ES2953815T3 - Derivados de indol y bencimidazol como antagonistas duales de los receptores 5-HT2A y 5-HT6 - Google Patents
Derivados de indol y bencimidazol como antagonistas duales de los receptores 5-HT2A y 5-HT6 Download PDFInfo
- Publication number
- ES2953815T3 ES2953815T3 ES19706271T ES19706271T ES2953815T3 ES 2953815 T3 ES2953815 T3 ES 2953815T3 ES 19706271 T ES19706271 T ES 19706271T ES 19706271 T ES19706271 T ES 19706271T ES 2953815 T3 ES2953815 T3 ES 2953815T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- trifluoromethyl
- piperazin
- indole
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/10—Radicals substituted by halogen atoms or nitro radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Psychology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
Abstract
La invención se refiere a nuevos 4-(piperazin-1-il)-2-(trifluorometil)-1H-indoles y 4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazoles representados por la fórmula (I). donde todos los símbolos y variables son como se definen en la descripción. Los compuestos pueden encontrar uso en un método de prevención y/o tratamiento de enfermedades seleccionadas del grupo que consiste en enfermedad de Alzheimer, enfermedad de Parkinson, demencia con cuerpos de Levy, psicosis relacionada con la demencia, esquizofrenia, síndromes delirantes y otras condiciones psicóticas relacionadas y no relacionadas con la toma. sustancias psicoactivas, depresión, trastornos de ansiedad de diversas etiologías, trastornos del sueño de diversas etiologías. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Derivados de indol y bencimidazol como antagonistas duales de los receptores 5-HT2A y 5-HT6
La presente invención se refiere a nuevos 4-(piperazin-1-il)-2-(trifluorometil)-1H-indoles y 4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazoles. La presente invención también se refiere a composiciones farmacéuticas que comprenden los compuestos, y estos compuestos y la composición farmacéutica para su uso como medicamento.
La demencia es un conjunto de deterioro progresivo de las funciones cognitivas asociado con trastornos conductuales y psicológicos y dificultades en el funcionamiento cotidiano (Hersch y Falzgraf, 2007). La categoría más importante de factores que dañan el cerebro hasta tal punto que se producen los síntomas de la demencia son las enfermedades neurodegenerativas, que conducen a la degeneración progresiva del tejido nervioso. La clasificación CIE-10 distingue, entre otras, la demencia de tipo Alzheimer (DAT, por sus siglas en inglés), así como la demencia en la enfermedad de Pick (frontotemporal), la demencia en la enfermedad de Huntington, la demencia en la enfermedad de Parkinson y la demencia muy similar con cuerpos de Lewy. La causa del daño cerebral que conduce a la demencia pueden ser enfermedades infecciosas tales como la enfermedad de Creutzfeldt-Jakob (incluida junto con las enfermedades neurodegenerativas), la infección por VIH/SIDA o la neuroborreliosis. Además de las enfermedades neurodegenerativas e infecciosas, los síntomas de la demencia también pueden estar relacionados con enfermedades vasculares tales como un accidente cerebrovascular, que puede causar la denominada demencia de inicio agudo o demencia vascular (después de una serie de accidentes cerebrovasculares). En los ancianos, la causa más común de demencia es la enfermedad de Alzheimer. La prevalencia global de la demencia se estima en aproximadamente el 3,9 % de la población mayor de 60 años (Ferri y col., 2005), lo que significa que actualmente hay aproximadamente 35,6 millones de personas con diferentes formas de demencia en el mundo. A la luz del aumento previsto en la esperanza de vida, este número se duplicará para 2030 y se triplicará para 2050. Por lo tanto, la demencia es un problema médico y social muy grave y creciente.
Además de los trastornos cognitivos axiales, hasta el 60 % de los pacientes con demencia también experimentan los denominados síntomas conductuales y psicológicos de la demencia (BPSD, por sus siglas en inglés). Entre ellos se pueden distinguir los siguientes: trastornos psicóticos (delirios y alucinaciones), depresión, apatía, desinhibición sexual, irritabilidad, agresividad verbal y física, agitación psicomotora y ansiedad (Carson y col., 2006; Jeste y col., 2008). Por ejemplo, del 40 al 60 % de los pacientes con demencia experimentan trastornos depresivos considerables en alguna fase de la enfermedad (Hersch y Falzgraf, 2007), mientras que la prevalencia de síntomas psicóticos puede alcanzar el 63 % de los pacientes en caso de delirios, y el 41 % en caso de alucinaciones (Jeste y col., 2008). Los BPSD pueden tener lugar en cualquier fase de la enfermedad, algunos síntomas son más comunes en la demencia leve (depresión, apatía, ansiedad, irritabilidad), mientras que otros son más comunes en las fases avanzadas de la demencia (delirios, alucinaciones, desinhibición) (Hersch y Falzgraf, 2007). Se ha demostrado muchas veces que solo los BPSD son la carga principal tanto para los pacientes con demencia como para sus cuidadores, y pueden experimentarse incluso de manera más aguda que el deterioro cognitivo básico. La aparición de BPSD también se asocia con un mal pronóstico de la enfermedad, una pérdida más rápida de la función cognitiva y un deterioro específico de la vida cotidiana. La psicosis, la agitación, la agresividad y la depresión que acompañan a la demencia son los principales predictores de la institucionalización del paciente, y son los principales objetivos en el tratamiento de los BPSD desde las perspectivas clínica y social (Amano y col., 2009; Gauthier y col., 2010; Hersch y Falzgraf, 2007).
Hasta mediados de la década de 1990, los fármacos de elección en el tratamiento de BPSD eran antipsicóticos de primera generación (es decir, neurolépticos típicos), especialmente en el caso de delirios y alucinaciones. Se demostró que el principal representante de esta clase de fármacos, el haloperidol, no afecta a la excitación ni a los síntomas conductuales en su conjunto, reduce la agresividad. Al mismo tiempo, un metanálisis de ensayos clínicos demostró la falta de diferencias entre los antipsicóticos de primera generación en cuanto a su eficacia para BPSD (Sink y col., 2005). En los años siguientes, los neurolépticos típicos fueron parcialmente reemplazados en el tratamiento de los BPSD por fármacos antipsicóticos de segunda generación (es decir, neurolépticos atípicos) (De Deyn y col., 2005), que se caracterizan por una menor tendencia a inducir trastornos extrapiramidales (síntomas extrapiramidales - EPS, por sus siglas en inglés) y mayor eficiencia en comparación con los fármacos de primera generación (Liperoti y col., 2008). Sin embargo, la eficacia y seguridad de los fármacos usados actualmente en el tratamiento de los BPSd no son satisfactorias (Nobili y col., 2009). La revisión de 16 ensayos clínicos con aplicación de antipsicóticos de segunda generación en el tratamiento de BPSD, realizada dentro de una actividad de la Cochrane (Cochrane Library) reveló que la risperidona y la olanzapina fueron eficaces en el tratamiento de la agresividad, y la risperidona también fue más eficaz que el placebo en el tratamiento de la psicosis asociada a la demencia (Ballard y Waite, 2006). Sin embargo, ambos fármacos causaron efectos secundarios significativos de carácter extrapiramidal y eventos cardiovasculares. Mientras tanto, el aripiprazol no mostró ninguna ventaja sobre el placebo en el tratamiento de delirios y alucinaciones en pacientes con psicosis relacionada con la enfermedad de Alzheimer (De Deyn y col., 2005). El uso de antipsicóticos en el tratamiento de los BPSD se complica adicionalmente por el hecho de que estos fármacos exacerban los deterioros cognitivos existentes, lo que es particularmente desventajoso en pacientes con demencia (Fasano y col., 2012; Jeste y col., 2008; Vigen y col., 2011).
A la luz de estos hechos, desde 2005, la Agencia de Alimentos y Medicamentos (FDA, por sus siglas en inglés) de los Estados Unidos exige que se coloquen advertencias especiales en los prospectos de los antipsicóticos de segunda generación. Estas advertencias (denominadas “ recuadros de advertencia” ) están asociadas con la aparición de
efectos secundarios graves y un mayor riesgo de muerte, en caso de uso de neurolépticos atípicos en pacientes con demencia (U.S. Food and Drug Administration, 2005). Desde 2008, el requisito de advertencias similares también se aplica en el caso de los antipsicóticos de primera generación (U.S. Food and Drug Administration, 2008).
A pesar de esto, los antipsicóticos todavía se usan ampliamente en pacientes con BPSD (Schneider y col., 2006; Schulze y col., 2013b), principalmente porque no existe una alternativa más favorable (Schulze y col., 2013a). Actualmente, no existen fármacos aprobados para el tratamiento de la psicosis asociada a la demencia, así como tampoco antidepresivos, ansiolíticos, fármacos antiagresividad, diseñados específicamente para cubrir las necesidades terapéuticas de las personas mayores.
La psicosis en la demencia puede tener un sustrato neurobiológico diferente al de la esquizofrenia. De hecho, los pacientes psicóticos de Alzheimer a menudo experimentan alucinaciones visuales e identificaciones erróneas de los cuidadores, síntomas que no se encuentran comúnmente en pacientes con esquizofrenia. Por el contrario, los delirios extraños o complejos que tienen lugar con frecuencia en pacientes con esquizofrenia no se observan a menudo en pacientes con demencia (Jeste y Finkel, 2000). La naturaleza distinta de los síntomas psicóticos en la demencia sugiere que están en juego diferentes sistemas de neurotransmisores. En particular, pueden estar involucrados los sistemas serotoninérgicos porque las alucinaciones en la demencia son similares a las causadas por agonistas serotoninérgicos tales como la mescalina o el ácido lisérgico (Marsh, 1979). Los antagonistas de los receptores NMDA tales como la ketamina o la fenciclidina también pueden provocar fuertes alucinaciones visuales (Siegel, 1978), pero son provocadas con menor frecuencia por los dopaminomiméticos tales como la anfetamina o la cocaína, que se usan ampliamente en la detección preclínica de nuevos fármacos para la esquizofrenia (Jones y col, 2011).
Hay datos sustanciales que respaldan la importancia del sistema de serotonina en el desarrollo de los BPSD. Por ejemplo, los polimorfismos del gen del receptor de serotonina están asociados con alucinaciones visuales y auditivas en pacientes con enfermedad de Alzheimer (EA) (Holmes y col., 1998). Un polimorfismo genético de la región promotora del transportador de serotonina (genotipo L/L) se ha asociado con el comportamiento agresivo (Sukonick y col., 2001). Otros estudios muestran la implicación de los receptores 5HT2A y 5HT6 en la patogenia de la EA (Lorke y col., 2006), así como la asociación de los receptores 5-HT6 con síntomas psicóticos en pacientes con EA (Marcos y col., 2008).
Se ha observado que las alucinaciones, principalmente visuales, provocadas por sustancias psicotomiméticas, tales como el LSD (dietilamida del ácido D-lisérgico) o el DOI (2,5-dimetoxi-4-yodoanfetamina), están asociadas a la activación de los receptores 5-HT2A en la corteza cerebral (Nichols, 2004). Teniendo en cuenta su similitud alucinógena clínica con las observadas en pacientes con demencia, se ha sugerido la implicación de mecanismos farmacológicos comunes, incluida la desregulación serotoninérgica. La implicación del bloqueo de los receptores de serotonina 5-HT2A en la actividad antipsicótica fue confirmada además por la actividad de los antagonistas de los receptores 5-HT2A en modelos glutamatérgicos de psicosis, asociados con la facilitación de la transmisión glutamatérgica en la corteza cerebral (Varty y col., 1999). En consonancia con lo anterior, la pimavanserina, un agonista inverso selectivo del receptor 5-HT2A, es el primer fármaco antipsicótico aprobado en 2016 para el tratamiento de la psicosis de la enfermedad de Parkinson. Sin embargo, es importante tener en cuenta que la pimavanserina tiene una afinidad significativa por los canales hERG (aproximadamente 210 nM), lo que puede causar cambios en el ECG, lo que podría provocar arritmias potencialmente mortales. Además, la pimavanserina no tiene afinidad por los receptores 5-HT6.
Líneas de evidencia convergentes indican que el bloqueo de los receptores de serotonina 5 HT6 (5 HT6R) puede estar implicado en: (i) efectos procognitivos debido a la facilitación de la transmisión colinérgica (Liu y Robichaud, 2009; Riemer y col., 2003), (ii) actividad antidepresiva por aumento del tono noradrenérgico y dopaminérgico, así como (iii) un efecto ansiolítico, mediado por la interacción con la transmisión GABA-érgica (Weso+owska, 2010; Weso'J'owska y Nikiforuk, 2007). Estos hallazgos están respaldados por la localización exclusiva de los receptores 5-HT6 en el sistema nervioso central (SNC), especialmente en las áreas límbicas y corticales del cerebro involucradas en el control del estado de ánimo y la cognición (Woolley y col., 2004).
El componente colinomimético del bloqueo de los receptores 5-HT6, además de su importancia para la actividad procognitiva, también parece ser significativo desde el punto de vista de los efectos antipsicóticos potencialmente beneficiosos. De hecho, se ha demostrado que los antagonistas de los receptores muscarínicos tienen propiedades antipsicóticas (Maehara y col., 2008). Por tanto, aunque el bloqueo selectivo del receptor 5-HT6 no induce por sí solo la actividad antipsicótica, puede contribuir a su potenciación. En línea con lo anterior, estudios recientes demostraron que una combinación del antagonismo de los receptores 5-HT2A y 5-HT6 puede producir un efecto antipsicótico más ra jfuerte que el uso independiente de un antagonista selectivo de cada uno de esos receptores (Fija y col., 2014).
El aumento de la eficacia terapéutica de los antagonistas duales de los receptores 5-HT2A y 5-HT6 en pacientes con demencia puede deberse no solo al aumento de la actividad antipsicótica resultante de la modulación sinérgica de la transmisión glutamatérgica y colinérgica, sino también a la actividad procognitiva mediada por el bloqueo del receptor 5-HT6, principalmente de naturaleza colinomimética. Estas propiedades son cruciales porque la presencia de psicosis en la demencia está indisolublemente ligada al deterioro cognitivo (Murray y col., 2014).
Por lo tanto, el antagonista dual de los receptores 5-HT2A y 5-HT6, que une la actividad antipsicótica y procognitiva en una molécula, aborda los desafíos terapéuticos más importantes en la psicosis relacionada con la demencia.
La Solicitud Internacional WO2007/006677 describe determinados derivados de bencimidazolona e hidroindolona como antagonistas selectivos de 5-HT6, antagonistas selectivos de 5-HT2A o ambos. La descripción no especifica qué compuestos son antagonistas duales de los receptores 5-HT2A y 5-HT6; sin embargo, en su respuesta a la opinión escrita, el Solicitante describió actividades para cuatro compuestos para ambos receptores. No obstante, todos los compuestos descritos que tienen derivados de bencimidazolona sustituidos por 4-piperazina tienen nitrógeno de piperazina sin sustituir como donante de enlaces de hidrógeno. Además, la presencia del grupo carbonilo en la posición 2 afecta negativamente a la estabilidad metabólica y química de dichos compuestos, y en el caso de los compuestos de bencimidazol, existe la posibilidad de formación de tautómeros lactama-lactima que es desfavorable debido a la ionización adicional en determinados intervalos de pH, y la formación de un sitio donante de enlaces de hidrógeno adicional. T odo esto afecta negativamente la penetración a través de las membranas biológicas y, por lo tanto, dificulta la absorción desde el tubo gastrointestinal y la penetración de la barrera hematoencefálica.
La Solicitud Internacional WO2008/055808 describe determinados compuestos como antagonistas selectivos de 5-HT6, antagonistas selectivos de 5-HT2A o ambos. Los compuestos descritos en esta solicitud internacional tienen un grupo amida opcionalmente sustituido en la posición 2. Los compuestos tienen una baja estabilidad química y metabólica debido a la hidrólisis del grupo amida con respecto a carboxilato. Además, la presencia del grupo amida no permite obtener compuestos con afinidad dual, es decir, no solo por 5-HT6, sino también por 5-HT2A.
Las Solicitudes Internacionales WO2010/056644 y WO2013/001499 describen compuestos que tienen sustitución en la posición 2 con un grupo alquilo o ninguna sustitución, es decir, hay un átomo de hidrógeno en la posición 2. De nuevo, los compuestos con una afinidad dual, es decir, no solo por 5-HT6, sino también por 5-HT2A, no se puede obtener.
Hasta el momento, no se ha identificado ningún compuesto que sea un fármaco potencial que una la actividad antipsicótica y procognitiva en una sola molécula, que actúe por antagonismo de los receptores 5-HT2A y 5-HT6 y, por otro lado, que tenga propiedades favorables en cuanto a, por ejemplo, biodisponibilidad y facilidad de penetración de la barrera hematoencefálica.
Por lo tanto, todavía existe la necesidad en la técnica de tales compuestos.
Por lo tanto, la presente invención proporciona nuevos compuestos con núcleo de 4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol o 4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol como antagonista dual de los receptores 5-HT2A y 5-HT6 que tienen características favorables tanto in vitro como in vivo y, por lo tanto, son candidatos prometedores en los ensayos clínicos.
La presente invención se define mediante las reivindicaciones adjuntas.
En un primer aspecto, la presente invención se refiere a un compuesto de fórmula general (I):

