims description 14
- 150000001721 carbon Chemical group 0.000 claims description 14
- 235000020824 obesity Nutrition 0.000 claims description 13
- 125000003342 alkenyl group Chemical group 0.000 claims description 12
- 125000002947 alkylene group Chemical group 0.000 claims description 12
- 230000002062 proliferating effect Effects 0.000 claims description 12
- 208000023275 Autoimmune disease Diseases 0.000 claims description 11
- 229910052760 oxygen Inorganic materials 0.000 claims description 11
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 10
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 9
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 9
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 9
- 125000006239 protecting group Chemical group 0.000 claims description 9
- 239000011593 sulfur Substances 0.000 claims description 9
- 208000005692 Bloom Syndrome Diseases 0.000 claims description 8
- 125000001188 haloalkyl group Chemical group 0.000 claims description 8
- 125000004429 atom Chemical group 0.000 claims description 7
- 230000027455 binding Effects 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- 201000001421 hyperglycemia Diseases 0.000 claims description 7
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 5
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 5
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 5
- 208000037803 restenosis Diseases 0.000 claims description 5
- 201000001320 Atherosclerosis Diseases 0.000 claims description 4
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 3
- 125000001624 naphthyl group Chemical group 0.000 claims description 3
- 125000005415 substituted alkoxy group Chemical group 0.000 claims description 3
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- 125000006163 5-membered heteroaryl group Chemical group 0.000 claims description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 2
- 108020000002 NR3 subfamily Proteins 0.000 claims 4
- 108020004021 3-ketosteroid receptors Proteins 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 37
- 101001091538 Homo sapiens Pyruvate kinase PKM Proteins 0.000 abstract 1
- 102100034911 Pyruvate kinase PKM Human genes 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 298
- 239000000203 mixture Substances 0.000 description 233
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 171
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 150
- 235000019439 ethyl acetate Nutrition 0.000 description 137
- 230000002829 reductive effect Effects 0.000 description 129
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 124
- 239000012044 organic layer Substances 0.000 description 107
- 239000000047 product Substances 0.000 description 104
- 230000015572 biosynthetic process Effects 0.000 description 101
- 238000003786 synthesis reaction Methods 0.000 description 101
- 239000011541 reaction mixture Substances 0.000 description 91
- 239000012267 brine Substances 0.000 description 80
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 80
- 239000000243 solution Substances 0.000 description 80
- 238000005160 1H NMR spectroscopy Methods 0.000 description 71
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 68
- 235000002639 sodium chloride Nutrition 0.000 description 64
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 61
- 239000011734 sodium Substances 0.000 description 60
- 239000007832 Na2SO4 Substances 0.000 description 52
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 52
- 229910052938 sodium sulfate Inorganic materials 0.000 description 52
- 235000011152 sodium sulphate Nutrition 0.000 description 52
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 50
- 210000004027 cell Anatomy 0.000 description 46
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 44
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 41
- 239000004698 Polyethylene Substances 0.000 description 40
- 238000012746 preparative thin layer chromatography Methods 0.000 description 40
- 235000019270 ammonium chloride Nutrition 0.000 description 39
- 229920006395 saturated elastomer Polymers 0.000 description 39
- 239000003480 eluent Substances 0.000 description 35
- 239000000741 silica gel Substances 0.000 description 35
- 229910002027 silica gel Inorganic materials 0.000 description 35
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 33
- 238000003818 flash chromatography Methods 0.000 description 30
- 238000010898 silica gel chromatography Methods 0.000 description 30
- 238000011282 treatment Methods 0.000 description 30
- 238000002953 preparative HPLC Methods 0.000 description 29
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 27
- 238000007792 addition Methods 0.000 description 27
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 27
- 238000006243 chemical reaction Methods 0.000 description 26
- 125000005842 heteroatom Chemical group 0.000 description 25
- 239000012258 stirred mixture Substances 0.000 description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 23
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 21
- 239000008194 pharmaceutical composition Substances 0.000 description 21
- 239000007858 starting material Substances 0.000 description 19
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 19
- 125000001424 substituent group Chemical group 0.000 description 18
- DWHIRSWNWDXFMQ-UHFFFAOYSA-N 4h-pyridazin-5-one Chemical compound O=C1CC=NN=C1 DWHIRSWNWDXFMQ-UHFFFAOYSA-N 0.000 description 17
- 229910052757 nitrogen Inorganic materials 0.000 description 17
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 16
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 16
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 208000035475 disorder Diseases 0.000 description 15
- 238000003756 stirring Methods 0.000 description 15
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 14
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 239000003795 chemical substances by application Substances 0.000 description 14
- 239000008103 glucose Substances 0.000 description 14
- 210000001519 tissue Anatomy 0.000 description 14
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 13
- 125000004122 cyclic group Chemical group 0.000 description 13
- 238000010511 deprotection reaction Methods 0.000 description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 12
- 125000000304 alkynyl group Chemical group 0.000 description 12
- 239000005457 ice water Substances 0.000 description 12
- 238000000746 purification Methods 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- 206010025323 Lymphomas Diseases 0.000 description 11
- RNBGYGVWRKECFJ-ARQDHWQXSA-N beta-D-fructofuranose 1,6-bisphosphate Chemical compound O[C@H]1[C@H](O)[C@@](O)(COP(O)(O)=O)O[C@@H]1COP(O)(O)=O RNBGYGVWRKECFJ-ARQDHWQXSA-N 0.000 description 11
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 125000004404 heteroalkyl group Chemical group 0.000 description 11
- 239000000543 intermediate Substances 0.000 description 11
- 229910052751 metal Inorganic materials 0.000 description 11
- 239000002184 metal Substances 0.000 description 11
- 239000012038 nucleophile Substances 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 11
- 239000011701 zinc Substances 0.000 description 11
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 239000012190 activator Substances 0.000 description 10
- 125000002619 bicyclic group Chemical group 0.000 description 10
- 125000004452 carbocyclyl group Chemical group 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 10
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 9
- 208000003174 Brain Neoplasms Diseases 0.000 description 9
- 108010029485 Protein Isoforms Proteins 0.000 description 9
- 102000001708 Protein Isoforms Human genes 0.000 description 9
- 230000004913 activation Effects 0.000 description 9
- 230000012010 growth Effects 0.000 description 9
- 239000012299 nitrogen atmosphere Substances 0.000 description 9
- 230000003647 oxidation Effects 0.000 description 9
- 238000007254 oxidation reaction Methods 0.000 description 9
- 239000001301 oxygen Substances 0.000 description 9
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical group CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- 102000004877 Insulin Human genes 0.000 description 8
- 108090001061 Insulin Proteins 0.000 description 8
- AIJULSRZWUXGPQ-UHFFFAOYSA-N Methylglyoxal Chemical compound CC(=O)C=O AIJULSRZWUXGPQ-UHFFFAOYSA-N 0.000 description 8
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 8
- 206010039491 Sarcoma Diseases 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 229910052794 bromium Inorganic materials 0.000 description 8
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 229940125396 insulin Drugs 0.000 description 8
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 8
- 125000003367 polycyclic group Chemical group 0.000 description 8
- 230000035935 pregnancy Effects 0.000 description 8
- 230000001681 protective effect Effects 0.000 description 8
- 108090000623 proteins and genes Proteins 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 150000003254 radicals Chemical class 0.000 description 8
- 239000003642 reactive oxygen metabolite Substances 0.000 description 8
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 7
- 206010006187 Breast cancer Diseases 0.000 description 7
- 208000026310 Breast neoplasm Diseases 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 7
- 125000005843 halogen group Chemical group 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 238000002626 targeted therapy Methods 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 7
- FSLWREAEYZFVPZ-UHFFFAOYSA-N 4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CC=1SC2=C(N(C=3C(NN=CC=32)=O)C)N=1 FSLWREAEYZFVPZ-UHFFFAOYSA-N 0.000 description 6
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 6
- 208000024172 Cardiovascular disease Diseases 0.000 description 6
- 206010027476 Metastases Diseases 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 230000002496 gastric effect Effects 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 229910052740 iodine Inorganic materials 0.000 description 6
- 208000032839 leukemia Diseases 0.000 description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 description 6
- 230000000269 nucleophilic effect Effects 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 125000004526 pyridazin-2-yl group Chemical group N1N(C=CC=C1)* 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 6
- 208000021309 Germ cell tumor Diseases 0.000 description 5
- 206010018338 Glioma Diseases 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 5
- 229910019142 PO4 Inorganic materials 0.000 description 5
- 230000002159 abnormal effect Effects 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 238000002512 chemotherapy Methods 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 238000004440 column chromatography Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 150000002430 hydrocarbons Chemical group 0.000 description 5
- 238000009169 immunotherapy Methods 0.000 description 5
- 201000007270 liver cancer Diseases 0.000 description 5
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 5
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 5
- 235000021317 phosphate Nutrition 0.000 description 5
- 238000007363 ring formation reaction Methods 0.000 description 5
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 5
- 229920002554 vinyl polymer Polymers 0.000 description 5
- VGISFWWEOGVMED-UHFFFAOYSA-N 1-(chloromethyl)-3-methoxybenzene Chemical compound COC1=CC=CC(CCl)=C1 VGISFWWEOGVMED-UHFFFAOYSA-N 0.000 description 4
- XAWAVQAMOQUIQF-UHFFFAOYSA-N 2-[[3-(benzenesulfonylmethyl)pyrazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound C1(=CC=CC=C1)S(=O)(=O)CC1=NN(C=C1)COCC[Si](C)(C)C XAWAVQAMOQUIQF-UHFFFAOYSA-N 0.000 description 4
- OEJLGUYHDVOECX-UHFFFAOYSA-N 4-[benzenesulfonyl(pyridin-2-yl)methyl]-10-benzyl-7-phenyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(C1=NC=CC=C1)S(=O)(=O)C1=CC=CC=C1)C1=CC=CC=C1 OEJLGUYHDVOECX-UHFFFAOYSA-N 0.000 description 4
- VFNAMFHLJBFXFZ-UHFFFAOYSA-N 7-methyl-4-methylsulfonyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)S(=O)(=O)C VFNAMFHLJBFXFZ-UHFFFAOYSA-N 0.000 description 4
- 206010060999 Benign neoplasm Diseases 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 4
- 208000032612 Glial tumor Diseases 0.000 description 4
- 208000017604 Hodgkin disease Diseases 0.000 description 4
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 4
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 229910002651 NO3 Inorganic materials 0.000 description 4
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 4
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 4
- 206010033307 Overweight Diseases 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 201000010769 Prader-Willi syndrome Diseases 0.000 description 4
- 206010060862 Prostate cancer Diseases 0.000 description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 239000002671 adjuvant Substances 0.000 description 4
- 238000005804 alkylation reaction Methods 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 125000003710 aryl alkyl group Chemical group 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 230000004663 cell proliferation Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 235000012000 cholesterol Nutrition 0.000 description 4
- 238000005859 coupling reaction Methods 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 150000001982 diacylglycerols Chemical class 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 150000002303 glucose derivatives Chemical class 0.000 description 4
- 230000002267 hypothalamic effect Effects 0.000 description 4
- 239000003446 ligand Substances 0.000 description 4
- 230000036210 malignancy Effects 0.000 description 4
- 201000001441 melanoma Diseases 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 201000005962 mycosis fungoides Diseases 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 210000001672 ovary Anatomy 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 201000009104 prediabetes syndrome Diseases 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 102000004196 processed proteins & peptides Human genes 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- UAWABSHMGXMCRK-UHFFFAOYSA-L samarium(ii) iodide Chemical compound I[Sm]I UAWABSHMGXMCRK-UHFFFAOYSA-L 0.000 description 4
- 201000000849 skin cancer Diseases 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 210000002784 stomach Anatomy 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 4
- 229910052725 zinc Inorganic materials 0.000 description 4
- 125000006708 (C5-C14) heteroaryl group Chemical group 0.000 description 3
- ZPIHRUCXNRKJIF-UHFFFAOYSA-N 1-(2-trimethylsilylethoxymethyl)imidazole-2-carbaldehyde Chemical compound C[Si](C)(C)CCOCN1C=CN=C1C=O ZPIHRUCXNRKJIF-UHFFFAOYSA-N 0.000 description 3
- GWQAWODFISMKSD-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C)C)C=CC=1 GWQAWODFISMKSD-UHFFFAOYSA-N 0.000 description 3
- ZWKFJGQTTNCGSF-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)SC)C)C=CC=1 ZWKFJGQTTNCGSF-UHFFFAOYSA-N 0.000 description 3
- OOFJBYVAZQMSQY-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-carbaldehyde Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C=O)C)C=CC=1 OOFJBYVAZQMSQY-UHFFFAOYSA-N 0.000 description 3
- DIUOELXIXSCFCR-UHFFFAOYSA-N 10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=NC(=CC=C2)N)=O)C)N=1 DIUOELXIXSCFCR-UHFFFAOYSA-N 0.000 description 3
- IFQHTKLLIUTGKO-UHFFFAOYSA-N 10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazole-5-carbonyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NC1=CC=CC(=N1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(=O)C1=NNC=C1)C IFQHTKLLIUTGKO-UHFFFAOYSA-N 0.000 description 3
- CRNZRFFYEZISDQ-UHFFFAOYSA-N 10-benzyl-7-ethyl-4-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1N=CC2=C(C1=O)N(C1=C2SC(=N1)CC1=NN(C=C1)COCC[Si](C)(C)C)CC CRNZRFFYEZISDQ-UHFFFAOYSA-N 0.000 description 3
- AYIYENZGDCIUSP-UHFFFAOYSA-N 10-benzyl-7-phenyl-4-(pyridin-2-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1N=CC2=C(C1=O)N(C1=C2SC(=N1)CC1=NC=CC=C1)C1=CC=CC=C1 AYIYENZGDCIUSP-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 3
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical group COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 3
- DQIDQJKJEIWQAD-UHFFFAOYSA-N 2-[[4-(chloromethyl)pyrazolo[4,3-c]pyridin-1-yl]methoxy]ethyl-trimethylsilane Chemical compound ClCC1=NC=CC2=C1C=NN2COCC[Si](C)(C)C DQIDQJKJEIWQAD-UHFFFAOYSA-N 0.000 description 3
- SNFGMJODMANUFI-UHFFFAOYSA-N 3-[(4,7-dimethyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzonitrile Chemical compound CC=1SC2=C(N(C=3C(N(N=CC=32)CC=2C=C(C#N)C=CC=2)=O)C)N=1 SNFGMJODMANUFI-UHFFFAOYSA-N 0.000 description 3
- LSLUHSRQWNXVSP-UHFFFAOYSA-N 3-[(4-bromo-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzonitrile Chemical compound BrC=1SC2=C(N(C=3C(N(N=CC=32)CC=2C=C(C#N)C=CC=2)=O)C)N=1 LSLUHSRQWNXVSP-UHFFFAOYSA-N 0.000 description 3
- HAHGIVSPBWEAKT-UHFFFAOYSA-N 4-[(5-chloro-1-tritylpyrazol-3-yl)methyl]-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound ClC1=CC(=NN1C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 HAHGIVSPBWEAKT-UHFFFAOYSA-N 0.000 description 3
- XYMXQEOFEGPFFX-UHFFFAOYSA-N 4-[benzenesulfonyl(pyridin-2-yl)methyl]-7-methyl-10-prop-2-ynyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC#C)=O)SC(=N2)C(C1=NC=CC=C1)S(=O)(=O)C1=CC=CC=C1 XYMXQEOFEGPFFX-UHFFFAOYSA-N 0.000 description 3
- LCEXYSFRDNNVPD-UHFFFAOYSA-N 4-[benzenesulfonyl-[6-(dimethylamino)pyridin-2-yl]methyl]-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN(C1=CC=CC(=N1)C(C=1SC2=C(N(C=3C(N(N=CC=32)CC2=NN(C=C2)COCC[Si](C)(C)C)=O)C)N=1)S(=O)(=O)C1=CC=CC=C1)C LCEXYSFRDNNVPD-UHFFFAOYSA-N 0.000 description 3
- RTPKMDVNLCBTHM-UHFFFAOYSA-N 6-(benzenesulfonylmethyl)-N,N-dimethylpyridin-2-amine Chemical compound CN(C1=NC(=CC=C1)CS(=O)(=O)C1=CC=CC=C1)C RTPKMDVNLCBTHM-UHFFFAOYSA-N 0.000 description 3
- LECLZVYANMESOD-UHFFFAOYSA-N 6-(chloromethyl)-n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC(CCl)=N1 LECLZVYANMESOD-UHFFFAOYSA-N 0.000 description 3
- OPAXNJUHNSHIRW-UHFFFAOYSA-N 7-methyl-10-[(1-methylpyrazol-3-yl)methyl]-4-[2-(1H-pyrazol-5-yl)ethyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CCC=1SC2=C(N(C=3C(N(N=CC=32)CC2=NN(C=C2)C)=O)C)N=1 OPAXNJUHNSHIRW-UHFFFAOYSA-N 0.000 description 3
- HNWIGKKYTJXUEQ-UHFFFAOYSA-N 7-methyl-10-prop-2-ynyl-4-(pyridin-2-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC#C)=O)SC(=N2)CC1=NC=CC=C1 HNWIGKKYTJXUEQ-UHFFFAOYSA-N 0.000 description 3
- OJINPWVFOQGLEV-UHFFFAOYSA-N 7-methyl-4-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)CC1=NN(C=C1)COCC[Si](C)(C)C OJINPWVFOQGLEV-UHFFFAOYSA-N 0.000 description 3
- OCDDDFJRPIVEAC-UHFFFAOYSA-N 7-methyl-4-methylsulfanyl-10-[[3-(trifluoromethoxy)phenyl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=CC(=CC=C1)OC(F)(F)F)=O)SC(=N2)SC OCDDDFJRPIVEAC-UHFFFAOYSA-N 0.000 description 3
- SAOYZJDJDQBKGG-UHFFFAOYSA-N 7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)S(=O)C SAOYZJDJDQBKGG-UHFFFAOYSA-N 0.000 description 3
- BXHUMPAZBOMBPR-UHFFFAOYSA-N 7-methyl-9-oxo-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-carbonitrile Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)C#N BXHUMPAZBOMBPR-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 3
- 125000005915 C6-C14 aryl group Chemical group 0.000 description 3
- 229920000858 Cyclodextrin Polymers 0.000 description 3
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 3
- 229910004373 HOAc Inorganic materials 0.000 description 3
- 102000001554 Hemoglobins Human genes 0.000 description 3
- 108010054147 Hemoglobins Proteins 0.000 description 3
- 206010022489 Insulin Resistance Diseases 0.000 description 3
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 208000034578 Multiple myelomas Diseases 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 208000006265 Renal cell carcinoma Diseases 0.000 description 3
- 208000000453 Skin Neoplasms Diseases 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 3
- ZQKHRAXLCGDJDL-UHFFFAOYSA-N [1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methanol Chemical compound C[Si](C)(C)CCOCN1C=CN=C1CO ZQKHRAXLCGDJDL-UHFFFAOYSA-N 0.000 description 3
- QFYKQJFZMMLNAW-UHFFFAOYSA-N [1-(2-trimethylsilylethoxymethyl)pyrazolo[4,3-c]pyridin-4-yl]methanol Chemical compound C[Si](CCOCN1N=CC=2C(=NC=CC=21)CO)(C)C QFYKQJFZMMLNAW-UHFFFAOYSA-N 0.000 description 3
- GBNLHIYJOSNQGZ-UHFFFAOYSA-N [6-(dimethylamino)pyridin-2-yl]methanol Chemical compound CN(C)C1=CC=CC(CO)=N1 GBNLHIYJOSNQGZ-UHFFFAOYSA-N 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 230000029936 alkylation Effects 0.000 description 3
- 125000004104 aryloxy group Chemical group 0.000 description 3
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 3
- 210000004204 blood vessel Anatomy 0.000 description 3
- 239000006227 byproduct Substances 0.000 description 3
- 208000002458 carcinoid tumor Diseases 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 210000003169 central nervous system Anatomy 0.000 description 3
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 229940097362 cyclodextrins Drugs 0.000 description 3
- 229940127089 cytotoxic agent Drugs 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- 238000003379 elimination reaction Methods 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 238000001794 hormone therapy Methods 0.000 description 3
- 230000028993 immune response Effects 0.000 description 3
- 230000001771 impaired effect Effects 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 125000002346 iodo group Chemical group I* 0.000 description 3
- 208000014018 liver neoplasm Diseases 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- CCERQOYLJJULMD-UHFFFAOYSA-M magnesium;carbanide;chloride Chemical compound [CH3-].[Mg+2].[Cl-] CCERQOYLJJULMD-UHFFFAOYSA-M 0.000 description 3
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 230000001394 metastastic effect Effects 0.000 description 3
- 206010061289 metastatic neoplasm Diseases 0.000 description 3
- 208000010125 myocardial infarction Diseases 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 125000006574 non-aromatic ring group Chemical group 0.000 description 3
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 3
- 238000005935 nucleophilic addition reaction Methods 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 201000002528 pancreatic cancer Diseases 0.000 description 3
- 208000008443 pancreatic carcinoma Diseases 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 3
- 201000008205 supratentorial primitive neuroectodermal tumor Diseases 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- WRSWIWOVJBYZAW-UHFFFAOYSA-M zinc;methanidylbenzene;bromide Chemical compound Br[Zn+].[CH2-]C1=CC=CC=C1 WRSWIWOVJBYZAW-UHFFFAOYSA-M 0.000 description 3
- QOWBXWFYRXSBAS-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C(OC)=C1 QOWBXWFYRXSBAS-UHFFFAOYSA-N 0.000 description 2
- HJOQLQQQMBTBRO-UHFFFAOYSA-N (5-chloro-1-tritylpyrazol-3-yl)methanol Chemical compound ClC1=CC(=NN1C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1)CO HJOQLQQQMBTBRO-UHFFFAOYSA-N 0.000 description 2
- HJRGGRKOIDKVCH-UHFFFAOYSA-N (5-chloro-1-tritylpyrazol-3-yl)methyl methanesulfonate Chemical compound CS(=O)(=O)OCC1=NN(C(=C1)Cl)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 HJRGGRKOIDKVCH-UHFFFAOYSA-N 0.000 description 2
- OPYHNLNYCRZOGY-UHFFFAOYSA-N 1,2,3,4,5-pentafluoro-6-iodobenzene Chemical compound FC1=C(F)C(F)=C(I)C(F)=C1F OPYHNLNYCRZOGY-UHFFFAOYSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 2
- ZXRLWHGLEJGMNO-UHFFFAOYSA-N 1,3-thiazole-5-carbaldehyde Chemical compound O=CC1=CN=CS1 ZXRLWHGLEJGMNO-UHFFFAOYSA-N 0.000 description 2
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 2
- COXCHFBJGOKFGS-UHFFFAOYSA-N 10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-carboxamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(=O)N)C COXCHFBJGOKFGS-UHFFFAOYSA-N 0.000 description 2
- GOCGNIXRVBVIPZ-UHFFFAOYSA-N 10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-sulfonamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)S(=O)(=O)N)C GOCGNIXRVBVIPZ-UHFFFAOYSA-N 0.000 description 2
- APZUODIIWKLSNZ-UHFFFAOYSA-N 10-[(2-amino-1,3-thiazol-5-yl)methyl]-7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NC=1SC(=CN=1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)S(=O)C)C APZUODIIWKLSNZ-UHFFFAOYSA-N 0.000 description 2
- FJXFVZUZBGYSOD-UHFFFAOYSA-N 10-[(2-bromo-1,3-thiazol-5-yl)methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound BrC=1SC(=CN=1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)SC)C FJXFVZUZBGYSOD-UHFFFAOYSA-N 0.000 description 2
- FPBDJQKLFUQJMK-UHFFFAOYSA-N 10-[(3-aminophenyl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C)C)C=CC=1 FPBDJQKLFUQJMK-UHFFFAOYSA-N 0.000 description 2
- RQHHPXNMLNUEPU-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC=N2)C)C=CC=1 RQHHPXNMLNUEPU-UHFFFAOYSA-N 0.000 description 2
- UFRKWOVBEFRLMU-UHFFFAOYSA-N 10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-[2-(1H-pyrazol-5-yl)ethyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CCC=1SC2=C(N(C=3C(N(N=CC=32)CC2=NC(=CC=C2)N)=O)C)N=1 UFRKWOVBEFRLMU-UHFFFAOYSA-N 0.000 description 2
- KFGHEWRUZSOAOF-UHFFFAOYSA-N 10-[[2-[(2,4-dimethoxyphenyl)methylamino]-1,3-thiazol-5-yl]methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC1=C(CNC=2SC(=CN=2)CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)SC)C)C=CC(=C1)OC KFGHEWRUZSOAOF-UHFFFAOYSA-N 0.000 description 2
- UIFCFQJZHAQVCV-UHFFFAOYSA-N 10-benzyl-7-ethyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC=CC=C2)=O)CC)N=1 UIFCFQJZHAQVCV-UHFFFAOYSA-N 0.000 description 2
- PPFKQEVWJGRSJC-UHFFFAOYSA-N 2-(benzenesulfonylmethyl)pyridine Chemical compound C=1C=CC=CC=1S(=O)(=O)CC1=CC=CC=N1 PPFKQEVWJGRSJC-UHFFFAOYSA-N 0.000 description 2
- OZMLUMPWPFZWTP-UHFFFAOYSA-N 2-(tributyl-$l^{5}-phosphanylidene)acetonitrile Chemical compound CCCCP(CCCC)(CCCC)=CC#N OZMLUMPWPFZWTP-UHFFFAOYSA-N 0.000 description 2
- NFBNRSPJIMVFNB-UHFFFAOYSA-N 2-[[2-(chloromethyl)imidazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCN1C=CN=C1CCl NFBNRSPJIMVFNB-UHFFFAOYSA-N 0.000 description 2
- SGIKHNDGGGZRQK-UHFFFAOYSA-N 2-[[3-(bromomethyl)phenyl]methyl]isoindole-1,3-dione Chemical compound BrCC1=CC=CC(CN2C(C3=CC=CC=C3C2=O)=O)=C1 SGIKHNDGGGZRQK-UHFFFAOYSA-N 0.000 description 2
- NMEFSJJGKNVMHV-UHFFFAOYSA-N 2-[[4-(benzenesulfonylmethyl)indazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound C1(=CC=CC=C1)S(=O)(=O)CC1=C2C=NN(C2=CC=C1)COCC[Si](C)(C)C NMEFSJJGKNVMHV-UHFFFAOYSA-N 0.000 description 2
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 2
- LSBDFXRDZJMBSC-UHFFFAOYSA-N 2-phenylacetamide Chemical group NC(=O)CC1=CC=CC=C1 LSBDFXRDZJMBSC-UHFFFAOYSA-N 0.000 description 2
- LAYQASGEOLDCQP-UHFFFAOYSA-N 3-(benzenesulfonylmethyl)-5-chloro-1-tritylpyrazole Chemical compound ClC1=CC(=NN1C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1)CS(=O)(=O)C1=CC=CC=C1 LAYQASGEOLDCQP-UHFFFAOYSA-N 0.000 description 2
- YKBBRISJFWONIK-UHFFFAOYSA-N 3-[(4,7-dimethyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzamide Chemical compound CC=1SC2=C(N(C=3C(N(N=CC=32)CC=2C=C(C(=O)N)C=CC=2)=O)C)N=1 YKBBRISJFWONIK-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- ZXTLVJTZVXUWED-UHFFFAOYSA-N 4-(aminomethyl)-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CN)C ZXTLVJTZVXUWED-UHFFFAOYSA-N 0.000 description 2
- DTFYLYYUHQGHNG-UHFFFAOYSA-N 4-(aminomethyl)-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NCC=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 DTFYLYYUHQGHNG-UHFFFAOYSA-N 0.000 description 2
- XMIAHPGIRWVZCR-UHFFFAOYSA-N 4-[(3-chloro-1H-pyrazol-5-yl)methyl]-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CC1=NNC(=C1)Cl)C XMIAHPGIRWVZCR-UHFFFAOYSA-N 0.000 description 2
- JAKOFRYSBQRAJT-UHFFFAOYSA-N 4-[benzenesulfonyl(pyridin-3-yl)methyl]-7-methyl-10-[2-(1-methylpyrazol-3-yl)ethyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CCC1=NN(C=C1)C)=O)SC(=N2)C(C=1C=NC=CC=1)S(=O)(=O)C1=CC=CC=C1 JAKOFRYSBQRAJT-UHFFFAOYSA-N 0.000 description 2
- XZQDQDHSHFJTGA-UHFFFAOYSA-N 4-benzoyl-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)(=O)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 XZQDQDHSHFJTGA-UHFFFAOYSA-N 0.000 description 2
- SVEHMTFJVUSNFX-UHFFFAOYSA-N 4-benzoyl-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)(=O)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 SVEHMTFJVUSNFX-UHFFFAOYSA-N 0.000 description 2
- DXJYSQGFNKMHMY-UHFFFAOYSA-N 4-benzyl-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 DXJYSQGFNKMHMY-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- NZXJBBMEDSFERF-UHFFFAOYSA-N 7-methyl-10-(1H-pyrazolo[4,3-c]pyridin-4-ylmethyl)-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=NC=CC3=C2C=NN3)=O)C)N=1 NZXJBBMEDSFERF-UHFFFAOYSA-N 0.000 description 2
- JHXBMLRMLQFXTA-UHFFFAOYSA-N 7-methyl-10-[2-(1-methylpyrazol-3-yl)ethyl]-4-(pyridin-3-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CCC1=NN(C=C1)C)=O)SC(=N2)CC=1C=NC=CC=1 JHXBMLRMLQFXTA-UHFFFAOYSA-N 0.000 description 2
- IFIMIFAJRJPFRR-UHFFFAOYSA-N 7-methyl-10-[2-(1-methylpyrazol-3-yl)ethyl]-4-methylsulfonyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CCC1=NN(C=C1)C)=O)SC(=N2)S(=O)(=O)C IFIMIFAJRJPFRR-UHFFFAOYSA-N 0.000 description 2
- UJAFSKHLTQVZDW-UHFFFAOYSA-N 7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)imidazol-2-yl]methyl]-4-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC=1N(C=CN=1)COCC[Si](C)(C)C)=O)SC(=N2)CC1=NN(C=C1)COCC[Si](C)(C)C UJAFSKHLTQVZDW-UHFFFAOYSA-N 0.000 description 2
- YNJALPNRJPTAPE-UHFFFAOYSA-N 7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC=N2 YNJALPNRJPTAPE-UHFFFAOYSA-N 0.000 description 2
- BJSLYOKSJMILBD-UHFFFAOYSA-N 7-methyl-4-methylsulfonyl-10-[[3-(trifluoromethoxy)phenyl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=CC(=CC=C1)OC(F)(F)F)=O)SC(=N2)S(=O)(=O)C BJSLYOKSJMILBD-UHFFFAOYSA-N 0.000 description 2
- PPVHLKXUVNWRQP-UHFFFAOYSA-N 7-methyl-4-methylsulfonyl-10-prop-2-ynyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC#C)=O)SC(=N2)S(=O)(=O)C PPVHLKXUVNWRQP-UHFFFAOYSA-N 0.000 description 2
- UJDSQASUDOYPNA-UHFFFAOYSA-N 7-methyl-4-methylsulfonyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)S(=O)(=O)C UJDSQASUDOYPNA-UHFFFAOYSA-N 0.000 description 2
- SRCFQTDFBOCFEM-UHFFFAOYSA-N 7-methyl-9-oxo-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-carbaldehyde Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)C=O SRCFQTDFBOCFEM-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 206010002383 Angina Pectoris Diseases 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010003571 Astrocytoma Diseases 0.000 description 2
- 206010060971 Astrocytoma malignant Diseases 0.000 description 2
- 206010004593 Bile duct cancer Diseases 0.000 description 2
- 206010005003 Bladder cancer Diseases 0.000 description 2
- 102000004506 Blood Proteins Human genes 0.000 description 2
- 108010017384 Blood Proteins Proteins 0.000 description 2
- 206010006143 Brain stem glioma Diseases 0.000 description 2
- FHBXBSNAKPDMNQ-UHFFFAOYSA-N COC(=O)C1=NN(C=C1)COCC[Si](C)(C)C Chemical compound COC(=O)C1=NN(C=C1)COCC[Si](C)(C)C FHBXBSNAKPDMNQ-UHFFFAOYSA-N 0.000 description 2
- 206010007275 Carcinoid tumour Diseases 0.000 description 2
- 206010007559 Cardiac failure congestive Diseases 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 101710088194 Dehydrogenase Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- 208000012468 Ewing sarcoma/peripheral primitive neuroectodermal tumor Diseases 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical group NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 208000002705 Glucose Intolerance Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000001204 Hashimoto Disease Diseases 0.000 description 2
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 238000007341 Heck reaction Methods 0.000 description 2
- 101000704557 Homo sapiens Sulfiredoxin-1 Proteins 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- 208000031226 Hyperlipidaemia Diseases 0.000 description 2
- 208000004044 Hypesthesia Diseases 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010061252 Intraocular melanoma Diseases 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 206010023825 Laryngeal cancer Diseases 0.000 description 2
- 206010025557 Malignant fibrous histiocytoma of bone Diseases 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 2
- 208000000172 Medulloblastoma Diseases 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- 238000006751 Mitsunobu reaction Methods 0.000 description 2
- BTYXZJYEGUZSKE-UHFFFAOYSA-N N-[[10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]-(1H-pyrazol-5-yl)methyl]acetamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(NC(C)=O)C1=NNC=C1)C BTYXZJYEGUZSKE-UHFFFAOYSA-N 0.000 description 2
- WEIRUKRLOQRRLP-UHFFFAOYSA-N N-[[10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]methyl]methanesulfonamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CNS(=O)(=O)C)C WEIRUKRLOQRRLP-UHFFFAOYSA-N 0.000 description 2
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 2
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical group CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N Pd(PPh3)4 Substances [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 208000001280 Prediabetic State Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 102100034909 Pyruvate kinase PKLR Human genes 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 201000000582 Retinoblastoma Diseases 0.000 description 2
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 2
- 206010061934 Salivary gland cancer Diseases 0.000 description 2
- 206010041067 Small cell lung cancer Diseases 0.000 description 2
- 102100031797 Sulfiredoxin-1 Human genes 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- 201000009365 Thymic carcinoma Diseases 0.000 description 2
- 208000024770 Thyroid neoplasm Diseases 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 2
- 201000005969 Uveal melanoma Diseases 0.000 description 2
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- WZELLTUAICWMCZ-UHFFFAOYSA-N [1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methanol Chemical compound C[Si](C)(C)CCOCN1C=CC(CO)=N1 WZELLTUAICWMCZ-UHFFFAOYSA-N 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 208000009956 adenocarcinoma Diseases 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 2
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 2
- 230000006536 aerobic glycolysis Effects 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 description 2
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 2
- 230000003281 allosteric effect Effects 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 230000001195 anabolic effect Effects 0.000 description 2
- 230000036528 appetite Effects 0.000 description 2
- 235000019789 appetite Nutrition 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- UENWRTRMUIOCKN-UHFFFAOYSA-N benzyl thiol Chemical compound SCC1=CC=CC=C1 UENWRTRMUIOCKN-UHFFFAOYSA-N 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 102000023732 binding proteins Human genes 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 208000035269 cancer or benign tumor Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 201000007335 cerebellar astrocytoma Diseases 0.000 description 2
- 239000013626 chemical specie Substances 0.000 description 2
- 208000011654 childhood malignant neoplasm Diseases 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000012039 electrophile Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 201000004101 esophageal cancer Diseases 0.000 description 2
- CIPMPQGRFNDLAP-UHFFFAOYSA-N ethyl 1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C1=CN=CS1 CIPMPQGRFNDLAP-UHFFFAOYSA-N 0.000 description 2
- AXDZSLPARMAAQN-UHFFFAOYSA-N ethyl 2-azido-3-hydroxy-3-(1,3-thiazol-5-yl)propanoate Chemical compound N(=[N+]=[N-])C(C(=O)OCC)C(C1=CN=CS1)O AXDZSLPARMAAQN-UHFFFAOYSA-N 0.000 description 2
- HVJJYOAPXBPQQV-UHFFFAOYSA-N ethyl 2-azidoacetate Chemical compound CCOC(=O)CN=[N+]=[N-] HVJJYOAPXBPQQV-UHFFFAOYSA-N 0.000 description 2
- KXIWQCRIZCNLEV-UHFFFAOYSA-N ethyl 2-bromo-4-methylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound CCOC(=O)c1cc2sc(Br)nc2n1C KXIWQCRIZCNLEV-UHFFFAOYSA-N 0.000 description 2
- RULBHHKYXGRXMM-UHFFFAOYSA-N ethyl 4-methyl-2-methylsulfanylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound CN1C(=CC2=C1N=C(S2)SC)C(=O)OCC RULBHHKYXGRXMM-UHFFFAOYSA-N 0.000 description 2
- QMDMJORSFLNIOB-UHFFFAOYSA-N ethyl 5-chloro-1-tritylpyrazole-3-carboxylate Chemical compound ClC1=CC(=NN1C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1)C(=O)OCC QMDMJORSFLNIOB-UHFFFAOYSA-N 0.000 description 2
- RENWADNBKJXNAU-UHFFFAOYSA-N ethyl 6-formyl-2,4-dimethylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(=O)C1=C(N(C=2N=C(SC=21)C)C)C(=O)OCC RENWADNBKJXNAU-UHFFFAOYSA-N 0.000 description 2
- LMPPOZVNUYXLCW-UHFFFAOYSA-N ethyl 6-formyl-4-methyl-2-methylsulfanylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(=O)C1=C(N(C=2N=C(SC=21)SC)C)C(=O)OCC LMPPOZVNUYXLCW-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 208000024519 eye neoplasm Diseases 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 238000007306 functionalization reaction Methods 0.000 description 2
- 206010061989 glomerulosclerosis Diseases 0.000 description 2
- 230000034659 glycolysis Effects 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 230000026030 halogenation Effects 0.000 description 2
- 238000005658 halogenation reaction Methods 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000031261 interleukin-10 production Effects 0.000 description 2
- 230000009545 invasion Effects 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 210000004153 islets of langerhan Anatomy 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 210000000244 kidney pelvis Anatomy 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 206010023841 laryngeal neoplasm Diseases 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 208000012987 lip and oral cavity carcinoma Diseases 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000005229 liver cell Anatomy 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 208000003747 lymphoid leukemia Diseases 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 208000006178 malignant mesothelioma Diseases 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- XSDUQPYIDQQPIY-UHFFFAOYSA-N methyl 1-(2-trimethylsilylethoxymethyl)pyrazolo[4,3-c]pyridine-4-carboxylate Chemical compound C[Si](CCOCN1N=CC=2C(=NC=CC=21)C(=O)OC)(C)C XSDUQPYIDQQPIY-UHFFFAOYSA-N 0.000 description 2
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 210000003470 mitochondria Anatomy 0.000 description 2
- 230000004065 mitochondrial dysfunction Effects 0.000 description 2
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 210000005170 neoplastic cell Anatomy 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 2
- 201000008106 ocular cancer Diseases 0.000 description 2
- 201000002575 ocular melanoma Diseases 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 201000002530 pancreatic endocrine carcinoma Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 2
- 208000010626 plasma cell neoplasm Diseases 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 230000004481 post-translational protein modification Effects 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 201000007271 pre-malignant neoplasm Diseases 0.000 description 2
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 2
- 230000000770 proinflammatory effect Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 208000000587 small cell lung carcinoma Diseases 0.000 description 2
- RMBAVIFYHOYIFM-UHFFFAOYSA-M sodium methanethiolate Chemical compound [Na+].[S-]C RMBAVIFYHOYIFM-UHFFFAOYSA-M 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- NECLQTPQJZSWOE-UHFFFAOYSA-N spiro[5.5]undecane Chemical compound C1CCCCC21CCCCC2 NECLQTPQJZSWOE-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 239000010414 supernatant solution Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 201000002510 thyroid cancer Diseases 0.000 description 2
- 239000011135 tin Substances 0.000 description 2
- 229960005267 tositumomab Drugs 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 206010044412 transitional cell carcinoma Diseases 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- DNEHHUXTVQXYOO-UHFFFAOYSA-N trimethyl-[2-[[4-(phenylsulfanylmethyl)indazol-1-yl]methoxy]ethyl]silane Chemical compound C1(=CC=CC=C1)SCC1=C2C=NN(C2=CC=C1)COCC[Si](C)(C)C DNEHHUXTVQXYOO-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 2
- 208000018417 undifferentiated high grade pleomorphic sarcoma of bone Diseases 0.000 description 2
- 210000000626 ureter Anatomy 0.000 description 2
- 201000005112 urinary bladder cancer Diseases 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 2
- ITHMAZGQCBSQFB-UHFFFAOYSA-M zinc;1-fluoro-4-methanidylbenzene;bromide Chemical compound [Zn+2].[Br-].[CH2-]C1=CC=C(F)C=C1 ITHMAZGQCBSQFB-UHFFFAOYSA-M 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- JRHPOFJADXHYBR-HTQZYQBOSA-N (1r,2r)-1-n,2-n-dimethylcyclohexane-1,2-diamine Chemical compound CN[C@@H]1CCCC[C@H]1NC JRHPOFJADXHYBR-HTQZYQBOSA-N 0.000 description 1
- YKZFSISAODWSQG-UHFFFAOYSA-N (6-chloropyridin-2-yl)methanol Chemical compound OCC1=CC=CC(Cl)=N1 YKZFSISAODWSQG-UHFFFAOYSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- 125000006648 (C1-C8) haloalkyl group Chemical group 0.000 description 1
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- GLUABPSZMHYCNO-UHFFFAOYSA-N 1,2,3,3a,4,5,6,6a-octahydropyrrolo[3,2-b]pyrrole Chemical compound N1CCC2NCCC21 GLUABPSZMHYCNO-UHFFFAOYSA-N 0.000 description 1
- 125000005904 1,2,3,4-tetrahydro-1,6-naphthyridinyl group Chemical group 0.000 description 1
- 125000001359 1,2,3-triazol-4-yl group Chemical group [H]N1N=NC([*])=C1[H] 0.000 description 1
- VYMPLPIFKRHAAC-UHFFFAOYSA-N 1,2-ethanedithiol Chemical compound SCCS VYMPLPIFKRHAAC-UHFFFAOYSA-N 0.000 description 1
- OXHOPZLBSSTTBU-UHFFFAOYSA-N 1,3-bis(bromomethyl)benzene Chemical compound BrCC1=CC=CC(CBr)=C1 OXHOPZLBSSTTBU-UHFFFAOYSA-N 0.000 description 1
- 125000005895 1,4,5,7-tetrahydropyrano[3,4-b]pyrrolyl group Chemical group 0.000 description 1
- QSIVWRRHVXSDNE-UHFFFAOYSA-N 1-(bromomethyl)-3-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=CC(CBr)=C1 QSIVWRRHVXSDNE-UHFFFAOYSA-N 0.000 description 1
- LNWXALCHPJANMJ-UHFFFAOYSA-N 1-(bromomethyl)-3-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC(CBr)=C1 LNWXALCHPJANMJ-UHFFFAOYSA-N 0.000 description 1
- NVNPLEPBDPJYRZ-UHFFFAOYSA-N 1-(bromomethyl)-4-fluorobenzene Chemical compound FC1=CC=C(CBr)C=C1 NVNPLEPBDPJYRZ-UHFFFAOYSA-N 0.000 description 1
- FJANNOJSTOGZHK-UHFFFAOYSA-N 1-adamantyl carbamate Chemical compound C1C(C2)CC3CC2CC1(OC(=O)N)C3 FJANNOJSTOGZHK-UHFFFAOYSA-N 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- RYIFAUNVBKLDLW-UHFFFAOYSA-N 10-(1H-imidazol-2-ylmethyl)-7-methyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(SC(CC3=NNC=C3)=N2)C2=C1C(=O)N(CC1=NC=CN1)N=C2 RYIFAUNVBKLDLW-UHFFFAOYSA-N 0.000 description 1
- QWHNLQBRMNPMIS-UHFFFAOYSA-N 10-(1H-indazol-4-ylmethyl)-7-methyl-4-(1-phenylcyclopropyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C1(CC1)C1=CC=CC=C1)C QWHNLQBRMNPMIS-UHFFFAOYSA-N 0.000 description 1
- QZMSSWQEVFNOAS-UHFFFAOYSA-N 10-(1H-indazol-4-ylmethyl)-7-methyl-4-(1-phenylethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CC(c1nc2n(C)c3c(cnn(Cc4cccc5[nH]ncc45)c3=O)c2s1)c1ccccc1 QZMSSWQEVFNOAS-UHFFFAOYSA-N 0.000 description 1
- WPJWAZMLXAQRIE-UHFFFAOYSA-N 10-(1H-indazol-4-ylmethyl)-7-methyl-4-(methylaminomethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CNC)C WPJWAZMLXAQRIE-UHFFFAOYSA-N 0.000 description 1
- SYECWMSNDDGKHZ-UHFFFAOYSA-N 10-(hydroxymethyl)-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OCN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C)C SYECWMSNDDGKHZ-UHFFFAOYSA-N 0.000 description 1
- FNEZASPJAXEBNR-UHFFFAOYSA-N 10-[(2-aminopyridin-4-yl)methyl]-7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NC1=NC=CC(=C1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)S(=O)C)C FNEZASPJAXEBNR-UHFFFAOYSA-N 0.000 description 1
- PJLQJTJHUUIGJC-UHFFFAOYSA-N 10-[(2-chloropyrimidin-4-yl)methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound ClC1=NC=CC(=N1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)SC)C PJLQJTJHUUIGJC-UHFFFAOYSA-N 0.000 description 1
- FEQBOBJVZANBKI-UHFFFAOYSA-N 10-[(3-acetylphenyl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C)(=O)C=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C)C)C=CC=1 FEQBOBJVZANBKI-UHFFFAOYSA-N 0.000 description 1
- LGKISNDMBVFWKF-UHFFFAOYSA-N 10-[(3-acetylphenyl)methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C)(=O)C=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)SC)C)C=CC=1 LGKISNDMBVFWKF-UHFFFAOYSA-N 0.000 description 1
- CJLLICAXRLKIDR-UHFFFAOYSA-N 10-[(3-hydroxyphenyl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound Cc1nc2n(C)c3c(cnn(Cc4cccc(O)c4)c3=O)c2s1 CJLLICAXRLKIDR-UHFFFAOYSA-N 0.000 description 1
- PDALVFYIJZWEDY-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-4-(2-phenyl-1,3-dithiolan-2-yl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COc1cccc(Cn2ncc3c4sc(nc4n(C)c3c2=O)C2(SCCS2)c2ccccc2)c1 PDALVFYIJZWEDY-UHFFFAOYSA-N 0.000 description 1
- GSCVUBFSPSHGBQ-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-4-(trifluoromethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COc1cccc(Cn2ncc3c4sc(nc4n(C)c3c2=O)C(F)(F)F)c1 GSCVUBFSPSHGBQ-UHFFFAOYSA-N 0.000 description 1
- QPPAISWQTMITES-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)S(=O)C)C)C=CC=1 QPPAISWQTMITES-UHFFFAOYSA-N 0.000 description 1
- PJRLARMGEHDZPH-UHFFFAOYSA-N 10-[(3-methoxyphenyl)methyl]-7-methyl-4-methylsulfonyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COc1cccc(Cn2ncc3c4sc(nc4n(C)c3c2=O)S(C)(=O)=O)c1 PJRLARMGEHDZPH-UHFFFAOYSA-N 0.000 description 1
- OQJMQTIOQSYNFQ-UHFFFAOYSA-N 10-[(6-aminopyridin-2-yl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound NC1=CC=CC(=N1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C)C OQJMQTIOQSYNFQ-UHFFFAOYSA-N 0.000 description 1
- RUAQNKKXFCKIET-UHFFFAOYSA-N 10-[(6-aminopyridin-2-yl)methyl]-7-methyl-4-(1H-pyrazol-5-yloxy)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound Cn1c2nc(Oc3ccn[nH]3)sc2c2cnn(Cc3cccc(N)n3)c(=O)c12 RUAQNKKXFCKIET-UHFFFAOYSA-N 0.000 description 1
- JENFKQFPMGPAAK-UHFFFAOYSA-N 10-[(6-fluoropyridin-2-yl)methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound FC1=CC=CC(=N1)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C)C JENFKQFPMGPAAK-UHFFFAOYSA-N 0.000 description 1
- OMJGEVXISLDSDA-UHFFFAOYSA-N 10-[[2-[(2,4-dimethoxyphenyl)methylamino]-1,3-thiazol-5-yl]methyl]-7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC1=C(CNC=2SC(=CN=2)CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)S(=O)C)C)C=CC(=C1)OC OMJGEVXISLDSDA-UHFFFAOYSA-N 0.000 description 1
- ALENXKIQRWWTKH-UHFFFAOYSA-N 10-[[3-(1-aminoethyl)phenyl]methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CC(N)C1=CC=CC(CN2N=CC3=C(N(C)C4=C3SC(C)=N4)C2=O)=C1 ALENXKIQRWWTKH-UHFFFAOYSA-N 0.000 description 1
- ZRXWJABHSOLOHY-UHFFFAOYSA-N 10-[[3-(2-hydroxypropan-2-yl)phenyl]methyl]-7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OC(C)(C)C=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)SC)C)C=CC=1 ZRXWJABHSOLOHY-UHFFFAOYSA-N 0.000 description 1
- NMHWBFJBHMPYOF-UHFFFAOYSA-N 10-[[3-(2-hydroxypropan-2-yl)phenyl]methyl]-7-methyl-4-methylsulfinyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound Cn1c2nc(sc2c2cnn(Cc3cccc(c3)C(C)(C)O)c(=O)c12)S(C)=O NMHWBFJBHMPYOF-UHFFFAOYSA-N 0.000 description 1
- VAIHZCUBAXDRHF-UHFFFAOYSA-N 10-[[3-(aminomethyl)phenyl]methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound Cc1nc2n(C)c3c(cnn(Cc4cccc(CN)c4)c3=O)c2s1 VAIHZCUBAXDRHF-UHFFFAOYSA-N 0.000 description 1
- VQXGMWMPIRGBHP-UHFFFAOYSA-N 10-[[4-(hydroxymethyl)phenyl]methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OCC1=CC=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C)C)C=C1 VQXGMWMPIRGBHP-UHFFFAOYSA-N 0.000 description 1
- SXGGJHOKIFXZCM-UHFFFAOYSA-N 10-[[6-[(2,4-dimethoxyphenyl)methylamino]pyridin-2-yl]methyl]-4,7-dimethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COc1ccc(CNc2cccc(Cn3ncc4c5sc(C)nc5n(C)c4c3=O)n2)c(OC)c1 SXGGJHOKIFXZCM-UHFFFAOYSA-N 0.000 description 1
- VGXNENQIVUDCFD-UHFFFAOYSA-N 10-benzyl-4-methylsulfonyl-7-phenyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1N=CC2=C(C1=O)N(C1=C2SC(=N1)S(=O)(=O)C)C1=CC=CC=C1 VGXNENQIVUDCFD-UHFFFAOYSA-N 0.000 description 1
- 125000005894 1H-benzo[e][1,4]diazepinyl group Chemical group 0.000 description 1
- XYHKNCXZYYTLRG-UHFFFAOYSA-N 1h-imidazole-2-carbaldehyde Chemical compound O=CC1=NC=CN1 XYHKNCXZYYTLRG-UHFFFAOYSA-N 0.000 description 1
- AAILEWXSEQLMNI-UHFFFAOYSA-N 1h-pyridazin-6-one Chemical group OC1=CC=CN=N1 AAILEWXSEQLMNI-UHFFFAOYSA-N 0.000 description 1
- UPQQXPKAYZYUKO-UHFFFAOYSA-N 2,2,2-trichloroacetamide Chemical group OC(=N)C(Cl)(Cl)Cl UPQQXPKAYZYUKO-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical group NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- 125000005899 2,3-dihydro-1H-pyrrolo[2,3-b]pyridinyl group Chemical group 0.000 description 1
- 125000005900 2,3-dihydrofuro[2,3-b]pyridinyl group Chemical group 0.000 description 1
- PXVUDLXXKGSXHH-UHFFFAOYSA-N 2,4,6-trimethoxybenzenesulfonamide Chemical compound COC1=CC(OC)=C(S(N)(=O)=O)C(OC)=C1 PXVUDLXXKGSXHH-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- QXLQZLBNPTZMRK-UHFFFAOYSA-N 2-[(dimethylamino)methyl]-1-(2,4-dimethylphenyl)prop-2-en-1-one Chemical compound CN(C)CC(=C)C(=O)C1=CC=C(C)C=C1C QXLQZLBNPTZMRK-UHFFFAOYSA-N 0.000 description 1
- SIOVWRVUOJJEKO-UHFFFAOYSA-N 2-[10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]acetamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CC(=O)N)C SIOVWRVUOJJEKO-UHFFFAOYSA-N 0.000 description 1
- QCXYQYZDPNDMER-UHFFFAOYSA-N 2-[7-methyl-9-oxo-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]-2-phenylacetonitrile Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)C(C#N)C1=CC=CC=C1 QCXYQYZDPNDMER-UHFFFAOYSA-N 0.000 description 1
- HTULHGICLRIZBC-UHFFFAOYSA-N 2-[[3-(iodomethyl)pyrazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound ICC1=NN(C=C1)COCC[Si](C)(C)C HTULHGICLRIZBC-UHFFFAOYSA-N 0.000 description 1
- PKNVLYJROLXRRX-UHFFFAOYSA-N 2-[[3-[(4,7-dimethyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]phenyl]methyl]isoindole-1,3-dione Chemical compound Cc1nc2n(C)c3c(cnn(Cc4cccc(CN5C(=O)c6ccccc6C5=O)c4)c3=O)c2s1 PKNVLYJROLXRRX-UHFFFAOYSA-N 0.000 description 1
- ZIQSXKVOGHYBAJ-UHFFFAOYSA-N 2-[[4-(bromomethyl)indazol-1-yl]methoxy]ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCn1ncc2c(CBr)cccc12 ZIQSXKVOGHYBAJ-UHFFFAOYSA-N 0.000 description 1
- PPXUUPXQWDQNGO-UHFFFAOYSA-N 2-azidoacetic acid Chemical compound OC(=O)CN=[N+]=[N-] PPXUUPXQWDQNGO-UHFFFAOYSA-N 0.000 description 1
- YYHRYECWWYONCH-UHFFFAOYSA-N 2-bromo-5-(bromomethyl)-1,3-thiazole Chemical compound BrCC1=CN=C(Br)S1 YYHRYECWWYONCH-UHFFFAOYSA-N 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M 2-methylbenzenesulfonate Chemical compound CC1=CC=CC=C1S([O-])(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- NYANCYPJKOOJRB-UHFFFAOYSA-N 3-(benzenesulfonylmethyl)pyridine Chemical compound C=1C=CC=CC=1S(=O)(=O)CC1=CC=CN=C1 NYANCYPJKOOJRB-UHFFFAOYSA-N 0.000 description 1
- MYHUFJLABUEBLV-UHFFFAOYSA-N 3-(bromomethyl)-1-methylpyrazole Chemical compound CN1C=CC(CBr)=N1 MYHUFJLABUEBLV-UHFFFAOYSA-N 0.000 description 1
- QXUABQCRQWEHGB-UHFFFAOYSA-N 3-[(7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzonitrile Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC=1C=C(C#N)C=CC=1)=O)SC=N2 QXUABQCRQWEHGB-UHFFFAOYSA-N 0.000 description 1
- UZFPOOOQHWICKY-UHFFFAOYSA-N 3-[13-[1-[1-[8,12-bis(2-carboxyethyl)-17-(1-hydroxyethyl)-3,7,13,18-tetramethyl-21,24-dihydroporphyrin-2-yl]ethoxy]ethyl]-18-(2-carboxyethyl)-8-(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-2-yl]propanoic acid Chemical compound N1C(C=C2C(=C(CCC(O)=O)C(C=C3C(=C(C)C(C=C4N5)=N3)CCC(O)=O)=N2)C)=C(C)C(C(C)O)=C1C=C5C(C)=C4C(C)OC(C)C1=C(N2)C=C(N3)C(C)=C(C(O)C)C3=CC(C(C)=C3CCC(O)=O)=NC3=CC(C(CCC(O)=O)=C3C)=NC3=CC2=C1C UZFPOOOQHWICKY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- 125000006201 3-phenylpropyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- CICPLKAJEGYYGM-UHFFFAOYSA-N 3h-pyridazin-4-one Chemical compound O=C1CN=NC=C1 CICPLKAJEGYYGM-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- 125000005901 4,5,6,7-tetrahydro-1H-pyrrolo[2,3-b]pyridinyl group Chemical group 0.000 description 1
- 125000005902 4,5,6,7-tetrahydrofuro[3,2-c]pyridinyl group Chemical group 0.000 description 1
- 125000005903 4,5,6,7-tetrahydrothieno[3,2-b]pyridinyl group Chemical group 0.000 description 1
- LIAVXZALGVZDJU-UHFFFAOYSA-N 4,7-dimethyl-10-[(3-nitrophenyl)methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)[N+](=O)[O-])=O)C)N=1 LIAVXZALGVZDJU-UHFFFAOYSA-N 0.000 description 1
- YPBZGIIMWBGRSX-UHFFFAOYSA-N 4,7-dimethyl-10-[[3-(methylamino)phenyl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CNc1cccc(Cn2ncc3c4sc(C)nc4n(C)c3c2=O)c1 YPBZGIIMWBGRSX-UHFFFAOYSA-N 0.000 description 1
- ZTEZUKHSZIPADJ-UHFFFAOYSA-N 4-(1-hydroxy-1-phenylethyl)-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OC(C)(C1=CC=CC=C1)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 ZTEZUKHSZIPADJ-UHFFFAOYSA-N 0.000 description 1
- WXVHGJMEKWKMFK-UHFFFAOYSA-N 4-(2-hydroxyethyl)-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)CCO)C WXVHGJMEKWKMFK-UHFFFAOYSA-N 0.000 description 1
- VFRFFXKQPYBDBJ-UHFFFAOYSA-N 4-(2-hydroxypropan-2-yl)-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OC(C)(C)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 VFRFFXKQPYBDBJ-UHFFFAOYSA-N 0.000 description 1
- QIZZFHXBGQYABP-UHFFFAOYSA-N 4-(hydroxymethyl)-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OCC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 QIZZFHXBGQYABP-UHFFFAOYSA-N 0.000 description 1
- PYVPFSJZGPVBPY-UHFFFAOYSA-N 4-[(4,7-dimethyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzoic acid Chemical compound Cc1nc2n(C)c3c(cnn(Cc4ccc(cc4)C(O)=O)c3=O)c2s1 PYVPFSJZGPVBPY-UHFFFAOYSA-N 0.000 description 1
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 1
- DNPWUUHBVZUOHK-UHFFFAOYSA-N 4-[(4-fluorophenyl)methyl]-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound FC1=CC=C(CC=2SC3=C(N(C=4C(N(N=CC=43)CC3=CC(=CC=C3)OC)=O)C)N=2)C=C1 DNPWUUHBVZUOHK-UHFFFAOYSA-N 0.000 description 1
- UFGVYROFSXDOGS-UHFFFAOYSA-N 4-[(7-methyl-4-methylsulfanyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzamide Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=CC=C(C(=O)N)C=C1)=O)SC(=N2)SC UFGVYROFSXDOGS-UHFFFAOYSA-N 0.000 description 1
- OXJZZVDCKHJGOB-UHFFFAOYSA-N 4-[(7-methyl-4-methylsulfanyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzonitrile Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=CC=C(C#N)C=C1)=O)SC(=N2)SC OXJZZVDCKHJGOB-UHFFFAOYSA-N 0.000 description 1
- VAPGDMXMPPYPLH-UHFFFAOYSA-N 4-[(7-methyl-4-methylsulfinyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]benzamide Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=CC=C(C(=O)N)C=C1)=O)SC(=N2)S(=O)C VAPGDMXMPPYPLH-UHFFFAOYSA-N 0.000 description 1
- UVJKLNBXZSRQFM-WQRHYEAKSA-N 4-[5-[(z)-(1-oxo-[1,3]thiazolo[3,2-a]benzimidazol-2-ylidene)methyl]furan-2-yl]benzenesulfonamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1C(O1)=CC=C1\C=C/1C(=O)N2C3=CC=CC=C3N=C2S\1 UVJKLNBXZSRQFM-WQRHYEAKSA-N 0.000 description 1
- QOHSAZWKWVCPCQ-UHFFFAOYSA-N 4-[[6-(dimethylamino)pyridin-2-yl]methyl]-7-methyl-10-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN(C)c1cccc(Cc2nc3n(C)c4c(cnn(Cc5ccn[nH]5)c4=O)c3s2)n1 QOHSAZWKWVCPCQ-UHFFFAOYSA-N 0.000 description 1
- SVDNKDDYFKLSHA-UHFFFAOYSA-N 4-[[6-(dimethylamino)pyridin-2-yl]methyl]-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN(C1=CC=CC(=N1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=NN(C=C2)COCC[Si](C)(C)C)=O)C)N=1)C SVDNKDDYFKLSHA-UHFFFAOYSA-N 0.000 description 1
- JFARIKMTOJELLP-UHFFFAOYSA-N 4-[amino(phenyl)methyl]-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(C1=CC=CC=C1)N)C JFARIKMTOJELLP-UHFFFAOYSA-N 0.000 description 1
- KGZSGYAUHCIXPN-UHFFFAOYSA-N 4-[benzenesulfonyl-(5-chloro-1-tritylpyrazol-3-yl)methyl]-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound ClC1=CC(=NN1C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1)C(C=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1)S(=O)(=O)C1=CC=CC=C1 KGZSGYAUHCIXPN-UHFFFAOYSA-N 0.000 description 1
- KSNSBPOJBOEVRC-UHFFFAOYSA-N 4-[benzenesulfonyl-[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-10-benzyl-7-ethyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(C1=NN(C=C1)COCC[Si](C)(C)C)S(=O)(=O)C1=CC=CC=C1)CC KSNSBPOJBOEVRC-UHFFFAOYSA-N 0.000 description 1
- SOXOGBFAHTVSDV-UHFFFAOYSA-N 4-[benzenesulfonyl-[2-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)C(C1=CC=NN1COCC[Si](C)(C)C)S(=O)(=O)C1=CC=CC=C1 SOXOGBFAHTVSDV-UHFFFAOYSA-N 0.000 description 1
- MGCDAASKGPGKES-UHFFFAOYSA-N 4-[difluoro(phenyl)methyl]-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COc1cccc(Cn2ncc3c4sc(nc4n(C)c3c2=O)C(F)(F)c2ccccc2)c1 MGCDAASKGPGKES-UHFFFAOYSA-N 0.000 description 1
- DWXJOMRBYHCXEZ-UHFFFAOYSA-N 4-[hydroxy(phenyl)methyl]-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(C1=CC=CC=C1)O)C DWXJOMRBYHCXEZ-UHFFFAOYSA-N 0.000 description 1
- CYZPYJHEPYWNTK-UHFFFAOYSA-N 4-[hydroxy(phenyl)methyl]-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OC(C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1)C1=CC=CC=C1 CYZPYJHEPYWNTK-UHFFFAOYSA-N 0.000 description 1
- ZKMAZEURPANVHO-UHFFFAOYSA-N 4-[hydroxy(phenyl)methyl]-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound OC(C=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1)C1=CC=CC=C1 ZKMAZEURPANVHO-UHFFFAOYSA-N 0.000 description 1
- BBWVOVISWICYOI-UHFFFAOYSA-N 4-acetyl-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C)(=O)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 BBWVOVISWICYOI-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- UBFUCBMBJCTGMX-UHFFFAOYSA-N 4-benzoyl-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(C1=CC=CC=C1)=O)C UBFUCBMBJCTGMX-UHFFFAOYSA-N 0.000 description 1
- NNBKVXFACWXMOW-UHFFFAOYSA-N 4-benzyl-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 NNBKVXFACWXMOW-UHFFFAOYSA-N 0.000 description 1
- NOOPXZCJTLZMQB-UHFFFAOYSA-N 4-benzyl-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)C=1SC2=C(N(C=3C(NN=CC=32)=O)C)N=1 NOOPXZCJTLZMQB-UHFFFAOYSA-N 0.000 description 1
- WGYDMPKGHCKWCG-UHFFFAOYSA-N 4-benzylsulfanyl-10-(1H-indazol-4-ylmethyl)-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)SCC1=CC=CC=C1)C WGYDMPKGHCKWCG-UHFFFAOYSA-N 0.000 description 1
- FISFBDVEULKQBP-UHFFFAOYSA-N 4-benzylsulfanyl-7-methyl-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)SC=1SC2=C(N(C=3C(N(N=CC=32)CC2=C3C=NN(C3=CC=C2)COCC[Si](C)(C)C)=O)C)N=1 FISFBDVEULKQBP-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- DJYSSYAHGFSRAT-UHFFFAOYSA-N 4-chloro-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound ClC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 DJYSSYAHGFSRAT-UHFFFAOYSA-N 0.000 description 1
- JVVRCYWZTJLJSG-UHFFFAOYSA-N 4-dimethylaminophenol Chemical compound CN(C)C1=CC=C(O)C=C1 JVVRCYWZTJLJSG-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-dimethylaminopyridine Substances CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 1
- FZHZKNQAQPFLCK-UHFFFAOYSA-N 4-ethenyl-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1C=C(CN2N=CC3=C(C2=O)N(C2=C3SC(=N2)C=C)C)C=CC=1 FZHZKNQAQPFLCK-UHFFFAOYSA-N 0.000 description 1
- XNTWVETUZLMRNJ-UHFFFAOYSA-N 4-ethyl-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C)C=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 XNTWVETUZLMRNJ-UHFFFAOYSA-N 0.000 description 1
- KKGMHBQYVPPVES-UHFFFAOYSA-N 4-iodo-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound IC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 KKGMHBQYVPPVES-UHFFFAOYSA-N 0.000 description 1
- BXQAZSQZGVBRPN-UHFFFAOYSA-N 4-methoxy-10-[(3-methoxyphenyl)methyl]-7-methyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound COC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC(=CC=C2)OC)=O)C)N=1 BXQAZSQZGVBRPN-UHFFFAOYSA-N 0.000 description 1
- 125000005896 5,6-dihydro-4H-furo[3,2-b]pyrrolyl group Chemical group 0.000 description 1
- 125000005898 5,7-dihydro-4H-thieno[2,3-c]pyranyl group Chemical group 0.000 description 1
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- 125000005897 6,7-dihydro-5H-furo[3,2-b]pyranyl group Chemical group 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- ZWKJWVSEDISQIS-UHFFFAOYSA-N 6-[(3-aminophenyl)methyl]-4-methyl-2-methylsulfinyl-5-thieno[3,4]pyrrolo[1,3-d]pyridazinone Chemical compound O=C1C=2N(C)C=3C=C(S(C)=O)SC=3C=2C=NN1CC1=CC=CC(N)=C1 ZWKJWVSEDISQIS-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- KWYKNFANWSPUMT-UHFFFAOYSA-N 7,10-dibenzyl-4-(1H-pyrazol-5-ylmethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CC2=CC=CC=C2)=O)CC2=CC=CC=C2)N=1 KWYKNFANWSPUMT-UHFFFAOYSA-N 0.000 description 1
- JQOPJFQRYAQCDR-UHFFFAOYSA-N 7,10-dibenzyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound C(C1=CC=CC=C1)N1C2=C(C3=C1C(N(N=C3)CC1=CC=CC=C1)=O)SC(=N2)SC JQOPJFQRYAQCDR-UHFFFAOYSA-N 0.000 description 1
- ROAUPWGUPMSTIZ-UHFFFAOYSA-N 7-methyl-10-[(1-methylindazol-4-yl)methyl]-4-(1-phenylethyl)-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)C)=O)SC(=N2)C(C)C1=CC=CC=C1 ROAUPWGUPMSTIZ-UHFFFAOYSA-N 0.000 description 1
- SAJAOKCWSOLSTE-UHFFFAOYSA-N 7-methyl-10-[(1-methylpyrazol-3-yl)methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=NN(C=C1)C)=O)SC=N2 SAJAOKCWSOLSTE-UHFFFAOYSA-N 0.000 description 1
- TWQMGYAQJYCCSA-UHFFFAOYSA-N 7-methyl-10-[(1-methylpyrazol-3-yl)methyl]-4-[2-[1-(oxan-2-yl)pyrazol-3-yl]ethyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=NN(C=C1)C)=O)SC(=N2)CCC1=NN(C=C1)C1OCCCC1 TWQMGYAQJYCCSA-UHFFFAOYSA-N 0.000 description 1
- CXQGCEFTEDZLPS-UHFFFAOYSA-N 7-methyl-4-(1-phenylcyclopropyl)-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)C1(CC1)C1=CC=CC=C1 CXQGCEFTEDZLPS-UHFFFAOYSA-N 0.000 description 1
- LLGOMGICZDOADO-UHFFFAOYSA-N 7-methyl-4-(1H-pyrazol-5-ylmethyl)-10-[2-(1,3-thiazol-4-yl)ethyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound N1N=C(C=C1)CC=1SC2=C(N(C=3C(N(N=CC=32)CCC=2N=CSC=2)=O)C)N=1 LLGOMGICZDOADO-UHFFFAOYSA-N 0.000 description 1
- SMLZYVDXSWRUKB-UHFFFAOYSA-N 7-methyl-4-(methylaminomethyl)-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)CNC SMLZYVDXSWRUKB-UHFFFAOYSA-N 0.000 description 1
- IRYVAPYWWHRFIQ-UHFFFAOYSA-N 7-methyl-4-[[2-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)CC1=CC=NN1COCC[Si](C)(C)C IRYVAPYWWHRFIQ-UHFFFAOYSA-N 0.000 description 1
- GRXKSTHCLCTZGY-UHFFFAOYSA-N 7-methyl-4-methylsulfanyl-10-[(2-oxo-1H-pyrimidin-6-yl)methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC=1NC(N=CC=1)=O)=O)SC(=N2)SC GRXKSTHCLCTZGY-UHFFFAOYSA-N 0.000 description 1
- FSHCGBVXYLWCBU-UHFFFAOYSA-N 7-methyl-4-methylsulfanyl-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(NN=C3)=O)SC(=N2)SC FSHCGBVXYLWCBU-UHFFFAOYSA-N 0.000 description 1
- BACCGGQSZGQWEY-UHFFFAOYSA-N 7-methyl-4-methylsulfonyl-10-[[1-(2-trimethylsilylethoxymethyl)pyrazol-3-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-9-one Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=NN(C=C1)COCC[Si](C)(C)C)=O)SC(=N2)S(=O)(=O)C BACCGGQSZGQWEY-UHFFFAOYSA-N 0.000 description 1
- YCJHCDCYZBENJF-UHFFFAOYSA-N 7-methyl-9-oxo-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-sulfonamide Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)S(=O)(=O)N YCJHCDCYZBENJF-UHFFFAOYSA-N 0.000 description 1
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 description 1
- ZZOKVYOCRSMTSS-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl carbamate Chemical group C1=CC=C2C(COC(=O)N)C3=CC=CC=C3C2=C1 ZZOKVYOCRSMTSS-UHFFFAOYSA-N 0.000 description 1
- 208000002008 AIDS-Related Lymphoma Diseases 0.000 description 1
- 206010000591 Acrochordon Diseases 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000003200 Adenoma Diseases 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 206010061424 Anal cancer Diseases 0.000 description 1
- 208000000058 Anaplasia Diseases 0.000 description 1
- 241000272525 Anas platyrhynchos Species 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 241000272814 Anser sp. Species 0.000 description 1
- 208000002267 Anti-neutrophil cytoplasmic antibody-associated vasculitis Diseases 0.000 description 1
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 1
- 208000007860 Anus Neoplasms Diseases 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000032841 Bulimia Diseases 0.000 description 1
- 206010006550 Bulimia nervosa Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 125000000041 C6-C10 aryl group Chemical group 0.000 description 1
- VSEIDZLLWQQJGK-CHOZPQDDSA-N CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O Chemical compound CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O VSEIDZLLWQQJGK-CHOZPQDDSA-N 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 1
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- 206010007733 Catabolic state Diseases 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 201000005262 Chondroma Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 206010073140 Clear cell sarcoma of soft tissue Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 208000008960 Diabetic foot Diseases 0.000 description 1
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 1
- 206010014476 Elevated cholesterol Diseases 0.000 description 1
- 206010014486 Elevated triglycerides Diseases 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 206010048554 Endothelial dysfunction Diseases 0.000 description 1
- SAMRUMKYXPVKPA-VFKOLLTISA-N Enocitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 SAMRUMKYXPVKPA-VFKOLLTISA-N 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical group CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 208000017259 Extragonadal germ cell tumor Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 201000003741 Gastrointestinal carcinoma Diseases 0.000 description 1
- 102000013382 Gelatinases Human genes 0.000 description 1
- 108010026132 Gelatinases Proteins 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000987493 Homo sapiens Phosphatidylethanolamine-binding protein 1 Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 1
- 206010020710 Hyperphagia Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 206010021042 Hypopharyngeal cancer Diseases 0.000 description 1
- 206010056305 Hypopharyngeal neoplasm Diseases 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 102000004195 Isomerases Human genes 0.000 description 1
- 108090000769 Isomerases Proteins 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 1
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 241000270322 Lepidosauria Species 0.000 description 1
- 206010062038 Lip neoplasm Diseases 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 206010024612 Lipoma Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 206010025312 Lymphoma AIDS related Diseases 0.000 description 1
- 210000004322 M2 macrophage Anatomy 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- 206010054805 Macroangiopathy Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000004059 Male Breast Neoplasms Diseases 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 208000030070 Malignant epithelial tumor of ovary Diseases 0.000 description 1
- 206010073059 Malignant neoplasm of unknown primary site Diseases 0.000 description 1
- 208000032271 Malignant tumor of penis Diseases 0.000 description 1
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 1
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 1
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 1
- XOGTZOOQQBDUSI-UHFFFAOYSA-M Mesna Chemical compound [Na+].[O-]S(=O)(=O)CCS XOGTZOOQQBDUSI-UHFFFAOYSA-M 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- CRZQGDNQQAALAY-UHFFFAOYSA-N Methyl benzeneacetate Chemical compound COC(=O)CC1=CC=CC=C1 CRZQGDNQQAALAY-UHFFFAOYSA-N 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 1
- 241000872931 Myoporum sandwicense Species 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- YFAOSYGFYXGGSL-UHFFFAOYSA-N N-[[10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]methyl]acetamide Chemical compound CC(=O)NCc1nc2n(C)c3c(cnn(Cc4cccc5[nH]ncc45)c3=O)c2s1 YFAOSYGFYXGGSL-UHFFFAOYSA-N 0.000 description 1
- HTYBGJKCVAYXGG-UHFFFAOYSA-N N-[[7-methyl-9-oxo-10-[[1-(2-trimethylsilylethoxymethyl)indazol-4-yl]methyl]-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-4-yl]methyl]acetamide Chemical compound CN1C2=C(C3=C1C(N(N=C3)CC1=C3C=NN(C3=CC=C1)COCC[Si](C)(C)C)=O)SC(=N2)CNC(C)=O HTYBGJKCVAYXGG-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- WFPSUUXUHGCSKA-UHFFFAOYSA-N N-hydroxy-10-(1H-indazol-4-ylmethyl)-7-methyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraene-4-carboxamide Chemical compound N1N=CC2=C(C=CC=C12)CN1N=CC2=C(C1=O)N(C1=C2SC(=N1)C(=O)NO)C WFPSUUXUHGCSKA-UHFFFAOYSA-N 0.000 description 1
- LYPFDBRUNKHDGX-SOGSVHMOSA-N N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 Chemical compound N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 LYPFDBRUNKHDGX-SOGSVHMOSA-N 0.000 description 1
- 241001274216 Naso Species 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 238000006411 Negishi coupling reaction Methods 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 206010029266 Neuroendocrine carcinoma of the skin Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 206010031096 Oropharyngeal cancer Diseases 0.000 description 1
- 206010057444 Oropharyngeal neoplasm Diseases 0.000 description 1
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061328 Ovarian epithelial cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 101150005879 PKM gene Proteins 0.000 description 1
- 229910019213 POCl3 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 201000011152 Pemphigus Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 206010034299 Penile cancer Diseases 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 102100028489 Phosphatidylethanolamine-binding protein 1 Human genes 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 1
- 101150025052 Pklr gene Proteins 0.000 description 1
- 201000008199 Pleuropulmonary blastoma Diseases 0.000 description 1
- 208000004880 Polyuria Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 208000033464 Reiter syndrome Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 206010038934 Retinopathy proliferative Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010048810 Sebaceous hyperplasia Diseases 0.000 description 1
- 206010039796 Seborrhoeic keratosis Diseases 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 1
- 206010068771 Soft tissue neoplasm Diseases 0.000 description 1
- 238000006619 Stille reaction Methods 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- WFWLQNSHRPWKFK-UHFFFAOYSA-N Tegafur Chemical compound O=C1NC(=O)C(F)=CN1C1OCCC1 WFWLQNSHRPWKFK-UHFFFAOYSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 206010043458 Thirst Diseases 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- 206010044407 Transitional cell cancer of the renal pelvis and ureter Diseases 0.000 description 1
- YCPOZVAOBBQLRI-WDSKDSINSA-N Treosulfan Chemical compound CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O YCPOZVAOBBQLRI-WDSKDSINSA-N 0.000 description 1
- UMILHIMHKXVDGH-UHFFFAOYSA-N Triethylene glycol diglycidyl ether Chemical compound C1OC1COCCOCCOCCOCC1CO1 UMILHIMHKXVDGH-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 206010047124 Vasculitis necrotising Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- WCYHIPUFLSSCFD-UHFFFAOYSA-N [3-[(4,7-dimethyl-9-oxo-3-thia-5,7,10,11-tetrazatricyclo[6.4.0.02,6]dodeca-1(8),2(6),4,11-tetraen-10-yl)methyl]phenyl]urea Chemical compound Cc1nc2n(C)c3c(cnn(Cc4cccc(NC(N)=O)c4)c3=O)c2s1 WCYHIPUFLSSCFD-UHFFFAOYSA-N 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 229930183665 actinomycin Natural products 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 150000001266 acyl halides Chemical class 0.000 description 1
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 1
- 229910001573 adamantine Inorganic materials 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 150000008055 alkyl aryl sulfonates Chemical class 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 230000008856 allosteric binding Effects 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229960002749 aminolevulinic acid Drugs 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- BVCZEBOGSOYJJT-UHFFFAOYSA-N ammonium carbamate Chemical compound [NH4+].NC([O-])=O BVCZEBOGSOYJJT-UHFFFAOYSA-N 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 238000012435 analytical chromatography Methods 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 201000011165 anus cancer Diseases 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 238000007080 aromatic substitution reaction Methods 0.000 description 1
- 125000005199 aryl carbonyloxy group Chemical group 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 125000005200 aryloxy carbonyloxy group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 150000001541 aziridines Chemical class 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- LASLVGACQUUOEB-UHFFFAOYSA-N bicyclo[1.1.0]butane Chemical compound C1C2CC21 LASLVGACQUUOEB-UHFFFAOYSA-N 0.000 description 1
- MKCBRYIXFFGIKN-UHFFFAOYSA-N bicyclo[1.1.1]pentane Chemical compound C1C2CC1C2 MKCBRYIXFFGIKN-UHFFFAOYSA-N 0.000 description 1
- JSMRMEYFZHIPJV-UHFFFAOYSA-N bicyclo[2.1.1]hexane Chemical compound C1C2CC1CC2 JSMRMEYFZHIPJV-UHFFFAOYSA-N 0.000 description 1
- NAVGAEZCCRCJQT-UHFFFAOYSA-N bicyclo[3.3.3]undecane Chemical compound C1CCC2CCCC1CCC2 NAVGAEZCCRCJQT-UHFFFAOYSA-N 0.000 description 1
- OOSCHYDNHHSIKU-UHFFFAOYSA-N bicyclo[4.3.2]undecane Chemical compound C1CC2CCCC1CCCC2 OOSCHYDNHHSIKU-UHFFFAOYSA-N 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 201000008873 bone osteosarcoma Diseases 0.000 description 1
- 208000012172 borderline epithelial tumor of ovary Diseases 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 125000005620 boronic acid group Chemical class 0.000 description 1
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- 208000030224 brain astrocytoma Diseases 0.000 description 1
- 201000007983 brain glioma Diseases 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 238000002619 cancer immunotherapy Methods 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- YAYRGNWWLMLWJE-UHFFFAOYSA-L carboplatin Chemical compound O=C1O[Pt](N)(N)OC(=O)C11CCC1 YAYRGNWWLMLWJE-UHFFFAOYSA-L 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 229960002115 carboquone Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 230000001925 catabolic effect Effects 0.000 description 1
- 229960002412 cediranib Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 208000030239 cerebral astrocytoma Diseases 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 208000010575 cherry hemangioma Diseases 0.000 description 1
- 208000018805 childhood acute lymphoblastic leukemia Diseases 0.000 description 1
- 201000011633 childhood acute lymphocytic leukemia Diseases 0.000 description 1
- 201000004677 childhood cerebellar astrocytic neoplasm Diseases 0.000 description 1
- 201000008522 childhood cerebral astrocytoma Diseases 0.000 description 1
- 208000015632 childhood ependymoma Diseases 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- VXIVSQZSERGHQP-UHFFFAOYSA-N chloroacetamide Chemical group NC(=O)CCl VXIVSQZSERGHQP-UHFFFAOYSA-N 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 210000003690 classically activated macrophage Anatomy 0.000 description 1
- 201000000292 clear cell sarcoma Diseases 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 208000003611 congenital autoimmune diabetes mellitus Diseases 0.000 description 1
- 208000012696 congenital leptin deficiency Diseases 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 239000000824 cytostatic agent Substances 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 125000005892 decahydro-1,8-naphthyridinyl group Chemical group 0.000 description 1
- 125000004652 decahydroisoquinolinyl group Chemical group C1(NCCC2CCCCC12)* 0.000 description 1
- 125000005891 decahydronaphthyridinyl group Chemical group 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 229960005052 demecolcine Drugs 0.000 description 1
- 229960002923 denileukin diftitox Drugs 0.000 description 1
- 108010017271 denileukin diftitox Proteins 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical compound [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004582 dihydrobenzothienyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 125000005054 dihydropyrrolyl group Chemical group [H]C1=C([H])C([H])([H])C([H])([H])N1* 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 229960001776 edrecolomab Drugs 0.000 description 1
- 229960000925 efaproxiral Drugs 0.000 description 1
- BNFRJXLZYUTIII-UHFFFAOYSA-N efaproxiral Chemical compound CC1=CC(C)=CC(NC(=O)CC=2C=CC(OC(C)(C)C(O)=O)=CC=2)=C1 BNFRJXLZYUTIII-UHFFFAOYSA-N 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- BKJIXTWSNXCKJH-UHFFFAOYSA-N elesclomol Chemical compound C=1C=CC=CC=1C(=S)N(C)NC(=O)CC(=O)NN(C)C(=S)C1=CC=CC=C1 BKJIXTWSNXCKJH-UHFFFAOYSA-N 0.000 description 1
- 229950003247 elesclomol Drugs 0.000 description 1
- MGQRRMONVLMKJL-KWJIQSIXSA-N elsamitrucin Chemical compound O1[C@H](C)[C@H](O)[C@H](OC)[C@@H](N)[C@H]1O[C@@H]1[C@](O)(C)[C@@H](O)[C@@H](C)O[C@H]1OC1=CC=CC2=C(O)C(C(O3)=O)=C4C5=C3C=CC(C)=C5C(=O)OC4=C12 MGQRRMONVLMKJL-KWJIQSIXSA-N 0.000 description 1
- 229950002339 elsamitrucin Drugs 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 230000008694 endothelial dysfunction Effects 0.000 description 1
- 229950011487 enocitabine Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- HCZKYJDFEPMADG-UHFFFAOYSA-N erythro-nordihydroguaiaretic acid Natural products C=1C=C(O)C(O)=CC=1CC(C)C(C)CC1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-UHFFFAOYSA-N 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- YJFLUNOXLXTRAK-UHFFFAOYSA-N ethyl 2,4-dimethylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound CC=1SC2=C(N=1)N(C(=C2)C(=O)OCC)C YJFLUNOXLXTRAK-UHFFFAOYSA-N 0.000 description 1
- WQHAXVIUJDYOLU-UHFFFAOYSA-N ethyl 2-benzyl-4-methylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(C1=CC=CC=C1)C=1SC2=C(N=1)N(C(=C2)C(=O)OCC)C WQHAXVIUJDYOLU-UHFFFAOYSA-N 0.000 description 1
- YWKZVZKHAQGEKL-UHFFFAOYSA-N ethyl 2-methylsulfanyl-4-phenylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound CSC=1SC2=C(N=1)N(C(=C2)C(=O)OCC)C1=CC=CC=C1 YWKZVZKHAQGEKL-UHFFFAOYSA-N 0.000 description 1
- LHUTUSROXQUHKA-UHFFFAOYSA-N ethyl 4-benzyl-6-formyl-2-methylsulfanylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(C1=CC=CC=C1)N1C(=C(C2=C1N=C(S2)SC)C=O)C(=O)OCC LHUTUSROXQUHKA-UHFFFAOYSA-N 0.000 description 1
- CZHCJYBPYVHWJI-UHFFFAOYSA-N ethyl 4-ethyl-2-methylsulfanylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(C)N1C(=CC2=C1N=C(S2)SC)C(=O)OCC CZHCJYBPYVHWJI-UHFFFAOYSA-N 0.000 description 1
- XWFJPGZVQINIEB-UHFFFAOYSA-N ethyl 4-methylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound CCOC(=O)c1cc2scnc2n1C XWFJPGZVQINIEB-UHFFFAOYSA-N 0.000 description 1
- BXACPZGBYFAFIX-UHFFFAOYSA-N ethyl 4h-pyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound S1C=NC2=C1C=C(C(=O)OCC)N2 BXACPZGBYFAFIX-UHFFFAOYSA-N 0.000 description 1
- WXRAGHWFXPRSLV-UHFFFAOYSA-N ethyl 5-chloro-1h-pyrazole-3-carboxylate Chemical compound CCOC(=O)C=1C=C(Cl)NN=1 WXRAGHWFXPRSLV-UHFFFAOYSA-N 0.000 description 1
- LJTHWECGIPQTIF-YIXHJXPBSA-N ethyl 6-[(E)-hydrazinylidenemethyl]-4-methylpyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound N(/N)=C\C1=C(N(C=2N=CSC=21)C)C(=O)OCC LJTHWECGIPQTIF-YIXHJXPBSA-N 0.000 description 1
- UIGFFJAGZAJDAU-UHFFFAOYSA-N ethyl 6-formyl-2-methylsulfanyl-4H-pyrrolo[2,3-d][1,3]thiazole-5-carboxylate Chemical compound C(=O)C1=C(NC=2N=C(SC=21)SC)C(=O)OCC UIGFFJAGZAJDAU-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960005237 etoglucid Drugs 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 201000008819 extrahepatic bile duct carcinoma Diseases 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 235000019152 folic acid Nutrition 0.000 description 1
- 239000011724 folic acid Substances 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 230000009395 genetic defect Effects 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 208000004104 gestational diabetes Diseases 0.000 description 1
- 201000007116 gestational trophoblastic neoplasm Diseases 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000004153 glucose metabolism Effects 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 230000002414 glycolytic effect Effects 0.000 description 1
- 230000006692 glycolytic flux Effects 0.000 description 1
- 201000009277 hairy cell leukemia Diseases 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 238000011134 hematopoietic stem cell transplantation Methods 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000004474 heteroalkylene group Chemical group 0.000 description 1
- 125000004475 heteroaralkyl group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 208000029824 high grade glioma Diseases 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- MHLPKAGDPWUOOT-UHFFFAOYSA-N housane Chemical compound C1CC2CC21 MHLPKAGDPWUOOT-UHFFFAOYSA-N 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 150000002429 hydrazines Chemical class 0.000 description 1
- 238000006197 hydroboration reaction Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 1
- 201000008980 hyperinsulinism Diseases 0.000 description 1
- 208000034783 hypoesthesia Diseases 0.000 description 1
- 201000006866 hypopharynx cancer Diseases 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 230000018276 interleukin-1 production Effects 0.000 description 1
- 201000002313 intestinal cancer Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- NICJCIQSJJKZAH-AWEZNQCLSA-N irofulven Chemical compound O=C([C@@]1(O)C)C2=CC(C)=C(CO)C2=C(C)C21CC2 NICJCIQSJJKZAH-AWEZNQCLSA-N 0.000 description 1
- 229950005254 irofulven Drugs 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-M lactobionate Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-M 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 229950005692 larotaxel Drugs 0.000 description 1
- SEFGUGYLLVNFIJ-QDRLFVHASA-N larotaxel dihydrate Chemical compound O.O.O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@@]23[C@H]1[C@@]1(CO[C@@H]1C[C@@H]2C3)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 SEFGUGYLLVNFIJ-QDRLFVHASA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 206010024217 lentigo Diseases 0.000 description 1
- 229950001845 lestaurtinib Drugs 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 201000006721 lip cancer Diseases 0.000 description 1
- 230000037356 lipid metabolism Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 1
- 229960003538 lonidamine Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- FBQPGGIHOFZRGH-UHFFFAOYSA-N lucanthone Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(C)=CC=C2NCCN(CC)CC FBQPGGIHOFZRGH-UHFFFAOYSA-N 0.000 description 1
- 229950005239 lucanthone Drugs 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000003265 lymphadenitis Diseases 0.000 description 1
- 201000000564 macroglobulinemia Diseases 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 1
- 201000003175 male breast cancer Diseases 0.000 description 1
- 208000010907 male breast carcinoma Diseases 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 208000030883 malignant astrocytoma Diseases 0.000 description 1
- 201000011614 malignant glioma Diseases 0.000 description 1
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- UUVIQYKKKBJYJT-ZYUZMQFOSA-N mannosulfan Chemical compound CS(=O)(=O)OC[C@@H](OS(C)(=O)=O)[C@@H](O)[C@H](O)[C@H](OS(C)(=O)=O)COS(C)(=O)=O UUVIQYKKKBJYJT-ZYUZMQFOSA-N 0.000 description 1
- 229960000733 mannosulfan Drugs 0.000 description 1
- 229960003951 masoprocol Drugs 0.000 description 1
- HCZKYJDFEPMADG-TXEJJXNPSA-N masoprocol Chemical compound C([C@H](C)[C@H](C)CC=1C=C(O)C(O)=CC=1)C1=CC=C(O)C(O)=C1 HCZKYJDFEPMADG-TXEJJXNPSA-N 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 210000000716 merkel cell Anatomy 0.000 description 1
- 229960004635 mesna Drugs 0.000 description 1
- 230000006371 metabolic abnormality Effects 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- 208000010658 metastatic prostate carcinoma Diseases 0.000 description 1
- 208000037970 metastatic squamous neck cancer Diseases 0.000 description 1
- HNQIVZYLYMDVSB-NJFSPNSNSA-N methanesulfonamide Chemical compound [14CH3]S(N)(=O)=O HNQIVZYLYMDVSB-NJFSPNSNSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- ORUCTBNNYKZMSK-UHFFFAOYSA-N methyl 1h-pyrazole-5-carboxylate Chemical compound COC(=O)C=1C=CNN=1 ORUCTBNNYKZMSK-UHFFFAOYSA-N 0.000 description 1
- GQJCAQADCPTHKN-UHFFFAOYSA-N methyl 2,2-difluoro-2-fluorosulfonylacetate Chemical compound COC(=O)C(F)(F)S(F)(=O)=O GQJCAQADCPTHKN-UHFFFAOYSA-N 0.000 description 1
- NLWBJPPMPLPZIE-UHFFFAOYSA-N methyl 4-(bromomethyl)benzoate Chemical compound COC(=O)C1=CC=C(CBr)C=C1 NLWBJPPMPLPZIE-UHFFFAOYSA-N 0.000 description 1
- YUUAYBAIHCDHHD-UHFFFAOYSA-N methyl 5-aminolevulinate Chemical compound COC(=O)CCC(=O)CN YUUAYBAIHCDHHD-UHFFFAOYSA-N 0.000 description 1
- 229960005033 methyl aminolevulinate Drugs 0.000 description 1
- NYXHSRNBKJIQQG-UHFFFAOYSA-N methyl n-methylcarbamate Chemical group CNC(=O)OC NYXHSRNBKJIQQG-UHFFFAOYSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- KTMKRRPZPWUYKK-UHFFFAOYSA-N methylboronic acid Chemical compound CB(O)O KTMKRRPZPWUYKK-UHFFFAOYSA-N 0.000 description 1
- 206010063344 microscopic polyangiitis Diseases 0.000 description 1
- 230000027939 micturition Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 229960003539 mitoguazone Drugs 0.000 description 1
- MXWHMTNPTTVWDM-NXOFHUPFSA-N mitoguazone Chemical compound NC(N)=N\N=C(/C)\C=N\N=C(N)N MXWHMTNPTTVWDM-NXOFHUPFSA-N 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000002625 monoclonal antibody therapy Methods 0.000 description 1
- 208000029077 monogenic diabetes Diseases 0.000 description 1
- 208000001022 morbid obesity Diseases 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 206010051747 multiple endocrine neoplasia Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- JIKUXBYRTXDNIY-UHFFFAOYSA-N n-methyl-n-phenylformamide Chemical compound O=CN(C)C1=CC=CC=C1 JIKUXBYRTXDNIY-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000005893 naphthalimidyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 210000003928 nasal cavity Anatomy 0.000 description 1
- 208000018795 nasal cavity and paranasal sinus carcinoma Diseases 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- 230000019261 negative regulation of glycolysis Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- 230000005937 nuclear translocation Effects 0.000 description 1
- 238000001668 nucleic acid synthesis Methods 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 238000007344 nucleophilic reaction Methods 0.000 description 1
- 231100000862 numbness Toxicity 0.000 description 1
- 229940078552 o-xylene Drugs 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 125000005889 octahydrochromenyl group Chemical group 0.000 description 1
- 125000005890 octahydroisochromenyl group Chemical group 0.000 description 1
- 125000005069 octynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 208000022982 optic pathway glioma Diseases 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 201000005443 oral cavity cancer Diseases 0.000 description 1
- 210000004789 organ system Anatomy 0.000 description 1
- 150000002894 organic compounds Chemical group 0.000 description 1
- 201000006958 oropharynx cancer Diseases 0.000 description 1
- BWKDAMBGCPRVPI-ZQRPHVBESA-N ortataxel Chemical compound O([C@@H]1[C@]23OC(=O)O[C@H]2[C@@H](C(=C([C@@H](OC(C)=O)C(=O)[C@]2(C)[C@@H](O)C[C@H]4OC[C@]4([C@H]21)OC(C)=O)C3(C)C)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)CC(C)C)C(=O)C1=CC=CC=C1 BWKDAMBGCPRVPI-ZQRPHVBESA-N 0.000 description 1
- 229950001094 ortataxel Drugs 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 125000005882 oxadiazolinyl group Chemical group 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- 125000005880 oxathiolanyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003551 oxepanyl group Chemical group 0.000 description 1
- 125000003585 oxepinyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 210000003695 paranasal sinus Anatomy 0.000 description 1
- 208000035824 paresthesia Diseases 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 201000000389 pediatric ependymoma Diseases 0.000 description 1
- 229960001744 pegaspargase Drugs 0.000 description 1
- 108010001564 pegaspargase Proteins 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 201000001976 pemphigus vulgaris Diseases 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 1
- 229940083251 peripheral vasodilators purine derivative Drugs 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 210000003800 pharynx Anatomy 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- NIXKBAZVOQAHGC-UHFFFAOYSA-N phenylmethanesulfonic acid Chemical compound OS(=O)(=O)CC1=CC=CC=C1 NIXKBAZVOQAHGC-UHFFFAOYSA-N 0.000 description 1
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 1
- 208000028591 pheochromocytoma Diseases 0.000 description 1
- 229930029653 phosphoenolpyruvate Natural products 0.000 description 1
- DTBNBXWJWCWCIK-UHFFFAOYSA-K phosphonatoenolpyruvate Chemical compound [O-]C(=O)C(=C)OP([O-])([O-])=O DTBNBXWJWCWCIK-UHFFFAOYSA-K 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 201000003113 pineoblastoma Diseases 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- 208000010916 pituitary tumor Diseases 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 206010036067 polydipsia Diseases 0.000 description 1
- 229940113116 polyethylene glycol 1000 Drugs 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 208000005987 polymyositis Diseases 0.000 description 1
- 208000022530 polyphagia Diseases 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229960004293 porfimer sodium Drugs 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- GKKCIDNWFBPDBW-UHFFFAOYSA-M potassium cyanate Chemical compound [K]OC#N GKKCIDNWFBPDBW-UHFFFAOYSA-M 0.000 description 1
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- 150000003141 primary amines Chemical group 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 229940095055 progestogen systemic hormonal contraceptives Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- OCAAZRFBJBEVPS-UHFFFAOYSA-N prop-2-enyl carbamate Chemical compound NC(=O)OCC=C OCAAZRFBJBEVPS-UHFFFAOYSA-N 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 229940048914 protamine Drugs 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004942 pyridazin-6-yl group Chemical group N1=NC=CC=C1* 0.000 description 1
- JVANLUCASVHWEW-UHFFFAOYSA-N pyridazine Chemical compound N1=C=C=C=C=N1 JVANLUCASVHWEW-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- 208000002574 reactive arthritis Diseases 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 208000030859 renal pelvis/ureter urothelial carcinoma Diseases 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- 229950006896 sapacitabine Drugs 0.000 description 1
- LBGFKUUHOPIEMA-PEARBKPGSA-N sapacitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](C#N)[C@H](O)[C@@H](CO)O1 LBGFKUUHOPIEMA-PEARBKPGSA-N 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 201000003385 seborrheic keratosis Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical group 0.000 description 1
- 208000011581 secondary neoplasm Diseases 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 1
- 229950000055 seliciclib Drugs 0.000 description 1
- 229950003647 semaxanib Drugs 0.000 description 1
- WUWDLXZGHZSWQZ-WQLSENKSSA-N semaxanib Chemical compound N1C(C)=CC(C)=C1\C=C/1C2=CC=CC=C2NC\1=O WUWDLXZGHZSWQZ-WQLSENKSSA-N 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- 201000005574 senile angioma Diseases 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229960000269 sitimagene ceradenovec Drugs 0.000 description 1
- 108010086606 sitimagene ceradenovec Proteins 0.000 description 1
- 201000008261 skin carcinoma Diseases 0.000 description 1
- 230000037380 skin damage Effects 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 229960001922 sodium perborate Drugs 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- CHLCPTJLUJHDBO-UHFFFAOYSA-M sodium;benzenesulfinate Chemical compound [Na+].[O-]S(=O)C1=CC=CC=C1 CHLCPTJLUJHDBO-UHFFFAOYSA-M 0.000 description 1
- RZWQDAUIUBVCDD-UHFFFAOYSA-M sodium;benzenethiolate Chemical compound [Na+].[S-]C1=CC=CC=C1 RZWQDAUIUBVCDD-UHFFFAOYSA-M 0.000 description 1
- YKLJGMBLPUQQOI-UHFFFAOYSA-M sodium;oxidooxy(oxo)borane Chemical compound [Na+].[O-]OB=O YKLJGMBLPUQQOI-UHFFFAOYSA-M 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- OGNAOIGAPPSUMG-UHFFFAOYSA-N spiro[2.2]pentane Chemical compound C1CC11CC1 OGNAOIGAPPSUMG-UHFFFAOYSA-N 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 208000023516 stroke disease Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 125000004354 sulfur functional group Chemical group 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 208000014794 superficial urinary bladder carcinoma Diseases 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 229950010924 talaporfin Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960002197 temoporfin Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 210000002435 tendon Anatomy 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- RKFMLIZYBJXJEB-UHFFFAOYSA-N tert-butyl 3-iodopyrazole-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1C=CC(I)=N1 RKFMLIZYBJXJEB-UHFFFAOYSA-N 0.000 description 1
- RRJIWPFOOZCGAO-UHFFFAOYSA-N tert-butyl n-[6-(bromomethyl)pyridin-2-yl]-n-[(2-methylpropan-2-yl)oxycarbonyl]carbamate Chemical compound CC(C)(C)OC(=O)N(C(=O)OC(C)(C)C)C1=CC=CC(CBr)=N1 RRJIWPFOOZCGAO-UHFFFAOYSA-N 0.000 description 1
- 229950009016 tesetaxel Drugs 0.000 description 1
- MODVSQKJJIBWPZ-VLLPJHQWSA-N tesetaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3CC[C@@]2(C)[C@H]2[C@@H](C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C(=CC=CN=4)F)C[C@]1(O)C3(C)C)O[C@H](O2)CN(C)C)C(=O)C1=CC=CC=C1 MODVSQKJJIBWPZ-VLLPJHQWSA-N 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 125000005887 tetrahydrobenzofuranyl group Chemical group 0.000 description 1
- 125000005886 tetrahydrobenzothienyl group Chemical group 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000005888 tetrahydroindolyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000005247 tetrazinyl group Chemical group N1=NN=NC(=C1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000005305 thiadiazolinyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000005458 thianyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000001583 thiepanyl group Chemical group 0.000 description 1
- 125000003777 thiepinyl group Chemical group 0.000 description 1
- 125000001730 thiiranyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- PIZNQHDTOZMVBH-UHFFFAOYSA-N thionylimide Chemical compound N=S=O PIZNQHDTOZMVBH-UHFFFAOYSA-N 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 208000008732 thymoma Diseases 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- LMYRWZFENFIFIT-UHFFFAOYSA-N toluene-4-sulfonamide Chemical compound CC1=CC=C(S(N)(=O)=O)C=C1 LMYRWZFENFIFIT-UHFFFAOYSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 229960003181 treosulfan Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- PXSOHRWMIRDKMP-UHFFFAOYSA-N triaziquone Chemical compound O=C1C(N2CC2)=C(N2CC2)C(=O)C=C1N1CC1 PXSOHRWMIRDKMP-UHFFFAOYSA-N 0.000 description 1
- 229960004560 triaziquone Drugs 0.000 description 1
- 125000005881 triazolinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 1
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 208000029387 trophoblastic neoplasm Diseases 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 208000037965 uterine sarcoma Diseases 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000002435 venom Substances 0.000 description 1
- 231100000611 venom Toxicity 0.000 description 1
- 210000001048 venom Anatomy 0.000 description 1
- 229960003895 verteporfin Drugs 0.000 description 1
- ZQFGRJWRSLZCSQ-ZSFNYQMMSA-N verteporfin Chemical compound C=1C([C@@]2([C@H](C(=O)OC)C(=CC=C22)C(=O)OC)C)=NC2=CC(C(=C2C=C)C)=NC2=CC(C(=C2CCC(O)=O)C)=NC2=CC2=NC=1C(C)=C2CCC(=O)OC ZQFGRJWRSLZCSQ-ZSFNYQMMSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- LVLANIHJQRZTPY-UHFFFAOYSA-N vinyl carbamate Chemical compound NC(=O)OC=C LVLANIHJQRZTPY-UHFFFAOYSA-N 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 201000005102 vulva cancer Diseases 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/08—Plasma substitutes; Perfusion solutions; Dialytics or haemodialytics; Drugs for electrolytic or acid-base disorders, e.g. hypovolemic shock
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/36—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains three hetero rings
- C07D513/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
En el presente documento se describen métodos para usar compuestos de Fórmula (I) para modular la actividad de PKM2 en un sujeto. Estos compuestos están representados por la Fórmula (I): en la que R1, R2, L1 y L2 son como se definen aquí. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Moduladores de piruvato quinasa y uso de los mismos
Antecedentes
La piruvato quinasa (PK) es una enzima metabólica que convierte el fosfoenolpiruvato en piruvato durante la glucólisis. Existen cuatro isoformas de la PK en mamíferos: las isoformas L y R (del gen PKLR) se expresan en el hígado y los glóbulos rojos respectivamente, y el gen PKM codifica dos variantes de empalme, la isoforma M1 que se expresa en la mayoría de los tejidos adultos y la Isoforma M2 que se expresa durante el desarrollo embrionario y en algunos tejidos adultos, incluido el riñón y las células madre hematopoyéticas. Muchas células tumorales también expresan PKM2. Esta isoforma tetrámera regulada alostéricamente está intrínsecamente diseñada para regular por disminución su actividad, a través de la modificación postraduccional, la modulación alostérica por ligandos moléculas pequeñas, incluidos algunos aminoácidos, y por la disociación de subunidades (en la forma dimérica), lo que da como resultado una inhibición parcial de la glucólisis en la última etapa. Esto acumula intermedios glucolíticos aguas arriba como una fuente de carbono anabólico para la síntesis de lípidos y ácidos nucleicos, mientras que la reasociación de PKM2 en el tetrámero activo repone el estado catabólico normal como retroalimentación después de la división celular (Protein Sci. noviembre de 2010; 19(11): 2031-2044). La modulación (p. ej., inhibición o activación) de PKM2 puede ser eficaz en el tratamiento de una serie de trastornos, p. ej., cáncer, obesidad, enfermedades diabéticas (p. ej., nefropatía diabética (DN)), enfermedad arterial coronaria (cA d), síndrome de Bloom (BS), afecciones autoinmunitarias y enfermedades dependientes de la proliferación (p. ej., hiperplasia prostática benigna (HPB)). Jiang, J-K. et al., Bioorg. & Med. Chem. Lett., 2010, 20, 3387 3393 se refiere a activadores de PKM2 basados en un armazón de tieno[3,2-6]pirrol[3,2-d]piridazinona sustituido.
Resumen
En el presente documento se describen métodos para modular la actividad de la piruvato quinasa M2 (PKM2) en un sujeto que lo necesite, que comprenden administrar una cantidad eficaz de un compuesto de fórmulas (I), (II), (III), (IV), (V-a), (V-b), (VI) o (IX) (denominados colectivamente en el presente documento "fórmulas (I)-(IX)") o una de sus sales farmacéuticamente aceptables, o un compuesto de fórmulas (I'), (II'), (III'), (IV'), (V'), (denominados colectivamente en el presente documento fórmulas (I')-(V')" o una de sus sales farmacéuticamente aceptables, que regulan enzimas PKM2, de tipo natural y/o mutante (tales como las descritas en el presente documento).
El alcance de la invención está definido por las reivindicaciones. Cualquier referencia en la descripción a métodos de tratamiento se refiere a los compuestos, composiciones farmacéuticas y medicamentos de la presente invención para usar en un método para el tratamiento del cuerpo humano (o animal) mediante terapia (o para diagnóstico).
En una realización, la invención proporciona un método para modular la actividad de la piruvato quinasa M2 (PKM2) en un sujeto, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables

