ES2953821T3 - Inhibidores de KIF18A - Google Patents
Inhibidores de KIF18A Download PDFInfo
- Publication number
- ES2953821T3 ES2953821T3 ES19845855T ES19845855T ES2953821T3 ES 2953821 T3 ES2953821 T3 ES 2953821T3 ES 19845855 T ES19845855 T ES 19845855T ES 19845855 T ES19845855 T ES 19845855T ES 2953821 T3 ES2953821 T3 ES 2953821T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- alq
- compound
- methyl
- nrara
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Treating Waste Gases (AREA)
Abstract
Compuestos de fórmula (I): (I), como se define en el presente documento, y sus intermedios sintéticos, que son capaces de modular la proteína KIF18A, influyendo así en el proceso del ciclo celular y la proliferación celular para tratar el cáncer y las enfermedades relacionadas con el cáncer. La invención también incluye composiciones farmacéuticas, incluidos los compuestos, y métodos para tratar estados patológicos relacionados con la actividad de KIF18A. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Inhibidores de KIF18A
La invención se refiere al campo de los agentes farmacéuticos y, más específicamente, se refiere a compuestos y composiciones útiles para modular KIF18A, y para usos y métodos para controlar la proliferación celular y para tratar el cáncer.
Antecedentes de la invención
El cáncer es una de las enfermedades más extendidas que afecta a la humanidad y la causa principal de muerte en el mundo. En un esfuerzo para encontrar un tratamiento eficaz o una cura de los muchos cánceres diferentes en el último par de décadas, numerosos grupos han invertido una cantidad tremenda de tiempo, esfuerzo y recursos financieros. Sin embargo, hasta la fecha, de los tratamientos y terapias disponibles contra el cáncer, solo unos pocos ofrecen algún grado considerable de éxito.
El cáncer está a menudo caracterizado por una proliferación celular desregulada. El daño a uno o más genes, responsable de las vías celulares, que controlan el avance de la proliferación a través del ciclo celular y el ciclo del centrosoma, puede producir la pérdida de la regulación normal de la proliferación celular. Estos genes desregulados pueden codificar diversos supresores tumorales o proteínas oncogénicas, que participan en una cascada de eventos, que conducen a un avance del ciclo celular y una proliferación celular no controladas. Se han identificado diversas proteínas cinasa y cinesina, que desempeñan funciones clave en el ciclo celular y la regulación mitótica y evolución de células en división normales y células cancerosas.
Las cinesinas son motores moleculares que desempeña funciones importantes en la división celular y en el transporte de orgánulos y vesículas intracelulares. La cinesina mitótica desempeña funciones en varios aspectos del ensamblaje del huso mitótico, segregación de cromosomas, separación y dinámica del centrosoma (revisado en O. Rath y F. Kozielski, Nature Review Cáncer, 12:527-39, 2012). Las cinesinas humanas se clasifican en 14 subfamilias basadas en la homología secuencial dentro del denominado “dominio motor”, esta actividad de la ATPasa de dominios impulsa el movimiento unidireccional a lo largo de los microtúbulos (MT). El dominio no motor de estas proteínas es responsable de la fijación de la carga; una “carga” puede incluir uno cualquiera de una variedad de diferentes orgánulos membranosos, sistemas de andamios de transducción de señales y cromosomas. Las cinesinas utilizan la energía de la hidrólisis del ATP para mover la carga a lo largo de los microtúbulos polarizados. Por tanto, las cinesinas se denominan a menudo motores dirigidos a “extremo más” o “extremo menos”.
El gen KIF18A pertenece a la subfamilia de la Cinesina-8 y es un motor dirigido a extremo más. Se cree que KIF18A influencia la dinámica en el extremo más de los microtúbulos del cinetocoro para controlar el posicionamiento correcto de los cromosomas y la tensión del huso mitótico. La deleción de KIF18A humano conduce a husos mitóticos más largos, un aumento de la oscilación del cromosoma en la metafase, y activación del punto de control del ensamblaje del huso mitótico en las células HeLa de cáncer de cuello uterino (MI Mayr et al., Current Biology 17, 488-98, 2007). KIF18A parece ser una diana viable para el tratamiento del cáncer. KIF18A está sobreexpresado en diversos tipos de cánceres, incluidos, sin carácter limitante, cánceres de colon, mama, pulmón, páncreas, próstata, vejiga, cabeza, cuello, cuello uterino y ovario. Además, la deleción genética o atenuación génica, o inhibición de KIF18A afecta al aparato del huso mitótico en líneas de células cancerosas. Particularmente, se ha observado que la inhibición de KIF18A induce la detención de las células mitóticas, una conocida vulnerabilidad que puede promover la muerte celular en la mitosis mediante apoptosis, catástrofe mitótica, o letalidad impulsada por multipolaridad o muerte tras el desfase mitótico en la interfase. Por consiguiente, existe un gran interés en el hallazgo de inhibidores de las proteínas KIF18A.
Por tanto, la inhibición de la actividad ATPasa de KIF18A es un enfoque prometedor para el desarrollo de agentes anticancerosos novedosos.
El documento WO2011085261 divulga derivados de benzamida como agentes anticancerosos.
Compendio de la invención
La presente invención proporciona una nueva clase de compuestos útiles para modular la proteína KIF18A sola o en un complejo unido a microtúbulos para tratar las afecciones y/o enfermedades mediadas por KIF18A, que incluyen cáncer, inflamación, o ciliopatologías.
El alcance de la invención está definido por las reivindicaciones. Cualesquiera referencias en la descripción de los métodos de tratamiento se refieren a los compuestos, composiciones farmacéuticas y medicamentos de la presente invención para su uso en un método para el tratamiento del cuerpo humano (o animal) por terapia (o para diagnóstico).
Los compuestos proporcionados por la invención tienen actividad moduladora de KIF18A basada en los MT y, en particular, actividad inhibidora de KIF18A. Con este fin, la invención proporciona también el uso de estos compuestos, así como también las sales farmacéuticamente aceptables de estos, en la preparación y fabricación de una composición o medicamento farmacéutico para el tratamiento terapéutico, profiláctico, agudo o crónico de enfermedades y trastornos mediados por KIF18A, que incluyen, sin carácter limitante, cáncer. Por tanto, los compuestos de la invención son útiles en la fabricación de medicamentos anticancerosos. La invención proporciona
también procesos para preparar compuestos de Fórmula I, así como también intermedios útiles en tales procesos. En la realización 1, la presente invención proporciona un compuesto de Fórmula (I), un compuesto de fórmula I:

o cualquier sal farmacéuticamente aceptable de este, donde:
X1 es N o -CR6;
R1 es un grupo -Z-R12 donde Z es -alq C0-4-, -NR11-, -NR11SO2-alq C0-4-, -SO2NR11-alq C0-4-, -NR11SO2NR11-, -NR11SO2NR11-C(=O)-O-, -alq C0-4-S(=O)(=NH)-, alq C0-4-NR11-S(=O)(=NH), -alq C0-4-S-, -alq C0-4-S(=O)-, -alq C0-4-SO2-, alq C0-4-O-, -P-, -P(=O), -P(=O)2, -(C=O)-, -(C=O)NR11-, -C=N(OH)-, o -NR11(C=O); o el grupo -Z-R12 es -N=S(=O)-(R12)2 , donde el par de los dos R12 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1,2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S;
R2 es halo o un grupo -Y -R 13, donde Y es -alq C0-4-, -N(alq C0-1)-alq C0-4-, -C(=O)NRaRa(alq C1-4)-, -O-alq C0-4-, -S-, -S=O, -S(=O)2-, -SO2N(alq C0-1)-alq C0-4-, -N(alq C0-1)-SO2-alq C0-4-, -alq C0-4-S(=O)(=NH)-, -(C=O)-, -alq C0-4-(C=O)-O-; o
el grupo -Y-R13 es -N=S(=O)-(R13)2 , donde el par de los dos R13 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1,2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S;
R3 es H, metilo o etilo;
R4 es H, halo, CN, alq C1-4 o haloalq C1-4;
R5 es H, halo, alq C1-8 o haloalq C1-4;
R6 es H, halo, CN, alq C1-8, haloalq C1-4, -O-alq C0-6-, o R6a;
R7 es H, halo, alq C1-8 o haloalq C1-4;
R8 es H, halo, alq C1-8 o haloalq C1-4;
R9 es H, halo, alq C1-8 o haloalq C1-4;
Rx se selecciona del grupo que consiste en y
y

Cada uno de R10a, R10b, R10c, R10d, R10e, R10f, R10g, R10h, R10i, y R10i es H, halo, R10k, o R1
o como alternativa, cada uno del par R10a y R10b, par R10c y R10d, par R10e y R10f, par R10g y R10h o par R10¡ y R10j, se puede combinar independientemente con el átomo de carbono unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado espiro con el anillo Rx; donde dicho anillo monocíclico de 3, 4, 5 o 6 miembros contiene 0, 1, 2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S, y además donde dicho anillo monocíclico de 3, 4, 5 o 6 miembros está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -NRaRa, u oxo;
R11 es H o alq C1-8;
R12 es H, R12a o R12b;
R13 es R13a o R13b;
R6a, R10k, R12a y R13a se seleccionan independientemente en cada caso del grupo que consiste en un anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 4, 5, 6, 7, 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1,2 o 3 átomos de N y 0, 1, o 2 átomos seleccionados entre O y S, que está sustituido con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -C(=O)Rb, -C(=O)ORa, -C(=O)NRaRa, -C(=NRa)NRaRa, -OC(=O)Rb, -OC(=O)NRaRa, -Oalq C2-6NRaRa, -Oalq C2-6ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRaRa, -NRaRa, -N(Ra)C(=O)Rb, -N(Ra)C(=O)ORb,-N(Ra)C(=O)NRaRa, -N(Ra)C(=NRa)NRaRa, -N(Ra)S(=O)2Rb, -N(Ra)S(=O)2NRaRa, -NRaalq C2-6NRaRa, -NRaalq C2-6ORa, -alq C1-6NRaRa, -alq C1-6ORa, -alq C1-6N(Ra)C(=O)Rb, -alq C1-6OC(=O)Rb, -alq C1-6C(=O)NRaRa, -alq C1-6C(=O)ORa, R14, y oxo;
R101, R12b y R13b se seleccionan independientemente en cada caso del grupo que consiste en alq C1-6 sustituido con 0, 1, 2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, -C(=O)ORa, -ORa, -haloalq C1-2, -Ohaloalq C1-4, CN, NH2, NH(CHa) o N(CHa)2;
R14 se selecciona independientemente en cada caso del grupo que consiste en anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 4, 5, 6, 7, 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1, 2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S, que está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -C(=O)Rb, -C(=O)ORa, -C(=O)NRaRa, -C(=NRa)NRaRa, -OC(=O)Rb, -OC(=O)NRaRa, -Oalq C2-sNRaRa, -Oalq C2-sORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRaRa, -NRaRa, -N(Ra)C(=O)Rb, -N(Ra)C(=O)ORb,-N(Ra)C(=O)NRaRa, -N(Ra)C(=NRa)NRaRa, -N(Ra)S(=O)2Rb, -N(Ra)S(=O)2NRaRa, -NRaalq C2-sNRaRa, -NRaalq C2-sORa, -alq C1-sNRaRa, -alq C1-sORa, -alq C1-sN(Ra)C(=O)Rb, -alq C1-sOC(=O)Rb, -alq C1-sC(=O)NRaRa, -alq C1-sC(=O)ORa, y oxo;
Ra es independientemente, en cada caso, H o Rb; y
Rb es independientemente, en cada caso, alq C1-6, fenilo o bencilo, donde el alq C1-6 está sustituido con 0, 1, 2 o 3 sustituyentes seleccionados entre halo, -OH, -Oalq C1-4, -NH2, -NHalq C1-4, -OC(=O)alq C1-4 o -N(alq C1-4)alq C1-4; y el fenilo o bencilo está sustituido con 0, 1,2 o 3 sustituyentes seleccionados entre halo, alq C1-4, halolaq C1-3, -OH, -Oalq C1-4, -NH2, -NHalq C1-4, -OC(=O)alq C1-4 o -N(alq C1-4)alq C1-4.
En la realización 2, la presente invención proporciona compuestos donde Rx es
En la realización 3, la presente invención proporciona compuestos donde X1 es -CR6; que tienen la fórmula (la):

En la realización 4, la presente invención proporciona compuestos donde X1 es N; que tienen la fórmula (Ib):

En la realización 5, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R3 es H o metilo.
En la realización 6, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde cada uno de R10c, R10d, R10e, R10f, R10g, R10h, R10i y R10j es H, halo, alq C1-6 o haloalq C1-4; y cada uno del par R10a y R10b se combina con el átomo de carbono unido a cada uno de ellos para formar un anillo monocíclico de 3, 4 o 5 miembros saturado espiro con el anillo Rx; donde dicho anillo contiene 0, 1, 2 o 3 átomos de N y 0 o 1 átomos seleccionado entre O y S.
En la realización 7, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde cada uno de R10c, R10d, R10e, R10f, R10g, R10h, R10i y R10j es H, metilo o etilo; y cada uno del par R10a y R10b se combina con el átomo de carbono unido a cada uno de ellos para formar un anillo ciclopropilo, ciclobutilo o ciclopentilo espiro con el anillo Rx.
En la realización 8, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde Rx se selecciona entre:

En la realización 9, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones
anteriores, o sales farmacéuticamente aceptables de estos, donde Rx es
En la realización 10, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde Z está ausente, es NH-, -NHSO2-(CH2)0-4-, -N(CH3)-SO2-(CH2)0-4-, -NCH3SO2NH, -NHSO2NH-C(=O)-O-, -SO2NH-(CH2)0-4-, -(CH2)0-2-S(=O)(=NH)-, -(CH2)0-2-S-,-(CH2)0-2-S(=O)-, (CH3CH)-S(=O)-, -(CH2)0-2-SO2-, -O-, -P(=O), -(C=O)- o -NH(C=O)-.
En la realización 11, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde el grupo -Z-R12 es -N=S(=O)-(R12)2 , donde el par de los dos R12 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1, 2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S; que se selecciona entre:

En la realización 12, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R1 es -Z-R12, donde Z está ausente, es NH-, -NHSO2-(CH2)0-4-, -N(CH3)-SO2-(CH2)0-4-, -NCH3SO2NH, -NHSO2NH-C(=O)-O-, -SO2NH-(CH2)0-4-, -(CH2)0-2-S(=O)(=NH)-, -(CH2)0-2-S-, -(CH2)0-2-S(=O)-, (CH3CH)-S(=O)-, -(CH2)0-2-SO2-, -O-, -P(=O), -(C=O)- o -NH(C=O); y R12 se selecciona entre:
(a) H;
(b) ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, oxiranilo, oxetanilo, tetrahidrofuranilo, azetidinilo, imidazolilo, morfolinilo, pirrolidinilo, piperazinilo,


seleccionados entre donde cada anillo está sustituido con 0, 1, 2 o 3 OH, F, metilo, -CH2OH, -C(=O)OCH3, -C(=O)OC(CH3)3, NH2, CN y oxo; o alq C1-6 sustituido con 0, 1, 2 o 3 OH, F, -C(=O)OCH3, -NH2, -NH(CH3) o -N(CH3)2.
En la realización 13, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R1 es un grupo -Z-R12, donde Z es -NHSO2- o -SO2NH-; y R12 es oxetanilo, ciclopropilo, o R12 es alq C1-6 sustituido con 0, 1,2 o 3 grupos OH.
En la realización 14, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R1 es un grupo -Z-R12, donde Z es -NHSO2- y R12 es -C H 2-CH2-OH.
En la realización 15, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde
R2 es halo o un grupo -Y -R 13, donde Y está ausente, es -SO2NH-(CH2)0-4-, NH-, -NH-SO2-(CH)2)0-4-, -O-(CH2)0-4, -O-(CH(CH3))-, -(CH2)0-4-S(=O)(=NH)-, -(C=O)-, -(CH2)0-4-(C=O)-O- o -(CH2)1-4;
R13 es un anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1, 2 o 3 átomos de N y 0 o 1 átomos seleccionados entre O y S, que está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -OH, -Ohaloalq C1-4, CN, R14 y oxo; o; R13 es alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, -OH, -Ohalolaq C1-4 o CN.
En la realización 16, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R2 es un anillo monocíclico de 5 o 6 miembros saturado donde cada uno de dichos anillos contiene 0, 1 o 2 átomos de N y 0 o 1 átomos de O, y donde cada uno de dichos anillos está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -OH, -OCH3, -Ohaloalq C1-4, CN, R14 y oxo.
En la realización 17, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R2 es:
(a) F, Br;
(b) un grupo -Y -R 13, donde Y está ausente; y R13 es morfolinilo, piperidinilo, azetidinilo, pirrolidinilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, piperazinilo, tetrahidrofuranilo, tetrahidropiranilo, piridinilo, pirimidinilo, 3,6-dihidro-2H-piranilo,

con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, metilo, CF3, CH2OH, CH(CH3)OH, C(CH3)2OH, -OH, -OCHF2, CN, oxo o ciclopropilo; o
(c) un grupo -Y -R 13, donde Y está ausente, es -SO2NH-, NH, -O-, S(=O)(=NH)- , -O-(CH2), -O-(CH(CH3))-, C(=O)-,
C(=O)-O-, -CH2C(=O)-O-, o -CH2-; y donde R13 es 

; donde cada anillo está sustituido con 0, 1, 2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo,
CF3, -OH o CN: o R13 es H o alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN.
En la realización 18, la presente invención proporciona compuestos de acuerdo con las realizaciones 1-17, o sales farmacéuticamente aceptables de estos, donde R2 es (a) halo; (b) un grupo -Y R 13, donde Y está ausente; y R13 es morfolinilo, piperidinilo, azetidinilo, pirrolidinilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, piperazinilo, tetrahidrofuranilo,


dichos anillos está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH, -OCHF2, CN y oxo; o (c) un grupo -Y -R 13, donde Y es NH, -O-, -0-(CH2)-, -O-(CH2)-(CH2)- o
-O-(CH2)-(CH2)-(CH2)-, y donde R13 es o R13 es alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN.
o R13 es alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN.
En la realización 19, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R2 es

En la realización 20, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R2 es morfolinilo o piperidinilo sustituido con 0, 1, 2 o 3 grupo seleccionados entre F, Cl, Br, metilo, CF3, -OH, -OCHF2, CN u oxo.
En la realización 21, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R2 es morfolinilo sustituido con 1, 2 o 3 grupos metilo.
En la realización 22, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R4 se selecciona entre H, F, metilo, CN o Br. En la realización 23, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R4 es H.
En la realización 24, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R5 es H.
En la realización 25, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R6 es H, metilo, ciclopropilo, CN, CF3 o azetidinilo. En la realización 26, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R7 es H.
En la realización 27, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R8 es H o F.
En la realización 28, la presente invención proporciona compuestos de acuerdo con cualquiera de las realizaciones anteriores, o sales farmacéuticamente aceptables de estos, donde R9 es H o F.
En la realización 29, la presente invención proporciona un compuesto, o sales farmacéuticamente aceptables de este, seleccionado entre:





o cualquier sal farmacéuticamente aceptable de estos.
En la realización 30, la presente invención proporciona composiciones farmacéuticas que comprenden un compuesto, o sales farmacéuticamente aceptables de este, de acuerdo con una cualquiera de las realizaciones 1 -29, y un diluyente o portador farmacéuticamente aceptable.
En la realización 31, la presente invención proporciona un método para tratar una afección que se puede tratar con inhibidores de KIF18a, comprendiendo el método administrar a un paciente que lo necesita una cantidad terapéuticamente eficaz del compuesto de acuerdo con las realizaciones 1-29 o el compuesto de acuerdo con la realización 30.
En la realización 32, la presente invención proporciona el método de la reivindicación 31, donde dicha afección es cáncer seleccionado del grupo que consiste en (a) un tumor sólido o derivado hematológicamente seleccionado entre
cáncer del cáncer de vejiga, de endometrio, escamocelular de pulmón, mama, colon, riñón, hígado, pulmón, cáncer pulmonar microcítico, de esófago, vesícula biliar, cerebro, cabeza y cuello, ovario, páncreas, estómago, cuello uterino, tiroides, próstata y piel, (b) un tumor hematopoyético de linaje linfoide seleccionado entre leucemia, leucemia linfocítica aguda, leucemia linfoblástica aguda, linfoma de linfocitos B, linfoma de linfocitos T, linfoma de Hodgkin, linfoma no hodgkiniano, linfoma de tricoleucocitos y linfoma de Burkitt, (c) un tumor hematopoyético de linaje mieloide seleccionado entre leucemias mielógenas aguda y crónica, síndrome mielodisplásico y leucemia promielocítica (d) un tumor de origen mesenquimatoso seleccionado entre fibrosarcoma y rabdomiosarcoma, (e) un tumor del sistema nervioso central y periférico seleccionado entre astrocitoma, neuroblastoma, glioma y schwannoma, o (f) un melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderomia pigmentosa, queratoctantoma, cáncer folicular tiroideo o sarcoma de Kaposi.
En la subrealización 32a, la presente invención proporciona el método de la realización 31, donde dicha afección es cáncer seleccionado del grupo que consiste en melanoma, cáncer de próstata, cáncer de cuello uterino, cáncer de mama, cáncer de colon, sarcoma, o leucemia. Véase: Zhang C. et. a l, "Kif18A is involved in human breast carcinogenesis", Carcinogenesis, septiembre de 2010; 31(9):1676-84. doi: 10.1093/carcin/bgq134. Publicación electrónica de 1 de julio de 2010. Véase también: (1) https://www.proteinatlas.org/ENSG00000121621-KIF18A/pathology; (2) Nagahara, M. et. al., ''Kinesin 18A expression: clinical relevance to colorectal cancer progression'', Int. J. Cancer: 129, 2543-2552 (2011) VC 2011 UIC; y (3) Yu, Y et. al., "The Role of Kinesin Family Proteins in Tumorigenesis and Progression - Potential Biomarkers and Molecular Targets for Cancer Therapy", Cancer 2010;116:5150-60. VC 2010 American Cancer Society.
En la realización 33, la presente invención proporciona un método para reducir el tamaño de un tumor sólido en un sujeto, comprendiendo el método administrar al sujeto que lo necesita una cantidad terapéuticamente eficaz del compuesto de acuerdo con las realizaciones 1-29, o la composición de acuerdo con la realización 30.
En la realización 34, la presente invención proporciona un método para tratar un trastorno de proliferación celular en un sujeto, comprendiendo el método administrar al sujeto que lo necesita una cantidad terapéuticamente eficaz del compuesto de acuerdo con las realizaciones las realizaciones de 1-29, o la composición de acuerdo con la reivindicación 30.
En la realización 35, la presente invención proporciona un método para inhibir KIF18A en una célula, que comprende poner en contacto la célula con un compuesto, o sales farmacéuticamente aceptables de este, de acuerdo con las realizaciones las realizaciones de 1-29, o la composición de acuerdo con la realización 30.
En la realización 36, la invención proporciona un método para preparar un compuesto de Fórmula (I) como se describe en la presente.
En la realización 37, la invención proporciona un compuesto intermedio utilizado en el método de preparación de un compuesto de Fórmula (I) como se describe en la presente.
Descripción detallada de la invención
La presente invención incluye todos los compuestos marcados con isótopos farmacéuticamente aceptables de la presente invención donde uno o más átomos son sustituidos por átomos que tienen el mismo número atómico, pero una masa atómica o un número másico diferente de la masa atómica o número másico que predomina en la naturaleza.
Los ejemplos de isótopos adecuados para la inclusión en los compuestos de la invención incluyen, sin carácter limitante, isótopos de hidrógeno, tales como 2H y 3H, carbono, tales como 11C, 13C y 14C, cloro, tales como 38Cl, flúor, tales como 18F, yodo, tales como 123I y 125I, nitrógeno, tales como 13N y 15N, oxígeno, tales como 15O, 17O y 18O, fósforo, tales como 32P, y azufre, tales como 35S.
Ciertos compuestos marcados con isótopos de la presente invención, por ejemplo, aquellos que incorporan un isótopo radioactivo, son útiles en los estudios de distribución tisulares de fármaco y/o sustrato. Los isótopos radiactivos tritio, es decir, 3H, y carbono-14, es decir, 14C, son particularmente útiles para este fin a la vista de su facilidad de incorporación y de los medios de detección disponibles.
La sustitución con isótopos más pesados tales como deuterio, es decir 2H, puede dar como resultado ciertas ventajas terapéuticas resultantes de una mayor estabilidad metabólica, por ejemplo, mayor semivida in vivo o requisitos posológicos reducidos, y por lo tanto, puede preferirse en algunas circunstancias.
La sustitución con isótopos que emiten positrones, tales como 11C, 18F, 15O y 13N, puede ser útil en estudios de topografía por emisión de positrones (PET, por sus siglas en inglés) para examinar la ocupación del receptor por el sustrato.
Los compuestos marcados con isótopos de la presente invención se pueden preparar generalmente mediante técnicas convencionales conocidas por los expertos en la técnica o mediante procesos análogos a los descritos en los Ejemplos y Preparaciones que se adjuntan utilizando un reactivo marcado con isótopos adecuado en lugar del reactivo no marcado empleado previamente.
Los solvatos farmacéuticamente aceptables de acuerdo con la invención incluyen aquellos en donde el disolvente de cristalización puede estar sustituido con isótopos, por ejemplo, D2O, d6-acetona, d6-DMSO.
Las realizaciones específicas de la presente invención incluyen los compuestos ejemplificados en los Ejemplos siguientes y sus sales, complejos, solvatos, polimorfos, estereoisómeros, metabolitos, profármacos, y otros derivados de estos farmacéuticamente aceptables.
Salvo que se especifique de otra forma, las siguientes definiciones se aplican a términos que se encuentran en la memoria descriptiva y en las reivindicaciones:
“Alq Ca-p” se refiere a un grupo alquilo que comprende un mínimo de átomos de carbono a y un máximo de átomos de carbono p en una relación ramificada o lineal o cualquier combinación de los tres, donde a y p representan números enteros. Los grupos alquilo descritos en esta sección pueden contener también uno o dos dobles o triples enlaces. Una designación de alq C0 indica un enlace directo. Los ejemplos de alq C1-6 incluyen, sin carácter limitante, los siguientes:

“Grupo benzo”, solo o combinado, se refiere al radical divalente C4H4=, una representación del cual es -CH=CH-CH=CH-, que cuando se une vecinalmente a otro anillo forma un anillo de tipo benceno, por ejemplo, tetrahidronaftileno, indol y similares.
Los términos “oxo” y “tioxo” representan los grupos =O (como en carbonilo) y =S (como en tiocarbonilo), respectivamente.
“Halo” o “halógeno” se refiere a un átomo de halógeno seleccionado entre F, Cl, Br y I.
“Haloalq Ca-p” se refiere a un grupo alq, como se ha descrito anteriormente, donde cualquier número, al menos uno, de los átomos de hidrógeno unidos a la cadena alq son reemplazados por F, Cl, Br o I.
El grupo N(Ra)Ra y similares incluyen sustituyentes donde los dos grupos Ra forman juntos un anillo, que incluye opcionalmente un átomo de N, O o S, e incluye grupos tales como:

El grupo N(alq Ca-p)alq Ca-p, donde a y p son como se han definido anteriormente, incluye sustituyentes donde los dos grupos alq Ca-p forman juntos un anillo, que incluye opcionalmente un átomo de N, O o S, e incluye grupos tales como:


“Anillo bicíclico” se refiere a un grupo que presenta dos anillos unidos. Un anillo bicíclico puede ser carbocíclico (todos los átomos del anillo son carbonos), o heterocíclico (los átomos del anillo consisten, por ejemplo, en 1, 2 o 3 heteroátomos, tales como N, O, o S, además de los átomos de carbono). Los dos anillos pueden ser alifáticos (por ejemplo, decalina y norbornano) o pueden ser aromáticos (por ejemplo, naftaleno), o una combinación de alifático y aromático (por ejemplo, tetralina). Los anillos bicíclicos incluyen (a) compuestos espirocíclicos, donde los dos anillo comparten solo un único átomo, el átomo espiro, que es habitualmente un carbono cuaternario. Los ejemplos de compuestos espirocíclicos incluyen, sin carácter limitante:

(b) compuestos bicíclicos condensados, donde dos anillos comparten dos átomos adyacentes. En otras palabras, los anillos comparten un enlace covalente, es decir, los átomos que son cabeza de puente están conectados directamente (por ejemplo, a-thujene y decalina). Los ejemplos de anillos bicíclicos condensados incluyen, sin carácter limitante:

y (c) compuestos bicíclicos con puente, donde los dos anillos comparten tres o más átomos, que separan los dos átomos que son cabeza de puente mediante un puente que contiene al menos un átomo. Por ejemplo, norbornano, conocido también como biciclo[2.2.1]heptano, se puede considerar como un par de anillos de ciclopentano que comparten cada uno tres de sus cinco átomos de carbono. Los ejemplos de anillos bicíclicos con puente incluyen, sin carácter limitante:
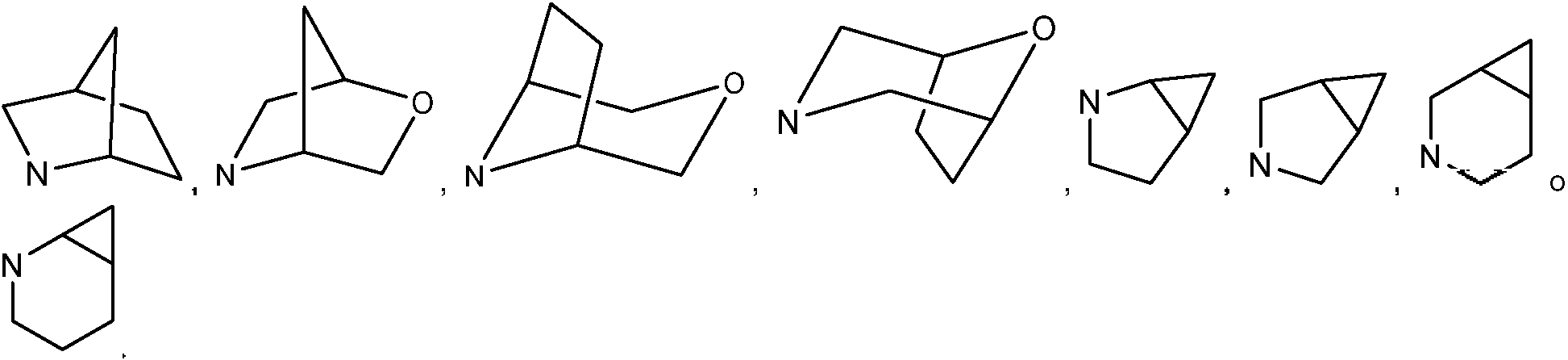
“Carbociclo” o “Carbocíclico” se refiere a un anillo que comprende por sí mismo o combinado con otros términos, representa, a menos que se indique de otro modo, una versión cíclica de “alq Ca-p”. Los ejemplos de carbociclo incluyen ciclopentilo, ciclohexilo, 1-ciclohexenilo, 3-ciclohexenilo, cicloheptilo, ciclobutileno, ciclohexileno y similares.
“Heterociclo” o “Heterocíclico” se refiere a un anillo que comprende al menos un átomo de carbono y al menos un átomo diferente seleccionado entre N, O y S. Los ejemplos de heterociclos que se pueden encontrar en las reivindicaciones incluyen, sin carácter limitante, los siguientes:


“Sal farmacéuticamente aceptable” se refiere a una sal preparada mediante medios convencionales, y son muy conocidas por los expertos en la técnica. Las “sales farmacológicamente aceptables” incluyen sales básicas de ácidos orgánicos e inorgánicos, que incluyen, sin carácter limitante, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido fosfórico, ácido metanosulfónico, ácido etanosulfónico, ácido málico, ácido acético, ácido oxálico, ácido tartárico, ácido cítrico, ácido láctico, ácido fumárico, ácido succínico, ácido maleico, ácido salicílico, ácido benzoico, ácido fenilacético, ácido mandélico y similares. Cuando los compuestos de la invención incluyen una función ácida tal como un grupo carboxi, entonces, los pares de cationes farmacéuticamente aceptables adecuados para el grupo carboxi son muy conocidos por los expertos en la técnica e incluyen cationes alcalinos, alcalinotérreos, de amonio, de amonio cuaternario y similares. Para consultar ejemplos adicionales de “sales farmacológicamente aceptables”, véase más abajo y Berge et. al., J. Pharm. Sci. 66:1 (1977).
“Saturados, parcialmente saturados o insaturados” incluye sustituyentes saturados con hidrógenos, sustituyentes completamente insaturados con hidrógenos y sustituyentes parcialmente saturados con hidrógenos.
“Grupo saliente” se refiere generalmente a grupos que se pueden desplazar fácilmente con un nucleófilo, tales como una amina, un tiol o un nucleófilo alcohólico. Tales grupos salientes son muy conocidos en la técnica. Los ejemplos de tales grupos salientes incluyen, sin carácter limitante, N-hidroxisuccinimida, N-hidroxibenzotriazol, haluros, triflatos, tosilatos y similares. Los grupos salientes preferidos se indican en la presente cuando es adecuado.
“Grupo protector” se refiere generalmente a grupos muy conocidos en la técnica que se utilizan para prevenir que los grupos reactivos seleccionados, tales como carboxi, amino, hidroxi, mercapto y similares, experimenten reacciones no deseadas, tales como nucleófilas, electrófilas, de oxidación, reducción y similares. Los grupos protectores preferidos se indican en la presente cuando es adecuado. Los ejemplos de grupos protectores de amino incluyen, aunque sin carácter limitante, aralquilo, aralquilo sustituido, cicloalquenilalquilo y cicloalquenilalquilo sustituido, alilo, alilo sustituido, acilo, alcoxicarbonilo, aralcoxicarbonilo, sililo y similares. Los ejemplos de aralquilo incluyen, sin carácter limitante, bencilo, orto-metilbencilo, tritilo y benchidrilo, que pueden estar sustituidos opcionalmente con halógeno, alquilo, alcoxi, hidroxi, nitro, acilamino, acilo y similares, y sales, tales como sales de amonio y de fosfonio.
Los ejemplos de grupos arilo incluyen fenilo, naftilo, indanilo, antracenilo, 9-(9-fenilfluorenilo), fenantrenilo, durenilo y similares. Los ejemplos de radicales cicloalquenilalquilo o cicloalquilenilalquilo sustituido, que tienen preferentemente 6-10 átomos de carbono, incluyen, sin carácter limitante, ciclohexenilmetilo y similares. Los grupos acilo, alcoxicarbonilo y aralcoxicarbonilo adecuados incluyen benciloxicarbonilo, f-butoxicarbonilo, isobutoxicarbonilo, benzoílo, benzαlo sustituido, butirilo, acetilo, trifluoroacetilo, tricloroacetilo, ftalαlo y similares. Es posible utilizar una mezcla de grupos protectores para proteger el mismo grupo amino, de modo que un grupo amino primario se puede proteger tanto con un grupo aralquilo como con un grupo aralcoxicarbonilo. Los grupos protectores de amino también pueden formar un anillo heterocíclico con el nitrógeno al cual están unidos, por ejemplo, 1,2-bis(metilen)benceno, ftalimidilo, succinimidilo, maleimidilo y similares y donde estos grupos heterocíclicos pueden incluir además anillos arilo y cicloalquilo adyacentes. Además, los grupos heterocíclicos pueden ser monosustituidos, disustituidos o trisustituidos, tales como nitroftalimidilo. Los grupos amino también se pueden proteger contra reacciones no deseadas, tales como oxidación, mediante la formación de una sal de adición, tal como clorhidrato, con ácido toluenosulfónico, ácido trifluoroacético y similares. Muchos de los grupos protectores de amino son también adecuados para la protección de grupos carboxi, hidroxi y mercapto. Por ejemplo, grupos aralquilo. Los grupos alquilo también son adecuados para la protección de los grupos hidroxi y mercapto, tales como ferf-butilo.
Los grupos protectores de tipo sililo son átomos de silicio sustituidos opcionalmente con uno o más grupos alquilo, arilo y aralquilo. Los grupos protectores de tipo sililo adecuados incluyen, sin carácter limitante, trimetilsililo, trietilsililo, triisopropilsililo, ferf-butildimetilsililo, dimetilfenilsililo, 1,2-bis(dimetilsilil)benceno, 1,2-bis(dimetilsilil)etano y difenilmetilsililo. La sililación de grupos amino proporciona grupos monosililamino o disililamino. La sililación de compuestos de aminoalcohol puede dar lugar a un derivado de W,W,0-tris¡lilo. La eliminación de la función sililo de una función éter de sililo se lleva a cabo fácilmente mediante tratamiento con, por ejemplo, un reactivo de tipo hidróxido metálico o fluoruro de amonio, ya sea en un paso de reacción discreto o in sifu durante una reacción con el grupo alcohol. Los agentes sililantes adecuados son, por ejemplo, cloruro de trimetilsililo, cloruro de ferf-butildimetilsililo, cloruro de fenildimetilsililo, cloruro de difenilmetilsililo, o sus productos combinados con imidazol o DMF. Los métodos para la sililación de aminas y la eliminación de grupos protectores de tipo sililo son muy conocidos por los expertos en la técnica. Los métodos de preparación de estos derivados de amina a partir de los correspondientes aminoácidos, amidas de aminoácidos, o ésteres de aminoácidos son muy conocidos por los expertos en la técnica de la química orgánica e incluyen la química de aminoácidos/ésteres de aminoácidos o la química de aminoalcoholes.
Los grupos protectores se eliminan en condiciones que no afectarán la porción restante de la molécula. Estos métodos son muy conocidos en la técnica e incluyen la hidrólisis ácida, hidrogenólisis y similares. Un método preferido conlleva la eliminación de un grupo protector, tal como la eliminación de un grupo benciloxicarbonilo mediante hidrogenólisis utilizando paladio sobre carbón en un sistema disolvente adecuado tal como un alcohol, ácido acético y similares o las mezclas de estos. Un grupo protector de f-butoxicarbonilo se puede eliminar con un ácido orgánico o inorgánico, tal como HCl o ácido trifluoroacético, en un sistema disolvente adecuado, tal como dioxano o cloruro de metileno. La sal de amino resultante se puede neutralizar fácilmente para generar la amina libre. El grupo protector de carboxi, tal como metilo, etilo, bencilo, ferf-butilo, 4-metoxifenilmetilo y similares, se puede eliminar en condiciones de hidrólisis e hidrogenólisis que son muy conocidas por los expertos en la técnica.
Cabe destacar que los compuestos de la invención pueden contener grupos que pueden existir en formas tautoméricas, tales como grupos guanidina y amidina cíclicos y acíclicos, grupos heteroarilo sustituidos con heteroátomos (Y' = O, S, NR), y similares, que se ilustran en los siguientes ejemplos:

y si bien en la presente se nombra, describe, presenta y/o reivindica una forma, se pretende que todas las formas tautoméricas se incluyan inherentemente en dicho nombre, descripción, presentación y/o reivindicación.
También se contemplan en la presente invención profármacos de los compuestos de esta invención. Un profármaco es un compuesto activo o inactivo que se modifica químicamente mediante la acción fisiológica in vivo, tal como hidrólisis, metabolismo y similares, para obtener un compuesto de la presente invención tras la administración del profármaco a un paciente. La idoneidad y las técnicas implicadas en la elaboración y el uso de profármacos son muy conocidas por los expertos en la técnica. Para una exposición general de profármacos que impliquen ésteres véase Svensson y Tunek Drug Metabolism Reviews 165 (1988) y Bundgaard Design of Prodrugs, Elsevier (1985). Los ejemplos de un anión carboxilato enmascarado incluyen varios ésteres, tales como alquilo (por ejemplo, metilo, etilo), cicloalquilo (por ejemplo, ciclohexilo), aralquilo (por ejemplo, bencilo, p-metoxibencilo) y alquilcarboniloxialquilo (por ejemplo, pivaloiloximetilo). Las aminas se han enmascarado como derivados sustituidos con arilcarboniloximetilo que son escindidos mediante esterasas in vivo que liberan el fármaco libre y formaldehído (Bungaard J. Med. Chem. 2503 (1989)). Asimismo, los fármacos que contienen un grupo NH ácido, tal como imidazol, imida, indol y similares, se han enmascarado con grupos N-aciloximetilo (Bundgaard Design of Prodrugs, Elsevier (1985)). Los grupos hidroxi se han enmascarado como ésteres y éteres. El documento EP 039051 (Sloan y Little, 11/4/81) divulga profármacos de ácido hidroxámico-base de Mannich, su preparación y su uso.
La memoria descriptiva y las reivindicaciones contienen una lista de especies que utiliza el lenguaje “seleccionado entre .. . y .. .” y “es .. . o .. .” (denominados a veces como grupos de Markush). Cuando se utiliza este lenguaje en la presente solicitud, a menos que se indique otra cosa, se pretende que se incluya el grupo como un total, o cualquiera de los miembros individuales de este, o cualquier subgrupo de este. El uso de este lenguaje tiene únicamente el objetivo de abreviar y no se pretende de ninguna manera que limite la eliminación de elementos o subgrupos individuales cuando sea necesario.
Composiciones farmacéuticas, dosificación y rutas de administración
También se proporcionan en la presente composiciones farmacéuticas que incluyen un compuesto tal como se divulga en la presente junto con un excipiente farmacéuticamente aceptable, tal como, por ejemplo, un diluyente o portador. Los compuestos y composiciones farmacéuticas adecuados para su uso en la presente invención incluyen aquellos donde el compuesto se puede administrar en una cantidad eficaz para lograr su fin deseado. La administración del compuesto se describe en más detalle a continuación.
El experto en la materia puede determinar las formulaciones farmacéuticas adecuadas de acuerdo con la ruta de administración y la dosis deseada. Véase, por ejemplo, Remington's Pharmaceutical Sciences, 1435-712 (18.a ed., Mack Publishing Co, Easton, Pensilvania, 1990). Las formulaciones pueden alterar el estado físico, la estabilidad, la velocidad de liberación in vivo y la velocidad de aclaramiento in vivo de los agentes administrados. Dependiendo de la ruta de administración, se puede calcular una dosis adecuada de acuerdo con el peso corporal, área superficial del cuerpo o tamaño del órgano. Los expertos en la técnica realizan de forma rutinaria el refinamiento adicional de los cálculos necesarios para determinar la dosis de tratamiento adecuada sin experimentación innecesaria, especialmente teniendo en cuenta la información posológica y los ensayos divulgados en la presente, así como los datos farmacocinéticos que se pueden obtener mediante ensayos clínicos en animales o seres humanos.
Las expresiones “farmacéuticamente aceptable” o “farmacológicamente aceptable” se refieren a entidades moleculares y composiciones que no producen reacciones adversas, alérgicas, u otras reacciones perjudiciales cuando se administran a un animal o a un ser humano. Tal como se utiliza en la presente, “farmacéuticamente aceptable” incluye todos y cada uno de los disolventes, medios de dispersión, recubrimientos, agentes antibacterianos y antifúngicos, agentes isotónicos y retardantes de la absorción y similares. En la técnica se conoce el uso de tales excipientes para sustancias farmacéuticamente activas. Excepto en la medida en que algún agente o medio convencional sea incompatible con las composiciones terapéuticas, se contempla su uso en las composiciones terapéuticas. Se pueden incorporar también principios activos complementarios a las composiciones. En realizaciones ejemplares, la formulación puede comprender sólidos de jarabe de maíz, aceite de cártamo alto oleico, aceite de coco, aceite de soya, L-leucina, fosfato de calcio tribásico, L-tirosina, L-prolina, acetato de L-lisina, DATEM (un emulsionante), L-glutamina, L-valina, fosfato de potasio dibásico, L-isoleucina, L-arginina, L-alanina, glicina, monohidrato de L-asparagina, L-serina, citrato de potasio, L-treonina, citrato de sodio, cloruro de magnesio, L-histidina, L-metionina, ácido ascórbico, carbonato de calcio, ácido L-glutámico, diclorhidrato de L-cistina, L-triptófano, ácido L-aspártico, cloruro de colina, taurina, m-inositol, sulfato ferroso, palmitato de ascorbilo, sulfato de zinc, L-carnitina, acetato de alfa-tocoferilo, cloruro sódico, niacinamida, tocoferoles mixtos, pantotenato de calcio, sulfato cúprico, clorhidrato de cloruro de tiamina, palmitato de vitamina A, sulfato de manganeso, riboflavina, clorhidrato de piridoxina, ácido fólico, beta-caroteno, yoduro de potasio, filoquinona, biotina, selenato de sodio, cloruro de cromo, molibdato de sodio, vitamina D3 y cianocobalamina.
El compuesto puede estar presente en una composición farmacéutica como una sal farmacéuticamente aceptable. Tal como se utiliza en la presente, las “sales farmacéuticamente aceptables” incluyen, por ejemplo, sales de adición de bases y sales de adición de ácidos.
Las sales de adición de bases farmacéuticamente aceptables se pueden formar con metales o aminas, tales como
metales alcalinos y alcalinotérreos o aminas orgánicas. Las sales farmacéuticamente aceptables de los compuestos también se pueden preparar con un catión farmacéuticamente aceptable. Los expertos en la técnica conocen bien los cationes farmacéuticamente aceptables adecuados, y estos incluyen cationes alcalinos, alcalinotérreos, de amonio y de amonio cuaternario. También son posibles carbonatos o hidrogenocarbonatos. Son ejemplos de metales utilizados como cationes el sodio, potasio, magnesio, amonio, calcio o férricos y similares. Los ejemplos de aminas adecuadas incluyen isopropilamina, trimetilamina, histidina, W,N-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, diciclohexilamina, etilendiamina, N-metilglucamina y procaína.
Las sales de adición de ácidos farmacéuticamente aceptables incluyen sales de ácidos inorgánicos u orgánicos. Los ejemplos de sales de ácidos adecuadas incluyen los clorhidratos, formiatos, acetatos, citratos, salicilatos, nitratos y fosfatos. Los expertos en la materia conocen bien otras sales farmacéuticamente aceptables adecuadas y estas incluyen, por ejemplo, ácido fórmico, acético, cítrico, oxálico, tartárico o mandélico, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico o ácido fosfórico; con ácidos orgánicos carboxílicos, sulfónicos, sulfo- o fosfoácidos o ácidos sulfámicos sustituidos en el N, por ejemplo, ácido acético, ácido trifluoroacético (TFA), ácido propiónico, ácido glicólico, ácido succínico, ácido maleico, ácido hidroximaleico, ácido metilmaleico, ácido fumárico, ácido málico, ácido tartárico, ácido láctico, ácido oxálico, ácido glucónico, ácido glucárico, ácido glucurónico, ácido cítrico, ácido benzoico, ácido cinámico, ácido mandélico, ácido salicílico, ácido 4-aminosalicílico, ácido 2-fenoxibenzoico, ácido 2-acetoxibenzoico, ácido embónico, ácido nicotínico o ácido isonicotínico; y con aminoácidos, tales como los 20 alfaaminoácidos involucrados en la síntesis de proteínas en la naturaleza, por ejemplo, ácido glutámico o ácido aspártico y también con ácido fenilacético, ácido metanosulfónico, ácido etanosulfónico, ácido 2-hidroxietanosulfónico, ácido etano-1,2-disulfónico, ácido bencenosulfónico, ácido 4-metilbencenosulfónico, ácido naftaleno-2-sulfónico, ácido naftaleno-1,5-disulfónico, 2- o 3-fosfoglicerato, glucosa-6-fosfato, ácido N-ciclohexilsulfámico (con formación de ciclamatos) o con otros compuestos orgánicos ácidos, tales como ácido ascórbico.
Las composiciones farmacéuticas que contienen los compuestos divulgados en la presente se pueden fabricar de manera convencional, por ejemplo, mediante procesos convencionales de mezcla, disolución, granulación, producción de grageas, levigado, emulsión, encapsulamiento, captura o liofilización. La formulación adecuada depende de la vía de administración escogida.
En el caso de administración oral, las composiciones adecuadas se pueden formular fácilmente mediante la combinación de un compuesto divulgado en la presente con excipientes farmacéuticamente aceptables, tales como portadores muy conocidos en la técnica. Tales excipientes y portadores hacen posible la formulación de los compuestos de la presente como comprimidos, pastillas, grageas, cápsulas, líquidos, geles, jarabes, suspensiones espesas y similares para ingesta oral por parte del paciente que se va a tratar. Los preparados farmacéuticos para uso oral se pueden obtener añadiendo un compuesto como se divulga en la presente con un excipiente sólido, opcionalmente moliendo la mezcla resultante, y procesando la mezcla de gránulos, tras la adición de agentes auxiliares adecuados, si se desea, para obtener comprimidos o núcleos de grageas. Los excipientes adecuados incluyen, por ejemplo, relleno y preparados de celulosa. Si se desea, se pueden añadir agentes disgregantes. Se conocen muy bien los ingredientes farmacéuticamente aceptables para los diversos tipos de formulación y estos pueden ser, por ejemplo, aglutinantes (por ejemplo, polímeros naturales o sintéticos), lubricantes, surfactantes, agentes edulcorantes y saborizantes, materiales de recubrimiento, conservantes, tintes, espesantes, adyuvantes, agentes antimicrobianos, antioxidantes y portadores para los diversos tipos de formulaciones.
Cuando se administra por vía oral una cantidad terapéuticamente eficaz de un compuesto divulgado en la presente, la composición normalmente está en forma de una formulación sólida (por ejemplo, comprimido, cápsula, pastilla, polvo o pastilla para chupar) o líquida (por ejemplo, jarabe, elixir, solución o suspensión acuosos).
Cuando se administra en forma de comprimido, la composición también puede contener un sólido funcional y/o un portador sólido, tal como una gelatina o un adyuvante. El comprimido, cápsula y polvo pueden contener de aproximadamente un 1 a aproximadamente un 95 % de compuesto y preferentemente, de aproximadamente un 15 a aproximadamente un 90 % de compuesto.
Cuando se administra en forma líquida o de suspensión, se puede añadir un líquido funcional y/o un portador líquido, tal como agua, vaselina o aceites de origen animal o vegetal. La forma líquida de la composición también puede contener solución salina fisiológica, soluciones de alditoles, dextrosa u otras soluciones de sacáridos o glicoles. Cuando se administra en forma líquida o de suspensión, la composición puede contener de aproximadamente un 0.5 a aproximadamente un 90 % en peso de un compuesto divulgado en la presente y preferentemente, de aproximadamente un 1 a aproximadamente un 50 % de un compuesto divulgado en la presente. En una realización contemplada, el portador líquido es no acuoso o sustancialmente no acuoso. Para su administración en forma líquida, la composición se puede suministrar como una formulación sólida de disolución rápida para su disolución o suspensión inmediatamente antes de la administración.
Cuando se administra una cantidad terapéuticamente eficaz de un compuesto divulgado en la presente mediante inyección intravenosa, cutánea o subcutánea, la composición se encuentra en forma de una solución acuosa aceptable para uso parenteral exenta de pirógenos. La preparación de dichas soluciones aceptables para uso parenteral, teniendo debidamente en cuenta el pH, la isotonicidad, la estabilidad y similares, está dentro de las competencias de la técnica. Una composición preferida para inyección intravenosa, cutánea o subcutánea normalmente contiene,
además de un compuesto divulgado en la presente, un vehículo isotónico. Tales composiciones se pueden preparar para administración como soluciones de base libre o sales farmacéuticamente aceptables en agua que se mezclan convenientemente con un surfactante, tal como hidroxipropilcelulosa. También se pueden preparar dispersiones en glicerol, polietilenglicoles líquidos y mezclas de estos y en aceites. En condiciones de almacenamiento y uso normales, estos preparados pueden contener opcionalmente un conservante para evitar el crecimiento de microorganismos.
Las composiciones inyectables pueden incluir soluciones, suspensiones o dispersiones acuosas estériles y polvos estériles para la preparación extemporánea de soluciones, suspensiones o dispersiones inyectables estériles. En todas las realizaciones, la forma debe ser estéril y debe ser fluida en un grado suficiente que sea posible su fácil administración con una jeringuilla. Debe ser estable en las condiciones de fabricación y almacenamiento y debe resistir a la acción contaminante de microorganismos, tales como bacterias y hongos, mediante la inclusión opcional de un conservante. El portador puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol líquido y similares), mezclas adecuadas de estos y aceites vegetales. En una realización contemplada, el portador es no acuoso o sustancialmente no acuoso. Es posible mantener la fluidez adecuada, por ejemplo, mediante el uso de un recubrimiento, tal como lecitina, manteniendo el tamaño de partícula requerido del compuesto en la realización de la dispersión y mediante el uso de tensioactivos. La prevención de la acción de microorganismos puede lograrse por medio de diversos agentes antibacterianos y antifúngicos, por ejemplo, parabenos, clorobutanol, fenol, ácido sórbico, tiomerosal y similares. En muchas realizaciones, resultará preferible incluir agentes isotónicos, por ejemplo, azúcares o cloruro de sodio. Se puede lograr la absorción prolongada de las composiciones inyectables mediante el uso en las composiciones de agentes que retrasan la absorción, por ejemplo, monoestearato de aluminio y gelatina.
Las soluciones inyectables estériles se preparan incorporando los compuestos activos en la cantidad necesaria en el disolvente adecuado con varios de los demás ingredientes enumerados anteriormente, según sea necesario, seguido de esterilización por filtración. Generalmente, las dispersiones se preparan incorporando los diversos principios activos esterilizados en un vehículo estéril que contiene un medio de dispersión básico y los demás ingredientes necesarios de entre los mencionados anteriormente. En la realización de polvos estériles para la preparación de soluciones inyectables estériles, los métodos de preparación preferidos son las técnicas de secado al vacío y liofilización que generan un polvo del principio activo más cualquier ingrediente adicional deseado de una solución de este esterilizada por filtración previamente.
También se pueden preparar formulaciones de liberación lenta o de liberación sostenida para lograr una liberación controlada del compuesto activo en contacto con los fluidos corporales en el tubo GI y para proporcionar un nivel sustancialmente constante y eficaz del compuesto activo en el plasma sanguíneo. Por ejemplo, se puede controlar la liberación mediante uno o más de disolución, difusión e intercambio iónico. Además, el enfoque de liberación lenta puede mejorar la absorción mediante vías saturables o limitantes en el tubo GI. Por ejemplo, el compuesto se puede incluir para tal fin en una matriz polimérica de un polímero biológico degradable, un polímero hidrosoluble o una mezcla de ambos y, opcionalmente, surfactantes adecuados. En este contexto, la inclusión puede significar la incorporación de micropartículas en una matriz de polímeros. Las formulaciones de liberación controlada también se obtienen mediante encapsulación de micropartículas dispersadas o microgotas emulsionadas mediante tecnologías de recubrimiento con emulsión o dispersión conocidas.
Para administración mediante inhalación, los compuestos de la presente invención se administran de manera conveniente en una forma de presentación de pulverización mediante aerosol en envases presurizados o un nebulizador con el uso de un propulsor adecuado. En la realización de un aerosol presurizado, se puede determinar la unidad posológica proporcionando una válvula para administrar una cantidad medida. Las cápsulas y cartuchos de, por ejemplo, gelatina, para su uso en un inhalador o insuflador se pueden formular conteniendo una mezcla en polvo del compuesto y una base en polvo adecuada, tal como lactosa o almidón.
Los compuestos divulgados en la presente se pueden formular para administración parenteral mediante inyección (por ejemplo, mediante inyección por bolo o infusión continua). Las formulaciones para inyección se pueden presentar en formas farmacéuticas unitarias (por ejemplo, en ampollas o en recipientes multidosis) con un conservante añadido. Las composiciones pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos y pueden contener agentes de formulación tales como agentes de suspensión, estabilizantes y/o de dispersión.
Las formulaciones farmacéuticas para administración parenteral incluyen soluciones acuosas de los compuestos en forma hidrosoluble. Adicionalmente, las suspensiones de los compuestos se pueden preparar como suspensiones para inyección oleosas apropiadas. Los vehículos o disolventes lipófilos adecuados incluyen aceites grasos o ésteres de ácidos grasos sintéticos. Las suspensiones para inyección acuosa pueden contener sustancias que aumentan la viscosidad de la suspensión. Opcionalmente, la suspensión también puede contener estabilizantes o agentes adecuados que aumentan la solubilidad de los compuestos y permiten la preparación de soluciones muy concentradas. Como alternativa, la presente composición puede estar en forma de polvo para reconstitución con un vehículo adecuado (por ejemplo, agua exenta de pirógenos estéril) antes de su uso.
Los compuestos divulgados en la presente también se pueden formular en composiciones rectales, tales como supositorios o enemas de retención (por ejemplo, que contienen bases de supositorio convencionales). Además de
las formulaciones descritas previamente, los compuestos también se pueden formular como una preparación de liberación lenta. Tales formulaciones de acción prolongada se pueden administrar mediante implante (por ejemplo, de forma subcutánea o intramuscular) o mediante inyección intramuscular. Por lo tanto, por ejemplo, los compuestos se pueden formular con materiales poliméricos o hidrófobos adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico o como derivados poco solubles, por ejemplo, como una sal poco soluble.
En particular, un compuesto divulgado en la presente se puede administrar por vía oral, bucal o sublingual en forma de comprimidos que contienen excipientes, tales como almidón o lactosa, o en cápsulas u óvulos ya sea solos o mezclados con excipientes, o en forma de elixires o suspensiones que contienen agentes saborizantes o colorantes. Tales preparados líquidos se pueden preparar con aditivos farmacéuticamente aceptables, tales como agentes de suspensión. Un compuesto también se puede inyectar por vía parenteral, por ejemplo, intravenosa, intramuscular, subcutánea o intracoronaria. Para la administración parenteral, es mejor utilizar el compuesto en forma de una solución acuosa estéril que puede contener otras sustancias, por ejemplo, sales o alditoles, tales como manitol o glucosa, para que la solución sea isotónica con la sangre.
Para el uso veterinario, un compuesto divulgado en la presente se administra como una formulación adecuada aceptable de acuerdo con la práctica veterinaria habitual. El veterinario puede determinar fácilmente la pauta posológica y la ruta de administración más adecuadas para un animal concreto.
En algunas realizaciones, se pueden empaquetar en un kit todos los componentes necesarios para el tratamiento de un trastorno relacionado con KIF18A utilizando un compuesto como se divulga en la presente ya sea solo o combinado con otro agente o intervención utilizada tradicionalmente para el tratamiento de tal enfermedad. Específicamente, la presente invención proporciona un kit para su uso en la intervención terapéutica de la enfermedad, que comprende un conjunto empaquetado de medicamentos que incluyen el compuesto divulgado en la presente, así como también tampones y otros componentes para preparar formas suministrables de dichos medicamentos, y/o dispositivos para suministrar tales medicamentos, y/o cualquier agente que se utilice en terapia combinada con el compuesto divulgado en la presente, y/o instrucciones para el tratamiento de la enfermedad empaquetadas con los medicamentos. Las instrucciones se pueden encontrar en cualquier medio tangible, tal como papel impreso o un medio magnético u óptico de lectura informática o instrucciones que remiten a una fuente de datos informática remota, tal como una página web a la que se accede a través de Internet.
Una “cantidad terapéuticamente eficaz” se refiere a una cantidad eficaz para tratar o prevenir el desarrollo, o para aliviar los síntomas existentes del sujeto que se está tratando. La determinación de las cantidades eficaces se encuentra dentro de las competencias de los expertos en la técnica, especialmente a la vista de la divulgación detallada proporcionada en la presente. En general, una “dosis terapéuticamente eficaz” se refiere a la cantidad del compuesto que da como resultado la consecución del efecto deseado. Por ejemplo, en una realización preferida, una cantidad terapéuticamente eficaz de un compuesto divulgado en la presente disminuye la actividad de KIF18A en al menos un 5 %, en comparación con el control, al menos un 10 %, al menos un 15 %, al menos un 20 %, al menos un 25 %, al menos un 30 %, al menos un 35 %, al menos un 40 %, al menos un 45 %, al menos un 50 %, al menos un 55 %, al menos un 60 %, al menos un 65 %, al menos un 70 %, al menos un 75 %, al menos un 80 %, al menos un 85 % o al menos un 90 %.
La cantidad de compuesto administrado puede depender del sujeto que se está tratando, la edad, el estado de salud, el sexo y el peso del sujeto, el tipo de tratamiento concomitante (si lo hubiera), la gravedad de la aflicción, la naturaleza del efecto deseado, la forma y la frecuencia de tratamiento y el criterio del médico que prescribe el tratamiento. La frecuencia de dosificación también puede depender de los efectos farmacodinámicos sobre las presiones arteriales de oxígeno. Si bien las necesidades individuales varían, la determinación de los intervalos óptimos de cantidades eficaces del compuesto está dentro de las competencias de la técnica. Tales dosis se pueden administrar en una sola dosis o se pueden dividir en múltiples dosis.
Los términos “cáncer” y “canceroso” cuando se utilizan en la presente, se refieren a o describen la afección fisiológica en mamíferos que está caracterizada normalmente por el crecimiento celular desregulado. Los ejemplos de cánceres incluyen, sin carácter limitante, carcinoma, linfoma, sarcoma, blastoma y leucemia. Los ejemplos más particulares de tales cánceres incluyen carcinoma escamocelular, cáncer de pulmón, cáncer de páncreas, cáncer de cuello uterino, cáncer de vejiga, hepatoma, cáncer de mama, carcinoma de colon, y cáncer de cabeza y cuello, cáncer de ovario, y cáncer de endometrio. Aunque el término “cáncer” tal como se utiliza en la presente no se limita a una forma específica cualquiera de la enfermedad, se cree que los métodos de la invención serán particularmente eficaces para los cánceres en los que se observa que están acompañados por niveles desregulados de KIF18A o dependientes de KIF18A para la segregación adecuada de los cromosomas y la supervivencia en el mamífero.
Los términos “tratar”, “que trata” y “tratamiento”, tal como se utilizan en la presente, se refieren a la terapia, incluidas, sin carácter limitante, terapia curativa, terapia profiláctica y terapia preventiva. El tratamiento profiláctico está constituido generalmente tanto por prevenir completamente el inicio de los trastornos como por retrasar el inicio de un estadio evidente desde un punto de vista preclínico de trastornos en individuos.
El término “paciente”, “sujeto” o “mamífero” tal como se utiliza en la presente se refiere a cualquier “paciente”, “sujeto” o “mamífero”, incluidos seres humanos, vacas, caballos, perros y gatos. En una realización de la invención, el mamífero
es un ser humano.
La expresión “que comprende” pretende ser abierta, e incluir el (los) componente(s) indicado(s) pero sin excluir otros elementos.
Los términos "Fórmula I" incluyen cualquier subfórmula.
Métodos de uso de inhibidores de kif18a
La presente divulgación proporciona compuestos que tienen actividad moduladora de KIF18A basada en MT en general, y actividad inhibidora en particular. En una realización de la invención, se proporciona un método para modular la proteína KIF18A en un sujeto, comprendiendo el método administrar al sujeto una cantidad posológica eficaz de un compuesto de Fórmulas I. Como tal, los compuestos de la invención se pueden utilizar para tratar trastornos de proliferación celular, incluido el crecimiento celular descontrolado, regulación del ciclo celular aberrante, anomalías del centrosoma (estructurales y/o numéricas, fragmentación). Otras enfermedades o trastornos asociados con la acumulación de centrosomas extra (>2) incluyen la infección por el virus del papiloma humano (VPH), incluidas neoplasias asociadas con el VPH. Los compuestos también son útiles para las enfermedades relacionadas con los cilios, así como también para la ablación de la población de células germinales haploides que se podría utilizar como anticonceptivo masculino.
Además, los compuestos de la invención son útiles para, sin carácter limitante, la prevención o el tratamiento del cáncer y otras enfermedades o trastornos mediados por KIF18A. Por ejemplo, los compuestos de la invención serían útiles para el tratamiento de diversos tumores sólidos y derivados hematológicamente, tales como carcinomas, incluidos, sin carácter limitante, cáncer de la vejiga, mama, colon, riñón, hígado, pulmón (incluido el cáncer de pulmón escamocelular y cáncer de pulmón microcítico), esófago, vesícula biliar, ovario, páncreas, estómago, cuello uterino, de tiroides, próstata, y piel (incluido el carcinoma escamocelular); tumores hematopoyéticos de linaje linfoide (incluidas la leucemia, leucemia linfocítica aguda, leucemia linfoblástica aguda, linfoma de linfocitos B, linfoma de linfocitos T, linfoma de Hodgkin, linfoma no hodgkiniano, linfoma de tricoleucocitos y linfoma de Burkitt); tumores hematopoyéticos de linaje mieloide (incluidas las leucemias mielógenas aguda y crónica, síndrome mielodisplásico y leucemia promielocítica); tumores de origen mesenquimatoso (incluidos el fibrosarcoma y rabdomiosarcoma, y otros sarcomas, por ejemplo, de tejidos blandos y de hueso); tumores del sistema nervioso central y periférico (incluidos astrocitoma, neuroblastoma, glioma y schwannomas); y otros tumores (incluidos el melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderomia pigmentosa, queratoctantoma, cáncer folicular de tiroides y sarcoma de Kaposi).
Los compuestos de la invención son también útiles en el tratamiento de indicaciones relacionadas con el cáncer tales como tumores sólidos, sarcomas (especialmente, sarcoma de Ewing y osteosarcoma) retinoblastoma, rabdomiosarcomas, neuroblastoma, neoplasias malignas hematopoyéticas, incluidos leucemia y linfoma, efusiones pericárdicas o pleurales inducidas por tumores y ascitis malignas.
Basándose en la capacidad de modular la quinesina que afecta la angiogénesis, los compuestos de la invención son también útiles en el tratamiento y la terapia de enfermedades proliferativas. Concretamente, estos compuestos se pueden utilizar para el tratamiento de una enfermedad inflamatoria, especialmente de las manifestaciones en el aparato locomotor, tales como diversas enfermedades reumatoides inflamatorias, especialmente la poliartritis crónica, incluida la artritis reumatoide, artritis juvenil, o artropatía psoriásica; síndrome paraneoplásico o enfermedades inflamatorias inducidas por tumores, efusiones turbias, colagenosis, tales como lupus eritematoso sistémico, polimiositis, dermatomiositis, esclerodermia sistémica, o colagenosis mixta; artritis posinfecciosa, (cuando se pueden encontrar organismos patógenos sin vida en o en el interior de la parte afectada del cuerpo), espondilartritis seronegativa, tal como espondilitis anquilosante; vasculitis, sarcoidosis, o artrosis; o adicionalmente cualquier combinación de estos.
Los compuestos de la invención se pueden utilizar también como agentes activos contra tales patologías como artritis, ateroesclerosis, psoriasis, hemangiomas, angiogénesis de miocardio, colaterales coronario y cerebral, angiogénesis de la extremidad isquémica, cicatrización de heridas, enfermedades relacionadas con Helicobacter en úlcera péptica, fracturas, fiebre por arañazo de gato, rubeosis, glaucoma neovascular y retinopatía tales como las asociadas con la retinopatía diabética o degeneración macular. Además, algunos de estos compuestos se pueden utilizar como agentes activos contra tumores sólidos, ascitis maligno, cánceres hematopoyéticos y trastornos hiperproliferativos tales como hiperplasia de tiroides (especialmente enfermedad de Graves), y quistes (tales como hipervascularidad del estroma ovárico, síndrome característico del ovario poliquístico (síndrome de Stein-Leventhal)) debido a que tales enfermedades requieren una proliferación de células de los vasos sanguíneos para el crecimiento y/o metástasis.
Además de ser útiles para el tratamiento en seres humanos, estos compuestos son útiles para el tratamiento veterinario de animales de compañía, animales exóticos y animales de granja, incluidos mamíferos, roedores y similares. Por ejemplo, animales que incluyen caballos, perros y gatos se pueden tratar con los compuestos que se proporcionan en la invención.
Combinaciones
Aunque los compuestos de la invención se pueden dosificar o administrar como un solo agente farmacéutico activo,
también se puede utilizar combinado con uno o más compuestos de la invención o junto con otros agentes. Cuando se administran como una combinación, los agentes terapéuticos se pueden formular como composiciones separadas que se administran simultánea o secuencialmente en diferentes momentos, o los agentes terapéuticos se pueden administrar como una sola composición.
La frase “coterapia” (o “terapia combinada”), al definir el uso de un compuesto de la presente invención y otro agente farmacéutico, pretende abarcar la administración de cada agente de manera secuencial en un régimen que proporcionará efectos beneficiosos de la combinación de medicamentos, y también pretende abarcar la coadministración de estos agentes de una manera sustancialmente simultánea, tal como en una cápsula única que tiene una proporción fija de estos agentes activos o en múltiples cápsulas separadas para cada agente.
Específicamente, la administración de compuestos de la presente invención se puede realizar junto con terapias adicionales conocidas por los expertos en la técnica en la prevención o tratamiento del cáncer, tales como con radioterapia, agentes dirigidos que son moléculas de bajo peso molecular (por ejemplo, inhibidores de PARP, inhibidores de cinasas), anticuerpos terapéuticos (por ejemplo, puros y conjugados con fármacos) anticuerpos para inmunoterapia (inhibidores del punto de control, acopladores de linfocitos T biespecíficos) con agentes neoplásicos o citotóxicos.
Si se formulan como una dosis fija, tales productos combinados emplean los compuestos de esta invención dentro de los intervalos posológicos aceptados. Los compuestos de Fórmula I también se pueden administrar secuencialmente con agentes anticancerosos o citotóxicos conocidos cuando una formulación de combinación es inadecuada. La invención no está limitada en la secuencia de administración: los compuestos de la invención se pueden administrar antes, simultáneamente o después de la administración del agente anticanceroso o citotóxico conocido.
Existen muchos agentes anticancerosos comercializados, en la evaluación clínica y en el desarrollo preclínico, que se seleccionarían para el tratamiento de neoplasias mediante la quimioterapia farmacológica combinada. Tales agentes se encuentran comprendidos en varias categorías principales, tales como agentes de tipo antibiótico, agentes alquilantes y análogos a alquilantes, agentes antimitóticos, agentes dirigidos que son moléculas de bajo peso molecular, agentes antimetabolitos, agentes hormonales, agentes inmunológicos, agentes antiangiogénicos, agentes de tipo interferón, y una categoría de diversos agentes.
La presente divulgación también proporciona métodos para terapias combinadas en las que se utilizan un agente conocido para modular otras vías, u otros componentes de la misma vía, o incluso conjuntos solapantes de enzimas dianas combinados con un compuesto de la presente divulgación o una sal farmacéuticamente aceptable de este. En un aspecto, dicha terapia incluye, sin carácter limitante, la combinación de uno o más compuestos de la divulgación con agentes quimioterápicos, anticuerpos terapéuticos, agentes que son moléculas dirigidas de bajo peso molecular y tratamiento con radiación para proporcionar un efecto terapéutico sinérgico o aditivo.
Actualmente, se conocen en la técnica muchos agentes quimioterápicos y se pueden utilizar combinados con los compuestos de la divulgación. En algunas realizaciones, el agente quimioterápico se selecciona del grupo que consiste en agentes antimitóticos, agentes alquilantes, antimetabolitos, antibióticos intercalantes, inhibidores de factores de crecimiento, inhibidores del ciclo celular, enzimas, inhibidores de topoisomerasas, modificadores de la respuesta biológica, antihormonas, inhibidores de la angiogénesis y antiandrógenos. Son ejemplos no limitantes los agentes quimioterápicos, agentes citotóxicos y moléculas de bajo peso molecular no peptídicas tales como Gleevec® (Mesilato de Imatinib), Kyprolis® (carfilzomib), Velcade® (bortezomib), Casodex (bicalutamida), Iressa® (gefitinib) y Adriamicina, así como también una miríada de agentes quimioterápicos. Los ejemplos no limitantes de agentes quimioterápicos incluyen agentes alquilantes tales como tiotepa y ciclofosfamida (CYTOXANTM); alquilsulfonatos tales como busulfán, improsulfán y piposulfán; aziridinas tales como benzodopa, carbocuona, meturedopa, y uredopa; etileniminas y metilmelaminas que incluyen altretamina, trietilenmelamina, trietilenfosforamida, trietilentiofosforamida y trimetilolomelamina; mostazas nitrogenadas, tales como clorambucilo, clornafazina, colofosfamida, estramustina, ifosfamida, mecloretamina, clorhidrato de óxido de mecloretamina, melfalán, novembiquina, fenesterina, prednimustina, trofosfamida, mostaza de uracilo; nitrosoureas, tales como carmustina, clorozotocina, fotemustina, lomustina, nimustina, ranimustina; antibióticos tales como aclacinomisinas, actinomicina, autramicina, azaserina, bleomicinas, cactinomicina, caliceamicina, carabicina, carminomicina, carcinofilina, CasodexTM, cromomicinas, dactinomicina, daunorrubicina, detorrubicina, 6-diazo-5-oxo-L-norleucina, doxorrubicina, epirrubicina, esorrubicina, idarrubicina, marcelomicina, mitomicinas, ácido micofenólico, nogalamicina, olivomicinas, peplomicina, potfiromicina, puromicina, quelamicina, rodorrubicina, estreptonigrina, estreptozocina, tubercidina, ubenimex, zinostatina, zorrubicina; antimetabolitos tales como metotrexato y 5-fluorouracilo (5-FU); análogos del ácido fólico tales como denopterina, metotrexato, pteropterina, trimetrexato; análogos de purina tales como fludarabina, 6-mercaptopurina, tiamiprina, tioguanina; análogos de pirimidina tales como ancitabina, azacitidina, 6-azauridina, carmofur, citarabina, didesoxiuridina, doxifluridina, enocitabina, floxuridina, andrógenos tales como calusterona, propionato de dromostanolona, epitiostanol, mepitiostano, testolactona; antisuprarrenales tales como aminoglutetimida, mitotano, trilostano; reabastecedor del ácido fólico tal como ácido frolínico; aceglatona; glucósido de aldofosfamida; ácido aminolevulínico; amsacrina; bestrabucilo; bisantreno; edatraxato; defofamina; demecolcina; diazicuona; elfomitina; acetato de eliptinio; etoglúcido; nitrato de galio; hidroxiurea; lentinán; lonidamina; mitoguazona; mitoxantrona; mopidamol; nitracrina; pentostatina; fenamet; pirarrubicina; ácido podofilínico; 2-etilhidrazida; procarbazina; PSK, razoxano; sizofirán; espirogermanio; ácido tenuazónico; triazicuona; 2,2',2"-triclorotrietilamina; uretano; vindesina;
dacarbazina; manomustina; mitobronitol; mitolactol; pipobromano; gacitosina; arabinósido (“Ara-C”); ciclofosfamida; tiotepa; taxanos, por ejemplo, paclitaxel y docetaxel; Nab-paclitaxel; ácido retinoico; esperamicinas; capecitabina; y sales farmacéuticamente aceptables, ácidos o derivados de cualquiera de los anteriores.
También se incluyen como acondicionadores quimioterapéuticos celulares adecuados los agentes antihormonales que actúan para regular o inhibir la acción hormonal sobre los tumores como los antiestrógenos que incluyen, por ejemplo, tamoxifeno, (NolvadexTM), raloxifeno, 4(5)imidazoles inhibidores de la aromatasa, 4-hidroxitamoxifeno, trioxifeno, keoxifeno, LY 117018, onapristona, y toremifeno (Fareston); y antiandrógenos tales como flutamida, nilutamida, bicalutamida, leuprolida y goserelina; clorambucilo; gemcitabina; 6 -tioguanina; mercaptopurina; metotrexato; análogos de platino tales como cisplatino, oxaliplatino, carboplatino; etopósido (VP-16); ifosfamida; mitomicina C; mitoxantrona; vinblastina, vincristina; vinorelbina; navelbina; novantrona; tenipósido; daunomicina; aminopterina; xeloda; ibandronato; topotecán; camptotecina-11 (CPT-11); inhibidor de la topoisomerasa RFS 2000; difluorometilornitina (DMFO).
En los casos en los que se desee, los compuestos o composiciones farmacéuticas de la presente divulgación se pueden utilizar combinados con fármacos anticancerosos indicados habitualmente, tales como Herceptin®, Avastin®, Erbitux®, Rituxan®, Taxol®, Abraxane, Arimidex®, Taxotere®, ABVD, AVICINE, Abagovomab, acridina carboxamida, Adecatumumab, 17-N-alilamino-17-desmetoxigeldanamicina, Alfaradin, Alvocidib, 3-aminopiridino-2-carboxaldehído tiosemicarbazona, amonafida, antracenodiona, inmunotoxinas anti-CD22, agentes antineoplásicos, hierbas antitumorigénicas, apazicuona, Atiprimod, azatioprina, belotecán, bendamustina, BIBW 2992, biricodar, brostalicina, briostatina, sulfoximina de butionina, CBV (quimioterapia), caliculina, agentes antineoplásicos no específicos para el ciclo celular, ácido dicloroacético, discodermolida, elsamitrucina, enocitabina, epotilona, eribulina, everolimus, exatecán, exisulind, ferruginol, forodesina, fosfestrol, régimen quimioterápico ICE, IT-101, imexón, imiquimod, indolocarbazol, irofulveno, laniquidar, larotaxel, lenalidomida, lucantona, lurtotecán, mafosfamida, mitozolomida, nafoxidina, nedaplatino, olaparib, talazoparib, niraparib, ortataxel, PAC-1, Pawpaw, pixantrona, inhibidor del proteasoma, rebecamicina, resiquimod, rubitecán, SN-38, salinosporamida A, sapacitabina, Stanford V, swainsonina, talaporfina, tariquidar, tegafur-uracilo, temodar, tesetaxel, tetranitrato de triplatino, tris(2 -cloroetil)amina, troxacitabina, uramustina, vadimezán, vinflunina, ZD6126 o zosuquidar, inhibidores de CDK4/6 (Palbociclib, Ibrance; Ribociclib, Kisqali; Abemaciclib, Verzenio).
Esta divulgación se refiere además a un método para utilizar los compuestos o composiciones farmacéuticas proporcionados en la presente, combinados con radioterapia para inhibir el crecimiento celular anómalo o tratar el trastorno hiperproliferativo en el mamífero. Se conocen en la materia técnicas para administrar radioterapia y estas técnicas se pueden utilizar en la terapia combinada descrita en la presente. Se puede determinar la administración del compuesto de la divulgación en esta terapia combinada tal como se describe en la presente.
La radioterapia se puede administrar a través de uno de varios métodos o una combinación de métodos, lo que incluye, sin carácter limitante, radioterapia externa, radioterapia interna, radiación de implante, radiocirugía estereotáctica, radioterapia sistémica, radioterapia y braquiterapia intersticial permanente o temporal. El término “braquiterapia”, tal como se utiliza en la presente, se refiere a una radioterapia proporcionada mediante un material radiactivo confinado espacialmente introducido en el cuerpo en o cerca de un tumor u otro sitio de una enfermedad con tejido proliferativo. Se pretende que el término incluya, sin carácter limitante, la exposición a isótopos radiactivos (por ejemplo, At-211, I-131, I-125, Y-90, Re-186, Re-188 , Sm-153, Bi-212, P-32 e isótopos radiactivos de Lu). Las fuentes de radiación adecuadas para su uso como acondicionador celular de la presente divulgación incluyen tanto sólidos como líquidos. A modo de ejemplo no limitante, la fuente de radiación puede ser un radionúclido, tal como I-125, I-131, Yb-169, Ir-192 como una fuente sólida, I-125 como una fuente sólida, u otros radionúclidos que emiten fotones, partículas beta, radiación gamma u otros rayos terapéuticos. El material radiactivo también puede ser un fluido constituido por cualquier solución de radionúclido(s), por ejemplo, una solución de I-125 o I-131, o se puede producir un fluido radiactivo utilizando una suspensión espesa de un fluido adecuado que contiene partículas pequeñas de radionúclidos sólidos, tales como Au-198, Y-90. Asimismo, el (los) radionúclido(s) puede(n) adoptar la forma de gel o de microesferas radiactivas.
Los compuestos o composiciones farmacéuticas de la divulgación se pueden utilizar combinados con una cantidad de una o más sustancias seleccionadas entre agentes antiangiogénicos, inhibidores de transducción de señal, agentes antiproliferativos, inhibidores de glucólisis o inhibidores de autofagia.
Los agentes antiangiogénicos, tales como inhibidores de MMP-2 (metaloproteinasa de la matriz 2), inhibidores de MMP-9 (metaloproteinasa de la matriz 9) e inhibidores de COX-11 (cicloooxigenasa 11), se pueden utilizar junto con un compuesto de la divulgación y composiciones farmacéuticas que se describen en la presente. Los agentes antiangiogénicos incluyen, por ejemplo, rapamicina, temsirolimus (CCI-779), everolimus (RAD001), sorafenib, sunitinib y bevacizumab. Los ejemplos de inhibidores de COX-II útiles incluyen alecoxib, valdecoxib y rofecoxib. Se describen ejemplos de inhibidores de metaloproteinasa de la matriz útiles en los documentos WO 96/33172, WO 96/27583, publicación de patente europea EP0818442, publicación de patente europea EP1004578, WO 98/07697, WO 98/03516, WO 98/34918, WO 98/34915, WO 98/33768, WO 98/30566, publicación de patente europea 606046, publicación de patente europea 931 788, WO 90/05719, WO 99/52910, WO 99/52889, WO 99/29667, WO1999007675, publicación de patente europea EP1786785, publicación de patente europea n.° EP1181017, publicación de EE. UU. n.° US20090012085, publicación de EE. UU. US5863 949, publicación de EE. UU. US5861 510 y publicación de
patente europea EP0780386. Los inhibidores de MMP-2 y MMP-9 preferidos son aquellos con poca o nula actividad de inhibición de MMP-1. Son más preferidos aquellos que inhiben de forma selectiva MMP-2 y/o AMP-9 respecto a otras metaloproteinasas de la matriz (es decir, MaP-1, MmP-3, MMP-4, MMP-5, MMP-6, MMP- 7, MMP- 8, MMP-10, MMP-11, mMp-12 y MMP-13). AG-3340, RO 32-3555 y RS 13-0830 son algunos ejemplos específicos de inhibidores de MMP útiles en la divulgación.
Los presentes compuestos también se pueden utilizar en terapias simultáneas con otros agentes antineoplásicos, tales como acemanano, aclarrubicina, aldesleucina, alemtuzumab, alitretinoína, altretamina, amifostina, ácido aminolevulínico, amrubicina, amsacrina, anagrelida, anastrozol, ANCER, ancestim, ARGLABIN, trióxido de arsénico, BAM 002 (Novelos), bexaroteno, bicalutamida, broxuridina, capecitabina, celmoleucina, cetrorelix, cladribina, clotrimazol, ocfosfato de citarabina, DA 3030 (Dong-A), daclizumab, denileucina diftitox, deslorrelina, dexrazoxano, dilazep, docetaxel, docosanol, doxercalciferol, doxifluridina, doxorrubicina, bromocriptina, carmustina, citarabina, fluorouracilo, HIT diclofenaco, interferón alfa, daunorrubicina, doxorrubicina, tretinoína, edelfosina, edrecolomab, eflornitina, emitefur, epirrubicina, epoetina beta, fosfato de etopósido, exemestano, exisulind, fadrozol, filgrastim, finasterida, fosfato de fludarabina, formestano, fotemustina, nitrato de galio, gemcitabina, gemtuzumab zogamicina, combinación de gimeracilo/oteracilo/tegafur, glicopina, goserelina, heptaplatino, gonadotropina coriónica humana, fetoproteína alfa de feto humano, ácido ibandrónico, idarrubicina, (imiquimod, interferón alfa, interferón alfa, natural, interferón alfa-2, interferón alfa-2a, interferón alfa-2b, interferón alfa-N1, interferón alfa-n3, interferón alfacón-1, interferón alfa, natural, interferón beta, interferón beta-1a, interferón beta-1b, interferón gamma, interferón gamma-1a natural, interferón gamma-1b, interleucina-1 beta, iobenguano, irinotecán, irsogladina, lanreotida, LC 9018 (Yakult), leflunomida, lenograstim, sulfato de lentinán, letrozol, interferón alfa leucocitario, leuprorelina, levamisol fluorouracilo, liarozol, lobaplatino, lonidamina, lovastatina, masoprocol, melarsoprol, metoclopramida, mifepristona, miltefosina, mirimostim, ARN bicatenario con emparejamiento erróneo, mitoguazona, mitolactol, mitoxantrona, molgramostim, nafarelina, naloxona pentazocina, nartograstim, nedaplatino, nilutamida, noscapina, proteína novedosa estimulante de la eritropoyesis, NSC 631570 octreotida, oprelvekina, osaterona, oxaliplatino, paclitaxel, ácido pamidrónico, pegaspargasa, peginterferón alfa-2b, polisulfato de pentosán de sodio, pentostatina, picibanilo, pirarrubicina, anticuerpo policlonal de conejo dirigido contra timocitos, polietilenglicol interferón alfa-2a, porfímero de sodio, raloxifeno, raltitrexed, rasburiembodiment, etidronato de renio Re 186, retinamida RII, rituximab, romurtida, samario (153 Sm) lexidronam, sargramostim, sizofirán, sobuzoxano, sonermin, cloruro de estroncio-89, suramin, tasonermina, tazaroteno, tegafur, temoporfina, temozolomida, tenipósido, tetraclorodecaóxido, talidomida, timalfasina, tirotropina alfa, topotecán, toremifeno, tositumomab-yodo 131, trastuzumab, treosulfán, tretinαna, trilostano, trimetrexato, triptorrelia, factor de necrosis tumoral alfa, natural, ubenimex, vacuna contra el cáncer de vejiga, vacuna de Maruyama, vacuna de lisado de melanoma, valrrubicina, verteporfina, vinorrelbina, VIRULIZIN, zinostatina stimalamer, o ácido zoledrónico; abarelix; AE 941 (Aeterna), ambamustina, oligonucleótido antisentido, bcl-2 (Genta), APC 8015 (Dendreon), cetuximab, decitabina, dexaminoglutetimida, diazicuona, EL 532 (Elan), EM 800 (Endorecherche), eniluracilo, etanidazol, fenretinida, filgrastim SD01 (Amgen), fulvestrant, galocitabina, inmunógeno de gastrina 17, terapia génica con HLA-B7 (Vical), factor estimulante de colonias de granulocitos y macrófagos, diclorhidrato de histamina, ibritumomab tiuxetano, ilomastat, IM 862 (Cytran), interleucina-2, iproxifeno, LDI 200 (Milkhaus), leridistim, lintuzumab, MAb contra CA 125 (Biomira), MAb contra cáncer (Japan Pharmaceutical Development), MAb contra HER-2 y Fc (Medarex), MAb 105AD7 idiotípico (CRC Technology), MAb contra CEA idiotípico (Trilex), MAb contra LYM-1 marcado con yodo 131 (Techniclone), MAb contra mucina epitelial polimórfica conjugado con itrio 90 (Antisoma), marimastat, menogarilo, mitumomab, motexafin gadolinio, MX 6 (Galderma), nelarabina, nolatrexed, proteína P 30, pegvisomant, pemetrexed, porfiromicina, prinomastat, RL 0903 (Shire), rubitecán, satraplatino, fenilacetato de sodio, ácido esparfósico, SRL 172 (SR Pharma), SU 5416 (SUGeN), TA 077 (Tanabe), tetratiomolibdato, taliblastina, thrombopoyetina, etiletiopurpurina de estaño, tirapazamina, vacuna contra el cáncer (Biomira), vacuna contra el melanoma (Universidad de Nueva York), vacuna contra el melanoma (Instituto Sloan Kettering), vacuna con oncolisados de melanoma (Facultad de Medicina de Nueva York), vacuna de lisados víricos de células de melanoma (Hospital Real de Newcastle), o valspodar.
Los compuestos de la invención también se pueden utilizar con inhibidores de VEGFR. Es posible utilizar en una terapia combinada otros compuestos descritos en las siguientes patentes y solicitudes de patente: US 6258812, US 2003/0105091, WO 01/37820, US 6235764, WO 01/32651, US 6630500, US 6515004, US 6713485, US 5 521 184, US 5770599, US 5747498, WO 02/68406, WO 02/66470, WO 02/55501, WO 04/05279, WO 04/07481, WO 04/07458, WO 04/09784, WO 02/59110, WO 99/45009, WO 00/59509, WO 99/61422, US 5,990,141, WO 00/12089 y WO 00/02871.
En algunas realizaciones, la combinación comprende una composición de la presente invención combinada con al menos un agente antiangiogénico. Los agentes incluyen, sin carácter limitante, composiciones químicas preparadas de forma sintética in vitro, anticuerpos, regiones de unión al antígeno, radionúclidos, y combinaciones y conjugados de estos. Un agente puede ser un agonista, antagonista, modulador alostérico, toxina o, más generalmente, puede actuar para inhibir o estimular su diana (por ejemplo, activación o inhibición del receptor o enzima) y así promueven la muerte celular o la detención del crecimiento celular.
Los agentes antiangiogénicos ilustrativos incluyen ERBITUX™ (IMC-C225), agentes inhibidores de KDR (receptor del dominio de cinasa) (por ejemplo, anticuerpos y regiones de unión al antígeno que se unen específicamente al receptor del dominio de cinasa), agentes anti-VEGF (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a VEGF, o receptores de VEGF solubles o una región de unión al ligando de este) tales como
AVASTIN™ o VEGF-TRAP™ y agentes anti-receptor de VEGF (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a estos), agentes inhibidores de EGFR (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a estos) tales como Vectibix (panitumumab), IRESSA™ (gefitinib), TARCEVA™ (erlotinib), agentes anti-Ang1 y anti-Ang2 (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a estos o a sus receptores, por ejemplo, Tie2/Tek) y agentes inhibidores anti-cinasa Tie2 (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a esta). Las composiciones farmacéuticas de la presente invención también pueden incluir uno o más agentes (por ejemplo, anticuerpos, regiones de unión al antígeno o receptores solubles) que se unen específicamente a factores de crecimiento e inhiben su actividad, tales como antagonistas del factor de crecimiento de hepatocitos (HGF, por sus siglas en inglés, también conocido como factor de dispersión), y anticuerpos o regiones de unión al antígeno que se unen específicamente a su receptor “c-met”.
Otros agentes antiangiogénicos incluyen Campath, IL-8, B-FGF, antagonistas de Tek (Ceretti et al., publicación de EE. UU. n.° 2003/0162712; patente de EE. UU. n.° 6413932), agentes anti-TWEAK (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen de forma específica, o antagonistas del receptor TWEAK soluble; véase Wiley, patente de EE. UU. n.° 6727225), dominio desintegrina de ADAM para antagonizar la unión de integrina a sus ligandos (Fanslow et al., publicación de EE. UU. n.° 2002/0042368), receptor anti-eph que se une de forma específica y/o anticuerpos o regiones de unión al antígeno antiefrina (patentes de EE. UU. n.os 5981 245; 5728813; 5969 110; 6 596852; 6232447; 6057 124 y miembros de la familia de patentes de estas) y antagonistas anti-PDGF-BB (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen de forma específica), así como también anticuerpos o regiones de unión al antígeno que se unen de forma específica a ligandos PDGF-BB y agentes inhibidores de la cinasa PDGFR (por ejemplo, anticuerpos o regiones de unión al antígeno que se unen específicamente a esta).
Los agentes antitumorales/antiangiogénicos adicionales incluyen: SD-7784 (Pfizer, EE. UU.); cilengitida (Merck KGaA, Alemania, EPO 770622); pegaptanib octasodio, (Gilead Sciences, EE. UU.); Alfastatina, (BioActa, Reino Unido); M-PGA, (Celgene, EE. UU., US 5712291); ilomastat, (Arriva, EE. UU., US 5892112); emaxanib, (Pfizer, EE. UU., US 5792783); vatalanib, (Novartis, Suiza); 2-metoxiestradiol, (EntreMed, EE. UU.); TLC ELL-12, (Elan, Irlanda); acetato de anecortave, (Alcon, EE. UU.); Mab alfa-D148, (Amgen, EE. UU.); CEP-7055, (Cephalon, Ee. UU.); Mab anti-Vn, (Crucell, Países Bajos) DAC: antiangiogénico, (ConjuChem, Canadá); Angiocidina, (InKine Pharmaceutical, EE. UU.); KM-2550, (Kyowa Hakko, Japón); SU-0879, (Pfizer, EE. UU.); CGP-79787, (Novartis, Suiza, EP 970070); Tecnología ARGENT, (Ariad, EE. UU.); YIGSR-Stealth, (Johnson & Johnson, EE. UU.); fragmento de fibrinógeno-E, (BioActa, Reino Unido); inhibidor de la angiogénesis, (Trigen, Reino Unido); TBC-1635, (Encysive Pharmaceuticals, EE. UU.); SC-236, (Pfizer, EE. UU.); ABT-567, (Abbott, EE. UU.); Metastatina, (EntreMed, Ee. Uu.); inhibidor de la angiogénesis, (Tripep, Suecia); maspin, (Sosei, Japón); 2-metoxiestradiol, (Oncology Sciences Corporation, EE. UU.); ER-68203-00, (IVAX, EE. UU.); Benefin, (Lane Labs, EE. UU.); Tz-93, (Tsumura, Japón); TAN-1120, (Takeda, Japón); FR-111142, (Fujisawa, Japón, JP 02233610); factor plaquetario 4, (RepliGen, EE. uU., EP 407122); antagonista del factor de crecimiento endotelial vascular, (Borean, Dinamarca); bevacizumab (pINN), (Genentech, EE. UU.); inhibidores de la angiogénesis, (SUGEN, EE. UU.); XL 784, (Exelixis, EE. UU.); XL 647, (Exelixis, EE. UU.); MAb, integrina alfa5beta3, segunda generación, (Applied Molecular Evolution, EE. UU. y MedImmune, EE. UU.); terapia génica, retinopatía, (Oxford BioMedica, Reino Unido); clorhidrato de enzastaurina (USAN), (Lilly, EE. UU.); CEP 7055, (Cephalon, EE. UU. y Sanofi-Synthelabo, Francia); BC 1, (Instituto de Investigación del Cáncer de Génova, Italia); inhibidor de la angiogénesis, (Alchemia, Australia); antagonista de VEGF, (Regeneron, EE. UU.); rBPI 21 y antiangiogénico derivado de BPI, (XOMA, EE. UU.); PI 88, (Progen, Australia); cilengitide (pINN), (Merck KGaA, Alemania; Universidad Técnica de Múnich, Alemania, Fundación Clínica y de Investigación Scripps, EE. UU.); cetuximab (DCI), (Aventis, Francia); AVE 8062, (Ajinomoto, Japón); AS 1404, (Laboratorio de Investigación del Cáncer, Nueva Zelanda); SG 292, (Telios, EE. UU.); Endostatina, (Hospital Infantil de Boston, EE. UU.); ATN 161, (Attenuon, EE. UU.); ANGIOSTATINA, (Hospital Infantil de Boston, eE. UU.); 2-metoxiestradiol, (Hospital Infantil de Boston, EE. UU.); ZD 6474, (AstraZeneca, Reino Unido); ZD 6126, (Angiogene Pharmaceuticals, Reino Unido); PPI 2458, (Praecis, EE. UU.); AZD 9935, (AstraZeneca, Reino Unido); AZD 2171, (AstraZeneca, Reino Unido); vatalanib (pINN), (Novartis, Suiza y Schering AG, Alemania); inhibidores de la vía del factor tisular, (EntreMed, EE. UU.); pegaptanib (Pinn), (Gilead Sciences, EE. UU.); xantorrizol, (Universidad de Yonsei, Corea del Sur); vacuna, basada en genes, VEGF-2, (Fundación Clínica y de Investigación Scripps, EE. UU.); SPV5.2, (Supratek, Canadá); SDX 103, (Universidad de California en San Diego, EE. UU.); PX 478, (ProlX, EE. UU.); METASTATINA, (EntreMed, EE. UU.); troponina I, (Universidad de Harvard, EE. UU.); SU 6668, (SUGEN, EE. UU.); OXI 4503, (OXiGENE, EE. UU.); o-guanidinas, (Dimensional Pharmaceuticals, EE. UU.); motuporamina C, (Universidad de la Columbia Británica, Canadá); CDP 791, (Celltech Group, Reino Unido); atiprimod (pINN), (GlaxoSmithKline, Reino Unido); E 7820, (Eisai, Japón); CYC 381, (Universidad de Harvard, eE. UU.); AE 941, (Aeterna, Canadá); vacuna, angiogénesis, (EntreMed, EE. UU.); inhibidor del activador del plasminógeno urocinasa, (Dendreon, EE. UU.); oglufanida (pINN), (Melmotte, EE. UU.); inhibidores de HIF-1 alfa, (Xenova, Reino Unido); CEP 5214, (Cephalon, EE. UU.); BAY RES 2622, (Bayer, Alemania); Angiocidina, (InKine, EE. UU.); A6, (Angstrom, EE. UU.); KR 31372, (Instituto de Investigación de Tecnología Química de Corea, Corea del Sur); GW 2286, (GlaxoSmithKline, Reino Unido); EHT 0101, (ExonHit, Francia); CP 868596, (Pfizer, EE. UU.); CP 564959, (OSI, EE. UU.); CP 547632, (Pfizer, EE. UU.); 786034, (GlaxoSmithKline, Reino Unido); KRN 633, (Kirin Brewery, Japón); sistema de suministro de fármacos, intraocular, 2-metoxiestradiol, (EntreMed, EE. UU.); anginex, (Universidad de Maastricht, Países Bajos y Universidad de Minnesota, EE. UU.); ABT 510, (Abbott, EE. UU.); AAL 993, (Novartis, Suiza); VEGI, (ProteomTech, Ee. UU.); inhibidores del factor alfa de necrosis tumoral, (Instituto Nacional sobre el Envejecimiento, EE. UU.); SU 11248, (Pfizer, EE. UU. y SUGEN EE. UU.); ABT 518, (Abbott, EE. UU.); YH16,
(Yantai Rongchang, China); S-3APG, (Hospital Infantil de Boston, EE. UU. y EntreMed, EE. UU.); MAb, KDR, (ImClone Systems, EE. UU.); MAb, alfa5 betal, (Protein Design, EE. UU.); inhibidor de la cinasa KDR, (Celltech Group, Reino Unido y Johnson & Johnson, EE. UU.); GFB 116, (Universidad de Florida del Sur, EE. UU. y Universidad de Yale, EE. UU.); CS 706, (Sankyo, Japón); profármaco combretastatina A4, (Universidad Estatal de Arizona, EE. UU.); condroitinasa AC, (IBEX, Canadá); BAY RES 2690, (Bayer, Alemania); AGM 1470, (Universidad de Harvard, EE. UU., Takeda, Japón y TAp, EE. UU.); aG 13925, (Agouron, EE. UU.); Tetratiomolibdato, (Universidad de Michigan, EE. UU.); GCS 100, (Universidad Estatal de Wayne, Ee. UU.) CV 247, (Ivy Medical, Reino Unido); CKD 732, (Chong Kun Dang, Corea del Sur); MAb, factor de crecimiento endotelial vascular, (Xenova, Reino Unido); irsogladina (INN), (Nippon Shinyaku, Japón); RG 13577, (Aventis, Francia); WX 360, (Wilex, Alemania); escualamina (pINN), (Genaera, eE. UU.); RPI 4610, (Sirna, EE. UU.); terapia contra el cáncer, (Marinova, Australia); inhibidores de heparanasa, (InSight, Israel); KL 3106, (Kolon, Corea del Sur); Honokiol, (Universidad de Emory, EE. UU.); ZK CDK, (Schering AG, Alemania); ZK Angio, (Schering AG, Alemania); ZK 229561, (Novartis, Suiza y Schering AG, Alemania); XMP 300, (XOMA, EE. Uu.); VGA 1102, (Taisho, Japón); moduladores del receptor de VEGF, (Farmacopea, EE.UU.); antagonistas de VE-cadherina-2, (ImClone Systems, EE. UU.); Vasostatina, (Institutos Nacionales de Salud, EE. UU.); vacuna, Flk-1, (ImClone Systems, EE. Uu.); TZ 93, (Tsumura, Japón); TumStatin, (Hospital Beth Israel, EE. UU.); FLT 1 soluble truncado (receptor 1 del factor de crecimiento endotelial vascular), (Merck & Co, EE. UU.); ligandos de Tie-2, (Regeneron, EE. UU.); e inhibidor de trombospondina 1, (Fundación de Salud, Educación e Investigación de Allegheny, EE. UU.).
Los inhibidores de la autofagia incluyen, sin carácter limitante, cloroquina, 3-metiladenina, hidroxicloroquina (Plaquenil ™), bafilomicina A1, ribósido de 5-amino-4-imidazolcarboxamida (AICAR), ácido okadaico, toxinas de algas supresoras de la autofagia que inhiben las proteínas fosfatasas de tipo 2A o tipo 1, análogos de cAMP y fármacos que elevan los niveles de cAMP tales como adenosina, LY204002, ribósido de W6-mercaptopurina y vinblastina. Además, también se puede utilizar ARNip o antisentido que inhibe la expresión de proteínas que incluyen, sin carácter limitante, ATG5 (que están involucradas en la autofagia).
Los agentes o compuestos farmacéuticamente activos adicionales que se pueden usar en el tratamiento de cánceres y que se pueden usar combinados con uno o más compuestos de la presente invención incluyen: epoetina alfa; darbepoetina alfa; panitumumab; pegfilgrastim; palifermina; filgrastim; denosumab; ancestim; AMG 102; AMG 386; AMG 479; AMG 655; AMG 745; aMg 951 y AMG 706 o una sal farmacéuticamente aceptable de estos.
En ciertas realizaciones, una composición proporcionada en la presente se administra junto con un agente quimioterápico. Los agentes quimioterápicos adecuados pueden incluir, productos naturales tales como los alcaloides de la vinca (por ejemplo, vinblastina, vincristina, y vinorrelbina), paclitaxel, epidipodofilotoxinas (por ejemplo, etopósido y tenipósido), antibióticos (por ejemplo, dactinomicina (actinomicina D), daunorrubicina, doxorrubicina, y idarrubicina), antraciclinas, mitoxantrona, bleomicinas, plicamicina (mitramicina), mitomicina, enzimas (por ejemplo, L-asparaginasa que metaboliza sistémicamente la L-asparagina y provoca la inanición de las células que no tienen capacidad para sintetizar su propia asparagina), agentes antiplaquetarios, agentes alquilantes antimitóticos/antiproliferativos tales como mostazas nitrogenadas (por ejemplo, mecloretamina, ciclofosfamida y análogos, melfalán y clorambucilo), etileniminas y metilmelaminas (por ejemplo, hexametilmelamina y tiotepa), inhibidores de CDK (por ejemplo, seliciclib, UCN-01, P1446A-05, PD-0332991, dinaciclib, P27-00, AT-7519, RGB286638, y SCH727965), alquilsulfonatos (por ejemplo, busulfán), nitrosoureas (por ejemplo, carmustina (BCNU) y análogos y estreptozocina), trazenosdacarbazinina (DTIC), antimetabolitos antimitóticos/antiproliferativos tales como análogos de ácido fólico (por ejemplo, metotrexato), análogos de pirimidina (por ejemplo, fluorouracilo, floxuridina, y citarabina), análogos de purina e inhibidores relacionados (por ejemplo, mercaptopurina, tioguanina, pentostatina y 2-clorodesoxiadenosina), inhibidores de la aromatasa (por ejemplo, anastrozol, exemestano, y letrozol), y complejos de coordinación de platino (por ejemplo, cisplatino y carboplatino), procarbazina, hidroxiurea, mitotano, aminoglutetimida, inhibidores de la desacetilasa de histonas (HDAC) (por ejemplo, tricostatina, butirato de sodio, apicidán, ácido suberoilanilidahidroxámico, vorinostat, LBH 589, romidepsina, ACY-1215, y panobinostat), inhibidores de mTor (por ejemplo, temsirolimus, everolimus, ridaforolimus, y sirolimus), inhibidores de KSP(Eg5) (por ejemplo, Array 520), agentes de unión al ADN (por ejemplo, Zalypsis), inhibidor de PI3K delta (por ejemplo, GS-1101 y TGR-1202), inhibidor de PI3K delta y gamma (por ejemplo, CAL-130), inhibidor de múltiples cinasas (por ejemplo, TG02 y sorafenib), hormonas (por ejemplo, estrógeno) y agonistas de hormonas tales como la hormona liberadora de hormona luteinizante (LHRH) (por ejemplo, goselerina, leuprolida y triptorelina), anticuerpo neutralizador de BAFF (por ejemplo, LY2127399), inhibidores de IKK, inhibidores de p38MAPK, anti-IL-6 (por ejemplo, CNTO328), inhibidores de la telomerasa (por ejemplo, GRN 163L), inhibidores de la cinasa aurora (por ejemplo, MLN8237, aMg 900, AZD-1152), anticuerpos monoclonales de la superficie celular (por ejemplo, anti-CD38 (HUMAX-CD38), anti-CS1 (por ejemplo, elotuzumab), inhibidores de HSP90 (por ejemplo, 17 AaG y KOS 953), inhibidores de P13K/Akt (por ejemplo, perifosina), inhibidor de Akt (por ejemplo, GSK-2141795), inhibidores de PKC (por ejemplo, enzastaurina), FTI (por ejemplo, Zarnestra™), anti-CD138 (por ejemplo, BT062), inhibidor de cinasas específico de Torc1/2 (por ejemplo, INK128), inhibidor de cinasas (por ejemplo, GS-1101), agente de direccionamiento a ER/UPR (por ejemplo, MKC-3946), inhibidor de cFMS (por ejemplo, ArRY-382), inhibidor de JAK1/2 (por ejemplo, CYT387), inhibidor de PARP (por ejemplo, olaparib, talazoparib, niraparib veliparib (ABT-888)), antagonista de BCL-2. Otros agentes quimioterápicos pueden incluir mecloretamina, camptotecina, ifosfamida, tamoxifeno, raloxifeno, gemcitabina, navelbina, sorafenib o cualquier análogo o variante derivada de los anteriores.
Los compuestos de la presente invención también se pueden utilizar junto con radioterapia, terapia hormonal, cirugía
e inmunoterapia, que son terapias muy conocidas por los expertos en la técnica.
En ciertas realizaciones, una composición farmacéutica proporcionada en la presente se administra junto con un esteroide. Los esteroides adecuados pueden incluir, pero sin carácter limitante, 21-acetoxipregnenolona, alclometasona, algestona, amcinonida, beclometasona, betametasona, budesonida, cloroprednisona, clobetasol, clocortolona, cloprednol, corticosterona, cortisona, cortivazol, deflazacort, desonida, desoximetasona, dexametasona, diflorasona, diflucortolona, difuprednato, enoxolona, fluazacort, flucloronida, flumetasona, flunisolida, acetónido de fluocinolona, fluocinonida, fluocortinbutilo, fluocortolona, fluorometolona, acetato de fluperolona, acetato de fluprednideno, fluprednisolona, flurandrenolida, propionato de fluticasona, formocortal, halcinonida, propionato de halobetasol, halometasona, hidrocortisona, etabonato de loteprednol, mazipredona, medrisona, meprednisona, metilprednisolona, furoato de mometasona, parametasona, prednicarbato, prednisolona, 25-dietilaminoacetato de predinisolona, fosfato de prednisolona de sodio, prednisona, prednival, prednilideno, rimexolona, tixocortol, triamcinolona, acetónido de triamcinolona, benetónido de triamcinolona, hexacetónido de triamcinolona y sales y/o derivados de estos. En una realización particular, los compuestos de la presente invención también se pueden utilizar combinados con agentes farmacéuticamente activos adicionales para el tratamiento de las náuseas. Los ejemplos de agentes que se pueden usar para tratar las náuseas incluyen: dronabinol; granisetrón; metoclopramida; ondansetrón; y proclorperazina; o una sal farmacéuticamente aceptable de estos.
También se pueden utilizar los compuestos o composiciones farmacéuticas de la divulgación combinados con una cantidad de una o más sustancias seleccionadas entre inhibidores de EGFR, inhibidores de MEK, inhibidores de PI3K, inhibidores de AKT, inhibidores de TOR e inmunoterapias, incluidos agentes anti-PD-1, anti-PDL1, anti-CTLA4, anti-LAG1 y anti-OX40, agonistas de GITR, linfocitos CAR-T y BiTE.
Los inhibidores de EGFR incluyen, sin carácter limitante, antagonistas que son moléculas de bajo peso molecular, inhibidores de anticuerpos o ARNip o nucleótidos antisentido específicos. Los anticuerpos inhibidores de EGFR útiles incluyen cetuximab (Erbitux), panitumumab (Vectibix), zalutumumab, nimotuzumab y matuzumab. Los antagonistas que son moléculas de bajo peso molecular de EGFR incluyen gefitinib, erlotinib (Tarceva) y más recientemente, lapatinib (TykerB). Véanse, por ejemplo, Yan L, et. al., Pharmacogenetics and Pharmacogenomics In Oncology Therapeutic Antibody Development, BioTechniques 2005; 39(4): 565-8, y Paez J G, et. al., EGFR Mutations In Lung Cancer Correlation With Clinical Response To Gefitinib Therapy, Science 2004; 304(5676): 1497-500.
Los ejemplos no limitantes de inhibidores de EGFR que son moléculas de bajo peso molecular incluyen cualquiera de los inhibidores de EGFR descritos en las siguientes publicaciones de patente y todas las sales y solvatos farmacéuticamente aceptables de dichos inhibidores de EGFR: solicitud de patente europea EP 520722 publicada el 30 de diciembre de 1992; solicitud de patente europea EP 566226 publicada el 20 de octubre de 1993; publicación internacional PCT WO 96/33980 publicada el 31 de octubre de 1996; patente de EE. UU. n.° 5747498 emitida el 5 de mayo de 1998; publicación internacional PCT WO 96/30347 publicada el 3 de octubre de 1996; solicitud de patente europea EP 787772 publicada el 6 de agosto de 1997; publicación internacional PCT WO 97/30034 publicada el 21 de agosto de 1997; publicación internacional PCT WO 97/30044 publicada el 21 de agosto de 1997; publicación internacional PCT WO 97/38994 publicada el 23 de octubre de 1997; publicación internacional PCT w O 97/49688 publicada el 31 de diciembre de 1997; solicitud de patente europea EP 837063 publicada el 22 de abril de 1998; publicación internacional PCT WO 98/02434 publicada el 22 de enero de 1998; publicación internacional PCT WO 97/38983 publicada el 23 de octubre de 1997; publicación internacional PCT WO 95/19774 publicada el 27 de julio de 1995; publicación internacional PCT WO 95/19970 publicada el 27 de julio de 1995; publicación internacional PCT WO 97/13771 publicada el 17 de abril de 1997; publicación internacional PCT WO 98/02437 publicada el 22 de enero de 1998; publicación internacional PCT WO 98/02438 publicada el 22 de enero de 1998; publicación internacional PCT WO 97/32881 publicada el 12 de septiembre de 1997; solicitud alemana DE 19629652 publicada el 29 de enero de 1998; publicación internacional PCT WO 98/33798 publicada el 6 de agosto de 1998; publicación internacional PCT WO 97/32880 publicada el 12 de septiembre de 1997; publicación internacional PCT w O 97/32880 publicada el 12 de setiembre de 1997; solicitud de patente europea EP 682027 publicada el 15 de noviembre de 1995; publicación internacional PCT WO 97/02266 publicada el 23 de enero de 197; publicación internacional PCT w O 97/27199 publicada el 31 de julio de 1997; publicación internacional PCT WO 98/07726 publicada el 26 de febrero de 1998; publicación internacional PCT WO 97/34895 publicada el 25 de setiembre de 1997; publicación internacional PCT WO 96/31510' publicada el 10 de octubre de 1996; publicación internacional PCT WO 98/14449 publicada el 9 de abril de 1998; publicación internacional PCT WO 98/14450 publicada el 9 de abril de 1998; publicación internacional PCT WO 98/14451 publicada el 9 de abril de 1998; publicación internacional PCT WO 95/09847 publicada el 13 de abril de 1995; publicación internacional PCT WO 97/19065 publicada el 29 de mayo de 1997; publicación internacional PCT WO 98/17662 publicada el 30 de abril de 1998; patente de EE. UU. n.° 5789427 emitida el 4 de agosto de 1998; patente de EE. UU. n.° 5650415 emitida el 22 de julio de 1997; patente de EE. UU. n.° 5656643 emitida el 12 de agosto de 1997; publicación internacional PCT WO 99/35146 publicada el 15 de julio de 1999; publicación internacional PCT WO 99/35132 publicada el 15 de julio de 1999; publicación internacional PCT WO 99/07701 publicada el 18 de febrero de 1999 y publicación internacional PCT WO 92/20642 publicada el 26 de noviembre de 1992. Los ejemplos no limitantes adicionales de inhibidores de EGFR que son moléculas de bajo peso molecular incluyen cualquiera de los inhibidores de EGFR descritos en Traxler, P., 1998, Exp. Opin. Ther. Patents 8(12):1599-1625.
Los inhibidores de EGFR basados en anticuerpos incluyen cualquier anticuerpo o fragmento de anticuerpo anti-EGFR que puede bloquear de forma parcial o total la activación de EGFR por su ligando natural. Los ejemplos no limitantes
de inhibidores de EGFR basados en anticuerpos incluyen los descritos en Modjtahedi, H., et al., 1993, Br. J. Cáncer 67:247-253; Teramoto, T., et al., 1996, Cáncer 77:639-645; Goldstein et al., 1995, Clin. Cáncer Res. 1:1311-1318; Huang, S. M., et al., 1999, Cáncer Res. 15:59(8):1935-40; y Yang, X., et al., 1999, Cáncer Res. 59:1236-1243. Por lo tanto, el inhibidor de EGFR puede ser un anticuerpo monoclonal Mab E7.6.3 (Yang, 1999 mencionado anteriormente) o Mab C225 (n.° de acceso ATCC HB-8508) o un anticuerpo o fragmento de anticuerpo que tiene la especificidad de unión de estos.
Los inhibidores de MEK incluyen, sin carácter limitante, CI-1040, AZD6244, PD318088, PD98059, PD334581, RDEA119, ARRY-142886, ARRY-438162 y PD-325901.
Los inhibidores de PI3K incluyen, sin carácter limitante, wortmanina, análogos de 17-hidroxiwortmanina descritos en el documento WO 06/044453, 4-[2-(1H-indazol-4-il)-6-[[4-(metilsulfonil)piperazin-1-il]metil]tieno[3,2-a]pirimidin-4-iljmorfolina (también conocida como gDc 0941 y descrita en las publicaciones PCT n.os Wo 09/036 082 y WO 09/055730), 2-metil-2-[4-[3-metil-2-oxo-8-(quinolin-3-il)-2,3-dihidroimidazo[4,5-c]quinolin-1-il]fenil]propionitrilo (también conocido como BEZ 235 o NVP-BEZ 235, y descrito en la publicación PCT n.° WO 06/122806), (S)-1 -(4-((2-(2-aminopirimidin-5-il)-7-metil-4-morfolinotieno[3,2-a]pirimidin-6-il)metil)piperazin-1-il)-2-hidroxipropan-1-ona (descrita en la publicación PCT n.° WO 2008/070740), LY294002 (2-(4-morfolinil)-8-fenil-4H-1-benzopiran-4-ona disponible de Axon Medchem), clorhidrato de PI 103 (clorhidrato de 3-[4-(4-morfolinilpirido-[3',2':4,5]furo[3,2-a(|pirimidin-2-il]fenol disponible de Axon Medchem), PIK 75 (clorhidrato de N-[(1 E)-(6-bromoimidazo[1 ,2-a]piridin-3-il)metileno]-N,2-dimetil-5-nitrobencenesulfonohidrazida disponible de Axon Medchem), PIK 90 (N-(7,8-dimetoxi-2,3-dihidroimidazo[1,2-c]quinazolin-5-il)nicotinamida disponible de Axon Medchem), bimesilato de GDC-0941 (bimesilato de 2-(1 H-indazol-4-il)-6-(4-metanosulfonilpiperazin-1-ilmetil)-4-morfolin-4-iltieno[3,2-adpirimidina disponible de Axon Medchem), AS-252424 (5-[1-[5-(4-fluoro-2-hidroxifenil)furan-2-il]met-(Z)ilideno]tiazolidino-2,4-diona disponible de Axon Medchem), y TGX-221 (7-metil-2-(4-morfolinil)-9-[1-(fenilamino)etil]-4H-pirido[1,2-a]pirimidin-4-ona disponible de Axon Medchem), XL-765, y Xl-147. Otros inhibidores de PI3K incluyen demetoxiviridina, perifosina, CAL101, PX-866 , BEZ235, SF1126 , INK1117, IPI-145, BKM120, XL147, XL765, Palomid 529, GSK1059615, ZSTK474, PWT33597, IC87114, TG100-115, CAL263, PI-103, GNE-477, CUDC-907 y AEZS-136.
Los inhibidores de AKT incluyen, sin carácter limitante, Akt-1 -1 (inhibe Akt1) (Barnett et al. (2005) Biochem. J., 385 (Pt.
2), 399-408); Akt-1 -1,2 (inhibe Ak1 y 2) (Barnett et al. (2005) Biochem. J. 385 (Pt. 2), 399-408); API-59CJ-Ome (por ejemplo, Jin et al. (2004) Br. J. Cancer 91, 1808-12); compuestos de 1-H-imidazo[4,5-c]piridinilo (por ejemplo, WO05011700); indol-3-carbinol y derivados de este (por ejemplo, patente de EE. UU. n.° 6656963; Sarkar y Li (2004) J Nutr. 134(Suμl. 12), 3493S-3498S); perifosina (por ejemplo, interfiere con la ubicación en la membrana de Akt; Dasmahapatra et al. (2004) Clin. Cancer Res. 10(15), 5242-52, 2004); análogos lipídicos de éter de fosfatidilinositol (por ejemplo, Gills y Dennis (2004) Expert. Opin. Investig. Drugs 13, 787-97); y triciribina (TCN, API-2 o identificador NCI: NSC 154020; Yang et al. (2004) Cancer Res. 64, 4394-9).
Los inhibidores de TOR incluyen, sin carácter limitante, los inhibidores incluyen AP-23573, CCI-779, everolimus, RAD-001, rapamicina, temsirolimus, inhibidores de TORC1/TORC2 que compiten con ATP, incluidos PI-103, PP242, PP30 y Torina 1. Otros inhibidores de TOR incluyen potenciador FκΒP12, rapamicinas y derivados de estos, incluidos: CCI-779 (temsirolimus), RAD001 (Everolimus; WO 9409010) y AP23573; rapálogos, por ejemplo, como los divulgados en los documentos WO 98/02441 y WO 01/14387, por ejemplo, AP23573, AP23464 o AP23841; 40-(2-hidroxietil)rapamicina, 40-[3-hidroxi(hidroximetil)metilpropanoato]rapamicina (también denominada CC1779), 40-epi-(tetrazolil)rapamicina (también denominada ABT578), 32-desoxorrapamicina, 16-pentiniloxi-32(S)-dihidrorrapanicina y otros derivados divulgados en el documento WO 05005434; derivados divulgados en la patente de EE. UU. n.° 5 258389, WO 94/090101, WO 92/05179, patente de EE. UU. n.25118677, patente de EE. UU. n.25118678, patente de EE. UU. n.25100883, patente de EE. UU. n.25151 413, patente de EE. UU. n.25120842, WO 93/111130, WO 94/02136, WO 94/02485, WO 95/14023, WO 94/02136, WO 95/16691, WO 96/41807, WO 96/41807 y patente de EE. UU. n.° 5256790; derivados de rapamicina que contienen fósforo (por ejemplo, WO 05016252); derivados de 4H-1-benzopiran-4-ona (por ejemplo, solicitud provisional de EE. UU. n.° 60/528340).
Las inmunoterapias incluyen, sin carácter limitante, agentes anti-PD-1, agentes anti-PDL-1, agentes anti-CTLA-4, agentes anti-LAG1 y agentes anti-OX40. Los anticuerpos anti-PD-1 ejemplares y sus métodos de uso se describen en Goldberg et al., Blood 110(1 ):186-192 (2007), Thompson et al., Clin. Cancer Res. 13(6):1757-1761 (2007) y Korman et al., solicitud internacional n.° PCT/JP2006/309606 (publicación n.° WO 2006/121168 A 1 ) e incluyen: Yervoy™ (ipilimumab) o Tremelimumab (contra CTLA-4), galiximab (contra B7.1), BMS-936558 (contra PD-1), mK-3475 (contra PD-1), AMP224 (contra B7DC), BMS-936559 (contra B7-H1), MPDL3280A (contra B7-H1), MEDI-570 (contra ICOS), AMG557 (contra B7H2), MGA271 (contra B7H3), IMP321 (contra LAG-3), BMS-663513 (contra CD137), PF-05082566 (contra CD137), CDX-1127 (contra CD27), anti-OX40 (Providence Health Services), huMAbOX40L (contra OX40L), Atacicept (contra TACI), CP-870893 (contra CD40), Lucatumumab (contra CD40), Dacetuzumab (contra CD40), Muromonab-CD3 (contra CD3), Ipilumumab (contra CTLA-4). Las inmunoterapias también incluyen linfocitos T con modificaciones genéticas (por ejemplo, linfocitos CAR-T) y anticuerpos biespecíficos (por ejemplo, BiTE).
Los antagonistas de GITR incluyen, sin carácter limitante, proteínas de fusión de GITR y anticuerpos anti-GITR (por ejemplo, anticuerpos anti-GITR bivalentes), tales como una proteína de fusión de GITR descrita en la patente de EE. Uu. n.° 6 111 090box.c, patente europea n.°: 090505B1, patente de EE. UU. n.° 8 586 023, publicaciones PCT n.os: WO 2010/003118 y 2011/090754, o un anticuerpo anti-GITR descrito, por ejemplo, en la patente de EE. UU. n.°
7 025962, patente europea n.°: 1947183B1, patente de EE. UU. n.° 7812 135, patente de EE. UU. n.° 8388967, patente de EE. UU. n.° 8591 886, patente europea n.°: EP 1866339, publicación PCT n.°: WO 2011/028683, publicación PCT n.2: WO 2013/039954, publicación PCT n.2: W02005/007190, publicación PCT n.2: WO 2007/133822, publicación PCT n.°: WO2005/055808, publicación PCT n.°: WO 99/40196, publicación PCT n.°: WO 2001/03720, publicación PCT n.°: WO99/20758, publicación PCT n.°: WO2006/083289, publicación PCT n.°: WO 2005/115451, patente de EE. UU. n.° 7618632, y publicación PCT n.°: WO 2011/051726.
Los compuestos descritos en la presente se pueden utilizar combinados con los agentes divulgados en la presente u otros agentes adecuados, dependiendo de la afección que se esté tratando. Por lo tanto, en algunas realizaciones, el uno o más compuestos de la divulgación se coadministrarán con otros agentes como se ha descrito anteriormente. Cuando se utilizan en terapias combinadas, los compuestos descritos en la presente se administran con el segundo agente de forma simultánea o separada. La administración combinada puede incluir la administración simultánea de los dos agentes en la misma forma farmacéutica, la administración simultánea en formas farmacéuticas separadas y administración por separado. Es decir, un compuesto descrito en la presente y cualquiera de los agentes descritos anteriormente se pueden formular juntos en la misma forma farmacéutica y administrar simultáneamente. Como alternativa, se puede administrar simultáneamente un compuesto de la divulgación y cualquiera de los agentes descritos anteriormente, donde ambos agentes están presentes en formulaciones separadas. En otra alternativa, un compuesto de la presente divulgación se puede administrar seguido de cualquiera de los agentes descritos anteriormente o viceversa. En algunas realizaciones del protocolo de administración separada, un compuesto de la divulgación y cualquiera de los agentes descritos anteriormente se administran con unos minutos de diferencia, con unas horas de diferencia o con unos pocos días de diferencia.
Debido a que un aspecto de la presente invención contempla el tratamiento de las enfermedades/afecciones con un combinado de compuestos farmacéuticamente activos que se pueden administrar por separado, la invención se refiere además a la combinación de composiciones farmacéuticas separadas en forma de kit. El kit comprende dos composiciones farmacéuticas separadas: un compuesto de la presente invención y un segundo compuesto farmacéutico. El kit comprende un recipiente para contener las composiciones separadas tal como una botella dividida o un envase de aluminio con divisiones. Los ejemplos adicionales de recipientes incluyen jeringuillas, cajas y bolsas. En algunas realizaciones, el kit comprende instrucciones para el uso de los componentes individuales. La forma del kit es particularmente ventajosa cuando los componentes individuales se administran preferentemente en distintas formas farmacéuticas (por ejemplo, oral y parenteral) se administran en intervalos posológicos diferentes, o cuando el profesional de la salud a cargo del tratamiento desea la titulación de los componentes individuales de la combinación.
Datos experimentales
Abreviaturas: En la presente se pueden utilizar las siguientes abreviaturas:



Salvo que se indique otra cosa, todos los materiales se obtuvieron de proveedores comerciales y se utilizaron sin purificación adicional. Todas las partes son en peso y las temperaturas están en grados centígrados salvo que se indique otra cosa. Todas las reacciones asistidas por microondas se realizaron con un Smith SynthesizerTM de Biotage™. Todos los compuestos mostraron espectros de RMN coherentes con sus estructuras asignadas. Se determinaron los puntos de fusión en un aparato Buchi y están sin corregir. Se determinaron los datos de espectros de masas mediante la técnica de ionización por electronebulización. Se purificaron todos los ejemplos hasta >90 % de pureza según se determinó mediante cromatografía líquida de alto rendimiento. Salvo que se indique otra cosa, las reacciones se realizaron a temperatura ambiente.
En la síntesis de los compuestos de la presente invención, puede ser deseable utilizar determinados grupos salientes. La expresión “grupos salientes” (“LG”) se refiere generalmente a grupos que pueden ser desplazados por un nucleófilo. Tales grupos salientes son conocidos en la técnica. Los ejemplos de grupos salientes incluyen, sin carácter limitante, haluros (por ejemplo, I, Br, F, Cl), sulfonatos (por ejemplo, mesilato, tosilato), sulfuros (por ejemplo, SCH3), N-hidroxisuccinimida, N-hidroxibenzotriazol, y similares. Los ejemplos de nucleófilos incluyen, sin carácter limitante, aminas, tioles, alcoholes, reactivos de Grignard, especies aniónicas (por ejemplo, alcóxidos, amidas, carbaniones) y similares.
Los ejemplos presentados a continuación ilustran realizaciones específicas de la presente invención. Estos ejemplos pretenden ser representativos y no se pretende que limiten el alcance de las reivindicaciones de forma alguna.
Cabe destacar que cuando se utiliza un porcentaje (%) con respecto a un líquido, es un porcentaje en volumen con respecto a la solución. Cuando se utiliza con un sólido, es el porcentaje con respecto a la composición sólida. Los materiales obtenidos de los proveedores comerciales se utilizaron normalmente sin purificación adicional. Las reacciones que utilizan reactivos sensibles a la humedad o al aire se realizaron normalmente en una atmósfera de nitrógeno o argón. Se midió la pureza con un sistema de cromatografía líquida de alta resolución (HPLC) con detección UV a 254 nm y 215 nm (Sistema A: Agilent Zorbax Eclipse XDB-C84.6 x 150 mm, 5 μm, CH3CN de un 5 a un 100 % en H2O con un 0.1 % de TFA durante 15 min a 1.5 mL/min; Sistema B: Zorbax SB-C8, 4.6 x 75 mm, CH3CN de un 10 a un 90 % en H2O con un 0.1 % de ácido fórmico durante 12 min a 1.0 mL/min) (Agilent Technologies, Santa Clara,
CA). La cromatografía sobre gel de sílice se realizó generalmente con cartuchos de gel de sílice preempaquetados (Biotage, Uppsala, Suecia o Teledyne-Isco, Lincoln, NE). Se registraron los espectros de 1H RMN en un espectrómetro Bruker AV-400 (400 MHz) (Bruker Corporation, Madison, WI) o un espectrómetro Varian (Agilent Technologies, Santa Clara, CA) de 400 MHz a temperatura ambiente. Se notificaron todos los protones observados como partes por millón (ppm) en campo bajo respecto a tetrametisilano (TMS) u otra referencia interna en el disolvente adecuado indicado. Se notificaron los datos de la siguiente manera: desplazamiento químico, multiplicidad (s = singulete, d = doblete, t = triplete, c = cuadruplete, a = amplio, m = multiplete), constantes de acoplamiento y número de protones. Se determinaron los datos espectrales de masas (MS) de baja resolución en un LC/MS Agilent serie 1100 (Agilent Technologies, Santa Clara, CA) con detección UV a 254 nm y 215 nm y un modo de electronebulización (ESI) de baja resonancia.
A efectos de claridad en esta sección de síntesis general, los Compuestos de Fórmula (I), como se definen en el compendio de las invenciones se pueden dibujar esquemáticamente para contener el Anillo Ar1 y el Anillo Ar2 de la siguiente manera:

del conector, y el anillo Ar2 se ubica a la derecha del conector. Generalmente, los compuestos de Fórmula (I) se pueden sintetizar mediante tres pasos generales, de la siguiente manera:
Paso 1: Preparación del compuesto con anillo Ar1.
Paso 2: Preparación del compuesto con anillo Ar2.
Paso 3: Acoplamiento del compuesto con anillo Ar1 con el compuesto con anillo Ar2:
Se pretende que los siguientes Esquemas genéricos A-E sirvan como guía para los expertos en síntesis química, quienes observarán fácilmente que es posible modificar el disolvente, concentración, reactivo, grupo protector, orden de los pasos de síntesis, tiempo, temperatura y similares según sea necesario, lo que está dentro de sus competencias, conforme al juicio del experto en la técnica.
ESQUEMAA
De acuerdo con el Esquema A, en una realización, un compuesto de Fórmula (I) como se divulga en la presente se puede sintetizar de la siguiente manera:
Paso 1a: Preparación del compuesto con anillo Ar1:

Paso 1a: Preparación del compuesto con anillo Ar1: : El compuesto A-1, donde W 1 es un halógeno, por ejemplo fluoro, cloro o bromo se puede hacer reaccionar con un agente que contiene un grupo R2 en presencia de una base adecuada, en un disolvente orgánico adecuado tal como NMP, dioxano, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno, y similares, para formar el compuesto A-2. El compuesto A-1 está comercializado o se puede sintetizar mediante métodos conocidos por los expertos en la técnica. Los ejemplos del compuesto A-1 incluyen, sin carácter limitante, 6-fluoropiridin-2-amina, 6-fluoro-4-metilpiridin-2-amina, 6-cloro-5-metilpiridin-2-amina, 6-bromo-5-metilpiridin-2- amina o 6-cloropirazin-2-amina. Los ejemplos de reactivos R2 incluyen, sin carácter limitante (1) (fl)-2-metilmorfolina, (2) clorhidrato de 4,4-difluoropiperidina, (3) clorhidrato de 3,3-difluoroazetidina, o (4) 3,3,3-trifluoropropan-1-ol. Los ejemplos de bases incluyen, sin carácter limitante, diisopropiletilamina, carbonato de potasio o hidruro de sodio.
P aso 1b: P rep a ra c ión de l co m p u e s to con an illo A r1:

Como alternativa, el compuesto A-1 como se ha definido en el paso 1a, se puede convertir en el compuesto A-2, como se ha definido en el paso 1 a, mediante una reacción de acoplamiento cruzado de Suzuki con un reactivo de organoboro R2 adecuado (R2-BY2, donde Y es un grupo funcional orgánico) tal como 2-(4,4-difluorociclohex-1-en-1-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano o 2-(4-fluorociclopent-1 -en-1 -il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano, y un catalizador de paladio y una base adecuados, tales como el aducto PdCl2(dppf)-DCM y fosfato de potasio tribásico. Este paso va seguido de una reducción con un catalizador de paladio adecuado y una fuente de hidrógeno, tal como Pd/C en presencia de hidrógeno gaseoso, para formar el compuesto A-2. Esta reacción de Suzuki alternativa se puede utilizar cuando el grupo R2 está ligado al anillo Ar1 mediante un enlace carbono-carbono.
Paso 2a: Preparación del compuesto con anillo Ar2:

En la Paso 2a, el compuesto A-3, donde cada uno de W2 y W3 es independientemente un halógeno, por ejemplo fluoro, cloro, bromo, o yodo, se puede hacer reaccionar con un reactivo Rx, tal como (1) clorhidrato de 6-azaespiro[2.5]octano, (2) clorhidrato de 4,4-dimetilpiperidina, (3) clorhidrato de 3,4,4-trimetilpiperidina, (4) clorhidrato de 4-metil-6-azaespiro[2.5]octano, o (5) clorhidrato de 7-azaespiro[3.5]nonano, en un disolvente orgánico adecuado tal como NMP, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno, DMSO, y similares, para formar el compuesto A-4.

En el Paso 3a, el compuesto A-4, que se obtuvo del Paso 2a, se puede hacer reaccionar con un agente activante tal como un cloruro de ácido (COCl)2 o SOCl2, en un disolvente orgánico adecuado tal como tetrahidrofurano, cloruro de metileno y similares, para formar un derivado de cloruro de ácido, que después se puede hacer reaccionar con un compuesto A-2 para formar el compuesto A-5. Como alternativa, el compuesto A-2 se puede acoplar directamente con el compuesto A-4, que se obtuvo del Paso 2a, en un disolvente orgánico adecuado tal como acetonitrilo,
tetrahidrofurano, DMF, cloruro de metileno, y similares, en presencia de un reactivo de acoplamiento tal como N, N-diisopropilcarbodiimida, N-(3-dimetilaminopropil)-N'-etilcarbodiimida, hexafluorofosfato de benzotriazol-1-iloxitripirrolidinofosfonio, hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio, carbonildiimidazol y anhídrido polifosfónico. Los expertos en síntesis química comprenderán fácilmente que se pueden utilizar otros reactivos de acoplamiento. Se puede utilizar la manipulación adicional del grupo halógeno W3 mediante reacciones de transformación tales como sulfoamidación, sulfinación o sulfonilación catalizada por metal en un disolvente orgánico adecuado tal como DMSO, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno y similares, en presencia de un catalizador metálico y de un reactivo R1, tal como (1) 1-metilciclopropano-1-sulfonamida, (2) 3-metiloxetan-3-amina, (3) 3-mercaptoazetidino-1-carboxilato de íerí-butilo, (4) 2-sulfamoilpropanoato de etilo, (5) 2-hidroxipropano-1-sulfonamida, (6) 2-hidroxietano-1-sulfonamida, (7) yodoacetato de etilo, (8) 2-mercaptopropan-1-ol, (9) 2-mercapto-2-metilpropan-1-ol, (10) 2-aminoetan-1-ol, o (11) ciclopropanotiol para formar el Compuesto (I). Los químicos expertos entenderán fácilmente que una reacción de acoplamiento tal como la mostrada en el Paso 3a se puede llevar a cabo en varias condiciones conocidas.
ESQUEMA B
Paso 1a o 1b: Preparación del compuesto con anillo Ring Ar1: véase el ESQUEMA A anterior
Paso 2b: Preparación del compuesto con anillo Ar2:

El Esquema B proporciona un método alternativo para la formación de compuestos de Fórmula (I), tal como se divulga en la presente. Después del Paso 1a o el Paso 1 b como se describe en el Esquema A, el grupo R1 se puede introducir como alternativa en el Anillo Ar2 en el Paso 2b en lugar de en el Paso 3a como en el Esquema A. De acuerdo con el Paso 2b, el compuesto B-1, donde cada uno de W4 y W5 es independientemente un halógeno, por ejemplo fluoro, cloro, bromo, o yodo, se puede hacer reaccionar con un grupo protector de ácido carboxílico adecuado (reactivo PG1), tal como yoduro de metilo en presencia de una base tal como carbonato de potasio para formar un éster metílico, u otro grupo protector adecuado, para formar otro éster tal como un éster bencílico, en un disolvente orgánico adecuado tal como NMP, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno y similares, para formar el compuesto B-2, donde cada uno de W4 y W5 son como se han definido en el compuesto B-1. El compuesto B-2 se puede hacer reaccionar a continuación con un reactivo Rx, tal como 6-azaespiro[2.5]octano, en un disolvente orgánico adecuado tal como NMP, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno, DMSO, y similares, para formar el compuesto B-3, donde W5 es como se ha definido en el compuesto B-1. El compuesto B-3 se puede hacer reaccionar a continuación con un reactivo R1 mediante una reacción de transformación tal como una sulfoamidación, sulfinación, o sulfonilación catalizada por metales, en un disolvente orgánico adecuado tal como DMSO, acetonitrilo, tetrahidrofurano, DMF, y similares, en presencia de un catalizador metálico, tal como yoduro de cobre, Pd2(dba)3 para formar el compuesto B-4 , que posteriormente se puede hacer reaccionar con un agente desprotector de ácido carboxílico adecuado para formar el compuesto B-5. Los expertos en la técnica conocen los grupos protectores de ácido carboxílico y los agentes de desprotección adecuados, por ejemplo, como se analiza en Greene's Protective Groups in Organic Synthesis.
P aso 3b : A co p la m ie n to de l co m p u e s to con an illo A r1 con el co m p u e s to de l A n illo A r2:

El Paso 3b es similar a la reacción de acoplamiento que se ha descrito anteriormente en el Paso 3a.
ESQUEMAC
El Esquema C provee otro método alternativo más para la formación de compuestos de Fórmula (I), como se divulgan en la presente. De acuerdo con el Esquema C, el Paso 1a se puede realizar como se describe en el Esquema A, seguido del Paso 2b como se describe en el Esquema B.
Paso 3c: Acoplamiento del compuesto con anillo Ar1 con el compuesto con Anillo Ar2:

En el Paso 3c, el compuesto A-1a, que es el compuesto A-1 del Esquema A, donde X1 es N o CR6 y W 1 es un halógeno, por ejemplo fluoro o cloro, se puede hacer reaccionar con un compuesto B-5 obtenido en el paso 2b del esquema B, en presencia de agentes activantes en condiciones similares a las de los pasos 3a y 3b anteriores, para formar el compuesto C-1, donde W 1 es como se ha definido en el compuesto A-1a, que a continuación se puede hacer reaccionar con un agente que contiene un grupo R2, tal como (1) (fi)-2-metilmorfolina, (2) clorhidrato de 4,4-difluoropiperidina, (3) clorhidrato de 3,3-difluoroazetidina o (4) 3,3,3-trifluoropropan-1-ol, en presencia de una base adecuada, tal como diisopropiletilamina, carbonato de potasio o hidruro de sodio, en un disolvente orgánico adecuado tal como NMP, dioxano, acetonitrilo, tetrahidrofurano, DMF, cloruro de metileno, y similares, para formar el compuesto de fórmula (I).
ESQUEMAD
El Esquema D proporciona otro método alternativo más para la formación de compuestos de Fórmula (I) como se divulgan en la presente. De acuerdo con el Esquema D, el Paso 1a o 1b se puede realizar como se describe en el Esquema A, seguido del Paso 2b como se describe en el Esquema B.
P aso 3d : A co p la m ie n to de l c o m p u e s to con an illo A r1 con el co m p u e s to con A n illo A r2:

En el Paso 3d, el compuesto A-1a, que es el compuesto A-1 del Esquema A, donde X1 es N o CR6 y W 1 es un halógeno, por ejemplo fluoro o cloro, se puede hacer reaccionar con un compuesto B-5 obtenido en el paso 2a del esquema A, en presencia de un agente activante en condiciones similares a las de los pasos 3a y 3b anteriores, para formar el compuesto D-1, donde W 1 es como se ha definido en el compuesto A-1 a y W3 es como se ha definido en el compuesto B-5, que después se puede hacer reaccionar con un agente que contiene un grupo R2, tal como (1) (R)-2-metilmorfolina, (2) clorhidrato de 4,4-difluoropiperidina, (3) clorhidrato de 3,3-difluoroazetidina, o (4) 3,3,3-trifluoropropan-1-ol, opcionalmente en presencia de una base adecuada, tal como diisopropiletilamina, carbonato de potasio o hidruro de sodio, en un disolvente orgánico adecuado tal como NMP, dioxano, acetonitrilo, tetrahidrofurano, DMF y similares, para formar el compuesto A-5a, que es el compuesto A-5, donde X1 es N y W3 es como se ha definido en el compuesto B-5, que se puede hacer reaccionar a continuación con un agente que contiene un grupo R1 mediante una reacción de transformación tal como una sulfoamidación, sulfinación, o sulfonilación catalizada por metales, en un disolvente orgánico adecuado tal como DMSO, dioxano, acetonitrilo, tetrahidrofurano, DMF y similares, en presencia de un catalizador metálico, para formar el compuesto formula (I).
ESQUEMAE
El Esquema E proporciona otro método alternativo más para la formación de compuestos de Fórmula (I), tal como se divulgan en la presente. De acuerdo con el Esquema E, el Paso 1a o 1b se puede realizar como se describe en el Esquema A para preparar el compuesto A-2. El compuesto E-1, donde W6 es un halógeno, por ejemplo fluoro o cloro, que incluye, sin carácter limitante ácido 2-fluoro-4-nitrobenzoico, ácido 2,5-difluoro-4-nitrobenzoico o ácido 2,6 difluoro-4-nitrobenzoico, está comercializado o se puede sintetizar de acuerdo con métodos conocidos por los expertos en la técnica.
Paso 3e: Acoplamiento del compuesto con anillo Ar1 con el compuesto con Anillo Ar2
En el paso 3e, el compuesto A-2 se puede hacer reaccionar con un compuesto E-1, en presencia de un reactivo activante en condiciones similares a las de los pasos 3a y 3b anteriores, para formar el compuesto E-2, que a continuación se puede hacer reaccionar con un reactivo Rx de una forma similar a la descrita en el Paso 2a para formar el compuesto E-3. El grupo nitro del compuesto E-4 se puede convertir a continuación en un grupo amino por reacción con un agente reductor que incluye, sin carácter limitante, paladio sobre carbón e hidrógeno gaseoso, para formar el compuesto E-4, que puede hacerse reaccionar a continuación con un reactivo R1, tales como (1) 1-metilciclopropano-1-sulfonamida, (2) 3-metiloxetan-3-amina, (3) 3-mercaptoazetidina-1-carboxilato de ferf-butilo, (4) 2-sulfamoilpropanoato de etilo, (5) 2-hidroxipropano-1-sulfonamida, (6) 2-hidroxietano-1-sulfonamida, (7) yodoacetato de etilo, (8) 2-mercaptopropan-1-ol, (9) 2-mercapto-2-metilpropan-1-ol, (10) 2-aminoetan-1-ol, o (11) ciclopropanotiol, en una reacción de transformación tal como sulfoamidación, sulfinación, o sulfonilación catalizada por metales, en un disolvente orgánico adecuado tal como DMSO, dioxano, acetonitrilo, tetrahidrofurano, DMF, y similares, en presencia de un catalizador metálico, para formar el compuesto (I).
EJEMPLOS
Preparación de Intermedios Sintéticos
Intermedios con Anillo Ar1:
Intermedio 1: 6-Amino-N-ífetf-butil)piridino-2-sulfonamida

Paso 1: Una solución enfriada con hielo de cloruro de 6-bromopiridino-2-sulfonilo (0.50 g, 1.9 mmol, Suzhou sibian, China) en diclorometano (10 mL), se añadieron sucesivamente trietilamina (0.543 mL, 3.90 mmol) y ferf-butilamina (0.310 mL, 2.92 mmol) en una atmósfera de nitrógeno. La mezcla de reacción se agitó a temperatura ambiente durante 1.5 h. Una vez completada, la mezcla de reacción se desactivó con agua (10 mL) y la mezcla bifásica se extrajo con diclorometano (3 x 15 mL). Los extractos orgánicos combinados se lavaron con una solución saturada de salmuera (15 mL) y se secaron con Na2SO4 anhidro. La solución se filtró y se concentró a presión reducida para obtener el material crudo como un aceite amarillo claro. El material crudo se absorbió en un tapón de gel de sílice y se purificó mediante Isolera-Biotage, eluido con de un 17 % a un 22 % de acetato de etilo en éter de petróleo, para proporcionar 6-bromo-N-(ferf-butil)piridino-2-sulfonamida (0.35 g, 1.19 mmol, 61 % de rendimiento) como un sólido blanquecino. 1H
RMN (400 MHz, Cloroformo-d) 57.98 (dd, J = 7.6, 0.9 Hz, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.65 (dd, J = 8.0, 0.9 Hz, 1H), 4.96 (s, 1 H), y 1.27 (s, 9H).
Paso 2: En un tubo sellado de 500 mL, a una mezcla de 6-bromo-N-(ferf-butil)piridino-2-sulfonamida (14.5 g, 49.5 mmol), N7,W2-dimetiletano-1,2-diamina (0.436 g, 4.95 mmol), K2CO3 (1.367 g, 9.89 mmol), y yoduro de cobre (I) (0.471 g, 2.47 mmol) se añadieron amoniaco acuoso (21 %, 100 mL, 970 mmol) y etilenglicol (100 mL). El tubo se selló con un tapón de rosca en atmósfera de nitrógeno. La mezcla de reacción resultante se agitó a 60 °C durante 18 h. Se permitió que la mezcla de reacción se enfriara hasta la temperatura ambiente, se diluyó con agua (150 mL) y se extrajo con EtOAc (3 x 100 mL). Los extractos orgánicos combinados se lavaron con una solución saturada de salmuera (50 mL) y se secaron con Na2SO4 anhidro. La solución se filtró y se concentró a presión reducida para obtener el material crudo como un aceite amarillo claro. El material crudo se absorbió sobre un tapón de gel de sílice y se purificó mediante Isolera-Biotage, eluido con de un 5 % a un 6 % de metanol en cloroformo, para proporcionar 6-amino-N-(íe/tbutil)piridino-2-sulfonamida (6.94 g, 30.3 mmol, 61 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-de) 57.54 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.21 (s, 1H), 7.03 (dt, J = 7.3, 0.9 Hz, 1H), 6.59 (dt, J = 8.4, 1.0 Hz, 1H), 6.33 (s, 2H), y 1.12 (d, J = 1.3 Hz, 9H). m /z (ESI): 230.1 (M+H)+.