o una sal farmacéuticamente aceptable del mismo,
en donde:
G es CH o N;
R1 es H, alquilo C1-C4, HO-alquilo C1-C4 o alquilo Ci-C4-O-alquilo C1-C4;
R2 se selecciona de un grupo que consiste en:
• un grupo fenilo sin sustituir o sustituido por al menos un sustituyente, o
• un grupo heteroarilo de 5 o 6 miembros sin sustituir o sustituido por al menos un sustituyente,
en donde el sustituyente se selecciona de F, Cl, Br, alquilo C1-C4 y alquilo C1-C4-O-.
En una primera realización, en los compuestos de fórmula (I), G es CH.
En una realización alternativa, en los compuestos de fórmula (I), G es N.
Preferiblemente, en los compuestos de fórmula (I), R1 es H, metilo o 2-hidroxietilo.
En una realización preferible de los compuestos de fórmula (I), R2 se selecciona de un grupo fenilo sin sustituir o sustituido por al menos un sustituyente.
En otra realización preferible de los compuestos de fórmula (I), R2 se selecciona de un grupo heteroarilo de 5 o 6 miembros sin sustituir o sustituido por al menos un sustituyente. En esta realización preferida, preferiblemente, el heteroarilo de 5 o 6 miembros se selecciona de furilo, tienilo, tiazolilo o piridilo.
En otra más, la realización mucho más preferible,
R2 se selecciona de un grupo que consiste en:
• un grupo fenilo sin sustituir o sustituido por al menos un sustituyente, o
• un grupo heteroarilo de 5 o 6 miembros sin sustituir o sustituido por al menos un sustituyente,
en donde el heteroarilo de 5 o 6 miembros se selecciona de furilo, tienilo, tiazolilo o piridilo,
en donde el sustituyente se selecciona de F, Cl, Br, alquilo C1-C4 y alquilo C1-C4-O-.
En todas las realizaciones, cuando R2 es un grupo sustituido, está sustituido por uno o dos sustituyentes. Más preferiblemente, está sustituido por un sustituyente.
Preferiblemente, en las definiciones del grupo R2, el sustituyente se selecciona de F, Cl, metilo o metoxi.
Como alternativa, también es preferible cuando el grupo R2 está sin sustituir.
Se pueden mencionar los siguientes compuestos específicos de fórmula (I) de la invención:
1-bencil-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
1- bencil-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
2- {4-[1-bencil-2-(trifluorometil)-1H-bencimidazol-4-il]piperazin-1-il}etanol,
1-(furan-2-ilmetil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
1-[(5-metilfuran-2-il)metil]-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
1-(3-clorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
1-(3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol,
1-(3,4-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1-(3-cloro-4-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1-(3,4-difluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1-(3,5-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1 -bencil-4-(piperazin-1 -il)-2-(trifluorometil)-1 H-indol,
1 -(3,4-diclorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-indol,
1-(4-cloro-3-fluorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol,
4-(piperazin-1 -il)-1 -(1,3-tiazol-2-ilmetil)-2-(trifluorometil)-1 H-indol,
1-(4-cloro-3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol,
1-(furan-2-Nmetil)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-metoxibendl)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-fluorobendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-dorobendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(furan-2-Nmetil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol,
1-(3,4-difluorobendl)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-metoxibendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-fluorobendl)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol,
1-(3,4-difluorobendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-bendl-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-dorobendl)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-[(5-metilfuran-2-N)metil]-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-[(5-metiltiofen-2-N)metil]-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol,
4-(piperazin-1-N)-1-(tiofen-2-Nmetil)-2-(trifluorometil)-1H-indol,
4-(piperazin-1-N)-1-(tiofen-3-Nmetil)-2-(trifluorometil)-1H-indol,
4-(4-metilpiperazin-1-N)-1-(tiofen-3-ilmetil)-2-(trifluorometil)-1H-indol,
4-(4-metilpiperazin-1-N)-1-(tiofen-2-ilmetil)-2-(trifluorometil)-1H-indol y
4-(4-metilpiperazin-1-il)-1-[(5-metil-1,3-tiazol-2-N)metil]-2-(trifluorometil)-1H-indol. En una realización especialmente preferible,
G es N, R2 es un grupo fenilo sin sustituir, y R1 es H,
y el compuesto es

o una sal farmacéuticamente aceptable del mismo.
En otra realización especialmente preferible,
G es CH, R2 es un grupo 2-furilo sin sustituir, y R1 es H,
y el compuesto es

o una sal farmacéuticamente aceptable del mismo.
En otra realización especialmente preferible,
G es CH, R2 es un grupo 2-furilo sin sustituir, y R1 es -CH3,
y el compuesto es

o una sal farmacéuticamente aceptable del mismo.
Y, en otra realización especialmente preferible,
G es CH, R2 es un grupo 5-metil-2-furilo, y R1 es H,
y el compuesto es

o una sal farmacéuticamente aceptable del mismo.
El mecanismo de acción de los compuestos de la invención se basa en el bloqueo selectivo de los receptores de serotonina 5 -HT2A y 5-HT6, cuyo papel en el mecanismo patológico y la farmacoterapia de los trastornos psicóticos y cognitivos ha sido bien confirmado tanto en estudios preclínicos como en estudios clínicos.
Por lo tanto, los compuestos de la invención pueden ser útiles en medicina como medicamentos para el tratamiento y/o la prevención de afecciones sensibles al control del sistema serotoninérgico, especialmente el antagonismo de los receptores 5-HT2A y 5-HT6, tales como: trastornos cognitivos de diversos tipos, por ejemplo, enfermedad de Alzheimer, enfermedad de Parkinson, demencia con cuerpos de Levy, psicosis relacionada con demencia, esquizofrenia, trastornos esquizoafectivos, trastornos esquizofreniformes, síndromes delirantes y otras afecciones psicóticas relacionadas y no relacionadas con el consumo de sustancias psicoactivas, trastorno afectivo, trastorno bipolar, manía, depresión, trastornos de ansiedad de diversa etiología, reacciones de estrés, trastornos de la conciencia, coma, delirio alcohólico y de diversa etiología, agresividad, agitación psicomotora y otros trastornos de conducta, trastornos del sueño de diversa etiología, síndromes de abstinencia de diversa etiología, adicción, síndromes de dolor de diversa etiología, intoxicación con sustancias psicoactivas, trastornos circulatorios cerebrales de diversa etiología, trastornos psicosomáticos de diversa etiología, trastornos de conversión, trastornos disociativos, trastornos urinarios, autismo y otros trastornos del desarrollo, por ejemplo, se entienden nocturia, tartamudeo, tics, síntomas psicopatológicos y trastornos neurológicos en el curso de otras enfermedades del sistema nervioso central y periférico.
Por lo tanto, en el segundo aspecto, la invención se refiere al compuesto de la presente invención para su uso como medicamento.
Preferiblemente, el compuesto de la presente invención se puede usar en el tratamiento de trastornos cognitivos de diversos tipos, es decir, enfermedad de Alzheimer, enfermedad de Parkinson, demencia con cuerpos de Levy, psicosis relacionada con demencia, esquizofrenia, síndromes delirantes y otras afecciones psicóticas relacionadas y no relacionadas con el consumo de sustancias psicoactivas, depresión, trastornos de ansiedad de diversa etiología, trastornos del sueño de diversa etiología.
En el tratamiento de trastornos del sistema nervioso central, los compuestos de fórmula (I) pueden administrarse en forma de una composición farmacéutica o formulación que los contenga.
Por lo tanto, en el tercer aspecto, la invención se refiere a una composición farmacéutica que comprende un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo y al menos un excipiente farmacéuticamente aceptable.
Descripción detallada
Los términos usados en la presente invención tienen los siguientes significados. Otros términos no definidos a continuación tienen los significados que entienden los expertos en la técnica.
El término “ alquilo C1-C4” es un hidrocarburo saturado de cadena lineal o ramificada que tiene de 1 a 4 átomos de carbono. Los ejemplos de alquilo C1-C4 son metilo, etilo, n-propilo, isopropilo, n-butilo, terc-butilo o sec-butilo. Más preferiblemente, alquilo C1-C4 es un alquilo C1-C3, alquilo C1-C2 o alquilo C1. La notación alquilo C1-C3, alquilo C1-C2 significa un hidrocarburo saturado de cadena lineal o ramificada que tiene de 1 a 3 o 2 átomos de carbono, respectivamente. Mucho más preferiblemente, el alquilo C1-C4 es alquilo C1-C2 que es un grupo metilo (abreviado como CH3) o un grupo etilo.
La expresión “ grupo heteroarilo de 5 o 6 miembros” es un grupo anular aromático monocíclico que tiene de 1 a 4 heteroátomos seleccionados de nitrógeno, azufre y oxígeno e incluyen, por ejemplo, un grupo furilo, un grupo tienilo, un grupo pirrolilo, un grupo imidazolilo, un grupo pirazolilo, un grupo tiazolilo, un grupo isotiazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo, un grupo tiadiazolilo, un grupo tetrazolilo, un grupo piridilo, un grupo pirimidilo, un grupo piridazinilo y un grupo pirazinilo. Preferiblemente, el grupo heteroarilo de 5 o 6 miembros se selecciona de un grupo furilo, un grupo tienilo, un grupo triazolilo o un grupo piridilo.
Dado que los compuestos de la invención son básicos, pueden formar sales de adición de ácidos adecuadas.
La sal de adición de ácidos farmacéuticamente aceptable se refiere a aquellas sales que conservan la eficacia biológica de las bases libres y que no son biológicamente indeseables. Las sales de adición de ácidos se pueden formar con ácidos inorgánicos (minerales) o ácidos orgánicos. Como ejemplos de ácidos, pueden mencionarse clorhídrico, bromhídrico, yodhídrico, fosfórico, sulfúrico, nítrico, carbónico, succínico, maleico, fórmico, acético, propiónico, fumárico, cítrico, tartárico, láctico, benzoico, salicílico, glutámico, aspártico, p-toluenosulfónico, bencenosulfónico, metanosulfónico, etanosulfónico, naftalenosulfónico tal como ácido 2-naftalenosulfónico, pamoico, xinafoico, hexanoico.
Los compuestos de fórmula (I) se pueden obtener usando los siguientes métodos.
Los compuestos de fórmula (I), cuando G es N, es decir, compuestos basados en un núcleo de 4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol pueden obtenerse según el siguiente Esquema de reacción 1.

Inicialmente, se trató 2,6-difluoronitrobenceno A-1 con un derivado de piperazina (R1 = Me, BOC) en presencia de una base (típicamente K2CO3). El producto resultante A-2 se hizo reaccionar posteriormente con bencilamina (R2 = arilo, heteroarilo) en presencia de la base (típicamente K2CO3), proporcionando el compuesto A-3. A continuación, se preparó bisanilina A-4 mediante una reducción del grupo nitro en A-3 con ditionito de sodio a temperatura elevada o hierro metálico. Finalmente, la reacción de la bisanilina A-4 con TFA y la posterior formación de la sal HCl dieron los bencimidazoles A-5 esperados.

El Esquema de reacción 2 ilustra un ejemplo representativo cuando se hizo reaccionar 1-(3-fluoro-2-nitrofenil)piperazina A-6 con acetato de 2-bromoetilo en presencia de K2CO3 para obtener el derivado de piperazina A-2C.

El Esquema de reacción 3 ilustra ejemplos cuando R2 = 2-furilo o 5-metil-2-firilo. En este caso, se hizo reaccionar la bisanilina A-4 con TFAA para dar una mezcla de los compuestos A-7 y A-8. El compuesto A-7 se convirtió
cuantitativamente en A-8 usando AcOH y dicho compuesto A-8 obtenido se trató con una solución al 36 % de HCl seguido de basificación, dando como resultado los bencimidazoles A-5 deseados, el compuesto de fórmula (I), en donde G=N.
Como alternativa, los compuestos de fórmula (I), cuando G es CH, es decir, compuestos basados en un núcleo de 4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol, pueden obtenerse según el siguiente Esquema de reacción 4.

Inicialmente, se trató la 3-bromo-2-metilanilina B-1 con anhídrido trifluoroacético para producir la amida B-2. El producto resultante B-2 se hizo reaccionar posteriormente con bromo en presencia de peróxido de benzαlo y luz, proporcionando el compuesto B-3. A continuación, el bromuro de bencilo B-4 se convirtió en un derivado de fosfonio y se cicló en DMF caliente para proporcionar el indol B-4. La protección del nitrógeno de indol con un grupo bencilo seguida de una reacción de acoplamiento con un derivado de piperazina (R1 = Me, BOC) dio el compuesto B-6. La desprotección de bencilo en presencia de oxígeno y terc-butóxido de potasio proporcionó el componente básico B-7. Finalmente, la reacción del indol B-7 con bromuro de bencilo (R2 = arilo, heteroarilo) y, en algunos casos, la posterior formación de la sal HCl dieron los trifluoroindoles B-9 finales, el compuesto de fórmula (I), en donde G=CH. Opcionalmente, las sales HCl de los trifluoroindoles B-9 pueden alcalinizarse y transformarse en otra sal farmacéuticamente aceptable o usarse como base libre.

El Esquema de reacción 6 ilustra un enfoque alternativo a los compuestos B-9 usando las condiciones de reacción de Mitsunobu. Se hizo reaccionar el indol B-7 con un alcohol bencílico apropiado en presencia de trifenilfosfina y azadicarboxilato de diisopropilo. El producto B-8 de la reacción se trató con TFA o HCl en el disolvente elegido seguido de basificación para producir el compuesto B-9 en forma de base libre.
Se puede preparar una sal de adición de ácidos de una manera sencilla haciendo reaccionar un compuesto de fórmula (I) en forma de base libre con un ácido inorgánico u orgánico adecuado en una cantidad sustancialmente equimolar al compuesto de fórmula (I), opcionalmente en un disolvente adecuado, tal como un disolvente orgánico, para formar una sal que normalmente se aísla, por ejemplo, por cristalización y filtración.
Por ejemplo, una base libre de un compuesto de fórmula (I) se puede convertir en la sal clorhidrato correspondiente tratando una solución del compuesto, por ejemplo, en metanol, con una cantidad estequiométrica de ácido clorhídrico o hidrogenocloruro en metanol, etanol, éter dietílico u otro disolvente adecuado, seguido de la evaporación de los disolventes.
Como alternativa, las sales de clorhidrato se pueden obtener durante la desprotección del grupo N-t-butoxicarbonilo en nitrógeno de piperidina usando hidrogenocloruro en metanol, etanol, éter dietílico u otro disolvente adecuado, seguido de la evaporación de los disolventes, como se ilustra en la transformación del compuesto B-8 en el compuesto B-9.
En el tratamiento de las enfermedades mencionadas anteriormente, los compuestos de fórmula (I) se pueden administrar como un compuesto químico, pero típicamente se usarán en forma de composiciones farmacéuticas, que comprenden un compuesto de fórmula (I) o una sal farmacéuticamente aceptable del mismo como se ha definido anteriormente como principio activo, junto con portadores y/o excipientes farmacéuticamente aceptables.
En el tratamiento de las enfermedades mencionadas anteriormente, el compuesto de fórmula (I) o una composición farmacéutica de la presente invención se puede administrar por cualquier vía, preferiblemente por vía oral o parenteral, y tendrá la forma de una formulación destinada para su uso en medicina, dependiendo de la vía de administración prevista.
Las formulaciones sólidas pueden adoptar la forma de, por ejemplo, comprimidos o cápsulas preparadas por medios convencionales con excipientes farmacéuticamente aceptables tales como agentes aglutinantes (por ejemplo, almidón de maíz pregelatinizado, polivinilpirrolidona o hidroxipropilmetilcelulosa); cargas (por ejemplo, lactosa, sacarosa, carboximetilcelulosa, celulosa microcristalina o hidrogenofosfato de calcio); lubricantes (por ejemplo, estearato de magnesio, talco o sílice); disgregantes (por ejemplo, crospovidona, almidón de patata o glicolato sódico de almidón); agentes humectantes (por ejemplo, lauril sulfato de sodio). Los comprimidos se pueden recubrir según métodos bien conocidos en la técnica con recubrimientos convencionales, recubrimientos para retardar/controlar la liberación o recubrimientos entéricos.
Las formulaciones líquidas para administración oral pueden adoptar la forma de, por ejemplo, soluciones, jarabes o suspensiones, o pueden presentarse como un producto seco para reconstitución con agua u otro vehículo adecuado antes de su uso. Dichas formulaciones líquidas se pueden preparar por medios convencionales con excipientes farmacéuticamente aceptables tales como agentes de suspensión (por ejemplo, jarabe de sorbitol, derivados de celulosa o grasas comestibles hidrogenadas); agentes emulsionantes (por ejemplo, lecitina o goma arábiga); vehículos no acuosos (por ejemplo, aceite de almendras, ésteres oleosos, alcohol etílico o aceites vegetales fraccionados); y conservantes (por ejemplo, hidroxibenzoato de metilo p- o propilo o ácido sórbico). Las formulaciones también pueden comprender tampones, agentes saporíferos, agentes colorantes y edulcorantes adecuados.
Las formulaciones para administración oral pueden formularse adecuadamente mediante métodos conocidos por los expertos en la técnica para obtener una liberación controlada del compuesto activo.
La administración parenteral incluye la administración por inyección o infusión intramuscular e intravenosa. Las formulaciones para administración parenteral pueden estar en forma farmacéutica unitaria, por ejemplo, en ampollas o en envases multidosis, con un conservante añadido. Las composiciones pueden adoptar formas de suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, y pueden contener agentes de formulación tales como agentes de suspensión, estabilización y/o dispersión.
Como alternativa, los compuestos de fórmula (I) pueden estar en forma de polvo para reconstitución con un vehículo adecuado, por ejemplo, agua estéril apirógena.
El método de tratamiento que usa los compuestos de la presente invención implicará la administración de una cantidad terapéuticamente eficaz de un compuesto de la invención, preferiblemente en forma de una composición farmacéutica a un sujeto que necesite tal tratamiento.
Una dosis propuesta de los compuestos de la presente invención es de aproximadamente 0,1 a aproximadamente 1000 mg al día, en dosis únicas o divididas. El experto en la técnica apreciará que la selección de la dosis requerida para lograr el efecto biológico deseado dependerá de una serie de factores, por ejemplo, el compuesto específico, el uso, el modo de administración, la edad y estado del paciente, y la dosificación precisa se determinará en última instancia a discreción del médico tratante.
Ejemplos
Abreviaturas
AcOEt acetato de etilo
AcOH ácido acético
ACN acetonitrilo
s a singlete ancho
CHCI3 cloroformo
d doblete
dd doblete de dobletes
ddd doblete de doblete de dobletes
dc doblete de cuadrupletes
DCM diclorometano
Et2O éter dietílico
DIPEA N,N'-diisopropil-N"-etilamina
DMSO dimetilsulfóxido
EtOH etanol
equiv. equivalentes
ESI ionización por electronebulización
h hora(s)
HCI hi drogen ocl oruro
HPLC Cromatografía líquida de alto rendimiento
LiOH hidróxido de litio
I litro(s)
m multiplete
MeOH metanol
MgSO4 sulfato de magnesio
ml mililitro(s)
NaHCO3 bicarbonato de sodio
Na2S2O4 ditionito de sodio
NaOH hidróxido de sodio
Na2SO4 sulfato de sodio
RMN Resonancia magnética nuclear
K2CO3 carbonato de potasio
i-PrOH 2-propanol, iso-propanol
c cuadruplete
RP-HPLC Cromatografía líquida de alto rendimiento de fase inversa
s singlete
sep septuplete
SQD MS Espectrómetro de masas con detector de cuadrupolo simple
t triplete
TFA ácido trifluoroacético
TFAA anhídrido trifluoroacético
THF tetrahidrofurano
TLC Cromatografía de capa fina
UPLCMS Espectrometría de masas-Cromatografía líquida de ultra rendimiento
Se realizaron TLC con gel de sílice 60 F254 sobre láminas de aluminio (Sigma-Aldrich, Merck) usando sistemas de disolventes apropiados. La visualización se realizó generalmente con luz ultravioleta (254 nm).
Método UPLC-MS:
Método A:
Los análisis UPLCMS se realizaron en un cromatógrafo de líquidos UPLC equipado con un detector PDA y un detector SQD MS, operando bajo ESI(+) o ESI(-) usando una columna C18, de 2,1 mm x 100 mm, 1,7 μm (AQUlTY UPLC BEH o equivalente). Se usaron metanol de calidad HPLC o LC/MS, agua de calidad HPLC, ácido fórmico de calidad HPLC o LC/MS, solución al 25 % de amoniaco de calidad p.a. y una mezcla de los mismos como fase móvil. Las condiciones operativas fueron las siguientes: flujo de la fase móvil 0,45 ml/min, longitud de onda 210-400 nm, volumen de inyección 1 μl, temperatura de la columna 60 °C, temperatura del automuestreador 5 °C.
El análisis se realizó durante 3,3 min 0,5 min para “ el retraso de la siguiente inyección” . Gradiente de elución con un curso lineal:
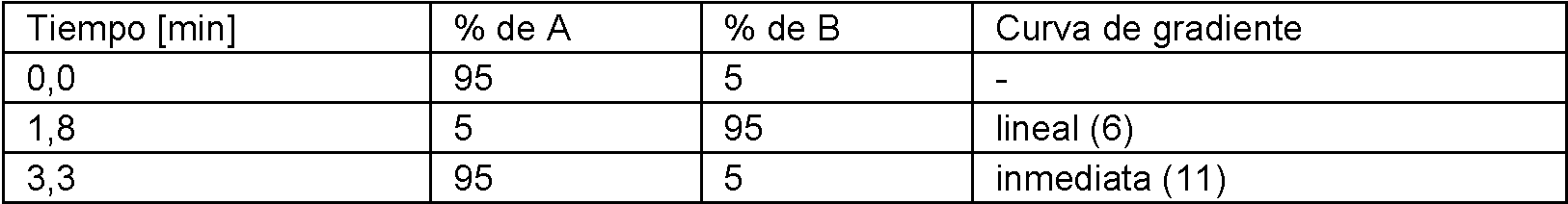
El análisis se realizó durante 5,5 min 1,5 min para “ el retraso de la siguiente inyección” . Gradiente de elución con un curso lineal:
Tiempo [min] % de A % de B Curva de gradiente