en donde Q, R1, R2, L1, L2yQ son como se definen en el presente documento.
En una realización, el compuesto o sal farmacéuticamente aceptable del mismo se selecciona de los compuestos de la Tabla 1 y las Figuras 1A-1C, 2A-2C y 3.
También se proporciona un método para modular la actividad de la piruvato quinasa M2 (PKM2) en un sujeto que lo necesite, que comprende administrar una composición farmacéutica que comprende una cantidad eficaz de un compuesto de fórmulas (I)-(IX) o una de sus sales farmacéuticamente aceptables, o un compuesto de fórmulas (I')-(V') o una de sus sales farmacéuticamente aceptables, y un vehículo farmacéuticamente aceptable.
En otra realización, se proporciona un método para modular (p. ej., aumentar o disminuir) el nivel de actividad de la PKM2 en un sujeto que lo necesite, que comprende administrar al sujeto una cantidad eficaz de un compuesto descrito en el presente documento. En algunas realizaciones, un compuesto o una composición descrita en el presente documento se usa para mantener la PKM2 en su conformación activa o activar la actividad de piruvato quinasa en células en proliferación como medio para desviar metabolitos de glucosa hacia procesos catabólicos en lugar de anabólicos en el paciente. En ciertas realizaciones, el método proporcionado aumenta el nivel de (es decir, activa) la actividad de la PKM2 en el sujeto. En ciertas realizaciones, el método proporcionado reduce el nivel de actividad de PKM2 en el sujeto.
En otra realización, se proporciona un método para modular (p. ej., aumentar o disminuir) el nivel de glucosa en plasma
en un sujeto que lo necesite, que comprende administrar al sujeto una cantidad eficaz de un compuesto descrito en el presente documento. En ciertas realizaciones, el método proporcionado aumenta el nivel de glucosa en plasma en el sujeto. En ciertas realizaciones, el método proporcionado reduce el nivel de glucosa en plasma en el sujeto.
En otra realización, se proporciona un método para inhibir la proliferación celular en un sujeto que lo necesite, que comprende administrar al sujeto una cantidad eficaz de un compuesto descrito en el presente documento. P. ej., este método puede inhibir el crecimiento de una célula transformada, p. ej., una célula cancerosa o, en general, inhibir el crecimiento en una célula dependiente de PKM2 que experimenta glucólisis aeróbica.
En otra realización, se proporciona un método para tratar a un sujeto que padece o es susceptible de padecer una enfermedad o trastorno asociado con la función de PKM2 que comprende administrar al sujeto una cantidad eficaz de un compuesto descrito en el presente documento. En cierta realización, el método comprende además identificar o seleccionar un sujeto que se beneficiaría de la modulación (p. ej., activación) de la PKM2 y/o glucosa en plasma. Por ejemplo, el paciente puede identificarse basándose en el nivel de actividad de la PKM2 en una célula del paciente para el tratamiento del cáncer asociado con la función de la PKM2. En otra realización, el paciente seleccionado es un sujeto que padece o es susceptible de padecer un trastorno o enfermedad identificado en el presente documento, p. ej., un trastorno caracterizado por el crecimiento o proliferación celular no deseado. En ciertas realizaciones, la enfermedad es un trastorno neoplásico. En ciertas realizaciones, la enfermedad es cáncer, obesidad, una enfermedad diabética (p. ej., nefropatía diabética (DN)), aterosclerosis, reestenosis, enfermedad arterial coronaria (CAD), síndrome de Bloom (b s ), hiperplasia prostática benigna (HPB) o una enfermedad autoinmunitaria. En ciertas realizaciones, la enfermedad es cáncer. En ciertas realizaciones, la enfermedad es una enfermedad diabética. En ciertas realizaciones, la enfermedad diabética es nefropatía diabética (DN). En ciertas realizaciones, la enfermedad es enfermedad arterial coronaria (CAD).
En una realización, se proporciona el uso de un compuesto descrito en el presente documento o una de sus sales farmacéuticamente aceptables o una composición farmacéutica que comprende el mismo en cualquiera de los métodos de la invención descritos anteriormente. En una realización, se proporciona un compuesto descrito en el presente documento o una de sus sales farmacéuticamente aceptables o una composición farmacéutica que comprende el mismo para usar en cualquiera de los métodos de la invención descritos anteriormente. En otra realización, se proporciona el uso de un compuesto descrito en el presente documento o una de sus sales farmacéuticamente aceptables o una composición farmacéutica que comprende el mismo para la fabricación de un medicamento para cualquiera de los métodos de la invención descritos.
Breve descripción de las figuras
Las Figuras 1A-1C son listas de las estructuras de otros compuestos de ejemplo usados en los métodos de la invención.
Las Figuras 2A-2C son listas de las estructuras de otros compuestos de ejemplo usados en los métodos de la invención.
La Figura 3 es una lista de las estructuras de otros compuestos de ejemplo usados en los métodos de la invención.
La Figura 4 muestra la síntesis de compuestos intermedios de ejemplo usados en los Ejemplos 1-10.
Descripción detallada de la invención
Definiciones
Los compuestos descritos en el presente documento, que se utilizan en los métodos de la invención, pueden comprender uno o más centros asimétricos y, por lo tanto, pueden existir en diversas formas estereoisómeras, p. ej., enantiómeros y/o diastereómeros. Por ejemplo, los compuestos descritos en el presente documento pueden estar en forma de un enantiómero, diastereómero o isómero geométrico individual, o pueden estar en forma de una mezcla de estereoisómeros, incluidas mezclas racémicas y mezclas enriquecidas en uno o más estereoisómeros. Los isómeros se pueden aislar de las mezclas mediante métodos conocidos por los expertos en la técnica, que incluyen cromatografía líquida de alta presión (HPLC) quiral y la formación y cristalización de sales quirales; o los isómeros preferidos pueden prepararse por síntesis asimétrica. Véase, por ejemplo, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, Nueva York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Eliel, E. L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); y Wilen, S. H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. de Notre Dame Press, Notre Dame, IN 1972).
En una realización, los compuestos descritos en el presente documento también pueden comprender una o más sustituciones isotópicas. Por ejemplo, compuestos que tienen las presentes estructuras excepto por el reemplazo de hidrógeno por deuterio o tritio, reemplazo de 19F con 18F, o el reemplazo de 12C con 13C o 14C están dentro del alcance de la descripción. Dichos compuestos son útiles, por ejemplo, como herramientas analíticas o sondas en ensayos biológicos.
Los compuestos descritos en el presente documento también se pueden representar en múltiples formas tautómeras, en tales casos, incluye expresamente todas las formas tautómeras de los compuestos descritos en el presente documento, incluso aunque pueda estar representada solo una sola forma tautómera (p. ej., la alquilación de un sistema de anillo
puede dar como resultado la alquilación en múltiples sitios; todos estos productos de reacción están expresamente incluidos). Todas las formas isoméricas de dichos compuestos están expresamente incluidas. Si un tautómero de un compuesto es aromático, este compuesto es aromático. De manera similar, si un tautómero de un sustituyente es un heteroarilo, este sustituyente es heteroarilo.
El término "alquilo" se refiere a un radical de un grupo hidrocarburo saturado de cadena lineal o ramificado que tiene de 1 a 10 átomos de carbono ("alquilo C1-10"). Los ejemplos de grupos alquilo C1-6 incluyen metilo (C1), etilo (C2), propilo (C3) (p. ej., n-propilo, isopropilo), butilo (C4) (p. ej., n-butilo, ferc-butilo, sec-butilo, isobutilo), pentilo (C5) (p. ej., n-pentilo, 3-pentanilo, amilo, neopentilo, 3-metil-2-butanilo, amilo terciario) y hexilo (C6) (p. ej., n-hexilo). A menos que se especifique lo contrario, cada caso de un grupo alquilo está independientemente no sustituido (un "alquilo no sustituido") o sustituido (un "alquilo sustituido") con uno o más sustituyentes (p. ej., halógeno, tal como F). En ciertas realizaciones, el grupo alquilo es -alquilo C1-10 no sustituido. En ciertas realizaciones, el grupo alquilo es -alquilo C1-10 sustituido.
El término "haloalquilo" se refiere a un grupo alquilo sustituido, en donde uno o más de los átomos de hidrógeno se reemplazan independientemente por un halógeno, p. ej., fluoro, bromo, cloro o yodo e incluye restos alquilo en los que todos los hidrógenos se han reemplazados por halógeno (p. ej., perfluoroalquilo). En algunas realizaciones, el resto haloalquilo tiene de 1 a 8 átomos de carbono ("haloalquilo C1-8").
El término "alcoxi" o "alcoxilo" se refiere a un radical -O-alquilo. P. ej., con entre 1 y 6 átomos de carbono.
El término "ariloxi" se refiere a un radical -O-arilo. En algunas realizaciones, el grupo ariloxi es fenoxi.
"Hidroxialquilo" o "hidroxilalquilo" puede incluir estructuras de alquilo que están sustituidas con uno o más grupos hidroxilo.
El término "heteroalquilo" se refiere a un grupo alquilo, que además incluye al menos un heteroátomo (p. ej., 1, 2, 3 o 4 heteroátomos) seleccionados de oxígeno, nitrógeno o azufre dentro (es decir, insertados entre átomos de carbono adyacentes) y/o colocado en una o más posiciones terminales de la cadena principal. En ciertas realizaciones, un grupo heteroalquilo se refiere a un grupo saturado que tiene de 1 a 10 átomos de carbono y 1 o más heteroátomos dentro de la cadena principal ("heteroalquilo C1-10"). A menos que se especifique lo contrario, cada caso de un grupo heteroalquilo está independientemente no sustituido (un "heteroalquilo no sustituido") o sustituido (un "heteroalquilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo heteroalquilo es un grupo heteroalquilo C1-10. En ciertas realizaciones, el grupo heteroalquilo es un grupo heteroalquilo C1-10 sustituido.
El término "alquenilo" se refiere a un radical de un grupo hidrocarburo de cadena lineal o ramificado que tiene de 2 a 10 átomos de carbono y uno o más dobles enlaces carbono-carbono (p. ej., 1, 2, 3 o 4 dobles enlaces). El uno o más dobles enlaces carbono-carbono pueden ser internos (tal como en 2-butenilo) o terminales (tal como en 1 -butenilo). Los ejemplos de grupos alquenilo C2-4 incluyen etenilo (C2), 1-propenilo (C3), 2-propenilo (C3), 1 -butenilo (C4), 2-butenilo (C4), butadienilo (C4), y similares. A menos que se especifique lo contrario, cada caso de un grupo alquenilo está independientemente no sustituido (un "alquenilo no sustituido") o sustituido (un "alquenilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo alquenilo es un -alquenilo C2-10 no sustituido. En ciertas realizaciones, el grupo alquenilo es un -alquenilo C2-10 sustituido. En un grupo alquenilo, un doble enlace C=C puede ser un enlace doble (E) o (Z).
El término "alquinilo" se refiere a un radical de un grupo hidrocarbonado de cadena lineal o ramificado que tiene de 2 a 10 átomos de carbono y uno o más enlaces triples carbono-carbono (p. ej., 1, 2, 3 o 4 enlaces triples) ("alquinilo C2-10"). Los ejemplos de grupos alquinilo incluyen etinilo (C2), 1 -propinilo (C3), 2-propinilo (C3), 1 -butinilo (C4), 2-butinilo (C4), pentinilo (C5), hexinilo (C6) heptinilo (C7), octinilo (C8), y similares. A menos que se especifique lo contrario, cada caso de un grupo alquinilo está independientemente no sustituido (un "alquinilo no sustituido") o sustituido (un "alquinilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo alquinilo es un -alquinilo C2-10 no sustituido. En ciertas realizaciones, el grupo alquinilo es un -alquinilo C2-10 sustituido.
El término "carbociclilo" o "carbocíclico" se refiere a un radical de un sistema de anillo de hidrocarburo monocíclico, bicíclico o tricíclico o policíclico no aromático que tiene de 3 a 14 átomos de carbono en el anillo ("carbociclilo C3-14") y cero heteroátomos en el sistema de anillos no aromáticos. Los grupos carbociclilos incluyen sistemas de anillos completamente saturados (p. ej., cicloalquilos) y sistemas de anillos parcialmente saturados. En algunas realizaciones, un grupo carbociclilo tiene de 3 a 10 átomos de carbono en el anillo ("carbociclilo C3-10").
El término "cicloalquilo" como se emplea en el presente documento incluye grupos hidrocarburos saturados cíclicos, bicíclicos, tricíclicos o policíclicos que tienen de 3 a 14 carbonos que contienen el número indicado de anillos y átomos de carbono (por ejemplo, un cicloalquilo monocíclico C3-C14, bicíclico C4-C14, tricíclico C5-C14 o policíclico C6-C14). En algunas realizaciones, "cicloalquilo" es un cicloalquilo monocíclico. Los ejemplos de grupos cicloalquilo monocíclicos incluyen ciclopentilo (C5), ciclohexilo (C5). ciclopropilo (C3) ciclobutilo (C4), cicloheptilo (C7) y ciclooctilo (C8). En algunas realizaciones, "cicloalquilo" es un cicloalquilo bicíclico. Los ejemplos de cicloalquilos bicíclicos incluyen biciclo[1.1.0]butano (C4), biciclo[1.1.1]pentano (C5), espiro[2.2]pentano (C5), biciclo[2.1.0]pentano (C5), biciclo[2.1.1]hexano (C6), biciclo[3.3.3]undecano (C11), decahidronaftaleno (C10), biciclo[4.3.2]undecano (C11), espiro[5.5]undecano (C11) y biciclo[4.3.3]dodecano (C12). En algunas realizaciones, "cicloalquilo" es un cicloalquilo tricíclico. Los ejemplos de cicloalquilos tricíclicos incluyen adamantina (C12). A menos que se especifique lo contrario,
cada caso de un grupo cicloalquilo está independientemente no sustituido (un "cicloalquilo no sustituido") o sustituido (un "cicloalquilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo cicloalquilo es un grupo cicloalquilo C3-14 no sustituido. En ciertas realizaciones, el grupo cicloalquilo es un cicloalquilo C3-14 sustituido.
El término "heterociclilo" o "heterocíclico" se refiere a un radical de un sistema de anillos no aromático de 3 a 14 miembros que tiene átomos de carbono en el anillo y de 1 a 4 heteroátomos en el anillo, en donde cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno y azufre ("heterociclilo de 3-14 miembros") . En los grupos heterociclilo que contienen uno o más átomos de nitrógeno, el punto de unión puede ser un átomo de carbono o de nitrógeno, según lo permita la valencia. Un grupo heterociclilo puede ser monocíclico ("heterociclilo monocíclico") o policíclico (p. ej., un sistema de anillos condensados, con puente o espiro tal como un sistema bicíclico ("heterociclilo bicíclico") o un sistema tricíclico ("heterociclilo tricíclico")), y puede estar saturado o puede contener uno o más enlaces dobles o triples carbonocarbono. Los sistemas de anillos policíclicos de heterociclilo pueden incluir uno o más heteroátomos en uno o ambos anillos. "Heterociclilo" también incluye sistemas de anillos en donde el anillo heterociclilo, como se define anteriormente, está condensado con uno o más grupos carbociclilo en donde el punto de unión está en el anillo de carbociclilo o heterociclilo, o sistemas de anillos en donde el anillo heterociclilo, como se define anteriormente, está condensado con uno o más grupos arilo o heteroarilo, en donde el punto de unión está en el anillo de heterociclilo y, en tales casos, el número de miembros del anillo continúa designando el número de miembros del anillo en el sistema de anillo de heterociclilo. A menos que se especifique lo contrario, cada caso de heterociclilo está independientemente no sustituido (un "heterociclilo no sustituido") o sustituido (un "heterociclilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo heterociclilo es un heterociclilo de 3-14 miembros no sustituido. En ciertas realizaciones, el grupo heterociclilo es un heterociclilo de 3-14 miembros sustituido. En algunas realizaciones, un grupo heterociclilo es un sistema de anillos no aromático de 5-10 miembros que tiene átomos de carbono en el anillo y 1-4 heteroátomos en el anillo, en donde cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno y azufre ("heterociclilo de 5-10 miembros").
Ejemplos de grupos heterociclilo incluyen aziridinilo, oxiranilo, tiiranilo, azetidinilo, oxetanilo, tietanilo, tetrahidrofuranilo, dihidrofuranilo, tetrahidrotiofenilo, dihidrotiofenilo, pirrolidinilo, dihidropirrolilo, pirrolil-2,5-dina, dioxolanilo, oxatiolanilo, ditiolanilo, triazolinilo, oxadiazolinilo, tiadiazolinilo, piperidinilo, tetrahidropiranilo, dihidropiridinilo, tianilo, piperazinilo, morfolinilo, ditianilo, dioxanilo, triazinanilo, azepanilo, oxepanilo, tiepanilo, azocanilo, oxecanilo, tiocanilo, indolinilo, isoindolinilo, dihidrobenzofuranilo, dihidrobenzotienilo, tetrahidrobenzotienilo, tetrahidrobenzofuranilo, tetrahidroindolilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, decahidroquinolinilo, decahidroisoquinolinilo, octahidrocromenilo, octahidroisocromenilo, decahidronaftiridinilo, decahidro-1,8-naftiridinilo, octahidropirrolo[3,2-b]pirrol, indolinilo, ftalimidilo, naftalimidilo, cromanilo, cromenilo, 1H-benzo[e][1,4]diazepinilo, 1,4,5,7-tetrahidropirano[3,4-b]pirrolilo, 5,6-dihidro-4H-furo[3,2-b]pirrolilo, 6,7-dihidro-5H-furo[3,2-b]piranilo, 5,7-dihidro-4H-tieno[2,3-c]piranilo, 2,3-dihidro-1H-pirrolo[2,3-b] piridinilo, 2,3-dihidrofuro[2,3-b]piridinilo, 4,5,6,7-tetrahidro-1H-pirrolo[2,3-b]piridinilo, 4,5,6,7-tetrahidrofuro[3,2-c] piridinilo, 4,5,6,7-tetrahidrotieno[3,2-b]piridinilo, 1,2,3,4-tetrahidro-1,6-naftiridinilo, y similares.
El término "arilo" se refiere a un radical de un sistema de anillos aromáticos 4n+2 (p. ej., que tiene 6, 10 o 14 electrones n compartidos en una matriz cíclica) monocíclico o policíclico (p. ej., bicíclico o tricíclico) que tiene 6-14 átomos de carbono en el anillo y cero heteroátomos proporcionados en el sistema de anillos aromáticos ("arilo C6-14"). En algunas realizaciones, un grupo arilo tiene 6 átomos de carbono en el anillo ("arilo C6"; p. ej., fenilo). En algunas realizaciones, un grupo arilo tiene 10 átomos de carbono en el anillo ("arilo C10"; p. ej., naftilo tal como 1-naftilo y 2-naftilo). En algunas realizaciones, un grupo arilo tiene 14 átomos de carbono en el anillo ("arilo C14"; p. ej., antracilo). "Arilo" también incluye sistemas de anillos en donde el anillo de arilo, como se define anteriormente, está condensado con uno o más grupos carbociclilo o heterociclilo en donde el radical o punto de unión está en el anillo de arilo y, en tales casos, el número de átomos de carbono continúa designando el número de átomos de carbono en el sistema de anillos de arilo. A menos que se especifique lo contrario, cada caso de un grupo arilo está independientemente no sustituido (un "arilo no sustituido") o sustituido (un "arilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo arilo es un arilo C6-14 no sustituido. En ciertas realizaciones, el grupo arilo es un arilo C6-14 sustituido.
"Arilalquilo" o "aralquilo" es un subconjunto de "alquilo" y se refiere a un grupo alquilo sustituido con un grupo arilo, en donde el punto de unión está en el resto alquilo. Los ejemplos de "arilalquilo" o "aralquilo" incluyen grupos bencilo, 2-feniletilo, 3-fenilpropilo, 9-fluorenilo, bencidrilo ytritilo.
El término "heteroarilo" se refiere a un radical de un sistema de anillo aromático 4n+2 (p. ej., que tienen 6, 10 o 14 electrones n compartidos en una matriz cíclica) monocíclico o policíclico de 5-14 miembros (p. ej., bicíclico, tricíclico) que tienen átomos de carbono en el anillo y 1-4 heteroátomos en el anillo proporcionados en el sistema de anillo aromático, en donde cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno y azufre ("heteroarilo de 5-14 miembros"). En algunas realizaciones, el heteroarilo puede ser un heteroarilo monocíclico de 5-8 miembros que contiene 1-4 heteroátomos. En algunas realizaciones, el heteroarilo puede ser un heteroarilo bicíclico de 8-12 miembros que tiene 1-6 heteroátomos. En algunas realizaciones, el heteroarilo puede ser un sistema de anillo de heteroarilo tricíclico de 11 14 miembros que tiene 1-9 heteroátomos. En los grupos heteroarilo que contienen uno o más átomos de nitrógeno, el punto de unión puede ser un átomo de carbono o nitrógeno, según lo permita la valencia. Los sistemas de anillos policíclicos de heteroarilo pueden incluir uno o más heteroátomos en uno o ambos anillos. "Heteroarilo" incluye sistemas de anillos en donde el anillo de heteroarilo, como se define anteriormente, está condensado con uno o más grupos carbociclilo o heterociclilo en donde el punto de unión está en el anillo de heteroarilo y, en tales casos, el número de
miembros del anillo continúa designando el número de miembros del anillo en el sistema de anillo de heteroarilo. "Heteroanlo" también incluye sistemas de anillos en donde el anillo de heteroarilo, como se define anteriormente, está condensado con uno o más grupos arilo en donde el punto de unión está en el anillo de arilo o heteroarilo, y en tales casos, el número de miembros del anillo designa el número de miembros del anillo en el sistema de anillos policíclicos condensados (arilo/heteroarilo). En los grupos heteroarilo policíclicos en donde un anillo no contiene un heteroátomo (p. ej., indolilo, quinolinilo, carbazolilo y similares), el punto de unión puede estar en cualquier anillo, es decir, el anillo que lleva un heteroátomo (p. ej., 2-indolilo) o el anillo que no contiene un heteroátomo (p. ej., 5-indolilo). En algunas realizaciones, un grupo heteroarilo es un sistema de anillo aromático monocíclico de 5-10 miembros que tiene átomos de carbono en el anillo y 1-4 heteroátomos en el anillo proporcionados en el sistema de anillo aromático, en donde cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno y azufre ("heteroarilo monocíclico de 5-10 miembros"). En algunas realizaciones, un grupo heteroarilo es un sistema de anillos aromáticos bicíclico de 8-12 miembros que tiene átomos de carbono en el anillo y 1-6 heteroátomos en el anillo proporcionados en el sistema de anillos aromáticos, en donde cada heteroátomo se selecciona independientemente de nitrógeno, oxígeno y azufre ("heteroarilo bicíclico de 8-12 miembros"). A menos que se especifique lo contrario, cada caso de un grupo heteroarilo está independientemente no sustituido (un "heteroarilo no sustituido") o sustituido (un "heteroarilo sustituido") con uno o más sustituyentes. En ciertas realizaciones, el grupo heteroarilo es un heteroarilo de 5-14 miembros no sustituido. En ciertas realizaciones, el grupo heteroarilo es un heteroarilo de 5-14 miembros sustituido. En ciertas realizaciones, el grupo heteroarilo es un heteroarilo monocíclico de 5 miembros opcionalmente sustituido. En ciertas realizaciones, el grupo heteroarilo es un heteroarilo monocíclico de 6 miembros opcionalmente sustituido.
Ejemplos de grupos heteroarilo monocíclicos incluyen pirrolilo, furanilo, tiofenilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, isotiazolilo, triazolilo, oxadiazolilo, tiadiazolilo, tetrazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, triazinilo, tetrazinilo, azepinilo, oxepinilo, tiepinilo, indolilo, isoindolilo, indazolilo, benzotriazolilo, benzotiofenilo, isobenzotiofenilo, benzofuranilo, benzoisofuranilo, bencimidazolilo, benzoxazolilo, bencisoxazolilo, benzoxadiazolilo, benztiazolilo, bencisotiazolilo, benztiadiazolilo, indolizinilo, purinilo, naftiridinilo, pteridinilo, quinolinilo, isoquinolinilo, cinolinilo, quinoxalinilo, ftalazinilo, quinazolinilfenantridinilo, dibenzofuranilo, carbazolilo, acridinilo, fenotiazinilo, fenoxazinilo y fenazinilo.
"Heteroaralquilo" o "heteroarilalquilo" se refiere a un grupo alquilo sustituido con un grupo heteroarilo, en donde el punto de unión está en el resto alquilo.
El término "saturado" se refiere a un resto que no contiene un doble o triple enlace, es decir, el resto solo contiene enlaces simples.
Poner el sufijo "-eno" a un grupo indica que el grupo es un resto divalente, p. ej., alquileno es el resto divalente de alquilo, alquenileno es el resto divalente de alquenilo, alquinileno es el resto divalente de alquinilo, heteroalquileno es el resto divalente de heteroalquilo, heteroalquenileno es el resto divalente de heteroalquenilo, heteroalquinileno es el resto divalente de heteroalquinilo, carbociclileno es el resto divalente de carbociclilo, el heterociclileno es el resto divalente de heterociclilo, arileno es el resto divalente de arilo y el heteroarileno es el resto divalente de heteroarilo.
La expresión "opcionalmente sustituido" se refiere a estar sustituido o no sustituido. En general, el término "sustituido" significa que al menos un hidrógeno presente en un grupo se reemplaza con un sustituyente permitido, p. ej., un sustituyente que tras la sustitución da como resultado un compuesto estable, p. ej., un compuesto que no sufre transformación espontánea, tal como por transposición, ciclación, eliminación u otra reacción. Amenos que se indique lo contrario, un grupo "sustituido" tiene un sustituyente (p. ej. alquilo C1-6, halógeno, nitro, ciano, hidroxilo, haloalquilo C1-6, haloalcoxi C1-6, acilo C1-6, cicloalquilo C3-6, arilo C6-10, heteroarilo monocíclico o bicíclico y heterociclilo monocíclico o bicíclico), en una o más posiciones sustituibles del grupo, y cuando está sustituida más de una posición en cualquier estructura dada, el sustituyente es el mismo o diferente en cada posición. Se contempla que el término "sustituido" incluye la sustitución con todos los sustituyentes permitidos de compuestos orgánicos, e incluye cualquiera de los sustituyentes descritos en el presente documento que dé como resultado la formación de un compuesto estable. La presente descripción contempla todas y cada una de dichas combinaciones para llegar a un compuesto estable. Para los fines de esta descripción, los heteroátomos tales como el nitrógeno pueden tener sustituyentes hidrógeno y/o cualquier sustituyente adecuado como se describe en el presente documento que satisface las valencias de los heteroátomos y da como resultado la formación de un resto estable. No se pretende que la descripción esté limitada de ninguna manera por los ejemplos de sustituyentes descritos en el presente documento.
El término "halo" o "halógeno" se refiere a flúor, cloro, bromo o yodo.
El término "acilo" se refiere a un grupo que tiene la fórmula general -C(=O)RX1, -C(=O)ORX1, -C(=O)-O-C(=O)RX1, -C(=O)SRX1, -C(=O)N(RX1)2, -C(=S)RX1, -C(=S)N(RX1)2 y -C(=S)S(RX1), -C(=NRX1)RX1, -C(=NRX1)ORX1, -C(=NRX1)SRX1 y-C(=NRX1)N(RX1)2, en donde RX1 es hidrógeno; halógeno; hidroxilo sustituido o no sustituido; tiol sustituido o no sustituido; amino sustituido o no sustituido; acilo sustituido o no sustituido, alquilo C1-10 cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; alquenilo C2-10 cíclico o acíclico, sustituido o no sustituido, ramificado o no ramificado; alquinilo C2-10 sustituido o no sustituido; arilo C6-12 sustituido o no sustituido, heteroarilo sustituido o no sustituido, cuando la valencia lo permita. Los grupos acilo de ejemplo incluyen aldehídos (-CHO), ácidos carboxílicos (-CO2H), cetonas, haluros de acilo, ésteres, amidas, iminas, carbonatos, carbamatos y ureas.
En ciertas realizaciones, el sustituyente presente en un átomo de nitrógeno, en un átomo de oxígeno o en un átomo de azufre es un grupo protector de nitrógeno, un grupo protector de oxígeno o un grupo protector de azufre, respectivamente. Los grupos protectores de nitrógeno, oxígeno y azufre son bien conocidos en la técnica e incluyen los descritos en detalle en Protecting Groups in Organic Synthesis, T. W. Greene y P. G. M. Wuts, 3.a edición, John Wiley & Sons, 1999.
Por ejemplo, los grupos protectores de nitrógeno incluyen, pero no se limitan a, formamida, acetamida, cloroacetamida, tricloroacetamida, trifluoroacetamida, fenilacetamida, metilcarbamato de metilo, carbamato de etilo, carbamato de 9-fluorenilmetilo (Fmoc), carbamato det-butilo (BOC o Boc), carbamato de 1-adamantilo (Adoc), carbamato de vinilo (Voc), carbamato de alilo (Alloc), 2-(trimetilsilil)etoxi]metilo (SEM), p-toluenosulfonamida (Ts), bencenosulfonamida, 2,3,6-trimetil-4-metoxibencenosulfonamida (Mtr), 2,4,6-trimetoxibencenosulfonamida (Mtb), derivado de fenotiazinil-(10)-acilo, derivado de N'-p-toluenosulfonilaminoacilo,
Los grupos protectores de oxígeno y azufre de ejemplos incluyen, pero no se limitan a, metilo, metoximetilo (MOM), metiltiometilo (MTM), t-butiltiometilo, (fenildimetilsilil)metoximetilo (SMOM), benciloximetilo (BOM), pmetoxibenciloximetilo (PMBM), (4-metoxifenoxi)metilo (p-AOM), tetrahidropiranilo (THP), metanosulfonato (mesilato), bencilsulfonato y tosilato (Ts).
A la expresión "grupo saliente" se le da su significado ordinario en la técnica de la química orgánica sintética y se refiere a un átomo o un grupo capaz de ser desplazado por un nucleófilo. Los ejemplos de grupos salientes adecuados incluyen, pero no se limitan a, halógeno (tal como F, Cl, Br o I (yodo)), alcoxicarboniloxi, ariloxicarboniloxi, alcanosulfoniloxi, arenosulfoniloxi, alquilcarboniloxi (p. ej., acetoxi), un éster de ácido sulfónico, tal como toluenosulfonato (tosilato, -OTs), metanosulfonato (mesilato, -OMs), p-bromobencenosulfoniloxi (brosilato, -OBs), -OS(=O)2(CF2)3CF3 (nonaflato, -ONf), o trifluorometanosulfonato (triflato, -OTf).
Como se usa en el presente documento, el término "sal" se refiere a todas y cada una de las sales, y abarca las sales farmacéuticamente aceptables.
La expresión "sal farmacéuticamente aceptable" se refiere a aquellas sales que son, dentro del alcance del buen criterio médico, adecuadas para usar en contacto con los tejidos de seres humanos y animales inferiores sin toxicidad indebida, irritación, respuesta alérgica y similares, y son acordes con una relación beneficio/riesgo razonable. Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Por ejemplo, Berge et al. describen sales farmacéuticamente aceptables en detalle en J. Pharmaceutical Sciences, 1977, 66, 1-19. Las sales farmacéuticamente aceptables de los compuestos de esta descripción incluyen las derivadas de ácidos y bases inorgánicos y orgánicos adecuados. Ejemplos de sales de adición de ácido no tóxicas farmacéuticamente aceptables son sales de un grupo amino formadas con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico, o usando otros métodos conocidos en la técnica tales como intercambio iónico. Otras sales farmacéuticamente aceptables incluyen sales de adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, canforato, canforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, hidroyoduro, 2-hidroxietanosulfonato, lactobionato, lactato, laurato, laurilsulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato, y similares. Las sales derivadas de bases apropiadas incluyen sales de metales alcalinos, metales alcalinotérreos, amonio y N+(alquilo C-m K. Las sales de metales alcalinos o alcalinotérreos representativas incluyen sodio, litio, potasio, calcio, magnesio y similares. Otras sales farmacéuticamente aceptables incluyen, cuando sea apropiado, cationes de amonio, amonio cuaternario y amina no tóxicos formados usando contraiones tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, sulfonato de alquilo inferior y sulfonato de arilo.
Los términos "composición" y "formulación" se usan indistintamente.
Un "sujeto" para el que se contempla la administración se refiere a un ser humano (es decir, hombre o mujer de cualquier grupo de edad, p. ej., sujeto pediátrico (p. ej., bebé, niño o adolescente) o sujeto adulto (p. ej., adulto joven, adulto de mediana edad o adulto mayor)) o animal no humano. En ciertas realizaciones, el animal no humano es un mamífero (p. ej., primate (p. ej., macaco cangrejero o macaco rhesus), mamífero comercialmente relevante (p. ej., vaca, cerdo, caballo, oveja, cabra, gato o perro), o aves (p. ej., ave comercialmente relevante, tal como pollo, pato, ganso o pavo)). En ciertas realizaciones, el animal no humano es un pez, reptil o anfibio. El animal no humano puede ser un macho o hembra en cualquier etapa de desarrollo. El animal no humano puede ser un animal transgénico o un animal modificado genéticamente. En ciertas realizaciones, el sujeto es un paciente. El término "paciente" se refiere a un sujeto humano que necesita tratamiento de una enfermedad. En ciertas realizaciones, el término "paciente" es un adulto humano mayor de 18 años que necesita tratamiento de una enfermedad. En ciertas realizaciones, el término "paciente" es un niño humano de no más de 18 años que necesita el tratamiento de una enfermedad. En ciertas realizaciones, el paciente no recibe transfusión de modo regular (p. ej., haber tenido no más de 4 episodios de transfusión en el período de 12 meses). En ciertas realizaciones, el paciente recibe transfusiones de modo regular (p. ej., ha tenido al menos 4 episodios de transfusiones en el período de 12 meses).
El término "administra", "administrar" o "administración" se refiere a implantar, absorber, ingerir, inyectar, inhalar o introducir de otro modo un compuesto descrito en el presente documento, o una composición del mismo, en o sobre un sujeto.
Los términos "tratamiento", "trata" y "tratar" se refieren a revertir, aliviar, retrasar el inicio o inhibir el progreso de una enfermedad descrita en el presente documento. En algunas realizaciones, el tratamiento puede administrarse después de que se hayan desarrollado o se hayan observado uno o más signos o síntomas de la enfermedad (es decir, tratamiento terapéutico). En otras realizaciones, el tratamiento puede administrarse en ausencia de signos o síntomas de la enfermedad. Por ejemplo, el tratamiento puede administrarse a un sujeto susceptible antes de la aparición de los síntomas (es decir, tratamiento profiláctico) (p. ej., a la luz de un historial de síntomas y/o a la luz de la exposición a un patógeno). El tratamiento también se puede continuar después de que los síntomas se hayan resuelto, por ejemplo, para retrasar o prevenir la recurrencia. En ciertas realizaciones, el tratamiento incluye retrasar el inicio de al menos un síntoma del trastorno durante un período de tiempo.
Los términos "afección", "enfermedad" y "trastorno" se usan indistintamente.
Un "cantidad eficaz" de un compuesto descrito en el presente documento se refiere a una cantidad suficiente para provocar la respuesta biológica deseada. Una cantidad eficaz de un compuesto descrito en el presente documento puede variar dependiendo de factores tales como el criterio de valoración biológico deseado, la farmacocinética del compuesto, la afección que se trata, el modo de administración y la edad y la salud del sujeto. En ciertas realizaciones, una cantidad eficaz es una cantidad terapéuticamente eficaz. En ciertas realizaciones, la cantidad eficaz es para generar una respuesta de hemoglobina del sujeto de >1,5 g/dl de aumento en la concentración de Hb desde el valor inicial. La concentración de Hb inicial del sujeto es el promedio de todas las concentraciones de Hb disponibles antes del tratamiento con el compuesto. En ciertas realizaciones, la cantidad eficaz es para generar una respuesta de hemoglobina del sujeto de >1,0 g/dl de aumento en la concentración de Hb desde el valor inicial. En ciertas realizaciones, la cantidad eficaz es para generar una respuesta de hemoglobina del sujeto de >2,0 g/dl de aumento en la concentración de Hb desde el valor inicial. En ciertas realizaciones, una cantidad eficaz es la cantidad de un compuesto descrito en el presente documento en una sola dosis. En ciertas realizaciones, una cantidad eficaz son las cantidades combinadas de un compuesto descrito en el presente documento en dosis múltiples.
A "cantidad terapéuticamente eficaz" de un compuesto descrito en el presente documento es una cantidad suficiente para proporcionar un beneficio terapéutico en el tratamiento de una afección o para retrasar o minimizar uno o más síntomas asociados con la afección. Una cantidad terapéuticamente eficaz de un compuesto significa una cantidad de agente terapéutico, solo o en combinación con otros tratamientos, que proporciona un beneficio terapéutico en el tratamiento de la afección. La expresión "cantidad terapéuticamente eficaz" puede abarcar una cantidad que mejora el tratamiento global, reduce o evita los síntomas, signos o causas de la afección y/o mejora la eficacia terapéutica de otro agente terapéutico. En ciertas realizaciones, una cantidad terapéuticamente eficaz es una cantidad suficiente para provocar una activación medible de PKM2 de tipo natural o mutante. En ciertas realizaciones, una cantidad terapéuticamente eficaz es una cantidad suficiente para regular (p. ej., reducir) la glucosa en plasma en un sujeto que lo necesite. En ciertas realizaciones, una cantidad terapéuticamente eficaz es una cantidad suficiente para producir una eficacia medible para tratar una enfermedad proliferativa (p. ej., cáncer, una enfermedad autoinmunitaria). En ciertas realizaciones, una cantidad terapéuticamente eficaz es una cantidad suficiente para producir una eficacia medible para tratar y/o prevenir una enfermedad diabética (p. ej., DN). En ciertas realizaciones, una cantidad terapéuticamente eficaz es una cantidad suficiente para producir una eficacia medible para tratar y/prevenir la CAD.
El término "activador" como se usa en el presente documento significa un agente que aumenta (de forma medible) la actividad de una piruvato quinasa (p. ej., PKM2) o hace que la actividad de la piruvato quinasa (p. ej., PKM2) aumente a un nivel que es mayor que los niveles basales de actividad de la PKM2. Por ejemplo, el activador puede imitar el efecto causado por un ligando natural (p. ej., FBP). El efecto activador causado por un compuesto proporcionado en el presente documento puede ser de la misma, mayor o menor magnitud que el efecto de activación causado por un ligando natural, pero se produce el mismo tipo de efecto. Un compuesto proporcionado en el presente documento puede evaluarse para determinar si es un activador midiendo directa o indirectamente la actividad de la piruvato quinasa cuando se somete a dicho compuesto. La actividad de un compuesto proporcionado en el presente documento se puede medir, por ejemplo, frente a una sustancia de control. En algunos casos, la actividad medida del compuesto de ensayo es para la activación de la PKM2. La actividad de la PKM2 se puede medir, por ejemplo, vigilando la concentración de un producto tal como ATP o los niveles de un cofactor tal como NADH usado en un sistema de ensayo de enzimas acopladas (véase PCT/US2010/040486).
El término "inhibidor" como se usa en el presente documento significa un agente que retarda, detiene, disminuye o inactiva (de forma medible) la actividad enzimática de una piruvato quinasa (p. ej., PKM2) para disminuir a un nivel que es menor que los niveles o actividad basales de las piruvato quinasas (p. ej., PKM2).
La expresión "ex vivo" en referencia a un método como se usa en el presente documento significa que el método tiene lugar fuera de un organismo. Por ejemplo, se puede extraer una célula o un tejido del organismo para ponerlo en contacto con uno o más compuestos proporcionados en el presente documento o una de sus sales farmacéuticamente aceptables
o una composición farmacéutica de los mismos, opcionalmente en condiciones controladas artificialmente (p. ej., temperatura).
La expresión "in vitro" en referencia a un método como se usa en el presente documento significa que el método tiene lugar fuera de un organismo y está contenido dentro de un entorno artificial. Por ejemplo, se puede extraer una célula o un tejido del organismo para ponerlo en contacto con uno o más compuestos proporcionados en el presente documento o una sal farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo, en un entorno artificial contenido (p. ej., un sistema de cultivo), tal como en un tubo de ensayo, en un cultivo, en un matraz, en una placa de microtitulación, en una placa de Petri, y similares.
Compuestos
En el presente documento se describen métodos relacionados con compuestos y composiciones farmacéuticas que modulan la PKM2, y el uso de estos compuestos y composiciones farmacéuticas para estos métodos. Específicamente, estos métodos incluyen un método para modularla actividad de la piruvato quinasa M2 (PKM2) en un sujeto, un método para modular (p. ej., aumentar o disminuir) el nivel de actividad de la PKM2 en un sujeto que lo necesite, un método para modular (p. ej., aumentar o disminuir) el nivel de glucosa en plasma en un sujeto que lo necesite, un método para inhibir la proliferación celular en un sujeto que lo necesita (una célula transformada, p. ej., una célula cancerosa, o generalmente inhibir el crecimiento de una célula dependiente de PKM2 que se somete a glucólisis aeróbica), un método para tratar a un sujeto que padece o es susceptible de padecer una enfermedad o trastorno asociado con la función de p Km 2. En una realización, los compuestos y composiciones descritos en el presente documento modulan la PKM2 uniéndose a un bolsillo de unión alostérico. En una realización, los compuestos y composiciones descritos en el presente documento inhiben la PKM2. En una realización, los compuestos y composiciones descritos en el presente documento activan la PKM2. En una realización, el compuesto descrito en el presente documento es un compuesto de fórmulas (I)-(IX), o un compuesto de fórmulas (I')-(V'), o una de sus sales farmacéuticamente aceptables, o una composición farmacéutica que comprende un compuesto de fórmulas (I)-(IX), o un compuesto de fórmulas (I')-(V'), o una de sus sales farmacéuticamente aceptables. En una realización, el compuesto utilizado en los métodos de la invención es un compuesto descrito en la solicitud de patente internacional N.° pCt /CN2017/09496 y solicitudes de patente provisional de EE. UU. N.° 62/673.533 y 62/673.526.
En una primera realización de la invención, se proporciona un método para modular la actividad de la piruvato quinasa M2 (PKM2) en un sujeto que lo necesite, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables, en donde:

Q es hidrógeno, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido;
R1 es hidrógeno, alquilo opcionalmente sustituido, haloalquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido, -ORo1, -C(=O)Rc1 , o un grupo protector de nitrógeno;
L1 es un enlace, alquileno opcionalmente sustituido, -O-, -S-, -S-CH2-, -S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(=O)-, -C(=O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, -C(R4)2O--NR3C(R4)2-, -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2-, -S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O-, -OS(=O)2NR3-, -NR3S(=O)O-, -OS(=O)NR3- o -S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, alquileno opcionalmente sustituido, -C(=O)-, -S(=O)2- o -S(=O)-, en donde el punto de unión a Q está en el lado derecho;
R2 es hidrógeno, halógeno, alquilo opcionalmente sustituido, alcoxi opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido, o un grupo protector de nitrógeno cuando L1 es -NR3-, - NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O- o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es -O-, -OC(=O)-, -OC(=O)NR3-, -OC(R4)2-, -OS(=O)2- -OS(=0)2Nr 3-, -OS(=O)NR3-, o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
cada caso de R3 es independientemente hidrógeno, -ORo2, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro1 y Ro2 es independientemente hidrógeno, alquilo opcionalmente sustituido o un grupo protector de oxígeno;
cada caso de Rc1 es independientemente alquilo opcionalmente sustituido o -N(Rcn)2, en donde cada caso de Rcn es independientemente hidrógeno, -alquilo C1-6, o un grupo protector de nitrógeno; y
cada caso de R4 es independientemente hidrógeno, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido.
En una segunda realización de la invención, se proporciona un método para modular el nivel de glucosa en plasma en un sujeto que lo necesite, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde la fórmula (I) es como se define en la primera realización.
En una tercera realización de la invención, se proporciona un método para inhibir la proliferación celular en un sujeto que padece o es susceptible de padecer una enfermedad o trastorno asociado con la función de la PKM2 que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde la fórmula (I) es como se define en la primera realización.
En una cuarta realización de la invención, se proporciona un método para tratar una enfermedad asociada con la actividad anómala de la PKM2 en un sujeto que lo necesite, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde la fórmula (I) es como se define en la primera realización.
En una quinta realización de la invención, se proporciona un método según la cuarta realización como se describe anteriormente, en donde la enfermedad es una enfermedad proliferativa.
En una sexta realización de la invención, se proporciona un método según la cuarta realización como se describe anteriormente, en donde la enfermedad es cáncer, obesidad, una enfermedad diabética (p. ej., nefropatía diabética (DN)), aterosclerosis, reestenosis, enfermedad arterial coronaria (CAD), síndrome de Bloom (BS), hiperplasia prostática benigna (HPB) o una enfermedad autoinmunitaria.
En una séptima realización de la invención, se proporciona un método para tratar la hiperglucemia en un sujeto que lo necesite, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde la fórmula (I) es como se define en la primera realización.
En una octava realización de la invención, se proporciona un método para tratar una enfermedad diabética en un sujeto que lo necesite, que comprende administrar una cantidad eficaz de un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo, en donde la fórmula (I) es como se define en la primera realización.
En una novena realización de la invención, se proporciona un método según la octava realización como se describe anteriormente, en donde la enfermedad diabética es nefropatía diabética.
En una décima realización de la invención, se proporciona un método según cualquiera de las realizaciones primera a novena como se describe anteriormente, en donde el método comprende además identificar un sujeto que se beneficiaría de la modulación de la PKM2.
En una undécima realización de la invención, se proporciona un método según la primera realización como se describe anteriormente, en donde la modulación es activación.
En una duodécima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a undécima descritas anteriormente, en donde:
Q es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo de 6 a 14 miembros opcionalmente sustituido o heteroarilo de 5 a 14 miembros opcionalmente sustituido;
R1 es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -haloalquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, -alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo de 6 a 12 miembros opcionalmente sustituido, -ORo1, -C(=O)Rc1, o un grupo protector de nitrógeno;
L1 es un enlace, alquileno C1-6 opcionalmente sustituido, -O-, -S-, -S-CH2-, - S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(=O)-, -C(=O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, - C(R4)2O-, -NR3C(R4)2- -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2- -S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O- -OS(=O)2NR3-, -NR3S(=O)O- -OS(=O)NR3- o -S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, alquileno C1-C6 opcionalmente sustituido, -C(=O)-, -S(=O)2- o -S(=O)-, en donde el punto de unión a Q está en el lado derecho;
R2 es hidrógeno, halógeno,- alquilo C1-C6 opcionalmente sustituido, -alcoxi C1-C6 opcionalmente sustituido, -cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo C6-C12 opcionalmente sustituido o heteroarilo de 3 a 14 miembros opcionalmente sustituido, o un grupo protector de nitrógeno cuando L1 es -NR3-, -NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O- o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es-O-, -OC(=O)-, -OC(=O)NR3-, -OC(R4)2-, -OS(=O)-, -OS(=O)2-, -OS(=O)2NR3-, -OS(=O)NR3- o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
cada caso de R3 es independientemente hidrógeno, -ORo2, -alquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, -alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C2-C12 opcionalmente sustituido, heterociclilo C3-C12 opcionalmente sustituido, arilo C6-C12 opcionalmente sustituido, heteroarilo C5-C12 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro1 y Ro2 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc1 es independientemente -alquilo C1-C6 sustituido opcionalmente o -N(Rcn)2, en donde cada caso de Rcn es independientemente hidrógeno, -alquilo C1-C6 o un grupo protector de nitrógeno;
cada caso de R4 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, -alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo C6-C12 opcionalmente sustituido, o heteroarilo de 5 a 14 miembros opcionalmente sustituido.
En una decimotercera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a duodécima descritas anteriormente, en donde:
Q es arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros o heteroarilo bicíclico de 8 a 12 miembros, cada uno de los cuales está sustituido con 0-3 casos de RC;
R1 se selecciona de hidrógeno, -alquilo C1-C6, -haloalquilo C1-C6, cicloalquilo C3-C7 monocíclico y heterociclilo de 3 a 14 miembros, -ORo1, -C(=O)Rc1, o un grupo protector de nitrógeno; en donde cada alquilo, cicloalquilo o heterociclilo está sustituido con 0-3 casos de Rd;
R2 se selecciona de hidrógeno, halógeno, -alquilo C1-C6, -alcoxi C1-C6, cicloalquilo C3-C7 monocíclico, cicloalquilo C6-C12 bicíclico, heterociclilo de 3 a 14 miembros, arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros, heteroarilo bicíclico de 8 a 12 miembros, en donde cada alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido con 0-3 casos de Re, o un grupo protector de nitrógeno cuando L1 es -NR3-, -NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O- o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es -O-, -OC(=O)-, -OC(=O)NR3-, -OC(R4)2-, -OS(=O)-, -OS(=O)2- -OS(=O)2NR3-, -OS(=O)NR3- o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
R3 se selecciona de hidrógeno, -ORo2, -alquilo C1-C6, cicloalquilo C3-C7 monocíclico, cicloalquilo C6-C12 bicíclico, heterociclilo de 3 a 14 miembros, arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros y heteroarilo bicíclico de 8 a 12 miembros, en donde cada alquilo, cicloalquilo, heterocicloalquilo, arilo y heteroarilo está sustituido con 0-3 casos de Rf;
R4 se selecciona de hidrógeno, -alquilo C1-C6, cicloalquilo C3-C7 monocíclico y heterociclilo de 3 a 14 miembros, en donde cada alquilo, cicloalquilo o heterociclilo está sustituido con 0-1 casos de R9;
L1 es un enlace, un alquileno sustituido con 0-3 casos de Rh, -O-, -S-, -S-CH2-, -S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(-O)-, -C(-O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, -C(R4)2O-, -NR3C(R4)2-, -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2-, -S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O-, -OS(=O)2NR3-, -NR3S(=O)O-, -OS(=O)NR3- o -S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, un alquileno sustituido con 0-3 casos de Rh, -C(=O)-, -S(=O)2- o -S(=O)-, en donde el punto de unión a Q está en el lado derecho;
cada RC se selecciona independientemente de halógeno, -alquilo Ci -C6, -haloalquilo C i-C6 , -hidroxialquilo Ci -C6 , -OH, -O-alquilo C1-C6, -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2 , -C(=O)O-(alquilo C1-C6), -C(=O)OH, -C(=O)-(alquilo C1-C6), -C(=O)NH2, -C(=O)NH(alquilo Cr Cs), -C(=O)N(alquilo C1-C6)2, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-Cs), -NH(C=O)N(alquilo Cr Cs)2, -NHC(=O)(alquilo C1-C6), -N(alquilo C1-C6)C(=O)(alquilo Cr Cs), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-C6)2 , -NHS(=O)2 (alquilo C1-C6), -NH2, -CN y -NO2; o dos casos de RC unidos al mismo átomo de carbono o a átomos de carbono adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclil-C(=O)OH;
cada Rd se selecciona independientemente de halógeno, -alquilo C1-C6, -OH, -O-alquilo C1-C6, -NH2 y-CN;
cada RC se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6 , -hidroxialquilo C1-C6 , -OH, -O-alquilo C1-C6 , -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2, -C(=O)O(alquilo C1-C6), -C(=O)OH, -C(=O)(alquilo Cr Cs), -C(=O)NH2, -C(=O)NH(alquilo Cr Cs), -C(=O)N(alquilo C rC sfe, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-C6), -NH(C=O)N(alquilo C^C sfc, -NHC(=O)(alquilo Cr Cs), -N(alquilo C1-Cs)C(=O)(alquilo Cr Cs), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-C6)2 , -NHS(=O)2(alquilo C1-C6), -NH2 , -Cn y-NO 2 ; o dos casos de Re unidos al mismo átomo de carbono o a átomos de carbono adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclilo;
cada Rf se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -alcoxi C1-C6, -OH, -NH2, -CN y -NO2;
cada R9 se selecciona independientemente de halógeno, -alquilo C1-C6 , -haloalquilo C1-C6, -alcoxi C1-C6, -OH, NH2, -CN y NO2 y;
cada Rh se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -hidroxialquilo C1-C6, -OH, -O-alquilo C1-C6 , -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2, -C(=O)O(alquilo C1-C6), -C(=O)OH, -C(=O)(alquilo C1-C6), -C(=O)NH2, -C(=O)NH(alquilo C1-C6), -C(=O)N(alquilo C rC sk, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-C6), -NH(C=O)N(alquilo C1-Cs)2, -NHC(=O)(alquilo C1-C6), -N(alquilo C1-Cs)C(=O)(alquilo C1-C6), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-Cs)2, -NHS(=O)2(alquilo C1-C6), -NH2, -CN y -NO2, S(=O)2arilo, s(=O)2heteroarilo y =NOH o dos casos de Rcn unidos al mismo átomo de carbono o a átomos adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclilo.
En una decimocuarta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimotercera como se describe anteriormente, en donde el compuesto es un compuesto representado por la fórmula (II):

o una de sus sales farmacéuticamente aceptables; en donde:
Ra y Rb son cada uno independientemente hidrógeno, halógeno, -CN, -NO2 ,-N3 , alquilo opcionalmente sustituido, -ORo3, -N(Rn1)2 , -C(=O)N(Rn1)2 o -C(=O)Rc2, o Ra y Rb se pueden considerar junto con el átomo de carbono para formar cicloalquilo opcionalmente sustituido o heterociclilo opcionalmente sustituido;
cada caso de Rn1 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro3 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno; y
cada caso de Rc2 es independientemente -alquilo C1-C6 opcionalmente sustituido;
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a duodécima. En una decimoquinta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimotercera como se describe anteriormente, en donde L1 es un enlace, -alquileno C1-6 opcionalmente sustituido, -C(=O)-, -S(=O)-, -S(=O)2-, -NR3C(=O)- o -C(=O)NR3-; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimocuarta.
En una decimosexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones
primera a decimocuarta como se describe anteriormente, en donde L1 es alquileno C i-6 sustituido con Rj y Rk;
en donde cada caso de Rj y Rk se selecciona independientemente de H, halógeno, -CN, -ORo7, -N(Rn5)2, -N(Rn5)C(=O)Rc5, -C(=O)N(Rn5)2, -C(=O)Rc5, -C(=O)ORo7 -SRjs, -S(=O)2Rjs, o -S(=O)Rjs, -alquilo C1-C6 opcionalmente sustituido; o se pueden considerar junto con el átomo de carbono para formar C=O, C=NRjn, un anillo de cicloalquilo monocíclico C3-C6 opcionalmente sustituido o un anillo de heterociclilo monocíclico C3-C6 opcionalmente sustituido;
cada uno de Rn5 y Rjn es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -ORo8, o un grupo protector de nitrógeno;
cada caso de Ro7 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc5 es independientemente -alquilo C1-C6 opcionalmente sustituido; y
cada caso de Rjs es independientemente -alquilo C1-C6 opcionalmente sustituido, arilo C6-12 opcionalmente sustituido, heteroarilo opcionalmente sustituido o un grupo protector de azufre; y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimoquinta.
En una decimoséptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimosexta como se describe anteriormente, en donde el compuesto representado por la fórmula (I'):

o una de sus sales farmacéuticamente aceptables, en donde:
R1 es hidrógeno, un alquilo opcionalmente sustituido, un haloalquilo opcionalmente sustituido, un alquenilo opcionalmente sustituido, un alquinilo opcionalmente sustituido, un cicloalquilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un arilo opcionalmente sustituido, -ORo1, -C(=O)Rc1, o un grupo protector de nitrógeno; en donde:
Ro1 es hidrógeno, alquilo opcionalmente sustituido o un grupo protector de oxígeno;
Rc1 es alquilo opcionalmente sustituido o -N(Rcn)2, en donde cada caso de
Rcn es independientemente hidrógeno, -alquilo C1-6, o un grupo protector de nitrógeno;
R2 y Q son cada uno independientemente un heteroarilo monocíclico de 5 o 6 miembros opcionalmente sustituido;
Ra y Rb son cada uno independientemente hidrógeno, un halógeno, -CN, -NO2,-N3, un alquilo opcionalmente sustituido, -ORo3, -N(Rn1)2, -C(=O)N(Rn1)2 o -C(=O)Rc2; o alternativamente Ray Rb pueden considerarse junto con el átomo de carbono al que están unidos para formar un cicloalquilo opcionalmente sustituido o un heterociclilo opcionalmente sustituido; en donde:
cada caso de Rn1 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
Ro3 es hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno; y
Rc2 es un -alquilo C1-C6 opcionalmente sustituido; y
Rj y Rk son cada uno independientemente hidrógeno, un halógeno, -CN, -ORo7, -N(Rn5)2, -N(Rn5)C(=O)Rc5, -C(=O)N(Rn5)2, -C(=O)Rc5, -C(=O)ORo7, -SRjs, -S(=O)2Rjs, - S(=O)Rjs, o un -alquilo C1-C6 opcionalmente sustituido; o alternativamente Rj y Rk se pueden considerar junto con el átomo de carbono al que están unidos para formar C=O, un anillo de cicloalquilo monocíclico C1-C6 opcionalmente sustituido, o un anillo de heterociclilo monocíclico C3-C6 opcionalmente sustituido; en donde:
cada caso de Rn5 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, -ORo8, o un grupo protector de nitrógeno, en donde Ro8 es hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de
oxígeno;
cada caso de Ro7 es independientemente hidrógeno, un -alquilo C i -C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc5 es independientemente un -alquilo C1-C6 opcionalmente sustituido; y
cada caso de Rjs es independientemente un -alquilo C1-C6 opcionalmente sustituido, un arilo C6-12 opcionalmente sustituido, un heteroarilo opcionalmente sustituido o un grupo protector de azufre.
En una decimoctava realización de la invención, se proporciona un método según la decimoséptima realización como se describe anteriormente, en donde el compuesto es un compuesto representado por la fórmula (I') o una de sus sales farmacéuticamente aceptables, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por R2 está opcionalmente sustituido en cada átomo de carbono del anillo sustituible con Rp y opcionalmente sustituido en cada átomo de nitrógeno del anillo sustituible con Rn6; en donde:
cada caso de Rp es independientemente hidrógeno, un halógeno, -CN, -NO2, -N3 , un alquilo opcionalmente sustituido, un alquenilo opcionalmente sustituido, un alquinilo opcionalmente sustituido, un cicloalquilo opcionalmente sustituido, un arilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un heteroarilo opcionalmente sustituido, -ORo6, -SRs2 , -N(Rn3)2 , -C(=O)N(Rn3)2, -N(Rn3)C(=O)Rc4 , -C(=O)Rc4, -C(=O)ORo6, -OC(=O)Rc4, -S(=O)Rs2, -S(=O)2Rs2, -S(=O)ORo6, -OS(=O)Rc4, -S(=O)2ORo6, -OS(=O)2Rc4, -S(=O)N(Rn3)2, -S(=O)2N(Rn3)2 , -N(Rn3)S(=O)Rs2, -N(Rn3)S(=O)2 Rs2, -N(Rn3)C(=O)ORo6, -OC(=O)N(Rn3)2 , -N(Rn3)C(=O)N(Rn3)2 , -N(Rn3)S(=O)N(Rn3)2 , -N(Rn3)S(=O)2N(Rn3)2, -N(Rn3)S(=O)ORo6, -N(Rn3)S(=O)2ORo6, -OS(=O)N(Rn3)2 o -OS(=O)2N(Rn3)2; o alternativamente dos casos de Rp unidos al mismo átomo de carbono o a átomos de carbono adyacentes, pueden considerarse junto con el(los) átomo(s) de carbono al que están unidos para formar un cicloalquilo o heterocicloalquilo opcionalmente sustituido; en donde:
cada caso de Rn3 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro6 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno; y
cada caso de Rc4 es un -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rs2 es independientemente un -alquilo C1-C6 opcionalmente sustituido o un grupo protector de azufre; y Rn6 es hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; y
en donde el resto de las variables son como se definen en la decimoséptima realización.
En una decimonovena realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimoctava como se describe anteriormente, en donde Q es heteroarilo monocíclico de 5 a 6 miembros opcionalmente sustituido; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimoctava.
En una vigésima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimonovena como se describe anteriormente, en donde Q es una de las siguientes fórmulas:


en donde:
cada caso de Rn es independientemente hidrógeno, un halógeno, -CN, -NO2, -N3, un alquilo opcionalmente sustituido, un alquenilo opcionalmente sustituido, un alquinilo opcionalmente sustituido, un cicloalquilo opcionalmente sustituido, un arilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un heteroarilo opcionalmente sustituido, -ORo4, -SRs1 , -N(Rn2)2 , -C(=O)N(Rn2)2, -N(Rn2)C(=O)Rc3 , -C(=O)Rc3, -C(=O)ORo4, -OC(=O)Rc3, -S(=O)Rs1, -S(=O)2Rs1, -S(=O)ORo4, -OS(=O)Rc3, -S(=O)2ORo4, -OS(=O)2Rc3, -S(=O)N(Rn2)2, -S(=O)2N(Rn2)2 , -N(Rn2)S(=O)Rs1, -N(Rn2)S(=O)2 Rs1, -N(Rn2)C(=O)ORo4, -OC(=O)N(Rn2)2 , -N(Rn2)C(=O)N(Rn2)2 , -N(Rn2)S(=O)N(Rn2)2 , -N(Rn2)S(=O)2N(Rn2)2, -N(Rn2)S(=O)ORo4, -N(Rn2)S(=O)2ORo4, -OS(=O)N(Rn2)2 o -OS(=O)2N(Rn2)2; o dos casos de Rcn unidos al mismo átomo de carbono o átomos de carbono adyacentes, considerados junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o heterocicloalquilo opcionalmente sustituido; en donde:
cada caso de Rn2 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro4 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc3 es independientemente un -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rcn es independientemente un -alquilo C1-C6 opcionalmente sustituido o un grupo protector de azufre; n es 0, 1,2 o 3, según lo permita la valencia; y
cada uno de Rna, Rnby Rnd es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimonovena. En una vigésima primera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por Q se selecciona de los siguientes:
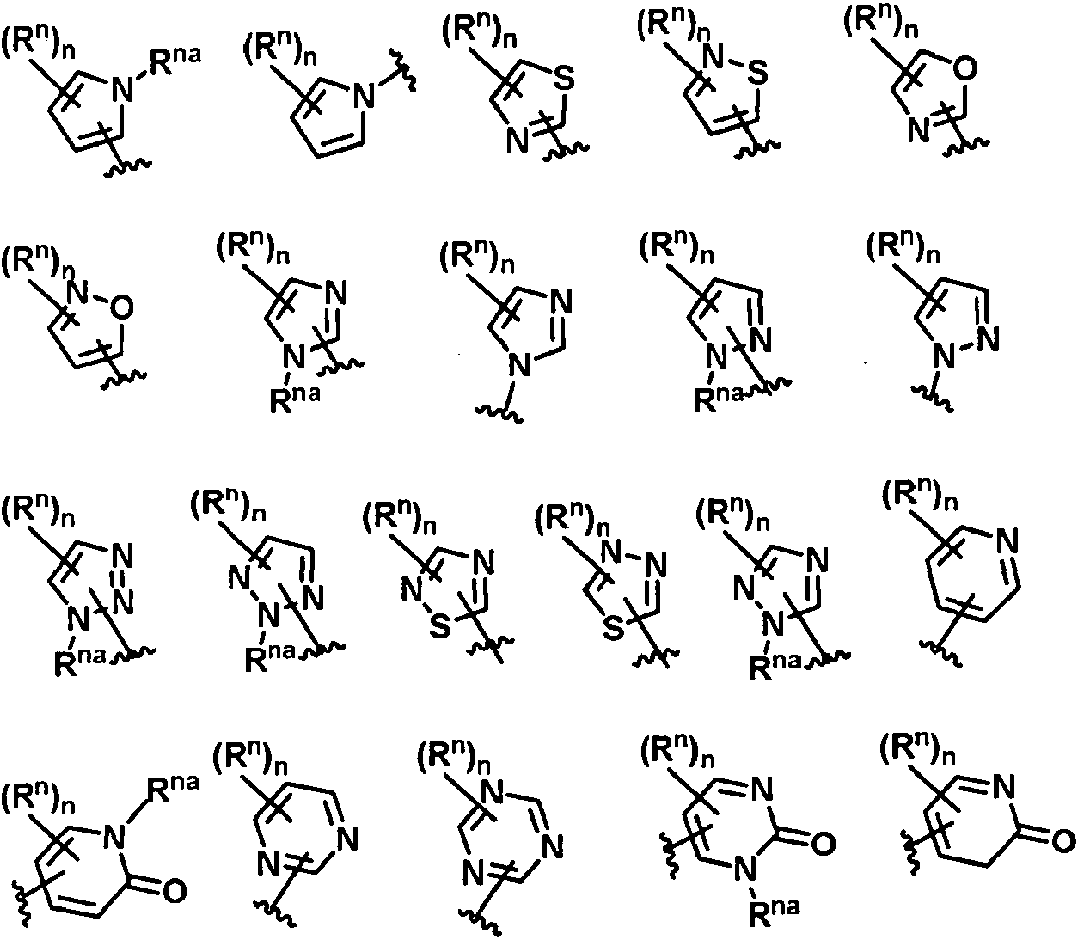
y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima. En una vigésima segunda realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésimo primera como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado porQ se selecciona de los siguientes:

y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima primera. En una vigésima tercera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima segunda como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por Q se selecciona de los siguientes:

en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima segunda. En una vigésima cuarta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima tercera como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por Q es

y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima tercera.
En una vigésima quinta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimosexta como se describe anteriormente, en donde Q es heteroarilo bicíclico de 8 a 12 miembros opcionalmente sustituido o heterociclilo bicíclico de 8 a 12 miembros opcionalmente sustituido; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimosexta.
En una vigésima sexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimosexta y vigésima quinta como se describe anteriormente, en donde Q es una de las siguientes fórmulas:
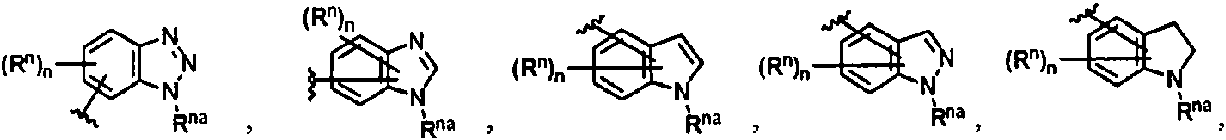

en donde cada caso de Rcn se selecciona independientemente de hidrógeno, halógeno, -CN, -NO2, -N3, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, arilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, -ORo4, -SRs1, - N(Rn2)2, -C(=O)N(Rn2)2, -N(Rn2)C(=O)Rc3, -C(=O)Rc3, -C(=O)ORo4, -OC(=O)Rc3, -S( S(=O)2Rs1, -S(=O)ORo4, -OS(=O)Rc3, -S(=O)2ORo4, -OS(=O)2Rc3, -S(=O)N(Rn2)2 , -S(=O)2 N(Rn2)2 ,-N(Rn2)S(=O)Rs1, -N(Rn2)S(=O)2Rs1 , -N(Rn2)C(=O)ORo4, -OC(=O)N(Rn2)2 , -N(Rn2)C(=O)N(Rn2)2 , -N(Rn2)S(=O)N(Rn2)2, -N(Rn2)S(=O)2N(Rn2)2, -N(Rn2)S(=O)ORo4, -N(Rn2)S(=O)2ORo4, -OS(=O)N(Rn2)2, -OS(=O)2N(Rn2)2, o dos casos de Rn unidos al mismo átomo de carbono o a átomos de carbono adyacentes, considerados junto con los átomos a los que están unidos forman un cicloalquilo o un heterocicloalquilo opcionalmente sustituido;
cada caso de Rna y Rnb es independientemente hidrógeno, -alquilo C i -C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Rn2 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro4 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc3 es independientemente -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rs1 es independientemente -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de azufre; n es 0, 1,2 o 3, según lo permita la valencia; y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimosexta y vigésima quinta.
En una vigésima séptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima sexta como se describe anteriormente, en donde R2 se selecciona de hidrógeno, hidroxilo, halógeno, -alquilo C1-C6, -alquenilo C2-C6, -alcoxilo C2-C6, fenilo, naftalenilo, cicloalquilo C3-6, heteroarilo de 5 miembros, heteroarilo de 6 miembros, heteroarilo bicíclico de 8 miembros, heteroarilo bicíclico de 9 miembros, en donde cada alquilo, alquenilo, fenilo y heteroarilo está sustituido con 0-3 casos de Re; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima sexta.
En una vigésima octava realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésimo séptima como se describe anteriormente, en donde R2 es de una de las siguientes fórmulas:



en donde cada caso de Rp se selecciona independientemente de hidrógeno, halógeno, -CN, -NO2 , -N3, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, arilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, -ORo6, -SRs2, - N(Rn3)2, -C(=O)N(Rn3)2, -N(Rn3)C(=O)Rc4, -C(=O)Rc4, -C(=O)ORo6, -OC(=O)Rc4, -S(=O)Rs2, -S(=O)2Rs2, -S(=O)ORo6, -OS(=O)Rc4, -S(=O)2ORo6, -OS(=O)2 Rc4, -S(=O)N(Rn3)2 , -S(=O)2 N(Rn3)2 , -N(Rn3)S(=O)Rs2, -N(Rn3)S(=O)2Rs2 , -N(Rn3)C(=O)ORo6, -OC(=O)N(Rn3)2 , -N(Rn3)C(=O)N(Rn3)2 , -N(Rn3)S(=O)N(Rn3)2, -N(Rn3)S(=O)2N(Rn3)2, -N(Rn3)S(=O)ORo6, -N(Rn3)S(=O)2ORo6, -OS(=O)N(Rn3)2 , -OS(=O)2 N(Rn3)2, o dos casos de Rp unidos al mismo átomo de carbono o a átomos de carbono adyacentes, considerados junto con los átomos a los que están unidos forman un cicloalquilo o un heterocicloalquilo opcionalmente sustituido;
cada caso de Rn3, Rnc y Rnd es independientemente hidrógeno, -alquilo Ci -C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro6 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc4 es independientemente -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rs2 es independientemente -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de azufre; p es 0, 1,2 o 3, según lo permita la valencia; y
en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima séptima. En una vigésima novena realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima octava como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por R2 se selecciona de uno de los siguientes:


en donde:
cada caso de Rnc y Rnd es independientemente hidrógeno, -alquilo Ci-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
p es 0, 1, 2, 3 o 4, según lo permita la valencia;
en donde las variables restantes son como se definen en una cualquiera de las realizaciones primera a vigésima octava. En una trigésima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima novena como se describe anteriormente, en donde el heteroarilo monocíclico de 5 o 6 miembros representado por R2 se selecciona de uno de los siguientes:

en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima novena.
En una trigésima primera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a trigésima como se describe anteriormente, en donde R2 se selecciona de uno de los siguientes:

y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima trigésima.
En una trigésima segunda realización de la invención, se proporciona un método según una cualquiera de las realizaciones vigésima séptima a trigésima primera como se describe anteriormente, en donde cada caso de Rp es independientemente hidrógeno, halógeno, alquilo C1-4 opcionalmente sustituido, -CN,- NO2, -N3, -ORo4, -N(Rn2)2, -C(=O)N(Rn2)2, - C(=O)Rc3, o -C(=O)ORo4; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones vigésima octava a trigésima primera.
En una trigésima tercera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimosexta y vigésima séptima a trigésima segunda como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (III):

o una de sus sales farmacéuticamente aceptables, en donde las variables son como se definen en una cualquiera de las realizaciones primera a decimosexta y vigésima séptima a trigésima segunda.
En una trigésima cuarta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda como se describe anteriormente, en el donde compuesto es un compuesto de fórmula (IV):

o una de sus sales farmacéuticamente aceptables, en donde las variables son como se definen en una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda.
En una trigésima quinta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (V-a):

o una de sus sales farmacéuticamente aceptables, en donde m es 0, 1 o 2; y el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda.
En una trigésima sexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (V-h):

o una de sus sales farmacéuticamente aceptables, en donde m es 0, 1 o 2; Rnc es independientemente hidrógeno, -alquilo Ci-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda. En una trigésima séptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a decimosexta y vigésima quinta a trigésima segunda como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (VT):

o una de sus sales farmacéuticamente aceptables, en donde Rn7 es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a decimosexta y vigésima quinta a trigésima segunda.
En una trigésima octava realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda como se describe anteriormente, en donde el compuesto es un compuesto de Fórmula (IX):

o una de sus sales farmacéuticamente aceptables, en donde Rnc es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a vigésima cuarta y vigésima séptima a trigésima segunda.
En una trigésima novena realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a trigésima octava como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (II'):
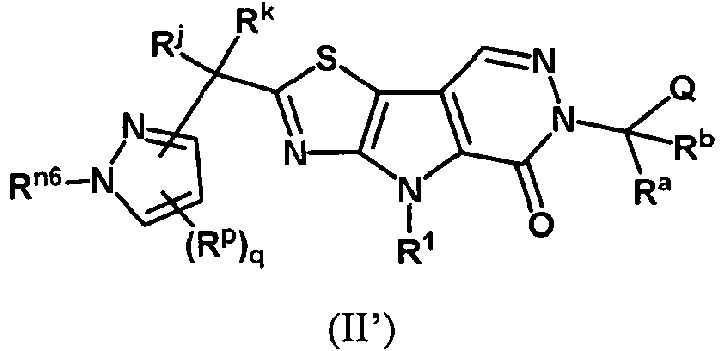
o una de sus sales farmacéuticamente aceptables, en donde Rn6 es hidrógeno, un -alquilo C1-C6 opcionalmente sustituido,
o un grupo protector de nitrógeno; q es 0, 1,2 o 3; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a trigésima octava. Alternativamente, las variables son como se describen en cualquiera de las realizaciones decimoséptima, decimoctava, vigésima novena, trigésima y trigésima primera.
En una cuadragésima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a trigésima octava como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (NI'):

o una de sus sales farmacéuticamente aceptable, en donde q es 0, 1,2o 3; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a trigésima octava.
En una cuadragésima primera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a trigésima octava como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (IV'):

o una de sus sales farmacéuticamente aceptables, en donde Rn6 es hidrógeno, un -alquilo Ci-C6 opcionalmente sustituido, o un grupo protector de nitrógeno; q es 0, 1, 2 o 3; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a trigésima octava.
En una cuadragésima segunda realización de la invención, se proporciona un método de acuerdo con una cualquiera de las realizaciones primera a trigésima octava como se describe anteriormente, en donde el compuesto es un compuesto de fórmula (V'):

o una de sus sales farmacéuticamente aceptable, en donde q es 0, 1,2o 3; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a trigésima octava.
En una cuadragésima tercera realización de la invención, se proporciona un método según una cualquiera de las realizaciones trigésima novena a cuadragésima segunda como se describe anteriormente, en donde Rn6 es hidrógeno o un -alquilo C1-4; y en donde el resto de variables son como se definen en una cualquiera de las realizaciones trigésima novena a cuadragésima segunda.
En una cuadragésima cuarta realización de la invención, se proporciona un método según una cualquiera de las realizaciones trigésima novena a cuadragésima segunda como se describe anteriormente, en donde cada caso de Rp es independientemente hidrógeno, halógeno, alquilo C1-C4 opcionalmente sustituido, -CN, -NO2, -N3, -ORo4, -N(Rn2)2, -C(=O)N(Rn2)2, -C(=O)Rc3 o -C(=O)ORo4; y en donde el resto de variables son como se definen en una cualquiera de las realizaciones trigésima novena a cuadragésima segunda.
En una cuadragésima quinta realización de la invención, se proporciona un método según una cualquiera de las realizaciones vigésima sexta a cuadragésima segunda como se describe anteriormente, en donde Rna es hidrógeno o alquilo C1-4; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones vigésima sexta a cuadragésima segunda.
En una cuadragésima sexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones vigésima a cuadragésima quinta como se describe anteriormente, en donde cada caso de Rn es independientemente hidrógeno, halógeno, alquilo C1-C4 opcionalmente sustituido, -CN, -NO2, -N3, -ORo4, -N(Rn2)2, -C(=O)N(Rn2)2, -C(=O)Rc3 o -C(=O)ORo4; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones vigésima a cuadragésimo quinta.
En una cuadragésima séptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a cuadragésima sexta como se describe anteriormente, en donde R1 es hidrógeno o un -alquilo C1-C4; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones primera a cuadragésima sexta.
En una cuadragésima octava realización de la invención, se proporciona un método según una cualquiera de las realizaciones decimosexta a cuadragésima séptima como se describe anteriormente, en donde Rj y Rk son cada uno independientemente hidrógeno, un halógeno, -ORo7 o un -alquilo C1-C4; o Rj y Rk se unen entre sí para formar =O; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones decimosexta a cuadragésimo séptima.
En una cuadragésima novena realización de la invención, se proporciona un método según una cualquiera de las realizaciones decimosexta a cuadragésima octava como se describe anteriormente, en donde Rj y Rk son cada uno hidrógeno; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones decimosexta a cuadragésimo octava.
En una quincuagésima realización de la invención, se proporciona un método según una cualquiera de las realizaciones decimocuarta a cuadragésima novena como se describe anteriormente, en donde Ra y Rb son cada uno hidrógeno; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones decimocuarta a cuadragésima novena.
En una quincuagésima primera realización de la invención, se proporciona un método según una cualquiera de las realizaciones trigésima quinta a quincuagésima como se describe anteriormente, en donde q es 0 o 1; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones trigésima quinta a quincuagésima.
En una quincuagésima segunda realización de la invención, se proporciona un método según una cualquiera de las realizaciones vigésima a quincuagésima primera como se describe anteriormente, en donde n es 0 o 1; y en donde el resto de las variables son como se definen en una cualquiera de las realizaciones vigésima a quincuagésima primera.
En una quincuagésima tercera realización de la invención, se proporciona un método según una cualquiera de las realizaciones primera a quincuagésima segunda como se describe anteriormente, en donde el compuesto es un compuesto seleccionado de la Tabla 1 o una de sus sales farmacéuticamente aceptables.
En una quincuagésima cuarta realización de la invención, se proporciona un método para modular la actividad de la piruvato quinasa M2 (PKM2), que comprende poner en contacto la PKM2 con una cantidad eficaz de un compuesto como se describe en una cualquiera de las realizaciones primera a quincuagésima tercera anteriores, o una de sus sales farmacéuticamente aceptables.
En una quincuagésima quinta realización de la invención, se proporciona un método para inhibir la proliferación de una célula que expresa la piruvato quinasa M2 (PKM2), que comprende poner en contacto la célula con una cantidad eficaz de un compuesto como se describe en una cualquiera de las realizaciones primera a quincuagésima tercera anteriores, o una de sus sales farmacéuticamente aceptables.
En una quincuagésima sexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta y quincuagésima quinta como se describe anteriormente, en donde el método es un método ex vivo.
En una quincuagésima séptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta y quincuagésima quinta como se describe anteriormente, en donde el método es un método in vitro.
En una quincuagésima octava realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta a quincuagésima séptima, en donde el método inhibe la actividad de la PKM2.
En una quincuagésima novena realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta a quincuagésima séptima, en donde el método activa la actividad de p KM2.
En una sexagésima realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta a quincuagésima novena, en donde la PKM2 se expresa en una célula que no es un glóbulo rojo. En una sexagésima primera realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima cuarta a sexagésima, en donde la célula se deriva o se obtiene de un sujeto que padece o es susceptible de padecer una enfermedad o trastorno asociado con la función de la PKM2.
En una sexagésima segunda realización de la invención, se proporciona un método según la sexagésima primera realización, en donde la enfermedad o trastorno está asociado con una actividad aberrante de la PKM2.
En una sexagésima tercera realización de la invención, se proporciona un método según la sexagésima primera realización, en donde la enfermedad o trastorno es cáncer, obesidad, una enfermedad diabética (p. ej., diabetes, nefropatía diabética (DN)), aterosclerosis, reestenosis, enfermedad arterial coronaria (CAD), síndrome de Bloom (BS), hiperplasia prostática benigna (HPB) o una enfermedad autoinmunitaria.
En una sexagésima cuarta realización de la invención, se proporciona un método según la sexagésima tercera realización, en donde la enfermedad diabética es nefropatía diabética.
En una sexagésima quinta realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima quinta a sexagésima cuarta, en donde la célula es una célula que sobreexpresa la PKM2. En una sexagésima sexta realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima quinta a sexagésima quinta, en donde la célula es una célula que expresa la PKM2 que tiene actividad aberrante.
En una sexagésima séptima realización de la invención, se proporciona un método según una cualquiera de las realizaciones quincuagésima quinta a sexagésima sexta, en donde la célula es una célula cancerosa, una célula pancreática, una célula hepática, una célula nerviosa o una célula renal.
En ciertas realizaciones de los compuestos de fórmulas (I)-(IX), (I')-(V') o sus sales farmacéuticamente aceptables, R2 y Q son cada uno independientemente heteroarilo monocíclico de 5 o 6 miembros opcionalmente sustituido. En ciertas realizaciones de los compuestos de fórmulas (I)-(IX), (I')-(V') o sus sales farmacéuticamente aceptables, R2 y Q son ambos heteroarilo monocíclico de 5 o 6 miembros opcionalmente sustituido; y L1 y L2 son cada uno independientemente alquileno C1-4 opcionalmente sustituido. En ciertas realizaciones, R2 y Q son iguales. En ciertas realizaciones, R2 y Q son diferentes.
En ciertas realizaciones, los compuestos descritos en el presente documento son útiles como activadores de PKM2 utilizados en los métodos y composiciones descritos en el presente documento y funcionan mediante o tienen uno o más de los siguientes mecanismos o propiedades:
a. es un activador alostérico de PKM2;
b. modula (p. ej., estabiliza) la unión de FBP en un bolsillo de unión de PKM2;
c. modula (p. ej., promueve) la liberación de FBP desde un bolsillo de unión de PKM2;
d. es un modulador (p. ej., un agonista), p. ej., un análogo, de FBP, p. ej., un agonista que se une a PKM2 con una afinidad menor, aproximadamente igual o mayor que la FBP;
e. modula (p. ej., promueve) la disolución de la PKM2 tetramérica;
f. modula (p. ej., promueve) el ensamblaje de la PKM2 tetramérica;
g. modula (p. ej., estabiliza) la conformación tetramérica de la PKM2;
h. modula (p. ej., promueve) la unión de un polipéptido que contiene fosfotirosina a la PKM2;
i. modula (p. ej., promueve) la capacidad de un polipéptido que contiene fosfotirosina para inducir la liberación de FBP de PKM2, p. ej., al inducir un cambio en la conformación de la PKM2, p. ej., en la posición de la Lys 433, lo que dificulta la liberación de FBP;
j. modula la propensión de la PKM2 a sufrir modificaciones postraduccionales (p. ej., oxidación en Cys358 o acetilación en Lys305) que afectan a la actividad de la enzima.
k. se une o cambia la posición de la Lys 433 en relación con el bolsillo de unión de FBP;
l. modula selectivamente (p. ej., activa) la PKM2 frente al menos otra isoforma de PK, p. ej., es selectivo para PKM2 frente a uno o más de PKR, Pk M1 o PKL;
m. tiene una afinidad por la PKM2 que es mayor que su afinidad por al menos otra isoforma de PK, p. ej., PKR, PKM1 o PKL.
Se puede ensayar un compuesto descrito en el presente documento en cuanto a su capacidad para activar la PKM2. Para simplificar, la actividad de activación de estos compuestos se representa como una AC50 en la Tabla 2. En las Tablas 2, un compuesto descrito en el presente documento puede tener una AC50 de PKM2 de tipo natural. "A" se refiere a una AC50 inferior a 0,300 ^M; "B" se refiere a una AC50 de 0,301 |jM a 0,800 ^M, y "C" se refiere a una AC50 mayor que 0,800 |jM.
Tabla 1. Compuestos de ejemplo como moduladores de PKM2






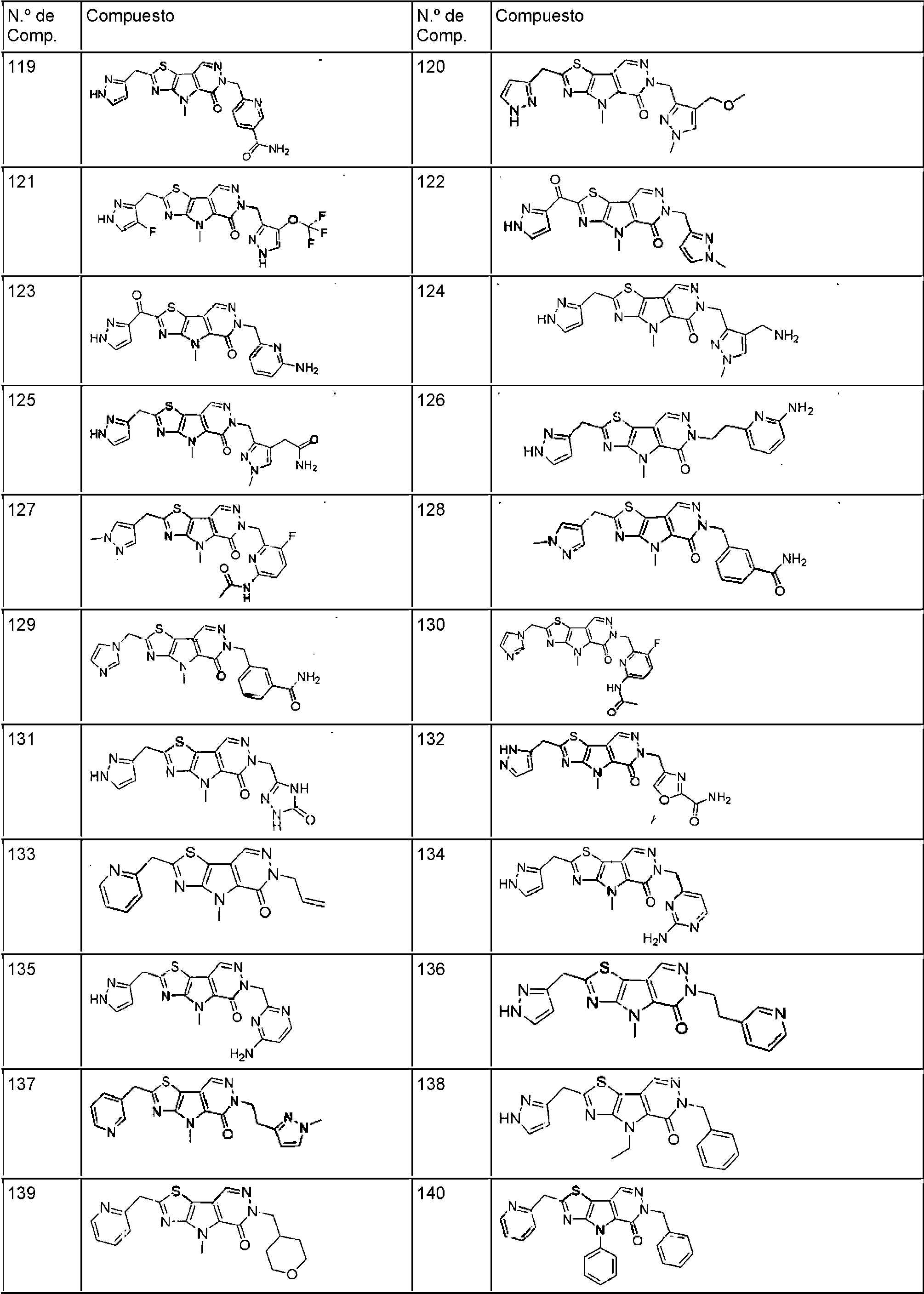

Tabla 2: AC50 de compuestos de ejemplo para PKM2 de tipo natural



En ciertas realizaciones, el compuesto de fórmulas (I)-(IX), (I')-(V') se selecciona de uno cualquiera de los compuestos expuestos en la Tabla 1 y en los ejemplos. En ciertas realizaciones, el compuesto de fórmulas (I)-(IX), (I')-(V') tiene la fórmula de una cualquiera de las siguientes:


en donde Rna, Rnc, Ro2y Ro6 son como se definen en el presente documento, y Rox es hidrógeno o un grupo protector de oxígeno. En ciertas realizaciones, Rna es un grupo protector de nitrógeno (p. ej., SEM o BOC). En ciertas realizaciones, Rnc es un grupo protector de nitrógeno (p. ej., SEM o BOC). En ciertas realizaciones, Ro2 es un grupo protector de oxígeno
(p. ej., THP). En ciertas realizaciones, Ro6 es un grupo protector de oxígeno (p. ej., TBS). En ciertas realizaciones, Rox es un grupo protector de oxígeno (p. ej., THP).
Los compuestos descritos en el presente documento se pueden preparar usando una variedad de técnicas sintéticas como se exponen en los ejemplos. Las transformaciones químicas sintéticas y las metodologías de grupos protectores (protección y desprotección) útiles para sintetizar los compuestos descritos en el presente documento son conocidas en la técnica e incluyen, por ejemplo, las que se describen en R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T. W. Greene y P.G.M. Wuts, Protective Groups in Organic Synthesis, 2a Ed., John Wiley and Sons (1991)); L. Fieser y M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994)); y L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995)), y ediciones posteriores de los mismos.
Ciertos compuestos activadores útiles como activadores mutantes y/o de tipo natural de PKM2 son aquellos que demuestran especificidad y activación de la enzima PKM2 (enzima de tipo natural y/o mutante) en ausencia de FBP en un nivel superior al de 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 99 o 100% en presencia de FBP.
En algunas realizaciones, los compuestos de fórmula (I) se pueden preparar utilizando métodos ilustrados en el Esquema 1. El tiazolil aldehído de fórmula S1 reacciona con azidoacetato de etilo en condiciones de adición nucleófila (p. ej., una base) en un disolvente apropiado (p. ej., etanol) para dar compuestos intermedios de fórmula S2. El grupo hidroxilo de la fórmula S2 puede convertirse en un grupo saliente y ser sometido a eliminación para dar la fórmula S3. La ciclación y posterior funcionalización del grupo amino proporciona el compuesto bicíclico de fórmula S5, que sufre desplazamiento nucleófilo con metanotiolato de sodio, seguido de oxidación para dar la fórmula S7. La ciclación adicional de la fórmula S7 en presencia de hidracina, seguida de desplazamiento nucleófilo con LG1-CH2-Q1 en presencia de una base proporciona compuestos intermedios de fórmula S9. El grupo de azufre en la fórmula S9 se puede oxidar a sulfinilo o sulfonilo para proporcionar la fórmula S10 o S11, que es un sustrato para un desplazamiento nucleófilo adicional para generar una fórmula general S12. Como se usa en el presente documento, X1 es un grupo saliente como se define en el presente documento. En ciertas realizaciones, X1 es halógeno, alcanosulfoniloxi, arenosulfoniloxi, diazonio, alquil diazenos, aril diazenos, alquil triazenos, aril triazenos, nitro, nitrato de alquilo, nitrato de arilo, fosfato de alquilo, fosfato de arilo, alquilcarboniloxi, arilcarboniloxi, alcoxcarboniloxi, arioxcarboniloxi amoníaco, alquilaminas, arilaminas, grupo hidroxilo, grupo alquiloxi, grupo ariloxi; LG1 es un grupo saliente como se define en el presente documento; Q1 es cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido; y Nu1 es un nucleófilo como se define en el presente documento. Nu1 del compuesto de fórmula S12 se puede convertir adicionalmente en otros grupos funcionales con transformaciones químicas convencionales. R1 es como se define en la primera realización.

En algunas realizaciones, los compuestos de fórmula (I) se pueden preparar utilizando los métodos que se muestran en el esquema 2. De forma similar al esquema 1, la fórmula S21 se puede preparar a partir del tiazol-aldehído de fórmula
S13. La halogenación de la fórmula S21 da la fórmula S22, que puede experimentar una reacción orgánica de acoplamiento con un alquilmetal, alquenilmetal, alquinilmetal, arilmetal, heteroarilmetal, heterociclilmetal o cicloalquilmetal para dar un compuesto de fórmula S23. Como se usa en el presente documento X3 es un halógeno; R1 es como se define en la primera realización de la invención; LG2 es un grupo saliente como se define en el presente documento; Q2 es cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido; M1 es un metal (p. ej., Li, Na, K, Mg, Zn, Sn, B, Pd, Si, Cu, etc.), X4 es halógeno o ésterde ácido alquilsulfónico o un éster de ácido arilsulfónico; Rr1 es alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido. En ciertas realizaciones, la reacción orgánica de acoplamiento es la reacción de Negishi; X3 es I; y M1 es Zn.
Los compuestos de fórmula S22 y S23 son compuestos intermediarios útiles para introducir más grupos funcionales en la posición X3 y/o Rr1 (Esquema 3). En ciertas realizaciones, el compuesto de fórmula 23-i puede oxidarse adicionalmente para formar la fórmula S24. La adición nucleófila de S24 con un nucleófilo apropiado genera un compuesto de S25. En otra realización, los compuestos de fórmula S22 pueden acoplarse con vinil-metal para introducir el grupo vinilo en el anillo de tiazol. La oxidación del grupo vinilo seguida de la adición nucleófila proporciona un compuesto de fórmula S28. Como se usa en el presente documento, Nu2 es un nucleófilo.