A un tubo resistente a la presión de 250 mL se añadieron 6-fluoro-4-metilpiridin-2-amina (10.0 g, 79 mmol, Sibian chemicals, China), morfolina (8.29 g, 95 mmol) y DIPEA (41.5 mL, 238 mmol). La mezcla se calentó a 150 °C durante 18 h. La mezcla de reacción se desactivó con agua (100 mL) y se extrajo con EtOAc (2 x 250 mL). Los extractos orgánicos combinados se lavaron con salmuera (200 mL), se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo crudo se absorbió en un tampón de gel de sílice y se purificó mediante cromatografía ultrarrápida a través de una columna de gel de sílice preempaquetada Redi-Sep (40 g), eluyendo con un gradiente de un 1 % a un 15 % EtOAc en hexanos, para obtener el compuesto del título (8.5 g, 44.0 mmol, 56 % de rendimiento) como un semisólido de color café. 1H RMN (400 MHz, DMSO-a6) 55.75 (s, 1H), 5.67 (s, 1H), 5.44 (s, 2H), 3.65 (t, J = 8.4 Hz, 4H), 3.30 (t, J = 8.4 Hz, 4H), 2.06 (s, 3H). m/z (ESI): 194.2 (M+H)+.

A un tubo resistente a la presión de 500-mL se añadieron 6-fluoropiridin-2-amina (30.0 g, 268 mmol, Combi-Blocks, San Diego, CA), clorhidrato de (R)-2-metilmorfolina (44.2 g, 321 mmol, F chemicals, China) y DIPEA (140 mL, 803 mmol) en agua (60 mL). La mezcla se calentó a 180 °C durante 18 h. La mezcla de reacción se enfrió hasta la temperatura ambiente y se diluyó con agua (100 mL). La mezcla se extrajo con EtOAc (2 x 250 mL) y se lavó con salmuera (200 mL). Los extractos orgánicos combinados se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo crudo se absorbió en un tampón de gel de sílice y se purificó mediante cromatografía ultrarrápida a través de una columna de gel de sílice preempaquetada Redi-Sep (330 g), eluyendo con un gradiente de un 1 % a un 20 % EtOAc en hexanos, para obtener el compuesto del título (38.0 g, 197 mmol, 74 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, DMSO-rá): 57.20-7.14 (m, 1H), 5.89 (d, J = 8.0 Hz, 1H), 5.79 (d, J = 7.8 Hz, 1H), 5.53 (s, 2H), 4.07 - 3.97 (m, 1H), 3.98 - 3.82 (m, 2H), 3.55 - 3.45 (m, 2H), 2.71 - 2.58 (m, 1H), 2.35 - 2.29 (m, 1H), 1.13 (d, J = 6.2 Hz, 3H). m/z (ESI): 194.2 (M+H)+.
Intermedio 4: (fl)-4-Metil-6-(2-metilmorfolino)piridin-2-amina
Una mezcla de 6-fluoro-4-metilpiridin-2-amina (75.0 g, 595 mmol), y clorhidrato de (R)- 2-metilmorfolina (84 g, 832 mmol) en DIPEA (312 mL, 11.78 mol) se calentó a 150 °C en un autoclave (600 mL) durante 48 h. La mezcla de reacción se desactivó con agua (1000 mL) y se extrajo con EtOAc (2 x 2500 mL). Las capas orgánicas combinadas se lavaron con una solución de salmuera (1000 mL), se secaron (Na2SO4), se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando de un 30 a un 100 % de acetato de etilo en hexanos para obtener el compuesto del título (62 g, 50 % de rendimiento) como un sólido blanquecino. 1H RMN (300 MHz, DMsO-afe): 5 5.74 (s, 1h), 5.63 (s, 1H), 5.43 (s, 2H), 3.98 (dt, J = 12.8, 2.5 Hz, 1H), 3.90 - 3.80 (m, 2H), 3.53 - 3.44 (m, 2H), 2.62 (ddd, J = 15.9, 8.0, 3.7 Hz, 1H), 2.29 (ddd, J = 13.4, 10.5, 3.1 Hz, 1H), 2.04 (s, 3H), 1.11 (d, J = 6.3 Hz, 3H). m/z (ESI): 208.1 (M+H)+.
Tabla 1: Los siguientes intermedios se prepararon siguiendo procedimientos similares a los de los Intermedios 2-4:


Intermedio 5: 6-(4,4-Difluoropiperidin-1 -il)-4-metilpiridin-2-amina

Paso 1: A un autoclave se añadieron 2,6-dicloro-4-metilpiridina (80 g, 490 mmol), clorhidrato de 4,4-difluoropiperidina (86 g, 540 mmol) y DIPEA (342 mL, 1980 mmol) en NMP (800 mL). La mezcla de reacción se calentó a 180 °C durante 24 h. La mezcla de reacción se enfrió hasta la temperatura ambiente y se basificó hasta pH ~9 utilizando una solución acuosa al 10 % de NaHCO3. La mezcla de reacción se extrajo con acetato de etilo (2 x 1500 mL), se lavó con agua (1500 mL), se secó (Na2SO4), se filtró y se concentró a presión reducida. El material crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando un 5-10 % de acetato de etilo en hexanos para obtener la mezcla de 2,6-dicloro-4-metilpiridina y 2-cloro-6-(4,4-difluoropiperidin-1-il)-4-metilpiridina en una proporción 1:3 (102 g) como un aceite de color café pálido. Esta mezcla (102 g) se purificó adicionalmente mediante cromatografía en fase inversa utilizando un 60 % de acetonitrilo en agua como diluyente para obtener 2-cloro-6-(4,4-difluoropiperidin-1-il)-4-metilpiridina (70 g, 58 % de rendimiento) como un líquido de color café pálido. 1H RMN (400 MHz, DMSO-afe): S 6.76 (s, 1H), 6.57 (s, 1H), 3.66 (t, J = 5.6 Hz, 4H), 2.22 (s, 3H), 2.03 - 1.91 (m, 4H). m /z (ESI): 247.1 (M+H)+.
Paso 2: A una solución de 2-cloro-6-(4,4-difluoropiperidin-1-il)-4-metipiridina (30.0 g, 122 mmol) en 1,4-dioxano (300 mL) se añadieron (4-metoxifenil)metanamina (23.8 mL, 182 mmol) y Cs2CO3 (79 g, 240 mmol). La mezcla de reacción se desgasificó y purgó con nitrógeno durante 30 min. Se añadieron BINAP (7.57 g, 12.2 mmol) y acetato de paladio (II) (2.73 g, 12.2 mmol), la mezcla de reacción y se agitó a 100 °C durante 16 h. La mezcla de reacción se enfrió hasta temperatura ambiente, se filtró a través de un lecho de CELITE® y se lavó con acetato de etilo (100 mL). El filtrado se concentró a presión reducida. El residuo se extrajo con EtOAc (2 x 500 mL) y se lavó con agua (500 mL) seguida de salmuera (500 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida. En el residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando un 5-8 % de acetato de etilo en hexanos para obtener 6-(4,4-difluoropiperidin-1-il)-N-(4-metoxibencil)-4-metilpiridin-2-amina (48 g, 76 % de rendimiento) como un aceite amarillo. 1H RMN (400 MHz, DMSO-afe): S 7.22 (d, J = 7.2 Hz, 2H), 6.85 (d, J = 7.2 Hz, 2H), 6.64 (t, J = 6.0 Hz, 1H), 5.84 (s, 1H), 5.68 (s, 1H), 4.31 (d, J = 6.0 Hz, 2H), 3.71 (s, 3H), 3.56 (t, J = 5.6 Hz, 4H), 2.05 (s, 3H), 1.90 - 1.80 (m, 4H). m/z (ESI): 348.1 (M+H)+.
Paso 3: A una solución de 6-(4,4-difluoropiperidin-1-il)-N-(4-metoxibencil)-4-metilpiridin-2-amina (48.0 g, 138 mmol) en diclorometano seco (480 mL) se añadieron anisol (30.2 mL, 276 mmol) y TFA (240 mL, 3120 mmol). La mezcla de reacción se agitó a 55 °C durante 4 h y se concentró a presión reducida. El residuo se disolvió en agua (200 mL) y se basificó con una solución acuosa al 10 % de bicarbonato de sodio hasta pH ~8 y se extrajo con acetato de etilo (2 x 500 mL). Las capas orgánicas combinadas se lavaron con agua (200 mL) seguida de salmuera (200 mL), se secaron (Na2SO4), se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice utilizando de un 25 % a un 35 % de acetato de etilo en hexanos para obtener 6-(4,4-difluoropiperidin-1 -il)-4-metilpiridin-2-amina (LCMS ~85 %) como un aceite de color café. Este material se purificó adicionalmente mediante cromatografía en fase inversa utilizando un 50-60 % de acetonitrilo en agua para obtener 6-(4,4-difluoropiperidin-1 -il)-4-metilpiridin-2-amina (16.5 g, 72 mmol, 53 % de rendimiento) como un aceite de color café.
1H RMN (400 MHz, DMSO-afe): 55.86 (s, 1 H), 5.65 (s, 1 H), 5.48 (s, 2H), 3.56 (t, J = 5.2 Hz, 4H), 2.06 (s, 3H), 1.96 -1.87 (m, 4H). m /z (ESI): 228.2 (M+H)+.
Tabla 2: Los siguientes intermedios se prepararon siguiendo un procedimiento similar al del Intermedio 5:

Intermedio 6: 6-(3,3.3-Trifluoropropoxi)piridin-2-amina

A una solución de 6-fluoropiridin-2-amina (50 g, 450 mmol, Combi-Blocks) en 1,4-dioxano (500 mL) se añadió 3,3,3-trifluoropropan-1-ol (102 g, 892 mmol, Apollo) en una atmósfera de nitrógeno y la reacción se enfrió hasta 0 °C. Se añadió NaH (60 % en aceite mineral, 42.8 g, 1780 mmol) a la mezcla de reacción a 0 °C y la mezcla resultante se agitó a 90 °C durante 2 h. La mezcla de reacción se desactivó con agua fría (500 mL) y se extrajo con acetato de etilo (2 x 1000 mL). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando un 10 % de acetato de etilo en hexanos para obtener el compuesto del título (45 g, 50 % de rendimiento) como un aceite de color café pálido. 1H RMN (400 MHz, DMSO-afe): 57.30 - 7.26 (t, J = 7.8 Hz, 1H), 6.02 - 6.00 (dd, J = 7.8, 0.8 Hz, 1H), 5.89 - 5.86 (m, 3H), 4.36 - 4.33 (t, J = 6.2 Hz, 2H), 2.79 - 2.67 (qt, J = 11.5, 6.2 Hz, 2H). m/z (ESI): 207.1 (M+H)+.
Intermedio 7: 6-(3,3-Difluorociclobutil)-4-metilpiridin-2-amina

Paso 1: A una solución de ácido 3,3-difluorociclobutano-1 -carboxílico (3.0 g, 22. mmol, Combi-Blocks) y DMAP (0.269
g, 2.20 mmol) en diclorometano (30 mL) se añadieron ferf-butanol (4.22 mL, 44.1 mmol) seguido de diciclohexilcarbodiimida (5.00 g, 24.2 mmol) a 0 °C. La mezcla de reacción se agitó a TA durante 18 h antes de añadir éter dietílico (20 mL) a la mezcla de reacción a 0 °C. El sólido precipitado se separó por filtración y el filtrado se lavó con HCl 1.5 N (100 mL), agua y salmuera, se secó con Na2SO4 y se concentró a presión reducida para proporcionar 3,3-difluorociclobutano-1-carboxilato de ferf-butilo (2.78 g, 14.5mmol, 65.6 % de rendimiento) como un aceite transparente. 1H RMN (300 MHz, DMSO-d3): 5 ppm 2.93 (dddd, J=14.8, 7.5, 3.8, 2.1 Hz, 1 H), 2.63 - 2.87 (m, 4 H), 1.42 (d, J=2.3 Hz, 9 H).
Paso 2: A una solución de 2-bromo-6-fluoro-4-metilpiridina (1.5 g, 7.9 mmol, TCI Chemicals) y 3,3-difluorociclobutano-1-carboxilato de ferf-butilo (1.82 g, 9.47 mmol) en tolueno (35 mL) se añadió gota a gota bis(trimetilsilil)amiduro de sodio en THF (11.84 mL, 11.84 mmol, 1 M, Symax laboratories) a 0 °C y la mezcla de reacción se agitó durante 20 min a 0 °C y 4 h a TA. La mezcla de reacción se desactivó con una solución acuosa saturada de NH4Cl (50 mL) y se extrajo con EtOAc (2 x 50 mL). Los extractos orgánicos combinados se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % a un 20 %, EtOAc en éter de petróleo para proporcionar 1-(6-bromo-4-metilpiridin-2-il)-3,3-difluorociclobutano-1-carboxilato de ferf-butilo (1.6 g, 3.2 mmol, 41 % de rendimiento) como un aceite amarillo transparente. 1H RMN (300 MHz, DMSO-afe): 5 ppm 7.48 (t, J=1.0 Hz, 1 H), 7.34 (t, J=1.0 Hz, 1 H), 3.15 - 3.31 (m, 2 H), 2.63 - 2.89 (m, 2 H), 2.34 (s, 3 H), 1.36 (s, 9 H). m/z (ESI): 362.0 (M+H)+.
Paso 3: A una solución de 1-(6-bromo-4-metilpiridin-2-il)-3,3-difluorociclobutano-1-carboxilato de ferf-butilo (1.4 g, 2.8 mmol) en diclorometano (24 mL) se añadió TFA (0.87 mL, 11.3 mmol) a TA. La mezcla de reacción se agitó durante 18 h antes de concentrarla para proporcionar ácido 1 -(6-bromo-4-metilpiridin-2-il)-3,3-difluorociclobutano-1 -carboxílico (0.90 g, 2.2 mmol, 77 % de rendimiento) como un líquido pegajoso de color café. 1H RMN (300 MHz, DMSO-afe): 5 ppm 7.47 (s, 1 H), 7.38 (s, 1 H), 3.25 (q, J=12.8 Hz, 2 H), 2.66 - 2.85 (m, 2 H), 2.33 (s, 3 H). m/z (ESI): 306.0 (M+H)+.
Paso 4: Una solución de ácido 1-(6-bromo-4-metilpiridin-2-il)-3,3-difluorociclobutano-1-carboxílico (0.60 g, 1.4 mmol) en 2-xileno (7.0 mL) se agitó a 120 °C durante 2 h. La mezcla de reacción se diluyó con agua (50 mL), se extrajo con EtOAc (2 x 50 mL), se lavó con agua (50 mL) y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % a un 5 %, EtOAc en éter de petróleo para proporcionar 2-bromo-6-(3,3-difluorociclobutil)-4-metilpiridina (0.41 g, 1.2 mmol, 85 % de rendimiento) como un aceite amarillo claro. 1H RMN (300 MHz, DMSO-afe): 5 ppm 7.39 (s, 1 H), 7.25 (s, 1 H), 3.47 (qt, J=9.1,4.5 Hz, 1 H), 2.73 - 2.97 (m, 4 H), 2.29 (s, 3 H). m/z (ESI): 262.0 (M+H)+.
Paso 5: Una mezcla de 2-bromo-6-(3,3-difluorociclobutil)-4-metilpiridina (0.400 g, 1.53 mmol), (4-metoxifenil)metanamina (0.314 g, 2.29 mmol), Cs2CO3 (1.49 g, 4.58 mmol), BINAP (0.095 g, 0.153 mmol) y Pd(OAc)2 (0.034 g, 0.15 mmol) en 1,4-dioxano (4 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se filtró y se diluyó con EtOAc. La solución resultante se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % a un 10 % de acetato de etilo en éter de petróleo para proporcionar 6-(3,3-difluorociclobutil)-N-(4-metoxibencil)-4-metilpiridin-2-amina (0.360 g, 1.131 mmol, 74 % de rendimiento) como un aceite amarillo. 1H RMN (400 MHz, DMSO-afe): 5 ppm 7.22 - 7.31 (m, 2 H), 6.85 (dt, J=8.5, 2.1 Hz, 2 H), 6.26 (s, 1 H), 6.15 (s, 1 H), 4.38 (d, J=6.0 Hz, 2 H), 3.71 (s, 3 H), 3.14 - 3.22 (m, 1 H), 2.71 - 2.86 (m, 4 H), 2.09 (s, 3 H). m/z (ESI): 319.1 (M+H)+.
Paso 6: A una mezcla de 6-(3,3-difluorociclobutil)-N-(4-metoxibencil)-4-metilpiridin-2-amina (0.355 g, 1.12 mmol) y anisol (0.244 mL, 2.23 mmol) en diclorometano (4 mL) se añadió ácido trifluoroacético (1.8 mL, 23 mmol) a 0 °C. La mezcla de reacción se agitó a 55 °C durante 16 h antes de concentrarla y a continuación se diluyó con DCM (20 mL). La capa orgánica se lavó con una solución acuosa saturada de NaHCO3 solución (3 mL), agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 10 % a un 20 %, EtOAc en éter de petróleo para proporcionar 6-(3,3-d ifluorociclobutil)-4-metilpiridin-2-amina (0.090 g, 0.45 mmol, 41 % de rendimiento) como un aceite amarillo claro. 1H RMN (300 MHz, DMSO-afe): 5 ppm 6.27 (s, 1 H), 6.12 (s, 1 H), 3.18 (td, J=8.8, 3.1 Hz, 1 H), 2.69 - 2.85 (m, 4 H), 2.10 (d, J=2.1 Hz, 3 H). m/z (ESI): 199.1 (M+H)+.
Intermedio 8: 5-Metil-6-morfolinopiridin-2-amina

Una mezcla de 6-cloro-5-metilpiridin-2-amina (0.1 g, 0.70 mmol), morfolina (0.092 g, 1.05 mmol), K2CO3 (0.145 g, 1.052 mmol), yoduro de cobre (I) (0.027 g, 0.140 mmol) y (1R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (0.020 g, 0.140 mmol) en DMF (2 mL) se calentó en microondas a 150 °C durante 4 h. A continuación, la mezcla de reacción se filtró a través de un tapón de celite y el filtrado se diluyó con EtOAc, se lavó con agua, salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un 15 % de acetato de etilo en éter de petróleo para proporcionar 5-metil-6-morfolinopiridin-2-amina (10 mg, 0.052 mmol, 7 % de rendimiento) como un sólido amarillo pálido. m/z (ESI): 194.2 (M+H)+.
Preparación de Intermedios con Anillo AR2:
Intermedio 9: Ácido 4-yodo-2-(6-azaespiro[2.5]octan-6-il)benzoico

se añadió clorhidrato de 6-azaespiro[2.5]octano (216 g, 1.47 mol, Wuxi AppTec) a 20 °C. A continuación, se añadió K2CO3 (468 g, 3.38 mol) y la solución de reacción se agitó a 140 °C durante 48 h en N2. La solución de reacción se vertió lentamente sobre hielo-agua (4.20 L) y a continuación se extrajo con hexanos (2 L x 3). Se tomó la fase acuosa y se ajustó el pH a 6 con HCl (2.00 mol/L, ac.). Se obtuvo un precipitado y se recogió por filtración. El sólido se lavó con agua (700 mL x 3) y se filtró. El sólido húmedo se extendió sobre un vidrio de reloj grande y se secó al aire a 25 °C. Se obtuvo 4-yodo-2-(6-azaespiro[2.5]octan-6-il)benzoico (280 g, 777 mmol, 68.9 % de rendimiento) como un sólido amarillo claro. 400 MHz DMSO-d65 ppm 8.07 (s, 1H), 7.76 - 7.66 (m, 2H), 3.10 (t, J = 5.2 Hz, 4H), 1.55 (s a, 4H), 0.41 (s, 4H).
Tabla 3: Los intermedios de 9-1 a 9-5 se prepararon siguiendo un procedimiento similar al del intermedio 9:


Intermedio 10: Ácido 4-(metilsulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoico

Paso 1: A una solución de ácido 2-fluoro-4-(metilsulfonil)benzoico (90.0 g, 412 mmol) en DMF (1 L) se añadieron bromuro de bencilo (78.1 g, 454 mmol) y carbonato de sodio (52.5 g, 495 mmol) a 0 °C. La mezcla de reacción se agitó durante 12 h a temperatura ambiente. La mezcla de reacción se desactivó con agua (1 L) y se extrajo con MTBE (3 x 1 L). Los extractos orgánicos combinados se lavaron con salmuera (1 L), se secaron con Na2SO4, se filtraron y se
concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna en gel de sílice (230 400 mesh) utilizando de un 0 a un 30 % de acetato de etilo en hexanos para obtener 2-fluoro-4-(metilsulfonil)benzoato de bencilo (100 g, 79 % de rendimiento) como un sólido blanco. 1H Rm N (300 MHz, DMSO-afe): 58.16 (dd, J = 8.2, 6.9 Hz, 1H), 7.98 - 7.86 (m, 2H), 7.48 - 7.31 (m, 5H), 5.40 (s, 2H), 3.33 (s, 3H).
Paso 2: A una solución de 2-fluoro-4-(metilsulfonil)benzoato de bencilo (55 g, 180 mmol) en sulfóxido de dimetilo (550 mL) se añadió DIPEA (57.6 g, 446 mmol) seguida de 6-azaespiro[2.5]octano (29.8 g, 268 mmol) y la mezcla de reacción se agitó a 100 °C durante 24 h. La mezcla de reacción se desactivó con agua (1 L) y se extrajo con MTBE (3 x 1 L). Los extractos orgánicos combinados se lavaron con salmuera (1 L), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida. El producto crudo se purificó mediante cromatografía en columna sobre gel de sílice (230-400 mesh) utilizando de un 0 a un 10 % de acetato de etilo en hexanos para obtener 4-(metilsulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (55 g, 77 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 57.76 (d, J = 8.0 Hz, 1H), 7.52 - 7.45 (m, 4H), 7.43 - 7.35 (m, 3H), 5.35 (s, 2H), 3.25 (s, 3H), 3.05 (t, J = 5.3 Hz, 4H), 1.36 (t, J = 5.3 Hz, 4H), 0.30 (s, 4H). m/z (ESI): 400.1 (M+H)+.
Paso 3: A una solución de 4-(metilsulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (65 g, 160 mmol) en THF (108 mL) y metanol (36 mL) se añadió hidróxido de sodio (1N, 407 mL) y la mezcla de reacción se agitó durante 12 h a 60 °C. La mezcla de reacción se concentró a presión reducida para eliminar el THF y metanol. La solución acuosa restante se acidificó hasta pH ~2 con una solución de HCl 1.5 N. El precipitado se filtró, se lavó con agua (200 mL) seguida de hexanos (200 mL), y se secó al vacío para obtener ácido 4-(metilsulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (42 g, 83 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-afe): 5 16.13 (s, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.99 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 3.29 (s, 3H), 3.17 (b s, 4H), 1.55 (b s, 4H), 0.41 (s, 4H). m/z (ESI): 308.1 (M+H)+.
Intermedio 11: Ácido 4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoico

Paso 1: A un tubo sellado de 250-mL se añadieron 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (9 g, 22.48 mmol, Int. 9-5), 1-metilciclopropano-1-sulfonamida (3.95 g, 29.2 mmol, Combi-Blocks, San Diego, CA) y K2CO3 (6.21 g, 45.0 mmol) en 1,4-dioxano (90 mL). La reacción se desgasificó y purgó con nitrógeno durante 5 min. A esta mezcla de reacción se añadió Xantphos (1.301 g, 2.248 mmol) seguido de Pd2(dba)3 (1.029 g, 1.12 mmol) y el tubo sellado se cerró y se agitó a 110 °C durante 18 h. La mezcla de reacción se desactivó con agua (250 mL) y se extrajo con acetato de etilo (2 x 150 mL). Los extractos orgánicos combinados se lavaron con agua (100 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice eluyendo con un gradiente de un 0 % a un 15 % de EtOAc en hexanos para obtener 4-((1 -metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (6.1 g, 59 % de rendimiento) como un aceite naranja. 1H RMN (400 MHz, DMSO-afe): 510.06 (s, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.48 - 7.31 (m, 5H), 6.98 (s, 1H), 6.81 (d, J = 8.5 Hz, 1H), 5.27 (s, 2H), 2.92 (t, J = 4.96 Hz, 4H), 1.40 - 1.30 (m, 7H), 1.16 (dd, J = 6.4, 4.7 Hz, 2H), 0.81 (dd, J = 6.4, 4.7 Hz, 2H), 0.28 (s, 4H). m/z (ESI): 455.2 (M+H)+.
Paso 2: A una solución de 4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (2.1 g, 4.6 mmol) en MeOH (20 mL) y acetato de etilo (10 mL) se añadió Pd-C al 10 % (1.05 g, 50 % p/p) en una atmósfera de nitrógeno. La mezcla de reacción se desgasificó y se agitó en hidrógeno (1 atm, presión de globo) durante 4 h. La mezcla de reacción se filtró a través de un lecho de celite y se lavó con metanol (20 mL). El filtrado se concentró a presión reducida. El residuo se purificó con éter dietílico (50 mL) para obtener ácido 4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoico (1.2 g, 71 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-afe): 513.20 (s, 1H), 10.33 (s, 1H), 7.95 (d, J = 8.6 Hz, 1H), 7.41 (s, 1H), 7.20 (d, J = 8.6 Hz, 1H), 2.99 (s, 4H), 1.56 (s, 4H), 1.39 (s, 3H), 1.18 (t, J = 4.8 Hz, 2H), 0.83 (t, J = 4.7 Hz, 2H), 0.42 (s, 4H). m/z (ESI): 363.2 (M-H)+.
Intermedio 12: Ácido 4-(N-(3-metiloxetan-3-il)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoico

Paso 1: A una solución de clorhidrato de 3-metil-3-oxetanamina (5.50 g, 44.5 mmol) y
DIPEA (23.3 mL, 134 mmol) en DCM (200 mL) a 0 °C, se añadió 4-(clorosulfonil)-2-fluorobenzoato de metilo (12.4 g, 49.0 mmol) y la mezcla se agitó de 0 °C a temperatura ambiente durante 1 h. La mezcla se diluyó con HCl 1.0 N (200 mL) y se extrajo con diclorometano (150 mL x 2). Las capas orgánicas combinadas se lavaron con salmuera, se secaron con sulfato de sodio anhidro, se filtraron y se concentraron a presión reducida para proporcionar el producto crudo. El producto crudo se purificó con una columna Biotage SNAP de 100 g eluyendo con un 0-30 % de EtOAc-EtOH (3:1) en heptano para proporcionar 2-fluoro-4-(N-(3-metiloxetan-3-il)sulfamoil)benzoato de metilo (13.6 g, 44.8 mmol, 100 % de rendimiento) como un sólido blanco. 1H RMN (500 MHz, DMSO-d6) 58.67 (s, 1H), 8.11 (t, J=7.37 Hz, 1H), 7.76-7.82 (m, 1 H), 7.69-7.76 (m, 1H), 4.56 (d, J=6.23 Hz, 2H), 4.18 (d, J=6.75 Hz, 2H), 3.90 (s, 3H), 1.42 (s, 3H).
Paso 2: Una mezcla de DIPEA (16 mL, 93 mmol), 6-azaespiro[2.5]octano (6.22 g, 55.9 mmol) y 2-fluoro-4-(N-(3-metiloxetan-3-il)sulfamoil)benzoato de metilo (14.1 g, 46.6 mmol) en dioxano anhidro se agitó a 100 °C durante 20 h. La mezcla se enfrió hasta la temperatura ambiente, se desactivó con agua y se extrajo con acetato de etilo. Los extractos orgánicos combinados se lavaron con salmuera, se secaron y se evaporaron hasta sequedad a presión reducida. El producto crudo se purificó utilizando la columna Biotage SNAP de 340 g eluyendo con un 0-40 % de EtOAc-EtOH (3:1) en heptano para obtener 4-(N-(3-metiloxetan-3-il)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (14.2 g, 35.9 mmol, 77 % de rendimiento) como un sólido blanquecino. 1H RMN (500 MHz, DMSO-Ó6) 58.42 (s, 1 H), 7.72 (d, J=8.04 Hz, 1H), 7.47 (d, J=1.56 Hz, 1 H), 7.36 (dd, J=1.82, 8.04 Hz, 1H), 4.55 (d, J=5.97 Hz, 2H), 4.14 (d, J=6.49 Hz, 2H), 3.85 (s, 3H), 3.02-3.09 (m, 4H), 1.44-1.50 (m, 4H), 1.42 (s, 3H), 0.35 (s, 4H).
Paso 3: Una mezcla de 4-(N-(3-metiloxetan-3-il)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (14.2 g, 35.9 mmol) e hidróxido de litio monohidratado (22.6 g, 538 mmol) en THF-agua-MeOH (1:1:1, 300 mL) se agitó a temperatura ambiente durante 16 h. La mezcla se concentró a presión reducida para eliminar parcialmente el disolvente orgánico. La solución se acidificó con HCl 2 N para alcanzar un pH < 3. El precipitado se filtró y se secó al aire para obtener ácido 4-(N-(3-metiloxetan-3-il)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (9.94 g, 26.1 mmol, 73 % de rendimiento) como un sólido blanco. 1H RMN (500 MHz, DMSO-Ó6) 58.51 (s, 1H), 8.04 (d, J=8.04 Hz, 1H), 7.89 (d, J = 1.30 Hz, 1H), 7.66 (dd, J=1.69, 8.17 Hz, 1H), 4.55 (d, J=6.23 Hz, 2H), 4.09-4.17 (m, 2H), 3.06-3.19 (m, 4H), 1.56 (t, J = 5.19 Hz, 4H), 1.40 (s, 3H), 0.36-0.46 (s, 4H).
In te rm e d io 13: Á c id o 4 -(N -(íe rí-b u til)s u lfa m o il)-2 -(6 -a z a e s p iro [2.5 ]o c ta n -6 - il)b e n z o ic o