Las soluciones se prepararon de la siguiente manera:
Preparación de la fase móvil A1 - gradiente básico: Se añadieron 25 μl de ácido fórmico y 250 μl de solución al 25 % de amoniaco a 250 ml de agua. Se desgasificó usando un baño de ultrasonido durante 10 min.
Preparación de la fase móvil A2 - gradiente ácido: Se añadieron 50 μl de ácido fórmico a 250 ml de agua. Se desgasificó usando un baño de ultrasonido durante 10 min.
Fase móvil B: Súper gradiente de metanol.
Método B:
Los análisis UPLC-MS o UPLC-MS/MS se realizaron en un sistema UPLC-MS/MS que comprendía Waters ACQUITY UPLC (Waters Corporation, Milford, MA, EE.UU.) acoplado con un espectrómetro de masas Waters TQD (modo de ionización por electronebulización ESI con cuadrupolo en tándem). Las separaciones cromatográficas se realizaron usando la columna Acquity UPLC BEH (híbrido etílico en puente) C18: 2,1 mm x 100 mm y 1,7 μm de tamaño de partícula. La columna se mantuvo a 40 °C y se eluyó en condiciones de gradiente usando del 95 % al 0 % de eluyente A durante 10 min, a un caudal de 0,3 ml/min. Eluyente A, agua/ácido fórmico (0,1 %, v/v); eluyente B, acetonitrilo/ácido fórmico (0,1 %, v/v). Se inyectaron un total de 10 μl de cada muestra y se registraron los cromatogramas usando un detector Waters eA PDA. Los espectros se analizaron en el intervalo de 200-700 nm con una resolución de 1,2 nm y a una velocidad de muestreo de 20 puntos/s. Los ajustes de detección de MS del espectrómetro de masas TQD de Waters fueron los siguientes: temperatura de fuente 150 °C, temperatura de desolvatación 350 °C, caudal de gas de desolvatación 600 l/h, flujo de gas de cono 100 l/h, potencial capilar 3,00 kV y potencial de cono 20 V. Se usó nitrógeno tanto para la nebulización como para el secado. Los datos se obtuvieron en un modo de barrido que variaba de 50 a 1000 m/z a intervalos de 0,5 s; se sumaron 8 barridos para obtener el espectro final. Se realizaron análisis de disociación activada por colisión (CAD, por sus siglas en inglés) con la energía de 20 eV, y se observaron todas las fragmentaciones en la fuente. En consecuencia, los espectros de iones se obtuvieron en el intervalo de 50 a 500 m/z. Se usó el software MassLynx V 4.1 (Waters) para la adquisición de datos. Se prepararon soluciones estándar (1 mg/ml) de cada compuesto en una mezcla que comprendía acetonitrilo/agua de calidad analítica (1/1, v/v).
Procedimientos sintéticos
A. Compuestos basados en núcleo de bencimidazol:
Compuesto A-2A: 4-(3-fluoro-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 1 l equipado con un agitador mecánico se añadió 2,6-difluoronitrobenceno (16,5 g, 104 mmol) y el matraz se llenó con DMSO (170 ml). A continuación, se añadieron K2CO3 seco (31,6 g, 229 mmol) y N-BOC-piperazina (21,2 g, 114 mmol). La mezcla de reacción se calentó a 40 °C y se agitó durante 2,5 h a esta temperatura. La reacción se vertió en agua (400 ml) y se diluyó con DCM (500 ml). Las fases se separaron y la fase orgánica se lavó con agua (2 x 150 ml), salmuera (100 ml), se secó en MgSO4 y el disolvente se eliminó al vacío. El residuo sólido se disolvió en MeOH (120 ml) a continuación se añadió gota a gota agua (15 ml) y toda la mezcla se enfrió a 5 °C y se almacenó a esta temperatura durante 2 h. Después de este tiempo, el producto sólido A-2A (21,9 g) se filtró y se lavó con la mezcla de MeOH:agua (10:1,20 ml). El filtrado se redujo a la mitad de su volumen y se almacenó a 5 °C durante 16 h. Una porción adicional del compuesto A-2A (6,3 g) se filtró y se combinó con el sólido obtenido previamente. Como resultado, el producto A-2A se obtuvo en forma del sólido de color amarillo (28,2 g, 83 % de rendimiento) con un 95 % de pureza, según el análisis UPLCMS (Método A). Compuesto A-3A: 4-[3-(bencilamino)-2-nitrofenil]piperazin-1-carboxilato de terc-butilo

En un matraz de 250 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (12 g, 45 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (100 ml). A continuación, se añadieron K2CO3 seco (9,31 g,
67,5 mmol) y bencilamina (5,82 g, 54 mmol), y la mezcla de reacción se calentó a 120 °C y se agitó durante 2 h a esta temperatura. Después de este tiempo, el análisis UPLCMS mostró un 1 % del área pico del sustrato. La reacción se vertió en hielo (aproximadamente 150 g) y se diluyó con AcOEt (300 ml). Las fases se separaron y la fase acuosa se extrajo con AcOEt (2 x 300 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. Como resultado, el producto A-3A se obtuvo en forma del sólido de color amarillo (11,9 g, 84 % de rendimiento) con un 95 % de pureza, según el análisis UPLCMS (Método A) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-4A: 4-[2-amino-3-(bencilamino)fenil]piperazin-1-carboxilato de terc-butilo

En un matraz de 500 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3A (4 g, 9,7 mmol) y EtOH (200 ml) y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (5,06 g, 29,1 mmol) en agua (50 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a la temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (30 ml. Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4 y el disolvente se eliminó al vacío. El producto en bruto A-4A se obtuvo en forma de un aceite de color pardo oscuro (3,01 g) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5A, Compuesto 1: 1-bencil-4-(piperazin-1-il')-2-(trifluorometil')-1H-bencimidazol, en forma de sal clorhidrato

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4A (382 mg, 1 mmol) y TFA (2 ml) y la mezcla de reacción se calentó a 80 °C. La mezcla de reacción se agitó durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con DCM (50 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 20 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4 y el disolvente se eliminó al vacío. El residuo se purificó usando cromatografía en columna (10 % al 20 % de MeOH en DCM). Las fracciones con producto se concentraron, se disolvieron de nuevo en 20 ml de i-PrOH y se añadieron 0,5 ml de una solución al 36 % de HCl. El disolvente se eliminó al vacío y el residuo se disolvió en 5 ml de i-PrOH, y a continuación se añadieron 20 ml de EtzO. El producto sólido se filtró y se lavó con EtzO (5 ml). Como resultado, el producto final A-5A , Compuesto 1 en forma de sal clorhidrato, se obtuvo en forma del sólido de color beige (141 mg, 39 % de rendimiento) con un 99,24 % de pureza, según el análisis UPLCMS (Método A).
1H RMN (500 MHz, DMSO-ds) 59,53 (d, J = 6,7 Hz, 2H), 7,37 - 7,25 (m, 4H), 7,19 (d, J = 8,2 Hz, 1H), 7,11 - 7,03 (m, 2H), 6,78 (d, J = 7,8 Hz, 1H), 5,67 (s, 2H), 3,80 (dd, J = 6,6, 3,8 Hz, 4H), 3,30 (m, J = 4,9 Hz, 4H).
13C RMN (125 MHz, DMSO-ds) 5 143,20, 137,63, 137,46, 137,31 (c, J = 38,2 Hz), 132,61, 129,22, 128,20, 127,05, 126,51, 119,40 (c, J = 271,1 Hz), 108,98, 104,74, 48,12, 46,39, 42,92.
Compuesto A-3B: N-bencil-3-(4-metilpiperazin-1-il)-2-nitroanilina

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadió 2,6-difluoronitrobenceno A-1 (5 g, 31 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (50 ml). A continuación, se añadieron K2CO3 seco (8,5 g, 62 mmol) y 1-metilpiperazina (3,3 g, 33 mmol). La mezcla de reacción se calentó a 30 °C y se agitó durante 16 h. Después de este tiempo, el análisis UPLCMS no mostró ningún pico de sustrato. Se añadió otra porción de K2CO3 (5,1 g, 37 mmol) a la mezcla de reacción seguido de bencilamina (3,96 g, 37 mmol). La mezcla de reacción se calentó a 70 °C y se agitó durante 16 h. Después de este tiempo, el análisis UPLCMS mostró un 70 % de conversión del
compuesto A-2B. Se añadió una porción más de K2CO3 (3 g, 22 mmol) y la agitación continuó durante una noche a 70 °C. Después de este tiempo, el análisis UPLCMS no mostró el compuesto A-2B en la mezcla de reacción. La reacción se vertió en hielo (aproximadamente 400 g), donde el producto comenzó a cristalizar. El sólido se filtró y se aclaró con agua. Dicho compuesto húmedo en bruto obtenido A-3B se usó en la siguiente etapa sin purificación adicional.
Compuesto A-4B: N1-bencil-3-(4-metilpiperazin-1-il)benceno-1,2-diamina

En un matraz de 1 l equipado con una barra de agitación magnética, se añadieron el compuesto húmedo A-3B de la etapa anterior y EtOH (500 ml) y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en 5 minutos una solución recién preparada de ditionito de sodio (16,2 g, 93 mmol) en agua (100 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se eliminó al vacío el EtOH y se añadió AcOEt (200 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4 y el disolvente se eliminó al vacío. El producto en bruto A-4B se obtuvo en forma de un aceite de color pardo oscuro (4,2 g) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5B, Compuesto 2: 1-bencil-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-bencimidazol, en forma de sal clorhidrato

En un matraz de 90 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4B (600 mg, 2 mmol) y TFA (5,8 g, 51 mmol) en una atmósfera de argón y una mezcla de reacción se calentó a 80 °C durante 2 h. El exceso de ácido se eliminó al vacío. El residuo se disolvió en 15 ml de IPA seco y se añadieron 5 ml de una solución al 36 % de HCl. Después de 1 hora de agitación, la mezcla se evaporó a sequedad. El residuo se calentó a reflujo con 5 ml de dioxano y algunas gotas de IPA durante 30 min. La solución se enfrió a 3 °C durante una noche sin agitación. A continuación, el sólido se filtró, se lavó con dioxano y se secó en un secador al vacío. Como resultado, el producto final A-5B , Compuesto 2 en forma de sal clorhidrato, se obtuvo en forma del sólido de color beige (100 mg, 12 % de rendimiento) con un 98,09 % de pureza, según el análisis UPLCMS (Método A).
1H RMN (500 MHz, DMSO-cfe) 5 11,41 (s, 1H), 7,39 - 7,25 (m, 4H), 7,21 (d, J = 8,2 Hz, 1H), 7,11 - 7,03 (m, 2H), 6,79 (d, J = 7,8 Hz, 1H), 5,68 (s, 2H), 4,55 - 4,24 (m, 2H), 3,65 - 3,44 (m, 2H), 3,46 - 3,19 (m, 4H), 2,82 (s, 3H). 13C RMN (125 MHz, DMSO-cfe) 5 142,82, 137,61, 137,36 (c, J = 37,9 Hz), 136,38, 132,63, 129,49, 129,22, 128,20, 127,04, 126,52, 120,48, 118,32, 109,14, 104,83, 52,48, 48,13, 46,51,42,49, 25,92.
Compuesto A-2C: Acetato de 2-[4-(3-fluoro-2-nitrofenil)piperazin-1-il]etilo

En un matraz de 250 ml equipado con una barra de agitación magnética, se añadió 1-(3-fluoro-2-nitrofenil)piperazina A-6 (5 g, 22,5 mmol) en una atmósfera de argón y el matraz se llenó con ACN seco (50 ml). A continuación, se añadieron K2CO3 seco (6,0 g, 45 mmol) y acetato de 2-bromoetilo (4,45 g, 26,6 mmol), y la mezcla de reacción se calentó a 60 °C y se agitó durante 20 h a esta temperatura. Después de este tiempo, el análisis UPLCMS no mostró ningún pico de sustrato. La reacción se enfrió a temperatura ambiente y el sólido se filtró. Como resultado, el producto A-2C se obtuvo en forma del sólido de color amarillo (6,8 g, 99 % de rendimiento) con un 99 % de pureza, según el análisis UPLCMS (Método A). Compuesto A-3C: Acetato de 2-{4-[3-(bencilamino)-2-nitrofenil]piperazin-1-il}-etilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2C (6,6 g, 22 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (60 ml). A continuación, se añadieron K2CO3 seco (6,07 g, 44 mmol) y bencilamina (2,59 g, 24,2 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 20 h a esta temperatura. Después de este tiempo, la reacción se vertió en hielo (aproximadamente 60 g) y se diluyó con AcOEt (300 ml). Las fases se separaron y la fase acuosa se extrajo con AcOEt (2 x 300 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. Como resultado, el producto A-3C se obtuvo en forma del sólido de color amarillo (6,3 g, 72 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5C, Compuesto 3: 2-{4-[1-bencil-2-(trifluorometil)-1H-bencimidazol-4-il]piperazin-1-il}etanol, en forma de sal clorhidrato

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió TFA (10 ml) y la mezcla se calentó a 70 °C. A continuación, se añadieron hierro metálico (1,12 g, 20 mmol) y el compuesto A-3C (2,0 g, 5 mmol). La mezcla de reacción se agitó durante 2 horas a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente y se diluyó con DCM (100 ml). Se añadió gota a gota una solución 2 M de NaHCO3 para lograr un pH de ~8 y a continuación las fases se separaron. La fase acuosa se extrajo con DCM (2 x 100 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4, y el disolvente se eliminó al vacío. El residuo se disolvió en 50 ml de THF y a continuación se añadieron 5 ml de agua y 1 g de LiOH. La mezcla de reacción se agitó durante 20 horas a temperatura ambiente. Después de este tiempo, el disolvente orgánico se eliminó al vacío y a la mezcla se le añadieron 100 ml de AcOEt. Las fases se separaron y la fase acuosa se extrajo con AcOEt (2 x 100 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. El residuo se purificó usando cromatografía en columna (10 % al 20 % de MeOH en DCM). Después de la eliminación de los disolventes, el residuo se disolvió en 20 ml de /-PrOH y se añadieron 0,5 ml de una solución al 36 % de HCl. Los disolventes se eliminaron al vacío y el residuo se disolvió de nuevo en 5 ml de /-PrOH y a continuación se añadieron 20 ml de EtzO. El producto sólido se filtró y se lavó con EtzO. Como resultado, el compuesto final A-5C, Compuesto 3 en forma de sal clorhidrato, se obtuvo en forma del sólido de color beige (81 mg, 3,7 % de rendimiento) con un 97 % de pureza, según el análisis UPLCMS (Método A).
1H RMN (500 MHz, DMSO-cfe) 5 10,78 (s, 1H), 7,37 - 7,25 (m, 4H), 7,20 (d, J = 8,2 Hz, 1H), 7,10 - 7,04 (m, 2H), 6,78 (d, J = 7,8 Hz, 1H), 5,67 (s, 2H), 4,39 (d, J = 11,9 Hz, 2H), 3,85 (dd, J = 6,2, 4,3 Hz, 2H), 3,67 (d, J = 11,2 Hz, 2H), 3,37 (m, 4H), 3,26 (c, J = 5,2 Hz, 2H).
13C RMN (125 MHz, DMSO-ds) 5 142,82, 137,62, 137,34 (c, J = 37,4), 136,39, 132,59, 129,23, 128,22, 127,05, 126,52, 119,40 (c, J = 270,7 Hz), 109,04, 104,82, 58,30, 55,45, 51,56, 48,12, 46,32.
Compuesto A-3D: 4-(3-(furan-2-ilmetilamino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,63 g, 5 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (6 ml). A continuación, se añadieron K2CO3 seco (2,07 g, 15 mmol) y furfurilamina (6,5 mmol, 631 mg), y la mezcla de reacción se calentó a 80 °C y se agitó durante 16 h a esta temperatura. Después de este tiempo, el análisis UPLCMS mostró un 5 % del área pico del sustrato A-2A. La reacción se vertió en hielo (aproximadamente 50 g) y se diluyó con AcOEt (30 ml). Las fases se separaron y la fase acuosa se extrajo con AcOEt (2 x 30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. Un residuo sólido se disolvió en una pequeña cantidad de MeOH con calentamiento suave y a continuación se almacenó a 5 °C durante una noche. Dicho sólido obtenido se filtró, se aclaró con MeOH frío (5 ml) y se secó a alto vacío.
El filtrado se concentró al vacío, se adsorbió previamente sobre gel de sílice y se purificó usando cromatografía en columna de gravedad (10 % de AcOEt en n-hexano). Después de la eliminación de los disolventes, el producto se combinó con el sólido obtenido previamente. Como resultado, el producto A-3D se obtuvo en forma del sólido de color rojo-pardo (1,29 g, 64 % de rendimiento) con un 98 % de pureza, según el análisis UPLCMS.
Compuesto A-4D: 4-(2-amino-3-(furan-2-ilmetilamino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3D (850 mg, 2,1 mmol) y EtOH (20 ml), y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,83 g, 10,5 mmol) en agua (12 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a la temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (30 ml. Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4 y el disolvente se eliminó al vacío. El producto en bruto A-4D se obtuvo en forma de un aceite de color pardo oscuro (705 mg) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-8A: 4-(1-(furan-2-ilmetil)-2-(trifluorometil)-1H-bencimidazol-4-il)piperazin-1-carboxilato de terc-butilo