Como se usa en el presente documento, Rr2 es alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido. Nu1 y Q2 es como se define en el esquema 2.
Como se usa en el presente documento, un nucleófilo es una especie química que dona un par de electrones a un electrófilo para formar un enlace químico en relación con una reacción. Todas las moléculas o iones con un par de electrones libres o al menos un enlace pi pueden actuar como nucleófilos. Los nucleófilos de ejemplo comprenden al menos un grupo que posee función nucleófila, por ejemplo, un carbono alfa (p. ej., el carbono adyacente a grupo carbonilo, sulfonilo, sulfinilo, arilo o heteroarilo), un grupo tiol, un grupo hidroxilo, un grupo amina primaria, un grupo amina secundaria, un haluro, cianuro, azida, alcóxido, metal orgánico o base inorgánica.
En algunas realizaciones, los compuestos de fórmula (I) se pueden preparar utilizando los métodos que se muestran en el esquema 4. El desplazamiento nucleófilo de las fórmulas S30 con una amina cíclica secundaria proporciona las fórmulas S31. Las reacciones de organoacoplamiento (p. ej., acoplamiento de Suzuki, acoplamiento de Pd, etc.) del compuesto S32 proporcionan un compuesto de fórmulas S33(i)-(iii). Además, el grupo sulfinilo de fórmula S34 se puede funcionalizar con carbamato de amonio para dar iminosulfanona de fórmula S35.

Como se usa en el presente documento,

representa el anillo A con un nitrógeno como átomo del anillo.
Como se usa en el presente documento,

representa el anillo B con el punto de unión en el átomo de carbono del anillo.
R1 es como se define en la primera realización. Cada caso de R35, R36y R37 es independientemente hidrógeno, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, heterociclilo opcionalmente sustituido o cicloalquilo opcionalmente sustituido.
X4 es halógeno o -OTf. M4 es un metal orgánico con ligandos apropiados si es necesario (orgánicos o inorgánicos) según lo permita la valencia. El M4 de ejemplo incluye, pero no se limita a Li, Sn, B orgánico (p. ej., ácidos borónicos y ésteres borónicos), Zn, Mg, Si, Pd y Cu.
Métodos de tratamiento
En una realización, se proporciona un método para tratar una enfermedad, afección o trastorno como se describe en el presente documento (p. ej., tratar) que comprende administrar un compuesto, una sal farmacéuticamente aceptable de un compuesto o composición farmacéutica que comprende un compuesto descrito en el presente documento (p. ej., un compuesto de fórmulas (I)-(IX), (I')-(V'), en los ejemplos, en la Tabla 1 y en las Figuras 1A-1C, 2A-2C, 3).
Los compuestos y composiciones descritos en el presente documento pueden administrarse a células en cultivo, p. ej. in vitro o ex vivo, o a un sujeto, p. ej., in vivo, para tratar y/o diagnosticar una variedad de trastornos, incluidos los que se describen a continuación en el presente documento.
Enfermedad proliferativa
En algunas realizaciones, se proporciona un método para tratar una enfermedad proliferativa que comprende administrar a un sujeto un compuesto, una de sus sales farmacéuticamente aceptables o una composición farmacéutica del mismo, como se describe en el presente documento. Como se usa aquí, "enfermedad proliferativa" se refiere a una enfermedad que ocurre debido al crecimiento o extensión anormal por la multiplicación de células (Walker, Cambridge Dictionary of Biology; Prensa de la Universidad de Cambridge: Cambridge, Reino Unido, 1990). Una enfermedad proliferativa puede estar asociada con: 1) la proliferación patológica de células normalmente quiescentes; 2) la migración patológica de las células desde su ubicación normal (p. ej., metástasis de células neoplásicas); 3) la expresión patológica de enzimas proteolíticas tales como las metaloproteinasas de matriz (p. ej., colagenasas, gelatinasas y elastasas); o 4) la angiogénesis patológica tal como en la retinopatía proliferativa y la metástasis tumoral. Ejemplos de enfermedades proliferativas incluyen cánceres (es decir, "neoplasias malignas"), neoplasias benignas, angiogénesis, enfermedades inflamatorias y enfermedades autoinmunitarias. En ciertas realizaciones, la enfermedad proliferativa es cáncer. En ciertas realizaciones, la enfermedad proliferativa es una enfermedad autoinmunitaria.
Los términos "neoplasia" y "tumor" se utilizan en el presente documento de forma intercambiable y se refieren a una masa anormal de tejido en donde el crecimiento de la masa supera y no está coordinado con el crecimiento de un tejido normal. Una neoplasia o tumor puede ser "benigno" o "maligno", dependiendo de las siguientes características: grado de diferenciación celular (incluyendo morfología y funcionalidad), tasa de crecimiento, invasión local y metástasis. Una "neoplasia benigna" generalmente está bien diferenciada, tiene un crecimiento característicamente más lento que una neoplasia maligna y permanece localizada en el sitio de origen. Además, una neoplasia benigna no tiene la capacidad de infiltrar, invadir o metastatizar a sitios distantes. Los ejemplos de neoplasias benignas incluyen, pero no se limitan a, lipoma, condroma, adenomas, acrocordón, angiomas seniles, queratosis seborreica, lentigos e hiperplasias sebáceas. En algunos casos, ciertos tumores "benignos" pueden dar lugar posteriormente a neoplasias malignas, que pueden resultar de cambios genéticos adicionales en una subpoblación de células neoplásicas del tumor, y estos tumores se denominan "neoplasias premalignas". Un ejemplo de neoplasia premaligna es un teratoma. Por el contrario, una "neoplasia maligna" generalmente se diferencia mal (anaplasia) y tiene un crecimiento rápido característico acompañado de infiltración, invasión y destrucción progresivas del tejido circundante. Además, una neoplasia maligna generalmente tiene la capacidad de metastatizar a sitios distantes. El término "metástasis", "metastásico" o "metastatizar" se refiere a la propagación o migración de células cancerosas desde un tumor primario u original a otro órgano o tejido y, por lo general, se identifica por la presencia de un "tumor secundario" o "masa celular secundaria" del tipo de tejido del tumor primario u original y no del órgano o tejido en donde se localiza el tumor secundario (metastásico). Por ejemplo, se dice que un cáncer de próstata que ha migrado al hueso es cáncer de próstata metastásico e incluye células cancerosas de cáncer de próstata que crecen en el tejido óseo.
El término "cáncer" se refiere a una clase de enfermedades caracterizadas por el desarrollo de células anormales que proliferan sin control y tienen la capacidad de infiltrarse y destruir tejidos corporales normales. Véase, p. ej. , Stedman's Medical Dictionary , 25a ed.; Hensyl ed.; Williams & Wilkins: Filadelfia, 1990. Los cánceres de ejemplo incluyen tumores sólidos, tumores de tejidos blandos y metástasis de los mismos. Los métodos descritos también son útiles en el tratamiento de cánceres no sólidos. Tumores sólidos de ejemplo incluyen malignidades (p. ej., sarcomas, adenocarcinomas y carcinomas) de los diversos sistemas de órganos, tales como los de pulmón, mama, linfoide, tractos gastrointestinal (p. ej., colon) y genitourinario (p. ej., tumores renales, uroteliales o testiculares), faringe, próstata y ovario. Los adenocarcinomas de ejemplo incluyen cánceres colorrectales, carcinoma de células renales, cáncer de hígado, carcinoma de pulmón no microcítico y cáncer de intestino delgado. Otros cánceres de ejemplo incluyen: leucemia linfoblástica aguda, adulto; leucemia linfoblástica aguda infantil; leucemia mieloide aguda, adulto; carcinoma corticosuprarrenal; carcinoma corticosuprarrenal, Infantil; linfoma relacionado con el SIDA; neoplasias malignas relacionadas con el SIDA; cáncer de ano; astrocitoma cerebeloso infantil; astrocitoma cerebral infantil; cáncer de las vías biliares, extrahepático; cáncer de vejiga; cáncer de vejiga, infantil; cáncer de hueso, osteosarcoma/histiocitoma fibroso maligno; glioma de tronco encefálico, infantil; tumor cerebral, adulto; tumor cerebral, glioma de tronco cerebral, infantil; tumor cerebral, astrocitoma cerebeloso, infantil; tumor cerebral, astrocitoma cerebral/glioma maligno, infantil; tumor cerebral, ependimoma, infantil; tumor cerebral, meduloblastoma, infantil; tumor cerebral, tumores neuroectodérmicos primitivos supratentoriales, infantiles; tumor cerebral, glioma vía óptica e hipotalámico, infantil; tumor cerebral, infantil (otro); cáncer de mama; cáncer de mama y embarazo; cáncer de mama, infantil; cáncer de mama, masculino; adenomas/carcinoides bronquiales, infantil; tumor carcinoide, infantil; tumor carcinoide, gastrointestinal; carcinoma, corticosuprarrenal; carcinoma, células de los islotes; carcinoma de primario desconocido; linfoma del sistema nervioso central, primario; astrocitoma cerebeloso, infantil; astrocitoma cerebral/glioma maligno, infantil; cáncer de cuello uterino; cánceres infantiles; leucemia linfocítica crónica; leucemia mielógena crónica; trastornos mieloproliferativos crónicos; sarcoma de células claras de las vainas tendinosas; cáncer de colon; cáncer colorrectal, infantil; linfoma cutáneo de células T; cáncer endometrial; ependimoma infantil; cáncer epitelial, ovario; cáncer de esófago; cáncer de esófago, infantil; familia de tumores de Ewing; tumor extracraneal de células germinales, infantil; tumor extragonadal de células germinales; cáncer de vías biliares extrahepáticas; cáncer de ojo, melanoma intraocular; cáncer de ojo, retinoblastoma; cáncer de vesícula biliar; cáncer gástrico (de estómago); cáncer gástrico (de estómago), infantil; tumor carcinoide gastrointestinal; tumor de células germinales, extracraneal, infantil; tumor de células germinales, extragonadal; tumor de células germinales, ovario; tumor trofoblástico gestacional; glioma, de tronco cerebral infantil; glioma, vía óptica e hipotalámico infantil; leucemia de células pilosas; cáncer de cabeza y cuello; cáncer hepatocelular (hígado), adulto (primario); cáncer hepatocelular (hígado), infantil (primario); linfoma de Hodgkin, adulto; Linfoma de Hodgkin, Infantil; linfoma de Hodgkin durante el embarazo; cáncer de hipofaringe; glioma hipotalámico y de la vía óptica, infantil; melanoma intraocular; carcinoma de células de los islotes (páncreas endocrino); sarcoma de Kaposi; cáncer de riñón; cáncer de laringe; cáncer de laringe, infantil; leucemia, linfoblástica aguda, adulto; leucemia,
linfoblástica aguda, infantil; leucemia, mieloide aguda, adulto; leucemia, mieloide aguda, infantil; leucemia, linfocítica crónica; leucemia, mielógena crónica; leucemia, de células pilosas; cáncer de labio y cavidad oral; cáncer de hígado, adulto (primario); cáncer de hígado, infantil (primario); cáncer de pulmón no microcítico; cáncer de pulmón microcítico; leucemia linfoblástica, aguda del adulto; leucemia linfoblástica, aguda infantil; leucemia linfocítica, crónica; linfoma, relacionado con el sida; linfoma, del sistema nervioso central (primario); linfoma, cutáneo de células t; linfoma, de hodgkin, adulto; linfoma, de hodgkin, infantil; linfoma, de hodgkin durante el embarazo; linfoma, no hodgkin, adulto; linfoma, no hodgkin, infantil; linfoma, no hodgkin durante el embarazo; linfoma, primario del sistema nervioso central; macroglobulinemia, enfermedad de waldenstrom; cáncer de mama masculino; mesotelioma maligno, adulto; mesotelioma maligno, infantil; timoma maligno; meduloblastoma, infantil; melanoma; melanoma, intraocular; carcinoma de células de Merkel; mesotelioma, maligno; cáncer de cuello escamoso metastásico con tumor primario oculto; síndrome de neoplasia endocrina múltiple, infantil; mieloma múltiple/neoplasia de células plasmáticas; micosis fungoide; síndromes mielodisplásicos; leucemia mielógena, crónica; leucemia mieloide, aguda infantil; mieloma múltiple; trastornos mieloproliferativos, crónicos; cáncer de cavidad nasal y senos paranasales; cáncer de nasofaringe; cáncer nasofaríngeo, infantil; neuroblastoma; linfoma no hodgkin, adulto; linfoma no hodgkin, infantil; linfoma no hodgkin durante el embarazo; cáncer de pulmón no microcítico; cáncer bucal, infantil; cáncer de cavidad oral y labio; cáncer orofaríngeo; osteosarcoma/histiocitoma fibroso maligno de hueso; cáncer de ovario, infantil; cáncer epitelial de ovario; tumor de células germinales, de ovario; tumor de ovario de bajo potencial maligno; cáncer de páncreas; cáncer de páncreas, infantil; cáncer de páncreas, de células de los islotes; cáncer de seno paranasal y cavidad nasal; cáncer de paratiroides; cáncer de pene; feocromocitoma; tumores neuroectodérmicos primitivos pineales y supratentoriales, infantil; tumor pituitario; neoplasia de células plasmáticas/mieloma múltiple; blastoma pleuropulmonar; embarazo y cáncer de mama; embarazo y linfoma de hodgkin; embarazo y linfoma no hodgkin; linfoma primario del sistema nervioso central; cáncer primario de hígado, adultos; cáncer primario de hígado, infantil; cáncer de próstata; cáncer de recto; cáncer de células renales (riñón); cáncer de células renales, infantil; pelvis renal y uréter, cáncer de células de transición; retinoblastoma; rabdomiosarcoma, infantil; cáncer de glándulas salivales; cáncer de glándulas salivales, infantil; sarcoma, familia de tumores de Ewing; sarcoma de Kaposi; sarcoma (osteosarcoma)/histiocitoma fibroso maligno de hueso; sarcoma, rabdomiosarcoma, infantil; sarcoma de tejido blando, adulto; sarcoma de tejido blando, infantil; síndrome de Sézary; cáncer de piel; cáncer de piel, infantil; cáncer de piel (melanoma); carcinoma de piel, células de merkel; cáncer de pulmón microcítico; cáncer de intestino delgado; sarcoma de tejido blando, adulto; sarcoma de tejido blando, infantil; cáncer escamoso de cuello con tumor primario oculto, metastásico; cáncer de estómago (gástrico); cáncer de estómago (gástrico), infantil; tumores neuroectodérmicos primitivos supratentoriales, infantil; linfoma cutáneo de células T; cáncer testicular; timoma, infantil; timoma, maligno; cáncer de tiroides; cáncer de tiroides, infantil; cáncer de células de transición de la pelvis renal y uréter; tumor trofoblástico, gestacional; sitio primario desconocido, cáncer de, infantil; cánceres inusuales de la infancia; uréter y pelvis renal, cáncer de células de transición; cáncer de uretra; sarcoma uterino; cáncer de vagina; glioma de vía óptica e hipotalámico, infantil; cáncer de vulva; macroglobulinemia de Waldenstrom; y tumor de Wilms. Las metástasis de los cánceres antes mencionados también se pueden tratar o prevenir de acuerdo con los métodos descritos en el presente documento.
Terapias combinadas contra el cáncer
En algunas realizaciones, el método proporcionado comprende además administrar uno o más tratamientos adicionales contra el cáncer. Los tratamientos para el cáncer de ejemplo incluyen, por ejemplo: quimioterapia, terapias dirigidas tales como terapias con anticuerpos, inmunoterapia y terapia hormonal. A continuación se proporcionan ejemplos de cada uno de estos tratamientos.
En algunas realizaciones, un compuesto descrito en el presente documento se administra con una o más quimioterapias. La quimioterapia es el tratamiento del cáncer con fármacos que pueden destruir las células cancerosas. "Quimioterapia" generalmente se refiere a fármacos citotóxicos que afectan a las células que se dividen rápidamente en general, en contraste con la terapia dirigida. Los fármacos de quimioterapia interfieren con la división celular de varias formas posibles, p. ej., con la duplicación del ADN o la separación de los cromosomas recién formados. La mayoría de las formas de quimioterapia se dirigen a todas las células que se dividen rápidamente y no son específicas para las células cancerosas, aunque cierto grado de especificidad puede provenir de la incapacidad de muchas células cancerosas para reparar el daño del ADN, mientras que las células normales generalmente pueden hacerlo.
Los ejemplos de agentes quimioterapéuticos utilizados en la terapia del cáncer incluyen, por ejemplo, antimetabolitos (p. ej., ácido fólico, purina y derivados de pirimidina) y agentes alquilantes (p. ej., mostazas nitrogenadas, nitrosoureas, platino, sulfonatos de alquilo, hidrazinas, triacenos, aziridinas, veneno del huso, agentes citotóxicos, inhibidores de toposimerasa y otros). Los ejemplos de agentes incluyen aclarubicina, actinomicina, alitretinon, altretamina, aminopterina, ácido aminolevulínico, amrubicina, amsacrina, anagrelida, trióxido de arsénico, asparaginasa, atrasentán, belotecán, bexaroteno, endamustina, bleomicina, bortezomib, busulfano, camptotecina, capecitabina, carboplatino, carbocuona, carmofur, carmustina, celecoxib, clorambucilo, clormetina, cisplatino, cladribina, clofarabina, crisantaspasa, ciclofosfamida, citarabina, dacarbazina, dactinomicina, daunorubicina, decitabina, demecolcina, docetaxel, doxorubicina, efaproxiral, elesclomol, elsamitrucina, enocitabina, epirubicina, estramustina, etoglúcido, etopósido, floxuridina, fludarabina, fluorouracilo (5FU), fotemustina, gemcitabina, implantes de gliadel, hidroxicarbamida, hidroxiurea, idarubicina, ifosfamida, irinotecán, irofulven, ixabepilona, larotaxel, leucovorina, doxorubicina liposomal, daunorubicina liposomal, lonidamina, lomustina, lucantona, manosulfano, masoprocol, melfalán, mercaptopurina, mesna, metotrexato, metil amino levulinato, mitobronitol, mitoguazona, mitotano, mitomicina, mitoxantrona, nedaplatino, nimustina, olimersen, omacetaxina, ortataxel, oxaliplatino, paclitaxel, pegaspargasa, pemetrexed, pentostatina, pirarubicina, pixantrona,
plicamicina, porfímero sódico, prednimustina, procarbazina, raltitrexed; ranimustina, rubitecán, sapacitabina, semustina, sitimagene ceradenovec, satraplatino, estreptozocina, talaporfina, tegafur-uracilo, temoporfina, temozolomida, tenipósido, tesetaxel, testolactona, tetranitrato, tiotepa, tiazofurina, tioguanina, tipifarnib, topotecán, trabectedina, triazicuona, trietilenmelamina, triplatino, tretinoína, treosulfano, trofosfamida, uramustina, valrubicina, verteporfina, vinblastina, vincristina, vindesina, vinflunina, vinorelbina, vorinostat, zorubicina y otros agentes citostáticos o citotóxicos descritos en el presente documento.
En algunas realizaciones, un compuesto descrito en el presente documento se administra con una o más terapias dirigidas. La terapia dirigida constituye el uso de agentes específicos para las proteínas desreguladas de las células cancerosas. Los fármacos de terapia dirigida de molécula pequeña son generalmente inhibidores de dominios enzimáticos en proteínas mutadas, sobreexpresadas o críticas de otra forma dentro de la célula cancerosa. Ejemplos destacados son los inhibidores de la tirosina quinasa tales como axitinib, bosutinib, cediranib, dasatinib, erlotinib, imatinib, gefitinib, lapatinib, lestaurtinib, nilotinib, semaxanib, sorafenib, sunitinib y vandetanib, y también inhibidores de quinasas dependientes de ciclina tales como alvocidib y seliciclib. La terapia con anticuerpos monoclonales es otra estrategia en donde el agente terapéutico es un anticuerpo que se une específicamente a una proteína en la superficie de las células cancerosas. Los ejemplos incluyen el anticuerpo anti-HER2/neu trastuzumab (HERCEPTIN®) que se usa típicamente en el cáncer de mama, y el anticuerpo anti-CD20 rituximab y tositumomab usados típicamente en una variedad de tumores malignos de células B. Otros anticuerpos de ejemplo incluyen cetuximab, panitumumab, trastuzumab, alemtuzumab, bevacizumab, edrecolomab y gemtuzumab. Ejemplos de proteínas de fusión incluyen aflibercept y denileukin diftitox. En algunas realizaciones, la terapia dirigida se puede usar en combinación con un compuesto descrito en el presente documento.
La terapia dirigida también puede implicar péptidos pequeños como "dispositivos de asentamiento" que pueden unirse a los receptores de la superficie celular o a la matriz extracelular afectada que rodea el tumor. Los radionucleidos que se unen a estos péptidos (p. ej., RGD) finalmente matan a la célula cancerosa si el nucleido se descompone en las proximidades de la célula. Un ejemplo de tal terapia incluye BEXXAR®.
En algunas realizaciones, un compuesto descrito en el presente documento se administra con una o más inmunoterapias. La inmunoterapia para el cáncer se refiere a un conjunto diverso de estrategias terapéuticas diseñadas para inducir al propio sistema inmunitario del paciente a combatir el tumor. Los métodos contemporáneos para generar una respuesta inmunitaria contra los tumores incluyen la inmunoterapia con BCG intravesicular para el cáncer de vejiga superficial y el uso de interferones y otras citoquinas para inducir una respuesta inmunitaria en pacientes con carcinoma de células renales y melanoma.
El trasplante alogénico de células madre hematopoyéticas puede considerarse una forma de inmunoterapia, ya que las células inmunitarias del donante a menudo atacarán el tumor en un efecto de injerto contra tumor. En algunas realizaciones, los agentes de inmunoterapia se pueden usar en combinación con un compuesto descrito en el presente documento.
En algunas realizaciones, un compuesto descrito en el presente documento se administra con una o más terapias hormonales. El crecimiento de algunos tipos de cáncer se puede inhibir proporcionando o bloqueando ciertas hormonas. Los ejemplos comunes de tumores sensibles a las hormonas incluyen ciertos tipos de cáncer de mama y de próstata. Eliminar o bloquear el estrógeno o la testosterona suele ser un tratamiento adicional importante. En ciertos tipos de cáncer, la administración de agonistas hormonales, como los progestágenos, puede ser terapéuticamente beneficiosa. En algunas realizaciones, los agentes de terapia hormonal se pueden usar en combinación con un compuesto descrito en el presente documento.
Obesidad y trastornos de la grasa
En algunas realizaciones, se proporciona un método para tratar o prevenir la obesidad en un sujeto humano (p. ej., un niño o un adulto) mediante la administración al sujeto humano de una cantidad eficaz del compuesto, sal farmacéuticamente aceptable o composición farmacéutica del mismo como se describe en el presente documento. "Obesidad" se refiere a una afección en donde un sujeto tiene un índice de masa corporal mayor o igual a 30. Muchos compuestos descritos en el presente documento se pueden usar para tratar o prevenir una afección de sobrepeso. "Sobrepeso" se refiere a una afección en donde un sujeto tiene un índice de masa corporal mayor o igual a 25,0. El índice de masa corporal (IMC) y otras definiciones son según el "NIH Clinical Guidelines on the Identification and Evaluation, and Treatment of Overweight and Obesity in Adults" (1998). El tratamiento con el compuesto puede ser en una cantidad eficaz para alterar el peso del sujeto, p. ej., en al menos 2, 5, 7, 10, 12, 15, 20, 25, 30, 25, 40, 45, 50 o 55%. El tratamiento con un compuesto puede ser en una cantidad eficaz para reducir el índice de masa corporal del sujeto, p. ej., a menos de 30, 28, 27, 25, 22, 20 o 18. Los compuestos pueden usarse para tratar o prevenir el aumento de peso, tasa metabólica o depósito de grasa anómalos o inadecuados, p. ej., anorexia, bulimia, obesidad, diabetes o hiperlipidemia (p. ej., triglicéridos elevados y/o colesterol elevado), así como trastornos del metabolismo de grasas o lípidos.
Un compuesto o composición descrito en el presente documento se puede administrar para tratar la obesidad asociada con el síndrome de Prader-Willi (PWS). El PWS es un trastorno genético asociado con la obesidad (p. ej., obesidad mórbida).
Un compuesto o composición descrito en el presente documento se puede usar para reducir la grasa corporal, prevenir el aumento de la grasa corporal, reducir el colesterol (p. ej., colesterol total y/o proporciones de colesterol total a colesterol HDL) y/o reducir el apetito en personas que tienen obesidad asociada con PWS y/o reducir comorbilidades tales como diabetes, enfermedades cardiovasculares y accidentes cerebrovasculares.
Hiperglucemia
Los niveles elevados de glucosa inducen anomalías metabólicas en las rutas metabólicas de la glucosa e inducen disfunción mitocondrial. Esto también sobreproduce especies reactivas de oxígeno (ROS). La glucosa intracelular elevada conduce a la acumulación de los metabolitos tóxicos de la glucosa sorbitol, metilglioxal (MG) y diacilglicerol (DAG), que se ha propuesto que contribuyen a la complicación microvascular, p. ej., DN. Se descubrió que los activadores de PKM2 de molécula pequeña invierten la elevación inducida por la hiperglucemia en los metabolitos tóxicos de la glucosa y la disfunción mitocondrial (Nat Med. 2017, 23(6): 753-762; patente de EE. UU. N.° 9921221).
En ciertas realizaciones, en el presente documento se proporciona un método para tratar la hiperglucemia en un sujeto que comprende administrar una cantidad terapéuticamente eficaz del compuesto, sal farmacéuticamente aceptable o composición farmacéutica del mismo.
En ciertas realizaciones, en el presente documento se proporciona un método para tratar una enfermedad diabética en un sujeto que comprende administrar una cantidad terapéuticamente eficaz del compuesto, sal farmacéuticamente aceptable o composición farmacéutica del mismo. Una "enfermedad diabética", como se usa en el presente documento, se refiere a diabetes y prediabetes, así como a implicaciones diabéticas. La diabetes se refiere a un grupo de enfermedades metabólicas en las que una persona tiene niveles altos de azúcar en la sangre, ya sea porque el cuerpo no produce suficiente insulina o porque las células no responden a la insulina que se produce. Este nivel alto de azúcar en la sangre produce los síntomas clásicos de poliuria (micción frecuente), polidipsia (aumento de la sed) y polifagia (aumento del hambre). Hay varios tipos de diabetes. La diabetes tipo I resulta de la incapacidad del cuerpo para producir insulina y actualmente requiere que la persona se inyecte insulina o lleve una bomba de insulina. La diabetes tipo II resulta de la resistencia a la insulina, una afección en la que las células no pueden usar la insulina adecuadamente, a veces combinada con una deficiencia absoluta de insulina. La diabetes gestacional ocurre cuando las mujeres embarazadas sin un diagnóstico previo de diabetes desarrollan un nivel alto de glucosa en sangre. Otras formas de diabetes incluyen la diabetes congénita, que se debe a defectos genéticos de la secreción de insulina, la diabetes relacionada con la fibrosis quística, la diabetes esteroidea inducida por altas dosis de glucocorticoides y varias formas de diabetes monogénica, p. ej., diabetes de inicio maduro en los jóvenes (p. ej., MODY 1,2, 3, 4, 5, 6, 7, 8, 9 o 10). La prediabetes indica una afección que ocurre cuando los niveles de glucosa en la sangre de una persona son más altos de lo normal pero no lo suficientemente altos como para diagnosticar diabetes. Todas las formas de diabetes aumentan el riesgo de complicaciones a largo plazo. Por lo general, estas se desarrollan después de muchos años, pero pueden ser el primer síntoma en aquellos que no han recibido un diagnóstico antes de ese momento. Las principales complicaciones a largo plazo están relacionadas con el daño a los vasos sanguíneos. Las implicaciones diabéticas de ejemplo incluyen enfermedad cardiovascular, enfermedades macrovasculares tales como la cardiopatía isquémica (angina, infarto de miocardio), accidente cerebrovascular y enfermedad vascular periférica, complicaciones microvasculares (p. ej., daño a los vasos sanguíneos pequeños), retinopatía diabética (es decir, el impacto de la diabetes en la formación de vasos sanguíneos en la retina del ojo), nefropatía diabética (es decir, el impacto de la diabetes en los riñones), neuropatía diabética (p. ej., el impacto de diabetes en el sistema nervioso, que lo más comúnmente causa entumecimiento, hormigueo y dolor en los pies y también aumenta el riesgo de daño en la piel debido a la sensibilidad alterada), úlceras del pie diabético y síndrome X. En ciertas realizaciones, una "enfermedad diabética" incluye una o más seleccionadas de hiperglucemia, hiperinsulinemia, diabetes, resistencia a la insulina, alteración del metabolismo de la glucosa, afecciones de alteración de la tolerancia a la glucosa (IGT), afecciones de alteración de la glucosa plasmática en ayunas, retinopatía diabética, nefropatía diabética, glomeruloesclerosis, neuropatía diabética y síndrome X.
En ciertas realizaciones, el compuesto o la composición descritos en el presente documento se pueden usar para reducir las especies reactivas de oxígeno (ROS) y/o al menos uno de los metabolitos de la glucosa (p. ej., sorbitol, metilglioxal (MG) y diacilglicerol (DAG)) en un sujeto.
En ciertas realizaciones, el compuesto o la composición descritos en el presente documento se pueden usar para tratar una complicación microvascular.
En ciertas realizaciones, el compuesto o la composición descritos en el presente documento se pueden usar para tratar la DN. En determinadas realizaciones, el tratamiento de la DN puede incluir la disminución de cualquier síntoma asociado con la DN, incluidos, pero no limitados a, cambios en el apetito, cambios en el sueño, proteína en suero, debilidad y/o náuseas.
En ciertas realizaciones, el método comprende además administrar al sujeto una cantidad terapéuticamente eficaz de uno o más agentes secundarios que aumentan el nivel o la actividad de uno o más de los factores protectores de DN. Los factores protectores de la DN de ejemplo incluyen, pero no se limitan a, SOD1-superóxido dismutasa; TPI1-triosafosfato isomerasa isoforma 2; SORD-sorbitol deshidrogenasa; ALDOA-aldolasa A, fructosa-bisfosfato; GAPDH-gliceraldehído-3
fosfato deshidrogenasa; isoenzimas M1/M2 de PKM-piruvato quinasa; ENOI-alfa-enolasa; FGB-cadena beta de fibrinógeno; SELENBP1-Proteína 1 de unión a selenio; PEBP1-proteína 1 de unión a fosfatidiletanolamina; homólogo de CRYLI-Lambda-cristalino (patente de EE. UU. N.° 9921221). Un agente secundario puede aumentar el nivel o la actividad de un factor protector o disminuir el nivel o la actividad de un factor de riesgo en al menos 50%, 100% (1 vez), 11^ veces, 2 veces, 3 veces, 4 -veces, 5 veces, 10 veces, 15 veces, 20 veces o más. En ciertas realizaciones, el método proporcionado comprende llevar el nivel o actividad de un factor protector esencialmente a su nivel o actividad en un sujeto que está protegido del desarrollo de una complicación microvascular. "Esencialmente dentro de su nivel" se refiere a dentro de menos del 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% o 100% del valor de control. El agente secundario puede ser una molécula pequeña, una proteína que comprende el factor protector o una variante biológicamente activa (p. ej., un fragmento) del mismo, o un ácido nucleico que codifica una proteína que comprende el factor protector o una variante biológicamente activa (p. ej., un fragmento) del mismo.
Las variantes biológicamente activas de las proteínas de los factores protectores también incluyen formas inmaduras y maduras de longitud completa o fragmentos de las mismas que comprenden una secuencia de aminoácidos que difiere de la secuencia o fragmento natural de la misma en como máximo 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50 o 100 deleciones, adiciones o sustituciones de aminoácidos, tales como sustituciones conservadoras de aminoácidos. Las variantes biológicamente activas de las proteínas de los factores protectores de la DN también pueden incluir variantes que son al menos 70%, 80%, 85%, 90%, 95%, 97%, 98% o 99% idénticas a la proteína PEBP1 humana madura o precursora de longitud completa (u otro biomarcador identificado en esta memoria descriptiva) o un fragmento de la misma.
En algunas realizaciones, el método proporcionado comprende además seleccionar un sujeto para el tratamiento. Por ejemplo, se puede seleccionar un sujeto si el sujeto tiene o está en riesgo de desarrollar DN, p. ej., un sujeto que tiene diabetes, por ejemplo, diabetes tipo 1 o tipo 2, o un sujeto que es prediabético, p. ej., tiene síndrome metabólico, resistencia a la insulina, hiperglucemia, hiperlipidemia o un sujeto que tiene sobrepeso u obesidad, p. ej., que tiene un IMC >25. En algunos casos, se puede seleccionar un sujeto si el sujeto tiene o está en riesgo de desarrollar diabetes tipo 1 y/o tipo 2. En algunos casos, se puede seleccionar un sujeto si el sujeto está tomando o tomará insulina, p. ej., para tratar la diabetes.
La enfermedad cardiovascular es una afección inflamatoria crónica. El aumento de la captación de glucosa y el flujo glucolítico promueven especies de oxígeno reactivas en las mitocondrias. Las ROS promueven la dimerización de la PKM2 y permiten su translocación nuclear. La PKM2 nuclear funciona como proteína quinasa y aumenta la producción de IL-6 e IL-1p. Esto da como resultado una inflamación sistémica y tisular. Se descubrió que reducir la glucólisis y forzar la tetramerización de la PKM2 corrigen el fenotipo proinflamatorio de los macrófagos de la enfermedad arterial coronaria (CAD) (J. Exp. Med. 2016, 213(3): 337-354).
En ciertas realizaciones, en el presente documento se proporciona un método para tratar una enfermedad cardiovascular en un sujeto que comprende administrar una cantidad terapéuticamente eficaz del compuesto, sal farmacéuticamente aceptable o composición farmacéutica del mismo. Los compuestos o la composición descritos en el presente documento pueden reducir el nivel de glucosa en plasma en un sujeto. Una "enfermedad cardiovascular", como se define en esta solicitud, comprende, pero no se limita a, hipertensión, insuficiencia cardíaca congestiva, diabetes, glomeruloesclerosis, insuficiencia renal crónica, enfermedad cardíaca coronaria, angina de pecho, infarto de miocardio, accidente cerebrovascular, reestenosis vascular disfunción endotelial, distensibilidad vascular alterada e insuficiencia cardiaca congestiva. En ciertas realizaciones, la enfermedad cardiovascular es enfermedad arterial coronaria (CAD). En ciertas realizaciones, el compuesto o la composición descritos en el presente documento se pueden usar para reducir las especies de oxígeno reactivas (ROS) en las mitocondrias de un sujeto.
En ciertas realizaciones, en el presente documento se proporciona un método para tratar una enfermedad autoinmunitaria en un sujeto que comprende administrar una cantidad terapéuticamente eficaz del compuesto, sal farmacéuticamente aceptable o composición farmacéutica del mismo. Se encontró que la activación de la PKM2 atenuaba un fenotipo de macrófago M1 proinflamatorio inducido por LPS mientras que promovía rasgos típicos de un macrófago M2. Además, se encontró que la activación de la PKM2 porTEPP-46 in vivo inhibía la producción de LPS e IL-1p, a la vez que aumentaba la producción de IL-10. (Cell Metab. 2015, 21(1): 65-80) En consecuencia, los activadores de PKM2 pueden ser útiles para tratar una enfermedad autoinmunitaria al promover la producción de IL-1p y/o IL-10.
Una "enfermedad autoinmunitaria" se refiere a una enfermedad que surge de una respuesta inmunitaria inapropiada del cuerpo de un sujeto contra sustancias y tejidos normalmente presentes en el cuerpo. Ejemplos de enfermedades autoinmunitarias incluyen, pero no se limitan a, glomerulonefritis, síndrome de Goodpasture, vasculitis necrosante, linfadenitis, periarteritis nodosa, lupus eritematoso sistémico, artritis reumatoide, artritis psoriásica, lupus eritematoso sistémico, psoriasis, colitis ulcerosa, esclerosis sistémica, dermatomiositis/polimiositis, síndrome de anticuerpos antifosfolípidos, esclerodermia, pénfigo vulgar, vasculitis asociada a ANCA (p. ej., granulomatosis de Wegener, poliangitis microscópica), uveítis, síndrome de Sjogren, enfermedad de Crohn, síndrome de Reiter, espondilitis anquilosante, enfermedad de Lyme, síndrome de Guillain-Barré, síndrome de Hashimoto tiroiditis y miocardiopatía.
Composiciones y vías de administración.
Las composiciones descritas en el presente documento incluyen los compuestos descritos en el presente documento (p.
ej., un compuesto descrito en el presente documento), así como agentes terapéuticos adicionales si están presentes, en cantidades eficaces para lograr una modulación de la enfermedad o los síntomas de la enfermedad, incluidos los descritos en el presente documento.
La expresión "vehículo o adyuvante farmacéuticamente aceptable" se refiere a un vehículo o adyuvante que se puede administrar a un paciente, junto con un compuesto proporcionado en este documento, y que no destruye su actividad farmacológica y no es tóxico cuando se administra en dosis suficientes para suministrar una cantidad terapéutica del compuesto.
Los vehículos, adyuvantes y portadores farmacéuticamente aceptables que se pueden usar en las composiciones farmacéuticas proporcionadas en este documento incluyen, pero no se limitan a, intercambiadores de iones, alúmina, estearato de aluminio, lecitina, sistemas de administración de fármacos autoemulsionantes (SEDDS) tales como succinato de d-a-tocoferol-polietilenglicol 1000, tensioactivos utilizados en formas farmacéuticas tales como Tweens u otras matrices de administración poliméricas similares, proteínas séricas, tales como albúmina sérica humana, sustancias de tamponamiento tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas parciales de glicéridos de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, hidrogenofosfato de disodio, hidrogenofosfato de potasio, cloruro de sodio, sales de zinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias a base de celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros de bloques de polietileno-polioxipropileno, polietilenglicol y grasa de lana. Las ciclodextrinas tales como a-, p- y Y-ciclodextrinas, o derivados químicamente modificados tales como hidroxialquilciclodextrinas, incluidas 2- y 3-hidroxipropil-p-ciclodextrinas, u otros derivados solubilizados, también pueden usarse ventajosamente para mejorar la administración de compuestos de las fórmulas descritas en el presente documento.
Las composiciones farmacéuticas proporcionadas en este documento pueden administrarse por vía oral, parenteral, por pulverización de inhalación, tópica, rectal, nasal, bucal, vaginal o mediante un depósito implantado, preferiblemente por administración oral o administración por inyección. Las composiciones farmacéuticas proporcionadas en este documento pueden contener cualquier vehículo, adyuvante o portador convencional no tóxico farmacéuticamente aceptable. En algunos casos, el pH de la formulación se puede ajustar con ácidos, bases o tampones farmacéuticamente aceptables para mejorar la estabilidad del compuesto formulado o su forma de administración. El término parenteral como se usa en el presente documento incluye técnicas de inyección o infusión subcutánea, intracutánea, intravenosa, intramuscular, intraarticular, intraarterial, intrasinovial, intraesternal, intratecal, intralesional e intracraneal.
Las composiciones farmacéuticas proporcionadas en este documento pueden administrarse por vía oral en cualquier forma farmacéutica aceptable por vía oral que incluyen, pero no se limitan a, cápsulas, comprimidos, emulsiones y suspensiones, dispersiones y soluciones acuosas. En el caso de comprimidos para uso oral, los vehículos que se usan comúnmente incluyen lactosa y almidón de maíz. También se añaden típicamente agentes lubricantes, tales como estearato de magnesio. Para la administración oral en forma de cápsula, los diluyentes útiles incluyen lactosa y almidón de maíz seco. Cuando las suspensiones y/o emulsiones acuosas se administran por vía oral, el principio activo puede suspenderse o disolverse en una fase oleosa combinada con agentes emulsionantes y/o de suspensión. Si se desea, se pueden añadir ciertos agentes edulcorantes y/o aromatizantes y/o colorantes.
Cuando las composiciones proporcionadas en este documento comprenden una combinación de un compuesto de las fórmulas descritas en el presente documento y uno o más agentes terapéuticos o profilácticos adicionales, tanto el compuesto como el agente adicional deben estar presentes en niveles de dosificación de entre aproximadamente 1 y 100%, y más preferiblemente entre aproximadamente 5 a 95% de la dosis normalmente administrada en un régimen de monoterapia. Los agentes adicionales se pueden administrar por separado, como parte de un régimen de dosis múltiples, de los compuestos proporcionados en este documento. Alternativamente, esos agentes pueden ser parte de una sola forma farmacéutica, mezclados junto con los compuestos proporcionados en este documento en una sola composición.
Los compuestos descritos en el presente documento pueden, por ejemplo, administrarse por inyección, por vía intravenosa, intraarterial, subdérmica, intraperitoneal, intramuscular o subcutánea; o por vía oral, bucal, nasal, transmucosa, tópica, en una preparación oftálmica, o por inhalación, con una dosis en el intervalo de aproximadamente 0,5 a aproximadamente 100 mg/kg de peso corporal, alternativamente dosis entre 1 mg y 1000 mg/dosis, cada 4 a 120 horas, o según los requisitos del fármaco en particular. Los métodos del presente documento contemplan la administración de una cantidad eficaz de compuesto o composición de compuesto para lograr el efecto deseado o declarado. Típicamente, las composiciones farmacéuticas proporcionadas en este documento se administrarán de aproximadamente 1 a aproximadamente 6 veces al día o, alternativamente, como una infusión continua. Dicha administración puede usarse como una terapia crónica o aguda. La cantidad de principio activo que se puede combinar con los materiales del vehículo para producir una única forma farmacéutica variará dependiendo del hospedante tratado y el modo particular de administración. Una preparación típica contendrá de aproximadamente 5% a aproximadamente 95% de compuesto activo (p/p). Alternativamente, dichas preparaciones contienen de aproximadamente 20% a aproximadamente 80% de compuesto activo.
Parte experimental
Lista de abreviaturas:

Parte experimental general
En los siguientes ejemplos, los reactivos químicos se adquirieron de fuentes comerciales (tales como Alfa, Acros, Sigma Aldrich, TCI y Shanghai Chemical Reagent Company) y se usaron sin purificación adicional. La cromatografía ultrarrápida se realizó en un Ez Purifier III mediante columna con partículas de gel de sílice de malla 200-300. Las placas de cromatografía en capa fina (TLC) analítica y preparativa fueron HSGF 254 (0,15-0,2 mm de espesor, Shanghai Anbang Company, China). Los espectros de resonancia magnética nuclear (RMN) se registraron utilizando Brucker AMX-300 o AMX-400 RMN(Brucker, Suiza). Los desplazamientos químicos se daban en partes por millón (ppm, 8) de etero(ESI) de un espectrómetro de masas Waters LCT TOF (Waters, EE. UU.). Las cromatografías HPLC se registraron en cromatografía líquida de Agilent 1200 (Agilent, EE. UU., columna: Ultimate 4,6 m * 50 mm, 5 M, fase móvil A: ácido fórmico al 0,1% en agua; fase móvil B: acetonitrilo). Las reacciones de microondas se realizaron en un sintetizador de microondas Initiator 2,5 (Biotage, Suecia).
Las condiciones de HPLC utilizadas en los experimentos descritos en el presente documento son las siguientes:
Método 1:
Instrumento: Shimadzu LC-2010AHT
Columna: YMC-Triart C18, 50 * 4,6 mm, 5 |jm
Fase móvil: Disolvente A: H2O/CH3OH/TFA = 90/10/0,1,
Disolvente B: H2O/CH3OH/TFA = 90/10/0,1
Caudal: 2,5 ml/min
Temperatura de la columna: 35°C
Longitud de onda: 220 nm/254 nm
Método 2:
Instrumento: Shimadzu LC-2010AHT
Columna: YMC-Triart C18, 50 * 4,6 mm, 5 jm
Fase móvil: Disolvente A: H2O/CH3OH/TFA = 90/10/0,1,
Disolvente B: H2O/CH3OH/TFA = 90/10/0,1
Caudal: 2,5 ml/min
Temperatura de la columna: 35°C
Longitud de onda: 220 nm/254 nm
Las condiciones de Prep-HPLC utilizadas en los experimentos descritos en el presente documento son las siguientes: Instrumento: Waters 2545B/2767
Columna: YMC-Triart C18, 50 * 4,6 mm, 5 jm
Fase móvil: Disolvente A: H2O (01.% FA),
Disolvente B: CH3OH o CH3CN
Caudal: 20 ml/min
Temperatura de la columna: 35°C
Longitud de onda: 220 nm/254 nm
Ejemplo 1. Preparación de los compuestos E1-vii con el Esquema E1
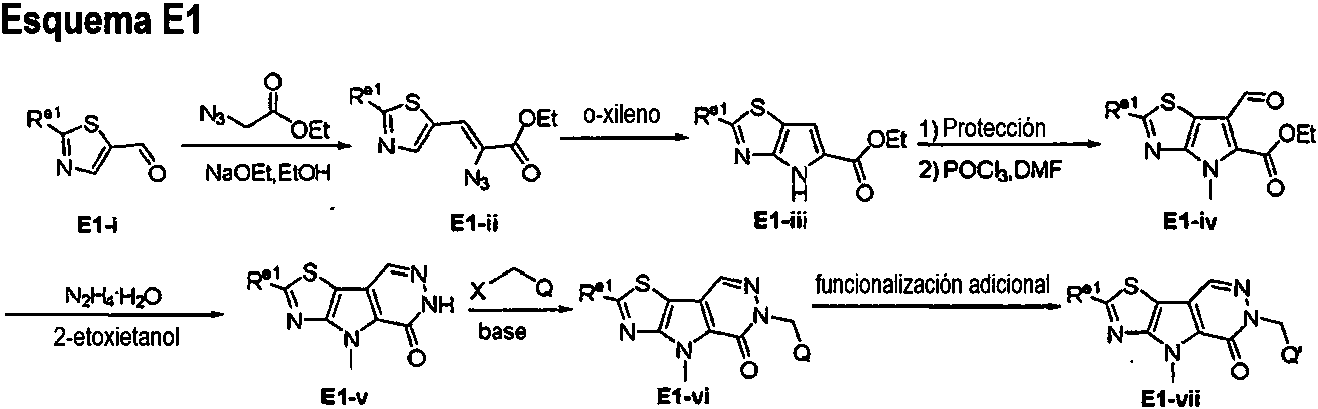
en donde Re1 es alquilo opcionalmente sustituido (p. ej., alquilo C1-3); Q es como se define en una cualquiera de las realizaciones primera a vigésima sexta de la invención; Q' es un Q funcionalizado adicionalmente, y X es un grupo saliente (p. ej., halógeno tal como Br o I; OMs u OTs). El tiazol-5-carbaldehído E1-i sufre condensación con 2-azidoacetato para dar un compuesto de fórmula E1-ii. El compuesto E1-ii sufre ciclación en o-xileno calentado para dar un sistema bicíclico de E1-iii, seguido de metilación del grupo amino y posterior oxidación para dar un compuesto E1-iv. El compuesto E1-iv reacciona con hidracina seguido de ciclación para dar un compuesto de E1-v. El compuesto E1-v puede reaccionar con un nucleófilo tal como X-CH2-Q para dar E1-vi, que se puede funcionalizar adicionalmente a E1-vii que tiene Q'.
Ejemplo 1A. Síntesis de 6-(3-metoxibencil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. (Z)-2-Azido-3-(2-metiltiazol-5-il)acrilato de etilo. A una solución de NaOEt (803 mg, 11,79 mmol) en EtOH (10 ml) entre aproximadamente -10°C y aproximadamente -5°C se añadió gota a gota una solución de 2-metiltiazol-5-carbaldehído (500 mg, 3,93 mmol) y 2-azidoacetato de etilo (1,53 g, 11,79 mmol) en EtOH anhidro (3 ml). La mezcla de reacción se agitó durante aproximadamente 1 h, mientras la temperatura se mantenía por debajo de 0°C, luego se calentó a t.a. y se agitó durante otras 2 h. La mezcla resultante se vertió en solución acuosa saturada de NH4Cl (50 ml) a 0°C y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (500 mg) que se usó directamente en la siguiente etapa sin ninguna purificación. LCMS: m/z 239 (M+H)+.
Etapa B. 2-Metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. Una mezcla de (Z)-2-azido-3-(2-metiltiazol-5-il)acrilato de etilo (500 mg, 2,1 mmol) en o-xileno (5 ml) se agitó a 140°C durante 2 h, luego se enfrió hasta t.a. y luego se purificó directamente por cromatografía en columna en gel de sílice (eluyente: pentano/EtOAc = 6/1 para dar el producto deseado (220 mg, 49,8% de rendimiento). LCMS: m/z 211 (M+H)+.
Etapa C. 2,4-Dimetil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. A una solución de 2-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (160 mg, 0,76 mmol) en DMF (3 ml) a 0°C se le añadió NaH (36,5 mg, 1,52 mmol). La mezcla de reacción se agitó a t.a. durante 0,5 h, seguido de la adición de CH3I (47 μl, 0,76 mmol). La mezcla resultante se agitó a t.a. durante 0,5 h, luego se vertió en solución acuosa saturada de NH4Cl a 0°C y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: pentano/acetato de etilo = 6/1) para dar el producto deseado (124 mg, 72,6% de rendimiento). LCMS: m/z 225 (M+H)+.
Etapa D 6-Formil-2,4-dimetil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. A una mezcla de 2,4-dimetil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (100 mg, 0,446 mmol) en DMF (1 ml) a 0°C se le añadió POCh (122,5 μl, 1,338 mmol). La mezcla de reacción se agitó a 100°C durante 2 h, luego se vertió en solución acuosa saturada de NaHCO3 a 0°C y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: pentano/acetato de etilo = 5/1) para dar el producto deseado (57 mg, 50,7% de rendimiento). LCMS: m/z 253 (M+H)+.
Etapa E. 2,4-Dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 6-formil-2,4-dimetil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (57 mg, 0,226 mmol) en 2-etoxietanol (2 ml) se le añadió N2H4.H2O (53,7 μl, 1,130 mmol). La mezcla de reacción se agitó a 100°C durante 1 h, luego se vertió en H2O y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: pentano/acetato de etilo = 5/1) para dar el producto deseado (49 mg, 98,4% de rendimiento). LCMS: m/z 221 (M+H)+.
Etapa F. 6-(3-Metoxibencil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (49 mg, 0,223 mmol) en DMF (1 ml) a 0°C se añadió t-BuOK (50,8 mg, 0,454 mmol). La mezcla de reacción se agitó a t.a. durante 0,5 h, seguido de la adición de 1-(clorometil)-3-metoxibenceno (34,9 mg, 0,223 mmol). La mezcla resultante se agitó a t.a. durante 1 h luego se vertió en solución acuosa saturada de NH4Cl a 0°C y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: pentano/acetato de etilo = 3/1) para dar el producto deseado. LCMS: m/z 341 (M+H)+. RMN 1H (400 MHz, DMSO-de) 88,56 (s, 1H), 7,23 (t, 1H), 6,92 - 6,72 (m, 3H), 5,32 (s, 2H), 4,26 (s, 3H), 3,72 (s, 3H), 2,85 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.



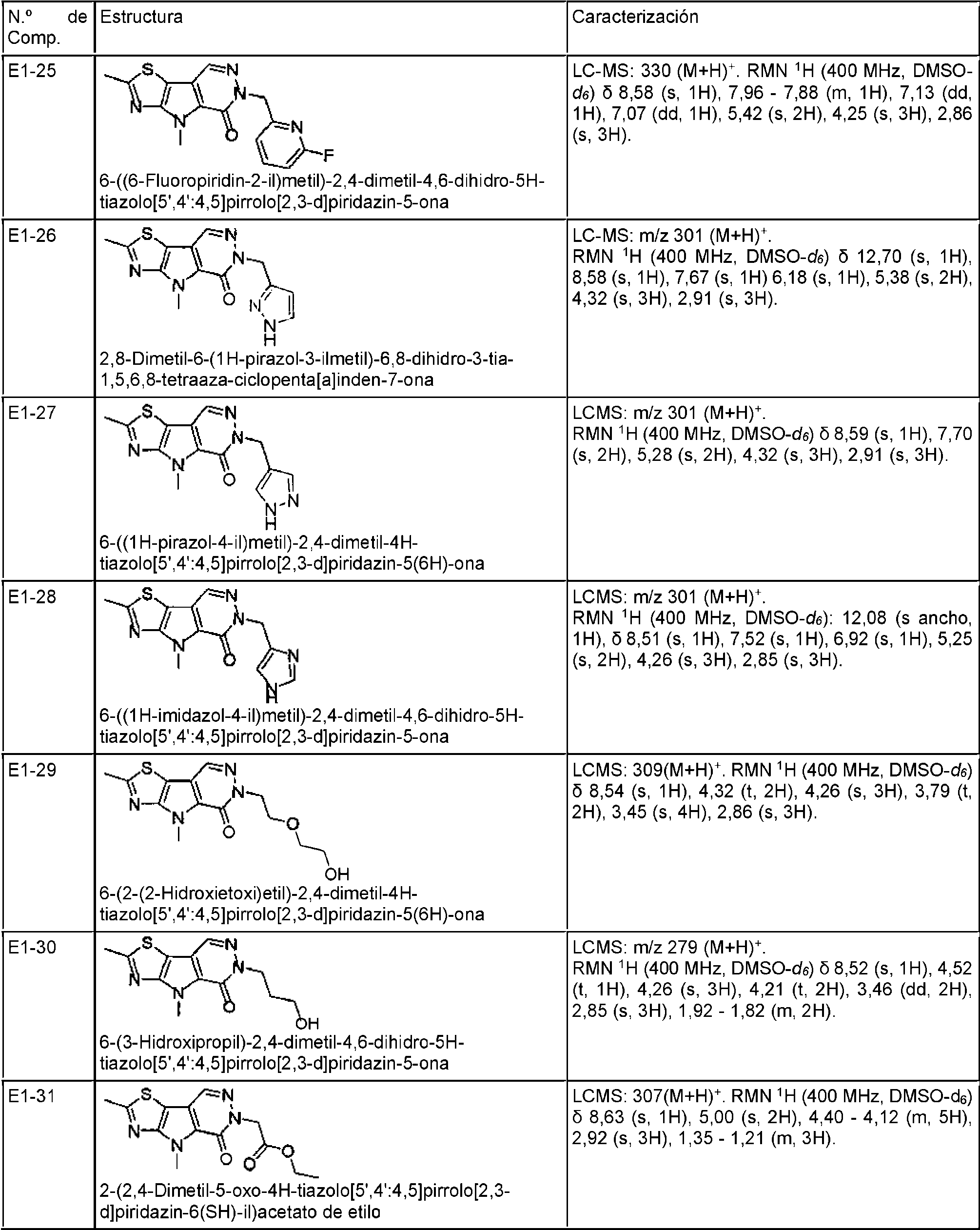
Ejemplo 1B. Síntesis de 1-(3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)fenil)urea
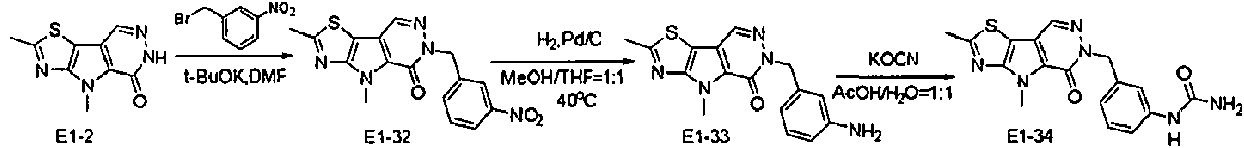
Etapa A. 2,4-Dimetil-6-(3-nitrobencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,45 mmol) en DMF (5 ml) se añadieron 1-(bromometil)-3-nitrobenceno (194 mg, 0,9 mmol) y t-BuOK (76 mg, 0,68 mmol). La reacción se agitó a t.a. durante 1 h, luego se vertió en agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (100 mg, 62,5% de rendimiento). LCMS: m/z 356 (M+H)+.
Etapa B. 6-(3-Aminobencil)-2,4-dimetil-4H-tiazob[5',4':4,5]pirrolo[2,3-d]pirídazin-5(6H)-ona. A una mezcla de 2,4-dimetil-6-(3-nitrobencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,28 mmol) en MeOH/THF (10 ml/10 ml) en atmósfera de N2 se añadió Pd/C (10%, 50 mg). La mezcla de reacción se agitó a 40°C en atmósfera de H2 durante 12 h, luego se filtró a través de Celite. El filtrado se concentró a presión reducida y el residuo se purificó por TLC preparativa para proporcionar el compuesto deseado (80 mg, 88% de rendimiento). LCMS: m/z 326 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,54 (s, 1H), 6,94 (t,, 1H), 6,57 - 6,32 (m, 3H), 5,19 (s, 2H), 5,04 (s, 2H), 4,26 (s, 3H), 2,85 (s, 3H).
Etapa C. 1-(3-((2,4-Dimetil-5-oxo-4H-tiazolo[5'.4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)fenil)urea. A una mezcla de 6-(3-aminobencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (65 mg, 0,2 mmol) en HOAc(2 ml) se añadió KOCN (160 mg, en HOAc:H2O=2 ml:4 ml). La mezcla de reacción se agitó a t.a. durante 2 h, luego se vertió en solución acuosa saturada de NaHCO3y se extrajo con EtOAc. Las capas orgánicas se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el compuesto deseado (4 mg, 5% de rendimiento). LCMS: m/z 369 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,60-8,50 (m, 2H), 7,39 (d, 1H), 7,23 (s, 1H), 7,16 (t, 1H), 6,85 (d, 1H), 5,78 (s, 2H), 5,28 (s, 2H), 4,27 (s, 3H), 2,86 (s, 3H).
Ejemplo 1C. Síntesis de 2,4-dimetil-6-(3-(metilamino)bencil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. (3-((2,4-Dimetil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)fenil)carbamato de tercbutilo. A una mezcla de 6-(3-aminobencil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (90 mg, 0,28 mmol) en 1,4-dioxano (10 ml) se añadió Boc2O (73 mg, 0,33 mmol). La mezcla de reacción se agitó a reflujo durante la noche y luego se concentró a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc - 3/1) para dar el producto deseado (90 mg, 76,3% de rendimiento). LCMS: m/z 426 (M+H)+.
Etapa. B (3-((2,4-Dimetil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)fenil)(metil)carbamato de terc-butilo. A una mezcla de (3-((2,4-dimetil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)fenil)carbamato de terc-butilo (90 mg, 0,21 mmol) en DMF anhidra (5 ml) a 0°C se añadió NaH (13 mg, 0,32 mmol, 60% en peso). La mezcla se agitó a 0°C durante 1 h, seguido de la adición gota a gota de Mel. La mezcla resultante se agitó a 0-5°C durante 3 h, luego se vertió en solución acuosa saturada de NH4Cl fría y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (70 mg, 75,2% de rendimiento). LCMS: m/z 440 (M+H)+.
Etapa C. 2,4-Dimetil-6-(3-(metilamino)bencil)-4,6-dihidro-5H-tiazolo[5,4 ':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de (3-((2,4-dimetil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)fenil)(metil)carbamato de terc-butilo (90 mg, 0,21 mmol) en DCM (3 ml) se añadió TFA (1 ml). La mezcla se agitó a t.a. durante 2 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (25 mg, 46,4% de rendimiento). LCMS: m/z 340 (M+H)+. RMN 1H (400 MHz, D M S O d) 58,54 (s, 1H), 7,12 (t, 1H), 6,66-6,61 (m, 3H), 5,26 (s, 2H), 4,26 (s, 3H), 2,85 (s, 3H), 2,68 (d, 3H).
Se usó el procedimiento expuesto anteriormente para producir el siguiente compuesto usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 1D. Síntesis de 6-(3-hidroxibencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

A una mezcla de 6-(3-metoxibencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (53 mg, 0,16 mmol) en DCM (4 ml) a 0°C se añadió BBr3 (195 mg, 0,778 mmol). La mezcla se agitó a t.a. durante 2 h, luego se inactivó con MeOH. La mezcla resultante se concentró a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (15,6 mg, 30,70% de rendimiento). LCMS: m/z 327 (M+H)+. RMN 1H (400 MHz, DMSO-de) 59,34 (s, 1H), 8,58 (s, 1H), 7,12 (t, 1H), 6,78-6,56 (m, 3H), 5,26 (s, 2H), 4,278 (s, 3H), 2,86 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 1E. Síntesis de 6-[3-(1-amino-etil)-bencil]-2,8-dimetil-6,8-dihidro-3-tia-1,5,6,8-tetraaza-ciclopenta[a]inden-7-ona

A una mezcla agitada de 6-(3-acetilbencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,142 mmol) en MeOH (4 ml) se añadieron NH4OAc (109 mg, 1,42 mmol) y NaBH3CN (18 mg, 0,284 mmol). La mezcla de reacción se agitó a 35°C durante 13 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (20 mg, 40,0% de rendimiento). LC-MS: m/z 354 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,48 (s, 1H), 7,25 (dd, 3H), 7,12 (d, 1H), 5,26 (s, 2H), 4,17 (s, 3H), 4,15-4,06 (m, 1H), 2,76 (s, 3H), 1,27 (d, 3H).
Ejemplo 1F. Síntesis de 6-(3-(aminometil)bencil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 2-(3-(Bromometil)bencil)isoindolin-1,3-diona. A una mezcla agitada de 1,3-bis(bromometil)benceno (1,3 g, 4,96 mmol) en DMF (20 ml) se añadieron 1,3-dioxoisoindolin-2-ida de potasio (0,918 g, 4,96 mmol) y K2CO3 (1,03 g, 7,44 mol). La mezcla de reacción se agitó a t.a. durante 2 h, luego se vertió en solución acuosa saturada de NH4Cl (30 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 30/1) para dar el producto deseado (1,1 g, 67,4% de rendimiento). LC-MS: m/z 330 (M+H)+.
Etapa B. 2-(3-((2,4-Dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)bendl)isoindolina-1,3-diona. A una mezcla agitada de 2-(3-(bromometil)bencil)isoindolina-1,3-diona (100 mg, 0,303 mmol) en DMF (4 ml) se le añadió 2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (66,7 mg, 0,303 mmol) y K2Co 3 (83,6 mg, 0,606 mol). La mezcla de reacción se agitó a t.a. durante 2 h, luego se vertió en solución acuosa saturada de NH4Cl (15 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (80 mg, 56,3% de rendimiento). LC-MS: m/z 470 (M+H)+.
Etapa C. 6-(3-(Aminometil)bencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirmlo[2,3-d]piridazin-5(6H)-ona. A una mezcla agitada de 2-(3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)bencil)isoindolina-1,3-diona (80 mg, 0,17 mmol) en EtOH (5 ml) se añadió N2H4.H2O (44 mg, 98% en peso, 0,85 mmol). La mezcla de reacción se agitó a 100°C durante 2 h, luego se vertió en solución acuosa saturada de NH4Cl (15 ml) y se extrajo dos veces con DCM. Las capas orgánicas se lavaron dos veces con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (30 mg, 52,1% de rendimiento). LC-MS: m/z 324 (M+H-NHs)+. RMN 1H (400 MHz, DMSO-da) 58,57 (s, 1H), 7,32 (m, 3H), 7,23 (d, 1H), 5,35 (s, 2H), 4,27 (s, 3H), 3,84 (s, 2H), 2,86 (s, 3H).
Ejemplo 1G. Síntesis de 6-(4-(hidroximetil)bencil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 4-((2,4-Dimetil-5-oxo-4H-tiazolo[5,4:4,5]pirrolo[2,3-d]piridazm-6(5H)-il)metil)benzoato de metilo. A una mezcla de 2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,4 mmol) en DMF (20 ml) se añadió K2CO3 (181 mg, 1,3 mmol). La mezcla se agitó a 60°C durante 30 min, seguido de la adición de 4-(bromometil)benzoato de metilo (100 mg, 0,4 mmol) a 0°C. La mezcla resultante se agitó a 60°C durante 18 h, luego se vertió en agua helada y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 50/1 a 10/1) para dar el producto deseado (120 mg, 74,61%). LCMS: m/z 369 (M+H)+.
Etapa B. 6-(4-(Hidroximetil)bencil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzoato (100 mg, 0,3 mmol) en THF (20 ml) a 0°C se añadió LAH (30 mg, 0,8 mmol). La reacción se agitó a 0°C en atmósfera de N2 durante 30 min y luego se inactivó
con NaSO4-10H2O y se filtró. El filtrado se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (3 mg, 3,25%). LCMS: m/z 341 (M+H)+. RMN 1H (400 MHz, CDCh) 58,12 (s, 1H), 7,36 (d, 2H), 7,25 (d, 2H), 5,37 (s, 2H), 4,59 (s, 2H), 4,31 (s, 3H), 2,80 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 1H. Síntesis de 6-((6-aminopiridin-2-il)metil)-2,4-dimetil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A: 6-((6-((2,4-Dimetoxibencil)amino)piridin-2-il)metil)-2,4-dimetil-4H-tiazolo[5,4:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 6-((6-fluoropiridin-2-il)metil)-2,4-dimetil-4H-tiazolo[5',4':4,5]-pirrolo[2,3-d]piridazin-5(6H)-ona (40 mg, 0,12 mmol) y (2,4-dimetoxifenil)metanamina (102 mg, 0,6 mmol) en NMP (1 ml) se agitó a 140°C hasta completarse. La mezcla resultante se concentró a presión reducida. El residuo se purificó por TLC preparativa para obtener el producto deseado (20 mg, 34,5% de rendimiento). LC-MS: 477 (M+H)+.
Etapa B: 6-((6-Aminopiridin-2-il)metil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 6-((6-((2,4-dimetoxibencil)amino)piridin-2-il)metil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,042 mmol) y TFA (45 mg, 0,42 mmol) en Dc M (1 ml) se agitó a t.a. hasta completarse. La mezcla resultante se concentró a presión reducida. El residuo se purificó por Hp l C preparativa para obtener el producto deseado (20 mg, 34,5% de rendimiento). LC-MS: 327(M+H)+. RMN 1H (400 MHz, DMSO-da) 58,55 (s, 1H), 7,26 (t, 1H), 6,30 (d, 1H), 6,09 (d, 1H), 5,90 (s, 2H), 5,19 (s, 2H), 4,25 (s, 3H), 2,85 (s, 3H).
Ejemplo 1I. Síntesis de 6-(hidroximetil)-2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Una mezcla de 2,4-dimetil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,14 mmol), formaldehído (1,5 ml, 40% en peso) y NH3 (0,75 ml, 33% en peso) en dioxano (2 ml) se agitó a 50°C durante 1 h, luego se vertió en agua y se extrajo con DCM. La capa orgánica se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (4,40 mg, 17% de rendimiento). LCMS: 251 (M+H). RMN 1H (400 MHz, DMSO-da) 58,55 (s, 1H), 6,63 (t, 1H), 5,44 (d, 2H), 4,27 (s, 3H), 2,85 (s, 3H).
Ejemplo 2. Preparación de compuestos de fórmula E2-vii con el Esquema E2

en donde Xa es un grupo saliente (p. ej., halógeno tal como Br o I; OMs u OTs); Xb es halógeno (p. ej., Cl, Br o 1); n i es 0 o 1; M es hidrógeno (por ejemplo, para la reacción de Heck) o un complejo de metal orgánico (p. ej., complejo de organoboro tal como ácido borónico o complejo de pinaco-boro; complejo de organoestaño tal como -Sn(Bu%; complejo de organozinc tal como -Zn (halógeno)); Q y R2 son como se definen en una cualquiera de las realizaciones primera a vigésima sexta de la invención. En ciertas realizaciones, Q y R2 son cada uno independientemente arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido, alquilo opcionalmente sustituido. De manera similar a la síntesis de los compuestos de fórmula E l-v en el ejemplo 1, el compuesto E2-iv se puede sintetizar a partir del tiazol aldehido E2-i con algunas modificaciones (p. ej., reacción del compuesto E2-ii con MsCl seguido de eliminación para dar el compuesto E2- iii; el sistema tricíclico se puede formar con AcOH cat. en 2-metoxietanol). La sustitución y halogenación (p. ej., CBr4 o ChCCCh en presencia de LiHMDS; o 1,2,3,4,5-pentafluoro-6-yodobenceno en presencia de t-BuOK y tolueno) del compuesto E2-iv proporciona el compuesto E2-vi. Las reacciones de acoplamiento del compuesto E2-vi con el organometal en presencia de un catalizador dan el compuesto E2-vii o E2-viii. La reacción nucleofílica directa de E2-vi con un nucleófilo (Nu) puede generar el compuesto E2-ix.
Ejemplo 2A. Síntesis de 2-(4-fluorobencil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 2-Azido-3-hidroxi-3-(tiazol-5-il)propanoato de etilo. Se añadió lentamente sodio (12,2 g, 0,531 mol) a t.a. a una solución agitada de EtOH seco (300 ml). Luego, la mezcla de reacción se enfrió a -20°C, seguido de la adición gota a gota de una solución de 2-azidoacetato de etilo (68,5 g, 0,531 mol) y tiazol-5-carbaldehído (20,0 g, 0,177 mol) en EtOH anhidro (100 ml) manteniendo la temperatura entre -20°C y -15°C. Después de la adición, la mezcla de reacción se agitó a -20°C durante 1 h adicional y luego se vertió en solución acuosa saturada de NH4Cl (1 L). La mezcla resultante se saturó con NaCl y se extrajo con EtOAc. La fase orgánica combinada se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice usando (eluyente: PE/EtOAc = 6/1 a 5/1 a 1/1) para proporcionar el producto deseado (34 g) de color pálido. LCMS: m/z= 243 (M+H)+.
Etapa B. (Z)-2-Azido-3-(tiazol-5-il)acrilato de etilo. A una mezcla agitada de 2-azido-3-hidroxi-3-(tiazol-5-il)propanoato de etilo (103 g, 0,426 mmol) en Dc M seco (1,5 L) a -35°C, se añadió MsCl (146 g, 1,28 mol), seguido de la adición gota a gota de TEA (301 g, 2,98 mol) mientras se mantenía la temperatura entre -35°C y -30°C. Después de la adición, la mezcla de reacción se agitó a -30°C durante otros 15 min y luego se vertió en solución acuosa saturada de NH4Cl (1,5 L). La mezcla resultante se saturó con NaCl y se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron en secuencia con HCl acuoso (1 M) y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice usando (eluyente: PE/EtOAc= 5/1) para proporcionar el producto deseado (82,0 g, 86,3% de rendimiento). LCMS: m/z= 225 (M+H)+.
Las etapas C-E para sintetizar el 4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo, 4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo y 6-formil-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo fueron similares a los procedimientos del Ejemplo 1A.
Etapa F. (E)-6-(Hidrazonometil)-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. A una mezcla agitada de N2H4.H2O (2,0 g, 98%, 40 mmol) en 2-metoxietanol (50 ml) a t.a. se añadió 6-formil-4-metil-4H-pirrolo [2,3-d]tiazol-5-carboxilato de etilo (4,8 g, 20 mmol), seguido de la adición de 20 gotas de AcOH. La mezcla de reacción se agitó a t.a. durante aproximadamente 30 min hasta que la mezcla se volvió transparente. La mezcla resultante se vertió en agua (100 ml) con agitación y se extrajo con DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado que se usó en la siguiente etapa sin más purificación. LCMS: m/z= 253 (M+H)+.
Etapa G. 4-Metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una suspensión agitada de (E)-6-(hidrazonometil)-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxiíato de etilo (4,8 g, 0,19 mol) en 2-metoxietanol (50 ml) a t.a. se añadió AcOH (20 gotas). La suspensión de reacción se agitó a 105°C durante 3 h y luego se filtró. La torta del filtración se lavó con agua y se secó con alto vacío para obtener el primer lote del producto deseado. El filtrado se diluyó con agua y se extrajo dos veces con DCM. Las capas orgánicas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el segundo lote del producto deseado. Los dos lotes combinados del producto deseado (2,5 g) se usaron directamente en la siguiente etapa sin purificación adicional. LCMS: m/z= 207 (M+H)+. RMN 1H (400 MHz, DMSO) 812,68 (s, 1H), 9,35 (s, 1H), 8,55 (s, 1H), 4,30 (s, 3H).
Etapa H. 6-(3-Metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla agitada de 4-metil-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (2 g, 10,0 mmol) y K2CO3 (2,7 g, 20 mmol) en DMF (15 ml) se añadió 1-(clorometil)-3-metoxibenceno (2,3 g, 15 mmol). La mezcla de reacción se agitó a 50°C durante 3 h, luego se vertió en agua y se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (2 g, 67% de rendimiento). LCMS: m/z= 327 (M+H)+. RMN 1H (400 MHz, DMSO-de) 89,36 (s, 1H), 8,64 (s, 1H), 7,24 (t, 1H), 6,88-6,80 (m, 3H), 5,33 (s, 2H),. 4,31 (s, 3H) 3,72 (s, 3H).
Etapa I. 2-Yodo-6-(3-metoxibendl)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla agitada de 6-(3-metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (1 g, 3 mmol) y t-BuOK (688 mg, 6 mmol) en tolueno (30 ml) a t.a. se añadió 1,2,3,4,5-pentafluoro-6-yodobenceno (3,6 g, 12 mmol). La mezcla de reacción se agitó a 135°C durante 4 h (el baño de aceite se precalentó) y luego se concentró a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 6/1) para proporcionar el producto deseado (1 g, 72% de rendimiento). LCMS: m/z= 453 (M+H)+. RMN 1H (400 MHz, DMSO-de) 88,61 (s, 1H), 7,23 (t, 1H), 6,88-6,80 (m, 3H), 5,31 (s, 2H),. 4,26 (s, 3H) 3,71 (s, 3H).
Etapa J. Bromuro de (4-fluorobencil)zinc(II). A un matraz de fondo redondo de tres bocas de 25 ml se le añadió Zn en polvo (1300 mg, 20 mmol). La mezcla se desgasificó con alto vacío y se volvió a purgar con N2 tres veces. Se añadieron THF seco (15 ml), TMSCl (108 mg, 1 mmol) y 1,2-dibromoetano (186 mg, 1 mmol) mediante jeringa a temperatura ambiente. La suspensión se calentó a 65°C durante 30 min, luego se enfrió a 0°C, seguido de la adición gota a gota de 1-(bromometil)-4-fluorobenceno (1,89 g, 10 mmol). La mezcla resultante se agitó a t.a. durante 1,5 h. La solución sobrenadante se usó directamente para la siguiente etapa.
Etapa K. 2-(4-Fluorobencil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A un matraz de fondo redondo de tres bocas de 25 ml se le añadió 2-yodo-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,22 mmol) y Pd(PPh3)4 (25,4 mg, 10% en moles). El matraz se desgasificó con alto vacío y se volvió a purgar con N2 tres veces. La solución sobrenadante de bromuro de (4-fluorobencil)zinc(II) (6 ml) se añadió al matraz mediante una jeringa. La mezcla resultante se agitó en atmósfera de N2 a 65°C durante 0,5 h, luego se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (6 mg). LCMS: m/z= 435 (M+H)+. RMN 1H (400 MHz, DMSO-da) 88,53 (s, 1H), 7,75-7,50 (m, 1H), 7,47 (s, 2H), 7,23 (s, 3H), 6,84 (s, 2H), 5,31 (s, 2H), 4,52 (s, 2H), 4,26 (s, 3H), 3,71 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales del grupo amino cuando sea apropiado. Los ejemplos de grupos protectores de amino incluyen, pero no se limitan a SEM. La desprotección de SEM se puede llevar a cabo en TFA y DCM.