Paso 1: A una solución de ácido 4-bromo-2-fluorobenzoico (40 g, 180 mmol) en DMF (600 mL) se añadieron carbonato de sodio (23.2 g, 219 mmol) y bromuro de bencilo (22.8 mL, 192 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h antes de desactivarla con agua (1.5 L) y se extrajo con acetato de etilo (3 x 500 mL). Los extractos orgánicos combinados se lavaron con agua (1500 mL) y salmuera (500 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna en gel de sílice (60-120 mesh) utilizando un 10 % de acetato de etilo en hexanos para obtener 4-bromo-2-fluorobenzoato de bencilo (50 g, 89 % de rendimiento) como un líquido amarillo pálido.1H RMN (400 MHz, Cloroformod): 57.86 (dd, J = 8.0, 7.4 Hz, 1H), 7.50 - 7.35 (m, 7H), 5.40 (s, 2H).
Paso 2: A una solución de 4-bromo-2-fluorobenzoato de bencilo (30 g, 97 mmol) en DMSO (300 mL) se añadieron 6-azaespiro[2.5]octano (15.1 g, 136 mmol) y DIPEA (33.9 mL, 194 mmol) a temperatura ambiente. La mezcla de reacción se agitó a 90 ° C durante 16 h. La mezcla de reacción se desactivó con agua (500 mL) y se extrajo con acetato de etilo (2 x 500 mL). Los extractos orgánicos combinados se lavaron con salmuera (600 mL), se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando de un 0 % a un 10 % de acetato de etilo en hexanos para obtener 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (25 g, 64 % de rendimiento) como un aceite amarillo pálido.
1H RMN (400 MHz, Cloroformo-d): 5 7.61 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.0 Hz, 2H), 7.43 - 7.35 (m, 3H), 7.22 (s, 1H), 7.09 (d, J = 8.4 Hz, 1H), 5.36 (s, 2H), 3.09 (t, J = 5.3 Hz, 4H), 1.49 (t, J = 5.3 Hz, 4H), 0.34 (s, 4H). m/z (ESI): 400.1 (M-H)+.
Paso 3: Una solución de 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (25 g, 62 mmol), DIPEA (21.8 mL, 125 mmol), xantphos (1.81 g, 3.12 mmol), Pd2(dba)3 (1.14 g, 1.25 mmol) y mercaptano de bencilo (10 g, 81 mmol) en 1,4 dioxano (250 mL) se desgasificó y purgó con nitrógeno durante 15 minutos. La mezcla de reacción se calentó a 100 °C durante 16 h en un recipiente resistente a la presión sellado. La mezcla de reacción se inactivó con agua (500 ml) y se extrajo con acetato de etilo (2 x 500 ml). Los extractos orgánicos combinados se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando de un 5 % a un 10 % de acetato de etilo en hexanos para obtener 4-(benciltio)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (20 g, 72 % de rendimiento) como un líquido amarillo pálido. 1H RMN (400 MHz, DMSO-ds): 57.57 - 7.53 (m, 1H), 7.48 - 7.26 (m, 10H), 6.93 - 6.87 (m, 2H), 5.27 (s, 2H), 4.32 (s, 2H), 2.95 - 2.87 (m, 4H), 1.35 (t, J = 5.3 Hz, 4H), 0.28 (s, 4H). m/z (ESI): 442.2 (M-H)+.
Pasos 4 y 5: A una solución de 4-(benciltio)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (20 g, 45 mmol) en DCM (160 mL) y agua (40 mL), se añadió cloruro de sulfurilo (18.3 mL, 225 mmol) a 0 °C. La mezcla de reacción se agitó durante 1 h antes de diluirla con agua (200 mL) y se extrajo con DCM (200 mL). El extracto orgánico se secó con Na2SO4 anhidro, se filtró y se enfrió hasta 0 °C. Se añadió fert-butilamina (47.8 mL, 451 mmol) a la solución anterior. La mezcla de reacción se agitó a temperatura ambiente durante 1 h antes de desactivarla con agua (200 mL) y se extrajo con DCM (2 x 100 mL). Los extractos orgánicos combinados se secaron con Na2SO4 anhidro, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice (60-120 mesh) utilizando un 15 % de acetato de etilo en hexanos para obtener 4-(N-(ferf-butil)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (12 g, 61 % de rendimiento) como un sólido amarillo pálido. 1H RMN (400 MHz, DMSO-ds): 57.71 (d, J = 8.0 Hz, 1H), 7.63 (s, 1H), 7.55 - 7.46 (m, 3H), 7.45 - 7.33 (m, 4H), 5.33 (s, 2H), 3.01 (b s, 4H), 1.38 (b s, 4H), 1.10 (s, 9H), 0.31 (2, 4H). m/z (ESI): 457.2 (M-H)+.
Paso 6: A una solución de 4-(N-(fe/t-butil)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (10 g, 21.9 mmol) en etanol (50 mL) y acetato de etilo (50 mL) se añadió paladio al 10 % sobre carbón (4.66 g, 4.38 mmol) a temperatura ambiente en una atmósfera de nitrógeno. La mezcla de reacción se desgasificó y se agitó en una atmósfera de hidrógeno (1 atm) a temperatura ambiente durante 16 h. La mezcla de reacción se filtró a través de un lecho de celite y el lecho del filtro se lavó con acetato de etilo (200 mL). El filtrado se concentró a presión reducida. El residuo crudo se purificó con éter dietílico (200 mL) para obtener el compuesto del título (6.0 g, 75% de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-da): 58.06 (d, J = 8.2 Hz, 1H), 7.98 (s, 1H), 7.73 - 7.68 (m, 2H), 3.12 (t, J = 5.3 Hz, 4H), 1.57 (t, J = 5.3 Hz, 4H), 1.10 (s, 9H), 0.43 (s, 4H). m /z (ESI): 367.2 (M-H)+.
Intermedio 14: Ácido 4-((metilsulfonil)metil)-2-(6-azaespiro[2.5]octan-6-il)benzoico

se añadieron NBS (11.6 g, 65.4 mmol) y AIBN (0.976 g, 5.95 mmol) a TA. La mezcla de reacción se agitó a 70 °C durante 3 h antes de desactivarla con agua (250 mL) y se extrajo con diclorometano (2 x 200 mL). Los extractos orgánicos combinados se lavaron con salmuera (150 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida para obtener 2-fluoro-4-metilbenzoato de metilo (14.0 g, crudo) como un aceite amarillo pálido.1H RMN (400 MHz, Cloroformo-d): 58.04 - 7.88 (m, 1H), 7.27 - 7.13 (m, 2H), 4.46 (s, 2H), 3.96 (s, 3H).
Paso 2: Una mezcla de 2-fluoro-4-metilbenzoato de metilo (14.0 g, 56.7 mmol) y metanosulfinato de sodio (12.15 g, 119 mmol) en DMF (42 mL) se irradió en un horno de microondas (Biotage initiator+) a 120 °C durante 30 min. La mezcla de reacción se desactivó con agua (150 mL) y se extrajo con acetato de etilo (2 x 200 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera saturada (150 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida para obtener 2-fluoro-4-((metilsulfonil)metil)benzoato de metilo (6 g, crudo) como un sólido blanquecino.
Paso 3: Una mezcla de 2-fluoro-4-((metilsulfonil)metil)benzoato de metilo (6.0 g, 24 mmol) y 6-azaespiro[2.5]octano (2.71 g, 24.4 mmol) en DMSO (30 mL) se irradió en un horno de microondas a 150 °C durante 1 h. La mezcla de reacción se desactivó con agua (50 mL) y se extrajo con acetato de etilo (2 x 50 mL). Los extractos orgánicos combinados se lavaron con salmuera (50 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida para obtener 4-((metilsulfonil)metil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (4.0 g, crudo) como un sólido blanquecino. El material tal cual se utilizó en el siguiente paso sin ninguna purificación adicional. 1H RMN (400 MHz, Cloroformo-d) 57.72 (d, J = 7.8 Hz, 1H), 7.11 (s, 1H), 6.96 (dd, J = 7.8, 1.6 Hz, 1H), 4.24 (s, 2H), 3.93 (s, 3H), 3.12 (t, J = 5.4 Hz, 4H), 2.78 (s, 3H), 1.55 (t, J = 5.5 Hz, 4H), 0.37 (s, 4H). m/z (ESI): 338.1 (M-H)+.
Paso 4: A una solución de 4-((metilsulfonil)metil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (2.0 g, 3.0 mmol) en THF (15 mL) se añadió hidróxido de sodio (0.474 g, 11.8 mmol) en agua (7 mL) y se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se acidificó con una solución de HCL 1.5 N hasta pH ~3 y se extrajo con acetato de etilo (5 x 20 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera (20 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida para obtener ácido 4-((metilsulfonil)metil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (1.0 g, 10 % de rendimiento a lo largo de 3 pasos) como un sólido blanco. m/z (ESI): 324.1 (M-H)+.
In te rm e d io 15: Á c id o 4-((1 -( fe /f-b u to x ic a rb o n il)a z e tid in -3 - il)s u lfo n il)-2 -(6 -a z a e s p iro [2.5 ]o c ta n -6 - il)b e n z o ic o

Paso 1: A través de una mezcla de 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (10.5 g, 32.4 mmol, Intermedio 9-2), DIPEA (8.37 mL, 64.8 mmol), Xantphos (1.87 g, 3.24 mmol) y Pd2(dba)3 (2.97 g, 3.24 mmol) en dioxano se burbujeó con flujo de argón, a continuación se añadió 3-mercaptoazetidino-1-carboxilato de te/t-butilo (7.66 mL, 40.5 mmol). La mezcla se agitó a 100 °C durante 18 h. La mezcla se enfrió hasta la temperatura ambiente, se concentró y se purificó a través de una columna Biotage SNAP de 340 g eluyendo con un gradiente de un 0 % a un 25 % de EtOAc-EtOH (3:1) en heptano para proporcionar 3-((4-(metoxicarbonil)-3-(6-azaespiro[2.5]octan-6-il)fenil)tio)azetidino-1-carboxilato de te/t-butilo (13.8 g, 32.0 mmol, 99 % de rendimiento) como un sólido pegajoso amarillo claro. 1H RMN (500 MHz, DMSO-afe) 5 ppm 7.55 (d, J=8.04 Hz, 1H), 6.79 (s, 1H), 6.76 (d, J=8.14 Hz, 1H), 4.34-4.43 (m, 2H), 4.27 4.34 (m, 1H), 3.79 (s, 3H), 3.70 (dd, J=4.67, 8.56 Hz, 2H), 2.97-3.04 (m, 4H), 1.41-1.51 (m, 4H), 1.38 (s, 9H), 0.29 0.37 (m, 1H).
Paso 2: A una solución de 3-((4-(metoxicarbonil)-3-(6-azaespiro[2.5]octan-6-il)fenil)tio)azetidino-1-carboxilato de tert-butilo (13.85 g, 32.0 mmol) en 1,4-dioxano (300 mL) se añadió Oxone, monopersulfato (39.4 g, 64.0 mmol) en 150 mL de agua. La mezcla de reacción se agitó a temperatura ambiente durante 5 h antes de añadir 150 mL de acetato de etilo y 150 mL de agua. Esta mezcla se agitó durante 10 min, la capa orgánica se separó y la capa acuosa se extrajo con acetato de etilo. Los extractos orgánicos combinados se lavaron con salmuera, se secaron, se filtraron y se concentraron. El material crudo se purificó a través de una columna Biotage SNAP de 340 g eluyendo con un gradiente de un 0 % a un 25 % de EtOAc-EtOH (3:1) en heptano para obtener 3-((4-(metoxicarbonil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfonil)azetidino-1 -carboxilato de te/t-butilo (12.8 g, 27.5 mmol, 86 % de rendimiento) como un sólido blanquecino. 1H RMN (500 MHz, DMSO-da) 57.75 (d, J=8.04 Hz, 1H), 7.44-7.50 (m, 2H), 4.48-4.55 (m, 1H), 4.09 (s a, 2H), 3.97-4.02 (m, 2H), 3.86 (s, 3H), 3.04-3.17 (m, 4H), 1.42-1.51 (m, 4H), 1.38 (s, 9H), 0.35 (s, 4H).
Paso 3: Una mezcla de 3-((4-(metoxicarbonil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfonil)azetidino-1-carboxilato de te/t-butilo (12.77 g, 27.5 mmol) e hidróxido de litio monohidratado (11.53 g, 275 mmol) en THF-agua-MeOH (1:1:1,230 mL) se agitó a temperatura ambiente durante 15 h. La mezcla se concentró a presión reducida para eliminar parte del disolvente orgánico. La solución se acidificó con HCl 2 N hasta pH < 3. El precipitado se filtró y se secó para obtener ácido 4-((1-(tert-butoxicarbonil)azetidin-3-il)sulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (10.6 g, 23.5 mmol, 86 % de rendimiento) como un sólido blanquecino. 1H RMN (500 m Hz , DMSO-afe) 5 ppm 8.03 (d, J=8.30 Hz, 1H), 7.93 (d, J=1.82 Hz, 1H), 7.72 (dd, J=1.69, 8.17 Hz, 1H), 4.48-4.60 (m, 1H), 4.10 (s a, 2H), 3.99-4.06 (m, 2H), 3.14-3.22 (m, 4H), 1.49-1.59 (m, 4H), 1.38 (s, 9H), 0.41 (s, 4H).
In te rm e d io 16: Á c id o 4 -(c ic lo p ro p a n o s u lfo n im id o il)-2 -(6 -a z a e s p iro [2.5 ]o c ta n -6 - il)b e n z o ic o

Paso 1: A una mezcla de 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (3.69 g, 11.4 mmol, Intermedio 9 2), DIPEA (2.94 mL, 22.8 mmol), Xantphos (0.659 g, 1.14 mmol) y Pd2(dba)3 (1.04 g, 1.14 mmol) en 1,4-dioxano se añadió ciclopropanotiol (0.928 mL, 12.5 mmol). La mezcla se agitó a 100 °C durante 16 h. La mezcla se diluyó con agua y se extrajo con acetato de etilo. Los extractos orgánicos se lavaron con salmuera, se secaron y se concentraron. La mezcla se purificó a través de una columna Biotage SNAP de 100 g eluyendo con un gradiente de un 0 % a un 20 % de EtOAc-EtOH (3:1) en heptano para proporcionar 4-(ciclopropiltio)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (3.42 g, 10.8 mmol, 95 % de rendimiento) como un aceite amarillo. 1H RMN (500 MHz, DMSO-d6) 67.57 (d, J=8.04 Hz, 1H), 7.00 (d, J=1.82 Hz, 1H), 6.93 (d, J=8.21 Hz, 1H), 3.79 (s, 3H), 2.95-3.04 (m, 4H), 2.26-2.36 (m, 1H), 1.43-1.49 (m, 4H), 1.07-1.16 (m, 2H), 0.55-0.66 (m, 2H), 0.33 (s, 4H).
Paso 2 : A una mezcla de 4-(ciclopropiltio)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (3.35 g, 10.6 mmol) en DCM a 0 °C, se añadió ácido 3-cloroperoxibenzoico (2.367 g, 13.72 mmol). La mezcla se agitó a 0 °C durante 30 min. La reacción se desactivó con NaHCO3 sat. y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó y se concentró. La mezcla se purificó a través de una columna Biotage SNAP de 100 g eluyendo con un gradiente de un 0 % a un 40 % de EtOAc-EtOH (3:1) en heptano para proporcionar 4-(ciclopropilsulfinil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (3.34 g, 10.0 mmol, 95 % de rendimiento) como un aceite amarillo. 1H RMN (500 MHz, DMSO-d6) 67.70 (d, J=8.04 Hz, 1H), 7.30-7.47 (m, 1H), 7.22 (d, J=8.02 Hz, 1H), 3.84 (s, 3H), 3.03 3.11 (m, 4H), 2.52-2.55 (m, 1H), 1.43-1.51 (m, 4H), 0.88-1.01 (m, 3H), 0.77-0.88 (m, 1H), 0.34 (s, 4H).
Paso 3: A una solución de 4-(ciclopropilsulfinil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (3.33 g, 9.99 mmol) en metanol se añadieron diacetato de yodobenceno (6.43 g, 20.0 mmol) y carbamato de amonio (3.12 g, 39.9 mmol) a 0 °C y la mezcla se agitó a 0 oC durante 1 h. La reacción se desactivó con agua y se extrajo con acetato de etilo y DCM. Los extractos orgánicos combinados se lavaron con salmuera, se secaron y se concentraron. La mezcla se purificó a través de una columna Biotage SNAP de 100 g eluyendo con un gradiente de un 0 % a un 60 % de EtOAc-EtOH (3:1) en heptano para proporcionar 4-(ciclopropanosulfonimidoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (1.89 g, 5.42 mmol, 54.3 % de rendimiento) como un sólido blanquecino. 1H RMN (500 MHz, DMSo-d6) 67.70 (d, J=8.04 Hz, 1H), 7.53 (d, J=1.56 Hz, 1H), 7.43 (dd, J=1.56, 8.04 Hz, 1H), 4.29 (s, 1H), 3.85 (s, 3H), 3.01-3.13 (m, 4H), 2.67-2.75 (m, 1H), 1.43-1.51 (m, 4H), 1.05-1.14 (m, 1H), 0.87-1.00 (m, 3H), 0.35 (s, 4H).
Paso 4: Una mezcla de 4-(ciclopropanosulfonimidoil)-2-(6-azaespiro[2.5]octan-6-il)benzoato de metilo (1.89 g, 5.42 mmol) e hidróxido de litio monohidratado (0.683 g, 16.3 mmol) en THF-agua-MeOH (2:1:1, 32 mL) se agitó a temperatura ambiente durante 15 h. La mezcla se concentró a presión reducida para eliminar parte del disolvente orgánico. La solución se acidificó con HCl 2 N hasta pH < 3. La mezcla se extrajo con DCM (2x). Los extractos combinados se lavaron con salmuera, se secaron y se concentraron para obtener ácido 4-(ciclopropanosulfonimidoil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (1.80 g, 5.38 mmol, 99 % de rendimiento) como un sólido blanquecino. 1H RMN (500 MHz, DMSO-d6) 68.08 (d, J=8.30 Hz, 1H), 8.01 (d, J=1.56 Hz, 1H), 7.78 (dd, J=1.56, 8.04 Hz, 1H), 4.44 (s a, 1H), 3.16 (t, J=5.32 Hz, 4H), 2.74-2.81 (m, 1H), 1.58 (t, J=5.19 Hz, 4H), 1.11-1.22 (m, 1H), 0.89-1.04 (m, 3H), 0.43 (s, 4H).
Compuestos que contienen Ar1 y Ar2
Intermedio 17: N-(6-Fluoropiridin-2-il)-4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida
A una solución de ácido 4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoico (1.0 g, 2.74 mmol, Intermedio 11) en d Cm (20 mL) se añadió 6-fluoropiridin-2-amina (0.308 g, 2.74 mmol, Combi-Blocks), T3P (50 % en acetato de etilo, 1.3 mL, 4.1 mmol) y DIPEA (0.958 mL, 5.49 mmol). La mezcla resultante se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se desactivó con una solución sat. de NaHCO3 (10 mL) y se extrajo con DCM (2 x 25 mL). Los extractos orgánicos combinados se lavaron con salmuera (20 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice utilizando un 20 % de acetato de etilo en hexanos para obtener el compuesto del título (0.65 g, 52 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-cfe): 5 13.09 (s, 1H), 10.28 (s, 1H), 8.20 (dd, J = 8.0, 2.4 Hz, 1H), 8.12 - 7.92 (m, 2H), 7.36 (d, J = 2.2 Hz, 1H), 7.17 (dd, J = 8.6 , 2.2 Hz, 1H), 6.89 (dd, J = 8.0, 2.4 Hz, 1H), 2.97 (t, J = 5.4 Hz, 4H), 1.79 - 1.56 (m, 4H), 1.41 (s, 3H), 1.32 - 1.14 (m, 2H), 0.90 - 0.79 (m, 2H), 0.41 (s, 4H). m/z (ESI): 459.1 (M-H)+.
Tabla 4: Los intermedios 18-36 se prepararon siguiendo un procedimiento similar al descrito para el intermedio 17.
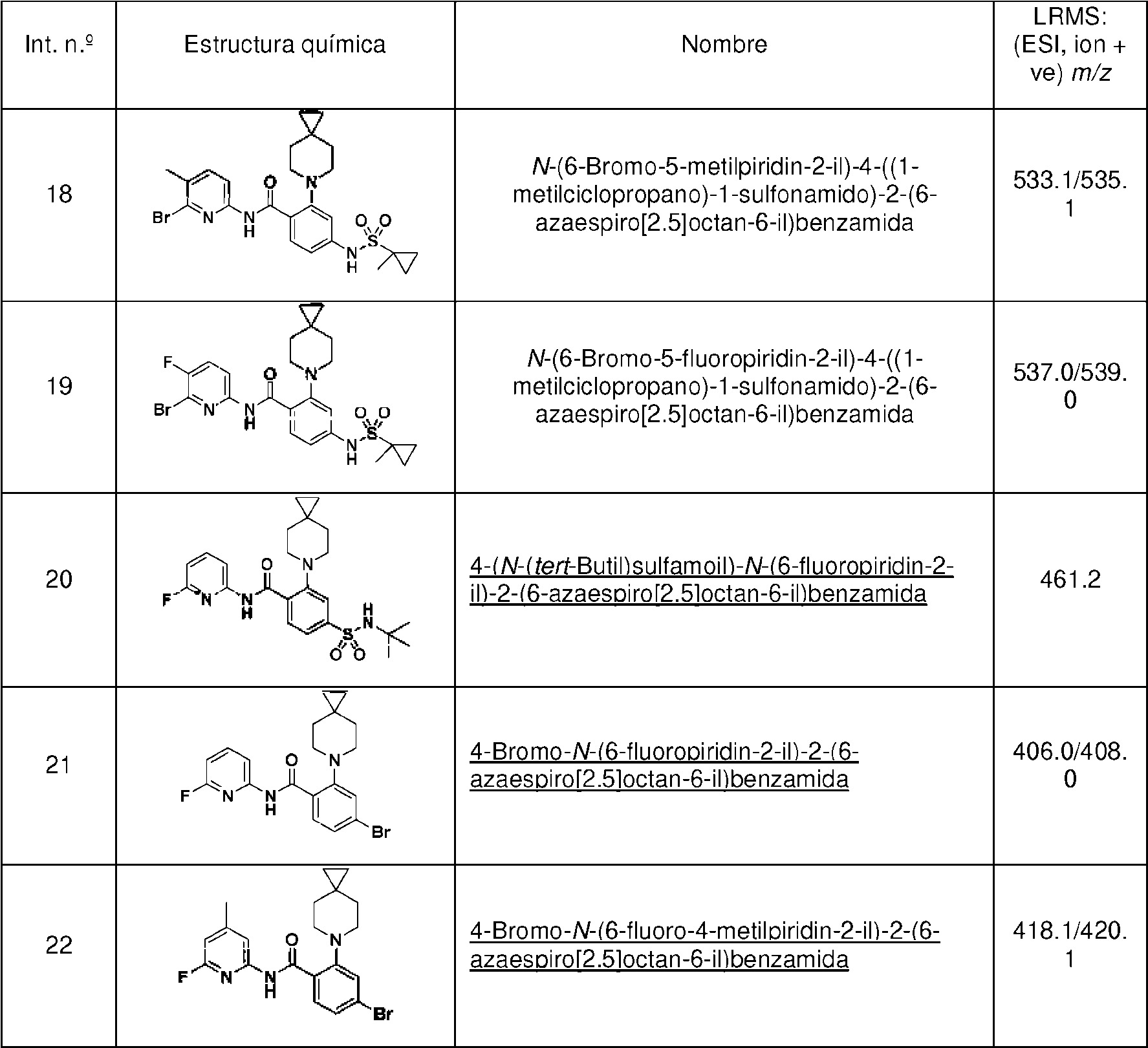


Intermedio 4-Yodo-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida

Se suspendió ácido 4-yodo-2-(6-azaespiro[2.5]octan-6-il)benzoico (3.0 g, 8.4 mmol, Intermedio 9) en DCM (10 mL) en argón. Se añadió cloruro de tionilo (1.463 g, 12.29 mmol, Sigma-Aldrich Corporation) y la mezcla se agitó a TA durante 15 min. La mezcla se evaporó hasta sequedad a presión reducida. El residuo crudo se destiló azeotrópicamente con tolueno (2 x 100 mL) y se suspendió en diclorometano (50 mL) en argón. Se añadió en una porción una solución de 6-(3,3,3-trifluoropropoxi)piridin-2-amina (2.0 g, 9.7 mmol, Intermedio 6) y 2,6-lutidina (3.68 g, 34.3 mmol, Sigma-Aldrich Corporation) en diclorometano (10 mL). La mezcla amarilla se agitó a TA durante 20 min y a continuación se evaporó hasta sequedad a presión reducida. Los sólidos crudos se purificaron en metanol (30 mL) durante 20 min y a continuación se filtraron a través de un fritado de vidrio sinterizado. Los sólidos se secaron en una corriente de nitrógeno para obtener 4-yodo-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (3.8 g, 7.0 mmol, 83 % de rendimiento). 1H RMN (400 MHz, DMSO-afe) 5 ppm 0.36 (s, 4 H) 1.58-180 (m, 4 H) 2.83 (dt, J=10.94, 5.42 Hz, 2 H) 2.91 - 3.13 (m, 4 H) 4.49 (t, J=5.29 Hz, 2 H) 6.58 (d, J=7.88 Hz, 1 H) 7.60 - 8.00 (m, 5 H) 13.20 (s a, 1 H)
Tabla 5: Los intermedios de 38-1 a 38-3 se prepararon siguiendo un procedimiento similar al descrito para el intermedio 38

Intermedio 39: 4-Bromo-N-(6-(3,3-difluoroazetidin-1 -il)-4-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida

Una mezcla de 4-bromo-N-(6-bromo-4-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (5.0 g, 10 mmol, Int.
22), clorhidrato de 3,3-difluoroazetidina (2.027 g, 15.65 mmol, Combi-Blocks) y CsF (9.51 g, 62.6 mmol) en DMSO (50 mL) se agitó a 145 °C durante 60 h. La mezcla de reacción se diluyó con hielo-agua (200 mL) y se agitó durante 30 min. El precipitado se filtró y se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % a un 5 % de EtOAc en éter de petróleo para proporcionar 4-bromo-N-(6-(3,3-difluoroazetidin-1 -il)-4-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (2.4 g, 4.8 mmol, 46 % de rendimiento) como un sólido blanco. 1H RMN (300 MHz, DMSO-cfe): 5 ppm 13.05 (s, 1 H), 8.03 (d, J=8.7 Hz, 1 H), 7.67 (s, 1 H), 7.53 - 7.56 (m, 2 H), 6.21 (s, 1 H), 4.32 - 4.41 (m, 4 H), 3.33 (t, J=5.1 Hz, 4 H), 2.28 (s, 3 H), 1.72 (s a, 4 H), 0.37 (s, 4 H). m/z (ESI): 491.1 (M+H)+.
Intermedio 40: 2-Sulfamoilpropanoato de etilo

Paso 1: A una solución de N,N-bis(4-metoxibencil)etanosulfonamida (200.0 g, 572.0 mmol) en tetrahidrofurano (4000 mL) se añadió nBuLi (1.6 M in hexane, 608.0 mL, 973.0 mmol) a -78 °C lentamente y se agitó durante 30 min. Se añadió carbonoclorohidrato de etilo (92.0 mL, 973.0 mmol) en THF (50 mL) a la mezcla de reacción y se agitó a -78 °C durante 1 h. La mezcla de reacción se desactivó con HCl (1.5 N, 3000 mL) y se extrajo con EtOAc (2 x 3000 mL). El extracto orgánico se secó con sulfato de sodio, se filtró y se concentró a presión reducida para obtener el material
crudo de 2-(W,N-bis(4-metoxibencil)sulfamoil)propanoato de etilo (250.0 g, puro en un 60 %) como un aceite amarillo. El 1H-RMN mostró los picos deseados y se procedió al siguiente paso sin purificación adicional.
Paso 2: A una solución de 2-(W,N-bis(4-metoxibencil)sulfamoil)propanoato de etilo (600.0 g, 1.4 mol) en ácido trifluoroacético (2.50 L, 32.45 mol) se añadió anisol (500.0 mL, 4.57 mol) y se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se concentró a presión reducida, se desactivó con una solución acuosa fría de NaHCO3 al 10 % (3 L) y se extrajo con EtOAc (2 x 3 L). La capa orgánica se secó con sulfato sódico, se filtró y se concentró a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice utilizando un 25 % de acetato de etilo en hexanos para obtener un sólido amarillo pálido (168 g) que se disolvió en DCM (1 L) y se hizo precipitar mediante la adición de hexanos (3000 mL). El sólido se filtró y se secó al vacío para obtener el compuesto del título (109.0 g, 42 % de rendimiento) como un sólido blanco. 1H r Mn (400 MHz, DMSO-afe) 5 ppm 7.14 (s, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.98 (q, J = 7.0 Hz, 1H), 1.45 (d, J = 7.0 Hz, 3H), 1.21 (t, J = 7.1 Hz, 3H). m/z (ESI): 180.1 (M-H)+.
EJEMPLOS
Métodos generales y ejemplos representativos
Método A (acoplamiento Br-S catalizado por Pd)
Ejemplo_____ 1 _____ 4-(N-(2-Hidroxietil)sulfamoil)-2-(6-azaespiro[2.51octan-6-il)-N-(6-(3.3.3-trifluoropropoxi)piridin-2-il)benzamida

Se cargó un vial de vidrio con 4-bromo-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (348 mg, 0.70 mmol Intermedio 38-1), aducto de 1,4-diazabiciclo[2.2.2]octano y bis(dióxido de azufre) (dAbSO) (101 mg, 0.42 mmol, Sigma-Aldrich), diacetoxipaladio (16 mg, 0.07 mmol, Strem), rac-((3R,5R,7R)-adamantan-1-il)((3S,5S,7S)-adamantan-1-il)(butil)fosfano (cataCXium® A) (38 mg, 0.11 mmol, Strem), trietilamina (194 uL, 1.40 mmol) e iPrOH (3 mL). El tubo se desgasificó durante 3 min, se selló y se calentó a 80 °C en un baño de aceite durante 3 h. La mezcla heterogénea se enfrió hasta TA y se trató con 2-aminoetan-1-ol (85 mg, 1.4 mmol, Sigma-Aldrich) seguido de una solución de hipoclorito de sodio (10 % p., 1040 mg, 1.40 mmol, Sigma-Aldrich) y se agitó a TA durante 18 h. Se añadieron EtOAc (20 mL) y agua (5 mL) a la mezcla heterogénea. Se separó por filtración el sólido insoluble. La masa retenida en el filtro se lavó con 2 x 2 mL de agua seguida de 2 x 4 mL de EtOAc. Los extractos orgánicos combinados se separaron y concentraron. El residuo se purificó mediante cromatografía en gel de sílice (de un 20 % a un 80 % EtOAc en heptano) para obtener 4-(N-(2-hidroxietil)sulfamoil)-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (106 mg, 0.19 mmol, 28 % de rendimiento) como un sólido amarillo claro. 1H RMN (400 MHz, DMSO-ds) 5 13.04 (s, 1H), 8.26 (d, J=8.09 Hz, 1H), 7.91 (d, J=7.67 Hz, 1H), 7.77-7.87 (m, 3H), 7.72 (d, J=7.26 Hz, 1H), 6.62 (d, J=8.09 Hz, 1H), 4.71 (t, J=5.08 Hz, 1H), 4.53 (t, J=6.01 Hz, 2H), 3.33-3.44 (m, 2H), 3.09 (m, 4H), 2.77-2.93 (m, 4H), 1.73 (s, 4H), 0.39 (s, 4H). m/z (ESI): (M+H)+ 543.2.
Tabla 6: Los Ejemplos de 1-1 a 1-13 se prepararon siguiendo un procedimiento similar al descrito para el Ejemplo 1


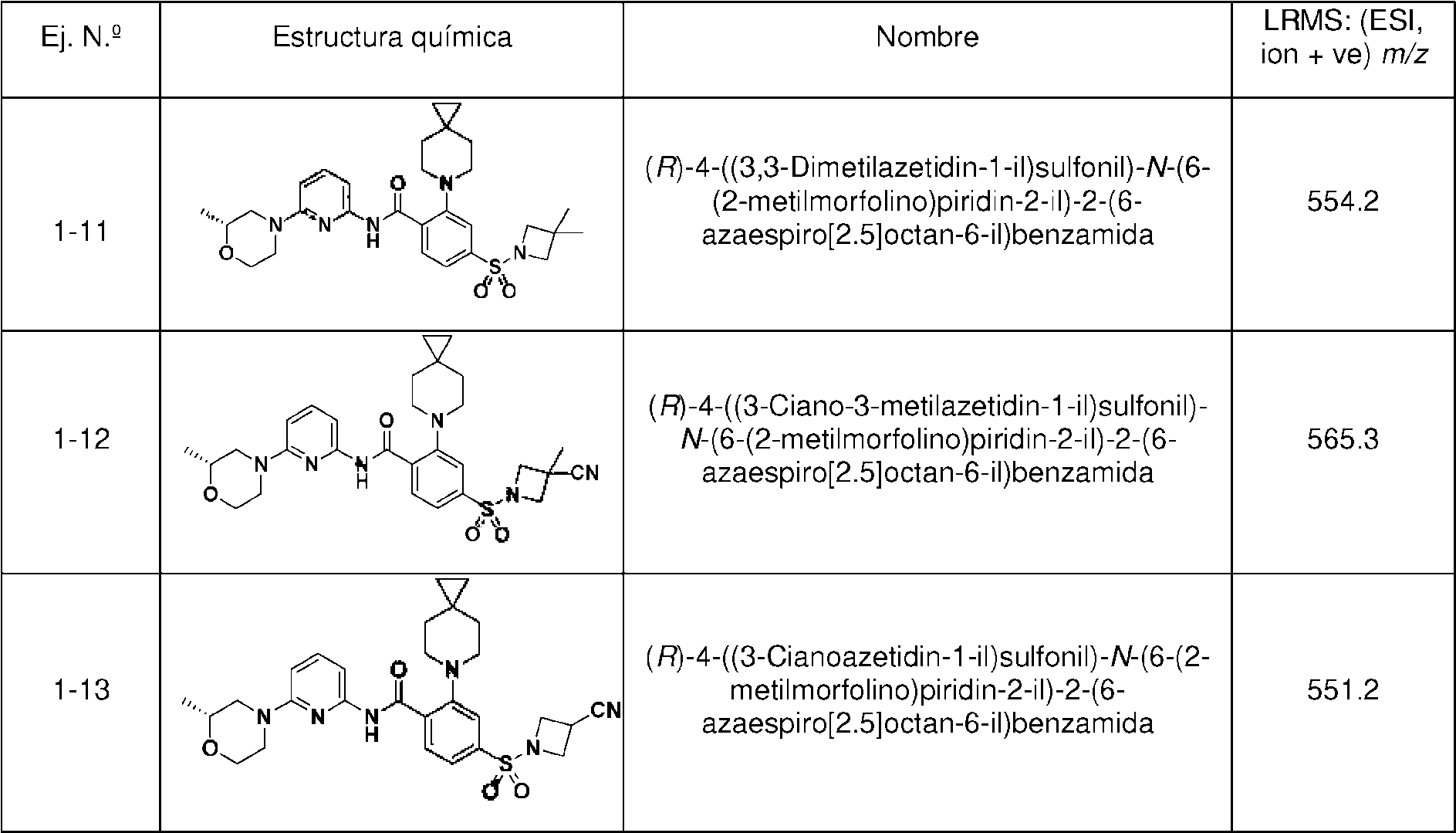
Método B (formación de sultanas)
Ejemplo 2: (fí)-4-(Isoprop¡lsulfon¡l)-M-(6-(2-metilmorfol¡no)p¡ridin-2-¡l)-2-(6-azaespiro[2.51octan-6-il)benzam¡da

Una mezcla de (fí)-4-yodo-N-(6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (89 mg, 0.17 mmol, Intermedio 29), trifenilfosfina (6 mg, 0.025 mmol, Aldrich St. Louis, MO EE. Uu .), 1,10-fenantrolina (24 mg, 0.132 mmol, Aldrich St. Louis, EE. UU.), acetato de paladio (ii) (5 mg, 0.025 mmol, Strem Chemicals, Inc. Newburyport, MA EE. UU.), formiato de sodio (25 mg, 0.37 mmol, Thermo Fisher Scientific, Grand Island, NY EE. UU.), y bromuro de de tetrabutilamonio (59 mg, 0.18 mmol, Aldrich St. Louis, MO USA ) en Dm So (2 mL) en N2 se agitó a 70 °C durante 3 h. A continuación, la mezcla se enfrió hasta la temperatura ambiente y se añadió yoduro de isopropilo (0.025 mL, 0.25 mmol, Aldrich St. Louis, MO EE. UU.). A continuación, la mezcla se agitó a temperatura ambiente durante 18 h. Después, la mezcla se diluyó con agua (20 mL) y después se extrajo con EtOAc (2 x 40 mL). Los extractos orgánicos combinados se secaron a continuación con Na2SO4 y se concentraron. La purificación cromatográfica de residuo (gel de sílice, 0 %-30 % de EtOAc/heptano) proporcionó (fí)-4-(isopropilsulfonil)-N-(6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (46 mg, 0.09 mmol, 36 % de rendimiento) como un sólido amarillo claro. 1H RMN (400 MHz, DMSO-d6) 5 12.63 (s, 1H), 8.31 (d, J = 8.22 Hz, 1H), 7.82 (d, J = 1.37 Hz, 1H), 7.77 (dd, J = 1.56, 8.22 Hz, 1H), 7.57-7.66 (m, 2H), 6.64 (d, J = 7.83 Hz, 1H), 4.17 (d a, J = 11.93 Hz, 1H), 4.08 (d a, J = 12.52 Hz, 1H), 3.93 (dd, J = 3.33, 11.35 Hz, 1H), 3.49-3.67 (m, 3H), 3.37 (quin, J = 6.65 Hz, 1H), 3.10 (t a, J = 5.18 Hz, 4H), 2.85 (dt, J = 3.52, 12.32 Hz, 1H), 1.72 (s a, 4H), 1.13-1.28 (m, 9H), 0.39 (s, 4H). m/z (ESI): 513.2 (M+H)+.
Tabla 7: Los Ejemplos de 2-1 a 2-7 se prepararon siguiendo un procedimiento similar al descrito para el Ejemplo 2