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4D (100 mg, 0,27 mmol) y ACN seco en una atmósfera de argón y una mezcla de reacción se enfrió a 0 °C. A continuación, se añadió DiPEa (243 mg, 1,88 mmol) seguido de la adición gota a gota (0,5 h) de una solución recién preparada de TFAA (216 mg, 1,03 mmol) en Ac N (1 ml). La mezcla de reacción se agitó a temperatura ambiente durante 16 h. Después de este tiempo, el análisis UPLCMS mostró un 15 % del área pico del producto de A-8A y un 35 % del área pico del producto no ciclado A-7A. La mezcla de reacción se diluyó con DCM y agua y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 20 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. El residuo se adsorbió previamente sobre gel de sílice y se purificó usando cromatografía en columna (10 % al 15 % de AcOEt en n-hexano). Después de la eliminación de los disolventes, se obtuvieron dos fracciones. La primera fracción, el producto A-8A esperado, se obtuvo en forma de un aceite incoloro (107 mg, 44 % de rendimiento) con un 99 % de pureza según el análisis UPLCMS (Método A). La segunda fracción (100 mg) era la mezcla (1:1) del producto A-8a esperado y el producto no ciclado A-7A. Es posible convertir cuantitativamente esta mezcla en el compuesto puro A-8A usando AcOH en EtOH a reflujo.
Compuesto A-5D, Compuesto 4: 1-(furan-2-ilmetil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol

En un matraz de 25 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-8A (100 mg, 0,22 mmol) y EtOH (2 ml) seguido de una solución al 36 % de HCl (0,5 ml) y la mezcla de reacción se agitó durante 40 h. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se diluyó con EtOH (5 ml), se enfrió a aproximadamente 5 °C y se añadió gota a gota una solución al 25 % de NH4OH (0,5 ml). A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 20 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en Na2SO4 y el disolvente
se eliminó al vacío. Como resultado, el producto final A-5D, Compuesto 4 , se obtuvo en forma del sólido de color pardo claro (59 mg, 76 % de rendimiento) con un 97,34 % de pureza, según el análisis UPLCMS (Método A).
1H RMN (500 MHz, DMSO-cfe) 57,59 (d, J = 1,6 Hz, 1H), 7,34 - 7,26 (m, 2H), 6,66 (ddd, J = 10,6, 5,6, 3,2 Hz, 1H), 6,55 (d, J = 3,2 Hz, 1H), 6,42 (dd, J = 3,3, 1,8 Hz, 1H), 5,60 (s, 2H), 3,42 (m, 4H), 2,90 (m, 4H).
13C RMN (125 MHz, DMSO-ds) 5 149,34, 145,53, 144,41, 137,69, 136,82 (c, J = 38,2 Hz), 133,01, 127,36, 119,94 (c, J = 271,5 Hz), 111,59, 110,35, 108,70, 104,01, 51,13, 46,42, 41,97.
Compuesto A-3E: 4-(3-((5-metilfuran-2-il)metilamino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (650 mg, 2 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (4 ml). A continuación, se añadieron K2CO3 seco (691 mg, 5 mmol) y 5-metilfurfurilamina (2,6 mmol, 289 mg), y la mezcla de reacción se calentó a 80 °C y se agitó durante 16 h. Después de esto, la mezcla de reacción se vertió en agua (aproximadamente 50 ml) y se diluyó con DCM (20 ml). Las fases se separaron y la fase acuosa se extrajo con d CM (2 x 20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El residuo se adsorbió previamente sobre gel de sílice y se purificó usando cromatografía en columna (10 % de AcOEt en nhexano). Como resultado, el producto final A-3E se obtuvo en forma del sólido de color rojo-pardo (450 mg, 54 % de rendimiento) con un 95 % de pureza, según el análisis UPLCMS (Método A).
Compuesto A-4E: 4-(2-amino-3-((5-metilfuran-2-il)metilamino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3E (492 mg, 1,18 mmol) y EtOH (17 ml) y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en una porción una solución recién preparada de ditionito de sodio (1,21 g, 5,9 mmol) en agua (4,3 ml). La mezcla de reacción se agitó durante 10 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se añadieron agua (20 ml) y AcOEt (30 ml) y las fases se separaron. La fase acuosa se extrajo con AcOEt (2 x 30 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4E se obtuvo en forma de un aceite de color pardo oscuro (402 mg) y se usó en la siguiente etapa sin purificación adicional (Método A).
Compuesto A-8B: 4-(1-((5-metilfuran-2-il)metil)-2-(trifluorometil)-1H-bencimidazol-4-il)piperazin-1-carboxilato de terc-butilo

En un matraz de 25 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4E (216 mg, 0,56 mmol) y ACN seco en una atmósfera de argón. A continuación, se añadió DIPEA (145 mg, 1,12 mmol) seguido de la adición gota a gota (20 min) de TFAA (130 mg, 0,62 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 16 h. Después de este tiempo, el análisis UPLCMS mostró un 15 % del área pico del producto A-8B y un 35 % del área pico del producto no ciclado A-7B. La mezcla de reacción se vertió en una solución saturada de NaHCO3 (20 ml), se diluyó con 30 ml de DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 20 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El residuo se disolvió en EtOH (6 ml) y se añadieron 0,5 ml de AcOH. A continuación, la mezcla se calentó a 80 °C y se agitó a esta temperatura durante 2 h. Después de este tiempo, todos los disolventes se eliminaron y el residuo se disolvió en AcOEt (10 ml). La fase orgánica se lavó con una solución saturada de NaHCOs, se secó en MgSO4 y el disolvente se eliminó al vacío. El residuo se adsorbió previamente sobre gel de sílice y se purificó usando
cromatografía en columna (20 % de AcOEt en n-hexano). Como resultado, el producto A-8B se obtuvo en forma del aceite incoloro (115 mg, 45 % de rendimiento) con un 99 % de pureza, según el análisis UPLCMS (Método A). Compuesto A-5E, Compuesto 5: 1-((5-metilfuran-2-il)metil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol

En un matraz de 25 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-8B (115 mg, 0,25 mmol) y EtOH (7 ml) seguido de una solución al 36 % de HCl (1,5 ml) y la mezcla de reacción se agitó durante 24 h. Después de este tiempo, se añadió otra porción de HCl concentrado (0,7 ml) y la mezcla de reacción se agitó durante 24 h más. La mezcla de reacción se diluyó con agua (10 ml), se enfrió a aproximadamente 5 °C y se añadió gota a gota una solución al 25 % de NH4OH (2 ml). A continuación, se añadió DCM (30 ml) y las fases se separaron. La fase acuosa se extrajo una vez más con DCM (30 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El residuo se adsorbió previamente sobre gel de sílice y se purificó usando cromatografía en columna (92:8:0,5 de DCM:MeOH:NH4OH). Como resultado, el producto final A-5E, Compuesto 5 , se obtuvo en forma del sólido de color pardo claro (60 mg, 66 % de rendimiento) con un 96 % de pureza, según el análisis UPLCMS (Método A).
1H RMN (500 MHz, CDCls) 57,29 (t, J = 8,0 Hz, 1H), 7,11 (d, J = 8,0 Hz, 1H), 6,66 (d, J = 8,0 Hz, 1H), 6,16 (m, 1H), 5,87 (m, 1H), 5,34 (s, 2H), 3,55 (m, 4H), 3,15 (m, 4H), 2,21 (s, 3H).
13C RMN (125 MHz, CDCls) 5 152,76, 146,23, 145,16, 137,33 (c, J = 38,9 Hz), 136,98, 133,23, 126,21, 119,17 (c, J = 271,1 Hz), 109,91, 108,16, 106,45, 103,11,51,00, 46,08, 41,60, 13,47.
Compuesto A-3F: 4-(3-((3-clorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,20 g, 3,69 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (0,97 g, 7,01 mmol) y 3-clorobencilamina (0,84 g, 5,91 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 48 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtOH (99,9 %, 10 ml) proporcionando el producto A-3F en forma de un sólido de color amarillo (0,77 g, 47 % de rendimiento) con un 100 % de pureza, según el análisis UPLCMS (Método B).
Compuesto A-4F: 4-(2-amino-3-((3-clorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3F (0,75 g, 1,68 mmol) y EtOH (28 ml) y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,31 g, 7,55 mmol) en agua (9 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (20 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4F se obtuvo en forma de un aceite cristalizante de color beige (0,63 g) y se usó en la siguiente etapa sin purificación adicional. Compuesto A-5F, Compuesto 6: 1-(3-clorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol, en forma de sal clorhidrato

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4F (0,31 g, 0,74 mmol) y TFA (1,48 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente y se diluyó con DCM (40 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 15 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto se disolvió de nuevo en 37 ml de i-PrOH y se añadieron 0,3 ml de una solución al 36 % de HCl. Los disolventes se eliminaron al vacío y el residuo se disolvió en 5 ml de i-PrOH y a continuación se añadieron 20 ml de EtzO. El producto sólido se filtró y se lavó con Et2O (5 ml). Como resultado, el producto final A-5F, Compuesto 6 en forma de sal clorhidrato, se obtuvo en forma de un sólido de color beige (104 mg, 33 % de rendimiento) con un 100 % de pureza, según el análisis UPLCMS (Método B).
1H RMN (300 MHz, DMSO-cfe) 59,41 (s a, 2H), 7,38 - 7,26 (m, 3H), 7,23 - 7,14 (m, 2H), 6,91 (d, J = 3,1 Hz, 1H), 6,77 (d, J = 7,7 Hz, 1H), 5,68 (s, 2H), 3,77 (s a, 4H), 3,28 (s a, 4H)
13C RMN (75 MHz, DMSO-cfe) 5 143,2, 139,0, 137,6, 137,0 (c, J = 2 Hz), 133,8, 132,5, 131,2, 128,2, 127,2, 126,5, 125,0, 119,3 (c, J = 271 Hz), 109,1, 104,6, 47,4, 46,4, 43,0
Compuesto A-3G: 4-(3-((3-fluorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,20 g, 3,69 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (0,97 g, 7,01 mmol) y 3-fluorobencilamina (0,74 g, 5,91 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 48 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtOH (99,9 %, 10 ml) proporcionando el producto A-3G en forma de un sólido de color amarillo (0,73 g, 46 % de rendimiento) con un 100 % de pureza, según el análisis UPLCMS (Método B).
Compuesto A-4G: 4-(2-amino-3-((3-fluorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3G (0,70 g, 1,63 mmol) y EtOH (27 ml) y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,27 g, 7,32 mmol) en agua (8 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (20 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4G se obtuvo en forma de un aceite de color beige pálido (0,60 g) y se usó en la siguiente etapa sin purificación adicional. Compuesto A-5G, Compuesto 7: 1-(3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-bencimidazol, en forma de sal clorhidrato

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4G (0,30 g, 0,75 mmol) y TFA (1,5 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente y se diluyó con DCM (40 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 15 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto se disolvió de nuevo en 37 ml de i-PrOH y se añadieron 0,3 ml de una solución al 36 % de HCl. Los disolventes se eliminaron al vacío y el residuo se disolvió en 5 ml de i-PrOH y a continuación se añadieron 20 ml de EtzO. El producto sólido se filtró y se lavó con EtzO (5 ml). Como resultado, el producto final A-5G, Compuesto 7 en forma de sal clorhidrato, se obtuvo en forma de un sólido de color beige (100 mg, 32 % de rendimiento) con un 98,84 % de pureza, según el análisis UPLCMS (Método B).
1H RMN (300 MHz, DMSO-cfe) 59,42 (s a, 2H), 7,39-7,27 (m, 2H), 7,18 (d, J = 8,0 Hz, 1H), 7,10 (dt, J = 2,6, 8,3 Hz, 1H), 6,93 (d, J = 10,0 Hz, 1H), 6,82-6,74 (m, 2H), 5,68 (s, 2H), 3,81-3,74 (m, 4H), 3,28 (s a, 4H)
13C RMN (75 MHz, DMSO-cfe) 5162,6 (d, J = 244 Hz), 143,2, 139,3 (d, J = 7,2 Hz), 137,6 (c, J = 2 Hz), 137,0, 132,6, 131,4 (d, J = 8,3 Hz), 127,2, 122,4 (d, J = 2,8 Hz), 119,3 (d, J = 271 Hz), 115,1 (d, J = 21 Hz), 113,6 (d, J = 22,5 Hz), 109,1, 104,6, 47,5, 46,4, 43,0
Compuesto A-3H: 4-(3-((3,4-diclorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,10 g, 3,38 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (0,7 g, 5,07 mmol) y 3,4-diclorobencilamina (0,65 g, 3,72 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 24 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtOH (99,9 %, 10 ml) proporcionando el producto A-3H en forma de un sólido de color rojo (0,7 g, 43 % de rendimiento) con un 94,4 % de pureza, según el análisis UPLCMS (Método B). Compuesto A-4H: 4-(2-amino-3-((3,4-diclorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3H (0,7 g, 1,54 mmol) y EtOH (22 ml), y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,08 g, 6,2 mmol) en agua (7 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (15 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (15 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4H se obtuvo en forma de un aceite de color amarillento (0,285 g) y se usó en la siguiente etapa sin purificación adicional. Compuesto A-5H, Compuesto 8: 1-(3,4-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4H (0,14 g, 0,33 mmol) y TFA (0,7 ml) y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con DCM (20 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 10 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto obtenido se purificó usando cromatografía en columna (n-hexano/DCM/metanol/NH3(ac.), 4,0/5,0/1,0/0,02, v/v/v/v) proporcionando el producto final A-5H, Compuesto 8 en forma de un aceite cristalizante de color amarillo pálido (120 mg, 85 % de rendimiento) con un 95,75 % de pureza, según el análisis UPLCMS (Método B). 1H RMN (300 MHz, DMSO-cfe) 57,60 - 7,49 (m, 2 H), 7,44 - 7,23 (m, 4 H), 7,17 - 7,06 (m, 2 H), 6,97 - 6,84 (m, 2 H), 6,72 - 6,63 (m, 2 H), 5,66 (s, 2 H), 3,57 - 3,50 (m, 4 H), 3,08 - 2,99 (m, 4 H)
13C RMN (75 MHz, CDsOD) 5 166,1, 160,9, 145,1, 140,3, 136,5 (c, J = 1,7 Hz), 136,7, 132,6, 126,7, 124,3, 120,2, 116,9 (c, J = 271 Hz), 108,7, 108,1, 102,8, 50,1,44,9, 43,8
Compuesto A-3I: 4-(3-((3-cloro-4-fluorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,1 g, 3,38 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (1,16 g, 8,45 mmol) y 3-cloro-4-fluorobencilamina (0,85 g, 5,4 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 24 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtOH (99,9 %, 10 ml) proporcionando el producto A-3I en forma de un sólido de color amarillento (0,5 g, 32 % de rendimiento) con un 93 % de pureza, según el análisis UPLCMS (Método B). Compuesto A-4I: 4-(2-amino-3-((3-cloro-4-fluorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3I (0,5 g, 1,07 mmol) y EtOH (15 ml), y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (0,75 g, 4,37 mmol) en agua (5 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a la temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (20 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4I se obtuvo en forma de un aceite de color amarillento (0,452 g) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5I, Compuesto 9: 1-(3-cloro-4-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4I (0,240 g, 0,55 mmol) y TFA (1,2 ml), y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con DCM (20 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 10 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto obtenido se purificó usando cromatografía en columna (n-hexano/DCM/metanol/NH3(ac.), 4,0/5,0/1,0/0,02, v/v/v/v) proporcionando el producto final A-5I, Compuesto 9 en forma de un aceite cristalizante de color amarillo (200 mg, 90 % de rendimiento) con un 97,50 % de pureza, según el análisis UPLCMS (Método B).
1H RMN (300 MHz, DMSO-cfe) 57,22 - 7,43 (m, 3H), 7,09 (d, J = 7,69 Hz, 1H), 6,97 (ddd, J = 2,18, 4,68, 8,53 Hz, 1H), 6,67 (d, J = 7,69 Hz, 1 H), 5,64 (s, 2H), 3,43 - 3,50 (m, 4H), 2,87 - 2,94 (m, 4H), protón NH no detectado
13C RMN (75 MHz, DMSO-cfe) 5 158,7, 155,3 (d, J = 271 Hz), 145,1, 136,5 (c, J = 1,7 Hz), 134,5 (d, J = 3,6 Hz) 132,6, 128,9, 127,2 (d, J = 7,6 Hz), 121,2, 120,6 (c, J = 271 Hz),117,9, 117,6, 108,4, 103,1, 50,5, 46,9, 45,9
Compuesto A-3J: 4-(3-((3,4-difluorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (1,10 g, 3,38 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (0,7 g, 5,07 mmol) y 3,4-difluorobencilamina (0,64 g, 3,72 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 24 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtOH (99,9 %, 10 ml) proporcionando el producto A-3J en forma de un sólido de color amarillento (0,8 g, 53 % de rendimiento) con un 94,4 % de pureza, según el análisis UPLCMS (Método B).
Compuesto A-4J: 4-(2-amino-3-((3,4-difluorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3J (0,8 g, 1,78 mmol) y EtOH (25 ml), y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,24 g, 7,14 mmol) en agua (8 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (15 ml).
Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (15 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4J se obtuvo en forma de un aceite de color amarillento (0,400 g) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5J, Compuesto 10: 1-(3,4-difluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4J (0,396 g, 1,0 mmol) y TFA (2,0 ml), y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLc Ms mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con DCM (30 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 20 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto obtenido se purificó usando cromatografía en columna (n-hexano/DCM/metanol/NH3(ac.),
4,0/5,0/1,0/0,02, v/v/v/v) proporcionando el producto final A-5J, Compuesto 10 en forma de un aceite cristalizante de color amarillo (350 mg, 88 % de rendimiento) con un 95,01 % de pureza, según el análisis UPLCMS (Método B).
1H RMN (300 MHz, DMSO-cfe) 57,44 - 7,17 (m, 3 H), 7,17 - 7,09 (m, 1 H), 6,88 - 6,77 (m, 1 H), 6,71 (d, J = 7,7 Hz, 1 H), 5,64 (s, 2 H), 3,65 - 3,51 (m, 4 H), 3,16 - 3,03 (m, 4 H), protones NH no detectados
13C RMN (75 MHz, DMSO-ds) 5151,43 (dd, J = 248 Hz y 12,7 Hz), 148,16 (dd, J = 248 Hz y 12,8 Hz), 144,30, 137,4, 136,8 (c, J = 38,2 Hz), 134,19 (dd, J = 5,7, 3,6 Hz), 132,5, 127,2, 123,33 (c, J = 3,4 Hz), 119,7 (c, J = 271 Hz), 118,38 (d, J = 17,5 Hz), 116,10 (d, J = 18,0 Hz), 108,7, 103,8, 79,6, 48,8, 47,0, 44,7
Compuesto A-3K: 4-(3-((3,5-diclorobencil)amino)-2-nitrofenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 50 ml equipado con una barra de agitación magnética, se añadió el compuesto A-2A (0,9 g, 2,76 mmol) en una atmósfera de argón y el matraz se llenó con DMSO seco (5 ml). A continuación, se añadieron K2CO3 seco (0,95 g, 6,9 mmol) y 3,5-diclorofluorobencilamina (0,78 g, 4,43 mmol), y la mezcla de reacción se calentó a 70 °C y se agitó durante 24 h a esta temperatura. Después de este tiempo, la mezcla de reacción se enfrió a temperatura ambiente, se vertió en una solución fría de salmuera (75 ml) y se diluyó con agua (75 ml). El precipitado obtenido se eliminó por filtración, se lavó con agua, se secó al aire y se cristalizó en EtoH (99,9 %, 10 ml) proporcionando el producto A-3K en forma de un sólido de color amarillento (0,97 g, 78 % de rendimiento) con un 95 % de pureza, según el análisis UPLCMS (Método B).
Compuesto A-4K: 4-(2-amino-3-((3,5-diclorobencil)amino)fenil)piperazin-1-carboxilato de terc-butilo