Ejemplo 2B. Síntesis de 2-bencil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 2-Bromo-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 2-bromo-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1,1 g, 4 mmol) en DMF anhidra (10 ml) se añadió NaH (320 mg, 60% en aceite, 8 mmol). La mezcla de reacción se agitó a t.a. durante 15 min, seguido de la adición de Mel (852 mg, 6 mmol). La mezcla resultante se agitó a t.a. durante otras 2 h, luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (950 mg, 82,2% de rendimiento). LCMS: m/z 289 (M+H)+.
Etapa B. 2-Bencil-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una mezcla de 2-bromo-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (720 mg, 2,5 mmol) y Pd(PPh3)4 (145 mg, 0,125 mmol) en THF seco en atmósfera de N2 se añadió bromuro de bencilzinc (20 ml, 0,5 M). La mezcla de reacción se agitó a 65°C durante 1 h, luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 4/1) para dar el producto deseado (600 mg, 80,0% de rendimiento). LCMS: m/z 301 (M+H)+.
Etapa C. 2-Bencil-6-formil-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 2-bencil-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (600 mg, 2 mmol) en 1,2-dicloroetano (6 ml) se añadió una mezcla de oxicloruro de fósforo (612 mg, 4 mmol) y N-metil-N-fenilformamida (540 mg, 4 mmol). La mezcla de reacción se calentó a reflujo durante la noche, luego se enfrió y se vertió en agua helada y luego se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para dar el producto deseado (140 mg) como un aceite amarillo. LCMS: m/z 329 (M+H)+.
Etapa D. 2-Bencil-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazm-5(6H)-ona A una solución de 2-bencil-6-formil-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (140 mg, bruto) en 2-etoxietanol (3 ml) se añadió hidrato de hidracina (0,5 ml, 98% en peso). La mezcla de reacción se agitó a 110°C durante 1 h, luego se enfrió a t.a. El precipitado se recogió por filtración y se lavó con MeOH para dar el producto deseado (60 mg, 10,1% de rendimiento en 2 etapas). LCMS: m/z 297 (M+H)+.
Etapa E. 2-Bencil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 2-bencil-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,169 mmol) y NaHCO3 (28 mg, 0,338 mmol) en DMA (1 ml) en atmósfera N2 se añadió 1-(clorometil)-3-metoxibenceno (40 mg, 0,254 mmol). La mezcla de reacción se agitó a 120°C durante 3 h, luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se concentraron a presión reducida y el residuo se purificó por HPLC preparativa para dar el producto deseado (8 mg, 11,4% de rendimiento). LCMS: m/z 417 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,53 (s, 1H), 7,36-7,43 (m, 4H), 7,30-7,32 (m, 1H), 7,20-7,25 (m, 1H), 6,82-6,85 (m, 3H), 5,31 (s, 2H), 4,51 (s, 2H), 4,27 (s, 3H), 3,71 (s, 3H).
Ejemplo 2C. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-2-(1-feniletil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. La 4-metil-6-((1-((2-(trimetilsMM)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4,:4,5]pirrolo[2,3-d]piridazin-5-ona se sintetizó utilizando un procedimiento similar al del ejemplo 2A. LCMS: m/z 467 (M+H)+.
Etapa B. La 2-yodo-4-metil-6-((1-((2-(trimetilsNN)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona se sintetizó utilizando un procedimiento similar al del ejemplo 2A. LCMS: 593 (M+H)+.
Etapa C. 2-Bencil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Se suspendió zinc en polvo (1,3 g, 20 mmol) en THF anhidro (5 ml) en atmósfera de N2, seguido de la adición de 1,2-dibromoetano (0,01 ml). La mezcla se calentó a 65°C durante 5 min, seguido de la adición de clorotrimetilsilano (0,01 ml). La mezcla resultante se calentó a 65°C durante otros 15 min y luego se enfrió a 0°C, seguido de la adición gota a gota de una solución de (bromometil)benceno (1,7 g, 10 mmol) en THF anhidro (5 ml). La mezcla resultante se agitó durante 1 h a 65°C, luego se enfrió para proporcionar el bromuro de bencilzinc (II) (aproximadamente 1 M en THF) que se usó directamente en la siguiente etapa. A una mezcla de 2-yodo-4-metil-6-((1-((2-(trimetilsNN)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg, 0,5 mmol) en THF seco (2 ml) en atmósfera de N2 se añadió secuencialmente Pd(Pph3)4 (58 mg, 0,05 mmol) y el bromuro de bencilzinc(II) anterior (5 ml, 1 M). La mezcla resultante se calentó a 65°C durante 1 h, luego se vertió en agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/2) para dar el producto deseado (230 mg, 82,7% de rendimiento). LCMS: 557 (M+H)+.
Etapa D. 2-Benzoil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4 ':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-bencil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,18 mmol) en DMF (5 ml) se añadió K2CO3 (74 mg, 0,53 mmol). La mezcla se agitó a 50°C en aire durante 4 h, luego se vertió en agua. El precipitado se recogió por filtración, se lavó con PE y se secó con alto vacío para dar el producto deseado (100 mg, 98% de rendimiento). LCMS: 571 (M+H)+.
Etapa E. 2-(1-Hidroxi-1-feniletil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de bromuro de metilmagnesio (0,6 ml, 1,5 M) en THF seco (2 ml) en un baño de hielo se le añadió una solución de 2-benzoil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,175 mmol) en THF seco. La mezcla se agitó durante 1 h y se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (30 mg, 29,4% de rendimiento) como aceite. LCMS: 587 (M+H)+.
Etapa F. 6-((1H-Indazol-4-il)metil)-4-metil-2-(1-feniletil)-4H-tiazolo[5',4':4,5]pirmto[2,3-d]piridazin-5(6H)-ona. Una solución de 2-(1-hidroxi-1-feniletil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,05 mmol) en una solución mixta de ufTFNTES (2 ml/0,5 ml) se agitó a t.a. durante 2 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (5 mg, 22,7% de rendimiento). LCMS: m/z 441 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,11 (s, 1H), 8,53 (s, 1H), 8,12 (s, 1H), 7,46 - 7,41 (m, 3H), 7,37 (t, 2H), 7,28 (m, 2H), 6,95 (d, 1H), 5,65 (s, 2H), 4,71 (q, 1H), 4,28 (s, 3H), 1,78 (d, 3H).
También se obtuvo un subproducto, la 4-metil-6-((1-metil-1H-indazol-4-il)metil)-2-(1-feniletil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona por HPLC preparativa:

LCMS: m/z 455 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,52 (s, 1H), 8,09 (s, 1H), 7,41 (d, 1H), 7,42 - 7,30 (m, 6H), 6,97 (d, 1H), 5,64 (s, 2H), 4,70 (q, 1H), 4,27 (s, 3H), 4,09 (s, 3H), 1,77 (d, 3H).

Ejemplo 2D. Síntesis de 6-((1H-indazol-4-il)metil)-2-(1-hidroxi-1 -feniletil)-4-metil-4,6-dihidro-5H-tiazolo[5,,4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 6-((1H-Indazol-4-il)metil)-2-benzoil-4-metil-4H-tiazolo[5',4':4,5]pirroto[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-benzoil-4-metil-6-((1-((2-(trimetilsNN)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (75 mg, 0,13 mmol) en DCM (2 ml) se añadió TFA (2 ml). La mezcla se agitó a t.a. durante 2 h, luego se concentró a presión reducida. El residuo se purificó por TLC preparativa para proporcionar el producto deseado (30 mg, 52,6% de rendimiento). LCMS: 441 (M+H)+.
Etapa B. 6-((IH-indazol-4-il)metil)-2-(1-hidroxi-1-feniletil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de bromuro de metilmagnesio (0,22 ml, 1,5 M) en THF seco (1 ml) en un baño de hielo se añadió una solución de 6-((1H-indazol-4-il)metil)-2-benzoil-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,068 mmol) en THF seco. La mezcla se agitó durante 30 min y luego se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (8 mg, 25,8% de rendimiento). LCMS: 457 (M+H)+. Rm N 1H (400 MHz, DMsO-d6) 5 13,11 (s, 1H), 8,56 (s, 1H), 8,13 (s, 1H), 7,62 (d, 2H), 7,45 (d, 1H), 7,34 (t, 2H), 7,29-7,23 (m, 2H), 6,99 - 6,93 (m, 2H), 5,65 (s, 2H), 4,26 (s, 3H), 2,01 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados.


Ejemplo 2E. Síntesis de 2-(hidroxi(fenil)metil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 2-Benzoil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 2-bencil-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (40 mg, 0,135 mmol) y K2CO3 (37 mg, 0,27 mmol) en DMF (1 ml) se añadió 1-(clorometil)-3-metoxibenceno (31 mg, 0,2 mmol). La mezcla de reacción se agitó a t.a. en el aire durante la noche y luego se vertió en agua. El precipitado se recogió por filtración y se lavó con EtOAc para dar el producto deseado (30 mg, 51,7% de rendimiento). LCMS: m/z 431 (M+H)+. RMN 1H (400 MHz, DMSOd>) 58,76 (s, 1H), 8,46-8,49 (m, 2H), 7,78 (t, 1H), 7,66 (t, 2H), 7,25 (t, 1H), 6,84-6,89 (m, 3H), 5,36 (s, 2H), 4,37 (s, 3H), 3,73 (s, 3H).
Etapa B. 2-(Hidroxi(fenil)metil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-benzoil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,047 mmol) en THF (1 ml) y MeOH (1 ml) se añadió NaBH4 (3,5 mg, 0,093 mmol). La mezcla se agitó a t.a. durante 15 min, se inactivó con agua y se extrajo con EtOAc. La capa orgánica se separó y se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (13 mg, 64,0% de rendimiento). LCMS: m/z 433 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,58 (s, 1H), 7,49-7,53 (m, 2H), 7,36-7,40 (m, 2H), 7,28-7,33 (m, 1H), 7,20-7,25 (m, 1H), 7,07 (d, 1H), 6,82-6,85 (m, 3H), 6,08 (d, 1H), 5,31 (d, 2H), 4,21 (s, 3H), 3,71 (s, 3H).
• Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando los materiales de partida apropiados.

Ejemplo 2F. Síntesis de 6-((1H-indazol-4-il)metil)-2-(hidroxi(fenil)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. La 2-(hidroxi(fenil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizó utilizando el procedimiento similar al del ejemplo 2e . lCm S: m/z 573 (M+H)+.
Etapa B. La 6-((1H-indazol-4-il)metil)-2-(hidroxi(fenil)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizó utilizando el procedimiento similar al del ejemplo 2D. LCMS: m/z 443 (M+h )+. Rm N 1H (400 MHz, Dm So -cÍ6) 5 13,12 (s, 1H), 8,59 (s, 1H), 8,13 (s, 1H), 7,56-7,42 (m, 3H), 7,37 (s, 2H), 7,28 (m, 2H), 7,07 (s, 1H), 6,95 (s, 1H), 6,08 (s, 1H), 5,65 (s, 2H), 4,21 (s, 3H).
Ejemplo 2G. Síntesis de 6-(3-metoxibencil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxilato de metilo

A una mezcla agitada de 2-yodo-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5prrolo[2,3-dpridazin-5(6H)-ona (40 mg, 0,088 mmol) en MeOH (5 ml) se añadieron Et3N (27 mg, 0,264 mmol) y Pd(dppf)Cl2 (6,5 mg, 0,009 milimoles). La mezcla de reacción se agitó a 85°C en CO durante 12 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (2 mg, 5,88% de rendimiento). LC-MS: m/z 385 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,72 (s, 1H), 7,24 (t, 1H), 6,95 - 6,81 (m, 3H), 5,34 (s, 2H), 4,29 (s, 3H), 3,99 (s, 3H), 3,72 (s, 3H). Se llevó a cabo una reacción similar con la 2-cloro-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona, que generó la 2-metoxi-6-(3-metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona como el subproducto principal. LC-MS: m/z 357 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,50 (s, 1H), 7,26-7,21 (m, 1H), 6,88 - 6,80 (m, 3H), 5,30 (s, 2H), 4,19 (s, 6H), 3,72 (s, 3H).

Ejemplo 2H. Síntesis de 2-(fluoro(fenil)metil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

A una solución de 2-(hidroxi(fenil)metil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,046 mmol) en DCM (2 ml) a -78°C se añadió DAST (0,3 ml). La mezcla se agitó a t.a. durante 30 min y luego se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (2,5 mg, 12,5% de rendimiento). LCMS: 435. (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,64 (s, 1H), 7,55-7,48 (m, 5H), 7,26-7,07 (m, 2H), 6,92-6,74 (m, 3H), 5,32 (s, 2H), 4,25 (s, 3H), 3,71 (s, 3H).
Ejemplo 2I. Síntesis de 6-((1H-indazol-4-il)metil)-2-(amino(fenil)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. (Z)-2-((Hidroxiimino)(fenil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-S(6H)-ona. Una mezcla de 2-benzoil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,175 mmol) y NH2OH.HCl (123 mg, 1,75 mmol). en MeOH anhidro (5 ml) se agitó a 100°C en un tubo sellado durante 12 h. Luego se vertió en solución acuosa saturada de NH4Cl (20 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EA = 2/1) para proporcionar el producto deseado (40 mg, 39,1% de rendimiento). LC-MS: m/z 586 (M+H)+.
Etapa B. 6-((1H-mdazol-4-il)metil)-2-(ammo(fem'l)metil)-4-metil-4H-tiazoto[5',4':4,5]pirmlo[2,3-d]piridazin-5(6H)-ona A una
mezcla agitada de (Z)-2-((hidroxMmino)(fenil)metil)-4-metil-6-((1-((2-(trimetilsiMl)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (4o mg, 0,068 mmol) en TFA (3 ml) se añadió Zn (44 mg, 0,68 milimoles). La mezcla de reacción se agitó a t.a. durante 16 h, luego se vertió en solución acuosa saturada de NaHCO3 (20 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para obtener el producto deseado (4,6 mg, 14,8% de rendimiento). LC-MS: m/z442 (M+H)+. RMN 1H (400 MHz, DMSO-da) 513,11 (s, 1H), 8,58 (s, 1H), 8,13 (s, 1H), 7,52-7,40 (m, 3H), 7,39-7,32 (m, 2H), 7,30 - 7,22 (m, 2H), 6,95 (d, 1H), 5,65 (s, 2H), 5,49 (s, 1H), 4,20 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados.

Ejemplo 2J. Síntesis de N-((6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)(1H-pirazol-3-il)metil)acetamida

Etapa A. N-((6-((1-Acetíl-1H-mdazol-4-il)metíl)-4-metíl-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirmlo[2,3-d]pirídazin-2-il)(1-acetil-1H-pirazol-3-il)metil)acetamida. A una mezcla agitada de 6-((1H-indazol-4-N)metil)-2-(amino(1H-pirazol-3-N)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,046 mmol) en DCM/MeCN (1 ml/1 ml) se añadió Et3N (14 mg, 0,139 mmol) y anhídrido acético (24 mg, 0,23 mmol). La mezcla resultante se agitó a 23°C durante 1 h, luego se inactivó con agua y se extrajo con DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto bruto (30 mg) que se usó directamente en la siguiente etapa sin purificación adicional. LCMS: m/z 558 (M+H)+.
Etapa B. N-((6-((1H-Indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)(1H-pirazol-3-il)metil)acetamida. A una mezcla agitada de N-((6-((1-acetil-1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)(1-acetil-1H-pirazo1-3-il)metil)acetamida (30 mg, 0,053 mmol) en MeOH (3 ml) en atmósfera de N2 se añadió K2CO3 (22 ml, 0,16 milimoles). La mezcla se agitó a 23°C durante 30 min y luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con DCM. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (2,0 mg, 9% de rendimiento). LC-MS: m/z: 474 (M+H)+. RMN 1H (400 MHz, DMSO-da): 58,57 (s, 1H), 8,13 (s, 1H), 7,46 (s, 1H), 7,48-7,60 (d, 1H), 7,31-7,27 (t, 1H), 7,00-6,98 (d, 1H), 6,51 (s, 1H), 6,28 (s, 1H), 5,66 (s, 2H), 4,33 (s, 3H), 1,97 (s, 3H).
Ejemplo 2K. Síntesis de 2-(difluoro(fenil)metil)-6-(3-metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 6-(3-Metoxibendl)-4-metíl-2-(2-fenil-1,3-ditíolan-2-il)-4,6-dihidro-5H-tíazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 2-benzoil-6-(3-metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (86 mg, 0,2 mmol) en tolueno (3 ml), se añadió p-TsOH (36 mg, 0,2 mmol) y etano-1,2-ditiol (39 mg, 0,4 mmol). La mezcla se agitó a 110°C durante 4 h, luego se vertió en agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía
en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para dar el producto deseado (80 mg, 80% de rendimiento). LCMS: m/z 507 (M+H)+.
Etapa B. 2-(Difluoro(fenil)metil)-6-(3-metoxibendl)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]pirídazin-5-ona. A una mezcla de 6-(3-metoxibencil)-4-metil-2-(2-fenil-1,3-ditiolan-2-il)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (65 mg, 0,13 mmol) y NIS (poco) en DCM (5 ml) se añadió Py.HF (1 ml). La mezcla de reacción se agitó a t.a. en atmósfera de N2 durante 2 h, luego se vertió en agua y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para dar un sólido (50 mg, 76% de rendimiento). LCMS: m/z 595 (M+H)+.
A una mezcla del sólido anterior (25 mg, 0,04 mmol) en THF/MeOH (3 ml/2 ml) se añadió Pd/C (5 mg). La mezcla se agitó a t.a. en atmósfera de H2 durante 40 min y luego se filtró. El filtrado se concentró a presión reducida y el residuo se purificó por TLC preparativa para proporcionar el producto deseado (7 mg, 36,8% de rendimiento). LCMS: 453 (M+H)+. RMN 1H (400 MHz, DMSO-da) 88,70 (s, 1H), 7,74-7,70 (m, 2H), 7,64-7,56 (m, 3H), 7,26-7,20 (m, 1H), 6,88-6,84 (m, 3H), 5,33 (s, 2H), 4,25 (s, 3H), 3,71 (s, 3H).
Ejemplo 2L. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-2-(1-fenilciclopropil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 4-Metil-2-(1-fenilcidopropil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-bencil-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg, 0,1 mmol) en DMF seco (2 ml) se añadieron 1,2-dibromoetano (10 |jl, 0,1 mmol) y TBAB (3 mg, 0,01 mmol). La mezcla se agitó a t.a. durante 30 min, seguido de la adición de NaH (8 mg, 0,2 mmol). La mezcla se agitó a t.a. durante 3 h, luego se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por TLC preparativa para proporcionar el producto deseado (26 mg, 44,7% de rendimiento). LCMS: 583 (M+H)+.
Etapa B. 6-((1H-Indazol-4-il)metil)-4-metil-2-(1-fenilddopropil)-4H-tiazolo[5',4’:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 4-metil-2-(1-fenilciclopropil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (26 mg, 0,044 mmol) en una solución de HCl en dioxano (4 M, 2 ml) se agitó a t.a. durante 3 h, luego se concentró a presión reducida. El residuo fue purificado por HPLC preparativa para obtener el producto deseado (3 mg, 15% de rendimiento). LCMS: 453 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 813,11 (s, 1H), 8,47 (s, 1H), 8,12 (s, 1H), 7,59 - 7,52 (m, 2H), 7,50 - 7,38 (m, 4H), 7,32 - 7,23 (m, 1H), 6,95 (d, 1H), 5,63 (s, 2H), 4,24 (s, 3H), 1,84-1,81 (m, 2H), 1,59-1,57 (m, 2H).
Ejemplo 2M. Síntesis de 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzamida .

Etapa A. Síntesis de 3-((2-bromo-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo A -40°C en atmósfera de N2, a una mezcla de 3-((4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo (200 mg, 0,62 mmol) y CBr4 (1,03 g, 3,11 mmol) en THF (15 ml) se añadió gota a gota LiHMDS (1,24 ml, 1 M en THF). La mezcla de reacción se agitó a -40 °C durante 2 h, se inactivó con solución sat. de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre agua Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna (gel de sílice, EtOAc en PE al 0-25%) para proporcionar el 3-((2-bromo-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo (200 mg, 80,6% de rendimiento). LC-MS (ESI): m/z 400 (M+H)+.
Etapa B. Síntesis de 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo A una mezcla de 3-((2-bromo-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo (100 mg, 0,25 mmol) y ácido metilborónico (45 mg, 0,75 mmol) en tolueno (2 ml) se añadió Na2CO2 (53 mg, 0,5 mmol), seguido de Pd2(dba)3 (23 mg, 0,025 mmol) y Xantophos (14 mg, 0,025 mmol). La mezcla de reacción se agitó a 100°C durante 15 h. La mezcla de reacción se filtró y el filtrado se evaporó. El residuo se purificó por cromatografía en columna (gel de sílice, EtOAc en éter de petróleo al 0-50%) para proporcionar el 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo (60 mg, 71,4% de rendimiento). LC-MS (ESI): m/z 336 (M+H)+.
Etapa C. Síntesis de 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzamida Una solución
de 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)benzonitrilo (60 mg, 0,18 mmol) en H2SO4 (1 ml) se agitó a 30°C durante la noche. La reacción se inactivó mediante solución sat. de NaHCO3 y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se evaporaron. El residuo se purificó por TLC preparativa (MeOH en DCM al 10%) para proporcionar la 3-((2,4-dimetil-5-oxo-4H-tiazolo[5',4':4,5]pirrol[2,3-d]piridazin-6(5H)-il)metil)benzamida (10 mg, 15,7% de rendimiento). lC-MS (ESI): m/z 354 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 88,59 (s, 1H), 7,98 (s, 1H), 7,81 (s, 1H), 7,77 (d, 1H), 7,46 (d, 1H), 7,41 (t, 1H), 7,35 (s, 1H), 5,40 (s, 2H), 4,26 (s, 3H), 2,86 (s, 3H).
Ejemplo 2N. Síntesis de 2-(2-(1H-pirazol-3-il)etil)-4-metil-6-((1 -metil-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Síntesis de 3-yodo-1-(tetrahidro-2H-piran-2-il)-1H-pirazol A una mezcla de 3-yodo-1H-pirazol (1 g, 5,16 mmol) y p-TsOH (88 mg, 0,52 mmol) en DCM (15 ml) se añadió DHP (0,56 ml, 6,19 mmol) y se agitó a t.a. durante 2 h. La mezcla de reacción se lavó con solución sat. de NaHCO3 y salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0 -10%) para dar el 3-yodo-1-(oxan-2-il)-1H-pirazol (1,4 g). LC-MS (m/z 279 (M+H)+.
Etapa B. Síntesis de (E)-1-(tetrahidro-2H-piran-2-il)-3-(2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)vinil)-1H-pirazol En atmósfera de nitrógeno, a una mezcla de 3-yodo-1-(tetrahidro-2H-piran-2-il)-1H-pirazol (150 mg, 0,54 mmol) en tolueno (3 ml) se le añadió 2-etenil-4,4,5,5-tetrametil-1,3,2-dioxaborolano (0,18 ml, 1,08 mmol), Et3N (0,37 ml, 2,7 mmol) y Pd(PBu3)2 (14 mg, 0,03 mmol). La reacción se agitó a 100°C durante 3 h. La mezcla se concentró y se purificó por TLC preparativa (EtOAc en PE al 35%) para dar el 1-(oxan-2-il)-3-[(E)-2-(tetrametil-1,3,2-dioxaborolan-2-il)etenil]-1H-pirazol (60 mg, 37% de rendimiento). LC-MS (ESI): m/z 305 (M+H)+.
Etapa C. Síntesis de 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (500 mg, 2,42 mmol) en DMF (15 ml) se añadió K2CO3 (335 mg, 2,42 mmol). Después de agitar a 50°C durante 30 min, se añadió una solución de 3-(bromometil)-1-metil-1H-pirazol (636 mg, 3,64 mmol) en DMF (2 ml). La reacción se agitó a 50°C durante la noche. La suspensión se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE 0 ~ 100) para dar la 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (280 mg). LC-MS (ESI): m/z 301 (M+H)+.
Etapa D. Síntesis de 2-yodo-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-6-((1-metil-1H-pirazo1-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (280 mg, 0,93 mmol) en tolueno (10 ml) se añadió pentafluoroyodobenceno (0,50 ml, 3,73 mmol) y t-BuOK (209 mg, 1,86 mmol). La reacción se agitó a 135°C durante 2 h en atmósfera de nitrógeno. La mezcla se enfrió a t.a. y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0 ~ 100%) para dar la 2-yodo-4-metil-6-((1-metil-1H-pirazo1-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg). LC-MS (ESI): m/z 427 (M+H)+.
Etapa E. Síntesis de (E)-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1-(tetrahidro-2H-piran-2-il)-1H-pirazol-3-il)vinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona En atmósfera de nitrógeno, a una mezcla de 2-yodo-4-metil-6-((1-metil-1H-pirazo1-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg, 0,71 mmol) y (E)-1-(tetrahidro-2H-piran-2-il)-3-(2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)vinil)-1H-pirazol (300 mg, 0,99 mmol) en Dm E (5 ml) y agua (1 ml) se añadieron Na2CO3 (149 mg, 1,41 mmol) y Pd (PPtb^Ch (49 mg, 0,071 mmol). La mezcla se agitó a 80°C durante 3 h. Luego, la mezcla se enfrió, se diluyó con EtOAc, se lavó con agua y salmuera. La capa orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0 ~ 100%) para dar la (E)-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1-(tetrahidro-2H-piran-2-il)-1H-pirazo1-3-il)vinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (170 mg). LC-MS (ESI): m/z 477 (M+H)+.
Etapa F. Síntesis de 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1-(tetrahidro-2H-piran-2-il)-1H-pirazol-3-il)etil)-4Htiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de (E)-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1-(tetrahidro-2H-piran-2-il)-1H-pirazol-3-il)vinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (80 mg, 0,17 mmol) en THF (3 ml) y MeOH (3 ml) se añadió Pd/C (10 mg). La reacción se agitó en H2 a t.a. durante 6 h. La mezcla se filtró y el filtrado se concentró. El residuo se purificó por TLC preparativa (EtOAc) para dar la 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1 -(tetrahidro-2H-piran-2-il)-1H-pirazol-3-il)etil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg). LC-MS (ESI): m/z479 (M+H)+.
Etapa G. Síntesis de 2-(2-(1H-pirazol-3-il)etil)-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(1-(tetrahidro-2H-piran-2-il)-1H-pirazo1-3-il)etil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,042 mmol) en etanol (2 ml) se añadió HCl (0,5 ml, 4 M en dioxano). La mezcla de reacción se agitó a 50°C durante 30 min. La mezcla de reacción se enfrió y se vertió en solución sat. de NaHCO3, se extrajo con EtOAc. La capa orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por TLC preparativa (MeOH en Dc M al 10%) para dar la 2-(2-(1H-pirazol-3-il)etil)-4-metil-6-((1-metil-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (10 mg, 61% de rendimiento). LC-MS (ESI): m/z 395 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 12,56 (s, 1H), 8,51 (s, 1H), 7,56 (d, 1H), 7,52 (s, 1H), 6,12 (d, 1H), 6,08 (d, 1H), 5,27 (s, 2H), 4,27 (s, 3H), 3,77 (s, 3H), 3,48 (t, 2H), 3,13 (t, 2H).

Ejemplo 20. Síntesis de 2-((1H-pirazol-5-il)oxi)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 5-hidroxi-1-(4-metoxibencil)-1H-pirazol-4-carboxilato de etilo A una mezcla de dihidrocloruro de (4-metoxibencil)hidrazina (2,25 g, 10 mmol) y K2CO3 (4.14 g, 30 mmol) en metanol (50 ml) y agua (10 ml) se añadió 2-(etoximetilen)malonato de dietilo (2,16 g, 10 mmol). La mezcla se agitó a 80°C durante 5 h. Después la mezcla se inactivó con solución ac. de NH4Cl y se extrajo con EtOAc. Las capas orgánicas se lavaron con salmuera, se secaron sobre Na2SO4 y concentraron. El residuo se purificó por cromatografía en columna (gel de sílice, EtOAc en PE al 0-50%) para proporcionar 1,1 g de 5-hidroxi-1-(4-metoxibencil)-1H-pirazol-4-carboxilato de etilo.
Etapa B. Síntesis de ácido 5-hidroxi-1-(4-metoxibencil)-1H-pirazol-4-carboxílico A una solución de 5-hidroxi-1 -(4-metoxibencil)-1H-pirazol-4-carboxilato de etilo (800 mg, 2,9 mmol) en metanol (10 ml) se añadió una solución de kOh (800 mg, 15 mmol) en agua (10 ml). La mezcla se agitó a temperatura ambiente durante 16 h. Después la mezcla se concentró y el residuo se usó directamente en la siguiente etapa. LC-MS (ESI): m/z 249 (M+H)+.
Etapa C. Síntesis de 1-(4-metoaibencil)-1H-pirazol-5-ol Una mezcla de ácido 5-hidroxi-1-(4-metoxibencil)-1H-pirazol-4-carboxílico se disolvió en HCl 2 M (50 ml) y se agitó a 60°C durante 16 h. Se eliminó el disolvente y se purificó el residuo por TLC preparativa para dar 120 mg de 1-(4-metoxibencil)-1H-pirazol-5-ol. LC-MS (ESI): m/z 205 (M+H)+.
Etapa D. Síntesis de N-(terc-butoxi)carbonil-(6-((2-((1-(4-metoxibencil)-1H-pirazol-5-il)oxi)-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo A una solución de 1-[(4-metoxifenil)metil]-1H-pirazol-5-ol (60 mg, 0,29 mmol) en DMF (8 ml) se añadió KOH (18 mg, 0,32 mmol) a 5°C. La mezcla de reacción se agitó a 5°C durante 30 min, se añadió una solución de N-(terc-butoxi)carbonil-(6-((2-bromo-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (174 mg, 0,29 mmol) en DMF (2 ml). La reacción se agitó a t.a. durante 16 h y luego se vertió en ácido cítrico acuoso 1 M, se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se evaporaron. El residuo se purificó por TLC preparativa (EA:PE=1:1) para proporcionar 80 mg del N-(tert-butoxi)carbonil-(6-((2-((1-(4-metoxibencil)1H-pirazol-5-il)oxi)-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de tercbutilo. LC-MS: m/z 715 (M+H)+.
Etapa E. Síntesis de 2-((1H-pirazol-5-il)oxi)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona Una solución de N-(terc-butoxi)carbonil-(6-((2-((1-(4-metoxibencil)-1H-pirazol-5-il)oxi)-4-metil-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (60 mg, 0,08 mmol) en TFA (2 ml) se agitó a t.a. durante la noche. La reacción se concentró y purificó por HPLC preparativa para dar 15 mg de la 2-((1H-pirazol-5-il)oxi)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 395 (M+H)+. RMN 1H (400 MHz, metanol-d4) 58,41 (s, 1H), 7,76 - 7,63 (m, 2H), 6,79 (d, 1H), 6,64 (d, 1H), 6,31 (d, 1H), 5,42 (s, 2H), 4,26 (s, 3H).
Ejemplo 3. Preparación de compuestos de fórmula E3-ii y derivados con el esquema E3
Esquema E3

El compuesto E3-ii se puede sintetizar mediante una reacción de Stille entre el compuesto E3-i y tributil(vinil)estannano. La reacción de Heck de E3-ii en presencia de un catalizador (p. ej., catalizador de paladio tal como Pd(Pt-Bu3)2, DMF) seguida de la reducción del grupo alquenilo puede generar el compuesto E3-ix. Alternativamente, la hidrogenación convencional del compuesto E3-ii genera el compuesto E3-iii. La hidroboración del compuesto E3-ii seguida de oxidación con perborato de sodio da el producto E3-vii. La oxidación directa del compuesto E3-ii con óxido de osmio (VIII) y peryodato de sodio proporciona el aldehido E3-iv. La adición nucleófila del aldehido E3-iv da el producto E3-v. La reducción convencional del compuesto E3-iv con borohidruro de sodio da el compuesto E3-vi. La aminación reductora del compuesto E3-iv da el compuesto E3-viii. En donde Q y R2 son como se definen en una cualquiera de las realizaciones primera a vigésima sexta de la invención. En ciertas realizaciones, Q y R2 son cada uno independientemente arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido.
Ejemplo 3A. Síntesis de 2-etil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 6-(3-Metoxibendl)-4-metíl-2-vmil-4-tíazolo[5',4':4,5]pirrolo[2,3-d]pirídazm-5(6H)-ona. A una mezcla de 2-cloro-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (600 mg, 1,67 mmol) y tributil(vinil)estannano (1 ml, 3,4 mmol) en DMF (6 ml) se añadió Pd(PPh3)4. La mezcla se agitó a 100°C durante la noche en atmósfera de N2 luego se vertió en agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/2) para dar el producto deseado (410 mg, 68% de rendimiento). LCMS: m/z 353 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,61 (s, 1H), 7,23 (t, 1H), 7,10 (dd, 1H), 6,89 - 6,80 (m, 3H), 6,28 (d, 1H), 5,75 (d, 1H), 5,32 (s, 2H), 4,27 (s, 3H), 3,72 (s, 3H).
Etapa B. 2-Etil-6-(3-metoxibendl)-4-metil-4H-tiazolo[5',4':4,5]pirmlo[2,3-d]pirídazin-S(6H)-ona. A una mezcla de 6-(3-metoxibencil)-4-metil-2-vinil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,88 mmol) en MeOH (1 ml) y THF (1 ml) en atmósfera de N2 se añadió Pd/C (10 mg). La mezcla se agitó en H2 a t.a. durante 1 h, luego se filtró a través de Celite. El filtrado se concentró a presión reducida y el residuo se purificó por TLC preparativa para proporcionar el
producto deseado (5 mg, 16,7% de rendimiento).
LCMS: m/z 355 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,56 (s, 1H), 7,23 (t, 1H), 6,89 - 6,78 (m, 3H), 5,32 (s, 2H), 4,26 (s, 3H), 3,72 (s, 3H), 3,17 (q, 2H), 1,38 (t, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados.

Ejemplo 3B. Síntesis de 2-(hidroximetil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 6-(3-Metoxibencil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído. A una mezcla de 6-(3-metoxibencil)-4-metil-2-vinil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (410 mg, 1,16 mmol) en dioxano (6 ml) y agua (2 ml) se añadieron NaIO4 (1 g, 4,6 mmol), 2,6-dimetilpiridina (0,27 ml, 2,32 mmol) y OsO4 (cat.). La mezcla se agitó a t.a. durante 4 h, luego se inactivó con solución acuosa saturada de Na2S2O3 y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc =1/1) para dar el producto deseado (130 mg, 31,7% de rendimiento). LCMS: m/z 387 (M+MeOH+H)+. RMN 1H (400 MHz, DMSO-d6) 5 10,08 (s, 1H), 8,75 (s, 1H), 7,24 (t, 1H), 6,88-6,83 (m, 3H), 5,34 (s, 2H), 4,34 (s, 3H), 3,72 (s, 3H).
Etapa B. 2-(Hidroximetil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5'.4'.4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 6-(3-metoxibencil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (30 mg, 0,08 mmol) en metanol (2 ml) se añadió NaBH4 (6 mg, 0,16 mmol). La mezcla se agitó a t.a. durante 10 min, luego se vertió en agua y se extrajo con EtOAc. La capa orgánica se concentró a presión reducida y el residuo se purificó por HPLC preparativa para proporcionar el producto deseado (10 mg, 35,7% de rendimiento). LCMS: m/z 357 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,59 (s, 1H), 7,23 (t, 1H), 6,86-6,82 (m, 3H), 6,34 (t, 1H), 5,32 (s, 2H), 4,89 (d, 2H), 4,26 (s, 3H), 3,72 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.


Ejemplo 3C: Síntesis de 2-(2-hidroxipropan-2-il)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 2-(1-Hidroxietil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5,4:4,5]pirrolo[2,3-d]piridazm-5(6H)-ona. A una mezcla de 6-(3-metoxibencil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (100 mg, 0,28 mmol) en THF (3 ml) a 0°C se añadió gota a gota cloruro de metilmagnesio (0,19 ml, 0,56 mmol). La mezcla se agitó a t.a. durante 10 min, luego se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/2) para dar el producto deseado (40 mg). LCMS: m/z 371 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,58 (s, 1H), 7,24 (t, 1H), 6 ,97-6,77 (m, 3H), 6,41 (d, 1H), 5,40 - 5,23 (m,2H), 5,18-5,02 (m, 1H), 4,26 (s, 3H), 3,72 (s, 3H), 1,55 (d, 3H).
Etapa B. 2-Acetil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-(1-hidroxietil)-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,08 mmol) en DCM (3 ml) se añadió óxido de manganeso (IV) (35 mg, 0,4 mmol). La mezcla se agitó a t.a. durante 1 h, luego se filtró a través de Celite. El filtrado se concentró a presión reducida para dar el producto deseado (25 mg). LCMS: m/z 369 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,83 (s, 1H), 7,35 (t, 1H), 6,99-6,94 (m, 3H), 5,45 (s, 2H), 4,45 (s, 3H), 3,83 (s, 3H), 2,85 (s, 3H).
Etapa C. 2-(2-Hidmxipmpan-2-il)-6-(3-metoxibencil)-4-metil-4H-tiazob[5',4':4,5]pirmlo[2,3-d]piridazm-5(6H)-ona. A una mezcla de 2-acetil-6-(3-metoxibencil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (20 mg, 0,05 mmol) en THF (1 ml) a 0°C se añadió gota a gota cloruro de metilmagnesio (0,08 ml, 0,15 mmol). La mezcla se agitó a t.a. durante 10, luego se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (10 mg). lCm S: m/z 385 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,58 (s, 1H), 7,24 (t, 1H), 6,89 - 6,81 (m, 3H), 6,28 (s, 1H), 5,33 (s, 2H), 4,26 (s, 3H), 3,72 (s, 3H), 1,60 (s, 6H).
Ejemplo 3D. Síntesis de 6-((1H-indazol-4-il)metil)-2-(2-hidroxietil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 2-(2-Hidroxietil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-mdazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirmb[2,3-d]piridazin-5-ona. A una mezcla de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-2-vinil-4,6-dihidro-5H-tiazolo[5',4,:4,5]pirrolo[2,3-d]piridazin-5-ona (100 mg, 0,20 mmol) en THF (2 ml) a 0°C en atmósfera de N2 se añadió BH3-THF (0,2 ml, 1 mol/l, 0,20 mmol). La mezcla se agitó a t.a. durante 2 h, luego se enfrió a 0°C, seguido de la adición de agua (1 ml) y NaBO3.4H2O (154 mg, 1,00 mmol). La mezcla se calentó lentamente a t.a. y se agitó a esa temperatura durante 3 h. La mezcla resultante se vertió en solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna sobre gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (30 mg). lC-MS: m/z 511 (M+H)+.
Etapa B. 6-((1H-Indazol-4-il)metil)-2-(2-hidroxietil)-4-metil-4,6-dihidro-5H-tiazolo[5,4 ':4,5]pirrob[2,3-d]pirídazm-5-ona. A una solución de 2 (30 mg, 0,18 mmol) en DCM (3 ml) a 0°C se añadió gota a gota TFA (1 ml). La mezcla resultante se agitó a t.a. durante 16 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para dar el
producto deseado (2,0 mg). LC-MS: m/z381 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,11 (s, 1H), 8,58 (s, 1H), 8,14 (s, 1H), 7,45 (d, 1H), 7,30-7,26 (m, 1H), 6,96 (d, 1H), 5,66 (s, 2H), 5,02-5,00 (m, 1H), 4,27 (s, 3H), 3,85-3,81 (m, 2H), 3,32 3,25 (m, 2H).
Ejemplo 3E. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-2-((metilamino)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 4-Metil-2-((metilamino)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (50 mg, 0,1 mmol) en THF (10 ml) a 0°C se añadió gota a gota MeNH2 (30% en MeOH, 21 mg, 0,2 mmol). La mezcla de reacción se agitó a t.a. durante 2 h, seguido de la adición de cianoborohidruro de sodio (19 mg, 0,3 mmol). La mezcla resultante se agitó a t.a. durante la noche y luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (35 mg). LCMS: m/z 511 (M+H)+.
Etapa B. 6-((1H-Indazol-4-il)metil)-4-metil-2-((metilamino)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-((metilamino)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (35 mg, 0,07 mmol) en DCM (10 ml) a 0°C se añadió gota a gota t Fa (3 ml). La mezcla de reacción se agitó a t.a. durante 2 h, luego se concentró a presión reducida. El residuo se purificó por HμLC preparativa para dar el producto deseado (5 mg). LCMS: m/z 380 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,13 (s, 1H), 8,59 (s, 1H), 8,16 (d, 1H), 7,45 (d, 1H), 7,31 - 7,25 (m, 1H), 6,96 (d, 1H), 5,66 (s, 2H), 4,26 (s, 3H), 4,09 (s, 2H), 2,40 (s, 3H).
Ejemplo 3F. Síntesis de 6-((1H-indazol-4-il)metil)-2-(aminometil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Oxima del (E)-4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-mdazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazma-2-carbaldehído. A una mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (120 mg, 0,24 mmol) en MeOH (10 ml) a 0°C se añadió hidrocloruro de hidroxilamina (50 mg, 0,73 mmol), seguido de la adición de KOAc (71 mg, 0,73 mmol). La mezcla de reacción se agitó a t.a. durante 8 h, luego se inactivó con solución acuosa saturada de NH4Cl y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (90 mg). LCMS: m/z 510 (M+H)+.
Etapa B. 2-(Ammometil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-mdazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de oxima del (E)-4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (90 mg, 0,18 mmol) en ácido acético (10 ml) se añadió Zn en polvo (58 mg, 0,88 mmol). La mezcla de reacción se agitó a t.a. durante la noche y luego se filtró a través de Celite. El filtrado se concentró a presión reducida y el residuo se purificó por HPLC preparativa para dar el producto deseado (70 mg). LCMS: m/z 496 (M+H)+.
Etapa C. La 6-((1H-indazol-4-il)metil)-2-(aminometil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizó utilizando el procedimiento del ejemplo 3D. LCMS: m/z 366 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 13,13 (s, 1H), 8,59 (s, 1H), 8,15 (s, 1H), 7,45 (d, 1H), 7,33- 7,20 (m, 1H), 6,97 (d, 1H), 5,66 (s, 2H), 4,24 (d, 3H), 4,17 (s, 2H). Ejemplo 3G. Síntesis de N-((6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metil)acetamida

Etapa A. N-((4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-mdazol-4-il)metil)-5,6-dihidro-4H- tíazolo[5',4':4,5]pirrolo[2,3-d]pirídazm-2-il)metíl)acetamida. A una mezcla de 2-(aminometil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (35 mg, 0,07 mmol) en DCM (10 ml) a 0°C se añadió anhídrido acético (22 mg, 0,21 mmol), seguido de la adición de trietilamina (22 mg, 0,21 mmol) y DMAP (0,8 mg, 0,007 mmol). La mezcla de reacción se agitó a t.a. durante 2 h, luego se inactivó con agua y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (25 mg) como aceite amarillo. LCMS: m/z 538 (M+H)+.
Etapa B. La N-((4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metil)acetamida se sintetizó utilizando el procedimiento del ejemplo 3D. LCMS: m/z 408 (M+H)+. RMN 1H (400 MHz, D M SO d) 513,12 (s, 1H), 8,67 (s, 1H), 8,29 (s, 1H), 8,16 (s, 1H), 7,46 (d, 1H), 7,31 7,21 (m, 1H), 6,97 (d, 1H), 5,67 (s, 2H), 4,31 (s, 3H), 1,93 (s, 3H).
Ejemplo 3H. Síntesis de N-((6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metil)metanosulfonamida

Etapa A. N-((4-metil-5-oxo-6-((1-((2-(trímetílsilil)etoxi)metíl)-1H-mdazol-4-il)metíl)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirmlo[2,3-d]piridazin-2-il)metil)metanosulfonamida. A una mezcla de 2-(aminometil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4’:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,1 mmol) en DCM (5 ml) a 0°C, se añadió EtaN (30,62 mg, 0,3 mmol), seguido de la adición de MsCl (9,24 mg, 0,081 mmol). La mezcla se agitó a 20°C durante 2 h, luego se concentró a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (20 mg).
Etapa B. La N-((6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metil)metanosulfonamida se sintetizó utilizando el procedimiento del ejemplo 3D. LCMS: m/z 444 (M+H)+. r Mn 1H (400 MHz, DMSO-d6) 5 13,11 (s, 1H), 8,62 (s, 1H), 8,22 (s, 1H), 8,17 (d, 1H), 7,45 (d, 1H), 7,34 - 7,20 (m, 1H), 6,97 (d, 1H), 5,66 (s, 2H), 4,64 (d, 2H), 4,27 (s, 3H), 3,04 (s, 3H).
Ejemplo 31. Síntesis de 2-(2-(1H-pirazol-3-il)etil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-dlpiridazin-5(6H)-ona

Etapa A. Síntesis de N-[(terc-butoxi)carbonil]-N-[6-({4-bromo-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo A -40°C en atmósfera de N2 atmósfera, a una mezcla de N-[(terc-butoxi)carbonil]-N-[6-({7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo (1,4 g, 2,73 mmol) y CBr4 (4.52 g, 13,65 mmol) en t Hf (20 ml) se añadió gota a gota LiHMDS (5,46 ml, 5,46 mmol). La mezcla de reacción se agitó a -40°C durante 30 min, luego se inactivó con agua (4 ml) y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, PE/EA = 10:1~3:1) para proporcionar 500 mg del N-[(tercbutoxi)carbonil]-N-[6-({4-bromo-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo. LC-MS: m/z 591 (M+H)+. Etapa B. Síntesis de N-[(terc-butoxi)carbonil]-N-[6-({4-vinil-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo A una solución de N-[(terc-butoxi)carbonil]-N-[6-({4-bromo-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo (500 mg, 0,85 mmol) en DMF (10 ml) se añadió tributil(etenil)estannano (536 mg, 1,69 mmol) y DIPEA (327 mg, 2,53 mmol), seguido de Pd(Pph3)4 (105 mg, 0,08 mmol). La mezcla de reacción se agitó en atmósfera de N2 a 80°C durante 3 h, luego se inactivó mediante H2O y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, PE/EA = 10:1-5:1) para proporcionar 300 mg del N-[(terc-butoxi)carbonil]-N-[6-({4-vinil-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo. LC-MS: m/z 539 (M+H)+.
Etapa C. Síntesis de (E)-3-(2-(6-((6-((terc-butoxicarbonil)[(terc-butoxi)carbonil]amino)piridin-2-il)metil)-4 -metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)vinil)-1H-pirazol-1-carboxilato de terc-butilo A una solución de N-[(terc-butoxi)carbonil]-N-[6-({4-vinil-7-metil-9-oxo-3-tia-5,7,10,1l-tetraazatricido[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo (300 mg, 0,56 mmol) en DMF (4 ml) se añadió 3-yodo-1H-pirazol-1-carboxilato de terc-butilo (180 mg, 0,61 mmol). La mezcla de reacción se agitó a 100°C durante la noche. Después de enfriar a t.a., la mezcla de reacción se inactivó con H2O y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (PE/EA=1:1) para proporcionar 200 mg de (E)-3-(2-(6-((6-((terc-butoxicarbonilo)[(tercbutoxi)carbonil]amino)piridin-2-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)vinil)-1H-pirazol-1-carboxilato de terc-butilo. LC-MS: m/z 705 (M+H)+.
Etapa D. Síntesis de 3-(2-(6-((6-((terc-butoxicarbonil)[(terc-butoxi)carbonil]amino)piridin-2-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)etil)-1H-pirazol-1-carboxilato de terc-butilo A una solución de (E)-3-(2-(6-((6-((terc-butoxicarbonil)[(terc-butoxi)carbonil]amino)piridin-2-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)vinil)-1H-pirazol-1 -carboxilato de terc-butilo (200 mg, 0,28 mmol) en THF/MeOH (4 ml, 10:1) se añadió Pd/C (6 mg, 10% en peso). La mezcla de reacción se agitó en hidrógeno a t.a. durante 12 h. La mezcla se filtró a través de una capa de Celite y el filtrado se concentró. El residuo se purificó por TLC preparativa (PE/EA=1:1) para proporcionar 100 mg de 3-(2-(6-((6-((terc-butoxicarbonN)[(terc-butoxi)carbonN]amino)piridin-2-N)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)etil)-1H-pirazol-1-carboxilato de terc-butilo. Lc -m S: m/z 707 (M+H)+.
Etapa E. Síntesis de 2-(2-(1H-pirazol-3-il)etil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A 0°C en atmósfera de N2, a una mezcla de 3-(2-(6-((6-((terc-butoxicarbonil)[(tercbutoxi)carbonil]amino)piridin-2-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)etil)-1H-pirazol-1-carboxilato de terc-butilo (100 mg, 0,14 mmol) en EtOH (2 ml) se añadió HCl (2 ml, 4 M en dioxano). Después de agitar a 80°C durante 1 h, la mezcla se vertió en solución sat. de NaHCO3, se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (DCM/MeOH=10:1) para producir 10 mg de 2-(2-(1H-pirazol-3-il)etil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS: m/z 407 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 812,52 (s, 1H), 8,55 (s, 1H), 7 ,60-7,10 (m, 2H), 6,30 (d, 1H), 6,18-6,02 (m, 2H), 5,90 (s, 2H), 5,19 (s, 2H), 4,26 (s, 3H), 3,61 - 3,41 (m, 2H), 3,19 - 3,12 (m, 2H).
Ejemplo 4. Síntesis de compuestos E4-vii y E4-viii

en donde Hal es halógeno (p. ej., Br o I); LG es un grupo saliente (p. ej., halógeno tal como Br o I; OMs u OTs); Q es como se define en una cualquiera de las realizaciones primera a vigésima sexta; y Q' es Q adicionalmente funcionalizado (p. ej., arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido). La reacción de sustitución aromática del compuesto E4-i con metanotiolato de sodio proporciona el compuesto E4-ii, que se puede convertir en el compuesto E4-v usando la síntesis del compuesto E1-iii a E1-vi. La oxidación del compuesto E4-v con mCPBA da el compuesto E4-vi y E4-vii respectivamente. El compuesto E4-viii se puede convertir a partir de E4-v mediante la funcionalización adicional de Q a Q'.
Ejemplo 4A. Síntesis de 6-(3-metoxibencil)-4-metil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A: 4-Metíl-2-(metíltío)-4H-pirrolo[2,3-d]tíazol-5-carboxilato de etilo. A una mezcla de 2-bromo-4-metil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (500,0 mg, 1,73 mmol) en EtOH (10,0 ml) se añadió NaSMe (240,0 mg, 3,5 mmol). La mezcla de reacción se agitó a 25°C durante 3 h, luego se inactivó con agua helada y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (460 mg) que se usó directamente en la siguiente etapa sin ninguna purificación. LC-MS: m/z 257 (M+H)+.
Etapa B: 6-Formil-4-metil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. A una solución de 4-metil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (460,0 mg, 1,8 mmol) y N-metil-N-fenilformamida (490 mg, 3,6 mmol) en DCE (10 ml) se añadió POCl3 (550,0 mg, 3,6 mmol). La mezcla resultante se agitó a 130°C durante 3 h, luego se inactivó con agua helada y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 8/1) para dar el producto deseado (320,0 mg). LC-MS: m/z 285 (M+H)+.
Etapa C: 4-Metil-2-(metiltio)-4,6-dihidro-5H-tiazolo[54':4,5]pirrolo[2,3-d]piridazin-5-ona. A una solución de 6-formil-4-metil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (300,0 mg, 1,06 mmol) en EtOH (5,0 ml) se añadió N2H4.H2O (2 ml, 98% en peso). La mezcla de reacción se agitó a t.a. durante 1 h, luego se calentó a 60°C durante la noche y luego se enfrió. El sólido se recogió por filtración y se secó con alto vacío para dar el producto deseado (180,0 mg). lC-MS: m/z 253 (M+H)+. RMN 1H (400 MHz, DMSO-da) 5 12,61 (s, 1H), 8,48 (s, 1H), 4,22 (s, 3H), 2,81 (s, 3H).
Etapa D: 6-(3-Metoxibencil)-4-metil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una solución de 4-metil-2-(metiltio)-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (180,0 mg, 0,7 mmol) en DMF (5,0 ml) se añadió carbonato de potasio (200 mg, 1,4 mmol). La mezcla se agitó a 60°C durante 1 h, seguido de la adición de 1-(clorometil)-3-metoxibenceno (170 mg, 1,07 mmol). La mezcla resultante se agitó a 60°C durante 3 h, luego se inactivó con agua helada (100,0 ml) y se extrajo con DCM. (10,0 m lx3). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: DCM/MeOH = 10/1) para dar el producto deseado (200,0 mg). LC-MS: m/z 373 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,57 (s, 1H), 7,24 (t, 1H), 6,88-6,82 (m, 3H), 5,32 (s, 2H), 4,24 (s, 3H), 3,72 (s, 3H), 2,82 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.


Ejemplo 4B. Síntesis de 4-metil-2-(metiltio)-6-((2-oxo-2,3-dihidropirimidin-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Una mezcla de 6-((2-cloropirimidin-4-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,132 mmol) en HCl (10 ml) se agitó a 100°C durante 1 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (5,0 mg, 10,51% de rendimiento). LCMS: m/z 361 (M+H)+. RMN 1H (400 MHz, DMSO-de) 511,82 (s, 1H), 8,60 (s, 1H), 7,82 (s, 1H), 6,14 (s, 1H), 5,22 (s, 2H), 4,24 (s, 3H), 2,82 (s, 4H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados.

Ejemplo 4C. Síntesis de 6-(3-metoxibencil)-4-metil-2-(metilsulfinil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

A una solución de 6-(3-metoxibencil)-4-metil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (30,0 mg, 0,08 mmol) en DCM (3,0 ml) a 0°C se añadió m-CPBA (14,0 mg, 0,08 mmol). La mezcla resultante se agitó a 0°C durante 1 h, luego se inactivó con agua helada y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: DCM/MeOH = 10/1) para dar el producto deseado (15,0 mg). LC-MS: m/z 389 (M+H)+. RMN 1H (400 MHz, D M S O d) 58,74 (s, 1H), 7,25 (t, 1H), 6,86 (dd, 3H), 5,35 (s, 2H), 4,30 (s, 3H), 3,73 (s, 3H), 3,11 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.