Ejemplo________3 _______ (fí)-N-(6-(3-Hidroxipiperidin-1-il)piridin-2-il)-4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.51octan-6-inbenzamida

A una solución de N-(6-fluoropiridin-2-il)-4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.44 mmol, Intermedio 17) en DMSO (2 mL) se añadió clorhidrato de (R)-piperidin-3-ol (0.060 g, 0.44 mmol, Essen Scientific) y fosfato de potasio tribásico (0.278 g, 1.31 mmol). La mezcla de reacción se agitó a 130 °C durante 16 h, antes de enfriarla, desactivarla con agua (5 mL) y extraer con acetato de etilo (2 x 10 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera (10 mL), se secaron con Na2SO4, se filtraron y se concentraron a presión reducida. En el residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice utilizando un 30-50 % de acetato de etilo en hexanos para obtener el compuesto del título (0.1 g, 43 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-cfe) 5 12.85 (s, 1H), 10.23 (s, 1H), 8.06 (d, J = 8.6 Hz, 1H), 7.52 (d, J = 6.1 Hz, 2H), 7.34 (s, 1H), 7.14 (d, J = 8.6 Hz, 1H), 6.65 - 6.34 (m, 1H), 4.78 (d, J = 4.6 Hz, 1H), 4.13 (d, J = 11.7 Hz, 1H), 4.05 - 3.85 (m, 1H), 3.63 - 3.46 (m, 1H), 3.10 - 2.74 (m, 6H), 1.99 - 1.58 (m, 5H), 1.42 (d, J = 13.8 Hz, 5H), 1.20 (s, 3H), 0.84 (d, J = 5.6 Hz, 2H), 0.38 (s, 4H). m/z (ESI): 540.2 (M+H)+.
Tabla 8: Los Ejemplos de 3-1 a 3-23 se prepararon siguiendo un procedimiento similar al descrito para el Ejemplo 3



Ejemplo 4: (fí)-N-(6-(3-Hidroxipiperidin-1-il)piridin-2-il)-4-((metilsulfonil)metil)-2-(6-azaespiro[2.5]octan-6-il)benzamida

14) en DMF (3 mL) se añadieron (R)-1-(6-aminopiridin-2-il)piperidin-3-ol (0.143 g, 0.742 mmol, Intermedio 4-5), HATU (353 mg, 0.928 mmol) y DIPEA (216 gL, 1.24 mmol) y se agitó a temperatura ambiente durante 12 h. La mezcla de reacción se desactivó con agua (20 mL) y se extrajo con acetato de etilo (3 x 20 mL). Los extractos orgánicos combinados se lavaron con salmuera (20 mL), se secaron con Na2SÜ4, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía en columna sobre gel de sílice utilizando un 50 % de acetato de etilo en hexanos para obtener el compuesto del título (0.20 g, 65 % de rendimiento) como un sólido amarillo.1H RMN (400 MHz, DMSO-ds): 513.06 (s, 1H), 8.15 - 8.13 (m, 1H), 7.58 - 7.50 (m, 3H), 7.37 (d, J = 7.8 Hz, 1H), 6.55 (d, J = 8.2 Hz, 1H), 4.80 (dd, J = 7.0 ,3.6 Hz, 1H), 4.59 (s, 2H), 4.13 (d, J = 12.4 Hz, 1H), 3.94 (d, J = 12.4 Hz, 1H), 3.60 - 2.80 (m, 7 H), 1.73 (b s, 4H), 1.42 (b s, 4H), 1.42 (q, J = 6.4 Hz, 3H), 0.39 (s, 4H). m/z (ESI): 499.1 (M+H)+.
Tabla 9: Los Ejemplos de 4-1 a 4-19 se prepararon siguiendo un procedimiento similar al descrito para el Ejemplo 4




Se combinaron 2-hidroxietano-1-sulfonamida (0.741 g, 5.92 mmol, Wuxi AppTec), sarcosina (0.172 g, 1.93 mmol, Ark Pharm, Inc.), yoduro de cobre (I) (0.241 g, 1.26mmol, Sigma-Aldrich Corporation), carbonato de potasio (2.78 g, 20.1 mmol, Thermo Fisher Scientific) y 4-yodo-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (2.74 g, 5.02 mmol, Intermedio 38) en N,N-dimetilformamida anhidra desgasificada (5 mL) en argón y se calentó a 130 oC durante 50 min. La reacción se enfrió hasta la temperatura ambiente, se añadieron agua (100 mL) y acetato de etilo (150 mL) y las fases se mezclaron y separaron. La capa orgánica se lavó con NH4Cl sat.: NH4ÜH:H2Ü (1:1:8, 2 x 75 mL) y se evaporó hasta sequedad a presión reducida. El producto crudo se suspendió en tolueno (30 mL) y se calentó a 90 °C durante 15 min. La mezcla se enfrió hasta la temperatura ambiente y los sólidos se separaron por filtración y se secaron en una corriente de nitrógeno. Los sólidos de color blanco se suspendieron en agua (100 mL) y se calentaron a 90 °C durante 20 minutos. La mezcla se enfrió hasta la temperatura ambiente y los sólidos se secaron en una corriente de nitrógeno para obtener 4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (2.41 g, 4.44 mmol, 88 % de rendimiento). 1H RMN (500 MHz, DMSO-de) S 13.18 (s, 1H), 10.19 (s a, 1H), 8.08 (d, J=8.72 Hz, 1H), 7.91 (d, J=7.80 Hz, 1H), 7.76 (t, J=7.96 Hz, 1H), 7.29 (d, J=1.99 Hz, 1H), 7.14 (dd, J=2.07, 8.64 Hz, 1H), 6.57 (d, J=7.96 Hz, 1H), 4.93 (s a, 1H), 4.52 (t, J=6.12 Hz, 2H), 3.77
(t, J=6.43 Hz, 2H), 3.37 (t, J=6.43 Hz, 2H), 3.00 (t a, J=4.74 Hz, 4H), 2.80-2.87 (m, 2H), 1.74 (s a, 4H), 0.39 (s, 4H). m/z (ESI): 543.2.2 (M+H)+.
Ejemplo 6: M-(6-(4.4-D¡fluorop¡per¡din-1-¡l)-4-metilpir¡din-2-¡l)-4-((2-h¡droxietil)sulfonam¡do)-2-(6-azaespiro[2.51octan-6-¡l benzamida

Una mezcla de 4-bromo-N-(6-(4,4-d¡fluorop¡per¡d¡n-1 -¡l)-4-met¡lp¡r¡d¡n-2-¡l)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzam¡da (1.0 g, 1.9 mmol, Intermediate 27), 2-sulfamoilacetato de metilo (0.361 g, 2.89 mmol, Wuxi AppTec), fosfato de potasio (i .23 g, 5.78 mmol), (1 fí,2fí)-N1,W2-d¡met¡lc¡clohexano-1,2-diamina (0.137 g, 0.963 mmol) y yoduro de cobre (I) (0.183 g, 0.963 mmol) en DMF (20 mL) se calentó a 90 °C durante 16 h. A continuación, la mezcla de reacción se filtró a través de un tapón de celite. El filtrado se diluyó con EtOAc, se lavó con agua, salmuera, se secó con Na2SÜ4, se filtró y se concentró. El residuo se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 40 % de EtOAc en éter de petróleo para proporcionar N-(6-(4,4-difluorop¡per¡d¡n-1 -il)-4-metilp¡r¡din-2-il)-4-((2-h¡drox¡et¡l)sulfonam¡do)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzam¡da (0.580 g, 1.02 mmol, 53 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-de) 5 ppm 12.85 (s, 1 H), 8.04 (d, J=8.6 Hz, 1 H), 7.51 (s, 1 H), 7.23 (d, J=2.2 Hz, 1 H), 7.09 (dd, J=8.7, 2.1 Hz, 1 H), 6.56 (s, 1 H), 3.74 (dt, J=12.5, 6.2 Hz, 6 H), 2.97 (t, J=5.2 Hz, 4 H), 2.26 (s, 3 H), 1.99 (tt, J=13.6, 5.4 Hz, 3 H), 1.79 (s, 4 H), 1.60 (s a, 4 H), 0.38 (s, 4 H). m/z (ESI): 564.2 (M+H)+.
Tabla 10: Los Ejemplos de 7-1 a 7-44 se prepararon siguiendo procedimientos similares a los descritos para los Ejemplos de 5 a 6.







Paso 1: Una mezcla de 4-bromo-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (0.63g, 1.3 mmol, Intermedio 38-1), 2-sulfamoilpropanoato de etilo (0.275 g, 1.52 mmol, Intermedio 40), fosfato de potasio tribásico (0.537 g, 2.53 mmol), (1 fi,2fi)-N7,N2-dimetilciclohexano-1,2-diamina (0.090 g, 0.63 mmol) y yoduro
de cobre (0.241 g, 1.26 mmol) en DMF (10 mL) se calentó a 90 °C durante 16 h. A continuación, la mezcla de reacción se filtró, se diluyó con EtOAc, y se lavó con agua, salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 50 % de EtOAc en éter de petróleo para proporcionar 2-(N-(3-(6-azaespiro[2.5]octan-6-il)-4-((6-(3,3,3-trifluoropropoxi)piridin-2-il)carbamoil)fenil)sulfamoil)propanoato de etilo (0.56 g, 0.94 mmol, 74 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe) 513.19 (s, 1H), 10.68 (s, 1H), 8.09 (d, J = 8.7 Hz, 1H), 7.91 (d, J=7.9 Hz, 1 H), 7.77 (t, J=7.9 Hz, 1 H), 7.32 (d, J=2.2 Hz, 1 H), 7.16 (dd, J=8.6, 2.2 Hz, 1 H), 6.58 (d, J=8.0 Hz, 1 H), 4.51 (t, J=6.1 Hz, 2 H), 4.32 (q, J=6.9 Hz, 1 H), 4.07 (qd, J=7.1,5.2 Hz, 2 H), 3.00 (d, J=5.0 Hz, 4 H), 2.84 (qt, J=11.5, 6.0 Hz, 2 H), 1.74 (s, 4 H), 1.49 (d, J=6.9 Hz, 3 H), 1.14 (t, J=7.1 Hz, 3 H), 0.39 (s, 4 H). m/z (ESI): 598.8 (M+H)+.
Paso 2: A una solución de 2-(N-(3-(6-azaespiro[2.5]octan-6-il)-4-((6-(3,3,3-trifluoropropoxi)piridin-2-il)carbamoil)fenil)sulfamoil)propanoato de etilo (0.56 g, 0.94 mmol) en THF(12 mL) se añadió LiBH4 (1.871 mL, 3.74 mmol) a -78 °C. A continuación, la mezcla de reacción se calentó lentamente hasta la temperatura ambiente durante 2 h antes de desactivarla con una solución saturada de NH4Cl (20 mL) y se extrajo con EtOAc (2 x 25 mL). Los extractos orgánicos combinados se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 50 % de EtOAc en éter de petróleo para proporcionar 4-((2-hidroxi-1-metiletil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida (0.45 g, 0.81 mmol, 86 % de rendimiento) como un sólido blanco. m/z (ESI): 557.2 (M+H)+. La mezcla racémica se sometió a separación quiral mediante SFC, utilizando una columna Lux A1 (250 x 21.2 mm, 5 |um), fase móvil: 80:20 (A: B), A = CO2 líquido, B = Metanol, tasa de flujo: 80 mL/min para generar:
Ejemplo_________ 8-1:_________ (fí)-4-((2-Hidroxi-1-metiletil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)-N-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida. Primer pico en eluir. 1H RMN (400 MHz, DMSO-cfe) 5 ppm 13.18 (s, 1 H), 10.22 (s, 1 H), 8.07 (d, J=8.6 Hz, 1 H), 7.90 (d, J=7.8 Hz, 1 H), 7.76 (t, J=8.0 Hz, 1 H), 7.31 (d, J=2.2 Hz, 1 H), 7.16 (dd, J = 8.6, 2.1 Hz, 1 H), 6.57 (dd, J = 8.1,0.8 Hz, 1 H), 5.03 (s, 1 H), 4.51 (t, J=6.1 Hz, 2 H), 3.85 (dd, J=11.1,4.4 Hz, 1 H), 3.49 (dd, J=11.1, 7.6 Hz, 1 H), 3.30 (m, 1 H), 2.87 - 3.24 (m, 4 H), 2.83 (tt, J=11.5, 6.0 Hz, 2 H), 1.74 (s a, 4 H), 1.30 (d, J=6.9 Hz, 3 H), 0.39 (s, 4 H). m/z (ESI): 557.1 (M+H)+.
Ejemplo_________ 8-2: (S)-4-((2-Hidroxi-1-metiletil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)-M-(6-(3,3,3-trifluoropropoxi)piridin-2-il)benzamida. Segundo pico en eluir 1H RMN (400 MHz, DMSO-cfe) 5 ppm 13.18 (s, 1 H), 10.22 (s, 1 H), 8.07 (d, J=8.6 Hz, 1 H), 7.78 - 8.03 (m, 1 H), 7.75 (d, J=7.9 Hz, 1 H), 7.31 (d, J=2.2 Hz, 1 H), 7.16 (dd, J=8.7, 2.2 Hz, 1 H), 6.57 (d, J=8.0 Hz, 1 H), 5.03 (s, 1 H), 4.51 (t, J=6.0 Hz, 2 H), 3.85 (dd, J=11.1,4.4 Hz, 1 H), 3.48 - 3.57 (m, 2 H), 3.30 (td, J=6.9, 4.3 Hz, 1 H), 2.99 (t, J=5.3 Hz, 4 H), 2.82 (tt, J=11.4, 5.9 Hz, 2 H), 1.74 (s a, 4 H), 1.29 (d, J=6.9 Hz, 3 H), 0.39 (s, 4 H). m/z (ESI): 557.1 (M+H)+.
La estereoquímica se asignó arbitrariamente.
Tabla 11: Los Ejemplos de 9-1 a 9-18 se prepararon siguiendo un procedimiento similar al descrito para los Ejemplos 8-1 y 8-2




Paso 1: A una solución de bis(4-metoxibencil)amina (8.95 g, 34.8 mmol) en DCM (50 mL) se añadieron DIPEA (6.06 mL, 43.5 mmol) y 2-(clorosulfonil)acetato de metilo (5.0 g, 29 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h antes de desactivarla con hielo-agua y extraer con DCM. La capa orgánica se lavó con una solución acuosa saturada de bicarbonato de sodio, agua y salmuera. El extracto orgánico se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 15 % de acetato de etilo en éter de petróleo para proporcionar 2-(N,N-bis(4-metoxibencil)sulfamoil)acetato de metilo (5.5 g, 14 mmol, 48 % de rendimiento) como una goma amarillo pálido. 1H RMN (400 MHz, DMSO-afe): 5 ppm 7.14 (d, J=8.4 Hz, 4 H), 6.86 (d, J=8.4 Hz, 4 H), 4.31 (s, 2 H), 4.25 (s, 4 H), 3.73 (s, 6 H), 3.70 (s, 3 H). m/z (ESI): 392.1 (M-H)-.
Paso 2: Una mezcla de 2-(N,N-bis(4-metoxibencil)sulfamoil)acetato de metilo (1.0 g, 2.5 mmol), K2CO3 (1.054 g, 7.62 mmol) y 1,2-dibromoetano (0.716 g, 3.81 mmol) en DMF (5 mL) se agitó a 65 °C durante 16 h. La mezcla de reacción se desactivó con hielo-agua y se extrajo con EtOAc. El extracto orgánico se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 5 % de acetato de etilo en éter de petróleo para proporcionar 1-(N,N-bis(4-metoxibencil)sulfamoil)ciclopropano-1-carboxilato de metilo (750 mg, 1.79 mmol, 70 % de rendimiento) como una goma amarillo pálido. 1H r Mn (300 MHz, DMSO-d6): 5 ppm 7.04 - 7.21 (m, 4 H), 6.83 - 6.94 (m, 4 H), 4.32 (s, 4 H), 3.72 (s, 6 H), 3.69 (s, 3 H), 1.67 (q, J=4.8, 3.6 Hz, 2 H), 1.56 (q, J=5.7, 4.8 Hz, 2 H).
Paso 3: Una mezcla de 1 -(N,N-bis(4-metoxibencil)sulfamoil)ciclopropano-1 -carboxilato de metilo (2.33 g, 5.55 mmol), TFA (23 mL) y anisol (3.03 mL, 27.8 mmol) se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se concentró y el concentrado se trató con una solución saturada acuosa de solución de bicarbonato de sodio y se extrajo con diclorometano. El extracto orgánico se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 50 % de acetato de etilo en éter de petróleo para proporcionar 1 - sulfamoilciclopropano-1 -carboxilato de metilo (0.59 g, 3.3 mmol, 59 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe) 5 ppm 7.08 (s a, 2 H), 3.70 (s, 3 H), 1.48 - 1.52 (m, 4 H). m/z (ESI): 180.0 (M+H)+.
Paso 4: Una mezcla de 4-bromo-N-(2-(4,4-difluoropiperidin-1-il)-6-metilpiridin-4-il)-2-(6- azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.38 mmol, Intermedio 27), 1-sulfamoilciclopropano-1-carboxilato de metilo (0.103 g, 0.578 mmol), yoduro de cobre (I) (0.073 g, 0.38mmol), fosfato de potasio tribásico (0.163 g, 0.770 mmol), (1R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (0.027 g, 0.19 mmol) en DMF (3 mL) se calentó a 90 °C durante 16 h. A continuación, la reacción se filtró a través de un tapón de celite y el filtrado se diluyó con EtOAc, se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando con un gradiente de un 28 % a un 35 % de EtOAc en éter de petróleo para proporcionar 1-(N-(4-((6-(4,4-difluoropiperidin-1-il)-4-metilpiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)ciclopropano-1-carboxilato de metilo (0.15 g, 0.24 mmol, 63.1 % de rendimiento) como un sólido blanco. m/z (ESI): 618.2 (M+H)+.
Paso 5: A una solución de 1 -(N-(4-((6-(4,4-difluoropiperidin-1 -il)-4-metilpiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)ciclopropano-1 -carboxilato de metilo (0.150 g, 0.243 mmol) en THF (8 mL) se añadió borohidruro de litio (0.486 mL, 0.971 mmol) a -78 °C. La mezcla de reacción se calentó lentamente hasta la temperatura ambiente durante 2 h antes de desactivarla con una solución acuosa saturada de cloruro de amonio y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 40 % a un 45 % de EtOAc en éter de petróleo para proporcionar N-(6-(4,4- difluoropiperidin-1 -il)-4-metilpiridin-2-il)-4-((1 -(hidroximetil)ciclopropano)-1-sulfonamido)-2-(6- azaespiro[2.5]octan-6-il)benzamida (0.090 g, 0.15mmol, 63 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 12.89 (s, 1 H), 10.13 (s, 1 H), 8.04 (d, J=8.6 Hz, 1 H), 7.51 (s, 1 H), 7.32 (d, J=2.2 Hz, 1 H), 7.13 (dd, J=8.6, 2.1 Hz, 1 H), 6.57 (s, 1 H), 4.98 (t, J=6.2 Hz, 1 H), 3.55 - 3.86 (m, 6 H), 2.96 (s, 4 H), 2.26 (s, 3 H), 1.98 (q, J=8.7, 7.5 Hz, 4 H), 1.65 - 1.75 (m, 4 H), 1.06 - 1.19 (m, 2 H), 0.93 - 1.06 (m, 2 H), 0.39 (s, 4 H). m/z (ESI): 590.2 (M+H)+.
Tabla 12: Los Ejemplos de 10-1 a 10-2 se prepararon siguiendo un procedimiento similar al descrito para el Ejemplo 10


Método E (formación de sulfonamida catalizada por Cu seguida de reducción)
Ejemplo 11: N-(6-(3.3-Difluoroazetidin-1 -il)-4-metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.51octan-6-il)benzamida

Paso 1: Una mezcla de 4-bromo-N-(6-(3,3-d¡fluoroazet¡d¡n-1-¡l)-4-met¡lp¡r¡d¡n-2-¡l)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzamlda (0.17 g, 0.35 mmol, Intermedio 39), 2-sulfamollacetato de metilo (0.079 g, 0.52 mmol), yoduro de cobre (I) (0.066 g. 0.35 mmol). fosfato de potasio tribásico (0.147 g. 0.692 mmol). (1R,2R)-N1,N2-dimetilciclohexano-1.2-diamina (0.025 g. 0.17 mmol) en DMF (4 mL) se calentó a 90 °C durante 16 h. A continuación. la mezcla de reacción se filtró a través de un tapón de celite. El filtrado se diluyó con EtOAc. se lavó con agua y salmuera. se secó con Na2SÜ4. se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % - 30 % de EtOAc en éter de petróleo para proporcionar 2-(N-(4-((6-(3.3-difluoroazetidin-1 -il)-4-metilpiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.16 g. 0.28 mmol. 82 % de rendimiento) como un semisólido. 1H RMN (400 MHz. DMSO-d6): δ ppm 13.12 (s a. 1 H). 10.65 (s. 1 H). 8.07 (dd. J=8.6. 1.8 Hz. 1 H). 7.57 (s. 1 H). 7.28 (t. J=2.0 Hz. 1 H). 7.15 (dt. J=8.6. 2.0 Hz. 1 H). 6.19 (s. 1 H). 4.32 - 4.46 (m. 6 H). 3.64 (s. 3 H). 2.98 (t. J=5.0 Hz. 4 H). 2.28 (s. 3 H). 1.76 (s a. 4 H). 0.39 (s. 4 H). m/z (ESI): 564.2 (M+H)+.
Paso 2: A una solución de 2-(N-(4-((6-(3.3-difluoroazetidin-1 -il)-4-metilpiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.16 g. 0.28 mmol) en t Hf (3.2 mL) se añadió gota a gota una solución 2 M de borohidruro de litio en THF (0.284 mL. 0.568 mmol) a 0 °C. La mezcla de reacción se calentó lentamente hasta la temperatura ambiente durante 1 h antes de desactivarla con una solución acuosa saturada de cloruro de amonio y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y salmuera. se secó con Na2SO4. se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna en fase inversa utilizando un gradiente de un 0 - 60 % de acetonitrilo en agua para proporcionar N-(6-(3.3-difluoroazetidin-1-il)-4- metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.51octan-6-il)benzamida (0.095 g. 0.18 mmol. 62 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz. DMSO-ds): 6 ppm 13.08 (s. 1 H). 10.20 (s. 1 H). 8.05 (d. J=8.6 Hz. 1 H). 7.56 (s. 1 H). 7.26 (d. J=2.2 Hz. 1 H). 7.12 (dd. J=8.6. 2.2 Hz. 1 H). 6.18 (s. 1 H). 4.93 (s a. 1 H). 4.36 (t. J=12.4 Hz. 4 H). 3.75 (q. J=6.0 Hz. 2 H). 3.35 (t. J=6.5 Hz. 2 H). 2.96 (t. J=5.3 Hz. 4 H). 2.27 (s. 3 H). 1.74 (s a. 4 H). 0.38 (s. 4 H). m/z (ESI): 564.2 (M+H)+.
- - - - . - - - - - - - - - - - - - -piridinil)benzamida y 2-(6-azaespiro[2.5]octan-6-il)-4-(R-ciclopropilsulfonimidoil)-N-(6-((2R)-2-metil-4-morfolinil)-2-piridinil)benzamida

Paso 1: A una solución de (F?)-4-bromo-N-(6-(2-metilmorfolino)piñdin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (1.0 g, 2.1 mmol, Intermedio 30) en THF (20 mL) se añadió gota a gota dibutilmagnesio (solución 1.0 M en THF, 1.24 mL, 1.24 mmol) a 0 °C y se agitó durante 10 min. La mezcla de reacción se enfrió hasta -78 °C y se añadió n-butillitio (solución 2.5 M in en hexanos, 0.989 mL, 2.47 mmol) y se agitó durante 10 min. Se añadió gota a gota 1,2-diciclopropildisulfano (0.301 g, 2.06 mmol, Wuxi AppTec) y se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se desactivó con HCl 2 N (20 mL) y se extrajo con acetato de etilo (2 x 20 mL). Los extractos orgánicos combinados se lavaron con salmuera (20 mL), se secaron con sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía ultrarrápida a través de una columna de gel de sílice preempaquetada Redi-Sep (40 g), eluyendo con un gradiente de un 30 % de acetato de etilo en hexanos para obtener (fi)-4-(ciclopropiltio)-N-(6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.90 g, 91 % de rendimiento) como un sólido amarillo pálido.1H RMN (400 MHz, DMSO-ofe): 512.91 (s, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.70 - 7.59 (m, 2H), 7.40 - 7.26 (m, 2H), 6.61 - 6.58 (m, 1 H), 4.11 (dd, J = 25.9, 12.9 Hz, 2H), 3.93 (dd, J = 11.5, 3.2 Hz, 1H), 3.62 - 3.55 (m, 2H), 3.03 (t, J = 5.5 Hz, 4H), 2.83 (td, J = 12.2, 3.4 Hz, 1 H), 2.38 (dq, J = 7.3, 3.8, 3.2 Hz, 1H), 1.72 (s, 4H), 1.20 - 1.16 (m, 6H), 0.66 - 0.62 (m, 2H), 0.38 (d, J = 1.6 Hz, 4H). m/z (ESI): 479.2 (M+H)+.
Paso 2: A una solución de (R)-4-(ciclopropiltio)-N-(6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.90 g, 1.9 mmol) en diclorometano (10 mL) se añadió m-CPBA (43.3 mg, 0.188 mmol) a 0 °C y se agitó a la misma temperatura durante 3 h. La mezcla de reacción se desactivó con una solución acuosa de bicarbonato de sodio al 10 % (15 mL) y se extrajo con DCM (2 x 20 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera (20 mL), se secaron con sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía ultrarrápida a través de una columna de gel de sílice preempaquetada Redi-Sep (40 g), eluyendo con un gradiente de un 1 % a un 50 % de acetato de etilo en hexanos para obtener 4-(ciclopropilsulfinil)-N-(6-((fl)-2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (650 mg, 70 % de rendimiento) como un sólido amarillo pálido. 1H RMN (400 MHz, Cloroformo-a): 512.89 (s, 1H), 8.23 (d, J = 8.3 Hz, 1H), 7.81 (dd, J = 7.9, 4.6 Hz, 1 H), 7.57 (td, J = 8.0, 3.3 Hz, 1H), 7.30 (dd, J = 8.3, 1.8 Hz, 1H), 7.25 (d, J = 1.8 Hz, 1 H), 6.42 (dd, J = 8.2, 3.7 Hz, 1H), 4.18 - 3.95 (m, 3H), 3.81 - 3.65 (m, 2H), 3.09 (q, J = 6.3, 5.6 Hz, 4H), 2.97 (tt, J = 12.8, 11.7, 2.7 Hz, 1H), 2.63 (ddd, J = 12.7, 10.4, 1.9 Hz, 1H), 2.23 (tt, J = 7.3, 4.4 Hz, 1H), 1.31 - 1.27 (m, 4H), 1.19 - 1.15 (m, 5H), 0.77 - 0.73 (m, 2H), 0.40 (d, J = 1.7 Hz, 4H). m/z (ESI): 495.2 (M+H)+.
Paso 3: A una solución de 4-(ciclopropilsulfinil)-N-(6-((fi)-2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida ( 0.65 g, 1.30 mmol) en MeOH (20 mL) se añadieron diacetato de yodobenceno (1.27 g, 3.94 mmol) y carbamato de amonio (410 mg, 5.26 mmol) a 0 °C durante 10 min, se calentó a temperatura ambiente y se agitó durante 12 h. La mezcla de reacción se desactivó con agua (10 mL) y se extrajo con acetato de etilo (2 x 10 mL). Los extractos orgánicos combinados se lavaron con una solución de salmuera (10 mL), se secaron con sulfato de sodio, se filtraron y se concentraron a presión reducida. El residuo crudo se purificó mediante cromatografía ultrarrápida a través de una columna de gel de sílice preempaquetada Redi-Sep (40 g), eluyendo con un gradiente de un 40 % de acetato de etilo en hexanos para obtener 4-(ciclopropanosulfonimidoil)-N-(6-((R)-2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (150 mg, racémica) como un sólido amarillo pálido. 1H RMN (400 MHz, Cloroformo-d): 512.67 (s, 1H), 8.45 (dd, J = 8.3, 1.2 Hz, 1H), 7.93 (t, J = 1.5 Hz, 1H), 7.84 (dt, J = 8.3, 1.5 Hz, 1H), 7.82 - 7.72 (m, 1H), 7.66 - 7.56 (m, 1 H), 6.46 (d, J = 8.2 Hz, 1H), 4.22 - 3.92 (m, 3H), 3.92 - 3.62 (m, 2H), 3.33 - 3.11 (m, 4H), 2.99 (td, J = 12.2, 3.5 Hz, 1H), 2.77 - 2.55 (m, 2H), 2.01 -1.69 (m, 4H), 1.47 (ddt, J = 9.9, 7.0, 5.0 Hz, 1H),
1.30 (dd, J = 6.2, 1.0 Hz, 3H), 1.28 - 1.20 (m, 2H), 1.18 - 1.08 (m, 1H), 1.00 (qd, J = 8.0, 6.0 Hz, 1H), 0.43 (d, J = 1.1 Hz, 4H). m/z (ESI): 510.2 (M+H)+. La mezcla racémica se separó mediante separación quiral mediante SFC, utilizando una columna (S,S)Whelk-01 (250 x 30 mm, 5 μm), fase móvil: 70:30 (A: B), A = CO2 líquido, B = Metanol, tasa de flujo: 70 mL/min para obtener:
Ejemplo____ 12-1: 2-(6-Azaesp¡ro[2.51octan-6-¡l)-4-(S-c¡cloprop¡lsulfon¡m¡do¡l)-M-(6-((2fí)-2-met¡l-4-morfol¡n¡l)-2-piridinil)benzamida. 1H RMN (400 MHz, Cloroformo-a) 5 12.64 (s, 1H), 8.44 (d, J = 8.1 Hz, 1H), 7.93 (d, J = 1.7 Hz, 1H), 7.83 (dd, J = 8.3, 1.8 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 6.46 (d, J = 8.1 Hz, 1H), 4.13 -4.01 (m, 3H), 3.80 - 3.64 (m, 2H), 3.19 - 3.13 (m, 4H), 2.98 (td, J = 12.2, 3.6 Hz, 1H), 2.67 - 2.60 (m, 2H), 2.01- 1.50 (m, 3H), 1.47 (ddt, J = 9.7, 6.8, 4.8 Hz, 1H), 1.32 - 1.26 (m, 6H), 1.15 (qd, J = 7.9, 4.9 Hz, 1H), 1.05 - 0.95 (m, 1H), 0.42 (s, 4H). m/z (ESI): 510.2 (M+H)+.
Ejemplo 12-2: 2-(6-Azaespiro[2.5loctan-6-il)-4-(fí-ciclopropilsulfonimidoil)-N-(6-((2fí)-2-metil-4-morfolinil)-2-piridinil)benzamida.1H RMN (400 MHz, Cloroformo-a): 5 12.65 (s, 1H), 8.44 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 1.7 Hz, 1H), 7.83 (dd, J = 8.2, 1.8 Hz, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.59 (t, J = 8.1 Hz, 1H), 6.45 (d, J = 8.3 Hz, 1H), 4.13 -3.95 (m, 3H), 3.73 (dddd, J = 17.1,9.9, 8.1,5.2 Hz, 2H), 3.19 - 3.13 (m, 4H), 2.98 (td, J = 12.3, 3.6 Hz, 1H), 2.63 (dq, J = 10.8, 3.9 Hz, 2H), 2.01 - 1.50 (m, 3H), 1.47 (dtd, J = 10.2, 4.8, 2.4 Hz, 1H), 1.32 - 1.29 (m, 2H), 1.29 (d, J = 6.2 Hz, 4H), 1.14 (ddd, J = 10.1,7.9, 3.8 Hz, 1H), 0.99 (dtd, J = 9.0, 7.4, 4.9 Hz, 1H), 0.42 (s, 4H). m/z (ESI): 510.2 (M+H)+.
La estereoquímica se asignó arbitrariamente.
Tabla 13: Los ejemplos de 13-1 a 13-4 se prepararon siguiendo un procedimiento similar al de los ejemplos 12-1 y 12 2.