En un matraz de 100 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-3K (0,97 g, 2,16 mmol) y EtOH (30 ml), y la mezcla de reacción se calentó a 80 °C. A continuación, se añadió en un minuto una solución recién preparada de ditionito de sodio (1,5 g, 8,66 mmol) en agua (8 ml). La mezcla de reacción se agitó durante 15 minutos más a 80 °C y a continuación se enfrió a la temperatura ambiente. Se eliminó el EtOH y se añadió AcOEt (20 ml). Las fases se separaron y la fase acuosa se extrajo una vez más con AcOEt (20 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto A-4K se obtuvo en forma de un aceite de color amarillento (0,890 g) y se usó en la siguiente etapa sin purificación adicional.
Compuesto A-5K, Compuesto 11: 1-(3,5-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol

En un matraz de 10 ml equipado con una barra de agitación magnética, se añadieron el compuesto A-4K (0,225 g, 0,538 mmol) y TFA (1,0 ml), y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. Después de este tiempo, el análisis UPLCMS mostró el consumo total del sustrato. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con DCM (20 ml) y se añadió gota a gota una solución saturada de NaHCO3 para lograr un pH de ~8. A continuación, se añadieron agua y DCM y las fases se separaron. La fase acuosa se extrajo con DCM (2 x 10 ml) y las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron en MgSO4 y el disolvente se eliminó al vacío. El producto en bruto obtenido se purificó usando cromatografía en columna (n-hexano/DCM/metanol/NH3(ac.), 4,0/5,0/1,0/0,02, v/v/v/v) proporcionando el producto final A-5K, Compuesto 11 en forma de un aceite cristalizante de color amarillo (180 mg, 78 % de rendimiento) con un 95 % de pureza, según el análisis UPLCMS (Método B).
1H RMN (300 MHz, CD3OD) 57,33 - 7,25 (m, 1 H), 6,97 (dd, J = 0,6, 8,3 Hz, 1 H), 6,89 - 6,80 (m, 1 H), 6,77 - 6,72 (m, 1 H), 6,62 (dd, J = 2,2, 8,1 Hz, 2 H), 5,60 (s, 2 H), 3,56 - 3,49 (m, 5 H), 3,08 (dd, J = 4,1,5,9 Hz, 5 H), protones NH no detectados
13C RMN (75 MHz, CD3OD) 5 145,4, 137,3, 136,0 (c, J = 1,7 Hz), 133,6, 132,4, 131,1, 129,1, 129,0, 127,1, 126,6, 126,3, 119,0 (c, J = 271 Hz), 108,2, 102,1, 51,7, 46,3, 45,1
B. Compuestos a base de un núcleo indol:
Compuesto B-2: N-(3-bromo-2-met¡lfenil)-2,2,2-trifluoroacetam¡da

En un matraz de 250 ml equipado con una barra de agitación magnética y cargado con 150 ml de DCM se añadieron 12,1 g (65 mmol) de 3-bromo 2-metilanilina. La mezcla de reacción se enfrió a 0 °C y se añadieron 16 ml (200 mmol) de piridina seguido de adición por goteo de 23 ml (165 mmol) de anhídrido trifluoroacético a esta temperatura. Después de la adición, la reacción se agitó durante 0,5 h a una temperatura <5 °C y durante 2,5 h a TA. Después de este tiempo, la reacción se interrumpió con 50 ml de NH4Clsat. y se diluyó con 50 ml de agua. Las fases se separaron y la fase acuosa se extrajo con DCM (2 x 100 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. Como resultado, el producto se obtuvo en forma del sólido de color blanco (14,7 g, 80 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional.
Compuesto B-3: N-[3-bromo-2-(bromometil)fenil]2,2,2-trifluoroacetamida

Un matraz de 250 ml equipado con una barra de agitación magnética, un condensador e iluminado con una lámpara de 100 W se llenó con 120 ml de CCU; 8,1 g (29 mmol) de B-2 y 0,38 g de peróxido de benzαlo. La mezcla de reacción se calentó a reflujo y se añadieron en pocas porciones mediante una jeringa 2,1 ml de bromo en 10 ml de CCk Después de la adición, la mezcla de reacción se calentó a reflujo durante una noche. Al día siguiente, el análisis por TLC mostró la falta de sustrato. La reacción se enfrió, se diluyó con 120 ml de DCM y se vertió en 100 ml de una solución 2 M de tiosulfato de sodio. Las fases se separaron y la fase acuosa se extrajo con DCM (2 x 60 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera y el disolvente se eliminó al vacío. El residuo se diluyó con una mezcla 1:3 de DCM:hexano, y el producto precipitó en forma de un sólido de color blanco, 9,1 g, 88 % de rendimiento.
Compuesto B-4: 4-bromo-2-(trifluorometil)-1H-indol

Un matraz de 500 ml equipado con una barra de agitación magnética se llenó con 200 ml de tolueno seco y 20,1 g (56 mmol) de sustrato B-3. Posteriormente, se añadieron 15,9 g (61 mmol) de PPH3. A continuación, la mezcla de reacción se calentó a 60 °C y se agitó durante 2 horas. Después de este tiempo, la reacción se enfrió a <5 °C, y el sólido de color blanco se filtró, se lavó con EtzO y se secó rápidamente en un flujo de aire. A continuación, el sólido se calentó a reflujo con 250 ml de DMF durante una noche y el análisis por UPLC mostró el fin de la reacción. El disolvente se evaporó, el residuo se diluyó con 50 ml de NaHCO3 ac. y se extrajo 3 veces con 50 ml de acetato de etilo. Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron con MgSO4 y el disolvente se eliminó al vacío. El producto en bruto se sometió a cromatografía con una mezcla de acetato de etilo:hexano, 3: 7, para dar 13,5 g de un producto oleoso, rendimiento del 92 %.
Compuesto B-5: 1-benc¡l-4-bromo-2-(tr¡fluoromet¡l)-1H-¡ndol

Un matraz de 250 ml equipado con una barra de agitación magnética se llenó con 80 ml de DMF seca y 13,1 g (0,05 mol) del sustrato B-4. A continuación, la mezcla de reacción se enfrió a 0 °C y se añadieron cuidadosamente 2,4 g (0,06 mol) de hidruro de sodio (60 % en aceite). 10 minutos después de la adición, se añadieron por goteo 5,95 ml (0,05 mol) de bromuro de bencilo a esta temperatura (0 °C). Después de la adición de todos los reactivos, la reacción se agitó durante
0,5 h a la temperatura de <5 °C, y durante 2,5 h a TA. Después de este tiempo, la reacción se interrumpió con 5 ml de agua y se evaporó. El residuo se diluyó con agua (100 ml) y se extrajo con DCM (3 x 70 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron con MgSO4 y se evaporaron. El producto se obtuvo en forma de un sólido de color blanco (17,5 g, ~100 % de rendimiento) y se usó en la siguiente etapa sin purificación adicional. Compuesto B-6A: 1 -bencil-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-indol

Un matraz seco de 250 ml equipado con una barra de agitación magnética, un tubo de CaCl2 y un condensador se llenó con 150 ml de dioxano seco, 17,9 g (50 mmol, 1 equiv.) del sustrato B-5, 5,64 ml (1 equiv.) de metilpiperazina, 1,4 g (0,03 equiv.) de Pd2(dba)3 y 33,6 g (2 equiv.) de Cs2CO3. El matraz se purgó completamente con argón. Posteriormente, se añadieron 2,24 g (0,07 equiv.) de BINAP y la mezcla de reacción se calentó a 100 °C y se agitó durante una noche. Al día siguiente, la mezcla de reacción se enfrió, se vertió en 200 ml de agua, se filtró a través de Celite y se extrajo con DCM (3 x 100 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y el disolvente se eliminó al vacío. El residuo se sometió a cromatografía con una mezcla de DCM:MeOH:NH3 (500:19:1) para dar 14 g del producto oleoso B-6A, 75 % de rendimiento.
Compuesto B-6B: 4-[1-bencil-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El Compuesto B-6B se preparó a partir de B-5 (7,1 g, 20 mmol), según el mismo procedimiento que para el compuesto B-6A, usando N-BOC-piperazina en lugar de metilpiperazina. Después de la purificación, se obtuvieron 8,7 g del compuesto B-6B en forma de un sólido de color pardo claro (66 % de rendimiento).
Compuesto B-7A: 4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-indol

Un matraz de 500 ml equipado con una barra de agitación magnética se llenó con 70 ml de DMSO seco y 14 g (37,5 mmol) del sustrato B-6A. A continuación, la mezcla de reacción se enfrió a 10 °C y se añadieron gota a gota 160 ml (160 mmol, 4,6 equiv.) de t-BuOK 1 M en THF. La mezcla de reacción se enfrió a aproximadamente 2 °C y se burbujeó oxígeno a través de la mezcla de reacción mediante un tubo de vidrio hasta que se observó el consumo completo del sustrato (aproximadamente 5 h, la temperatura de reacción se mantuvo a aproximadamente 5 °C). Después de este tiempo, la mezcla de reacción se vertió en agua con hielo (200 ml) y se extrajo con acetato de etilo (3 x 100 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y el disolvente se evaporó. El residuo se sometió a cromatografía con una mezcla de DCM:MeOH (95:5), para dar 6,5 g del producto B-7A, 61 % de rendimiento.
Compuesto B-7B: 4-[2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

Un matraz de 250 ml equipado con una barra de agitación magnética se llenó con 120 ml de THF seco, 45 ml de DMSO seco y 5,3 g (11,5 mmol) del sustrato B-6B. A continuación, la mezcla de reacción se enfrió a 0 °C y se añadieron 12 g (107 mmol, 10 equiv.) de t-BuOK. Posteriormente, se burbujeó oxígeno a través de la mezcla de reacción mediante un tubo de vidrio hasta que se observó el consumo completo del sustrato (normalmente 2-4 h, la temperatura de reacción se mantuvo a aproximadamente 5 °C). Después de este tiempo, la mezcla de reacción se vertió en agua con hielo (200 ml) y se extrajo con acetato de etilo (3 x 70 ml). Las fases orgánicas combinadas se lavaron con agua, salmuera, se secaron sobre MgSO4 y el disolvente se evaporó. El residuo se sometió a cromatografía con una mezcla de AcOEt:hexano (1:9), para dar 3,8 g del producto B-7B, 90 % de rendimiento. Procedimiento general A para la preparación de los compuestos B-8:
En un matraz seco y lleno de gas inerte se añadieron indol B-7 (1 equiv.) y DMF seca (0,1 M), y la mezcla de reacción se enfrió a 0 °C. Se añadió hidruro de sodio (60 % en aceite mineral) (1,5 equiv.) y la mezcla de reacción se agitó durante 10 min a 0-5 °C, y durante 1 h a temperatura ambiente. Después de este tiempo, la reacción se enfrió a 0 °C y se añadió gota a gota un derivado de bencilo (1,2 equiv.). La mezcla de reacción se agitó a temperatura ambiente hasta el consumo completo del material de partida. Se añadieron DCM y agua y las fases se separaron. La fase acuosa se extrajo con DCM (3 x 10 ml) y las fases orgánicas combinadas se lavaron con agua, se secaron sobre Na2SO4 y el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna. Compuesto B-8B, Compuesto 13: 1-(3,4-diclorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8B, Compuesto 13, se obtuvo usando el procedimiento general A, a partir de B-7A (50 mg, 0,17 mmol), en forma de un aceite de color pardo claro (29 mg, 37 % de rendimiento, 97,72 % de pureza según el análisis UPLCMS). Compuesto B-8C, Compuesto 14: 1-(4-cloro-3-fluorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8C, Compuesto 14, se obtuvo usando el procedimiento general A, a partir de B-7A (50 mg, 0,17 mmol), en forma de un aceite de color pardo claro (37 mg, 49 % de rendimiento, 96,5 % de pureza según el análisis UPLCMS). Compuesto B-8D: 4-[1-(tiazol-2-ilmetil)-2-(trifluorometil)indol-4-il]piperazin-1-carboxilato de terc-butilo

El producto B-8D se obtuvo usando el procedimiento general A, a partir de B-7B (63 mg, 0,17 mmol), en forma de un aceite de color pardo claro (36 mg, 45 % de rendimiento).
Compuesto B-8E: 4-[1-[(4-doro-3-fluoro-fenil)metil]-2-(trifluorometil)indol-4-il]piperazin-1-carboxilato de terc-butilo

El producto B-8E se obtuvo usando el procedimiento general A, a partir de B-7B (63 mg, 0,17 mmol), en forma de un aceite de color pardo claro (46 mg, 53 % de rendimiento).
Compuesto B-8F: 4-[1-(furan-2-ilmetil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

Se añadió hidruro de sodio (22 mg, 60 % en aceite mineral, 0,54 mmol) a la solución de B-7B (200 mg, 0,54 mmol) en DMF seca en una atmósfera de argón a temperatura ambiente. La mezcla de reacción se agitó durante 30 min y a continuación se añadió 2-(bromometil)furan (105 mg, 0,65 mmol). Después de 1 h, se añadieron las siguientes porciones de hidruro de sodio (22 mg, 60 % en aceite mineral, 0,54 mmol) y 2-(bromometil)furan (31 mg, 0,19 mmol) y la reacción continuó durante 2 h. La mezcla de reacción se vertió en agua (20 ml) y se extrajo con DCM (2 x 20 ml). Los extractos combinados se lavaron con salmuera, se secaron sobre MgSO4 y se evaporaron a presión reducida. El producto en bruto se purificó por cromatografía en columna (AcOEt/hexano, 7/93 v/v). Como resultado, el producto final B-8F se obtuvo en forma de un sólido de color gris (170 mg, 70 % de rendimiento).
Compuesto B-8G: 1-(3-metoxibencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8G se obtuvo usando el procedimiento general A, a partir de B-7A (100 mg, 0,35 mmol), en forma de un aceite de color pardo claro (89 mg, 63 % de rendimiento, 96 % de pureza según el análisis UPLCMS).
Compuesto B-8H, Compuesto 19: 1-(3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8H, Compuesto 19, se obtuvo usando el procedimiento general A, a partir de B-7B (100 mg, 0,27 mmol), seguido de la desprotección del grupo BOC con 200 μl de TFA en 1 ml de d Cm a TA para dar 58 mg de un sólido, 57 % de rendimiento, 99 % de pureza según el análisis UPLCMS.
Compuesto B-8I: 4-[1-(3-clorobencil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El producto B-8I se obtuvo usando el procedimiento general A, a partir de B-7B (150 mg, 0,41 mmol), en forma de un aceite de color amarillo (190 mg, 94 % de rendimiento, 98,5 % de pureza según el análisis UPLCMS).
Compuesto B-8J, Compuesto 21: 1-(furan-2-ilmetil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol

Se añadió hidruro de sodio (16 mg, 60 % en aceite mineral, 0,40 mmol) a la solución de B-7A (100 mg, 0,37 mmol) en DMF seca en una atmósfera de argón a temperatura ambiente. La mezcla de reacción se agitó durante 30 min y a continuación se añadió 2-(bromometil)furan (64 mg, 0,40 mmol). Después de 18 h, se añadieron las siguientes porciones de hidruro de sodio (16 mg, 60 % en aceite mineral, 0,40 mmol) y 2-(bromometil)furan (64 mg, 0,40 mmol) y la reacción continuó durante 2 h. La mezcla de reacción se vertió en agua (20 ml) y se extrajo con acetato de etilo (2 x 20 ml). Los extractos combinados se lavaron con salmuera, se secaron sobre MgSO4 y se evaporaron a presión reducida. El producto en bruto se purificó por cromatografía en columna (DCM/MeOH/NH3ac., 98/2/0,5 v/v/v) y a continuación por HPLC preparativa. Como resultado, el producto final B-8J, Compuesto 21, se obtuvo en forma de un aceite de color amarillo claro (22 mg, 16 %, 99,7 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-ds) 57,55 (m, 1H), 7,32 (d, J = 8,4 Hz, 1H), 7,24 (dd, J = 8,2, 7,8 Hz, 1H), 7,04 (s, 1H), 6,61 (d, J = 7,6 Hz, 1H), 6,38 (m, 2H), 5,46 (s, 2H), 3,12 (m, 4H), 2,54 (m, 4H), 2,25 (s, 3H).
Compuesto B-8K, Compuesto 22: 1-(3,4-difluorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8K, Compuesto 22, se obtuvo usando el procedimiento general A, a partir de B-7A (100 mg, 0,37 mmol). El producto en bruto se purificó por cromatografía en columna (DCM/MeOH/NH3ac., 98/2/0,5 v/v/v) y a continuación por TLC preparativa. Como resultado, el producto final se obtuvo en forma de un aceite de color amarillo claro (40 mg, 13 % de rendimiento, 94,7 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-cfe) 57,34 (m, 1H), 7,21 (dd, J = 8,5, 7,6 Hz, 1H), 7,14 (s, 1H), 7,08 (d, J = 8,4 Hz, 1H), 7,06 (m, 1H), 6,66 (m, 1H), 6,62 (d, J = 7,6 Hz, 1H), 5,54 (s, 2H), 3,17 (m, 4H), 2,56 (m, 4H), 2,26 (s, 3H).
Compuesto B-8L: 4-[1-(3-metoxibencil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El producto B-8L se obtuvo usando el procedimiento general A, a partir de B-7B (150 mg, 0,41 mmol), en forma de un aceite de color amarillo (180 mg, 90 % de rendimiento, 99,5 % de pureza según el análisis UPLc Ms ).
Compuesto B-8M: 1 -(3-fluorobencil)-4-(4-metilpiperazin-1 -il)-2-(trifluorometil)-1 H-indol