Ejemplo 4D. Síntesis de 4-((4-metil-2-(metilsulfinN)-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6
il)metil)benzamida

Etapa A: 4-((4-Metíl-2-(metíltío)-5-oxo-4,5-dihidro-6H-tíazolo(5';4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)benzamida. Una mezcla de 4-((4-metil-2-(metiltio)-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)benzonitrilo (50,0 mg, 0,13 mmol) en H2SO4 (1,0 ml) se agitó a 0°C durante 1 h, luego se neutralizó con solución acuosa saturada de NaHCO3 y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: DCM/MeOH = 10/1) para dar el producto deseado (20,0 mg). LC-MS: m/z 386 (M+H)+.
Etapa B: 4-((4-Metil-2-(metilsulfinil)-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)benzamida. A una solución de 4-((4-metil-2-(metiltio)-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)benzamida (20,0 mg, 0,052 mmol) en DCM (2,0 ml) a 0°C se añadió m-CPBA (10,0 mg, 0,052 mmol). La mezcla resultante se agitó a 0°C durante 1 h, luego se inactivó con agua helada (10,0 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: DCM/MeOH = 10/1) para dar el producto deseado (5,0 mg). LC-MS: m/z 402 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,75 (s, 1H), 7,93 (s, 1H), 7,82 (d, 2H), 7,38-7,33 (m, 3H), 5,42 (s, 2H), 4,29 (s, 3H), 3,11 (s, 3H).
Ejemplo 4E. Síntesis de 6-(3-(2-hidroxipropan-2-il)bencil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. a-(3-(2-Hidroxipropan-2-il)bencil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 6-(3-acetilbencil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (120 mg, 0,31 mmol) en THF seco (5 ml) a 0°C se añadió gota a gota cloruro de metilmagnesio (0,3 ml, 0,9 mmol). La mezcla se agitó a t.a. durante 30 min, luego se vertió en solución ac. saturada de NH4Cl y se extrajo con EtOAc. La capa orgánica se separó y se concentró a presión reducida. El residuo se purificó por TLC preparativa para proporcionar el producto deseado (70 mg). LCMS: m/z 401 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,56 (s, 1H), 7,49 (s, 1H), 7,33 (d, 1H), 7,23 (t, 1H), 7,09 (d, 1H), 5,33 (s, 2H), 4,98 (s, 1H), 4,24 (s, 3H), 2,81 (s, 3H), 1,39 (s, 6H).
Etapa B. 6-(3-(2-Hidroxipropan-2-il)bencil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 6-(3-(2-hidroxipropan-2-il)bencil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (46 mg, 0,115 mmol) en DCM (3 ml) a 0°C se añadió m-CPBA (20 mg, 0,1 mmol, 85% en p/p). La mezcla se agitó a t.a. durante 30 min, luego se inactivó con solución acuosa saturada de Na2S2O3 y se extrajo con DCM. La capa orgánica se separó y se concentró a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (10 mg). LCMS: m/z 417 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,73 (s, 1H), 7,49 (s, 1H), 7,34 (d, 1H), 7,24 (t, 1H), 7,09 (d, 1H), 5,37 (s, 2H), 4,98 (s, 1H), 4,30 (s, 3H), 3,11 (s, 3II), 1,39 (s, 6II).
Ejemplo 4F. Síntesis de 6-((2-aminopiridin-4-il)metil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 2-Bromo-4-(bromometil)piridina. Una mezcla de 2-bromo-4-metilpiridina (1 g, 5,81 mmol), NBS (1,1 g, 6,39 mmol) y una cantidad catalítica de AIb N (100 mg) en CCl4 (10 ml) se agitó a 80°C durante la noche. La mezcla resultante se concentró a presión reducida y el residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 200/1) para dar el producto deseado (500 mg).
Etapa B. 6-((2-Bromopiridin-4-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,40 mmol) y K2Co 3 (164 mg, 1,19 mmol) en DMF (8 ml) se agitó a 60°C durante 2 h, seguido de la adición de una solución de 2-bromo-4-(bromometil)piridina (199 mg, 0,80 mmol) en DMF (2 ml) y una cantidad catalítica de TBAB (13 mg). La mezcla se agitó a 60°C durante la noche, luego se inactivó con agua (20 ml) y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con solución acuosa saturada de NH4Cl, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: pE/EtOAc = 10/1) para dar el producto deseado (150 mg). LCMS: m/z 423 (M+H)+.
Etapa C. (4-((4-Metil-2-(metiltio)-5-oxo-4H-tiazoto[5’,4 ’:4,5]pirrolo[2,3-d]piridazm-6(5H)-il)metil)piridm-2-il)carbamato de terc-butilo. Una mezcla de 6-((2-bromopiridin-4-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,24 mmol), carbamato de terc-butilo (83 mg, 0,71 mmol), KíPO4 (201 mg, 0,95 mmol), Pd2(dba)3 (18 mg, 0,02 mmol) y Xantphos (11 mg, 0,02 mmol) en dioxano (10 ml) se agitó a 100°C en atmósfera de nitrógeno durante la noche. La mezcla resultante se inactivó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para dar el producto deseado (100 mg). LCMS: m/z 459 (M+H)+.
Etapa D-E: El (4-((4-metil-2-(metilsulfinil)-5-oxo-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo se sintetizó usando un procedimiento similar al del ejemplo 4C y la 6-((2-aminopiridin-4-il)metil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizó utilizando un procedimiento similar al del ejemplo 3G. LCMS: m/z 375 (M+H)+. RMN 1H (400 MHz, D M S O d) 58,67 (s, 1H), 7,73 (d, 1H), 6,30 (d, 1H), 6,12 (s, 1H), 5,89 (s, 2H), 5,24 - 5,03 (m, 2H), 4,29 (s, 3H), 3,03 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 4G. Síntesis de 6-((2-aminotiazol-5-il)metil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A-B. El 2-bromo-5-(bromometil)tiazol y la 6-((2-bromotiazol-5-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizaron de manera similar al ejemplo 4F.
Etapa C. 6-((2-((2,4-Dimetoxibencil)amino)tiazol-5-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 6-((2-bromotiazol-5-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (130 mg, 0,30 mmol) y DlPEA (0,1 ml) en NMP (0,1 ml) y (2,4-dimetoxifenil)metanamina (0,1 ml) se agitó a 150°C durante 4 h. Luego, la mezcla de tracción se inactivó con agua (10 ml), se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluida con PE/EtOAc = 5/1) para dar el producto deseado (60 mg, 38,4% de rendimiento). LC-MS: m/z 515 (M+H)+.
Etapa D. 6-((2-((2,4-Dimetoxibencil)amino)tiazol-5-il)metil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 6-((2-((2,4-dimetoxibencil)amino)tiazol-5-il)metil)-4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,10 mmol) en THF (3 ml) a 0°C se añadió Oxone (61 mg, 0,10 mmol). La mezcla se agitó a 0°C durante 1 h, luego se inactivó con solución acuosa saturada de Na2S2O3 (5 ml) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (30 mg, 50,1% de rendimiento) que se usó directamente en la siguiente etapa sin más purificación. LC-MS: m/z 531 (M+H)+.
Etapa E. La 6-((2-aminotiazol-5-il)metil)-4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se sintetizó de manera similar al ejemplo 3G. LC-MS: m/z 381 (M+H)+. RMN 1H (400 MHz, Dm SO-cÍ6) 58,70 (s, 1H), 6,97 (s, 1H), 6,87 (s, 2H), 5,28 (s, 2H), 4,29 (s, 3H), 3,11 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 4H. Síntesis de 6-(3-metoxibencil)-4-metil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

A una solución de 6-(3-metoxibencil)-4-metil-2-(metilsulfinil)-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (30,0 mg, 0,08 mmol) en DCM (3,0 ml) a 0°C, era m-CPBA (35,0 mg, 0,2 mmol). La mezcla resultante se agitó a 0°C durante 1 h, luego se inactivó con agua helada y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: DCM/MeOH = 10/1) para dar el producto deseado (10,0 mg, 32% de rendimiento). LC-MS: m/z 405 (M+H)+. RMN 1H (400 MHz, DMSO-de) 58,78 (s, 1H), 7,24 (t, 1H), 6,92 - 6,82 (m, 3H), 5,35 (s, 2H), 4,33 (s, 3H), 3,72 (s, 3H), 3,57 (s, 3H).

Ejemplo 5. Síntesis de compuestos de fórmula E5-ii y derivados con el esquema E5. Esquema E5

En donde Q es como se define en una cualquiera de las realizaciones primera a vigésima sexta de la invención; Nu es un nucleófilo (es decir, una especie química que dona un par de electrones a un electrófilo para formar un enlace químico en relación con una reacción). Nu' es alcoxilo opcionalmente sustituido, tiol opcionalmente sustituido, amino opcionalmente sustituido o nucleófilos de carbono opcionalmente funcionalizados.
Ejemplo 5A. Síntesis de 6-((1H-indazol-4-il)metil)-2-(benciltio)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 2-(Benciltio)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de fenilmetanotiol (91,21 mg, 734,32 |jmol) en DCM (5 ml) a 0°C se añadió DIPEA (142,3 mg, 1,10 mmol) y 4-metil-2-(metilsulfonil)-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (200 mg, 0,37 mmol). La mezcla se agitó a 20°C durante 1 h, luego se inactivó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc= 5/1 a 3/1) para dar el producto deseado (200 mg). lCm S: m/z 589 (M+H)+
Etapa B. 6-((1H-Indazol-4-il)metil)-2-(benciltio)-4-metil-4,6-dihidro-5H-tiazolo[5';4':4,5]pirrolo[2,3-d]piridazin-5-ona. Una mezcla de 2-(benciltio)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 50,95 ^mol) en DCM/TFA (V:V = 3:1) se agitó a 20°C durante 2 h, luego se concentró a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (5 mg). LCMS: m/z 459 (M+H)+. RMN 1H (400 MHz, DMSO-da) 513,11 (s, 1H), 8,57 (s, 1H), 8,14 (s, 1H), 7,49 (d, 2H), 7,45 (d, 1H), 7,34 (t, 2H), 7,28 (dd, 2H), 6,95 (d, 1H), 5,65 (s, 2H), 4,62 (s, 2H), 4,26 (s, 3H).
Ejemplo 5B. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-sulfonamida
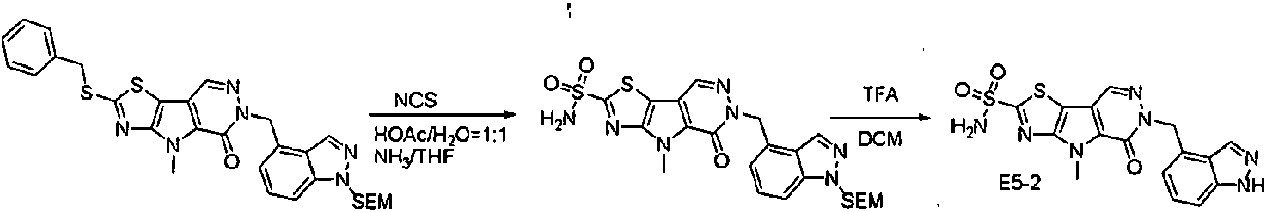
Etapa A. 4-Metil-5-oxo-6-((1-((2-(trímetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidm-4H-tíazolo[5',4':4,5]pirrob[2,3-d]piridazina-2-sulfonamida. A una solución de 2-(benciltio)-4-metil-6-((1 -((2-(trimetilsiMl)etoxi)metil)-lH-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,169 mmol) en HOAc/H2O (V:V= 1:1, 10 ml) se añadió NCS (45 mg, 0,34 mmol). La mezcla se agitó a 40°C durante 3 h, luego se enfrió a 0°C, seguido de adición lenta de NH3/THF (5 ml) hasta pH 9 a esa temperatura. La mezcla resultante se agitó a 20°C durante 0,5 h, luego se extrajo con EtOAc. Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por TLC preparativa (eluyente: PE/EtOAc = 1/1) para dar el producto deseado (20 mg). LCMS: m/z 546 (M+H)+.
Etapa B. 6-((1H-Indazol-4-il)metíl)-4-metíl-5-oxo-5,6-dihidro-4H-tíazolo[5',4 ':4,5]pirrolo[2,3-d]piridazina-2-sulfonamida. Mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-sulfonamida (20 mg, 36,65 ^mol) en DCM/TFA (V/V=3/1) se agitó a 20°C durante 2 h, luego se concentró a presión reducida. El residuo se purificó por Hp LC preparativa para dar el producto deseado (1,7 mg). LCMS: m/z 416 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,13 (s, 1H), 8,73 (s, 1H), 8,31 (s ancho, 2H), 8,17 (s, 1H), 7,47 (d, 1H), 7,36 -7,20 (m, 1H), 6,99 (d, 1H), 5,68 (s, 2H), 4,30 (s, 3H).


Ejemplo 5C. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)malonato de dimetilo

Etapa A. 2-(4-Metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)malonato de dimetilo. A una mezcla de t-BuOK (103 mg, 0,92 mmol) y malonato de dimetilo (9l mg, 0,69 mmol) en THF (5 ml) en atmósfera de N2 se añadió 4-metil-2-(metilsulfonil)-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (250 mg, 0,46 mmol). La mezcla de reacción se agitó a 60°C durante 16 h, luego se vertió en agua con hielo y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice para dar el producto deseado (100 mg, 36,48% de rendimiento). LC-MS: m/z 597 (M+H)+.
Etapa B. El 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)malonato se sintetizó de manera similar al ejemplo 5A. LC-MS: m/z 467 (M+H)+. RMN 1H (400 m Hz , DMSO-d6) 513,12 (s, 1H), 8,65 (s, 1H), 8,15 (s, 1H), 7,45 (d, 1H), 7,30-7,26 (m, 1H), 6,96 (d, 1H), 5,84 (s, 1H), 5,67 (s, 2H), 4,27 (s, 3H), 3,77 (s, 6H).
Ejemplo 5D. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetato de metilo
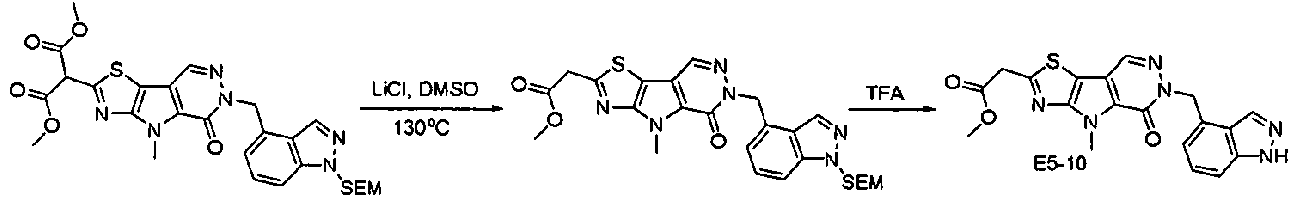
Etapa A. 2-(4-Metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetato de metilo. A una solución de 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)malonato de dimetilo (130 mg, 0,22 mmol) en DMSO (2 ml) en atmósfera de N2 se añadió solución acuosa saturada de LiCl (0,1 ml). La mezcla resultante se agitó a 130°C durante 10 min, luego se vertió en agua con hielo y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. Luego, el residuo se purificó por cromatografía en columna en gel de sílice para proporcionar el producto deseado (100 mg). LC-MS: m/z 539 (m H)+.
Etapa B. El 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetato de metilo se sintetizó como en el ejemplo 5A. LC-MS: m/z 409 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,12 (s, 1H), 8,62 (s, 1H), 8,15 (s, 1H), 7,45 (d, 1H), 7,30-7,26 (m, 1H), 6,96 (d, 1H), 5,67 (s, 2H), 4,39 (s, 2H), 4,27 (s, 3H), 3,70 (s, 3H).
Ejemplo 5E. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetamida

Etapa A. Ácido 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pir^rolo[2,3-d]piridazin-2-il)acético. A una mezcla de 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetato de metilo (100 mg, 0,186 mmol) en MeOH/H2O (3 ml/1 ml) a 0°C en atmósfera de N2 se añadió LiOH (23 mg, 0,558 mmol). La mezcla resultante se agitó a t.a. durante 16 h, luego se concentró a presión reducida. El residuo se acidificó con HCl acuoso (1 M) y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se usó directamente en la siguiente etapa sin más purificación. LC-MS: m/z 525 (M+H)+.
Etapa B. 2-(4-Metil-5-oxo-6-((1-((2-(trímetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetamida. A una mezcla de ácido 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acético (50 mg, 0,095 mmol), Ed CI (37 mg, 0,190 mmol), HOBT (26 mg, 0,190 mmol) y DIp EA (0,05 ml, 0,286 mmol) en DCM (5 ml) a 0°C se añadió NH4Cl (26 mg, 0,477 mmol). La mezcla resultante se agitó a t.a. durante 16 h, luego se vertió en agua helada y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice para proporcionar el producto deseado (20 mg). LC-MS: m/z 524. (M+H)+.
Etapa C. La 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)acetamida se sintetizó utilizando el procedimiento como el del ejemplo 5A. LCMS: m/z 394 (M+H)+. r Mn 1H (400 MHz, DMSO-d6) 5 13,12 (s, 1H), 8,59 (s, 1H), 8,15 (s, 1H), 7,77 (s, 1H), 7,45 (d, 1H), 7,33-7,22 (m, 2H), 6,96 (d, 1H), 5,66 (s, 2H), 4,27 (s, 3H), 4,07 (s, 2H).
Ejemplo 5F. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxamida

Etapa A. 4-Metil-2-(metiltio)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (1 g, 3,96 mmol) en d Mf (25 ml) a 0°C se añadió NaH (318 mg, 7,93 mmol). La mezcla se agitó a t.a. durante 30 min, seguido de la adición de una solución de 4-(bromometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (2 g, 5,94 mmol) en DMF (10 ml) a 0°C. La mezcla se agitó a t.a. durante 2 h, luego se vertió en agua helada y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 50/1 a 10/1) para dar el producto deseado (1,85 g). LCMS: m/z 513 (M+H)+.
Etapa B. 4-Metil-2-(metilsulfonil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5 ',4 ':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona.
A una mezcla de 4-metil-2-(metiltio)-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (700 mg, 1,37 mmol) en DCM (20 ml) a 0°C se añadió m-CPBA (831 mg, 4,01 mmol). La mezcla se agitó a t.a. durante la noche, luego inactivó con solución acuosa saturada de Na2SO3y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice para dar el producto deseado (360 mg). LCMS: m/z 545 (M+H)+.
Etapa C. 4-Metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbonitrilo. A una mezcla de 4-metil-2-(metilsulfonil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg, 0,552 mmol) en DMF (10 ml) a 0°C se añadió KCN (72 mg, 1,10 mmol). La mezcla se agitó a t.a. durante 2 h, luego se inactivó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (210 mg). LCMS: m/z 492 (M+H)+.
Etapa D. 6-((1H-Indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5 ',4 ':4,5]pirrolo[2,3-d]piridazina-2-carboxamida. Una mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbonitrilo (50 mg, 0,1 mmol) en H2SO4 conc. (3 ml) se agitó a t.a. durante 12 h, luego se inactivó con solución acuosa saturada de NaHCO3(ac.) y se extrajo con DCM. Las capas orgánicas combinadas
se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (6 mg). Lc MS: m/z 380 (M+H)+. RMN 1H (400 MHz, DMSO) 513,12 (s, 1H), 8,71 (s, 1H), 8,39 (s, 1H), 8,16 (s, 1H), 8,07 (s, 1H), 7,46 (d, 1H), 7,33 - 7,23 (m, 1H), 6,98 (d, 1H), 5,68 (s, 2H), 4,33 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producir los siguientes compuestos usando materiales de partida apropiados. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.

Ejemplo 5G. Síntesis de 6-((1H-indazol-4-il)metil)-N-hidroxi-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxamida

Etapa A. 4-Metil-5-oxo-6-((1-((2-(tnmetilsihl)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5,4:4,5]pirrolo[2,3-d]piridazina-2-carboxilato de metilo. A una mezcla de 4-metil-5-oxo-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbonitrilo (100 mg, 0,20 mmol) en MeOH (10 ml) a 0°C se añadió MeONa (110 mg, 0,61 mmol, 30% en peso). La mezcla de reacción se agitó a t.a. durante 1,5 h, luego se inactivó con solución saturada de HCl (1 M) y se extrajo con DCM (30 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (85 mg) como un aceite amarillo que se usó directamente en la siguiente etapa sin purificación adicional. LCMS: m/z 525 (M+H)+.
Etapa B. Ácido 4-metil-5-oxo-6((1-((2-(tn'metilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxílico. A una mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5’,4’:4,5]pirrolo[2,3-d]piridazina-2-carboxilato de metilo (90 mg, 0,18 mmol) en THF (10 ml) a 0°C se añadió una solución de LiOH (12 mg, 0,48 mmol) en H2O (10 ml). La mezcla de reacción se agitó a t.a. durante la noche, luego se ajustó lentamente a pH 5 con HCl acuoso (1 M) y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (70 mg, 84% de rendimiento bruto) como un aceite blanco que se usó directamente en la siguiente etapa sin purificación adicional. LCMS: m/z 511 (M+H)+.
Etapa C. 4-Metil-5-oxo-N((tetrahidm-2H-piram-2-il)oxi)-6-((1-((2-(trimetílsilil)etoxi)metil)-1H-mdazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxamida. A una mezcla de ácido 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carboxílico (70 mg, 0,14 mmol) en Dc M (10 ml) a 0°C se añadió O-(tetrahidro-2H-piran-2-il)hidroxilamina (24 mg, 0,21 mmol), EDCI (39 mg, 0,21 mmol) y HOBT (28 mg, 0,21 mmol). La mezcla de reacción se agitó a t.a. durante la noche, luego se inactivó con agua y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (35 mg) como aceite amarillo. LCMS: m/z 610 (M+H)+.
Etapa D. La 6-((1H-indazol-4-il)metil)-N-hidroxi-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2 ,3-d]piridazina-2-carboxamida se sintetizó utilizando el procedimiento del ejemplo 5A. LCMS: m/z 396 (M+H)+. Rm N 1H (400 MHz, DMSOde) 5 13,12 (s, 1H), 8,67 (s, 1H), 8,29 (s, 1H), 8,16 (s, 1H), 7,46 (d, 1H), 7,31 - 7,25 (m, 1H), 6,97 (d, 1H), 5,67 (s, 2H), 4,31 (s, 3H).
Ejemplo 5H. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetamida

Etapa A. 2-(4-Metíl-5-oxo-e-((1-((2-(trímetílsilil)etoxi)metíl)-1H-mdazol-4-il)metíl)-5,e-dihidro-4H-tíazolo[5',4-4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetonitrilo. A una mezcla de 2-fenilacetonitrilo (43 mg, 0,36 mmol) en THF (3 ml) se añadió NaH (14 mg, 0,36 mmol). La mezcla se agitó a t.a. durante 30 min, seguido de la adición de 4-metil-2-(metilsulfonil)-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5,,4':4,5]pirrolo[2,3-d]piridazin-5-ona (100 mg, 0,18 mmol). La mezcla de reacción se agitó a t.a. durante otras 3 h, luego se inactivó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para dar el producto (63 mg, 59% de rendimiento). LCMS: 582 (M+H)+.
Etapa B. La 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-dJpiridazin-2-il)-2-fenilacetamida se sintetizó de manera similar al ejemplo 5F. LCMS: m/z 470 (M+H)+. Rm N 1H (400 MHz, DMSO-de) 5 13,11 (s, 1H), 8,58 (s, 1H), 8,13 (s, 1H) 8,01 (s, 1H), 7,54-7,24 (m, 8H), 6,94 (d, 1H), 5,65 (s, 2H), 5,51 (s, 1H), 4,24 (s, 3H).

Ejemplo 51. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-hidroxi-2-fenilacetamida
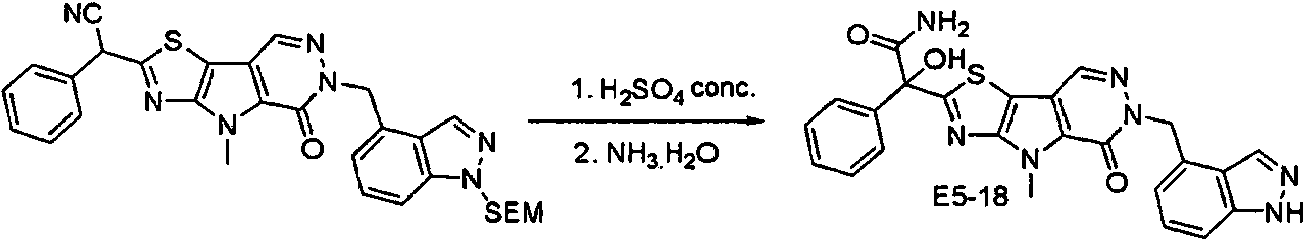
Una mezcla de 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetonitrilo (100 mg, 0,18 mmol) en H2SO4 conc. (1 ml) se agitó a t.a. durante 2 h, luego se vertió en agua y se extrajo con DCM. Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se disolvió en MeOH (3 ml), seguido de la adición de NH3.H2O (3 ml). La mezcla de reacción se agitó a t.a. durante 2 h, luego a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (2 mg). LCMS: 486 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 13,12 (s, 1H), 8,61 (s, 1H), 8,14 (s, 1H), 7,71 (d, 2H), 7,64 (d, 2H), 7,52 (s, 1H), 7,45 (d, 1H), 7,40-7,24 (m, 4H), 6,95 (d, 1H), 5,66 (s, 2H) 4,27 (s, 3H).
Ejemplo SJ. Síntesis de 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetato de metilo

Etapa A. 2-(4-Metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5'.4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetato de metilo. A una mezcla de 2-fenilacetato de metilo (43 mg, 0,29 mmol) en THF (3 ml) se le añadió NaH (11 mg, 0,29 mmol). La mezcla se agitó a t.a. durante 30 min, seguido de la adición de 4-metil-2-(metilsulfonil)-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazo1-4-il)metil)-4,6-dihidro-5H-tiazolo[5,,4,:4,5]pirrolo[2,3-d]piridazin-5-ona (82 mg, 0,15 mmol). La mezcla resultante se agitó a t.a. durante 3 h, luego se inactivó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 3/1) para proporcionar el producto deseado (20 mg). LCMS: 615 (M+H)+.
Etapa B. 2-(6-((1H-indazol-4-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5'4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetato de metilo. Una mezcla de 2-(4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)-2-fenilacetato de metilo (40 mg, 0,07 mmol) en DCM/TFA (V:V = 1:1, 2 ml) se agitó a t.a. durante 2 h, luego se vertió en agua y se extrajo con dCm . Las capas orgánicas combinadas se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (5 mg). LCMS: 485 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 13,11 (s, 1H), 8,58 (s, 1H), 8,13 (s, 1H) 7,54-7,50 (m, 2H), 7,46-7,36 (m, 4H), 7,30-7,24 (m, 1H), 6,94 (d, 1H), 5,87 (s, 1H), 5,65 (s, 2H), 4,26 (s, 3H), 3,73 (s, 3H).
Se usó el procedimiento expuesto anteriormente para producirlos siguientes compuestos usando los materiales de partida apropiados.

Ejemplo 6. Síntesis de 6-(3-metoxibencil)-4-metil-2-(trifluorometil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

A una mezcla agitada de 2-yodo-6-(3-metoxibencil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (90 mg, 0,2 mmol) y Cul (cat.) en DMF (5 ml) se añadió 2,2-difluoro-2-(fluorosulfonil)acetato de metilo (58 mg, 0,3 mmol). La mezcla de reacción se agitó a 70°C durante 4 h, luego se vertió en agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: PE/EtOAc = 5/1) para dar el producto deseado (10 mg). LCMS: m/z= 395 (M+H)+. RMN 1H (400 MHz, DMSO-da) 58,78 (s, 1H), 7,24 (t, 1H), 6,90-6,82 (m, 3H), 5,34 (s, 2H), 4,32 (s, 3H) 3,72 (s, 3H).
Ejemplo 7. Síntesis de los compuestos E7-v y E7-viii

La sustitución nucleófila aromática entre el compuesto E7-iii y el compuesto E7-i y/o el compuesto E7-ii da el compuesto intermedio E7-iv. La reducción del grupo fenilsulfonilo del compuesto E7-iv produce el compuesto intermedio E7-v. El uso
de la reacción de alquilación estándar de E7-vi y base (p. ej., K2CO3, K3PO4, t-BuOK o Cs2CO3) da el compuesto E7-viii, en donde Xa es un grupo saliente tal como Cl, Br, I, oMs, OTs; An y Ar2 son cada uno independientemente grupos arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido; alquilo opcionalmente sustituido, alquilarilo opcionalmente sustituido, alquilheteroarilo opcionalmente sustituido, alquenilo opcionalmente sustituido y alquinilo opcionalmente sustituido. El compuesto E7-viii también se puede sintetizar a partir del compuesto intermedio E7-v a través de la reacción de Mitsunobu usando cianometilentributilfosforano (CMBP) en tolueno. En determinadas realizaciones, Ar1 y Ar2 son cada uno independientemente heteroarilo opcionalmente sustituido.
Ejemplo 7A. Síntesis de 4-metil-2-(metilsulfinil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (compuestos intermedios E7-i)

A una suspensión agitada de 4-metil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (1,01 g, 4,0 mmol) en DCM (20 ml) se añadió ácido 3-cloro-benzoperoxoico (0,77 g, 3,8 mmol) a t.a. La mezcla se agitó a t.a. durante 2 h. Después la mezcla se filtró, se lavó con EtOAc y se trituró con MeOH para dar la 4-metil-2-(metilsulfinil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (600 mg). LCMS: m/z 269 (M+H)+. RMN 1H (400 MHz, DMSO) 812,78 (s, 1H), 8,64 (s, 1H), 4,28 (s, 3H), 3,11 (s, 3H).
Ejemplo 7B. Síntesis de 4-metil-2-(metilsulfonil)-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (E7-ii)

En un matraz de tres bocas cargado con 4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (30 g, 0,119 mol, 1,0 eq) en DCM (600 ml) se añadió m-CPBA (61,5 g, 3 eq) a 20°C en tres porciones. La mezcla se agitó a 30°C durante la noche, la LC-MS indicó un consumo de 100% del material de partida. La mezcla se enfrió a t.a., se añadió otra porción de m-CPBA (1,0 eq). La mezcla de reacción se agitó a 30°C durante 2 h, la LC-MS indicó E7-ii (LCMS: m/z 269 (M+H)+). La mezcla se enfrió a t.a. y se filtró. La torta filtrada se suspendió en MeOH (500 ml) y se agitó a t.a. por 1 h. El sólido se recogió por filtración, se lavó con acetato de etilo, se secó al vacío para proporcionar 28 g de una mezcla de 5% de E7-i y 95% de E7-ii. La mezcla (28 g) se suspendió en DMSO (600 ml), se calentó a 120°C - 130°C para formar una solución transparente. Luego se enfrió a t.a., precipitó un sólido. La mezcla se filtró y se secó para proporcionar 23 g de E5-1 puro, LCMS: m/z 285 (M+H)+. RMN 1H (400 MHz, DMSO) 812,87 (s, 1H), 8,69 (s, 1H), 4,32 (s, 3H), 3,56 (s, 3H).
Ejemplo 7C. Síntesis de 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol

1-((2-(Trimetilsilil)etoxi)metil)-1H-pirazol-3-carboxilato de metilo A 0°C en atmósfera de N2, a una solución agitada de 1H-pirazol-3-carboxilato de metilo (90 g, 0,72 mol) en THF (1 L) se añadió NaH (20,7 g, 0,864 mol, 60%). La mezcla resultante se calentó lentamente hasta la t.a. y se agitó durante 1 h. A continuación, la mezcla de reacción se enfrió de nuevo a 0°C y se añadió gota a gota SEMCl (151,5 ml, 0,842 mol). La agitación se continuó durante otras 2 h antes de inactivar con solución sat. de NH4Cl y se extrajo con acetato de etilo (3 x). Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre Na2SO4. Los disolventes se separaron al vacío para proporcionar 210 g de producto bruto que se utilizó en la siguiente etapa sin purificación.
(1-((2-(Trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metanol A 0°C en atmósfera de N2, a la suspensión de LAH (16,9 g, 0,44 mol) en THF (760 ml) se le añadió el 1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carboxilato de metilo bruto (76 g). La mezcla resultante se calentó lentamente hasta t.a. y se agitó durante 1 h. La mezcla de reacción se enfrió de nuevo a 0°C y se añadió sucesivamente H2O (15,6 ml), NaOH al 10% (15,6 ml), H2O (15,6 ml). La mezcla resultante se filtró a través de una capa de Celite y se lavó con MTBE (4 x). Las fracciones orgánicas combinadas se secaron sobre Na2SO4. Los disolventes se separaron a presión reducida para proporcionar 69,4 g de producto bruto que se utilizó en la siguiente etapa sin purificación. LC-m S: m/z 229 (M+H)+.
3-(Yodometil)-1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol A 0°C en atmósfera de N2, a una solución agitada de (1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metanol (61,5 g, teóricamente 0,262 mol) en THF (310 ml) se añadió TEA (55,42 ml, 0,393 mol) seguido de MsCl (24 ml, 0,314 mol). La reacción se calentó a t.a. y se agitó durante 1 h antes de la introducción de NaI (196,5 g, 1,31 mol, en 310 ml de DMF). La mezcla resultante se agitó durante otra hora y se inactivó con agua helada, se extrajo con MTBE (3 x). Las capas orgánicas combinadas se lavaron con solución sat. de Na2S2O3y salmuera, se secaron sobre Na2SO4 y se concentraron para proporcionar 77,5 g de producto bruto usado en la siguiente etapa sin purificación. LC-MS: m/z 339 (M+H)+.
3-((Fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol A 0°C en atmósfera de N2, a una solución agitada de (1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metanol (77,5 g, teóricamente 0,229 mol) en DMF (600 ml) se añadió bencenosulfinato de sodio (53,5 g, 0,32 mol) y se agitó durante 1 h a 0°C. Después de calentar a t.a., la mezcla de reacción se inactivó con agua helada y solución sat. de Na2S2O3, se extrajo con acetato de etilo (3 x). Las capas orgánicas combinadas se lavaron con solución sat. de NaHCO3 y salmuera sucesivamente, se secaron sobre Na2SO4. Los disolventes se separaron al vacío y el residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 20% ~ 70%) para proporcionar 56,7 g. LCMS: [M H]+ 353. RMN 1H (400 MHz, DMSO) 57,85-7,77 (m, 4H), 7,62 (dd, 2H), 6,19 (d, 1H), 5,35 (d, 2H), 4,70 (d, 2H), 3,44- 3,38 (m, 2H), 0,88-0,77 (m, 2H), -0,01 (s, 9H).
Ejemplo 7D. Síntesis de 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

4-Metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-5-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (1,8 g, 5,1 mmol) en THF seco (30 ml) a -40°C se añadió gota a gota LiHMDS (7,5 ml, 7,5 mmol). La mezcla se agitó a temperatura ambiente durante 30 min, seguido de la adición de una suspensión de 4-metil-2-(metilsulfinil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (580 mg, 2,7 mmol) en THF seco (30 ml) a temperatura ambiente. La mezcla se agitó a t.a. durante 1 h más y se vertió en solución acuosa saturada de NH4Cl (20 ml) enfriada con hielo y se extrajo con EtOAc (3 x 100 ml). Las capas orgánicas combinadas se lavaron con agua (60 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, metanol en diclorometano al 0 - 2,5%) para dar el producto deseado (800 mg). LC-MS (ESI) encontrado: 557 (M+H)+. RMN 1H (400 MHz, DMSO) 512,78 (s, 1H), 8,65 (s, 1H), 8,03 (d, 1H), 7,84-7,78 (m, 3H), 7,67-7,59 (m, 2H), 6,94 (s, 1H), 6,72 (d, 1H), 5,48 (d, 2H), 4,29 (s, 3H), 3,56 (dd, 2H), 0,88 (dd, 2H), 0,00 (s, 9H).
4-Metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-5-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-5-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (0,8 g, 1,41 mmol) en THF (5 ml) y MeOH (10 ml) en atmósfera de N2 se añadió gota a gota SmI2 (0,1 M/THF, 45 ml) en un baño de hielo Después de agitar durante 10 min, la reacción se inactivó con solución acuosa saturada de NH4Cl (50 ml) y se extrajo con eAo Ac (50 ml x 3). Las capas orgánicas combinadas se lavaron con agua (60 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, metanol en diclorometano al 0 - 3%) para dar el producto deseado (310 mg). lC-MS) encontrado: 417 (M+H)+. RMN 1H (400 MHz, CDCl3) 58,31 (s, 1H), 7,60 (d, 1H), 6,39 (d, 1H), 5,49 (s, 2H), 4,58 (s, 2H), 4,43 (s, 3H), 3,62 (t, 2H), 0,95 (t, 2H), 0,0 (s, 9H).
Ejemplo 7E. Síntesis de 2-((1H-pirazol-3-il)metil)-6-((1H-pirazolo[4,3-c]piridin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 1-((2-(Trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-carboxilato de metilo. A una solución de 1H-pirazol[4,3-c]piridin-4-carboxilato de metilo (900 mg, 5,1 mmol) en DMF seca (10 ml) se añadió NaH (305 mg, 7,6 mmol, 60%) a 0° C en porciones. La suspensión se agitó durante 15 min en un baño de hielo antes de la introducción gota a gota de (2-(clorometoxi)etil)trimetilsilano (1,07 ml, 6,0 mmol) y se agitó durante 1 h más a t.a. Luego la mezcla se vertió en solución sat. de NH4Cl (ac.), se extrajo con acetato de etilo. Las capas orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 30%) para producir el 1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridina- 4-carboxilato de metilo (1,32 g). LCMS: m/z 308 (M+H)+.
Etapa B. (1-((2-(Trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-il)metanol. A una solución de 1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-carboxilato de metilo (1 g, 3,2 mmol) en THF seco (10 ml) se añadió LiHAl4 (146 mg, 3,8 mmol) en pociones con baño de hielo. La mezcla se agitó durante 30 min a 0°C. Luego la suspensión en se vertió solución sat. de NH4Cl (ac.), se extrajo con acetato de etilo (2x). Las capas orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 60%) para producir el (1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin- 4-il)metanol (500 mg). LCMS: m/z 280 (M+H)+.
Etapa C. 4-(Clorometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridina A una solución de (1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-il)metanol (120 mg, 0,43 mmol) en diclorometano (2 ml) se añadió Pph3 (225 mg, 0,86 mmol). La mezcla se enfrió a 0°C y se añadió NCS (114 mg, 0,86 mmol). La suspensión se calentó a t.a. y se agitó durante 1 h más. Después la reacción se vertió en solución sat. de NaHCO3 (ac.). La fase acuosa se extrajo con diclorometano. Las capas orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 30%) para producir la 4-(clorometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3 -c]piridina (70 mg) como un aceite. LCMS: m/z 298 (M+H)+. Etapa D. 4-Metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-6-((1-((2-(trimetilsilil)etoxi))metil)-1H-pirazolo[4,3-c] piridin-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de K2CO3 (41 mg, 0,3 mmol) en DMF anhidra (2 ml) se añadió 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d] piridazin-5(6H)-ona (41 mg, 0,1 mmol) y se agitó a 50°C durante 30 min en atmósfera de argón. Se añadió una solución de 4-(clorometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridina (30 mg, 0,1 mmol) en DMF (1 ml) y se agitó durante otras 4 h. La suspensión se enfrió a t.a. y se vertió en Hcl 0,5 N (ac.). Las capas se separaron y la capa acuosa se extrajo con acetato de etilo. Las capas orgánicas combinadas se secaron con Na2SO4 anhidro, se filtraron y se concentraron. El residuo se purificó porTLC preparativa para proporcionar la 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (40 mg, 60%). LCMS: m/z 678 (M+H)+.
Etapa E. 2-((1H-Pirazol-3-il)metil)-6-((1H-pirazoto[4,3-c]piridm-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirroto[2,3-d]piridazm-5(6H)-ona. Una solución de 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazolo[4,3-c]piridin-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,044 mmol) en TFA al 35% (1 ml, en diclorometano) se agitó a t.a. durante la noche. La mezcla se concentró y el residuo se purificó por HPLC preparativa para proporcionar la 2-((1H-pirazol-3-il)metil)-6-((1H-pirazolo[4,3-c]piridin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (3 mg). LCMS: m/z 418 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 13,51 (s, 1H), 12,78 (s, 1H), 8,55 (s, 1H), 8,18 (d, 1H), 8,00 (s, 1H), 7,68 (s, 1H), 7,43 (d, 1H), 6,27 (d, 1H), 5,78 (s, 2H), 4,51 (s, 2H), 4,25 (s, 3H).
Ejemplo 7F. Síntesis de 6-((1H-imidazol-2-il)metil)-2-((1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. 1-((2-(Trimetilsilil)etoxi)metil)-1H-imidazol-2-carbaldehído: Una muestra de NaH se lavó con hexano (2 x10 ml) en atmósfera de N2. El matraz se cargó con DMF seca (20 ml) y se añadió en pequeñas porciones 1H-imidazol-2-carbaldehído (500 mg, 5,2 mmol). Después de agitara temperatura ambiente durante 1,5 h, se añadió gota a gota SEMCl (864 mg, 5,2 mmol). La mezcla de reacción se agitó a t.a. durante 30 min. La mezcla de reacción se vertió en agua y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4, se concentró a presión reducida para proporcionar el 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-carbaldehído bruto (800 mg). LCMS: 227 (M+H)+.
Etapa B. (1-((2-(Trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol: A una mezcla agitada de 1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-carbaldehído (1,6 g, 7 mmol) en THF (20 ml) se añadió NaBH4 (1.34 g, 35 mmol) a 0°C. La mezcla de reacción se agitó a t.a. durante 30 min. La mezcla de reacción se vertió en solución ac. de NH4Cl y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4, se concentró a presión reducida para proporcionar el (1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol bruto (1,3 g).
Etapa C. 2-(Clorometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol: A una mezcla agitada de (1-((2-(Trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metanol (400 mg, 1,75 mmol) en Dc M (20 ml) se añadieron NCS (466 mg, 3,5 mmol) y PPh3 (920 mg, 3,5 mmol) a t.a. La mezcla se agitó a t.a. durante 2 h. La mezcla de reacción se vertió en agua y se extrajo con DCM. La mezcla se lavó con agua y la capa orgánica se concentró a presión reducida. El residuo se purificó porTLC preparativa (PE:EtOAc=1:1) para proporcionar el 2-(clorometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol. LCMS: 247 (M+H)+. A una mezcla agitada de 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,12 mmol) en Dm F seca (5 ml) se añadió K2CO3 (66 mg, 0,48 mmol) a 60°C en atmósfera de N2.
Después de 20 min, se añadió el compuesto E7-3 (60 mg, 0,24 mmol), en DMF seca (2 ml) a 60°C en atmósfera de N2. La mezcla se agitó a 60°C durante 1,5 h en atmósfera de N2. La mezcla de reacción se enfrió a t.a. y se ajustó a pH= 5-6 con solución ac. de HCl. Luego, la mezcla se extrajo con EtOAc, se lavó con agua y salmuera. La capa orgánica se secó sobre Na2SO4, se concentró a presión reducida y se purificó por TLC preparativa (PE:EtOAc=1:1,5) para producir la 4 metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metil)-2-((1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (25 mg). lCm S: 627 (M+H)+. Una mezcla de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-imidazol-2-il)metil)-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (25 mg, 0,04 mmol) en DCM/TFA (2 ml/2 ml) se agitó a t.a. durante 1 hora. La mezcla de reacción se concentró. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (1,3 mg). LCMS: 367 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 58,51 (s, 1H), 7,66 (d, 1H), 6,9 (s, 2H), 6,27 (d, 1H), 5,34 (s, 2H), 4,51 (s, 2H), 4,27 (s, 3H).
Los siguientes compuestos se sintetizaron de acuerdo con el Esquema E7 y el procedimiento del Ejemplo 7C-7E usando el material de partida apropiado.











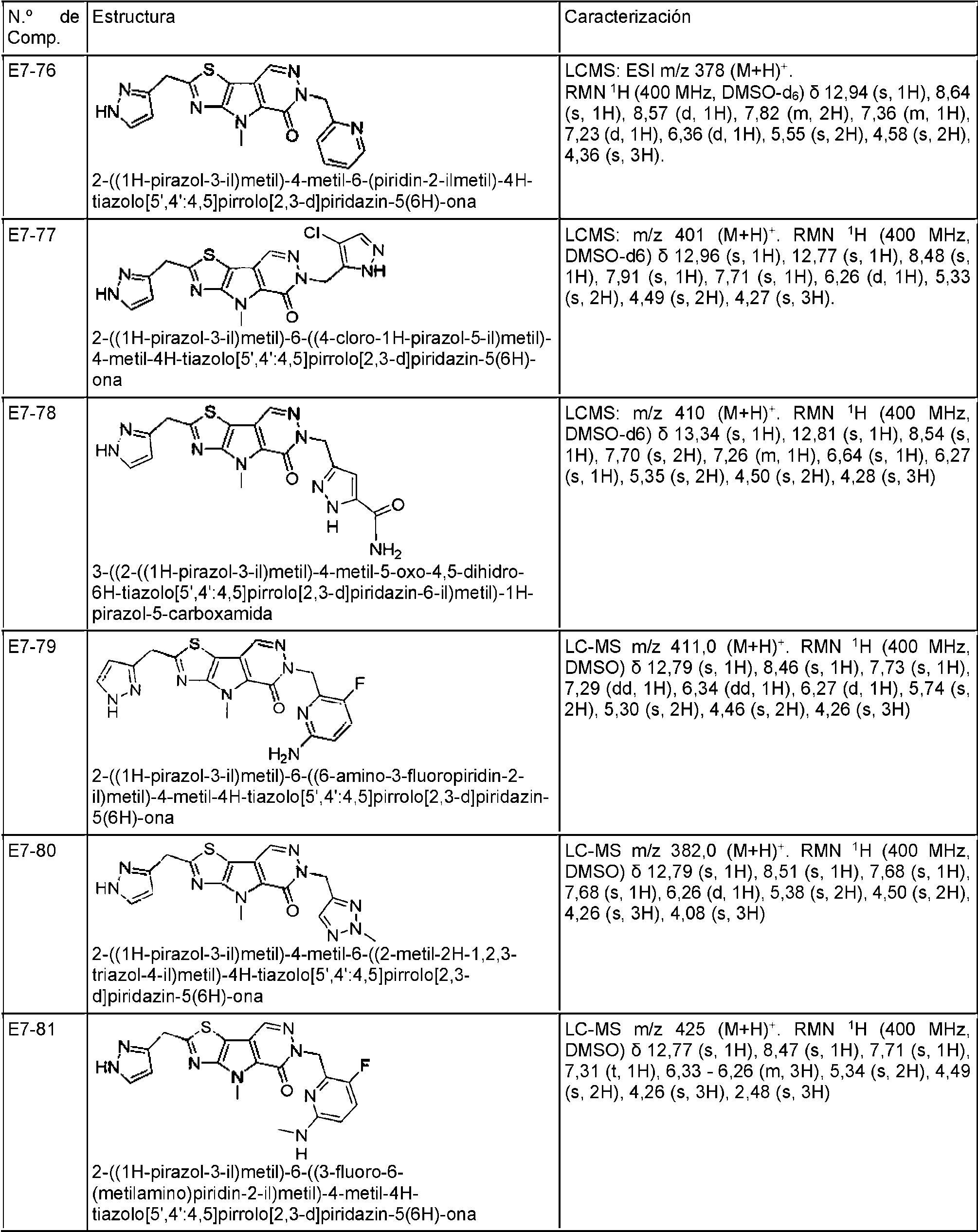




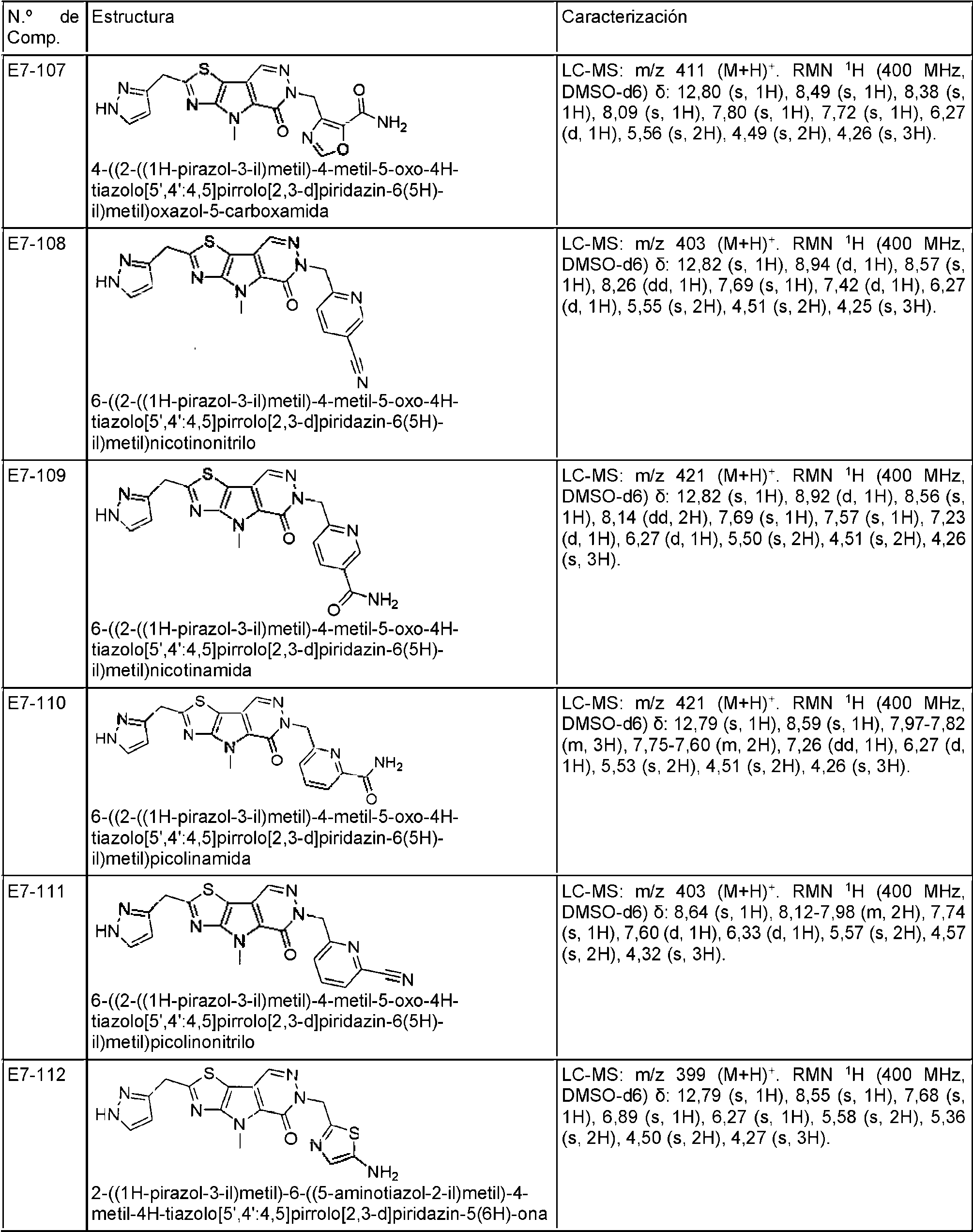



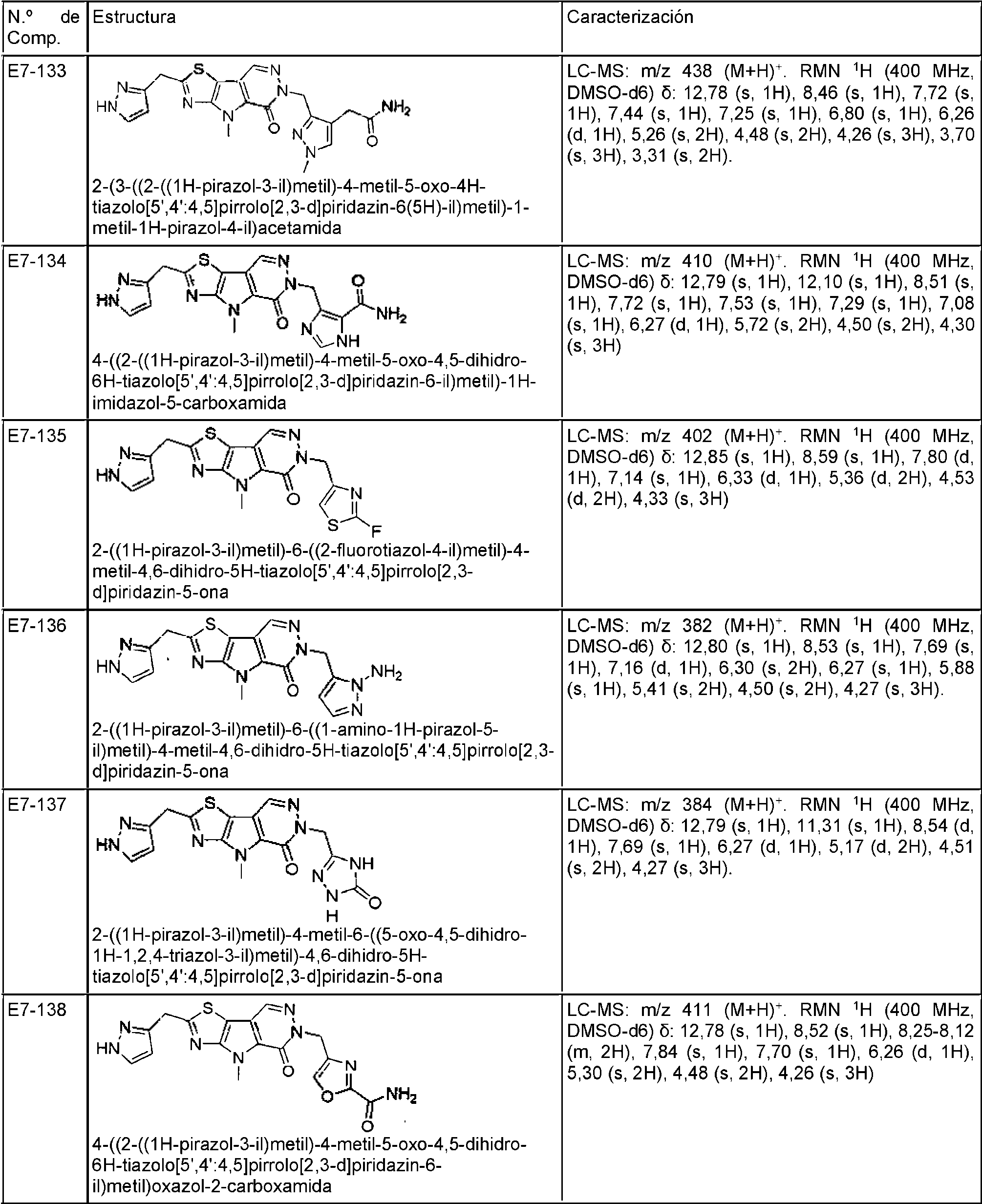

Ejemplo 7F. Síntesis de 2-((1H-pirazol-3-il)metil)-4-metil-6-(2-(tiazol-4-il)etil)-4H-tiazolo[5',4,:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 2-(tiazol-4-il)acetato de etilo A una solución de 2-(2-aminotiazol-4-il)acetato de etilo (2 g, 10,7 mmol) en THF (30 ml) se añadió t-BuONo (1,6 g, 16,1 mmol). La mezcla de reacción se agitó a 50°C durante l6 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 80-100%) para proporcionar el 2-(tiazol-4-il)acetato de etilo (400 mg). LC-MS (ESI): m/z 172 (M+1)+.
Etapa B. Síntesis de 2-(tiazol-4-il)etanol A una solución agitada de 2-(tiazol-4-il)acetato de etilo (400 mg, 2,3 mmol) en DCM (20 ml) se añadió DlBAL-H (4,7 ml, 7,0 mmol). La mezcla de reacción se agitó a temperatura ambiente en atmósfera de N2 durante 3 h. La reacción se inactivó con solución sat. de NaHCO3, se extrajo con DCM y la capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 50-100%) para proporcionar el 2-(tiazol-4-il)etanol (200 mg). LC-MS (ESI): m/z 130 (M+1)+.
Etapa C. Síntesis de 4-metil-6-(2-(tiazol-4-il)etil)-2-((1-((2-(trimetilsMil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg, 0,14 mmol) y 2-(tiazol-4-il)etanol (55 mg, 0,4 mmol) en tolueno (5 ml) se añadió Cm Bp (104 mg, 0,4 mmol). La mezcla de reacción se agitó a 110°C en atmósfera de N2 durante 3 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 80-100%) para proporcionar 60 mg del 4-metil-6-(2-(tiazol-4-il)etil)-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5,,4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Lc -MS: m/z 528 (M+1)+.
Etapa D. Síntesis de 2-((1H-pirazol-3-il)metil)-4-metil-6-(2-(tiazol-4-il)etil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de 4-metil-6-(2-(tiazol-4-il)etil)-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg, 0,1 mmol) en DCM (1 ml) se añadió TFA (0,5 ml). La mezcla de reacción se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se ajustó a pH = 7,5 con solución sat. de NaHCO3, se extrajo con DCM, se lavó con salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por TLC preparativa (MeOH en DCM al 10%) para proporcionar 10 mg de la 2-((1H-pirazol-3-il)metil)-4-metil-6-(2-(tiazol-4-il)etil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS: m/z 398 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 59,03 (s, 1H), 8,47 (s, 1H), 7,67 (s, 1H), 7,41 (s, 1H), 6,27 (s, 1H), 4,58 - 4,40 (m, 4H), 4,26 (s, 3H), 3,23 (t, 2H).

Ejemplo 8. Síntesis de los compuestos E8-v, E8-vi y E8-viii


El compuesto E8-i se puede convertir en el compuesto intermedio E8-ii mediante alquilación o reacción de Mitsunobu como en el ejemplo E7-v a E7-viii. La oxidación de E8-ii con mCPBA u Oxone genera compuestos E8-v y E8-vi. Ambos compuestos de E8-v y E8-vi también se pueden formar a partir de E8-i primero por oxidación seguida de alquilación o reacción de Mitsumobu. En donde Xa es un grupo saliente (p. ej., Cl, Br, I, OMs, OTs); Los compuestos E8-v y E8-vi se pueden convertir en el compuesto intermedio E8-viii mediante reacción de sustitución nucleófila aromática con el compuesto E8-vii, usando LiHMDS o t-BuOK como base. El compuesto E8-xi se puede sintetizar a partir del compuesto E8-viii usando SmI2 o Zn en AcOH o TES con AlCl3. Como se usa en el presente documento, R1 es alquilo opcionalmente sustituido, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido; Ar1 y Ar2 son cada uno independientemente arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido; alquilo opcionalmente sustituido, alquilarilo opcionalmente sustituido, alquilheteroarilo opcionalmente sustituido, alquenilo opcionalmente sustituido y alquinilo opcionalmente sustituido. En ciertas realizaciones, R1 es alquilo C1-6 opcionalmente sustituido (p. ej., metilo o etilo). En ciertas realizaciones, R1 es alquilo C1-6; y Ar1 y Ar2 son cada uno independientemente heteroarilo opcionalmente sustituido
Ejemplo 8A. Síntesis de 6-((1H-indazol-4-il)metil)-2-((5-cloro-1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