El producto B-8M se obtuvo usando el procedimiento general A, a partir de B-7A (100 mg, 0,35 mmol), en forma de un aceite de color pardo claro (97 mg, 71 % de rendimiento, 96 % de pureza según el análisis UPLCMS).
Compuesto B-8N: 4-[1-(3,4-difluorobencil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El producto B-8N se obtuvo usando el procedimiento general A, a partir de B-7B (200 mg, 0,54 mmol), en forma de un aceite incoloro (210 mg, 78 % de rendimiento).
Compuesto B-8P: 1-(3-clorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol

El producto B-8P se obtuvo usando el procedimiento general A, a partir de B-7A (100 mg, 0,35 mmol), en forma de un aceite de color amarillo claro (74 mg, 52 % de rendimiento, 98 % de pureza según el análisis UPLCMS).
Compuesto B-8S: 4-[1-(tiofen-2-ilmetil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El compuesto B-8S se obtuvo usando el procedimiento general A (se usó mesilato en lugar de bromuro, 1,5 equiv., 16 h a 60 °C), a partir de B-7B (507 mg, 1,37 mmol), en forma de un sólido de color pardo claro (420 mg, 65 % de rendimiento, 95 % de pureza según el análisis UPLCMS).
Compuesto B-8T: 4-[1-(tiofen-3-ilmetil)-2-(trifluorometil)-1H-indol-4-il]piperazin-1-carboxilato de terc-butilo

El compuesto B-8T se obtuvo usando el procedimiento general A (se usó mesilato en lugar de bromuro, 3 equiv., 16 h a 60 °C), a partir de B-7B (500 mg, 1,35 mmol), en forma de un sólido de color pardo claro (250 mg, 40 % de rendimiento, 95 % de pureza según el análisis UPLCMS.
Compuesto B-8V, Compuesto 34: 4-(4-metilpiperazin-1-il)-1-[(5-metil-1,3-tiazol-2-il)metil]-2-(trifluorometil)-1H-indol

E compuesto B-8V, Compuesto 34, se obtuvo usando el procedimiento general A, a partir de B-7B (142 mg, 0,5 mmol), en forma de un sólido amorfo (120 mg, 61 % de rendimiento, 99,7 % de pureza según el análisis UPLCMS. Procedimiento general B para la preparación de los compuestos B-8 y B-9:
En un matraz seco y lleno de gas inerte se añadieron indol B-7 (1 equiv.) y THF seco (0,1 M), y la mezcla de reacción se enfrió a 0 °C. Se añadieron (5-metil-2-furil)metanol (2 equiv.), trifenilfosfina (1,5 equiv.) y DIa D (1,5 equiv.), y la mezcla de reacción se agitó durante 10 min a 0-5 °C y durante 1 h a temperatura ambiente. Se añadieron DCM y agua y las fases se
separaron. La fase acuosa se extrajo con DCM (3 x 10 ml) y las fases orgánicas combinadas se lavaron con agua, se secaron sobre Na2SO4 y el disolvente se eliminó al vacío. El residuo se purificó por cromatografía en columna y por HPLC preparativa. Compuesto B-9Q, Compuesto 28: 1-[(5-metilfuran-2-il)metil]-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol

El compuesto B-9Q, Compuesto 28, se obtuvo usando el procedimiento general B, a partir de B-7B (184 mg, 0,5 mmol) seguido de la desprotección del grupo BOC con 200 μl de TFA en 1 ml de DCM a TA, en forma de un sólido amorfo (16 mg, 9 % de rendimiento, 99 % de pureza según el análisis UPLCMS).
Compuesto B-9R, Compuesto 29: 1 -[(5-metiltiofen-2-il)metil]-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol

El compuesto B-9R, Compuesto 29, se obtuvo usando el procedimiento general B, a partir de B-7B (184 mg, 0,5 mmol) seguido de la desprotección del grupo BOC con 400 μl de TFA en 4 ml de DCM a TA, en forma de un sólido de color pardo claro (22 mg, 12 % de rendimiento, 96,6 % de pureza según el análisis UPLCMS).
Compuesto B-9F, Compuesto 17: 1 -(furan-2-ilmetil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol

Se añadió ácido trifluoroacético (2 ml) a la solución agitada del compuesto B-8F (170 mg, 0,38 mmol) en 5 ml de DCM a 0 °C. La mezcla resultante se agitó durante 2 h y a continuación se concentró a presión reducida. El residuo se disolvió en 30 ml de DCM, se lavó con NaHCO3 saturado (2 x 20 ml), salmuera (20 ml) y se secó sobre MgSO4. El disolvente se eliminó al vacío y el producto en bruto se purificó por cromatografía en columna (DCM/MeOH/NH3ac., 95/5/0,5 v/v/v). Como resultado, el producto final B-9F, Compuesto 17, se obtuvo en forma de un aceite de color amarillo claro (41 mg, 31 % de rendimiento, 98,9 % de pureza según el análisis UPLCMS). 1H RMN (500 MHz, DMSO-ds) 57,58 (m, 1H), 7,35 (d, J = 8,4 Hz, 1H), 7,27 (dd, J = 8,2, 7,8 Hz, 1H), 7,07 (s, 1H), 6,63 (d, J = 7,6 Hz, 1H), 6,41 (m, 2H), 5,50 (s, 2H), 3,07 (m, 4H), 2,95 (m, 4H).
Compuesto B-9N, Compuesto 25: 1-(3,4-difluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol

Se añadió gota a gota una solución 4 M de HCl en dioxano (1,0 ml) a una solución agitada de B-8N (105 mg, 0,26 mmol) en 3 ml de THF. La mezcla de reacción se agitó a temperatura ambiente durante 2 h, a continuación se añadieron 2 ml de EtzO y la reacción se agitó adicionalmente durante 0,5 h. El sólido de color blanco se filtró, se lavó con EtzO (2 x 5 ml) y se secó al vacío. El sólido se suspendió en 20 ml de AcOEt, se añadió NaOH 1 M (10 ml) y la mezcla se agitó vigorosamente durante 10 min. La fase orgánica se separó, se lavó con salmuera y se secó sobre MgSO4. El disolvente se eliminó al vacío y el producto en bruto se purificó por cromatografía en columna (DCM/MeOH/NH3ac., 93/H0,5 v/v/v). Como resultado, el producto final B-9N, Compuesto 25, se obtuvo en forma de un aceite de color amarillo claro (40 mg, 39 % de rendimiento, 96,7 % de pureza según el análisis UPLCMS). 1H RMN (500 MHz, CDCls) 57,21 (m, 1H), 7,09-7,04 (m, 1H), 7,02 (s, 1H), 6,85 (d, J = 8,3 Hz, 1H), 6,81-6,73 (m, 2H), 6,67 (d, J = 7,7 Hz, 1H), 5,39 (s, 2H), 3,31 (m, 4H), 3,23 (m, 4H).
Procedimiento general C para la preparación de los compuestos B-9:
En un matraz de 25 ml se añadió el compuesto B-8 seguido de THF (5 ml) y HCl 4 M en dioxano (0,5 ml). La mezcla de reacción se agitó a temperatura ambiente hasta el consumo completo del material de partida, a continuación se añadieron 10 ml de EtzO y la reacción se agitó adicionalmente durante 0,5 h. El sólido de color blanco se filtró, se lavó con EtzO (2 x 10 ml) y se secó al vacío.
Compuesto B-9D, Compuesto 15: 4-(piperazin-1-il)-1-(1,3-tiazol-2-ilmetil)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

El producto B-9D, Compuesto 15 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C, a partir de B-8D (36 mg, 0,08 mmol), en forma de un sólido de color blanco (19 mg, 61 % de rendimiento, 99 % de pureza según el análisis UPLCMS).
Compuesto B-9E, Compuesto 16: 1-(4-cloro-3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

El producto B-9E, Compuesto 16 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C, a partir de B-8E (46 mg, 0,09 mmol), en forma de un sólido de color blanco (9 mg, 22 % de rendimiento, 98 % de pureza según el análisis UPLCMS).
Compuesto B-9G, Compuesto 18: 1-(3-metoxibencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol en forma de sal clorhidrato

El producto B-9G, Compuesto 18 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C, a partir de B-8G (89 mg, 0,22 mmol), en forma de un sólido de color blanco (92 mg, 92 % de rendimiento, 99,5 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-cfe) 5 11,22 (s a, 1H), 7,30 (s, 1H), 7,26 - 7,11 (m, 3H), 6,81 (dd, J = 8,1, 2,5 Hz, 1H), 6,69 (d, J = 7,5 Hz, 1H), 6,54 (m, 1H), 6,45 (d, J = 7,6 Hz, 1H), 5,52 (s, 2H), 3,71 (d, J = 12,7 Hz, 2H), 3,51 (d, J = 11,8 Hz, 2H), 3,38 - 3,27 (m, 2H), 3,27 - 3,17 (m, 2H), 2,84 (d, J = 4,7 Hz, 3H).
Compuesto B-9I, Compuesto 20: 1-(3-clorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato
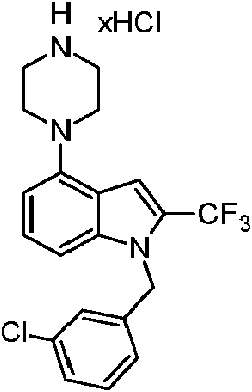
El producto B-9I, Compuesto 20 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C (2,5 ml de HCl 4 M durante 24 h), a partir de B-8I (190 mg, 0,39 mmol), en forma de un sólido de color blanco (137 mg, 82 % de rendimiento, 97,8 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-cfe) 59,55 (s a, 2H), 7,38 (s, 1H), 7,34 - 7,29 (m, 2H), 7,25 (m, 1H), 7,18 (d, J = 8,4 Hz, 1H), 7,04 (d, J = 2,0 Hz, 1H), 6,82 (ddd, J = 5,8, 3,0, 1,9 Hz, 1H), 6,71 (d, J = 7,6 Hz, 1H), 5,59 (s, 2H), 3,46 - 3,25 (m, 8H). Compuesto B-9L, Compuesto 23: 1-(3-metoxibencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

El producto B-9L, Compuesto 23 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C (2,5 ml de HCl 4 M durante 24 h), a partir de B-8L (180 mg, 0,37 mmol), en forma de un sólido de color blanco (126 mg, 81 % de rendimiento, 98,7 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-cfe) 59,53 (s a, 2H), 7,34 (s, 1H), 7,25 - 7,12 (m, 3H), 6,81 (dd, J = 8,0, 2,5 Hz, 1H), 6,69 (d, J = 7,6 Hz, 1H), 6,54 (s a, 1H), 6,45 (d, J = 7,6 Hz, 1H), 5,52 (s, 2H), 3,67 (s, 3H), 3,40 (m, 4H), 3,32 (m, 4H). Compuesto B-9M, Compuesto 24: 1-[(3-fluorofenil)metil]-4-(4-metilpiperazin-1 -il)-2-(trifluorometil)-1 H-indol, en forma de sal clorhidrato

El producto B-9M, Compuesto 24 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C, a partir de B-8M (97 mg, 0,25 mmol), en forma de un sólido de color blanco (102 mg, 92 % de rendimiento, 99,2 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-cfe) 5 11,30 (s a, 1H), 7,37 - 7,28 (m, 2H), 7,28 - 7,21 (m, 1H), 7,18 (d, J = 8,5 Hz, 1 H), 7,08 (td, J = 8,7, 2,6 Hz, 1H), 6,77 (dt, J = 10,1,2,0 Hz, 1H), 6,71 (dd, J = 7,9, 5,8 Hz, 2H), 5,59 (s, 2H), 3,72 (d, J = 12,5 Hz, 2H), 3,52 (d, J = 11,7 Hz, 2H), 3,39 - 3,19 (m, 4H), 2,84 (d, J = 4,7 Hz, 3H).
Compuesto B-9P, Compuesto 27: 1-(3-clorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

El producto B-9P, Compuesto 27 en forma de sal clorhidrato, se obtuvo usando el procedimiento general C, a partir de B-8P (74 mg, 0,18 mmol), en forma de un sólido de color blanco (80 mg, 99 % de rendimiento, 97,95 % de pureza según el análisis UPLCMS).
1H RMN (500 MHz, DMSO-ds) 5 11,39 (s a, 1H), 7,35 - 7,29 (m, 3H), 7,28 - 7,23 (m, 1H), 7,18 (d, J = 8,4 Hz, 1 H), 7,04 (c, J = 1,3 Hz, 1 H), 6,82 (ddd, J = 5,6, 3,5, 1,7 Hz, 1H), 6,71 (d, J = 7,6 Hz, 1H), 5,59 (s, 2H), 3,76 - 3,68 (m, 2H), 3,52 (d, J = 11,7 Hz, 2H), 3,39 - 3,19 (m, 4H), 2,84 (d, J = 4,7 Hz, 3H).
Procedimiento general D para la preparación de los compuestos B-9:
En un matraz de 25 ml se añadió el compuesto B-8 seguido de dioxano (10 ml) y HCl concentrado (1 ml). La mezcla de reacción se agitó a 60 °C durante 10 minutos. El disolvente se evaporó y el residuo se recristalizó en i-PrOH. El sólido se filtró, se lavó con i-PrOH (2 x 5 ml) y se secó al vacío.
Compuesto B-9S, Compuesto 30: 4-(piperazin-1-il)-1-(tiofen-2-ilmetil)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

El producto B-9S, Compuesto 30 en forma de sal clorhidrato, se obtuvo usando el procedimiento general D, a partir de B-8S (420 mg, 0,90 mmol), en forma de un sólido de color pardo claro (180 mg, 40 % de rendimiento, 95 % de pureza según el análisis UPLCMS).
Compuesto B-9T, Compuesto 31: 4-(piperazin-1-il)-1-(tiofen-3-ilmetil)-2-(trifluorometil)-1 H-indol, en forma de sal clorhidrato

El producto B-9T, Compuesto 31 en forma de sal clorhidrato, se obtuvo usando el procedimiento general D, a partir de B-8T (250 mg, 0,54 mmol), en forma de un sólido de color pardo claro (130 mg, 48 % de rendimiento, 96,7 % de pureza según el análisis UPLCMS).
Compuesto B-9U, Compuesto 32: 4-(4-metilpiperazin-1-il)-1-(tiofen-3-ilmetil)-2-(trifluorometil)-1H-indol, en forma de sal clorhidrato

En un matraz de fondo redondo se añadió el compuesto B-9T (250 mg, 0,62 mmol) seguido de MeOH (5,5 ml), AcOH (40 μl) y formaldehído (600 μl, solución al 37 % de agua). La mezcla de reacción se agitó a 40 °C durante 0,5 h y después de este tiempo todos los disolventes se evaporaron. El residuo se disolvió en dioxano (10 ml) y HCl concentrado (1 ml). La mezcla de reacción se agitó a 60 °C durante 10 minutos. El disolvente se evaporó y el residuo se recristalizó en i-PrOH. El sólido se filtró, se lavó con i-PrOH (2 x 5 ml) y se secó al vacío. Como resultado, el compuesto B-9U, Compuesto 32 en forma de sal clorhidrato, se obtuvo en forma de un sólido de color pardo claro (48 mg, 19 % de rendimiento, 95 % de pureza según el análisis UPLCMS).
Compuesto B-9W, Compuesto 33: 4-(4-metilpiperazin-1-il)-1-(tiofen-2-ilmetil)-2-(trifluorometil)-1 H-indol, en forma de sal clorhidrato

El producto B-9W, Compuesto 33 en forma de sal clorhidrato, se obtuvo usando la misma cantidad de reactivos que para el compuesto B-9U. Como resultado, el producto B-9W, Compuesto 33, se obtuvo, a partir de B-9S (65 mg, 0,16 mmol), en forma de un sólido de color pardo claro (39 mg, 59 % de rendimiento, 99 % de pureza según el análisis UPLCMS). Los siguientes ejemplos se han sintetizado según los procedimientos descritos en el presente documento o métodos conocidos de la bibliografía usando los materiales de partida apropiados y métodos conocidos por los expertos en la técnica:







Ejemplos biológicos
Ejemplo biológico 1. Ensayos de unión a receptor
Preparación de soluciones de compuestos de prueba y de referencia. Se prepararon soluciones madre 1 mM de los compuestos ensayados en DMSO. Se prepararon diluciones en serie de los compuestos en microplacas de 96 pocillos en tampones de ensayo usando el sistema de pipeteo automatizado epMotion 5070 (Eppendorf). Cada compuesto se ensayó en 10 concentraciones de 1,0E-6 a 1,0E-11 M (concentración final).
Ensayo de unión al receptor 5-HT2A. La unión de radioligandos se realizó usando membranas de células CHO K1 transfectadas de forma estable con el receptor 5-HT2A humano (PerkinElmer). Todos los ensayos se realizaron por duplicado. Se transfirieron 50 μl de solución de trabajo de los compuestos ensayados, 50 μl de [3H]-ketanserina (concentración final 1 nM) y 150 μl de membranas diluidas (7 μg de proteína por pocillo) preparadas en tampón de ensayo (Tris 50 mM, pH 7,4, CaCl24 mM, ácido ascórbico al 0,1 %) a una microplaca de polipropileno de 96 pocillos usando una estación de pipeteo de 96 pocillos Rainin Liquidator (Mettler Toledo). Se usó mianserina (10 μM) para definir la unión no específica. La microplaca se cubrió con una cinta selladora, se mezcló y se incubó durante 60 minutos a 27 °C. La reacción se terminó por filtración rápida a través de material filtrante GF/B previamente empapado con polietilenimina al 0,5 % durante 30 minutos. Se realizaron diez lavados rápidos con 200 μl de tampón Tris 50 mM (4 °C, pH 7,4) usando un sistema de recolección automatizado Harvester-96 MACH III FM (Tomtec). Los materiales filtrantes se secaron a 37 °C en una incubadora con ventilador de aire forzado y a continuación un centelleador sólido MeltiLex se fundió en los materiales filtrantes a 90 °C durante 5 minutos. La radiactividad se contó en un contador de centelleo MicroBeta2 (PerkinElmer). Los datos se ajustaron a una ecuación de ajuste de curvas de un sitio con Prism 6 (GraphPad Software) y los valores de Ki se estimaron a partir de la ecuación de Cheng-Prusoff.
Ensayo de unión al receptor 5-HT6. La unión de radioligandos se realizó usando membranas de células CHO-K1 transfectadas de forma estable con el receptor 5-HT6 humano (PerkinElmer). Todos los ensayos se realizaron por duplicado. Se transfirieron 50 μl de solución de trabajo de los compuestos ensayados, 50 μl de [3H]-LSD (concentración final 1 nM) y 150 μl de membranas diluidas (8 jg de proteína por pocillo) preparadas en tampón de ensayo (Tris 50 mM, pH 7,4, MgCL 10 mM, EDTA 0,1 mM) a una microplaca de polipropileno de 96 pocillos usando una estación de pipeteo de 96 pocillos Rainin Liquidator (Mettler Toledo). Se usó metiotepina (10 μM ) para definir la unión no específica. La microplaca se cubrió con una cinta selladora, se mezcló y se incubó durante 60 minutos a 37 °C. La reacción se terminó por filtración rápida a través de material filtrante GF/A previamente empapado con polietilenimina al 0,5 % durante 30 minutos. Se realizaron diez lavados rápidos con 200 μl de tampón Tris 50 mM (4 °C, pH 7,4) usando un sistema de recolección automatizado Harvester-96 MACH III FM (Tomtec). Los materiales filtrantes se secaron a 37 °C en una incubadora con ventilador de aire forzado y a continuación un centelleador sólido MeltiLex se fundió en los materiales filtrantes a 90 °C durante 5 minutos. La radiactividad se contó en un contador de centelleo MicroBeta2 (PerkinElmer). Los datos se ajustaron a una ecuación de ajuste de curvas de un sitio con Prism 6 (GraphPad Software) y los valores de Ki se estimaron a partir de la ecuación de Cheng-Prusoff.

Los resultados presentados anteriormente confirman que todos los compuestos ensayados poseen una alta afinidad por los receptores 5-HT2A y 5-HT6, lo que confirma sus propiedades de ligando de receptor dual.
Ejemplo biológico 2. Ensayos de actividad funcional
Preparación de soluciones de compuestos de prueba y de referencia. Se prepararon soluciones madre 1 mM de los compuestos ensayados en DMSO. Se prepararon diluciones en serie en microplacas de 96 pocillos en tampones de ensayo usando el sistema de pipeteo automatizado eμMotion 5070 (Eppendorf). Se realizaron dos experimentos independientes por duplicado y se ensayaron de 6 a 10 concentraciones.
Ensayos de la actividad funcional de 5-HT2A y 5-HT6. Se realizaron ensayos funcionales basados en aecuorina celular con células CHO-K1 recombinantes irradiadas con y que expresaban aecuorina dirigida mitocondrialmente, GPCR humano (5-HT2A o 5-HT6) y la proteína G promiscua a16 (PerkinElemer). Los ensayos se realizaron según el protocolo estándar proporcionado por el fabricante. Después de la descongelación, las células se transfirieron a tampón de ensayo (DMEM/HAM's F12 con BSA sin proteasa al 0,1 %) y se centrifugaron. El sedimento celular se resuspendió en tampón de ensayo y se añadió coelenterazina h a concentraciones finales de 5 jM . La suspensión celular se incubó a 21 °C, se protegió de la luz con agitación constante durante 4 horas y a continuación se diluyó con tampón de ensayo hasta una
concentración de 250.000 células/ml. Después de 1 hora de incubación, se dispensaron 50 μl de suspensión celular usando inyectores automáticos integrados en el contador de placas radiométrico y de luminiscencia MicroBeta2 LumiJET (PerkinElmer, EE.UU.) en una microplaca de color blanco opaca de 96 pocillos precargada con compuestos ensayados. Se registró la emisión de luz inmediata generada después de la movilización de calcio durante 30-60 segundos. En modo antagonista, después de 15-30 minutos de incubación, se añadió el agonista de referencia a la mezcla de ensayo anterior y se registró de nuevo la emisión de luz. La concentración final del agonista de referencia fue igual a CE80: serotonina 40 nM para el receptor 5-HT6 y a-metilserotonina 30 nM para el receptor 5-HT2A. Los ensayos se realizaron en modo agonista (5-HT6 AGO y 5-HT2A AGO), así como en modo antagonista (5-HT6 ANT y 5-HT2A ANT).
Se determinaron la CI50 y CE50 mediante análisis de regresión no lineal usando el software GraphPad Prism 6.0. Se usó la CI50 log. para obtener la Kb aplicando la aproximación de Cheng-Prusoff.

Los resultados presentados anteriormente confirman que todos los compuestos ensayados poseen altas propiedades antagonistas en los receptores 5-HT2A y 5-HT6, lo que confirma sus propiedades antagonistas duales de receptores.
Ejemplo biológico 3. Efectos de los compuestos 1 y 17 sobre las sacudidas de la cabeza inducidas por un agonista del receptor 5-HT2a/c , clorhidrato de 1-(2,5-dimetoxi-4-yodofenil)-2-aminopropano (DOI) en ratas Wistar
A. Sujetos
Se usaron ratas Wistar macho sin tratamiento previo con fármacos (Charles River, Sulzfeld, Alemania). Las ratas se alojaron cuatro por jaula de plástico estándar y se mantuvieron en una sala con condiciones ambientales constantes (22 ± 1 °C, humedad relativa al 60 %, un ciclo de luz-oscuridad de 12:12 con las luces encendidas a las 07:00 a. m.). Los animales se suministraron por el criador 2 semanas antes del inicio de los procedimientos conductuales. Durante este tiempo, los sujetos se pesaron y se manipularon varias veces. Las ratas también se habituaron a la administración p.o. de los compuestos ensayados en forma de sal clorhidrato mediante dosificación por sonda nasogástrica de agua destilada (1-2 ml). Estaban disponibles a voluntad agua del grifo y comida de laboratorio estándar (Labofeed H, WPIK, Kcynia, Polonia).
El tratamiento de las ratas en el presente estudio estuvo en completo acuerdo con los estándares éticos establecidos en las respectivas regulaciones polacas y europeas (Directiva n.° 2010/63/UE). Todos los procedimientos fueron revisados y aprobados por un comité de ética.
B. Sacudidas de la cabeza inducidas por DOI
Todas las pruebas se realizaron en una sala experimental con atenuación de sonido entre las 10:00 a. m. y las 4:00 p. m. Las sacudidas de la cabeza inducidas por DOI se calificaron según lo descrito por Millan y col. (2000). A las ratas se les inyectó DOI (2,5 mg/kg, i.p.) y se colocaron en jaulas de observación de vidrio (25 x 25 x 40 cm, ancho x alto x largo) con lecho de virutas de madera en el suelo. Cinco minutos más tarde, se contaron las sacudidas de la cabeza durante 5 min (300 s) por un observador entrenado. Los compuestos ensayados en forma de sal clorhidrato se administraron p.o.
180 minutos antes del inicio del período de observación a diferentes grupos de sujetos sin tratamiento previo con fármacos.
C. Fármacos
Se disolvió DOI en una solución salina fisiológica estéril (Baxter, Varsovia, Polonia) y se administró i.p. en un volumen de 1,0 ml/kg. Los compuestos ensayados en forma de sal clorhidrato se disolvieron en tween al 0,5 % y se administraron p.o. en un volumen de 2,0 ml/kg. Todas las soluciones se prepararon inmediatamente antes de su uso y se protegieron de la luz.
D. Análisis de los datos:
Se analizó el número total de sacudidas de la cabeza (n/5 min) con la ayuda del análisis de varianza de Kruskal-Wallis (ANOVA). Se usó la prueba U de Mann-Whitney para las comparaciones post hoc individuales (Tabla 1). Los valores de p inferiores a 0,05 se consideraron significativos. Se usó el paquete de software Statistica 12.0 para Windows (StatSoft, Tulsa, OK, EE.UU.) para analizar todos los datos.
Resultados
Ambos compuestos ensayados, sacudidas de la cabeza inducidas por DOI (2,5 mg/kg) atenuadas de forma dependiente de la dosis con una dosis eficaz mínima (MED, por sus siglas en inglés) de 3,0 mg/kg para el Compuesto 1 y de 1,0 mg/kg para el Compuesto 17.
Ejemplo biológico 4. Efectos de los Compuestos 1 y 17, sobre los déficits de aprendizaje y memoria inducidos por escopolamina en la prueba de evitación pasiva en ratas Wistar
A. Sujetos
Se usaron ratas Wistar macho sin tratamiento previo con fármacos (Charles River, Sulzfeld, Alemania). Las ratas se alojaron cuatro por jaula de plástico estándar y se mantuvieron en una sala con condiciones ambientales constantes (22 ± 1 °C, humedad relativa al 60 %, un ciclo de luz-oscuridad de 12:12 con las luces encendidas a las 07:00 a. m.). Los animales se suministraron por el criador 2-3 semanas antes del inicio de los procedimientos conductuales. Durante este tiempo, los sujetos se pesaron y se manipularon varias veces. Las ratas también se habituaron a la administración p.o. de los compuestos ensayados en forma de sal clorhidrato mediante dosificación por sonda nasogástrica de agua destilada (1-2 ml). Estaban disponibles a voluntad agua del grifo y comida de laboratorio estándar (Labofeed H, WPIK, Kcynia, Polonia).
El tratamiento de las ratas en el presente estudio estuvo en completo acuerdo con los estándares éticos establecidos en las respectivas regulaciones polacas y europeas (Directiva n.° 2010/63/UE). Todos los procedimientos fueron revisados y aprobados por un comité de ética local.
B. Prueba de evitación pasiva paso a paso
Los efectos de los compuestos ensayados sobre el aprendizaje y la función de la memoria se evaluaron usando una prueba de evitación pasiva paso a paso (Ishiyama y col., 2007). El aparato de evasión pasiva (PACS-30, Columbus Instruments, Columbus, OH, EE.UU.) comprendía cuatro jaulas idénticas de acero inoxidable con cubiertas de plexiglás de color negro. Cada jaula consistía en un compartimento iluminado y oscuro (23 x 23 x 23 cm) y un suelo de rejilla de acero inoxidable. Los dos compartimentos estaban separados por la puerta corredera automatizada (PACS-30, Columbus).
En la sesión de entrenamiento (adquisición), los animales se colocaron individualmente en el compartimento iluminado y se les permitió explorarlo libremente durante 10 s. A continuación, se abrió la puerta corredera y se midió la latencia paso a paso para que los animales entraran en el compartimento oscuro con un tiempo límite de 300 s. Tan pronto como los animales entraron en el compartimento oscuro, la puerta se cerró. Se suministró una descarga eléctrica ineludible en las patas (0,5 mA durante 3 s) 3 s más tarde a través del suelo de rejilla con un generador de descarga de corriente constante (Columbus). Se administró i.p. escopolamina (0,3 mg/kg) 30 min antes de la sesión de entrenamiento. Los compuestos ensayados en forma de sal clorhidrato, o su vehículo, se administraron p.o. 180 min antes del inicio de la sesión de entrenamiento.
La sesión de prueba (expresión) se realizó 24 h después de la sesión de entrenamiento usando el mismo paradigma pero sin ningún tipo de descarga eléctrica en las patas ni inyecciones de fármacos. Las latencias paso a paso para que los animales entraran en el compartimiento oscuro se midieron con un tiempo límite de 300 s. Los cambios inducidos por los compuestos ensayados en las latencias paso a paso para entrar en el compartimiento oscuro en la sesión de prueba se trataron como una medida de sus efectos promnésicos o amnésicos (Ishiyama y col., 2007).
C. Fármacos
La escopolamina (proporcionada por Adamed) se disolvió en una solución salina fisiológica estéril (NaCl al 0,9 %; Baxter, Varsovia, Polonia) y se administró i.p. en un volumen de 2,0 ml/kg. Los compuestos ensayados en forma de sal clorhidrato se disolvieron en Tween al 0,5 % y se administraron p.o. en un volumen de 2,0 ml/kg. Todas las soluciones se prepararon inmediatamente antes de su uso y se protegieron de la luz.
D. Presentación y análisis de datos
Los pesos corporales (g) y las latencias de entrenamiento/prueba (s) se analizaron con la ayuda de un análisis de varianza unidireccional (ANOVA). Como los datos de evitación pasiva no se distribuyeron normalmente, también se analizaron las latencias paso a paso con la ayuda de la prueba ANOVA de Kruskal-Wallis y de la U de Mann-Whitney. Los valores de p inferiores a 0,05 se consideraron significativos. Se usó el paquete de software Statistica 12.0 para Windows (StatSoft, Tulsa, OK, EE.UU.) para analizar todos los datos.
Resultados
Ambos compuestos ensayados, administrados en combinación con escopolamina (0,3 mg/kg), prolongaron significativamente las latencias paso a paso para entrar en el compartimento oscuro en la sesión de prueba. La dosis eficaz mínima (MED) fue de 3,0 mg/kg para el Compuesto 1 y de 1,0 mg/kg para el Compuesto 17.
Bibliografía:
Amano, N., Inuzuka, S., Ogihara, T., 2009. Behavioral and psychological symptoms of dementia and medical treatment. Psychogeriatr. Off. J. Jpn. Psychogeriatr. Soc. 9, 45-49. https://doi.org/101111/j.1479-8301.2009.00284.x
Ballard, C., Waite, J., 2006. The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer's disease. Cochrane Database Syst. Rev. CD003476. https://doi.org/10.1002/14651858.CD003476.pub2
Carson, S., McDonagh, M.S., Peterson, K., 2006. A systematic review of the efficacy and safety of atypical antipsychotics in patients with psychological and behavioral symptoms of dementia. J. Am. Geriatr. Soc. 54, 354 361. https://doi.org/10.1111/j.1532-5415.2005.00566.x
De Deyn, P., Jeste, D.V., Swanink, R., Kostic, D., Breder, C., Carson, W.H., Iwamoto, T., 2005. Aripiprazole for the treatment of psychosis in patients with Alzheimer's disease: a randomized, placebo-controlled study. J. Clin. Psychopharmacol. 25, 463-467.
Fasano, A., Plotnik, M., Bove, F., Berardelli, A., 2012. The neurobiology of falls. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 33, 1215-1223. https://doi.org/10.1007/s10072-012-1126-6
Ferri, C.P., Prince, M., Brayne, C., Brodaty, H., Fratiglioni, L., Ganguli, M., Hall, K., Hasegawa, K., Hendrie, H., Huang, Y., Jorm, A., Mathers, C., Menezes, P.R., Rimmer, E., Scazufca, M., Alzheimer's Disease International, 2005. Global prevalence of dementia: a Delphi consensus study. Lancet Lond. Engl. 366, 2112-2117. https://doi.org/10.1016/S0140-6736(05)67889-0
F i j a , K., Popik, P., Nikiforuk, A., 2014. C0-administration of 5-HT6 receptor antagonists with clozapine, risperidone, and a 5-HT2A receptor antagonist: effects on prepulse inhibition in rats. Psychopharmacology (Berl.) 231, 269-281. https://doi.org/10.1007/s00213-013-3234-2
Gauthier, S., Cummings, J., Ballard, C., Brodaty, H., Grossberg, G., Robert, P., Lyketsos, C., 2010. Management of behavioral problems in Alzheimer's disease. Int. Psychogeriatr. 22, 346-372. https://doi.org/10.1017/S1041610209991505
Hersch, E.C., Falzgraf, S., 2007. Management of the behavioral and psychological symptoms of dementia. Clin. Interv. Aging 2, 611-621.
Holmes, C., Arranz, M.J., Powell, J.F., Collier, D.A., Lovestone, S., 1998. 5-HT2A and 5-HT2C receptor polymorphisms and psychopathology in late onset Alzheimer's disease. Hum. Mol. Genet. 7, 1507-1509.
Home | Cochrane Library [WWW Document], n.d. URL http://www.cochranelibrary.com/ (accessed 2.7.18).
Jeste, D.V., Blazer, D., Casey, D., Meeks, T., Salzman, C., Schneider, L., Tariot, P., Yaffe, K., 2008. ACNP White Paper: update on use of antipsychotic drugs in elderly persons with dementia. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 33, 957-970. https://doi.org/10.1038/sj.npp.1301492
Jeste, D.V., Finkel, S.I., 2000. Psychosis of Alzheimer's disease and related dementias. Diagnostic criteria for a distinct syndrome. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 8, 29-34.
Jones, C.A., Watson, D.J.G., Fone, K.C.F., 2011. Animal models of schizophrenia. Br. J. Pharmacol. 164, 1162 1194. https://doi.org/10.1111/j.1476-5381.2011.01386.x
Liperoti, R., Pedone, C., Corsonello, A., 2008. Antipsychotics for the treatment of behavioral and psychological symptoms of dementia (BPSD). Curr. Neuropharmacol. 6, 117-124. https://doi.org/10.2174/157015908784533860 Liu, K.G., Robichaud, A.J., 2009. 5-HT6 antagonists as potential treatment for cognitive dysfunction. Drug Dev. Res.
70, 145-168. https://doi.org/10.1002/ddr.20293
Lorke, D.E., Lu, G., Cho, E., Yew, D.T., 2006. Serotonin 5-HT2A and 5-HT6 receptors in the prefrontal cortex of Alzheimer and normal aging patients. BMC Neurosci. 7, 36. https://doi.org/10.1186/1471-2202-7-36
Maehara, S., Hikichi, H., Satow, A., Okuda, S., Ohta, H., 2008. Antipsychotic property of a muscarinic receptor agonist in animal models for schizophrenia. Pharmacol. Biochem. Behav. 91, 140-149. https://doi.org/10.1016/j.pbb.2008.06.023 Marcos, B., Garcia-Alloza, M., Gil-Bea, F.J., Chuang, T.T., Francis, P.T., Chen, C.P., Tsang, S.W.T.Y., Lai, M.K.P., Ramirez, M.J., 2008. Involvement of an altered 5-HT -{6} receptor function in behavioral symptoms of Alzheimer's disease. J. Alzheimers Dis. JAD 14, 43-50.
Marsh, A., 1979. Visual hallucinations during hallucinogenic experience and schizophrenia. Schizophr. Bull. 5, 627-630. Murray, P.S., Kirkwood, C.M., Gray, M.C., Fish, K.N., Ikonomovic, M.D., Hamilton, R.L., Kofler, J.K., Klunk, W.E., Lopez, O.L., Sweet, R.A., 2014. Hyperphosphorylated tau is elevated in Alzheimer's disease with psychosis. J. Alzheimers Dis. JAD 39, 759-773. https://doi.org/10.3233/JAD-131166
Nichols, D.E., 2004. Hallucinogens. Pharmacol. Ther. 101, 131-181. https://doi.org/10.1016/j.pharmthera.2003.11.002 Nobili, A., Pasina, L., Trevisan, S., Riva, E., Lucca, U., Tettamanti, M., Matucci, M., Tarantola, M., 2009. Use and misuse of antipsychotic drugs in patients with dementia in Alzheimer special care units. Int. Clin. Psychopharmacol. 24, 97-104. Riemer, C., Borroni, E., Levet-Trafit, B., Martin, J.R., Poli, S., Porter, R.H.P., Bos, M., 2003. Influence of the 5-HT6 receptor on acetylcholine release in the cortex: pharmacological characterization of 4-(2-bromo-6-pyrrolidin-1-ylpyridine-4-sulfonyl)phenylamine, a potent and selective 5-HT6 receptor antagonist. J. Med. Chem. 46, 1273-1276. https://doi.org/10.1021/jm021085c
Schneider, L.S., Tariot, P.N., Dagerman, K.S., Davis, S.M., Hsiao, J.K., Ismail, M.S., Lebowitz, B.D., Lyketsos, C.G., Ryan, J.M., Stroup, T.S., Sultzer, D.L., Weintraub, D., Lieberman, J.A., CATIE-AD Study Group, 2006. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. N. Engl. J. Med. 355, 1525-1538. https://doi.org/10.1056/NEJMoa061240
Schulze, J., Glaeske, G., van den Bussche, H., Kaduszkiewicz, H., Koller, D., Wiese, B., Hoffmann, F., 2013a. Prescribing of antipsychotic drugs in patients with dementia: a comparison with age-matched and sex-matched nondemented controls. Pharmacoepidemiol. Drug Saf. 22, 1308-1316. https://doi.org/10.1002/pds.3527
Schulze, J., van den Bussche, H., Glaeske, G., Kaduszkiewicz, H., Wiese, B., Hoffmann, F., 2013b. Impact of safety warnings on antipsychotic prescriptions in dementia: nothing has changed but the years and the substances. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 23, 1034-1042. https://doi.org/10.1016/j.euroneuro.2013.02.001
Siegel, R.K., 1978. Phencyclidine and ketamine intoxication: a study of four populations of recreational users. NIDA Res. Monogr. 119-147.
Sink, K.M., Holden, K.F., Yaffe, K., 2005. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 293, 596-608. https://doi.org/10.1001/jama.293.5.596
Sukonick, D.L., Pollock, B.G., Sweet, R.A., Mulsant, B.H., Rosen, J., Klunk, W.E., Kastango, K.B., DeKosky, S.T., Ferrell, R.E., 2001. The 5-HTTPR*S/*L polymorphism and aggressive behavior in Alzheimer disease. Arch. Neurol. 58, 1425-1428. Varty, G.B., Bakshi, V.P., Geyer, M.A., 1999. M100907, a serotonin 5-HT2A receptor antagonist and putative antipsychotic, blocks dizocilpine-induced prepulse inhibition deficits in Sprague-Dawley and Wistar rats. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 20, 311-321. https://doi.org/10.1016/S0893-133X(98)00072-4
Vigen, C.L.P., Mack, W.J., Keefe, R.S.E., Sano, M., Sultzer, D.L., Stroup, T.S., Dagerman, K.S., Hsiao, J.K., Lebowitz, B.D., Lyketsos, C.G., Tariot, P.N., Zheng, L., Schneider, L.S., 2011. Cognitive effects of atypical
antipsychotic medications in patients with Alzheimer's disease: outcomes from CATIE-AD. Am. J. Psychiatry 168, 831-839. https://doi.org/10.1176/appi.ajp.2011.08121844
Wesotowska, A., 2010. Potential role of the 5-HT6 receptor in depression and anxiety: an overview of preclinical data. Pharmacol. Rep. PR 62, 564-577.
Wesotowska, A., Nikiforuk, A., 2007. Effects ofthe brain-penetrant and selective 5-HT6 receptor antagonist SB-399885 in animal models of anxiety and depression. Neuropharmacology 52, 1274-1283. https://doi.org/10.10167j.neuropharm.2007.01.007
Woolley, M.L., Marsden, C.A., Fone, K.C.F., 2004. 5-ht6 receptors. Curr. Drug Targets CNS Neurol. Disord. 3, 59-79.
Claims (15)
1. Un compuesto de fórmula general