5-Cloro-1-tritil-1H-pirazol-3-carboxilato de etilo: A una mezcla agitada de 5-cloro-1H-pirazol-3-carboxilato de etilo (100 mg, 0,575 mmol) y TeA (0,24 ml, 1,44 mmol) en DCM seco (10 ml) se añadió TrtCl (192 mg, 0,689 mmol) a t.a. La mezcla de reacción se agitó a t.a. durante 2 h y luego se vertió en H2O. La mezcla resultante se extrajo con DCM. La capa orgánica se lavó con salmuera (30 ml), se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0-4%) para dar el producto deseado (bruto, 240 mg, 100%).
(5-Cloro-1-tritil-1H-pirazol-3-il)metanol: A una mezcla agitada de 5-cloro-1-tritil-1H-pirazol-3-carboxilato de etilo (1,20 g, 2,88 mmol) en THF seco (10 ml) se añadió LAH (400 mg, 10,5 mmol) a -30°C. La mezcla de reacción se agitó a -30°C durante 0,5 h. Se añadió lentamente Na2SO410H2O (2 g) para inactivar la reacción. La mezcla resultante se diluyó con EtOAc y se filtró a través de una capa de Celite. El filtrado se concentró y el residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 10% -15%) para dar el producto deseado (540 mg). Metanosulfonato de (5-cloro-1-tritil-1H-pirazol-3-il)metilo: A una mezcla agitada de (5-cloro-1-tritil-1H-pirazol-3-il)metanol (100 mg, 0,267 mmol) y DIPEA (0,14 ml, 0,801 mmol) en DCM seco (10 ml) se añadió MsCl (46 mg, 0,401 mmol) a 10°C. La mezcla de reacción se agitó a t.a. durante 1 h. La mezcla de reacción se inactivó con agua. La mezcla resultante se extrajo con DCM (2X). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto deseado (bruto, 150 mg) como un aceite pegajoso. 5-Cloro-3-((fenilsulfonil)metil)-1-tritil-1H-pirazol: A una mezcla agitada de metanosulfonato de (5-cloro-1-tritil-1H-pirazol-3-il)metilo (150 mg bruto, 0,267 mmol) en DMF seca (10 ml) se añadió PhSO2Na (100 mg, 0,610 mmol) a t.a. La mezcla de reacción se agitó a t.a. durante 20 h. La mezcla de reacción se diluyó con agua. La mezcla resultante se extrajo con EtOAc (2X). Las capas orgánicas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (eluyente: PE/EtOAc = 3/1) para dar el producto deseado (80 mg). LCMS: m/z 521 (M+Na)+.
2-((5-Cloro-1-tritil-1H-pirazol-3-il)(fenilsulfonil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona: A una mezcla de 5-cloro-3-((fenilsulfonil)metil)-1-tritil-1H-pirazol (100 mg, 0,2 mmol) y 4-metil-2-(metilsulfonil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,2 mmol) en THF seco (10 ml) se añadió gota a gota LiHMDS (1 ml, 10 mmol, 1 M en THF) a 10°C. La mezcla de reacción se agitó a t.a. durante 10 min y se vertió en solución acuosa de NH4Cl. La siguiente mezcla se extrajo con EtOAc (2X). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 30%) para dar el producto deseado de E8-3 (75 mg) como aceite amarillo. LCMS: m/z 985 (M+Na)+.
2-((5-Cloro-1-tritil-1H-pirazol-3-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de compuestos E8-3 (75 mg, 0,0779 mmol) en THF (5 ml) y MeOH (5 ml) a t.a. en atmósfera de N2 se añadió SmI2 (5 ml, 0,1 M en THF). La mezcla de reacción se agitó a t.a. durante 10 min y luego se inactivó con agua. La siguiente mezcla se extrajo con EtOAc (2X). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (PE/EtOAc = 2/1) para dar la 2-((5-cloro-1-tritil-1H-pirazol-3-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg). LCMS: m/z 845 (M+23)+.
6-((1H-Indazol-4-il)metil)-2-((5-cloro-1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-((5-cloro-1-tritil-1H-pirazol-3-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,0608 mmol) en DCM (6 ml) a t.a. en atmósfera de N2 se añadió TFA (2 ml). La mezcla de reacción se agitó a t.a. durante 1 h. La siguiente mezcla se ajustó a pH=8 con solución acuosa de NaHCO3, se extrajo con DCM/iPrOH al 80% (2X). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa (C18, acetonitrilo en H2O con ácido fórmico al 0,1%, 0 - 50%) para dar el producto deseado (10 mg). LCMS: m/z 451 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 513,10-13,2 (s ancho, 2H), 8,58 (s, 1H), 8,14 (s, 1H), 7,43-7,46 (m, 1H), 7,25-7,30 (m, 1H), 6,94 6,97 (m, 1H), 6,32 (s, 1H), 5,65 (s, 2H), 4,55 (s, 2H), 4,27 (s, 3H).
Ejemplo 8B. Síntesis de 2-((1H-1,2,3-triazol-4-il)metil)-4-metil-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 4-metil-2-(metiltio)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de NaH (130 mg, 3,2 mmol) en DMF (4 ml) se le añadió 4-metil-2-(metiltio)-411-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (270 mg, 1,1 mmol) a 0°C en atmósfera de N2. Después de 5 min, se añadió la mezcla de 1-(bromometil)-3-(trifluorometoxi)benceno (420 mg, 1,65 mmol) en DMF (2 ml) a la mezcla de reacción. La mezcla se agitó a t.a. durante 2 h. La reacción se inactivó con solución saturada de NH4Cl y se extrajo con EA. La capa orgánica se lavó con solución saturada de NaCl (3x), se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 30%) para dar la 4-metil-2-(metiltio)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (420 mg). Lc MS: 427 (M+H)+ Etapa B. Síntesis de 4-metil-2-(metilsulfonil)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 4-metil-2-(metiltio)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (420 mg, 0,99 mmol) en DCM (10 ml) se añadió mCPBA (520 mg, 3,0 mmol) a 0°C en atmósfera de N2. La mezcla de reacción continuó agitando durante la noche. La solución se inactivó con solución saturada de Na2S2O3 y se extrajo con DCM (3x). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 50%) para dar la 4-metil-2-(metilsulfonil)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (330 mg). LCMS: m/z 459 (M+H)+.
Etapa C. Síntesis de 4-metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,3-triazol-4-il)metil)-6-(3-(trifluorometoxi)bencil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 4-metil-2-(metilsulfonil)-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (165 mg, 0,36 mmol) y 4
((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,3-triazol (50 mg, 0,54 mmol) en DMF seca (10 ml) se añadió Cs2CO3 (351 mg, 1,08 mmol) a 65°C. La mezcla de reacción se agitó a 65°C durante 2 h y se vertió en solución acuosa de NH4Cl. La mezcla resultante se extrajo con EtOAc (2x). Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 35%) para dar el producto deseado la 4-metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-1,2,3-triazol-4-il)metil)-6-(3-(trifluorometoxi)bencil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (E8-6) (200 mg). LCMS: m/z 732 (M+H)+.
Etapa D. Síntesis de 2-((1H-1,2,3-triazol-4-il)metil)-4-metil-6-(3-(trifluorometoxi)bencil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Similar al ejemplo 8A, el compuesto E8-6 se hizo reaccionar con SmI2, seguido de desprotección con TFA, para dar el producto deseado. LCMS: 462 (M+H)+. RMN 1H (400 MHz, DMSO) 58,52 (s, 1H), 7,88 (s, 1H), 7,46 (dd, 1H), 7,39-7,2 (m, 3H), 5,40 (s, 2H), 4,63 (s, 2H), 4,26 (s, 3H).
Ejemplo 8C. Síntesis de 2,6-bis((1H-indazol-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A: 4-((Feniltio)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol. A una solución de 4-(bromometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (340 mg, 1,0 mmol) en DMF (10 ml) se añadió bencenotiolato de sodio (265 mg, 2 mmol). La mezcla se agitó a t.a. durante 2 h, luego se inactivó con agua helada (10,0 ml) y se extrajo con EtOAc. (3 x 50,0 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida para dar el producto bruto (370 mg) que se usó directamente en la siguiente etapa. LCMS: m/z 371 (M+H)+.
Etapa B: 4-((Fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol. A una solución de 4-((feniltio)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (370 mg) en THF (20 ml) a 0°C se añadió una solución de Oxone (2,15 g, 3,5 mmol) en H2O (20 ml). La mezcla se agitó a t.a. durante 1 h, luego se inactivó con agua helada (50 ml) y se extrajo con AcOEt (3 x 50,0 ml). Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía en columna en gel de sílice (eluyente: ENPE = 1/5) para proporcionar el producto deseado (300 mg). LCMS: m/z 403 (M+H)+.
Etapa C 4-Metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 4-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-indazol (163 mg, 0,40 mmol, 2,2 eq) en THF seco (5 ml) a -40°C se añadió gota a gota LíHm Ds (0,46 ml, 0,46 mmol, 2,5 eq). La mezcla se agitó a esta temperatura durante 10 min, seguido de la adición gota a gota de una solución de 4-metil-2-(metilsulfonil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,18 mmol, 1,0 eq) en t Hf seco (3 ml) a -40°C. La mezcla se agitó a esta temperatura durante otros 30 min hasta que se completó. La mezcla resultante se vertió en solución saturada de NH4Cl helada (20 ml) y se extrajo con EtOAc ( 3x 10 ml). Las capas orgánicas combinadas se lavaron con agua (20 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa para dar el producto deseado (100 mg). LCMS: m/z 867 (M+H)+.
Etapa D: 2,6-Bis((1H-indazol-4-il)metil)-4-metil-4H-tiazoto[5,4':4,5]pirrolo[2,3-d]pirídazin-5(6H)-ona. A una solución de 4-metil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,06 mmol, 1,0 eq) en Dc E seco (2 ml) en atmósfera de N2 se añadieron AlCl3 (38 mg, 0,30 mmol, 5,0 eq) y TES (34 mg, 0,30 mmol, 5,0 eq). La mezcla se calentó a 60°C durante 30 min, luego se enfrió a t.a., se vertió en agua (10 ml) y se extrajo con DCM/MeOH (V:V = 20: 1, 3x10 ml). Las capas orgánicas combinadas se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC preparativa para dar el producto deseado (5 mg). LCMS: m/z 467 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 5 13,15 (s, 1H), 13,10 (s, 1H), 8,50 (s, 1H), 8,18 (s, 1H), 8,13 (s, 1H), 7,49 (d, 1H), 7,44 (d, 1H), 7,38-7,30 (m, 1H), 7,29 - 7,21 (m, 1H), 7,14 (d, 1H), 6,93 (d, 1H), 5,64 (s, 2H), 4,84 (s, 2H), 4,28 (s, 3H).
Ejemplo 8D. Síntesis de 2-((1H-pirazol-3-il)metil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y 6-((6-aminopiridin-2-il)metil)-4-metil-2-(1H-pirazol-3-carbonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. N-[(terc-butoxi)carbonil]-N-[6-({4-metanosulfonil-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridin-2-il]carbamato de terc-butilo Una mezcla de 4-metil-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (7,5 g, 26,4 mmol) y K3PO4 (8,3 g, 39,3 mmol) en MeCN anhidro (300 ml) se agitó a 70°C durante 1 h en atmósfera de N2. A continuación, se añadió una solución de N-[(terc-butoxi)carbonil]-N-[6-(bromometil)piridin-2-il]carbamato de terc-butilo (11,2 g, 29,0 mmol) en MeCN (30 ml). Después de agitar a 70°C durante 2,5 h en atmósfera de N2, la mezcla de reacción se inactivó con solución sat. de NH4Cl y se extrajo con EA (300 ml X 3). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre Na2SO4, se filtraron y la fase orgánica se concentró. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 50%) para dar el N-[(terc-butoxi)carbonil]-N-[6-({4-metanosulfonil-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-l0-il}metil)piridin-2-il]carbamato de terc-butilo (5,5 g). LC-MS (ESI) encontrado: 591,1 (M+H)+.
Etapa B. (6-((4-Metil-5-oxo-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4’:4,5)pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo. A una mezcla agitada de 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (11,9 g, 33,8 mmol) en THF anhidro (200 ml) se añadió LiHMDS (50 ml, 1 M en THF) a -40°C en atmósfera de argón. Después de 10 min, la mezcla se calentó a 10°C y se agitó durante 1 h, luego se añadió N-[(terc-butoxi)carbonil]-N-[6-({4-metanosulfonil-7-metil-9-oxo-3-tia-5,7,10,11-tetraazatriciclo[6.4.0.0{2,6}]dodeca-1(8),2(6),4,11-tetraen-10-il}metil)piridina-2-il]carbamato de terc-butilo (9,1 g, 15,4 mmol en 35 ml de THF). La reacción se agitó a 10°C durante otros 30 min. La mezcla de reacción se vertió en solución ac. de NH4O, se extrajo con EtOAc (200 ml x 3). Las capas orgánicas combinadas se lavaron con agua y salmuera, se secaron sobre Na2SO4 anhidro y concentraron. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 - 50%) para dar el (6-((4-metil-5-oxo-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (6,6 g). LC-MS (ESI) encontrado: 763,2 (M+H)+.
Etapa C. (6-((4-Metil-5-oxo-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H- tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo Una solución de (6-((4-metil-5-oxo-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (6,0 g, 7,86 mmol) en EtOH/AcOH (35 ml 150 ml) se calentó a 50°C con agitación vigorosa en presencia de Zn (2,55 g, 117,9 mmol) durante 40 min. Se añadió zinc adicional cada 40 min (2,55 g, dos veces, controlar la reacción por TLC/LC-MS para evitar el subproducto y el producto sobre-reducido). La solución se filtró y la torta de filtración se lavó con DCM. El filtrado se evaporó parcialmente, se neutralizó con solución saturada de NaHCO3, se secó sobre MgSO4 y el disolvente se eliminó al vacío. El producto bruto se purificó por cromatografía ultrarrápida (gel de sílice, DCM:MeOH= 40:1) para dar el (6-((4-metil-5-oxo-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (3,1 g). Lc -MS (ESI) encontrado: 623,3 (M+H)+.
Etapa D. 2-((1H-Pirazol-3-il)metil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de (6-((4-metil-5-oxo-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (3,0 g, 4,8 mmol) en etanol (30 ml) se añadió HCl (30 ml, 4 M en dioxano). La mezcla de reacción se agitó a 80°C durante 40 min. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y el sólido se recogió, se suspendió en agua y se neutralizó con solución acuosa de NaHCO3 a 10°C. Se filtró para dar el compuesto deseado 2-((1H-pirazol-3-il)metil)-6-((6-aminopiridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (1,5 g). LC-MS (ESI) encontrado: 393,2 (M+H)+. RMN 1H (400 MHz, DMSO-d6)5 12,78 (s, 1H), 8,53 (s, 1H), 7,72(s, 1H), 7,25 (dd, 1H), 6,33-6,24 (m, 2H), 6,08 (d, 1H), 5,90 (s, 2H), 5,19 (s, 2H), 4,49 (s, 2H), 4,26 (s, 3H).
Etapa E. Síntesis de (6-((4-metil-5-oxo-2-(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carbonil)-4H
tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo
A una solución de (6-((4-metil-5-oxo-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (100 mg, 0,16 mmol) en DMF (2 ml) se añadió K2CO3 (88 mg, 0,64 milimoles). La mezcla se agitó a 70°C durante 8 h. La mezcla se vertió en agua, el precipitado se recogió por filtración y se purificó por TLC preparativa (MeOH al 2% en DCM) para proporcionar el (6-((4-metil-5-oxo-2-(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carbonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (20 mg). LC-MS (ESI): m/z 637 (M+H)+.
Etapa F. Síntesis de 6-((6-aminopiridin-2-il)metil)-4-metil-2-(1H-pirazol-3-carbonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de (6-((4-metil-5-oxo-2-(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carbonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6(5H)-il)metil)piridin-2-il)carbamato de terc-butilo (20 mg, 0,03 mmol) en EtOH (1 ml) se añadió HCl ( i ml, 4 mol/l en dioxano). La mezcla se agitó a 80°C durante 1 h y se enfrió. El precipitado se recogió por filtración y se neutralizó con solución sat. de NaHCO3, se lavó con agua y se secó para proporcionar 5 mg de 6-((6-aminopiridin-2-il)metil)-4-metil-2-(1H-pirazol-3-carbonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-Ms (ESI): m/z 407 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 8: 8,75 (s, 1H), 7,96 (s, 1H), 7,50 (s, 1H), 7,31-7,22 (m, 1H), 6,31 (d, 1H), 6,14 (d, 1H), 5,91 (s, 2H), 5,23 (s, 2H), 4,38 (s, 3H).
Ejemplo 8E. Síntesis de 6-((1H-pirazol-3-il)metil)-2-((6-(dimetilamino)piridin-2-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. (6-(Dimetilamino)piridin-2-il)metanol. A una solución de (6-cloropiridin-2-il)metanol (500 mg, 2,67 mmol) en dimetilamina en THF (35 ml) se añadió Pd(OAc)2 (78 mg, 0,35 mmol), Xantphos (170 mg, 0,29 mmol) y f-BuONa (385 mg, 4.01 mmol). La mezcla de reacción se agitó a 100°C durante 18 h. La mezcla de reacción se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 35%) para proporcionar el (6-(dimetilamino)piridin-2-il)metanol (180 mg). LCMS: 153 (M+H)+.
Etapa B. 6-(Clorometil)-N,N-dimetilpiridin-2-amina. A una mezcla agitada de (6-(dimetilamino)piridin-2-il)metanol (170 mg, 1.1 mmol) en DCM (10 ml) se añadió SOCh (665 mg, 5,6 mmol) a 0°C. La mezcla de reacción se agitó a t.a. durante 1 h. La mezcla de reacción se ajustó a pH= 7-8 con solución ac. de NaHCO3. Luego, la mezcla se extrajo con DCM, se lavó con agua y salmuera. La capa orgánica se secó sobre Na2SO4, se concentró a presión reducida para proporcionar la 6-(clorometil)-N,N-dimetilpiridin-2-amina (70 mg). LCMS: 171 (M+H)+.
Etapa C. N,N-dimetil-6-((fenilsulfonil)metil)piridin-2-amina. A una mezcla agitada de 6-(clorometil)-N,N-dimetilpiridin-2-amina (500 mg, 2,94 mmol) en DMSO (10 ml) se añadió PhSO2Na (1,44 g, 8,82 mmol) a t.a. La mezcla se agitó a t.a. durante 18 h. La mezcla de reacción se vertió en agua y se extrajo con DCM. La mezcla se lavó con agua y la capa orgánica se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 20%) para proporcionar la N,N-dimetil-6-((fenilsulfonil)metil)piridin-2-amina (380 mg). LCMS: 277 (M+H)+.
Etapa D. 2-((6-(Dimetilamino)piridin-2-il)(fenilsulfonil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla agitada de 4-metil-2-(metilsulfonil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (180 mg, 0,36 mmol), que se sintetizó de manera similar al compuesto E8-1 en el ejemplo 8A, en THF seco (10 ml) se añadió N,N-dimetil-6-((fenilsulfonil)metil)piridin-2-amina (120 mg, 0,44 mmol) y t-BuOK (122 mg, 1,1 mmol) a 60°C en atmósfera de N2. La mezcla se agitó a 60°C durante 2 h en atmósfera de N2. Luego, la mezcla se vertió en agua y se extrajo con EtOAc, se lavó con agua y salmuera. La capa orgánica se secó sobre Na2SO4, se concentró a presión reducida y se purificó por TLC preparativa (PE: EtOAc=1: 1,5) para proporcionar la 2-((6-(dimetilamino)piridin-2-il)(fenilsulfonil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg). LCm S: 691 (M+H)+.
Etapa E. 2-((6-(Dimetilamino)piridin-2-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-((6-(dimetilamino)piridin-2-il)(fenilsulfonil)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg,
0,07 mmol) en THF (5 ml) y MeOH (5 ml) a t.a. en atmósfera de N2 se añadió SmI2 (5 ml, 0,1 M en THF) a -40°C. La mezcla de reacción se agitó a -40°C durante 10 min y luego se inactivó con agua. La siguiente mezcla se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera (30 ml), se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (PE/EtOAc = 1/1,5) para dar el producto deseado (10 mg). LCMS: m/z 551 (M+H)+.
Etapa F. 6((1H-Pirazol-3-il)metil)-2-((6-(dimetilamino)piridin-2-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de 2-((6-(dimetilamino)piridin-2-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-dJpiridazin-5(6H)-ona (10 mg, 0,018 mmol) en DCM/TFA (2 ml/ 2 ml) se agitó a t.a. durante 1 h. La mezcla de reacción se concentró. El residuo se purificó por HPLC preparativa para proporcionar el producto deseado (1,4 mg). LCMS: 421 (M+H)+. RMN 1H (400 MHz, DMSO) 58,51 (s, 1H), 7,55 (s, 1H), 7,48 (dd, 1H), 6,64 (d, 1H), 6,54 (d, 1H), 6,11 (d, 1H), 5,32 (s, 2H), 4,44 (s, 2H), 4,26 (s, 3H), 3,05 (s, 6H).
Los siguientes compuestos se sintetizaron según el esquema E8 y el ejemplo 8C usando el material de partida apropiado. Se puede utilizar la protección y desprotección convencionales cuando sea necesario.
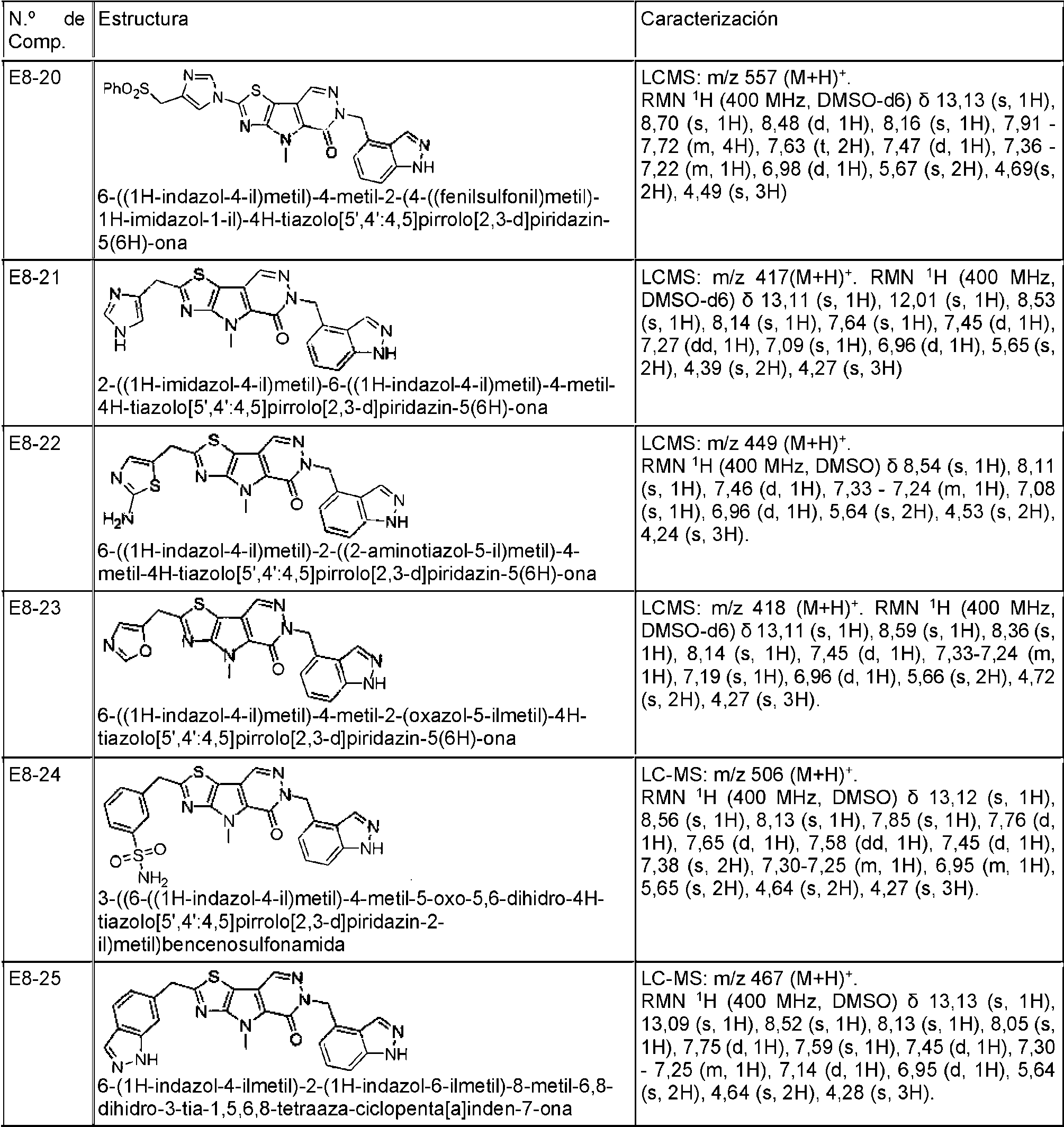















Ejemplo 8F. Síntesis de 4-metil-6-(2-(1 -metil-1H-pirazol-3-il)etil)-2-(piridin-3-Mmetil)-4H-tiazolo[5,,4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (360 mg, 1,4 mmol) y 2-(1-metil-1H-pirazol-3-il)etanol (300 mg, 2,4 mmol) en tolueno (10 ml) se añadió CMBp (600 mg, 2,1 mmol). La mezcla de reacción se agitó a 110°C en atmósfera de N2 durante 3 h. Después de enfriar a temperatura ambiente, la mezcla de reacción se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 80-100%) para producir la 4-metil-6-(2-(1-metil-lH-pirazol-3-il)etil)-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (500 mg). LC-MS (ESI): m/z 361 (M+1)+.
Etapa B. Síntesis de 4-metil-6-(2-(1 -metil-1H-pirazol-3-il)etil)-2-(metilsulfonil)-4H-tiazolo[5',4,:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-6-(2-(1-metiMH-pirazol-3-il)etil)-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (600 mg, 1,7 mmol) en DCM (20 ml) se añadió m-CpBA (1,01 g, 5,0 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se diluyó con EtOAc, se lavó con solución sat. de NaHCO3 y salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 80-100%) para producir la 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (600 mg). LC-MS (ESI): m/z 393 (M+1)+.
Etapa C. Síntesis de 4-metil-6-(2-(1 -metil-1H-pirazol-3-il)etil)-2-((fenilsulfonil)(piridin-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 3-((fenilsulfonil)metil)piridina (300 mg, 1,3 mmol) en THF
(20 ml) se añadió LiHMDS (2 ml, 2,0 mmol) a temperatura ambiente. Después de agitar a temperatura ambiente en atmósfera de N2 durante 15 min, se añadió 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg, 0,8 mmol) a la mezcla de reacción y la solución resultante se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se inactivó con solución sat. de NH4Cl, se extrajo con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 80-100%) para proporcionar 110 mg de 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-((fenilsulfonil)(piridin-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS: m/z 546 (M+1)+.
Etapa D. Síntesis de 4-metil-6-(2-(1 -metil-1H-pirazol-3-il)etil)-2-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-((fenilsulfonil)(piridin-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (110 mg, 0,2 mmol) en THF (5 ml) y MeOH (5 ml) se añadió Smh (5 ml, 0,1 M en THF) a t.a. Luego, la mezcla de reacción se agitó en atmósfera de N2 a t.a. durante 10 min. La solución de reacción se inactivó con agua, se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 80-100%) para proporcionar 6 mg de 4-metil-6-(2-(1-metil-1H-pirazol-3-il)etil)-2-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 406 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 88,66 (s, 1H), 8,52 (d, 2H), 7,84 (d, 1H), 7,54 (d, 1H), 7,41 (dd, 1H), 6,04 (d, 1H), 4,58 (s, 2H), 4,48 - 4,30 (m, 2H), 4,26 (s, 3H), 3,75 (s, 3H), 3,12-2,75 (m, 2H).
Ejemplo 8G. Síntesis de 4-metil-6-(prop-2-in-1-il)-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de_4-metil-2-(metilsulfonil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (200 mg, 0,70 mmol) en DMF (5 ml) se añadió t-BuOK (157 mg, 1,4 mmol), seguido de 3-bromoprop-1-ino (0,12 ml, 1,4 mmol). La reacción se agitó a temperatura ambiente durante 15 min. Después la suspensión se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0~50%) para proporcionar 60 mg de 4-metil-2-(metilsulfonil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 323 (M+H)+.
Etapa B. Síntesis de 4-metil-2-((fenilsulfonil)(piridin-2-il)metil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona._A una mezcla de 4-metil-2-(metilsulfonil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg, 0,18 mmol) y 2-((fenilsulfonil)metil)piridina (87 mg, 0,37 mmol) en Th F (5 ml) se añadió t-BuOK (63 mg, 0,57 mmol). Después de agitar a temperatura ambiente durante 1 h en atmósfera de nitrógeno, la reacción se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por TLC preparativa (eluyente: EtOAc en éter de petróleo al 70%) para proporcionar 50 mg de 4-metil-2-((fenilsulfonil)(piridin-2-il)metil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 476 (M+H)+.
Etapa C. Síntesis de 4-metil-6-(prop-2-in-1-il)-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona_A una solución de 4-metil-2-((fenilsulfonil)(piridin-2-il)metil)-6-(prop-2-in-1-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,11 mmol) en THF/MeOH (4 ml, 1:1) se añadió Sml2 (4,2 ml, 0,1 M en THF) a -70°C en atmósfera de nitrógeno. Después de agitar durante 5 min, la reacción se inactivó con agua. La mezcla se diluyó con EtOAc y se lavó con solución sat. de NH4CL La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por HPLC preparativa para proporcionar 5 mg de 4-metil-6-(prop-2-in-1-il)-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS: m/z 336 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 88,60 - 8,52 (m, 2H), 7,81 (td, 1H), 7,51 (d, 1H), 7,33 (dd, 1H), 4,93 (d, 2H), 4,67 (s, 2H), 4,26 (s, 3H), 3,27 (t, 1H).
Ejemplo 8H. Síntesis de 2-((1H-pirazol-3-il)metil)-6-bencil-4-etil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Síntesis de 4-etil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una mezcla de 2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (500 mg, 2,06 mmol) en DMF (5 ml) se añadió K2CO3 (856 mg, 6,19 milimoles). Después de agitara 70°C durante 1,5 h, se añadió EtI (483 mg, 3,10 mmol). La mezcla se agitó a 70°C durante 1 h más. La mezcla de reacción se diluyó con H2O y se extrajo con EtOAc. Las fases orgánicas combinadas se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0-30%) para dar el 4-etil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg). Lc -MS (ESI): m/z 271 (M+1)+.
Etapa B. Síntesis de 4-etil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo Una mezcla de POCh (3,6 ml) y PhNMeCHO (5 ml) se agitó a t.a. durante 1 h, luego se añadió a una solución de 4-etil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg, 1,48 mmol) en DCE (10 ml). Después de agitara 100°C durante 2 h, la mezcla de reacción se diluyó con H2O y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 anhidro y se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0~30%) para dar el 4-etil-6-formil-2-(metiltio)-4H-.pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg). LC-Ms (ESI): m/z 299 (M+1)+.
Etapa C. Síntesis de 6-bencil-4-etil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 4-etil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato (400 mg, 1,34 mmol) en AcOH (4 ml) se añadió dihidrocloruro de bencilhidracina (260 mg, 1,34 mmol) en atmósfera de N2. La mezcla se agitó a 100°C durante 3 h. La mezcla de reacción se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0-50%) para dar 250 mg de 6-bencil-4-etil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. LC-m S: m/z 357 (M+1)+. Etapa D. Síntesis de 6-bencil-4-etil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-etil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (250 mg, 0,70 mmol) en DCM (5 ml) se añadió mCPBA (657,5 mg, 3,8 mmol) a 0°C. Después de agitar durante 1,5 h, la mezcla de reacción se evaporó a presión reducida. El residuo se purificó por TLC preparativa (MeOH en DCM al 10%) para dar la 6-bencil-4-etil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (90 mg). LC-Ms : m/z 389 (M+1)1.
Etapa E. Síntesis de 6-bencil-4-etil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-etil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (90 mg, 0,23 mmol) y 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (123 mg, 0,35 mmol) en THF (3 ml) se añadió y KOfBu (85 mg, 0,76 mmol) en atmósfera de N2 a t.a. Después de agitar a t.a. durante 30 min, la mezcla de reacción se inactivó con solución sat. de NH4Cl y se extrajo con EtOAc. Las fases orgánicas combinadas se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0-50%) para dar la 6-bencil-4-etil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (140 mg, 92,2% de rendimiento) . LC-MS: m/z 661 (M+1)+.
Etapa F. Síntesis de 6-bencil-4-etil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-etil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4’:4,5]pirrolo[2,3-d]piridazin-5-ona (140 mg, 0,21 mmol) en EtOH (2 ml) y DCE (1 ml) se añadió ácido acético (0,2 ml, 2,8 mmol) y zinc (360 mg, 5,5 mmol). La mezcla se agitó a 80°C durante 3 h. La mezcla de reacción se filtró y el filtrado se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0-50%) para dar 30 mg de 6-bencil-4-etil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. LC-MS (ESI): m/z 521 (M+1)+.
Etapa G. Síntesis de 2-((1H-pirazol-3-il)metil)-6-bencil-4-etil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-etil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (30 mg, 0,06 mmol) en DCM (4 ml) se añadió TFA (4 ml). Después de agitar a t.a. durante 1,5 h, la mezcla de reacción se evaporó a presión reducida. El residuo se purificó por t Lc preparativa (MeOH en Dc M al 20%) para dar la 2-((1H-pirazol-3-il)metil)-6-bencil-4-etil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3
d]piridazin-5-ona (5 mg). LC-MS (ESI): m/z 391 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 512,79 (s, 1H), 8,53 (s, 1H), 7,72 (s, 1H), 7,42-7,11 (m, 5H), 6,27 (d, 1H), 5,36 (s, 2H), 4,78 (q, 2H), 4,48 (s, 2H), 1,44 (t, 3H).
Ejemplo 8I. Síntesis de 6-bencil-4-fenil-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg, 1,7 mmol) y Phl (265 mg, 2,55 mmol) en dioxano (5 ml) se añadió Cs2COa (1,1 g, 3,4 mmol), seguido de CuI (65 mg, 0,34 mmol) y (1R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (48 mg, 0,34 mmol). La mezcla se agitó en atmósfera de nitrógeno a 100°C durante 7 horas. La mezcla se vertió en agua y se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0 ~ 10%) para dar el 2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg). LC-MS (ESI): m/z 319 (M+1)+.
Etapa B. Síntesis de 6-formil-2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de PhNMeCHO (1,5 ml) en DCE (5 ml) se le añadió POCh (1,2 ml, 12,5 mmol) a 0°C. La mezcla se agitó a t.a. durante 30 min. Luego se añadió a la mezcla 2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (400 mg, 1,25 mmol), la mezcla se agitó a 50°C durante la noche. La mezcla se inactivó con solución sat. de NaHCO3 y se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0 ~ 50%) para dar el 6-formil-2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (280 mg). LC-MS (ESI): m/z 347 (M+1)+.
Etapa C. Síntesis de 6-bencil-2-(metiltio)-4-fenil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 6-formil-2-(metiltio)-4-fenil-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (280 mg, 0,81 mmol) en AcOH (6,0 ml) se añadió dihidrocloruro de bencilhidrazina (468 mg, 2,4 mmol). La mezcla de reacción se agitó a 100°C durante 2 h. El filtrado se evaporó y se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~70%) para dar 100 mg de 6-bencil-2-(metiltio)-4-fenil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. lC-MS: m/z 405 (M+1)+.
Etapa D. Síntesis de 6-bencil-2-(metilsulfonil)-4-fenil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de 6-bencil-2-(metiltio)-4-fenil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,25 mmol) en DCM (5 ml) se añadió mCPBA (151 mg, 0,75 mmol). La mezcla se agitó a t.a. durante 4 h. La mezcla se inactivó con solución ac. de Na2S2O3 y se extrajo dos veces con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0 ~ 100%) para dar la 6-bencil-2-(metilsulfonil)-4-fenil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg). LC-MS (ESI): m/z 437 (M+1)+.
Etapa E. Síntesis de 6-bencil-4-fenil-2-((fenilsulfonil)(piridin-2-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 6-bencil-4-fenil-2-((fenilsulfonil)(piridin-2-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (60 mg, 0,14 mmol) y 2-((fenilsulfonil)metil)piridina (50 mg, 0,21 mmol) en DMF seca (5 ml) se añadió t-BuOK (31 mg, 0,28 mmol) en atmósfera de N2. La mezcla de reacción se agitó a 60°C durante 1,5 h. Después de enfriar a t.a., la mezcla se vertió en solución sat. de NH4CL La siguiente mezcla se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por TLC preparativa (éter de petróleo/EtOAc = 1/4) para dar 40 mg de 6-bencil-4-fenil-2-((fenilsulfonil)(piridin-2-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 590 (M+1)+.
Etapa F. Síntesis de 6-bencil-4-fenil-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 6-bencil-4-fenil-2-((fenilsulfonil)(piridin-2-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,05 mmol) en THF (1,5 ml) y MeOH (1,5 ml) se añadió Smh (2,5 ml, 0,1 M en THF) a -78°C en atmósfera de N2. La mezcla de reacción se agitó a -78°C durante 10 min y luego se inactivó con agua. La mezcla se extrajo dos veces con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por HPLC para dar la 6-bencil-4-fenil-2-(piridin-2-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (5 mg). LC-MS (ESI): m/z 450 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 58,67 (s, 1H), 8,56 (d, 1H), 7,82-7,76 (m, 1H), 7,59-7,45 (m, 6H), 7,34-7,23 (m, 6H), 5,32 (s, 2H), 4,63 (s, 2H).
Ejemplo 8J. Síntesis de 2-((1H-pirazol-3-il)metil)-4,6-dibencil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 4-bencil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo. A una mezcla de 6-formil-2- (metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (300 mg, 1,1 mmol) en DMF (3 ml) se añadió K2CO3 (460 mg, 3,3 mmol). Después de agitar a 70°C durante 1,5 h, se añadió BnBr (0,2 ml, 1,6 mmol). La mezcla se agitó a 70°C durante 1 h. La mezcla de reacción se diluyó con H2O y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 anhidro y se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0-30%) para dar el 4-bencil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (370 mg). LC-MS (ESI): m/z 361 (M+1)+.
Etapa B. Síntesis de 4,6-dibencil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-bencil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (370 mg, 1,0 mmol) en AcOH (4 ml) se añadió dihidrocloruro de bencilhidracina (390 mg, 2,0 mmol) en atmósfera de N2. La mezcla se agitó a 100°C durante 3 h. La mezcla de reacción se diluyó con H2O y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 anhidro y se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar la 4,6-dibencil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (320 mg). LC-MS (ESI): m/z 419 (M+1)+.
Etapa C. Síntesis de 4,6-dibencil-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4,6-dibencil-2-(metiltio)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (320 mg, 0,76 mmol) en DCM (5 ml) se añadió mCPBA (657,5 mg, 3,2 mmol). Después de agitar a 0°C durante 1,5 h, la mezcla de reacción se inactivó con solución sat. de Na2S2O3, se extrajo con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se evaporó a presión reducida. El residuo se purificó por TLC preparativa (MeOH en DCM al 10%) para dar la 4,6-dibencil-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (170 mg). LC-MS (ESI): m/z 451 (M+1)+.
Etapa D. Síntesis de 4,6-dibencil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoaci)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4,6-dibencil-2-(metilsulfonil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (170 mg, 0,38 mmol) y 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (160 mg, 0,46 mmol) en THF (3 ml) se añadió KO'Bu (85 mg, 0,76 mmol) en atmósfera de N2 a t.a. Después de agitar a t.a. durante 3 h, la mezcla de reacción se inactivó con solución sat. de NH4Cl y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 anhidro y se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar la 4,6-dibencil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (200 mg). LC-MS (ESI): m/z 723 (M+1)+.
Etapa E. Síntesis de 4,6-dibencil-2-((1-((2-(trimetilsilil)etoario)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 6-((4-aminopirimidin-2-il)metil)-4-metil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3- il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (200 mg, 0,28 mmol) en EtOH (2 ml) y DCE (1 ml) se añadieron ácido acético (0,2 ml, 2,8 mmol) y zinc (360 mg, 5,5 mmol). Después de agitar a 100°C durante 3 h, la mezcla de reacción se filtró y el filtrado se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar la 4,6-dibencil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (120 mg). LC-MS: m/z 583 (M+1)+.
Etapa F. Síntesis de 2-((1H-pirazol-3-il)metil)-4,6-dibencil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4,6-dibencil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (120 mg, 0,21 mmol) en DCM (4 ml) se añadió TFA (4 ml). La mezcla se agitó a t.a. durante 1,5 h. La mezcla de reacción se evaporó a presión reducida. El residuo se purificó por TLC preparativa (MeOH en DCM al 20%) para dar 9 mg de 2-((1H-pirazol-3-il)metil)-4,6-dibencil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS: m/z 453 (M+1)+. RMN 1H (400 MHz, DMSO-CÍ6) 512,78 (s, 1H), 8,57 (s, 1H), 7,71 (s, 1H), 7,42 - 7,04 (m, 10H), 6,25 (d, 1H), 5,98 (s, 2H), 5,37 (s, 2H), 4,48 (s, 2H).
Ejemplo 8K. Síntesis de 2-((1H-pirazol-3-il)metil)-6-bencil-4-ciclopropil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Síntesis de 4-ciclopropil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una suspensión de ácido ciclopropilborónico (687 mg, 8 mmol) y 2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (970 mg, 4 mmol) en DCE (10 ml) se añadido Na2CO3 (848 mg, 8 mmol), seguido de Cu(OAc)2 (727 mg, 4 mmol) y bipiridina (625 mg, 4 mmol). La mezcla se agitó a 70°C durante 2 h en aire. La mezcla resultante se enfrió a temperatura ambiente y se inactivó con solución sat. de NH4Cl, se extrajo con DCM. Las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~30%) para dar el 4-ciclopropil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1,0 g, 88,5% de rendimiento). LC-MS (ESI): m/z 283 (M+1)1.
Etapa B. Síntesis de 4-ciclopropil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo Una mezcla de POCl3 (8,6 ml) y PhNMeCHO (12 ml) se agitó a t.a. durante 1 h, luego se añadió a una solución de 4-ciclopropil--2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1,0 g, 3,54 mmol) en DCE (10 ml). Después de agitara 100°C durante 2 h, la mezcla de reacción se diluyó con H2O y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 anhidro y se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~30%) para dar el 4-ciclopropil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (620 mg). LC-MS (ESI): m/z 311 (M+1)+.
Etapa C. Síntesis de 6-bencil-4-ciclopropil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 4-etil-6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato (620 mg, 2 mmol) en AcOH (4 ml) se le añadió dihidrocloruro de bencilhidracina (390 mg, 2 mmol) en atmósfera de N2. La mezcla se agitó a 100°C durante 3 h. La mezcla de reacción se evaporó a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar la 6-bencil-4-etil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (320 mg). LC-MS (ESI): m/z 369 (M+1)+.
Etapa D. Síntesis de 6-bencil-4-ciclopropil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-ciclopropil-2-(metiltio)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (320 mg, 0,87 mmol) en DCM (5 ml) se añadió mCPBA (704 mg, 3,5 mmol). Después de agitar a 0°C durante 1,5 h, la mezcla de reacción se inactivó con solución sat. de Na2S2O3, se extrajo con EtOAc. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se evaporó a presión reducida. El residuo se purificó por TLC preparativa (MeOH en DCM al 10%) para dar 110 mg de la 6-bencil-4-ciclopropil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. LC-MS: m/z 401 (M+1)+.
Etapa E. Síntesis de 6-bencil-4-ciclopropil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-etil-2-(metilsulfonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (110 mg, 0,27 mmol) y 3-((fenilsulfonil)metil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (123 mg, 0,35 mmol) en THF (3 ml) se añadió KO'Bu (85 mg, 0,76 mmol) en atmósfera de N2 a t.a. La mezcla se agitó a t.a. durante 3 h. La mezcla de reacción se inactivó con solución sat. de Nh4CI y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar 100 mg de la 6-bencil-4-ciclopropil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. LC-MS: m/z 673 (M+1)+.
Etapa F. Síntesis de 6-bencil-4-ciclopropil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-ciclopropil-2-((fenilsulfonil)(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (100 mg, 0,15 mmol) en MeOH (3 ml) y THF (3 ml) añadido Sml2 (4,5 ml, 0,45 mmol) a -60°C en atmósfera de N2. La mezcla se agitó a -60°C durante 10 min, se inactivó con H2O y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre Na2SO4 y se evaporaron a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 0~50%) para dar 40 mg de la 6-bencil-4-ciclopropil-2-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. lC-MS: m/z 533 (M+1)+.
Etapa G. Síntesis de 2-((1H-pirazol-3-il)metil)-6-bencil-4-cidopropil-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona A una mezcla de 6-bencil-4-ciclopropil-2-((1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona (40 mg, 0,07 mmol) en DCM (4 ml) se añadió TFA (4 ml). La mezcla se agitó a t.a. durante 1,5 h. La mezcla de reacción se evaporó a presión reducida. El residuo se purificó por TLC preparativa (MeOH en DCM al 20%) para dar 5 mg de 2-((1H-pirazol-3-il)metil)-6-bencil-4-ciclopropil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. LC-MS(ESI): m/z 403 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 812,77 (s, 1H), 8,50 (s, 1H), 7,68 (s, 1H), 7,35 - 7,23 (m, 5H), 6,26 (d, 1H), 5,35 (s, 2H), 4,48 (s, 2H), 4,19 (tt, 1H), 1,40 (td, 2H), 1,15 (td, 2H).
Ejemplo 8L. Síntesis de 4-metil-6-((1-metil-1H-pirazol-3-il)metil)-2-(2-(piridin-3-il)etil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 6-(hidroximetil)-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 6-formil-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1,3 g, 4,8 mmol) en MeOH (20 ml) se añadió NaBH4 (274 mg, 7,2 mmol) a 0°C. Después de agitar a 0°C durante 20 min, la solución de reacción se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4, se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0~10%) para dar el 6-(hidroximetil)-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1 g). LC-MS (ESI): m/z 273(M+1)+.
Etapa B. Síntesis de 6-(hidroximetil)-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 6-(hidroximetil)-2-(metiltio)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1 g, 3,7 mmol)y2-bromotiazol (3 g, 18,4 mmol) en dioxano (30 ml) se añadió Cs2CO3 (3 g, 9,2 mmol), seguido de Cul (700 mg, 3,7 mmol) y N1,N2-dimetilciclohexano-1,2-diamina (520 mg, 3,7 mmol). La mezcla de reacción se agitó a 110°C en atmósfera de N2 durante 16 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró. El filtrado se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 30-50%) para dar el 6-(hidroximetil)-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1 g). LC-MS (ESI): m/z 356 (M+1)+.
Etapa C. Síntesis de 6-formil-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo A una solución de 6-(hidroximetil)-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (1 g, 2,8 mmol) en DCM (20 ml) se añadió Dess-martin (1,3 g, 3,4 mmol). Después de agitar a t.a. durante 2 h, la solución de reacción se diluyó con DCM, se lavó con solución sat. de NaHCO3 y salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en éter de petróleo al 20~30%) para dar 6-formil-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (800 mg). LC-MS (ESI): m/z 354 (M+1)+. Etapa D. Síntesis de 6-bencil-2-(metiltio)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de 6-formil-2-(metiltio)-4-(tiazol-2-il)-4H-pirrolo[2,3-d]tiazol-5-carboxilato de etilo (800 mg, 2,3 mmol) en AcOH (10 ml) se añadió dihidrocloruro de bencilhidracina (449 mg, 2,3 mmol). Después de agitar a t.a. durante 1 h, la mezcla de reacción se calentó a 100°C durante 2 h. La mezcla de reacción se enfrió a 0°C, se neutralizó con solución ac. de NaOH 1M, se extrajo con DCM. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0~5%) para dar la 6-bencil-2-(metiltio)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg). LC-MS (ESI): m/z 412 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 8 8,72 (s, 1H), 7,98 (d, 1H), 7,87 (d, 1H), 7,40 - 7,15 (m, 5H), 5,31 (s, 2H), 2,76 (s, 3H).
Etapa E. Síntesis de 6-bencil-2-(metilsulfonil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 6-bencil-2-(metiltio)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (300 mg, 0,73 mmol) en MeOH (5 ml) y THF (5 ml) se añadió una solución de Oxone (2,2 g, 3,6 mmol) en H2O (5 ml) a 0°C. La mezcla de reacción se agitó a t.a. durante 2 h. La mezcla de reacción se diluyó con DCM, se lavó con solución sat. de NaHCO3 y salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice,
MeOH en DCM al 0~5%) para dar la 6-bencil-2-(metilsulfonil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (260 mg). LC-MS (ESI): m/z 444 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 88,93 (s, 1H), 8,04 (d, 1H), 7,91 (d, 1H), 7,40 - 7,18 (m, 5H), 5,35 (s, 2H), 3,53 (s, 3H).
Etapa F. Síntesis de 6-bencil-2-((fenilsulfonil)(piridin-2-il)metil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución agitada de 2-((fenilsulfonil)metil)piridina (120 mg, 0,51 mmol) en THF (3 ml) se añadió LiHMDS (0,7 ml, 0,7 mmol) a 0°C en atmósfera de N2. Después de agitar a 0°C durante 30 min, se añadió una solución de 6-bencil-2-(metilsulfonil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (150 mg, 0,34 mmol) en THF (3 ml). La mezcla resultante se agitó a t.a. durante 1 h. La reacción se inactivó con solución sat. de NH4Cl, se extrajo con DCM. La fase orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0~5%) para dar la 6-bencN-2-((fenNsulfonN)(piridin-2-N)metil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg). LC-MS (ESI): m/z 597 (M+1)+.
Etapa G. Síntesis de 6-bencil-2-(piridin-2-ilmetil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una solución de 6-bencil-2-((fenilsulfonil)(piridin-2-il)metil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,17 mmol) en AcOH (5 ml) se añadió Zn (110 mg, 1,7 mmol). La mezcla de reacción se agitó a t.a. durante 15 min. La mezcla de reacción se diluyó con DCM, se lavó con solución sat. de NaHCO3 y salmuera, se secó sobre Na2SO4 anhidro y se concentró al vacío. El residuo se purificó por TLC preparativa (EtOAc en éter de petróleo al 65%) para dar 5 mg de 6-bencil-2-(piridin-2-ilmetil)-4-(tiazol-2-il)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 457 (M+1)+. RMN 1H (400 MHz, DMSO-d6) 88,72 (s, 1H), 8,61 (d, 1H), 7,98 (d, 1H), 7,92 - 7,84 (m, 2H), 7,55 (d, 1H), 7,41 (dd, 1H), 7,35-7,23 (m, 5H), 5,32 (s, 2H), 4,70 (s, 2H).
Ejemplo 9. Síntesis de los compuestos E9-vi y E9-vii
Esquema E9

El compuesto E9-iv se puede sintetizar mediante dos enfoques, (i) y (ii) del esquema 9. Para el enfoque (i), el compuesto E9-ii se puede sintetizar a partir del compuesto E9-i mediante una reacción de alquilación como se muestra en el ejemplo 7 o ejemplo 8. Como se usa en el presente documento, Xa es un grupo saliente (p. ej., Br, I, OMs u OTs). La reacción de formilación del compuesto E9-ii con LiHMDS y DMF proporciona el compuesto intermedio E9-iii. E9-iii reacciona con un agente reductor (p. ej., NaBH4) para proporcionar el compuesto E9-iv. Alternativamente, en el enfoque (ii), la halogenación del compuesto E9-i genera el compuesto E9-ix. El compuesto E9-ix se somete a reacción de Stille, ozonólisis y reducción para proporcionar el compuesto E9-x. El compuesto E9-x se puede alquilar con E9-viii para proporcionar el compuesto E9-iv. En el Esquema 9, (iii), el compuesto E9-iv se somete a halogenación para dar el compuesto intermedio E9-v (Xb es halógeno tal como Cl o Br). Un acoplamiento catalizado por metal (p. ej., Pd o Cu) de E9-v con estaño, boro, zinc o magnesio orgánicos proporciona el compuesto E9-vi. Como se usa en el presente documento; el compuesto E9-vtambién puede reaccionar con algunos nucleófilos tales como el nitrógeno en un heterociclo para dar el producto E9-vii. Como se usa en el presente documento, M es un complejo de metal orgánico (p. ej., complejo de organoboro tal como ácido borónico o complejo de pinacoboro, complejo de organoestaño tal como -Sn(Bu%; complejo de organozinc tal como -Zn(halógeno)); An y Ar2 son cada uno independientemente arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, carbociclo opcionalmente sustituido o heterociclilo opcionalmente sustituido. En ciertas realizaciones, An yAr2 son cada uno independientemente heteroarilo opcionalmente sustituido.
Ejemplo 9A. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-2-(tiazol-4-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído. A una mezcla de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (2,6 g, 5,57 mmol, 1 eq) en THF seco (30 ml) se añadió LiHMDS ( i M, 11,14 ml, 2,0 eq) a -78°C. °C La mezcla se agitó a -78°C durante 2 h. Luego se añadió gota a gota DMF (2,04 g, 27,86 mmol, 2,14 ml, 5,0 eq) a la mezcla anterior. La mezcla se agitó a -78°C durante 2 h. La TLC (PE: EA=2:1, UV=254 nm) mostró que se había formado una nueva mancha principal. La mezcla se vertió en solución sat. de NH4Cl (20 ml) fría. Luego la mezcla se calentó a temperatura ambiente. La mezcla se extrajo con EtOAc (40 ml x 3). La capa orgánica se lavó con agua (20 ml x 3) y se concentró al vacío para dar el producto deseado (2,6 g, bruto). LCMS: m/z 495,2 [M+H]+
Etapa B. Síntesis de 2-(hidroximetil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (1,0 g, 2,02 mmol, 1 eq) en THF (10 ml) y MeOH (10 ml) se añadió NaBH4 (152,97 mg, 4,04 mmol, 2 equiv.). La mezcla se agitó a 30°C durante 14 h. La TLC (DCM:MeOH=10:1, UV=254 nm) mostró que el material de partida se había consumido por completo y se había formado una nueva mancha principal. La reacción se inactivó mediante la adición de agua (20 ml) y se extrajo con EtOAc (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml). La fase orgánica se concentró al vacío. El residuo se purificó por cromatografía ultrarrápida en gel de sílice (ISCO®; Columna ultrarrápida de sílice SepaFlash® 40 g, eluyente de MeOH/DCM al 0~5% a 30 ml/min). El eluyente se concentró al vacío para dar el producto deseado (382 mg). LCMS: m/z 497,1 [M+H]+. RMN 1H (400 MHz, DMSO-cfe) 8 ppm 8,61 (s, 1H), 8,25 (s, 1H), 7,66 (d, 1H), 7,38 (t, 1H), 7,05 (d,), 6,36 (t, 1H), 5,74 (s, 2H), 5,68 (s, 2H), 4,89 (d, 2H), 4,26 (s, 3H), 3,50 (t, 2H), 0,78 (t, 2H), -0,12 (s, 9H).
Etapa C. Síntesis de 2-(clorometil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-(hidroximetil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (150,0 mg, 302,02 umol, 1 eq) y EtsN (61,12 mg, 604,04 umol, 84,08 ul, 2,0 eq) en DCM (5 ml) se añadió cloruro de 4-metilbencenosulfonilo (75,0 mg, 393,40 umol, 1,30 eq). La mezcla se agitó a 30°C durante 5 h. La TLC (PE: EA=4:1, UV=254 nm) mostró que el material de partida se había consumido por completo. Se añadieron a la mezcla agua (10 ml) y DCM (20 ml). Las capas orgánicas se concentraron al vacío para dar una goma amarilla (0,1 g). El residuo se purificó por cromatografía ultrarrápida en gel de sílice (ISCO®; Columna ultrarrápida de sílice SepaFlash® 4 g, eluyente de gradiente de acetato de etilo/éter de petróleo al 0~20% a 30 ml/min). La fracción deseada se concentró al vacío para dar el producto deseado (40,0 mg, 76,88 umol). LCMS: m/z 515,1 [M+H]+. RMN 1H (400 MHz, cloroformo-d) 8 ppm 8,27 (d, 1H), 8,26 (s, 1H), 7,52 (d, 1 H), 7,39 (dd, 1 H), 7,25 (s, 1H), 5,76 (s, 2H), 5,72 (s, 2H), 4,96 (s, 2H), 4,40 (s, 3H), 3,50-3,57 (m, 2H), 0,84-0,90 (m, 2H), -0,09 a -0,06 (m, 9H).
Etapa D. Síntesis de 4-metil-2-(tiazol-4-ilmetil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una solución de 2-(clorometil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,97 mmol) y 4-(tributilestannil)tiazol (114 mg, 2,91 mmol) en tolueno (4 ml) se añadió Pd(PPh3)4 (402 mg, 2,91 mmol). Luego, la mezcla se calentó en un reactor de MW a 120°C durante 30 min en atmósfera de N2. La solución se vertió en agua y se extrajo con EtOAc, se secó sobre Na2SO4 anhidro. La capa orgánica se concentró a presión reducida. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, acetato de etilo en éter de petróleo al 0 ~ 50%) para dar la 4-metil-2-(tiazol-4-ilmetil)-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (compuesto E9-4) (30 mg). LCMS: 564 (M+H)+.
Etapa E. Síntesis de 6-((1H-indazol-4-il)metil)-4-metil-2-(tiazol-4-ilmetil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla del compuesto E9-4 (30 mg, 0,05 mmol) en DCM (3 ml) a t.a. en atmósfera de N2 se añadió TFA (3 ml). La mezcla de reacción se agitó a t.a. durante 1 h. La mezcla se concentró a presión reducida. El residuo se purificó por HPLC preparativa (C18, acetonitrilo en H2O con ácido fórmico al 0,1% al 0 ~ 90%) para dar el producto deseado (3,9 mg). LCMS: 434 (M+H)+. RMN 1H (400 MHz, DMSO) 8 13,12 (s, 1H), 9,12 (d, 1H), 8,56 (s, 1H), 8,14 (s, 1H), 7,71 (d, 1H), 7,45 (d, 1H), 7,35 - 7,24 (m, 1H), 6,96 (d, 1H), 5,65 (s, 2H), 4,70 (s, 2H), 4,27 (s, 3H).
Ejemplo 9B. Síntesis de 2-((1H-imidazol-1-il)metil)-6-((1H-indazol-4-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Síntesis de 2-((1H-imidazol-1-il)metil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una mezcla de 2-(clorometil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (25,0 mg, 48,53 umol, 1 eq) y KI (8,06 mg, 48,53 umol, 1 eq) en THF (0,3 ml) se añadió imidazol (33,04 mg, 485,34 umol, 10,0 eq). La mezcla se calentó a 60°C y se agitó a 60°C durante 12 h. La LCMS mostró que se había generado el producto deseado y el material de partida se había consumido por completo. La mezcla de reacción se combinó con otro lote (25 mg). La mezcla se concentró al vacío para dar una goma amarilla bruta (50,0 mg). El producto se usaría para la siguiente etapa de reacción sin ninguna purificación. LCMS: m/z 547,2 [M+H]+.
Etapa B. Síntesis de 2-((1H-imidazol-1-il)metil)-6-((1H-indazol-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 2-((1H-imidazol-1-il)metil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50,0 mg, 64,02 umol, 1 eq) en dioxano (1 ml) se añadió HCl/dioxano (4 M, 2 ml, 124,96 eq) y 2 gotas de agua. La mezcla se agitó a 30°C durante 12 h. La TLC (PE: eA=1:1, UV=254 nm) mostró que el material de partida se había consumido por completo. La mezcla se concentró al vacío para dar una goma amarilla (0,2 g) que se purificó por HPLC preparativa (básica) para dar el producto del título (4,0 mg). LCMS: m/z 417,0 [M+H]+. RMN 1H (400 MHz, metanol-ck) 8 ppm 8,43 (s, 1H), 8,22 (s, 1H), 7,89 (s, 1H), 7,43-7,49(m, 1H), 7,29-7,36(m, 2H), 7,12(d, 1H), 7,04 (s, H), 5,74 (s, 2H), 5,73 (s, 2H), 4,34 (s, 3H).
Ejemplo 9C. Síntesis de 4-metil-2-(piridin-2-ilmetil)-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina 5(6H)-ona

Etapa A. Síntesis de 2-(hidroximetil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5 (6H)-ona_A -78°C, a una mezcla de 4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (640 mg, 2,15 mmol) en THF (10 ml) se añadió LiHMDS (4,3 ml, 1 M en THF). Después de 30 min, se añadió a la mezcla DMF seca (0,84 ml, 10,8 mmol). Después del consumo completo del material de partida, se añadió una mezcla de NaBH4 (164 mg, 4,3 mmol) en EtOH (4 ml) y se agitó durante 5 min. Después la mezcla se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0~5%) para producir la 2-(hidroximetil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (220 mg). LC-MS (ESI): m/z 328 (M+H)+.
Etapa B. Síntesis de 2-(clorometil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 2-(hidroximetil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (100 mg, 0,31 mmol) en DCM (5 ml) se añadió Et3N (0,43 ml, 3,1 mmol) y MsCl (0,12 ml, 1,5 mmol). La reacción se agitó a temperatura ambiente durante 6 h. Después la mezcla se lavó con solución sat. de NH4Cl (ac.), se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, EtOAc en PE al 0~50%) para proporcionar 65 mg de 2-(clorometil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC MS (ESI): m/z 346 (M+H)+.
Etapa C. Síntesis de 4-metil-2-(piridin-2-ilmetil)-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona En atmósfera de nitrógeno, a una mezcla de 2-(clorometil)-4-metil-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (50 mg, 0,14 mmol) y 2-(tributilestannil)piridina (0,l4 ml, 0,43 mmol) en tolueno (3 ml) se añadió Pd(PPh3)4 (17 mg, 0,014 mmol). La mezcla de reacción se agitó a 100°C durante la noche. Luego, la mezcla se enfrió, se concentró a presión reducida y el residuo se purificó por TLC preparativa (eluyente: MeOH en DCM al 10%) para producir 2 mg de la 4-metil-2-(piridin-2-ilmetil)-6-(piridin-3-ilmetil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 389 (M+H)+. RMN 1H (400 MHz, DMSO-d6) 88,56 (m, 3H), 8,48 (dd, 1H), 7,81 (td, 1H), 7,71 (d, 1H), 7,50 (d, 1H), 7,37 - 7,30 (m, 2H), 5,38 (s, 2H), 4,67 (s, 2H), 4,25 (s, 3H).
Ejemplo 9D. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-6-((1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y 6-((lH-pirazol-3-il)metil)-2-((4H-1,2,4-triazol-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carbaldehído. A una suspensión de 1H-pirazol-3-carbaldehído (10,0 g, 104,07 mmol, 1 eq) y DIPEA (33,63 g, 260,18 mmol, 45,32 ml, 2,5 eq) en DCM (500 ml) se le añadió gota a gota 2-(clorometoxi)etil-trimetil-silano (26,03 g, 156,11 mmol, 27,63 ml, 1,5 eq) a -40°C. Luego, la mezcla de reacción se calentó a temperatura ambiente y se agitó durante 16 h. La TLC (éter de petróleo:EtOAc = 5:1) mostró que el material de partida se había consumido por completo y se habían formado dos nuevos puntos. La mezcla de reacción se concentró al vacío. El residuo se combinó con otros 2 lotes (10,0 g cada uno) y se purificó mediante Combiflash (de 100% de éter de petróleo a 40% de EtOAc en éter de petróleo) para dar 60,0 g del producto deseado. (nota: mezcla de 2 regioisómeros con una relación ~5/4). RMN 1H (400 MHz, cloroformo-d) 6 ppm 10,06 (s, 1H), 10,00 (s, 1H), 7,68 (d, 1H), 7,67 (d, 1H), 7,03 (d, 1H), 6,92 (d, 1H), 5,87 (s, 2H), 5,56 (s, 2H), 3,61-3,67 (m, 4H), 0,91-1,01 (m, 4H), -0,09-0,05 (m, 18H).
Etapa B. Síntesis de (1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metanol. A una solución de 1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-carbaldehído (30 g, 132,54 mmol, 1 eq, mezcla de 2 regioisómeros con relación ~5/4) en THF (200 ml)/MeOH (100 ml) se añadió NaBH4 (7,52 g, 198,81 milimoles, 1,50 eq) en porciones a 0°C, la mezcla de reacción se agitó de 0°C a temperatura ambiente durante 18 h. La TLC (éter de petróleo:EtOAc = 2:1) mostró que los materiales de partida se habían consumido por completo y se habían formado dos nuevos puntos. El disolvente se concentró al vacío. El residuo se purificó por Combiflash (100% de éter de petróleo a 100% de EtOAc) para dar 25 g del producto deseado. (Nota: mezcla de 2 regioisómeros en relación ~3/2). RMN 1H (400 MHz, cloroformo-d) 6 ppm 7,55 (d, 1H), 7,47 (s ancho, 1H), 6,36 (d, 1H), 6,34 (d, 1H), 5,57 (s, 2H), 5,42 (s, 2H), 4,74-4,76 (m, 4H), 3,55-3,60 (m, 4H), 0,85-0,96 (m, 4H), 0,00-0,06 (m, 18H).
Etapa C. Síntesis de 3-(bromometil)-1-((2-(trimetilsilil)etoari)metil)-1H-pirazol. A una solución de (1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metanol (23 g, 100,72 mmol, 1 eq, mezcla de 2 regioisómeros, relación ~3/2) y PPh3 (36,98 g, 141,00 mmol, 1,4 eq) en DCM (200 ml) se añadió CBr4 (46,76 g, 141,00 mmol, 1,4 eq) a 0°C y la mezcla de reacción se agitó a 0°C durante 3 h. La TLC (éter de petróleo:EtOAc = 5:1) mostró que los materiales de partida se habían consumido por completo y se había formado una nueva mancha. La mezcla de reacción se concentró al vacío. El residuo se combinó con otro lote (2,0 g) y se purificó por Combiflash (del 100% de éter de petróleo al 50% de EtOAc en éter de petróleo) para dar 22,0 g (75,53 mmol) del producto deseado. RMN 1H (400 MHz, cloroformo-d) 6 ppm 7,51 (d, 1H), 6,39 (d, 1H), 5,38 (s, 2H), 4,50 (s, 2H), 3,52-3,57 (m, 2H), 0,86-0,96 (m, 2H), - 0,03-0,02 (m, 9H).
Etapa D. Síntesis de 4-metil-6-((1-((2-(trimetilsilil)etoari)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una suspensión de 4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (1,0 g, 4,85 mmol, 1 eq), 3-(bromometil)-1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol (2,12 g, 7,27 mmol, 1,5 eq), K3PO4 (2,57 g, 12,12 mmol, 2,5 eq) y Nal (218,05 mg, 1,45 mmol, 0,3 eq) en DMF (15 ml) se agitó a 60°C durante 18 h en N2. La TlC (éter de petróleo:EtOAc =1:1) mostró que el material de partida se había consumido por completo y se había formado una nueva mancha. La mezcla de reacción se combinó con otros 3 lotes (1,0 g cada uno) y se vertió en agua helada (250 ml). La mezcla se extrajo con EtOAc (150 ml x 3). Las capas orgánicas combinadas se lavaron con agua (120 ml x 2), salmuera (120 ml) y se secaron sobre Na2SO4. El disolvente se concentró al vacío. El producto bruto se purificó mediante combiflash (de 100% de éter de petróleo a 80% de EtOA en éter de petróleo) para dar el producto deseado (3,6 g). RMN 1H (400 MHz, cloroformo-d) 6 ppm 8,91 (s, 1H), 8,27 (s, 1H), 7,49 (d, 1H), 6,36 (d, 1H), 5,51 (s, 2H), 5,40 (m, 2H), 4,45 (s, 3 H), 3,52-3,58 (m, 2H), 0,85-0,90 (m, 2H), -0,05 (s, 9H). Etapa E. Síntesis de 4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído. En atmósfera de argón, a una solución de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (1,7 g, 4,09 mmol, 1 eq) en Th F (30 ml) se añadió lentamente LíHm d S (1,0 M, 8,18 ml, 2 eq) a -78°C, la mezcla de reacción se agitó a -70°C durante 1 h. Luego se añadió gota a gota a la mezcla una solución de DMF (1,49 g, 20,45 mmol, 1,57 ml, 5 eq) en THF (3 ml). La mezcla resultante se agitó a -70°C durante 1 h. La TLC (éter de petróleo: EtOAc = 1:1) mostró que se había formado una nueva mancha. La mezcla de reacción se añadió gota a gota a solución ac. de NH4Cl (50 ml) a 0°C, luego la mezcla se extrajo con EtOAc (30 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (40 ml) y se secaron sobre Na2SO4. Se eliminó el disolvente al vacío para proporcionar el producto bruto deseado (1,8 g) que se usó para la siguiente etapa sin más purificación.
Etapa F. Síntesis de 2-(hidroximetil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazo1-3-il)metil)-4H
tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 4-metil-5-oxo-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (1,8 g, 3,24 mmol, 1 eq) en THF (20 ml) MeOH (10 ml) se añadió NaBH4 (245,08 mg, 6,48 mmol, 2 eq) a 0°Cy la mezcla de reacción se agitó a temperatura ambiente durante 18 h. La TLC (éter de petróleo:EtOAc = 1:2) mostró que el material de partida se había consumido por completo y se había formado una nueva mancha. La mezcla de reacción se concentró al vacío, el residuo se purificó por combiflash (de 100% de DCM a 5% de MeOH en DCM). Se obtuvo el producto deseado (1,1 g). RMN 1H (400 MHz, DMSO-CÍ6) 5 ppm 8,63 (s, 1H), 7,86 (d, 1H), 6,44 (t, 1H), 6,27 (d, 1H), 5,40-5,42 (m, 4H), 4,98 (d, 2H), 4,34 (s, 3H), 3,55 3,61 (m, 2H), 0,86-0,91 (m, 2H), 0,00 (s, 9H).
Etapa G. Síntesis de metanosulfonato de (4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metilo. A una solución de 2-(hidroximetil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (700 mg, 1,57 mmol, 1 eq) y EfeN (317,21 mg, 3,13 mmol, 436,33 ul, 2,0 eq) en Dc M (15 ml) se añadió gota a gota MsCl (269,32 mg, 2,35 mmol, 181,97 ul, 1,5 eq) a 0°C y la mezcla de reacción se agitó a temperatura ambiente durante 1 h. La TLC (éter de petróleo:EtOAc = 1:1) mostró que el material de partida se había consumido por completo y se había formado una nueva mancha. La mezcla de reacción se diluyó con EtOAc (80 ml) y se lavó con agua (30 ml x 4), salmuera (40 ml) y se secó sobre Na2SO4. Se eliminó el disolvente al vacío para proporcionar el producto bruto (700 mg). LCMS: (m/z 525,5 (M+H). Etapa H. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y 2-((4H-1,2,4-triazol-4-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. Una mezcla de metanosulfonato de (4-metil-5-oxo-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-il)metilo (150 mg, 285,88 umol, 1 eq) 1H-1,2,4-triazol (197,45 mg, 2,86 mmol, 10 eq) y CsF (86,85 mg, 571,77 umol, 21,08 ul, 2 eq) en MeCN (8 ml) se agitó a 60°C en N2 durante 18 h. La LCMS mostró que el material de partida se había consumido por completo y se habían formado dos nuevos picos. La mezcla de reacción se concentró al vacío, y el residuo se purificó por Combiflash (de 100% de DCM a 8% de MeOH en DCM). Se obtuvieron el producto 2-((1H-1,2,4-triazol-1-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (55 mg) y 2-((4H-1,2,4-triazol-4-il)metil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4,:4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg).
Etapa I. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-6-((1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una suspensión de 2-((1H-1,2,4-triazol-1-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (55 mg, 110,52 umol, 1 eq) y HCl/dioxano (4 M, 1 ml, 36,19 eq) en DCM (3 ml) se añadió H2O (1,99 mg, 110,52 umol, 0,05 ml, 1 eq) y la mezcla de reacción se agitó a temperatura ambiente durante 18 h. La LCMS mostró que el material de partida se había consumido por completo y se encontró 84% de producto deseado. La mezcla de reacción se concentró al vacío, y el residuo se purificó por HPLC preparativa para dar el producto deseado (24,1 mg, 65,60 umol, 59,35% de rendimiento). Columna: Xtimate c 18150*25 mm*5 um, fase móvil: [agua(0,225%FA)-ACN]; B%: 13%-43%, 11,2 min. LCMS: m/z 367,9 (M+H)+ RMN 1H (400 MHz, metanol-^) 5 ppm 8,72 (s, 1H), 8,35 (s, 1H), 8,05 (s, 1H), 7,52 (s ancho, 1H), 6,26 (d, 1H), 5,92 (s, 2H), 5,43 (s, 2H), 4,30 (s, 3H).
Etapa J. Síntesis de 6-((1H-pirazol-3-il)metil)-2-((4H-1,2,4-triazol-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una suspensión de 2-((4H-1,2,4-triazol-4-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 60,28 umol, 1 eq) y HCl/dioxano (4 M, 1 ml, 66,35 eq) en d Cm (3 ml) se añadió H2O (50,00 mg, 2,78 mmol, 0,05 ml, 46,04 eq) y la mezcla de reacción se agitó a temperatura ambiente durante 18 h. La LCMS mostró que el material de partida se había consumido por completo, se encontró el producto deseado. La mezcla de reacción se concentró al vacío, y el residuo se purificó por HPLC preparativa para dar el producto deseado (2,1 mg, 5,72 umol). Columna: Xtimate C18 150*25 mm*5 um; fase móvil: [agua (0,225% FA)-ACN]; B%: 13%-43%,11,2 min. LCMS: m/z 368,0 (M+H)+. RMN 1H (400 MHz, metanol-^) 5 ppm 8,74 (s, 2H), 8,38 (s, 1H), 7,52 (d, 1H), 6,26 (d, 1H), 5,84 (s, 2H), 5,44 (s, 2H), 4,31 (s, 3H).
Los siguientes compuestos se sintetizaron de acuerdo con el esquema E9 y el procedimiento de los ejemplos 9A-9B usando el material de partida apropiado.