o una sal farmacéuticamente aceptable del mismo,
en donde:
G es CH o N;
R1 es H, alquilo C1-C4, HO-alquilo C1-C4 o alquilo C1-C4-O-alquilo C1-C4;
R2 se selecciona de un grupo que consiste en:
-un grupo fenilo, sin sustituir o sustituido por al menos un sustituyente, y
-un grupo heteroarilo de 5 o 6 miembros, sin sustituir o sustituido por al menos un sustituyente,
en donde el sustituyente se selecciona de F, Cl, Br, alquilo C1-C4 y alquilo C1-C4-O-.
2. El compuesto de la reivindicación 1, en donde G es CH.
3. El compuesto de la reivindicación 1, en donde G es N.
4. El compuesto de una cualquiera de las reivindicaciones 1-3, en donde R1 es H, metilo o 2-hidroxietilo.
5. El compuesto de una cualquiera de las reivindicaciones 1-4, en donde R2 es un grupo fenilo, sin sustituir o sustituido por al menos un sustituyente.
6. El compuesto de una cualquiera de las reivindicaciones 1-4, en donde R2 es un grupo heteroarilo de 5 o 6 miembros, sin sustituir o sustituido por al menos un sustituyente, en donde el heteroarilo de 5 o 6 miembros se selecciona preferiblemente de furilo, tienilo, tiazolilo o piridilo.
7. El compuesto de una cualquiera de las reivindicaciones 5-6, en donde el sustituyente se selecciona de F, Cl, metilo o metoxi.
8. El compuesto de una cualquiera de las reivindicaciones 1-7, en donde el compuesto se selecciona del grupo que consiste en:
1 -bencil-4-(piperazin-1 -il)-2-(trifluorometil)-1 H-bencimidazol,
1- bencil-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-bencimidazol,
2- {4-[1-bencil-2-(trifluorometil)-1H-bencimidazol-4-il]piperazin-1-il}etanol, 1-(furan-2-ilmetil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-bencimidazol,
1-[(5-metilfuran-2-il)metil]-4-(piperazin-1-il)-2-(trifluorometil)-1 H-bencimidazol,
1-(3-clorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-bencimidazol,
1-(3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-bencimidazol,
1-(3,4-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1-(3-cloro-4-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol, 1-(3,4-difluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1-(3,5-diclorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1H-benzo[d]imidazol,
1 -bencil-4-(piperazin-1 -il)-2-(trifluorometil)-1 H-indol,
1 -(3,4-diclorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-indol,
1-(4-cloro-3-fluorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1 H-indol,
4-(piperazin-1-il)-1-(1,3-tiazol-2-ilmetil)-2-(trifluorometil)-1 H-indol,
1-(4-cloro-3-fluorobencil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol,
1-(furan-2-ilmetil)-4-(piperazin-1-il)-2-(trifluorometil)-1 H-indol,
1-(3-metoxibendl)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-fluorobendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol, 1-(3-dorobencN)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol, 1-(furan-2-Nmetil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol,
1-(3,4-difluorobencN)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol, 1-(3-metoxibendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol, 1-(3-fluorobencil)-4-(4-metilpiperazin-1-il)-2-(trifluorometil)-1H-indol,
1-(3,4-difluorobendl)-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol, 1-bendl-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-(3-dorobencN)-4-(4-metilpiperazin-1-N)-2-(trifluorometil)-1H-indol,
1-[(5-metilfuran-2-N)metil]-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
1-[(5-metiltiofen-2-N)metil]-4-(piperazin-1-N)-2-(trifluorometil)-1H-indol,
4-(piperazin-1-N)-1-(tiofen-2-Nmetil)-2-(trifluorometil)-1H-indol, 4-(piperazin-1-il)-1-(tiofen-3-ilmetil)-2-(trifluorometil)-1H-indol, 4-(4-metilpiperazin-1-N)-1-(tiofen-3-ilmetil)-2-(trifluorometil)-1H-indol,
4-(4-metilpiperazin-1-il)-1-(tiofen-2-ilmetil)-2-(trifluorometil)-1H-indol y
4-(4-metilpiperazin-1-il)-1-[(5-metil-1,3-tiazol-2-il)metil]-2-(trifluorometil)-1H-indol.
9. El compuesto de la reivindicación 1, en donde el compuesto es

o una sal farmacéuticamente aceptable del mismo.
10. El compuesto de la reivindicación 1, en donde el compuesto es
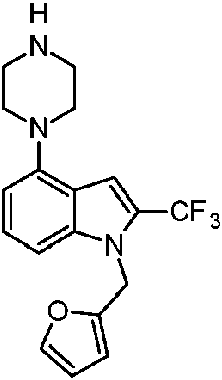
o una sal farmacéuticamente aceptable del mismo.
11. El compuesto de la reivindicación 1, en donde el compuesto es

o una sal farmacéuticamente aceptable del mismo.
12. El compuesto de la reivindicación 1, en donde el compuesto es

o una sal farmacéuticamente aceptable del mismo.
13. El compuesto de una cualquiera de las reivindicaciones 1 a 12 para su uso como un medicamento.
14. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1 a 12 y al menos un excipiente farmacéuticamente aceptable.
15. El compuesto de una cualquiera de las reivindicaciones 1 a 12 para su uso en el tratamiento de la enfermedad de Alzheimer, enfermedad de Parkinson, demencia con cuerpos de Levy, psicosis relacionada con demencia, esquizofrenia, síndromes delirantes y otras afecciones psicóticas relacionadas y no relacionadas con el consumo de sustancias psicoactivas, depresión, trastornos de ansiedad de diversa etiología o trastornos del sueño de diversa etiología.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18461519.3A EP3530651A1 (en) | 2018-02-21 | 2018-02-21 | Indole and benzimidazole derivatives as dual 5-ht2a and 5-ht6 receptor antagonists |
| PCT/EP2019/054171 WO2019162306A1 (en) | 2018-02-21 | 2019-02-20 | Indole and benzimidazole derivatives as dual 5-ht2a and 5-ht6 receptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2953815T3 true ES2953815T3 (es) | 2023-11-16 |
Family
ID=61256878
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19706271T Active ES2953815T3 (es) | 2018-02-21 | 2019-02-20 | Derivados de indol y bencimidazol como antagonistas duales de los receptores 5-HT2A y 5-HT6 |
Country Status (25)
| Country | Link |
|---|---|
| US (3) | US11034688B2 (es) |
| EP (2) | EP3530651A1 (es) |
| JP (1) | JP7200258B2 (es) |
| KR (1) | KR102688938B1 (es) |
| CN (1) | CN112041300B (es) |
| AU (1) | AU2019223036B2 (es) |
| BR (1) | BR112020016972B1 (es) |
| CA (1) | CA3088827A1 (es) |
| DK (1) | DK3755688T3 (es) |
| EA (1) | EA202091737A1 (es) |
| ES (1) | ES2953815T3 (es) |
| FI (1) | FI3755688T3 (es) |
| HR (1) | HRP20230793T1 (es) |
| HU (1) | HUE063564T2 (es) |
| IL (1) | IL276762B2 (es) |
| LT (1) | LT3755688T (es) |
| MX (1) | MX2020008745A (es) |
| PL (1) | PL3755688T3 (es) |
| PT (1) | PT3755688T (es) |
| RS (1) | RS64459B1 (es) |
| SG (1) | SG11202007091XA (es) |
| SI (1) | SI3755688T1 (es) |
| UA (1) | UA126252C2 (es) |
| WO (1) | WO2019162306A1 (es) |
| ZA (1) | ZA202005747B (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3530651A1 (en) | 2018-02-21 | 2019-08-28 | Adamed sp. z o.o. | Indole and benzimidazole derivatives as dual 5-ht2a and 5-ht6 receptor antagonists |
| CN115850204B (zh) * | 2022-11-26 | 2024-01-19 | 烟台大学 | 四氢苯并噻吩衍生物及其制备方法和应用 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0613430A2 (pt) * | 2005-07-13 | 2011-01-11 | Hoffmann La Roche | compostos derivados de benzimidazol, uso dos mesmos, método para a preparação destes e composição farmacêutica |
| CA2668959A1 (en) * | 2006-11-09 | 2008-05-15 | F. Hoffmann-La Roche Ag | Indole and benzofuran 2-carboxamide derivatives |
| US20100041669A1 (en) * | 2007-01-08 | 2010-02-18 | Venkata Satya Nirogi Ramakrishna | 4-(heterocyclyl)alkyl-n-(arylsulfonyl)indole compounds and their use as 5-ht6 ligands |
| UA100192C2 (en) * | 2008-11-11 | 2012-11-26 | УАЙТ ЭлЭлСи | 1-(arylsulfonyl)-4-(piperazin-1-yl)-1h-benzimidazoles as 5-hydroxytryptamine-6 ligands |
| PL395469A1 (pl) * | 2011-06-29 | 2013-01-07 | Adamed Spólka Z Ograniczona Odpowiedzialnoscia | Pochodne indoloamin do leczenia chorób osrodkowego ukladu nerwowego |
| RU2014153672A (ru) * | 2012-06-22 | 2016-08-10 | Мап Фармасьютикалс, Инк. | Новые каберголиновые производные |
| EP3530651A1 (en) * | 2018-02-21 | 2019-08-28 | Adamed sp. z o.o. | Indole and benzimidazole derivatives as dual 5-ht2a and 5-ht6 receptor antagonists |
-
2018
- 2018-02-21 EP EP18461519.3A patent/EP3530651A1/en not_active Withdrawn
-
2019
- 2019-02-20 WO PCT/EP2019/054171 patent/WO2019162306A1/en not_active Ceased
- 2019-02-20 SG SG11202007091XA patent/SG11202007091XA/en unknown
- 2019-02-20 FI FIEP19706271.4T patent/FI3755688T3/fi active
- 2019-02-20 CA CA3088827A patent/CA3088827A1/en active Pending
- 2019-02-20 HU HUE19706271A patent/HUE063564T2/hu unknown
- 2019-02-20 SI SI201930608T patent/SI3755688T1/sl unknown
- 2019-02-20 LT LTEPPCT/EP2019/054171T patent/LT3755688T/lt unknown
- 2019-02-20 HR HRP20230793TT patent/HRP20230793T1/hr unknown
- 2019-02-20 RS RS20230676A patent/RS64459B1/sr unknown
- 2019-02-20 JP JP2020544234A patent/JP7200258B2/ja active Active
- 2019-02-20 US US16/970,871 patent/US11034688B2/en active Active
- 2019-02-20 ES ES19706271T patent/ES2953815T3/es active Active
- 2019-02-20 EP EP19706271.4A patent/EP3755688B1/en active Active
- 2019-02-20 KR KR1020207022525A patent/KR102688938B1/ko active Active
- 2019-02-20 PT PT197062714T patent/PT3755688T/pt unknown
- 2019-02-20 EA EA202091737A patent/EA202091737A1/ru unknown
- 2019-02-20 UA UAA202005367A patent/UA126252C2/uk unknown
- 2019-02-20 DK DK19706271.4T patent/DK3755688T3/da active
- 2019-02-20 MX MX2020008745A patent/MX2020008745A/es unknown
- 2019-02-20 BR BR112020016972-2A patent/BR112020016972B1/pt active IP Right Grant
- 2019-02-20 CN CN201980027105.7A patent/CN112041300B/zh active Active
- 2019-02-20 IL IL276762A patent/IL276762B2/en unknown
- 2019-02-20 PL PL19706271.4T patent/PL3755688T3/pl unknown
- 2019-02-20 AU AU2019223036A patent/AU2019223036B2/en active Active
-
2020
- 2020-09-16 ZA ZA2020/05747A patent/ZA202005747B/en unknown
-
2021
- 2021-05-20 US US17/326,159 patent/US11981668B2/en active Active
-
2024
- 2024-04-16 US US18/637,083 patent/US12319682B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2319642T3 (es) | Derivados 1,4-di-piperidina-4-il-piperazina sustituidos y su uso como antagonistas de la neuroquinina. | |
| DK2491045T3 (en) | 2-oxo-1-pyrrolidinylimidazothiadiazolderivativer | |
| AU2017232906A1 (en) | Small molecules against cereblon to enhance effector T cell function | |
| US12319682B2 (en) | Indole and benzimidazole derivatives as dual 5-HT2A and 5-HT6 receptor antagonists | |
| AU2010256360A1 (en) | Aminopyrrolidinone derivatives and uses thereof | |
| BR112019011825A2 (pt) | composto, enantiômero isolado, composição farmacêutica, método para inibir a atividade do transportador de monocarboxilato mct4, ou um mutante do mesmo, em uma amostra biológica, método para inibir a atividade do transportador de monocarboxilato mct4, ou um mutante do mesmo, em relação ao transportador de monocarboxilato mct1, ou um mutante do mesmo, em um paciente, método para tratar um distúrbio mediado por transportador de monocarboxilato mct4 em um sujeito que necessite do mesmo, uso de um composto e método para obter um efeito em um paciente | |
| TW202142261A (zh) | 治療遺傳性疾病之方法及化合物 | |
| ES2247408T3 (es) | 1-arilpiperazinas 2-sustituidas como antangonistas de taquiquinina y/o inhibidores de la recaptacion de serotonina. | |
| KR20070038503A (ko) | 신규 헤테로시클릭 카르복시산 아미드 유도체 | |
| ES2346147T3 (es) | Derivados de oxa-diaza-espiro-(5.5)-undecanona sustituidos y su uso como antagonistas de las neuroquininas. | |
| CN112074511B (zh) | 取代的苯并二唑及其在治疗中的用途 | |
| ES2310260T3 (es) | Derivados sustituidos de 4-(4-piperidin-4-il-piperazin-1-il)-azepan y su uso como antagonistas de la neuroquinina. | |
| CN112088160B (zh) | 1-咪唑并噻二唑并-2h-吡咯-5-酮衍生物 | |
| KR20070043965A (ko) | Nr2b 수용체 길항물질인 키뉴렌산 아미드 유도체 | |
| WO2015163431A1 (ja) | 片頭痛治療剤 | |
| EA040709B1 (ru) | Производные индола и бензимидазола в качестве двойных антагонистов рецепторов 5-ht2a и 5-ht6 | |
| HK40033115A (en) | Indole and benzimidazole derivatives as dual 5-ht2a and 5-ht6 receptor antagonists | |
| HK40033115B (zh) | 作为双重5-ht2a和5-ht6受体拮抗剂的吲哚和苯并咪唑衍生物 | |
| EP1707206A1 (de) | Piperazinderivate zur inhibition von Beta-Sekretase, Cathepsin D, Plasmepsin ll und HIV-Protease und zur Behandlung von Malaria, Alzheimer und AIDS |