Ejemplo 9E. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-aminopirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de_2-doro-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (3 g, 14,5 mmol) en THF seco (80 ml) se añadió gota a gota LiHMDS (30,5 ml,) a -65°C. Después de agitar durante 1 h, se añadió una solución de hexacloroetano (1,8 ml, 16 mmol) en THF seco (20 ml). La mezcla de reacción se elevó a -20°C a lo largo de 3 h. Entonces la mezcla se inactivó con solución sat. de NH4Cl y se agitó a t.a. durante 20 min. El precipitado se recogió por filtración y se lavó con EtOAc para dar 3,5 g de 2-cloro-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. lC-MS (ESI): m/z241 (M+H)+.
Etapa B. Síntesis de 4-metil-2-vinil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona En atmósfera de nitrógeno, a una mezcla de 2-cloro-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (1,5 g, 6,2 mmol) y tributil(etenil)estannano (5,5 ml, 18,7 mmol) en DMF (30 ml) se añadió Pd(Pph3)4 (0.36 g, 0,31 mmol). La mezcla de reacción se agitó a 100°C durante 2 h. Luego, la mezcla se enfrió y se diluyó con EtOAc, se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0 ~ 10%) para dar
1,4 g de 4-metil-2-vinil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 233 (M+H)+. Etapa C. Síntesis de_4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído_Por debajo de -60°C, una mezcla de 4-metil-2-vinil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (500 mg, 2,15 mmol) en DCM/MeCN (500 ml, volumen 1:1) se purgó con O3 durante 20 min. Luego, la reacción se inactivó con dimetilsulfano y se concentró para dar 500 mg de 4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído. LC-MS (ESI): m/z 235 (M+H)+.
Etapa D. Síntesis de_2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona_A una mezcla de 4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazina-2-carbaldehído (500 mg, 2,13 mmol) en EtOH (3 ml) se añadió NaBH4 (81 mg, 2,13 mmol) a 0°C. La reacción se agitó a temperatura ambiente durante 5 min. Entonces la mezcla se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0 ~ 8%) para dar 120 mg de 2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 237 (M+H)+.
Etapa E. Síntesis de_6-((2-cloropirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona_A una mezcla de 2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (200 mg, 0,85 mmol) en DMF (8 ml) se añadió K2CO3 (351 mg, 2,54 mmol). Después de agitar a 60°C durante 30 min, se añadió una solución de 2-cloro-4-(clorometil)pirimidina (276 mg, 1,7 mmol) en DMF (2 ml). La mezcla de reacción se agitó durante otras 4 h, se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, MeOH en DCM al 0 ~ 8%) para dar 160 mg de 6-((2-cloropirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 363 (M+H)+. Etapa F. Síntesis de_6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona_A una mezcla de 6-((2-cloropirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (160 mg, 0,44 mmol) en MeCN (5 ml), se añadió bis(2,4-dimetoxibencil)amina (280 mg, 0,88 mmol) y AcOH (1 gota). La mezcla de reacción se agitó a 80°C durante la noche. La mezcla de reacción se evaporó y el residuo se purificó por TLC preparativa (eluyente: MeOH en DCM al 5%) para dar 75 mg de 6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 644 (M+H)+.
Etapa G. Síntesis de 6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(clorometil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A 0°C, a una solución de 6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(hidroximetil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (75 mg, 0,12 mmol) en DCM (5 ml) se añadió Et3N (0,16 ml, 1,16 mmol) y MsCl (0,05 ml, 0,58 mmol). La mezcla se agitó at.a. durante la noche. La mezcla de reacción se diluyó con DCM, se lavó con solución sat. de NH4Cl y salmuera, se secó sobre Na2SO4 anhidro y se concentró para dar 70 mg de producto bruto de 6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(clorometil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 662 (M+H)+.
Etapa H. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona Una mezcla de 4H-1,2,4-triazol (39 mg, 0,57 mmol) y K2CO3 (78 mg, 0,57 mmol) en DMF (3 ml) se agitó a 60°C durante 30 min. Se añadió 6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-2-(clorometil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (75 mg, 0,11 mmol) y se agitó durante otros 30 min. La suspensión se vertió en solución sat. de NH4Cl, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por cromatografía ultrarrápida (gel de sílice, DCM en MeOH al 0 ~ 10%) para dar 60 mg de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5 (6H)-ona. LC-MS (ESI): m/z 695 (M+H)+.
Etapa I. Síntesis de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-aminopirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona A una mezcla de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-(bis(2,4-dimetoxibencil)amino)pirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (30 mg, 0,043 mmol) en EtOH (2 ml) se añadió HCl (0,5 ml, 4 M en dioxano). La mezcla de reacción se agitó a 80°C durante la noche. Después la mezcla se enfrió y se vertió en solución sat. de NaHCO3, se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó sobre Na2SO4 anhidro y se concentró. El residuo se purificó por HPLC preparativa para dar 8 mg de 2-((1H-1,2,4-triazol-1-il)metil)-6-((2-aminopirimidin-4-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. LC-MS (ESI): m/z 395 (M+H)+. r Mn 1H (400 MHz, DMSO-d6) 88,80 (s, 1H), 8,62 (s, 1H), 8,12-8,10 (m, 2H), 6,60 (s, 2H), 6,19 (d, 1H), 6,01 (s, 2H), 5,19 (s, 2H), 4,26 (s, 3H).
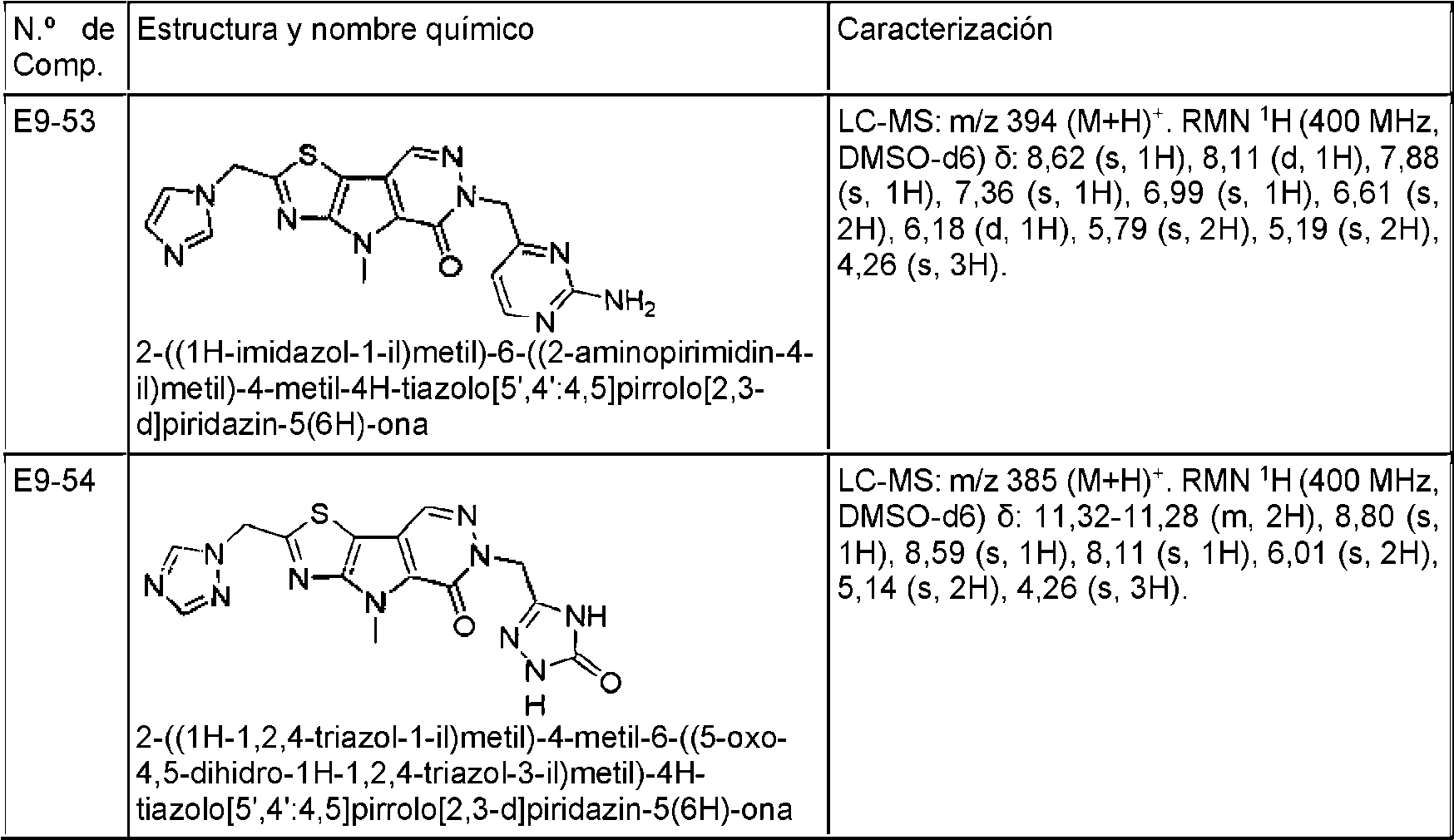
Ejemplo 10. Síntesis del compuesto E10-ii

Como se muestra en el esquema E10, la reacción de E10-i con un aldehido en presencia de una base (p. ej., LiHMDS) genera el compuesto E10-ii, que se puede separar con HPLC quiral o SFC para dar dos enantiómeros. Como se usa en el presente documento, Ar1 y Ar2 son cada uno independientemente alquilo opcionalmente sustituido, haloalquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido.
Ejemplo 10A. Síntesis de (S)-6-((1H-indazol-4-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona y (R)-6-((1H-indazol-4-il)met6il)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. Síntesis de 2-(hidroxi(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4-metil-6-((1 -((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. En atmósfera de argón, a una solución de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (0,5 g, 1,07 mmol, 1 eq) en THF (15 ml) se añadió lentamente LiHMDS (1,0 M, 2,14 ml, 2,0 eq) a -78° C, y la mezcla de reacción se agitó a -78 °C durante 1 h. Luego se añadió gota a gota a la mezcla de reacción una solución de 1-(2-trimetilsililetoximetil)pirazol-3-carbaldehído (1,21 g, 5,36 mmol, 5 eq) en THF (2 ml). La mezcla resultante se agitó a -78°C durante 1 h. La TLC (éter de petróleo:EtOAc = 2:1) mostró la formación de dos nuevas manchas. La mezcla de reacción se inactivó con solución ac. de NH4Cl (15 ml) a -70°C, y se diluyó con agua (20 ml). La mezcla se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml) y se secaron sobre Na2SO4. El disolvente se concentró al vacío. El residuo se purificó mediante Combiflash (de 100% de éter de petróleo a 100% de EtOAc) para proporcionar el producto deseado (70 mg, 101,01 umol). LCMS: m/z 693,2 (M+H)+
Etapa B. Síntesis de 6-((1H-indazol-4-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 2-(hidroxi(1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4-metil-6-((1 -((2-(trimetilsilil))etoxi)metil)-1H-indazol-4-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (0,04 g, 57,72 umol, 1 eq) en Dc M (3 ml) se añadió TFA (65,82 mg, 577,22 umol, 42,74 ul, 10 eq) a 0°C y la mezcla de reacción se agitó a temperatura ambiente durante 36 h. La LCMS mostró que el material de partida se había consumido por completo y se formó el 57% del producto deseado. La mezcla de reacción se concentró al vacío, y el residuo se purificó por HPLC preparativa para dar el producto deseado (11 mg). LCMS: m/z 432,9 [M+H]+. RMN 1H (400 MHz, DMSO-cfe) 5 ppm 13,10 (s ancho, 2H), 8,58 (s, 1H), 8,12 (s, 1H), 7,60 (s, 1H), 7,43 (d, 1H), 7,26 (dd, 1H), 6,93 (d, 2H), 6,19 (d, 1H), 6,07 (s, 1H), 5,63 (s, 2H), 4,20 (s, 3H).
Etapa C. Síntesis de (S)-6-((1H-indazol-4-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y (R)-6-((1H-indazol-4-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. El compuesto 6-((1H-indazol-4-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se separó por SFC. Condiciones de SFC: la columna es DAICEL CHIRALCEL OJ-H (250 mm * 30 mm, 5 um); fase móvil: A: 55% de CO2; B: 45% [NH3H2O en EtOH al 0,1%]/min. La separación por SFC proporcionó dos enantiómeros. Un enantiómero (2,6 mg): LCMS: m/z 433,0 [M+H]+. RMN 1H (400 MHz, DMSOCs) 5 ppm 8,63 (s, 1H), 8,16 (s, 1H), 7,64 (s ancho, 1H), 7,47 (d ancho, 1H), 7,27-7,32 (m, 1H), 6,97 (d ancho, 1H), 6,22 (d, 1H), 6,11 (s, 1H), 5,64 (s, 2H), 5,34 (s ancho, 1H), 4,24 (s, 3H). Y 3,5 mg de otro enantiómero: LCMS: m/z 433,0 (M+H)+. RMN 1H (400 MHz, DMSO-cfe) 5 ppm 8,63 (s, 1H), 8,17 (s, 1H), 7,64 (s ancho, 1H), 7,48 (d ancho, 1H), 7,28-7,32 (m, 1H), 6,98 (d ancho, 1H), 6,23 (d, 1H), 6,11 (s, 1H), 5,64 (s, 2H), 5,34 (s ancho, 1H), 4,25 (s, 3H).
Ejemplo 10B. Síntesis de (R)-6-((1H-pirazol-3-N)metil)-2-(hidroxi(1H-pirazol-3-N)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y (S)-6-((1H-pirazol-3-il)metil)-2-(hidroaci(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona

Etapa A. Síntesis de 2-(hidroxi(1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. En atmósfera de argón, a una solución de 4-metil-6-((1-((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazoío[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (0,5 g, 1,20 mmol, 1 eq) en THF (10 ml) se añadió lentamente LiHMDS (1,0 M, 2,41 ml, 2 eq) a -78°C, y la mezcla de reacción se agitó a -70°C durante 1 h. Luego se añadió a la mezcla de reacción una solución de 1-(2-trimetilsililetoximetil)pirazol-3-carbaldehído (816,97 mg, 3,61 mmol, 3 eq) en THF (1 ml). La mezcla resultante se agitó a -70°C durante 1 h. La TLC (éter de petróleo:EtOAc =1:1) mostró que se habían formado dos puntos nuevos. La mezcla de reacción se inactivó con solución ac. de NH4Cl (5 ml) a -70°C, y luego se calentó a temperatura ambiente. La mezcla se diluyó con agua (10 ml) y se extrajo con EtOAc (8 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml) y se secaron sobre Na2SO4. El disolvente se concentró al vacío. El residuo se purificó mediante Combiflash (de 100% de éter de petróleo a 100% de EtOAc) para dar el producto bruto (130 mg) como una goma de color marrón pálido, que se usó para la siguiente etapa sin más purificación. LCMS: m/z 643,2 [M+H]+
Etapa B. Síntesis de 6-((1H-pirazol-3-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. A una solución de 2-(hidroxi(1 -((2-(trimetilsilil)etoxi)metil)-1H-pirazol-3-il)metil)-4-metil-6-((1 -((2-(trimetilsilil))etoxi)metil)-1H-pirazol-3-il)metil)-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona (0,13 g, 202,20 umol, 1 eq) en DCM (8 ml) se añadió TFA (4,62 g, 40,52 mmol, 3 ml, 200,38 eq), seguido de H2O (500,00 mg, 27,75 mmol, 0,5 ml, 137,26 eq), luego la mezcla de reacción se agitó a temperatura ambiente durante 18 h, luego se calentó a -40°C durante 18 h. La LCMS mostró que el material de partida se había consumido por completo. La mezcla de reacción se concentró al vacío, y el residuo se purificó por HPLC preparativa para dar el producto deseado (20,5 mg). LCMS: m/z 382,9 [M+H]+. RMN 1H (400 MHz, DMSO-cfe) 5 ppm 12,72 (s ancho, 1H), 12,61 (s ancho, 1H), 8,52 (s, 1H), 7,56-7,62 (m, 2H), 6,82 (s ancho, 1H), 6,18 (d, 1H), 5,99-6,09 (m, 2H), 5,24-5,32 (m, 2H), 4,19 (s, 3H).
Etapa C. Síntesis de (R)-6-((1H-pirazol-3-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona y (S)-6-((1H-pirazol-3-il)metil)-2-(hidroxi(1H-pirazol-3-il)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona. El compuesto 6-((1H-pirazol-3-N)metil)-2-(hidroxi(1H-pirazol-3-N)metil)-4-metil-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5(6H)-ona se separó por SFC. Condiciones de SFC: la columna es DAICEL CHIRALCEL OJ-H (250 mm * 30 mm, 5 um); fase móvil: A: 55% de CO2; B: 45% [NH3H2O en EtOH al 0,1%]/min. La separación por SFC proporcionó la 2-[(R)-hidroxi(1H-pirazol-3-il)metil]-4-metil-6-(1H-pirazol-3-ilmetil)tiazolo[3,4]pirrolo[1,3-d]piridazin-5-ona y 2-[(S)-hidroxi(1H-pirazol-3-il)metil]-4-metil-6-(1H-pirazol-3-ilmetil)tiazolo[3,4]pirrolo[1,3-d]piridazin-5-ona. Un isómero (4,4 mg): LCMS: m/z 383 [M+H]+. RMN 1H (400 MHz, metanol-c/4) 5 ppm 8,39 (s, 1H), 7,61 (s ancho, 1H), 7,55 (s ancho, 1H), 6,34 (s ancho, 1H), 6,25 (s ancho, 1H), 6,18 (s ancho, 1H), 5,44 (s, 2H), 4,29 (s, 3H). Otro isómero (4,1 mg): LCMS: m/z 383 (M+H)+. RMN 1H (400 MHz, metanol-c/4) 5 ppm 8,40 (s, 1H), 7,59 (s ancho, 1H), 7,54 (s ancho, 1H), 6,34 (s ancho, 1H), 6,26 (s ancho, 1 H), 6,19 (s ancho, 1 H), 5,44 (s, 2H), 4,29 (s, 3H).
Ejemplo 10C: Síntesis de 6-((1H-pirazol-3-il)metil)-2-(difluoro(1H-pirazol-3-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 3-(6-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-carbonil)-N,N-dimetil-1H-pirazol-1-sulfonamida. A una mezcla de 3-((2-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)(hidroxi)metil)-4-metil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)-N,N-dimetil-1H-pirazol-1-sulfonamida (50 mg, 83,80 umol, hecho de manera similar a E10-1) en DCM (1,5 ml) se añadió MnO2 (72,85 mg, 838,00 umol) y la mezcla se agitó a 15°C durante 1,5 horas. La mezcla de reacción se filtró y se concentró a presión reducida para dar un producto bruto (60 mg, bruto). LCMS: m/z 595,1 (M+H)+. RMN 1H (400 MHz, CDCl3) 58,34 (s, 1H), 8,12 (d, 1H), 7,90 (d, 1H), 7,41 (d, 1H), 6,38 (d, 1H), 5,54 (s, 2H), 4,49 (s, 3H), 3,10 (s, 6H) 2,93 (s, 6H).
Etapa B. 3-((2-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-N)difluorometil)-4-metil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)-N,N-dimetil-1H-pirazol-1-sulfonamida. A una solución de 3-(6-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-carbonil)-N,N-dimetil-1H-pirazol-1-sulfonamida (240 mg, 403,60 umol) en DCE (4 ml) se añadió Ba ST (1,34 g, 6,05 mmol, 1,33 ml) y la mezcla se agitó a 50°C durante 12 h. La mezcla de reacción se diluyó con diclorometano (20 ml) y se lavó con solución saturada de NaHCO3 (10 ml*2), secado sobre Na2SO4 anhidro, se filtró y se concentró a presión reducida para dar un producto bruto (300 mg, bruto) que se usó en la etapa siguiente sin purificación adicional. LCMS: m/z 617,1 (M+H)+.
Etapa C 6-((1H-pirazol-3-N)metil)-2-(difluoro(1H-pirazol-3-N)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 3-((2-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)difluorometil)-4-metil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)-N,N-dimetil-1H-pirazol-1-sulfonamida (240 mg, 163,47 umol) en DCM (2 ml) se añadió TFA (2,07 g, 18,15 mmol, 1,34 ml) y la mezcla se calentó hasta 50°C durante 5 h. La mezcla de reacción se concentró a presión reducida. El residuo se purificó por HPLC preparativa (columna: Agela ASB 150 x 25 mm x 5 um; fase móvil: [agua (HCl al 0,05%)-ACN]; B%: 30%-60%, 8 min) para dar el producto deseado (3,9 mg, 5,45% de rendimiento, 92% de pureza) como un sólido blanco. LCMS: m/z 403,1 (M+H)+. Rm N 1H (400 MHz, DMs O-cí6) 58,67 (s, 1H), 7,95 (s, 1H), 7,61 (s, 1H), 6,72 (s, 1H), 6,16 (s, 1H), 5,36 (s, 2H), 4,28 (s, 3 H ).
Ejemplo 10D: Síntesis de 2-(1-(1H-pirazol-3-il)etil)-6-((1H-pirazol-3-il)metil)-4-metil-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A: 3-((2-(1-(1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)-1-hidroxietil)-4-metil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)-N,N-dimetil-1H-pirazol-1-sulfonamida. A una solución de 3-(6-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-carbonil)-N,N-dimetil-1H-pirazol-1-sulfonamida (160 mg, 269,07 umol) en THF (3 ml) se añadió CH3MgBr (3 M, 179,38 ul) y la mezcla de reacción se agitó a 0°C durante 3 h. La mezcla de reacción se vertió en solución saturada de NH4Cl (10 ml) a 0°C, se extrajo con acetato de etilo (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (10 ml), se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía ultrarrápida en gel de sílice (ISCO®; Columna ultrarrápida de sílice Sepa Flash® 12 g, eluyente de gradiente de acetato de etilo/ éter de petróleo al 0~90% a 40 ml/min) para dar el producto deseado (50 mg, 81,87 umol). LCMS: m/z 611,1 (M+H)+.
Etapa B: 2-(1-(1H-pirazol-3-il)etil)-6-((1H-pirazol-3-il)metil)-4-metil-4,6-dihidro-SH-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona. A una mezcla de 3-((2-(1-(1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)-1-hidroxietil)-4-metil-5-oxo-4,5-dihidro-6H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-6-il)metil)-N,N-dimetil-1H-pirazol-1-sulfonamida (50 mg, 81,87 umol) en DCE (0,5 ml) se añadió Et3SiH (19,04 mg, 163,75 umol, 26,15 ul) a 0°C, seguido de TFA (1,54 g, 13,51 mmol, 1 ml, 164,96 eq) y se agitó a 0°C durante 1 h. La mezcla se calentó a 50°C durante 1 h más. La mezcla de reacción se concentró a presión reducida. El residuo se purificó por HPLC preparativa (columna: Agela ASB 150*25 mm*5 um; fase móvil: [agua (HCl al 0,05%)-ACN]; B%: 25%-55%, 7 min) para dar 7,0 mg de producto deseado. LCMS: m/z 381,2 (M+H)+. RMN 1H (400 MHz,
CD3OD) 58,46 (s, 1H), 8,15 (d,2H), 6,78 (d, 1H), 6,74 (d, 1H), 5,60 (s, 2H), 5,00 (q, 1H), 4,33 (s, 3H), 1,94 (d, 3H).
Ejemplo 10E: Síntesis de 6-((1H-pirazol-3-il)metil)-4-metil-2-(1H-pirazol-3-carbonil)-4,6-dihidro-5H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-5-ona

Etapa A. 4-metil-2-(1H-pirazol-3-carbonil)-6-(1H-pirazol-3-ilmetil)tiazolo[3,4]pirrolo[1,3-d]piridazin-5-ona. A una solución de 3-(6-((1-(N,N-dimetilsulfamoil)-1H-pirazol-3-il)metil)-4-metil-5-oxo-5,6-dihidro-4H-tiazolo[5',4':4,5]pirrolo[2,3-d]piridazin-2-carbonil)-N,N-dimetil-1H-pirazol-1-sulfonamida (50 mg, 84,08 umol) en DCE (1,5 ml) se añadió TFA (2,31 g, 20,26 mmol, 1,5 ml) y la mezcla de reacción se calentó hasta 50°C durante 12 h. La mezcla de reacción se concentró al vacío. El residuo se purificó por HPLC preparativa para dar 6,0 mg del producto deseado. LCMS: m/z 381,1 (M+H). RMN 1H (400 MHz, DMSO-d6) 58,72 (s, 1H), 7,96 (d, 1H), 7,64 (d, 1H), 7,49 (d, 1H),6,19(d, 1H), 5,37 (s, 2H), 4,38 (s, 3H).
Ejemplo 11. Ensayo de PKM2
Procedimiento:
Se diluyó solución madre de enzima PKM2 para preparar un 1,11x Mezcla de reacción (sin ADP). Primero se añadió 1 μl del compuesto de ensayo a los pocillos seguido de 40 μl de 1,11x Mezcla de reacción (sin ADP) y se incubó a temperatura ambiente (25°C) durante 60 min. La reacción se inició con 10 μl de ADP (concentración final 0,4 mM), llevándose la Mezcla de reacción final a 1x, y el progreso de la reacción se midió como cambios en la absorbancia a una longitud de onda de 340 nm a temperatura ambiente.
Preparación del compuesto de ensayo: los compuestos de ensayo se prepararon a una concentración final de 50x en DMSO. Se hicieron de 1 a 3 diluciones para 11 puntos (por ejemplo, se añadieron 50 μl de compuesto 5000 μMa 100 μl de DMSO al 100% para dar 1667 μM, 50 μl de este se añadieron a 100 μl de d Ms O para producir 556 μM, y así sucesivamente). Los compuestos se añadieron al ensayo como una dilución de 1 a 50 (1 μl en 50 μl) para producir una concentración máxima de 100 μM, disminuyendo 3 veces para 11 puntos.
Mezcla de reacción: PKM2 (5 ng/pocillo, 0,1 |jg/ml), ADP (0,4 mM), PEP (0,11 mM), NADH (180 |jM), LDH (0,005 U/μl, Sigma n.° L3888), DTT 1 mM, Bs A al 0,03% en 1x Tampón de reacción
Tampón de reacción: KCI 100 mM, Tris 50 mM a pH 7,5, MgCh 5 mM.
Claims (16)
1. Un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables para usaren un método de tratamiento de una enfermedad proliferativa, obesidad, enfermedad diabética, aterosclerosis, reestenosis, enfermedad arterial coronaria, síndrome de Bloom o una enfermedad autoinmunitaria; en donde el método comprende administrar una cantidad eficaz del compuesto de fórmula (I) o su sal farmacéuticamente aceptable a un paciente, en donde:

Q es hidrógeno, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido;
R1 es hidrógeno, alquilo opcionalmente sustituido, haloalquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido, -ORo1, - C(=O)Rc1, o un grupo protector de nitrógeno;
L1 es un enlace, alquileno opcionalmente sustituido, -O-, -S-, -S-CH2-, -S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(=O)-, -C(=O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, -C(R4)2O--NR3C(R4)2-, -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2-, -S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O-, -OS(=O)2NR3-, -NR3S(=O)O-, -OS(=O)NR3- o -S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, alquileno opcionalmente sustituido, -C(=O)-, -S(=O)2- o -S(=O)-, en donde el punto de unión a Q está en el lado derecho;
R2 es hidrógeno, halógeno, alquilo opcionalmente sustituido, alcoxi opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido, o un grupo protector de nitrógeno cuando L1 es -NR3-, - NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O-, o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es -O-, -OC(=O)-, -OC(=o )n R3-, -OC(R4)2-, -OS(=O)2-, -OS(=O)2NR3-, -OS(=O)NR3- o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
cada caso de R3 es independientemente hidrógeno, -ORo2, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro1 y Ro2 es independientemente hidrógeno, alquilo opcionalmente sustituido o un grupo protector de oxígeno;
cada caso de Rc1 es independientemente alquilo opcionalmente sustituido o -N(Rcn)2, en donde cada caso de Rcn es independientemente hidrógeno, -alquilo C1-6, o un grupo protector de nitrógeno; y
cada caso de R4 es independientemente hidrógeno, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, arilo opcionalmente sustituido o heteroarilo opcionalmente sustituido.
2. El compuesto o sal farmacéuticamente aceptable del mismo para usar según la reivindicación 1, en donde la enfermedad es cáncer o hiperplasia prostática benigna.
3. Un compuesto de fórmula (I) o una de sus sales farmacéuticamente aceptables, en donde la fórmula (I) es como se define en la reivindicación 1 , para usar en un método para tratar la hiperglucemia; en donde el método comprende administrar una cantidad eficaz del compuesto de fórmula (I) o la sal farmacéuticamente aceptable del mismo a un paciente.
4. El compuesto o una de sus sales farmacéuticamente aceptables para usar según la reivindicación 1, en donde la enfermedad es una enfermedad diabética, tal como la nefropatía diabética.
5. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde:
(a) Q es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo de 6 a 14 miembros opcionalmente sustituido o heteroarilo de 5 a 14 miembros opcionalmente sustituido;
R1 es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -haloalquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, -alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo de 6 a 12 miembros opcionalmente sustituido, -ORo1, -C(=O)Rc1, o un grupo protector de nitrógeno;
L1 es un enlace, alquileno C1-6 opcionalmente sustituido, -O-, -S-, -S-CH2-, - S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(=O)-, -C(=O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, -C(R4)2O-, -NR3C(R4)2-, -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2-, - S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O-, -OS(=O)2NR3-, -NR3S(=O)O-, -OS(=O)NR3- o -S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, alquileno C1-C6 opcionalmente sustituido, -C(=O)-, -S(=O)2- o - S(=O)-, en donde el punto de unión a Q está en el lado derecho;
R2 es hidrógeno, halógeno, -alquilo C1-C6 opcionalmente sustituido, -alcoxi C1-C6 opcionalmente sustituido, -cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, -arilo C6-C12 opcionalmente sustituido o heteroarilo de 3 a 14 miembros opcionalmente sustituido, o un grupo protector de nitrógeno cuando L1 es -NR3-, -NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O- o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es -O-, -OC(=O)-, -Oc (=O)n R3-, -OC(R4)2-, -OS(=O)-, -OS(=O)2-, -OS(=O)2Nr 3-, -OS(=O)NR3- o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
cada caso de R3 es independientemente hidrógeno, -ORo2, -alquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, -alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo C3-C12 opcionalmente sustituido, arilo C6-C12 opcionalmente sustituido, heteroarilo C5-C12 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro1 y Ro2 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido o un grupo protector de oxígeno;
cada caso de Rc1 es independientemente -alquilo C1-C6 opcionalmente sustituido o -N(Rcn)2, en donde cada caso de Rcn es independientemente hidrógeno, -alquilo C1-C6, o un grupo protector de nitrógeno;
cada caso de R4 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -alquenilo C2-C6 opcionalmente sustituido, alquinilo C2-C6 opcionalmente sustituido, cicloalquilo C3-C12 opcionalmente sustituido, heterociclilo de 3 a 14 miembros opcionalmente sustituido, arilo C6-C12 opcionalmente sustituido, o heteroarilo de 5 a 14 miembros opcionalmente sustituido;
o
(b) Q es arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros o heteroarilo bicíclico de 8 a 12 miembros, cada uno de los cuales está sustituido con 0-3 casos de RC;
R1 se selecciona de hidrógeno, -alquilo C1-C6, -haloalquilo C1-C6, cicloalquilo monocíclico C3-C7 y heterociclilo de 3 a 14 miembros, -ORo1, -C(=O)Rc1, o un grupo protector de nitrógeno; en donde cada alquilo, cicloalquilo o heterociclilo está sustituido con 0-3 casos de Rd;
R2 se selecciona de hidrógeno, halógeno, -alquilo C1-C6, -alcoxi C1-C6, cicloalquilo monocíclico C3-C7, cicloalquilo bicíclico C6-C12, heterociclilo de 3 a 14 miembros, arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros, heteroarilo bicíclico de 8 a 12 miembros, en donde cada alquilo, cicloalquilo, heterociclilo, arilo y heteroarilo está sustituido con 0-3 casos de Re, o un grupo protector de nitrógeno cuando L1 es -NR3-, -NR3C(=O)-, -NR3C(=O)O-, -NR3C(R4)2-, -NR3S(=O)2-, -NR3S(=O)-, -NR3C(=O)NR3-, -NR3S(=O)2O- o -NR3S(=O)O-, un grupo protector de oxígeno cuando L1 es -O-, -OC(=O)-, -OC(=O)NR3-, -OC(R4)2-, -OS(=O)-, -OS(=O)2-, - OS(=O)2NR3-, -OS(=O)NR3- o -OS(=O)-, o un grupo protector de azufre cuando L1 es -S-;
R3 se selecciona de hidrógeno, -ORo2, -alquilo C1-C6, cicloalquilo monocíclico C3-C7, cicloalquilo bicíclico C6-C12, heterociclilo de 3 a 14 miembros, arilo C6-C12, heteroarilo monocíclico de 5 a 6 miembros y heteroarilo bicíclico de 8 a 12 miembros, en donde cada alquilo, cicloalquilo, heterocicloalquilo, arilo y heteroarilo está sustituido con 0-3 casos de Rf;
R4 se selecciona de hidrógeno, -alquilo C1-C6, cicloalquilo monocíclico C3-C7 y heterociclilo de 3 a 14 miembros, en donde cada alquilo, cicloalquilo o heterociclilo está sustituido con 0-1 casos de R9;
L1 es un enlace, un alquileno sustituido con 0-3 casos de Rh, -O-, -S-, -S-CH2-, -S(=O)CH2-, -S(=O)2CH2-, -NR3-, -NR3C(=O)-, -C(=O)NR3-, -C(=O)-, -OC(=O)-, -C(=O)O-, -NR3C(=O)O-, -OC(=O)NR3-, -NR3C(=O)NR3-, -OC(R4)2-, -C(R4)2O-, -NR3C(R4)2-, -C(R4)2NR3-, -S(=O)2-, -S(=O)-, -S(=O)2O-, -OS(=O)2-, - S(=O)O-, -OS(=O)-, -S(=O)2NR3-, -NR3S(=O)2-, -S(=O)NR3-, -NR3S(=O)-, -NR3S(=O)2O-, -OS(=O)2NR3-, -NR3S(=O)O-, -OS(=O)NR3- o-S(=O)(=NR3)-, en donde el punto de unión a R2 está en el lado izquierdo;
L2 es un enlace, un alquileno sustituido con 0-3 casos de Rh, -C(=O)-, -S(=O)2- o -S(=O)-, en donde el punto de unión a Q está en el lado derecho;
cada RC se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -hidroxialquilo C1-C6, -OH, -O-alquilo C1-C6, -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2, -C(=O)O(alquilo C1-C6), -C(=O)OH, -C(=O)-(alquilo C1-C6), -C(=O)NH2, -C(=O)NH(alquilo C1-C6), -C(=O)N(alquilo C1-C6)2, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-C6), -NH(C=O)N(alquilo C ^ h , -NHC(=O)(alquilo C1-C6), -N(alquilo C1-C6)C(=O)(alquilo C1-C6), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-C6)2, -NHS(=O)2(alquilo C1-C6), -NH2, -CN y -NO2; o dos casos de RC unidos al mismo átomo de carbono o a átomos de carbono adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclil-C(=O)OH;
cada Rd se selecciona independientemente de halógeno, -alquilo C1-C6, -OH, -O-alquilo C1-C6, -NH2 y-CN;
cada Re se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -hidroxialquilo C1-C6, -OH, -O-alquilo C1-C6, -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2, -C(=O)O(alquilo C1-C6), -C(=O)OH, -C(=O)(alquilo C1-C6), -C(=O)NH2, -C(=O)NH(alquilo C1-C6), -C(=O)N(alquilo C1-C6)2, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-C6), - NH(C=O)N(alquilo C1-C6)2, -NHC(=O)(alquilo C1-C6), -N(alquilo C1-C6)C(=O)(alquilo C1-C6), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-C6)2, -NHS(=O)2(alquilo C1-C6), -NH2, -CN y-NO2; o dos casos de Re unidos al mismo átomo de carbono o a átomos de carbono adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclilo;
cada Rf se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -alcoxi C1-C6, -OH, -NH2, -CN y -NO2;
cada R9 se selecciona independientemente de halógeno, -alquilo C1-C6, -C1-C6 haloalquilo,-alcoxi C1-C6, -OH, NH2, -CN y NO2 y;
cada Rh se selecciona independientemente de halógeno, -alquilo C1-C6, -haloalquilo C1-C6, -hidroxialquilo C1-C6, -OH, -O-alquilo C1-C6, -aminoalquilo C1-C6, -NH(alquilo C1-C6), -N(alquilo C1-C6)2, -C(=O)O(alquilo C1-C6), -C(=O)OH, -C(=O)(alquilo C1-C6), -C(=O)NH2, -C(=O)NH(alquilo C1-C6), -C(=O)N(alquilo C1-C6)2, -NHC(=O)NH2, -NHC(=O)NH(alquilo C1-C6), -NH(C=O)N(alquilo C1-C6K -NHC(=O)(alquilo C1-C6), -N(alquil C1-C6)C(=O)(alquilo C1-C6), -S(=O)2NH2, -S(=O)2NH(alquilo C1-C6), -S(=O)2N(alquilo C1-C6)2, -NHS(=O)2(alquilo C1-C6), -NH2, -CN y -NO2, S(=O)2arilo, S(=O)2heteroarilo y =NOH o dos casos de Rh unidos al mismo átomo de carbono o a átomos de carbono adyacentes, se consideran junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o un heterociclilo.
6. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde el compuesto es de fórmula (II):

o una de sus sales farmacéuticamente aceptables, en donde:
Ra y Rb son cada uno independientemente hidrógeno, halógeno, -CN, -NO2, -N3, alquilo opcionalmente sustituido, -ORo3, -N(Rn1)2, -C(=O)N(Rn1)2 o -C(=O)Rc2, o Ra y Rb se pueden considerar junto con el átomo de carbono para formar cicloalquilo opcionalmente sustituido o heterociclilo opcionalmente sustituido;
cada caso de Rn1 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro3 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno; y
cada caso de Rc2 es independientemente -alquilo C1-C6 opcionalmente sustituido.
7. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde L1 es alquileno C i-6 sustituido con Rj y Rk;
en donde cada caso de Rj y Rk se selecciona independientemente de H, halógeno, -CN, -ORo7, -N(Rn5)2, -N(Rn5)C(=O)Rc5, -C(=O)N(Rn1)2, -C(=O)Rc5, -C(=O)ORo7, -SRjs, -S(=O)2Rjs o -S(=O)Rjs, -alquilo C1-C6 opcionalmente sustituido; o Rj y Rk se pueden considerar junto con el átomo de carbono para formar C=O, C=NRjn, un anillo de cicloalquilo monocíclico C3-C6 opcionalmente sustituido o un anillo de heterociclilo monocíclico C3-C6 opcionalmente sustituido;
cada uno de Rn5 y Rjn es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, -ORo8, o un grupo protector de nitrógeno;
cada caso de Ro7 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Rc5 es independientemente -alquilo C1-C6 opcionalmente sustituido; y
cada caso de Rjs es independientemente -alquilo C1-C6 opcionalmente sustituido, arilo C6-12 opcionalmente sustituido, heteroarilo opcionalmente sustituido o un grupo protector de azufre.
8. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde Q es de una de las siguientes fórmulas:

en donde:
cada caso de Rn es independientemente hidrógeno, un halógeno, -CN, -NO2, -N3, un alquilo opcionalmente sustituido, un alquenilo opcionalmente sustituido, un alquinilo opcionalmente sustituido, un cicloalquilo opcionalmente sustituido, un arilo opcionalmente sustituido, un heterociclilo opcionalmente sustituido, un heteroarilo opcionalmente sustituido, -ORo4, -SRs1, -N(Rn2)2, -C(=O)N(Rn2)2, -N(Rn2)C(=O)Rc3, -C(=O)Rc3, -C(=O)ORo4, -OC(=O)Rc3, -S(=O)Rs1, -S(=O)2Rs1, -S(=O)ORo4, -OS(=O)Rc3, -S(=O)2ORo4, -OS(=O)2Rc3, -S(=O)N(Rn2)2, -S(=O)2N(Rn2)2, -N(Rn2)S(=O)Rsl, -N(Rn2)S(=O)2Rs1, -N(Rn2)C(=O)ORo4, -OC(=O)N(Rn2)2, -N(Rn2)C(=O)N(Rn2)2, -N(Rn2)S(=O)N(Rn2)2, -N(Rn2)S(=O)2N(Rn2)2, -N(Rn2)S(=O)ORo4, -N(Rn2)S(=O)2OR04, -OS(=O)N(Rn2)2 o -OS(=O)2N(Rn2)2; o dos casos de Rn unidos al mismo átomo de carbono o átomos de carbono adyacentes, considerados junto con los átomos de carbono a los que están unidos para formar un cicloalquilo o heterocicloalquilo opcionalmente sustituido; en donde:
cada caso de Rn2 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro4 es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Ro3 es independientemente un -alquilo Ci-C6 opcionalmente sustituido;
cada caso de Rs1 es independientemente un -alquilo C1-C6 opcionalmente sustituido o un grupo protector de azufre; n es 0, 1,2 o 3, según lo permita la valencia; y
cada uno de Rna, Rnby Rnd es independientemente hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno.
9. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones 1 7, en donde Q es de una de las siguientes fórmulas:


en donde cada caso de Rn se selecciona independientemente de hidrógeno, halógeno, -CN, -NO2, -N3, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, arilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, -ORo4, -SRs1, - N(Rn2)2, -C(=O)N(Rn2)2, -N(Rn2)C(=O)Rc3, -C(=O)Rc3, -C(=O)ORo4, -OC(=O)Rc3, -S(=O)Rs1, -S(=O)2Rs1, -S(=O)ORo4, -OS(=O)Rc3, -S(=O)2ORo4, -OS(=O)2Rc3,-S(=O)N(Rn2)2, -S(=O)2N(Rn2)2,-N(Rn2)S(=O)Rs1, -N(Rn2)S(=O)2Rs1, -N(Rn2)C(=O)ORo4, -OC(=O)N(Rn2)2, -N(Rn2)C(=O)N(Rn2)2, -N(Rn2)S(=O)N(Rn2)2, -N(Rn2)S(=O)2N(Rn2)2, -N(Rn2)S(=O)ORo4, -N(Rn2)S(=O)2ORo4, -OS(=O)N(Rn2)2, -OS(=O)2N(Rn2)2, o dos casos de Rn unidos al mismo átomo de carbono o a átomos de carbono adyacentes, considerados junto con los átomos a los que están unidos forman un cicloalquilo o un heterocicloalquilo opcionalmente sustituido;
cada caso de Rna y Rnb es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Rn2 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro4 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Ro3 es independientemente -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rs1 es independientemente -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de azufre; y n es 0, 1, 2 o 3, según lo permita la valencia.
10. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde R2 se selecciona de hidrógeno, hidroxilo, halógeno, -alquilo C1-C6, -alquenilo C2-C6, -alcoxilo C2-C6, fenilo, naftalenilo, cicloalquilo C3-6, heteroarilo de 5 miembros, heteroarilo de 6 miembros, heteroarilo bicíclico de 8 miembros, heteroarilo bicíclico de 9 miembros, en donde cada alquilo, alquenilo, fenilo y heteroarilo está sustituido con 0 3 casos de Re
11. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde R2 es de una de las siguientes fórmulas:



en donde cada caso de Rp se selecciona independientemente de hidrógeno, halógeno, -CN, -NO2, -N3, alquilo opcionalmente sustituido, alquenilo opcionalmente sustituido, alquinilo opcionalmente sustituido, cicloalquilo opcionalmente sustituido, arilo opcionalmente sustituido, heterociclilo opcionalmente sustituido, heteroarilo opcionalmente sustituido, -ORo6, -SRs2, -N(Rn3)2, -C(=O)N(Rn3)2, -N(Rn3)C(=O)Rc4, -C(=O)Rc4, -C(=O)ORo6, -OC(=O)Rc4, -S(=O)Rs2, -S(=O)2Rs2, -S(=O)ORo6, -OS(=O)Rc4, -S(=O)2ORo6, -OS(=O)2Rc4, -S(=O)N(Rn3)2, -S(=O)2N(Rn3)2, -N(Rn3)S(=O)Rs2, -N(Rn3)S(=O)2Rs2, -N(Rn3)C(=O)ORo6, -OC(=O)N(Rn3)2, -N(Rn3)C(=O)N(Rn3)2, -N(Rn3)S(=O)N(Rn3)2, -N(Rn3)S(=O)2N(Rn3)2, -N(Rn3)S(=O)ORo6, -N(Rn3)S(=O)2ORo6, -OS(=O)N(Rn3)2, -OS(=O)2N(Rn3)2, o dos casos de Rp unidos al mismo átomo de carbono o a átomos de carbono adyacentes, considerados junto con los átomos a los que están unidos forman un cicloalquilo o un heterocicloalquilo opcionalmente sustituido;
cada caso de Rn3, Rnc y Rnd es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
cada caso de Ro6 es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de oxígeno;
cada caso de Ro4 es independientemente -alquilo C1-C6 opcionalmente sustituido;
cada caso de Rs2 es independientemente -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de azufre; y p es 0, 1,2 o 3, según lo permita la valencia; opcionalmente
en donde cada caso de Rp es independientemente hidrógeno, halógeno, alquilo C1-4 opcionalmente sustituido, -CN-NO2-N3, -ORo4, -N(Rn2)2, -C(=O)N(Rn2)2, -C(=O)Rc3 o -C(=O)ORo4
12. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones 1-7 y 10 u 11, en donde el compuesto es de fórmula (III):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; y Rn y n son como se definen en la reivindicación 8 o reivindicación 9.
13. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones 1-8 y 10 u 11, en donde
(a) el compuesto es de fórmula (IV):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; y Rn y n son como se definen en la reivindicación 8 o reivindicación 9;
o
(b) el compuesto es de fórmula (V-a):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rn es como se define en la reivindicación 8 o reivindicación 9; y m es 0, 1 o 2;
o
(c) el compuesto es de fórmula (V-b):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rn es como se define en la reivindicación 8 o reivindicación 9; m es 0, 1 o 2; y Rnc es independientemente hidrógeno, -alquilo Ci-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
o
(d) el compuesto es de fórmula (IX):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rn y n son como se definen en la reivindicación 8 o reivindicación 9; y Rnc es independientemente hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno.
14. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones 1-7 y 9-11, en donde el compuesto es de fórmula (VI):

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rn y n son como se definen en la reivindicación 8 o reivindicación 9; y Rn7 es hidrógeno, -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno.
15. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde
(a) el compuesto se representa por la siguiente fórmula estructural:

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rj y Rk son como se definen en la reivindicación 7; Rp es como se define en la reivindicación 11; y q es 0, 1,2 o 3;
o
(b) el compuesto se representa por la siguiente fórmula estructural:

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rj y Rk son como se definen en la reivindicación 7; Rp es como se define en la reivindicación 11; y q es 0, 1,2 o 3;
o
(c) el compuesto se representa por la siguiente fórmula estructural:

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rj y Rk son como se definen en la reivindicación 7; Rp es como se define en la reivindicación 11; q es 0, 1,2 o 3; y Rn6 es
hidrógeno, un -alquilo C1-C6 opcionalmente sustituido, o un grupo protector de nitrógeno;
o
(d) el compuesto se representa por la siguiente fórmula estructural:

o una de sus sales farmacéuticamente aceptables, en donde Ra y Rb son como se definen en la reivindicación 6; Rj y Rk son como se definen en la reivindicación 7; Rp es como se define en la reivindicación 11; y q es 0, 1, 2 o 3.
16. El compuesto o sal farmacéuticamente aceptable del mismo para usar según una cualquiera de las reivindicaciones anteriores, en donde el compuesto se selecciona de los siguientes compuestos:








Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2017097496 | 2017-08-15 | ||
| US201862673533P | 2018-05-18 | 2018-05-18 | |
| US201862673526P | 2018-05-18 | 2018-05-18 | |
| PCT/US2018/000129 WO2019035865A1 (en) | 2017-08-15 | 2018-08-15 | MODULATORS OF PYRUVATE KINASE AND THEIR USE |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2944545T3 true ES2944545T3 (es) | 2023-06-22 |
Family
ID=63524352
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18779068T Active ES2905100T3 (es) | 2017-08-15 | 2018-08-15 | Activadores de piruvato quinasa para su uso en el tratamiento de trastornos sanguíneos |
| ES18766063T Active ES2966825T3 (es) | 2017-08-15 | 2018-08-15 | Activadores de piruvato quinasa para su uso en el tratamiento de trastornos sanguíneos |
| ES18766400T Active ES2944545T3 (es) | 2017-08-15 | 2018-08-15 | Moduladores de piruvato quinasa y uso de los mismos |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18779068T Active ES2905100T3 (es) | 2017-08-15 | 2018-08-15 | Activadores de piruvato quinasa para su uso en el tratamiento de trastornos sanguíneos |
| ES18766063T Active ES2966825T3 (es) | 2017-08-15 | 2018-08-15 | Activadores de piruvato quinasa para su uso en el tratamiento de trastornos sanguíneos |
Country Status (34)
| Country | Link |
|---|---|
| US (8) | US11464775B2 (es) |
| EP (4) | EP3668881B1 (es) |
| JP (5) | JP7301040B2 (es) |
| KR (3) | KR102642408B1 (es) |
| CN (3) | CN111032045B (es) |
| AU (4) | AU2018316588B2 (es) |
| BR (1) | BR112020003254A2 (es) |
| CA (1) | CA3072455A1 (es) |
| CL (1) | CL2020000375A1 (es) |
| CO (2) | CO2020002959A2 (es) |
| CR (1) | CR20200123A (es) |
| CY (1) | CY1124953T1 (es) |
| DK (2) | DK3668513T3 (es) |
| EC (1) | ECSP20018487A (es) |
| ES (3) | ES2905100T3 (es) |
| FI (1) | FI3668512T3 (es) |
| HR (2) | HRP20220039T1 (es) |
| HU (2) | HUE061910T2 (es) |
| IL (3) | IL272599B (es) |
| LT (2) | LT3668513T (es) |
| MD (1) | MD3668513T2 (es) |
| MX (2) | MX2020001833A (es) |
| MY (1) | MY204421A (es) |
| PE (1) | PE20200724A1 (es) |
| PH (1) | PH12020500343B1 (es) |
| PL (2) | PL3668513T3 (es) |
| PT (2) | PT3668513T (es) |
| RS (2) | RS62871B1 (es) |
| SG (2) | SG11202001262QA (es) |
| SI (2) | SI3668512T1 (es) |
| SM (2) | SMT202200023T1 (es) |
| TW (3) | TWI796353B (es) |
| UA (1) | UA126292C2 (es) |
| WO (3) | WO2019035864A1 (es) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| LT3483164T (lt) | 2017-03-20 | 2020-05-11 | Forma Therapeutics, Inc. | Pirolpirolo kompozicijos kaip piruvato kinazės (pkr) aktyvatoriai |
| TWI796353B (zh) * | 2017-08-15 | 2023-03-21 | 美商阿吉歐斯製藥公司 | 丙酮酸激酶調節劑及其用途 |
| EP3697354A4 (en) | 2017-10-20 | 2021-06-30 | The Regents of The University of Michigan | EYE DISEASE COMPOSITIONS AND METHODS |
| EP3852791B1 (en) | 2018-09-19 | 2024-07-03 | Novo Nordisk Health Care AG | Activating pyruvate kinase r |
| EP3853206B1 (en) | 2018-09-19 | 2024-04-10 | Novo Nordisk Health Care AG | Treating sickle cell disease with a pyruvate kinase r activating compound |
| BR112021015996A2 (pt) * | 2019-02-13 | 2021-11-09 | Agios Pharmaceuticals Inc | Derivados de tieno[3,2-b]pirrol[3,2-d]piridazinona e seu uso como derivados de pkm2 para o tratamento de câncer, obesidade e distúrbios relacionados a diabetes |
| WO2020262419A1 (ja) * | 2019-06-26 | 2020-12-30 | 日産化学株式会社 | 電荷輸送性ワニス |
| HRP20251148T1 (hr) | 2019-09-19 | 2025-11-21 | Novo Nordisk Health Care Ag | Pripravci koji aktiviraju piruvat-kinazu r (pkr) |
| EP4185293A4 (en) * | 2020-07-21 | 2025-01-15 | The Regents of The University of Michigan | COMPOSITIONS AND METHODS FOR ACTIVATING PYRUVATE KINASE |
| CN112490502B (zh) * | 2020-12-04 | 2022-01-21 | 广州天赐高新材料股份有限公司 | 一种电解液及锂二次电池 |
| US11566030B2 (en) | 2021-02-08 | 2023-01-31 | Global Blood Therapeutics, Inc. | Substituted 2,6-dihydropyrrolo[3,4-c]pyrazoles as pyruvate kinase activators |
| US12128035B2 (en) | 2021-03-19 | 2024-10-29 | Novo Nordisk Health Care Ag | Activating pyruvate kinase R |
| WO2023052783A1 (en) * | 2021-09-30 | 2023-04-06 | Sitryx Therapeutics Limited | Novel compounds |
| IL312679A (en) * | 2021-11-16 | 2024-07-01 | Agios Pharmaceuticals Inc | Compounds for the treatment of anemia associated with MDS and other conditions |
| CN116159061A (zh) * | 2023-03-09 | 2023-05-26 | 上海市第十人民医院 | 丙酮酸激酶m2激活剂在制备预防或治疗心力衰竭的药物中的应用 |
| EP4671102A1 (en) | 2023-03-28 | 2025-12-31 | Honda Motor Co., Ltd. | METHOD FOR PROVIDING INFORMATION, PROGRAM FOR PROVIDING INFORMATION, STORAGE MEDIA AND INFORMATION PROCESSING APPARATUS |
| TW202509043A (zh) | 2023-05-15 | 2025-03-01 | 美商阿吉歐斯製藥公司 | 2-((1h-吡唑-3-基)甲基)-6-((6-胺基吡啶-2-基)甲基)-4-甲基-4,6-二氫-5h-噻唑基[5',4':4,5]吡咯并[2,3-d]嗒𠯤-5-酮之結晶形式 |
| KR20260035223A (ko) * | 2023-07-05 | 2026-03-12 | 아지오스 파마슈티컬스 아이엔씨. | 피루브산 키나아제 활성화제의 합성 |
| TW202539675A (zh) | 2024-03-27 | 2025-10-16 | 美商阿吉歐斯製藥公司 | Pk活化劑在治療鐮狀細胞疾病中之用途 |
| WO2026076363A1 (en) | 2024-10-04 | 2026-04-09 | Agios Pharmaceuticals, Inc. | Compounds for treating mds-associated anemias and other conditions |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4405635A (en) | 1979-06-18 | 1983-09-20 | Merrell Dow Pharmaceuticals Inc. | 4-Aroylimidazol-2-ones and their use as pharmaceuticals |
| US4883914A (en) | 1987-08-17 | 1989-11-28 | American Cyanamid Company | Benzenesulfonyl carboxamide compounds useful as herbicidal agents |
| CA2380644A1 (en) | 1999-07-30 | 2001-02-08 | Basf Aktiengesellschaft | 2-pyrazolin-5-ones |
| CA2381581A1 (en) | 1999-09-04 | 2001-03-15 | Astrazeneca Ab | Substituted n-phenyl 2-hydroxy-2-methyl-3,3,3-trifluoropropanamide derivatives which elevate pyruvate dehydrogenase activity |
| EP1972629A1 (en) * | 2007-03-23 | 2008-09-24 | Mutabilis SA | New imidazolo-heteroaryl derivatives with antibacterial properties |
| FR2933983B1 (fr) * | 2008-07-15 | 2010-08-27 | Servier Lab | Nouveaux derives tricycliques,leur procede de preparation et les compositions pharmaceutiques qui les contiennent. |
| MX2011000843A (es) * | 2008-07-23 | 2011-04-11 | Vertex Pharma | Inhibidores de pirazolopiridin cinasa triciclicos. |
| EP2334673A1 (en) * | 2008-08-29 | 2011-06-22 | Amgen Inc. | PYRIDO[3,2-d]PYRIDAZINE-2(1H)-ONE COMPOUNDS AS P38 MODULATORS AND METHODS OF USE THEREOF |
| WO2010042867A2 (en) | 2008-10-09 | 2010-04-15 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Activators of human pyruvate kinase |
| CA2758071C (en) * | 2009-04-06 | 2018-01-09 | Agios Pharmaceuticals, Inc. | Pyruvate kinase m2 modulators, therapeutic compositions and related methods of use |
| TWI598337B (zh) | 2009-06-29 | 2017-09-11 | 阿吉歐斯製藥公司 | 治療化合物及組成物 |
| US20130109672A1 (en) | 2010-04-29 | 2013-05-02 | The United States Of America,As Represented By The Secretary, Department Of Health And Human Service | Activators of human pyruvate kinase |
| KR101861883B1 (ko) | 2010-12-06 | 2018-05-28 | 글락소 그룹 리미티드 | Lp-PLA₂에 의해 매개되는 질환 또는 상태의 치료에 사용하기 위한 피리미디논 화합물 |
| TWI549947B (zh) | 2010-12-29 | 2016-09-21 | 阿吉歐斯製藥公司 | 治療化合物及組成物 |
| CN103608016A (zh) * | 2011-05-03 | 2014-02-26 | 安吉奥斯医药品有限公司 | 丙酮酸激酶激活剂在治疗中的用途 |
| US9388164B2 (en) * | 2011-05-03 | 2016-07-12 | Agios Pharmaceuticals, Inc | Methods of using pyruvate kinase activators |
| WO2012151450A1 (en) | 2011-05-03 | 2012-11-08 | Agios Pharmaceuticals, Inc. | Pyruvate kinase activators for use for increasing lifetime of the red blood cells and treating anemia |
| SMT202400081T1 (it) * | 2011-05-03 | 2024-03-13 | Agios Pharmaceuticals Inc | Attivatori della piruvato chinasi per uso in terapia |
| HK1198443A1 (en) * | 2011-07-19 | 2015-04-24 | 无限药品股份有限公司 | Heterocyclic compounds and uses thereof |
| WO2013056153A1 (en) * | 2011-10-13 | 2013-04-18 | Kung Charles | Activators of pyruvate kinase m2 and methods of treating disease |
| US9921221B2 (en) | 2012-07-26 | 2018-03-20 | Joslin Diabetes Center, Inc. | Predicting and treating diabetic complications |
| EA032007B1 (ru) | 2012-11-08 | 2019-03-29 | Аджиос Фармасьютикалз, Инк. | Терапевтические соединения и композиции и их использование в качестве модуляторов пк-m2 |
| WO2014118135A1 (en) * | 2013-01-30 | 2014-08-07 | Bayer Pharma Aktiengesellschaft | Amidoimidazopyridazines as mknk-1 kinase inhibitors |
| EP2968323A4 (en) | 2013-03-13 | 2016-12-14 | Flatley Discovery Lab | PYRIDAZINONE COMPOUNDS AND METHODS FOR THE TREATMENT OF CYSTIC FIBROSIS |
| WO2014139144A1 (en) | 2013-03-15 | 2014-09-18 | Agios Pharmaceuticals, Inc. | Therapeutic compounds and compositions |
| AR097543A1 (es) * | 2013-09-06 | 2016-03-23 | Lexicon Pharmaceuticals Inc | COMPUESTOS BASADOS EN IMIDAZO[1,2-b]PIRIDAZINA, COMPOSICIONES QUE LOS COMPRENDEN Y SUS MÉTODOS DE USO |
| US11465992B2 (en) | 2017-07-07 | 2022-10-11 | Inflazome Limited | Sulfonamide carboxamide compounds |
| MX2020001776A (es) | 2017-08-15 | 2020-03-24 | Inflazome Ltd | Sulfonilureas y sulfoniltioureas como inhibidores de nlrp3. |
| AR112475A1 (es) | 2017-08-15 | 2019-10-30 | Agios Pharmaceuticals Inc | Moduladores de piruvato quinasas y uso de los mismos |
| US10676485B2 (en) | 2017-08-15 | 2020-06-09 | Abbvie Inc. | Macrocyclic MCL-1 inhibitors and methods of use |
| TWI796353B (zh) | 2017-08-15 | 2023-03-21 | 美商阿吉歐斯製藥公司 | 丙酮酸激酶調節劑及其用途 |
| BR112021015996A2 (pt) * | 2019-02-13 | 2021-11-09 | Agios Pharmaceuticals Inc | Derivados de tieno[3,2-b]pirrol[3,2-d]piridazinona e seu uso como derivados de pkm2 para o tratamento de câncer, obesidade e distúrbios relacionados a diabetes |
-
2018
- 2018-08-15 TW TW107128431A patent/TWI796353B/zh active
- 2018-08-15 MX MX2020001833A patent/MX2020001833A/es unknown
- 2018-08-15 RS RS20220003A patent/RS62871B1/sr unknown
- 2018-08-15 PL PL18779068T patent/PL3668513T3/pl unknown
- 2018-08-15 RS RS20230354A patent/RS64221B1/sr unknown
- 2018-08-15 SM SM20220023T patent/SMT202200023T1/it unknown
- 2018-08-15 PE PE2020000245A patent/PE20200724A1/es unknown
- 2018-08-15 CN CN201880053315.9A patent/CN111032045B/zh active Active
- 2018-08-15 EP EP18766063.4A patent/EP3668881B1/en active Active
- 2018-08-15 EP EP18766400.8A patent/EP3668512B1/en active Active
- 2018-08-15 SM SM20230146T patent/SMT202300146T1/it unknown
- 2018-08-15 MY MYPI2020000757A patent/MY204421A/en unknown
- 2018-08-15 US US16/639,086 patent/US11464775B2/en active Active
- 2018-08-15 FI FIEP18766400.8T patent/FI3668512T3/fi active
- 2018-08-15 CA CA3072455A patent/CA3072455A1/en active Pending
- 2018-08-15 ES ES18779068T patent/ES2905100T3/es active Active
- 2018-08-15 US US16/639,075 patent/US11364240B2/en active Active
- 2018-08-15 SG SG11202001262QA patent/SG11202001262QA/en unknown
- 2018-08-15 PL PL18766400.8T patent/PL3668512T3/pl unknown
- 2018-08-15 PT PT187790688T patent/PT3668513T/pt unknown
- 2018-08-15 PT PT187664008T patent/PT3668512T/pt unknown
- 2018-08-15 AU AU2018316588A patent/AU2018316588B2/en active Active
- 2018-08-15 KR KR1020207007346A patent/KR102642408B1/ko active Active
- 2018-08-15 HU HUE18766400A patent/HUE061910T2/hu unknown
- 2018-08-15 AU AU2018316587A patent/AU2018316587B2/en active Active
- 2018-08-15 DK DK18779068.8T patent/DK3668513T3/da active
- 2018-08-15 SG SG11202001353PA patent/SG11202001353PA/en unknown
- 2018-08-15 EP EP21207073.4A patent/EP4026838A1/en not_active Withdrawn
- 2018-08-15 WO PCT/US2018/000128 patent/WO2019035864A1/en not_active Ceased
- 2018-08-15 CN CN202310962497.4A patent/CN117186119B/zh active Active
- 2018-08-15 CR CR20200123A patent/CR20200123A/es unknown
- 2018-08-15 BR BR112020003254-9A patent/BR112020003254A2/pt not_active Application Discontinuation
- 2018-08-15 SI SI201830914T patent/SI3668512T1/sl unknown
- 2018-08-15 CN CN201880053325.2A patent/CN111032046B/zh active Active
- 2018-08-15 WO PCT/US2018/000127 patent/WO2019035863A1/en not_active Ceased
- 2018-08-15 TW TW107128429A patent/TWI790271B/zh active
- 2018-08-15 PH PH1/2020/500343A patent/PH12020500343B1/en unknown
- 2018-08-15 KR KR1020207007344A patent/KR102717594B1/ko active Active
- 2018-08-15 LT LTEPPCT/US2018/000128T patent/LT3668513T/lt unknown
- 2018-08-15 JP JP2020508455A patent/JP7301040B2/ja active Active
- 2018-08-15 HR HRP20220039TT patent/HRP20220039T1/hr unknown
- 2018-08-15 IL IL272599A patent/IL272599B/en unknown
- 2018-08-15 UA UAA202001747A patent/UA126292C2/uk unknown
- 2018-08-15 US US16/639,081 patent/US11590132B2/en active Active
- 2018-08-15 DK DK18766400.8T patent/DK3668512T3/da active
- 2018-08-15 LT LTEPPCT/US2018/000129T patent/LT3668512T/lt unknown
- 2018-08-15 JP JP2020508498A patent/JP7275107B2/ja active Active
- 2018-08-15 ES ES18766063T patent/ES2966825T3/es active Active
- 2018-08-15 EP EP18779068.8A patent/EP3668513B1/en active Active
- 2018-08-15 WO PCT/US2018/000129 patent/WO2019035865A1/en not_active Ceased
- 2018-08-15 HR HRP20230452TT patent/HRP20230452T1/hr unknown
- 2018-08-15 MX MX2020001832A patent/MX2020001832A/es unknown
- 2018-08-15 MD MDE20200663T patent/MD3668513T2/ro unknown
- 2018-08-15 SI SI201830558T patent/SI3668513T1/sl unknown
- 2018-08-15 ES ES18766400T patent/ES2944545T3/es active Active
- 2018-08-15 HU HUE18779068A patent/HUE057178T2/hu unknown
- 2018-08-15 TW TW111135803A patent/TWI823580B/zh active
- 2018-08-15 KR KR1020237028222A patent/KR102764124B1/ko active Active
-
2020
- 2020-02-11 IL IL272597A patent/IL272597B/en unknown
- 2020-02-13 CL CL2020000375A patent/CL2020000375A1/es unknown
- 2020-03-12 CO CONC2020/0002959A patent/CO2020002959A2/es unknown
- 2020-03-12 CO CONC2020/0002961A patent/CO2020002961A2/es unknown
- 2020-03-13 EC ECSENADI202018487A patent/ECSP20018487A/es unknown
- 2020-11-19 US US16/952,257 patent/US11040036B2/en active Active
-
2022
- 2022-01-18 CY CY20221100039T patent/CY1124953T1/el unknown
- 2022-01-27 US US17/586,777 patent/US11872225B2/en active Active
- 2022-04-26 IL IL292530A patent/IL292530B2/en unknown
- 2022-05-16 US US17/744,806 patent/US11957680B2/en active Active
- 2022-12-14 JP JP2022199462A patent/JP2023027276A/ja active Pending
-
2023
- 2023-01-18 JP JP2023005925A patent/JP7530453B2/ja active Active
- 2023-01-20 US US18/099,311 patent/US12133853B2/en active Active
- 2023-05-03 AU AU2023202772A patent/AU2023202772B2/en active Active
-
2024
- 2024-07-26 JP JP2024121066A patent/JP2024147805A/ja active Pending
- 2024-08-16 AU AU2024205818A patent/AU2024205818A1/en not_active Abandoned
- 2024-09-12 US US18/882,884 patent/US20250144093A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2944545T3 (es) | Moduladores de piruvato quinasa y uso de los mismos | |
| ES2967866T3 (es) | Compuestos de pirrolotriazina como inhibidores de TAM | |
| RU2537945C2 (ru) | Триазиновые, пиримидиновые и пиридиновые аналоги и их применение в качестве терапевтических агентов и диагностических проб | |
| CN112236429A (zh) | 用作ii型irak抑制剂的杂芳基化合物及其用途 | |
| CN107108561A (zh) | 用作irak抑制剂的杂芳基化合物及其用途 | |
| KR20210128435A (ko) | 티에노[3,2-b] 피롤[3,2-d]피리다지논 유도체, 및 암, 비만증, 및 당뇨병 관련 장애의 치료를 위한 pkm2 유도체로서 이의 용도 | |
| EP3350185A1 (en) | 1-phenylpyrrolidin-2-one derivatives as perk inhibitors | |
| CA2885783A1 (en) | Substituted indazol-pyrrolopyrimidines useful in the treatment of hyperproliferative diseases | |
| CN119894872A (zh) | 杂芳烃、含有杂芳烃的药物组合物及其使用方法 | |
| TW202325288A (zh) | 新穎化合物 | |
| RU2797518C2 (ru) | Модуляторы пируваткиназы и их применение | |
| HK40030322B (en) | Pyruvate kinase modulators and use thereof | |
| HK40030322A (en) | Pyruvate kinase modulators and use thereof | |
| CA3259052A1 (en) | HETEROCYCLIC COMPOUNDS USED AS PI3K.ALPHA INHIBITORS. | |
| WO2026047509A1 (en) | Substituted triazine compound derivatives and their pharmaceutical use | |
| HK40043591A (en) | Heteroaryl compounds as type ii irak inhibitors and uses thereof | |
| HK1242303A1 (en) | Heteroaryl compounds as irak inhibitors and uses thereof |