Paso 1: Una solución de ácido 4-((1-metilc¡clopropano)-1-sulfonam¡do)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzo¡co (5.0 g, 14 mmol, Intermedio 11) en DCM (50 mL) se trató con DIPEA (7.19 mL, 41.2 mmol), 6-fluoropiridin-2-amina (1.84 g, 16.5 mmol) y después T3P (17.46 g, 27.4 mmol, 50 % en EtOAc, Spectrochem) a TA. A continuación, la mezcla de
reacción se agitó durante 16 h antes de desactivarla con agua y se extrajo con DCM. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 15 % a un 20 % de EtOAc en éter de petróleo para proporcionar N-(6-fluoropiridin-2-il)-4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (3.0 g, 6.5 mmol, 48 % de rendimiento) como un sólido blanquecino. 1H Rm N (400 MHz, DMSO-afe): ó ppm 13.09 (s, 1 H), 10.28 (s, 1 H), 8.20 (d, J=7.7 Hz, 1 H), 8.03 (t, J=9.0 Hz, 2 H), 7.36 (s, 1 H), 7.17 (d, J=8.5 Hz, 1 H), 6.89 (d, J=8.0 Hz, 1 H), 2.97 (t, J=5.3 Hz, 4 H), 1.67 (s, 4 H), 1.41 (s, 3 H), 1.21 (s, 2 H), 0.81 - 0.89 (m, 2 H), 0.41 (s, 4 H). m/z (ESI): 459.1 (M+H)+.
Paso 2: Una solución de ciclopropilmetanol (23.59 mg, 0.327 mmol) en DMF (1.5 mL) se trató con hidruro de sodio (13.08 mg, 0.327 mmol, 60 % en aceite mineral) a 0 °C en una atmósfera de N2 y se agitó durante 10 min. A continuación, se añadió una solución de N-(6-fluoropiridin-2-il)-4-((1-metilciclopropano)-1-sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (100 mg, 0.218 mmol) en DMF (0.5 mL) a la mezcla de reacción. La solución resultante se agitó a TA durante 2 h antes de desactivarla con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na¿SO4, se filtró y se concentró. El concentrado se purificó mediante HPLC de fase inversa para proporcionar N-(6-(ciclopropilmetoxi)piridin-2-il)-4-((1-metilciclopropano)-1 -sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida como un sólido blanco. 1H RMN (400 MHz, DMSO-de): ó ppm 13.20 (s, 1 H), 10.22 (s a, 1 H), 8.08 (d, J=8.6 Hz, 1 H), 7.85 (d, J=7.8 Hz, 1 H), 7.72 (t, J=7.9 Hz, 1 H), 7.36 (d, J=2.2 Hz, 1 H), 7.17 (dd, J=8.7, 2.2 Hz, 1 H), 6.56 (d, J=8.0 Hz, 1 H), 4.12 (d, J=7.2 Hz, 2 H), 2.98 (d, J=5.3 Hz, 4 H), 1.71 (s a, 4 H), 1.41 (s, 3 H), 1.24 - 1.35 (m, 1 H), 1.17 - 1.27 (m, 2 H), 0.80 - 0.88 (m, 2 H), 0.55 - 0.64 (m, 2 H), 0.39 (s, 4 H), 0.28 - 0.36 (m, 2 H). m/z (ESI): 511.2 (M+H)+.
Ejemplo 15: N-(6-(Ciclopropilmetoxi)piridin-2-il)-4-((1 -metilciclopropano)-1 -sulfonamido)-2-(6-azaespiro[2.51octan-6-¡Dbenzamida

Paso 1: A una solución de ácido 6-hidroxipicolínico (5.0 g, 36 mmol) en MeOH (83 mL) se añadió gota a gota H2SO4 (4.79 mL, 90 mmol) a TA. A continuación, la mezcla de reacción se calentó a reflujo durante 18 h antes de que se
evaporaran los volátiles y el concentrado se neutralizó con una solución de NaHCÜ3 (100 mL) lentamente para mantener el pH de 7-8. A continuación, la mezcla de reacción se extrajo con CH2Cl2. La capa orgánica se lavó con agua y salmuera, se secó con Na2SÜ4, se filtró y se concentró. El concentrado se purificó con éter de petróleo para proporcionar 6-hidroxipicolinato de metilo (4.2 g, 27 mmol, 76 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe) 5 ppm 11.54 (s, 1 H), 7.64 (dd, J=8.9, 7.0 Hz, 1 H), 7.16 (dd, J=7.0, 1.0 Hz, 1 H), 6.73 (dd, J=8.9, 1.0 Hz, 1 H), 3.84 (s, 3 H). m/z (ESI): 154.1 (M+H)+.
Paso 2: Una solución de 6-hidroxipicolinato de metilo (1.0 g, 6.4 mmol) y 2-cloro-2,2-difluoroacetato de sodio (1.971 g, 12.93 mmol, TCI India) en acetonitrilo (25 mL) se calentó a 85 °C durante 72 h. A continuación, la mezcla de reacción se enfrió hasta TA y se evaporó hasta sequedad para obtener 6-(difluorometoxi)picolinato de metilo (2.1 g, 6.3 mmol, 98 % de rendimiento) como un aceite amarillo que se utilizó en el siguiente paso sin purificación adicional. 1H RMN (400 MHz, DMSO-ds) 5 ppm 8.12 (ddd, J=11.3, 6.7, 2.7 Hz, 1 H), 7.92 - 7.97 (m, 1 H), 7.50 - 7.91 (m, 1 H), 7.34 -7.42 (m, 1 H), 3.89 (s, 3 H). m/z (ESI): 204.1 (M+H)+
Paso 3: A una solución de 6-(difluorometoxi)picolinato de metilo (2.0 g, 5.9 mmol) en THF (90 mL) se añadió LiOH (11.81 mL, 23.63 mmol, solución 2 M en H2O). La mezcla de reacción se agitó a TA durante 16 h antes de desactivarla con HCl 1 N (pH=2-3) y se extrajo con CH2Cl2. La capa orgánica se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró para proporcionar ácido 6-(difluorometoxi)picolínico como un sólido amarillo claro que se utilizó en el siguiente paso sin purificación. 1H RMN (300 MHz, DMSO-afe) 5 ppm 13.34 (s, 1 H), 8.02 - 8.19 (m, 1 H), 7.91 (d, J=7.4 Hz, 1 H), 7.79 (t, J=72.6 Hz, 1 H), 7.34 (d, J = 8.2 Hz, 1 H). m/z (ESI): 190.0 (M+H)+.
Paso 4: A una solución de ácido 6-(difluorometoxi)picolínico (0.80 g, 4.1 mmol) en una mezcla de ferf-butanol (2.5 mL) y tolueno (28 mL) se añadieron Et3N (2.30 mL, 16.51 mmol) y después fosforazidato de difenilo (1.31 mL, 6.19 mmol) a TA en una atmósfera de N2. La mezcla de reacción se calentó a 100 °C durante 16 h antes de neutralizarla con una solución acuosa de NaHCO3 (100 mL) y se extrajo con EtOAc (2 x 50 mL). Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 2 % a un 5 % de EtOAc en éter de petróleo para proporcionar (6-(difluorometoxi)piridin-2-il)carbamato de ferf-butilo (0.51 g, 1.9 mmol, 47 % de rendimiento) como un aceite amarillo claro. 1H RMN (400 MHz, DMSO-afe) 5 ppm 9.85 (s, 1 H), 7.83 (t, J=8 Hz, 1 H), 7.73 - 7.36 (m, 2 H), 6.66 (d, J=12 Hz, 1 H), 1.47 (s, 9 H). m/z (ESI): 204.1 (M-fBu)+, 161.1 (M-Boc)+.
Paso 5: A una solución de (6-(difluorometoxi)piridin-2-il)carbamato de ferf-butilo (0.50 g, 1.9 mmol) en 1,4-dioxano (3.0 mL) se añadió HCl 4.0 M en dioxano (9.98 mL, 39.9 mmol) a TA durante 5 min. La mezcla de reacción se agitó durante 6 h antes de desactivarla con una solución acuosa saturada de una solución de NaHCO3 (100 mL) y se extrajo con CH2Cl2. Los extractos orgánicos se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron para proporcionar 6-(difluorometoxi)piridin-2-amina (0.31 g, 1.8 mmol, 95 % de rendimiento) como un aceite naranja que se utilizó en el siguiente paso sin purificación. 1H RMN (300 MHz, DMSO-afe) 5 ppm 7.21 - 7.79 (m, 2 H), 6.23 (d, J=7.7 Hz, 3 H), 6.05 (d, J=7.7 Hz, 1 H). m/z (ESI): 161.1 (M+H)+.
Paso 6: A una solución de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.40 g, 1.3 mmol, Intermedio 9-1) en DCM (5.0 mL) se añadieron DIPEA (0.676 mL, 3.87 mmol) y después T3P (2.28 mL, 3.87 mmol, 50 % en acetato de etilo) a TA y se agitó durante 15 min. A continuación, se añadió una solución de 6-(difluorometoxi)piridin-2-amina (0.327 g, 1.93 mmol) en diclorometano (2.0 mL) a la mezcla de reacción. La mezcla resultante se agitó a TA durante 16 h antes de desactivarla con agua (50 mL) y se extrajo con CH2Cl2. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 0 % a un 17 % de EtOAc en éter de petróleo para proporcionar 4-bromo-N-(6-(difluorometoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.30 g, 0.62 mmol, 48 % de rendimiento) como un líquido gomoso amarillo claro. 1H RMN (300 MHz, DMSO-afe) 5 ppm 13.45 (s, 1 H), 7.92 - 8.17 (m, 3 H), 7.70 - 7.80 (m, 1 H), 7.57 (d, J=6.7 Hz, 2 H), 6.84 (d, J=7.9 Hz, 1 H), 3.05 (t, J=5.3 Hz, 4 H), 1.69 (s, 4 H), 0.38 (s, 4 H). m/z (ESI): 452.0, 454.0 (M+H)+.
Paso 7: Una mezcla de 4-bromo-N-(6-(difluorometoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.30 g, 0.66 mmol), 2-hidroxietano-1-sulfonamida (0.125 g, 0.995 mmol), fosfato de potasio (0.422 g, 1.99 mmol), (1R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (0.094 g, 0.66 mmol) y yoduro de cobre (I) (0.126 g, 0.663 mmol) en DMF (3.0 mL) se calentó a 90 °C durante 16 h. La mezcla de reacción se filtró a través de un tapón de celite y el filtrado se diluyó con EtOAc. La capa orgánica se lavó con agua y salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con de un 5 % a un 44 % de EtOAc en éter de petróleo para proporcionar N-(6-(difluorometoxi)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.18 g, 0.36 mmol, 54 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe) 5 ppm 13.50 (s, 1 H), 10.26 (s, 1 H), 8.11 (t, J=8.0 Hz, 2 H), 7.96 (t, J=8.0 Hz, 1 H), 7.30 (d, J= 2.2 Hz, 1 H), 7.16 (dd, J=8.7, 2.2 Hz, 1 H), 6.81 (d, J=8.0 Hz, 1 H), 4.96 (t, J=5.6 Hz, 1 H), 3.77 (q, J=6.0 Hz, 2 H), 3.38 (d, J=6.4 Hz, 2 H), 2.99 (t, J=5.5 Hz, 4 H), 1.72 (s a, 4 H), 0.40 (s, 4 H). m/z (ESI): 497.1 (M+H)+.

Paso 1: A una solución de 2-hidroxiacetato de etilo (5.0 g, 48 mmol, Combi-Blocks) en THF (100 mL) se añadió cloruro de metilmagnesio (48.0 mL, 3.0 M en THF, 144 mmol) y la mezcla de reacción se agitó a 0 °C durante 30 min. La mezcla de reacción se desactivó con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró para proporcionar 2-metilpropano-1,2-diol (2.3 g, 26 mmol 53 % de rendimiento) como un aceite amarillo claro. 1H RMN (300 MHz, DMSO-afe) 5 ppm 4.49 (t, J=5.8 Hz, 1 H), 4.10 (s a, 1 H), 3.12 - 3.16 (m, 2 H), 1.03 (s, 6 H).
Paso 2: A una solución de 6-fluoropiridin-2-amina (0.700 g, 6.24 mmol) y 2-metilpropano-1,2-diol (0.844 g, 9.37 mmol) en dioxano (7 mL) a TA se añadió NaH (60 % en aceite mineral, 0.749 g, 18.7 mmol). La mezcla de reacción se agitó a 80 ° C durante 4 h, se enfrió hasta TA, se desactivó con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 10 % a un 80 % de acetato de etilo en éter de petróleo para proporcionar 1-((6-aminopiridin-2-il)oxi)-2-metilpropan-2-ol (0.60 g, 3.3 mmol, 53 % de rendimiento) como un aceite naranja. 1H RMN (400 MHz, DMSO-afe) 5 ppm 7.25 - 7.29 (m, 1 H), 5.97 (d, J=7.8 Hz, 1 H), 5.87 (d, J=7.8 Hz, 1 H), 5.82 (s, 2 H), 4.56 (s, 1 H), 3.88 (s, 2 H), 1.15 (s, 6 H). m/z (ESI): 183.2 (M+H)+.
Paso 3: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.30 g, 0.97 mmol, Intermedio 9-1), DIPEA (0.203 mL, 1.16 mmol), T3P (50 % en acetato de etilo, 0.691 mL, 1.16 mmol) y 1-((6-aminopiridin-2-il)oxi)-2-metilpropan-2-ol (0.211 g, 1.16 mmol) en DCM (3 mL) se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 5 % a un 30 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(6-(2-hidroxi-2-metilpropoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.42 mmol, 44 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-afe) 5 ppm 13.08 (s, 1 H), 8.05 (d, J=8.4 Hz, 1 H), 7.85 (d, J=7.8 Hz, 1 H), 7.69 - 7.76 (m, 2 H), 7.55 - 7.59 (m, 1 H), 6.59 (d, J=8.0 Hz, 1 H), 4.60 (s, 1 H), 4.06 (s, 2 H), 3.06 (t, J=5.3 Hz, 4 H), 1.73 (s a, 4 H), 1.22 (s, 6 H), 0.39 (s, 4 H). m/z (ESI): 476.1 (M+H)+.
Paso 4: Una mezcla de 4-bromo-N-(6-(2-hidroxi-2-metilpropoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.27 g, 0.57 mmol), 2-sulfamoilacetato de metilo (0.131 g, 0.854 mmol), fosfato de potasio (0.302 g, 1.42 mmol), (1 R,2R)-W1,W2-dimetilciclohexano-1,2-diamina (0.040 g, 0.28 mmol) y cloruro de cobre (I) (0.108 g, 0.569 mmol) en DMF (4 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 10 % a un 80 % de acetato de etilo en éter de petróleo para proporcionar 2-(N-(4-((6-(2-hidroxi-2-metilpropoxi)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6
il)fenil)sulfamoil)acetato de metilo (0.25 g, 0.46 mmol, 80 % de rendimiento) como un aceite amarillo claro. m/z (ESI): 547.2 (M+H)+.
A una solución de 2-(N-(4-((6-(2-hidroxi-2-metilpropoxi)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.24 g, 0.44 mmol) en THF (3.75 mL) se añadió L¡BH4 (0.439 mL, 0.878 mmol) a -30°C y la mezcla resultante se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se desactivó con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 30 % a un 90 % de acetato de etilo en éter de petróleo para proporcionar N-(6-(2-hidroxi-2-metilpropoxi)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.091 g, 0.18 mmol, 40 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-ds): 5 ppm 13.10 (s a, 1 H), 8.07 (d, J=8.7 Hz, 1 H), 7.84 (d, J=7.6 Hz, 1 H), 7.72 (t, J=7.9 Hz, 1 H), 7.28 (d, J=2.2 Hz, 1 H), 7.12 (dd, J=8.6, 2.1 Hz, 1 H), 6.55 (d, J=8.0 Hz, 1 H), 4.58 (s, 1 H), 4.05 (s, 2 H), 3.76 (t, J=6.5 Hz, 2 H), 3.35 (t, J=6.5 Hz, 2 H), 2.99 (t, J=5.2 Hz, 4 H), 1.75 (s a, 4 H), 1.21 (s, 6 H), 0.39 (s, 4 H). m/z (ESI): 519.2 (M+H)+.
Tabla 14: Los Ejemplos de 16-1 a 16-9 se prepararon siguiendo un procedimiento similar al del Ejemplo 16.



Paso 1: A una solución de 3,3,3-trifluoropropan-1-ol (0.550 g, 4.83 mmol) en 1,4-dioxano (10 mL) se añadió hidruro de sodio (0.386 g, 9.64 mmol, 60 % en aceite) a 10 °C y se agitó durante 30 min a temperatura ambiente. Se añadió una solución de 2-bromo-6-fluoro-4-metilpiridina (0.61 g, 3.2 mmol, Combi-Blocks) en dioxano (4 mL) a la mezcla de reacción. La solución resultante se agitó a temperatura ambiente durante 2 h antes de desactivarla con hielo-agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 4 % de acetato de etilo en éter de petróleo para proporcionar 2-bromo-4-metil-6-(3,3,3-trifluoropropoxi)piridina (0.76 g, 85 %
de rendimiento) como un aceite transparente. 1H RMN (400 MHz, Cloroformo-d): 5 ppm 6.96 (s, 1 H), 6.53 (s, 1 H), 4.51 - 4.56 (m, 2 H), 2.56 - 2.66 (m, 2 H), 2.29 (s, 3 H). m/z (ESI): 284.0 (M+H)+.
Paso 2: Una mezcla de 2-bromo-4-metil-6-(3,3,3-trifluoropropoxi)piridina (0.71 g, 2.5 mmol), (4-metoxifenil)metanamina (0.514 g, 3.75 mmol), carbonato de cesio (2.443 g, 7.50 mmol), xantphos (0.289 g, 0.500 mmol) y Pd2(dba)3 (0.023 g, 0.025 mmol) en 1,4-dioxano (14 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se filtró a través de un lecho de celite y el filtrado se lavó con agua, salmuera, se secó con Na2SO4 y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un 6 % de acetato de etilo en éter de petróleo para proporcionar N-(4-metoxibencil)-4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-amina (0.65 g, 1.91 mmol, 76 % de rendimiento) como un sólido amarillo. 1H RMN (300 MHz, DMSO-afe): 5 ppm 7.23 (d, J=8.7 Hz, 2 H), 6.94 (t, J=5.9 Hz, 1 H), 6.82 - 6.90 (m, 2 H), 5.89 (s, 1 H), 5.71 (s, 1 H), 4.33 (t, J=6.1 Hz, 4 H), 3.71 (s, 3 H), 2.56 - 2.66 (m, 2 H), 2.07 (s, 3 H). m/z (ESI): 341.1 (M+H)+.
Paso 3 : Una solución de N-(4-metoxibencil)-4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-amina (0.30 g, 0.88 mmol), anisol (0.193 mL, 1.76 mmol), TFA (1.5 mL, 19 mmol) en diclorometano (3 mL)se agitó a 55 °C durante 1.5 h. A continuación, la mezcla de reacción se concentró y el residuo se disolvió en agua y se basificó hasta pH~ 8 con bicarbonato de sodio al 10 % y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 10 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar 4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-amina (0.16 g, 0.73mmol, 82 % de rendimiento) como un aceite amarillo. 1H RMN (300 MHz, DMSO-afe): 5 ppm 5.88 (s, 1 H), 5.74 (s, 1 H), 4.30 (t, J=6.1 Hz, 2 H), 2.60 - 2.68 (m, 2 H), 2.05 (s, 3 H). m/z (ESI): 221.1 (M+H)+.
Paso 4: Una mezcla de 4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-amina (0.16 g, 0.73 mmol), ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.270 g, 0.872 mmol, Intermedio 9-1), Et3N (0.304 mL, 2.18 mmol) y T3P (solución al 50 % en acetato de etilo, 1.386 g, 2.180 mmol) en 1,2-dimetoxietano (4 mL) se calentó a 80 °C durante 16 h. La mezcla de reacción se desactivó con agua helada y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 8 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.39 mmol, 54 % de rendimiento) como un sólido blanco. 1H RMN (300 MHz, d Ms O-o6): 5 ppm 13.04 (s, 1 H), 8.02 (d, J=8.5 Hz, 1 H), 7.77 (s, 1 H), 7.68 (d, J=2.0 Hz, 1 H), 7.56 (dd, J=8.4, 1.9 Hz, 1 H), 6.45 (s, 1 H), 4.49 (t, J=6.1 Hz, 2 H), 3.05 (t, J=5.3 Hz, 4 H), 2.82 (dt, J=11.5, 5.8 Hz, 2 H), 2.31 (s, 3 H), 1.66 - 1.78 (m, 4 H), 0.37 (s, 4 H). m/z (ESI): 512.1 (M+H)+.
Paso 5: Una mezcla de 4-bromo-N-(4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.39 mmol), 2-hidroxietano-1-sulfonamida (0.073 g, 0.59 mmol), fosfato de potasio tribásico (0.166 g, 0.781 mmol), yoduro de cobre (I) (0.074 g, 0.39 mmol) y (1 R,2R)-N,N-dimetil-1,2-ciclohexanodiamina (0.028 g, 0.20 mmol) en DMF (3 mL) se agitó a 95 °C durante 16 h. La mezcla de reacción se desactivó con agua helada y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna de fase inversa utilizando un gradiente de un 65 % de acetonitrilo en agua para proporcionar 4-((2-hidroxietil)sulfonamido)-N-(4-metil-6-(3,3,3-trifluoropropoxi)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.166 g, 0.298 mmol, 76 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 13.09 (s, 1 H), 10.21 (s a, 1 H), 8.06 (d, J=8.6 Hz, 1 H), 7.78 (s, 1 H), 7.27 (d, J=2.1 Hz, 1 H), 7.13 (dd, J=8.6, 2.1 Hz, 1 H), 6.42 (s, 1 H), 4.96 (s a, 1 H), 4.49 (t, J=6.1 Hz, 2 H), 3.76 (t, J=6.4 Hz, 2 H), 3.38 - 3.40 (m, 2 H), 2.96 - 3.20 (m, 4 H), 2.76 - 2.88 (m, 2 H), 2.31 (s, 3 H), 1.60 - 1.80 (m, 4 H), 0.38 (s, 4 H). m/z (ESI): 557.2 (M+H)+.
Tabla 15: Los ejemplos de 17-1 a 17-3 se prepararon siguiendo un procedimiento similar al descrito para el ejemplo 17


Eiemplo 18: 4-((2-Hidroxietil)Sulfonamido)-N-(5-metil-6-morfohnopiridin-2-il)-2-(6-azaespiro[2.51octan-6-il)benzamida
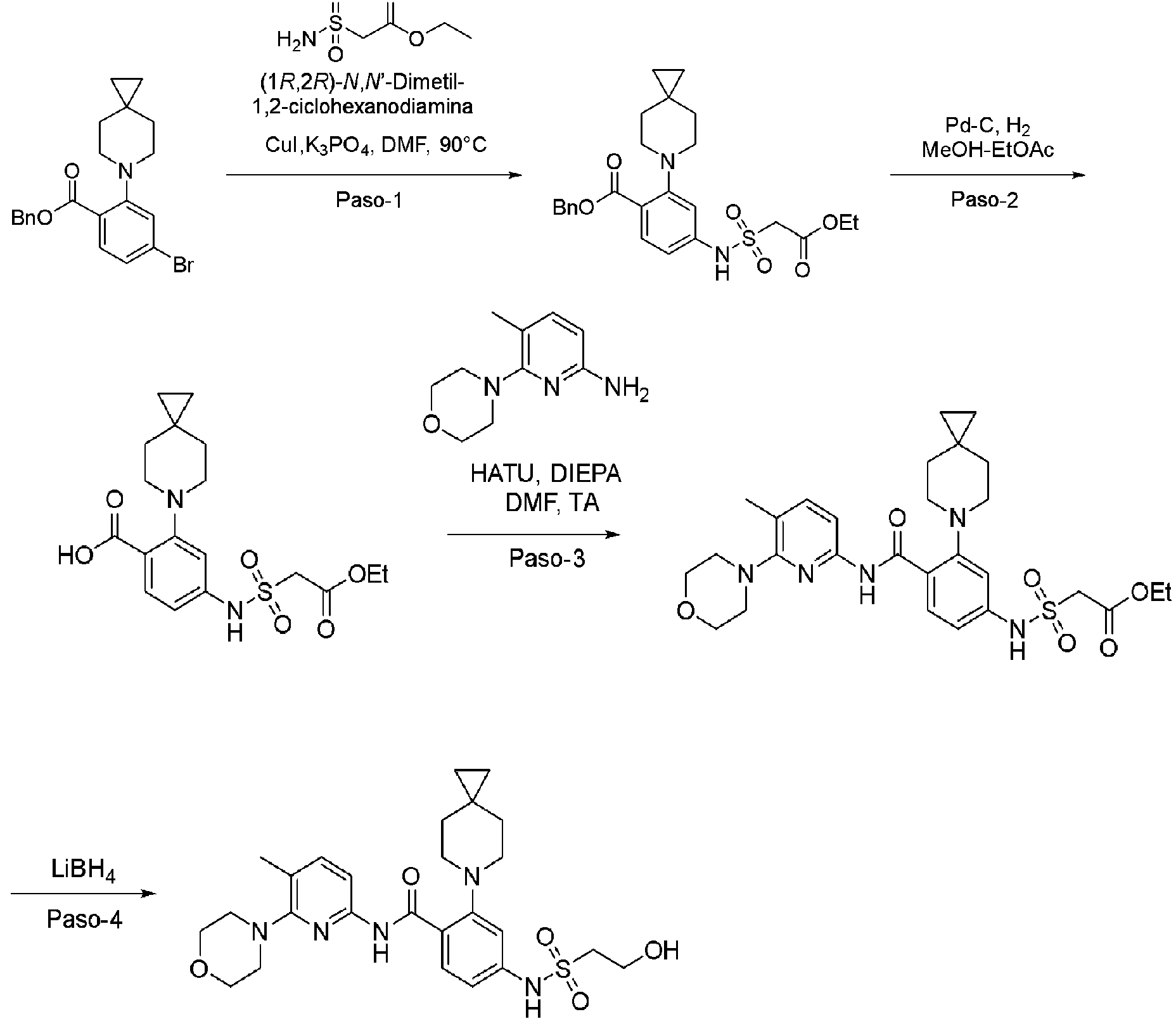
Paso 1: Una mezcla de 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (1.0 g, 2.5 mmol, Intermedio 9-4), 2-sulfamoilacetato de etilo (0.626 g, 3.75 mmol), fosfato de potasio (1.06 g, 5.00 mmol), yoduro de cobre (I) (0.476 g, 2.50 mmol), (1R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (0.178 g, 1.25 mmol) en Dm F (15 mL) se calentó a 90 °C durante 16 h. A continuación, la mezcla de reacción se filtró a través de un tapón de celite y el filtrado se diluyó con EtOAc, se lavó con agua, salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 25 % de EtOAc en éter de petróleo
para proporcionar 4-((2-etoxi-2-oxoetil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (0.75 g, 1.5 mmol, 53 % de rendimiento) como un aceite naranja. 1H RMN (400 m Hz , DMSO-afe): 5 ppm 10.45 (s, 1 H), 7.64 (d, J=8.5 Hz, 1 H), 7.29 - 7.48 (m, 5 H), 6.93 (d, J=2.1 Hz, 1 H), 6.79 (dd, J=8.5, 2.0 Hz, 1 H), 5.28 (s, 2 H), 4.31 (s, 2 H), 4.05 (dq, J=19.5, 7.1 Hz, 2 H), 2.94 (t, J=5.3 Hz, 4 H), 1.37 (t, J=5.3 Hz, 4 H), 1.16 (dt, J=11.2, 7.1 Hz, 3 H), 0.28 (s, 4 H). m/z (ESI): 487.2 (M+H)+.
Paso 2: A una solución de 4-((2-etoxi-2-oxoetil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoato de bencilo (0.55 g, 1.1 mmol) en acetato de etilo (3 mL) y metanol (6 mL) se añadió paladio sobre carbón (10 %, 0.28 g, 0.26 mmol) y se siguió agitando en una atmósfera de hidrógeno (1 atm) durante 2 h. A continuación, la mezcla de reacción se filtró a través de un lecho de celite y se lavó con metanol (150 mL). El filtrado se concentró y se purificó con Et2O para proporcionar ácido 4-((2-etoxi-2-oxoetil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.55 g, 1.4 mmol, 90 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMsO-ds): 5 ppm 10.69 (s, 1 H), 7.93 (d, J=8.6 Hz, 1 H), 7.31 (d, J=2.1 Hz, 1 H), 7.12 (dd, J=8.7, 2.1 Hz, 1 H), 4.31 (s, 2 H), 4.07 (q, J=7.1 Hz, 2 H), 3.00 (t, J=5.4 Hz, 4 H), 1.57 (s, 4 H), 1.14 (t, J=7.1 Hz, 3 H), 0.42 (s, 4 H). m/z (ESI): 397.1 (M+H)+.
Paso 3: A una solución de 5-metil-6-morfolinopiridin-2-amina (45.0 mg, 0.232 mmol, Intermedio 8) en DMF (3 mL) se añadieron ácido 4-((2-etoxi-2-oxoetil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzoico (92 mg, 0.23 mmol), DIPEA (48.8 μL, 0.279 mmol) y HATU (106 mg, 0.279 mmol) a temperatura ambiente. A continuación, la mezcla de reacción se agitó durante 12 h antes de desactivarla con agua y se extrajo con EtOAc. La capa orgánica se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un 15 % de acetato de etilo en éter de petróleo para proporcionar 2-(N-(4-((5-metil-6-morfolinopiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de etilo (50 mg, 0.087 mmol, 38 % de rendimiento) como un sólido amarillo pálido. m/z (ESI): 572.2 (M+H)+.
Paso 4: A una solución de 2-(N-(4-((5-metil-6-morfolinopiridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de etilo (50 mg, 0.087 mmol) en THF (10 mL) se añadió borohidruro de litio (65.6 μL, 0.131 mmol, 2 M en THF) a 0 °C. A continuación, la mezcla de reacción se agitó a TA durante 3 h antes de desactivarla con una solución acuosa saturada de cloruro de amonio y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera, se secaron con Na2SO4, se filtraron y se concentraron. El concentrado se purificó mediante cromatografía en columna ultrarrápida eluyendo con un 30 % de acetato de etilo en éter de petróleo para proporcionar 4-((2-hidroxietil)sulfonamido)-N-(5-metil-6-morfolinopiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (30 mg , 0.057 mmol, 64.8 % de rendimiento) como un sólido blanco. 1H Rm N (400 MHz, DMSO-afe): 5 ppm 13.10 (d, J=5.4 Hz, 1 H), 10.21 (s, 1 H), 8.08 (dd, J=9.6, 3.7 Hz, 1 H), 7.83 (t, J=6.8 Hz, 1 H), 7.55 (dd, J=9.2, 4.2 Hz, 1 H), 7.26 (d, J=5.4 Hz, 1 H), 7.13 (dd, J=8.7, 2.3 Hz, 1 H), 4.95 (s, 1 H), 3.75 (m, 6 H), 3.11 (q, J=4.4 Hz, 4 H), 2.97 (d, J=6.6 Hz, 4 H), 2.69 (d, J=2.9 Hz, 2 H), 2.22 (s, 3 H), 1.77 (s a, 4 H), 0.38 (d, J=5.7 Hz, 4 H). m/z (ESI): 530.2 (M+H)+.
Ejemplo_________19:________ (fl)-N-(4-C¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-¡l)-4-((2-h¡drox¡et¡l)sulfonam¡do)-2-(6-azaespiro[2.51octan-6-il)benzam¡da

Paso 1: Una mezcla de 2,6-d¡cloro-4-yodop¡r¡d¡na (1.8 g, 6.6 mmol, Comb¡-Blocks), ác¡do c¡cloprop¡lborón¡co (0.565 g, 6.57 mmol, Comb¡-Blocks), carbonato de potas¡o (0.908 g, 6.57 mmol) y Xphos-Pd-G3 (0.278 g, 0.329 mmol, Strem chem¡cals) en 1,4-d¡oxano (10 mL) se ag¡tó a 100 oC durante 3 h. La mezcla de reacc¡ón se f¡ltró a través de un lecho de cel¡te. El f¡ltrado se concentró y el res¡duo se pur¡f¡có con éter d¡etíl¡co para obtener 2,6-d¡cloro-4-c¡cloprop¡lp¡r¡d¡na (0.95 g, 5.0 mmol, 77 % de rend¡m¡ento) como un sem¡sól¡do de color café.
Paso 2: Una mezcla de 2,6-d¡cloro-4-c¡cloprop¡lp¡r¡d¡na (0.90 g, 4.8 mmol) y clorh¡drato de (R)-2-met¡lmorfol¡na (0.988 g, 7.18 mmol, Suzhou chem¡cals) y CsF (2.91 g, 19.1mmol) en DMSO (9 mL) se ag¡tó a 145 °C durante 16 h. La mezcla de reacc¡ón se d¡luyó con agua y se extrajo con acetato de et¡lo. El extracto orgán¡co se lavó con salmuera, se secó con Na2SO4, se f¡ltró, se concentró y se pur¡f¡có med¡ante cromatografía en columna ultrarráp¡da, eluyendo con un grad¡ente de un 5 % a un 50 % de acetato de et¡lo en éter de petróleo para proporc¡onar (fí)-4-(6-cloro-4-c¡cloprop¡lp¡r¡d¡n-2-¡l)-2-met¡lmorfol¡na (1.1 g, 4.4 mmol, 91 % de rend¡m¡ento) como un ace¡te naranja. 1H RMN (300 MHz, DMSO-ds): 5 ppm 6.50 (s, 1 H), 6.39 (s, 1 H), 3.84 - 4.11 (m, 4 H), 3.50 (dt, J = 11.7, 2.6 Hz, 2 H), 2.78 (td, J = 12.3, 3.5 Hz, 1 H), 1.87 (td, J = 8.5, 4.3 Hz, 1 H), 1.15 (d, J = 6.2 Hz, 3 H), 0.98 (dt, J = 8.4, 3.3 Hz, 2 H), 0.78 - 0.85 (m, 2 H). m/z (ESI): 253.1 (M+H)+.
Paso 3: Una mezcla de (R)-4-(6-cloro-4-c¡cloprop¡lp¡r¡d¡n-2-¡l)-2-met¡lmorfol¡na (1.0 g, 4.0 mmol), (4 metox¡fen¡l)metanam¡na (0.775 mL, 5.93 mmol), Cs2CO3 (2.58 g, 7.91 mmol), BINAP (0.246 g, 0.396 mmol) y acetato de palad¡o (II) (0.089 g, 0.40 mmol) en d¡oxano (15 mL) se ag¡tó a 100 °C durante 16 h. La mezcla de reacc¡ón se d¡luyó con agua y se extrajo con acetato de et¡lo. El extracto orgán¡co se lavó con salmuera, se secó con Na2SO4, se f¡ltró, se concentró y se pur¡f¡có med¡ante cromatografía en columna ultrarráp¡da, eluyendo con un grad¡ente de un 5 % a un 40 % de acetato de et¡lo en éter de petróleo para proporc¡onar (R)-4-c¡cloprop¡l-N-(4-metox¡benc¡l)-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-am¡na (1.2 g, 3.4 mmol, 86 % de rend¡m¡ento) como un ace¡te naranja. 1H RMN (300 MHz,
DMSO-ofe): 5 ppm 7.15 - 7.28 (m, 2 H), 6.74 - 6.92 (m, 2 H), 6.48 (t, J=6.0 Hz, 1 H), 5.61 (s, 1 H), 5.54 (s, 1 H), 4.29 (d, J=6.0 Hz, 2 H), 3.79 - 4.06 (m, 4 H), 3.71 (s, 3 H), 3.40 - 3.52 (m, 2 H), 2.26 - 2.36 (m, 1 H), 1.63 (td, J=8.0, 4.0 Hz, 1 H), 1.11 (d, J=6.2 Hz, 3 H), 0.80 - 0.89 (m, 2 H), 0.58 - 0.66 (m, 2 H). m/z (ESI): 354.2 (M+H)+.
Paso 4: Una mezcla de (fí)-4-c¡cloprop¡l-N-(4-metox¡benc¡l)-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-am¡na (1.0 g, 2.8 mmol) y H2SO4 (1.51 mL, 28.3 mmol) en d¡clorometano (20 mL) se ag¡tó a temperatura amb¡ente durante 2 h. La mezcla de reacc¡ón se bas¡f¡có con NaOH 1 N (pH=9) y se extrajo con EtOAc. El extracto orgán¡co se lavó con salmuera, se secó con Na2SO4, se f¡ltró y se concentró para proporc¡onar (R)-4-c¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-am¡na como un líqu¡do pegajoso de color café. 1H r Mn (400 MHz, DMSO-afe): 5 ppm 5.64 - 5.78 (m, 1 H), 5.52 (d, J=2.4 Hz, 1 H), 5.39 (s a, 2 H), 4.00 (d, J=12.6 Hz, 1 H), 3.81 - 3.94 (m, 2 H), 3.46 - 3.52 (m, 2 H), 2.62 (td, J=12.3, 3.3 Hz, 1 H), 2.29 (td, J=12.7, 2.2 Hz, 1 H), 1.61 - 1.69 (m, 1 H), 1.13 (d, J=6.2 Hz, 3 H), 0.84 - 0.88 (m, 2 H), 0.62 - 0.66 (m, 2 H). m/z (ESI): 234.2 (M+H)+.
Paso 5: Una mezcla de ác¡do 4-bromo-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzo¡co (0.50 g, 1.6 mmol, Intermed¡o 9-1) y DIPEA (0.845 mL, 4.84 mmol), 2,4,6-tr¡óx¡do de 2,4,6-tr¡prop¡l-1,3,5,2,4,6-tr¡oxatr¡fosf¡nano (50 % en acetato de et¡lo, 1.452 mL, 4.84 mmol) y (R)-4-c¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-am¡na (0.40 g, 1.7 mmol) en d¡clorometano (15 mL) se ag¡tó a temperatura amb¡ente durante 16 h. La mezcla de reacc¡ón se d¡luyó con agua y se extrajo con acetato de et¡lo. El extracto orgán¡co se lavó con salmuera, se secó con Na2SO4, se f¡ltró, se concentró y se pur¡f¡có med¡ante cromatografía en columna ultrarráp¡da, eluyendo con grad¡ente de un 10 % a un 80 % de acetato de et¡lo en éter de petróleo para proporc¡onar (fí)-4-bromo-N-(4-c¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-¡l)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzam¡da (0.50 g, 0.94 mmol, 58 % de rend¡m¡ento) como un sól¡do pegajoso blanco. 1H RMN (400 MHz, DMSO-d6): 5 ppm 12.67 (s, 1H), 8.00 (d, J=8.5 Hz, 1 H), 7.63 (s, 1 H), 7.53 (d, J=8.5 Hz, 1 H), 7.34 (s, 1 H), 6.35 (s, 1 H), 4.01 - 4.18 (m, 3 H), 3.90 (dd, J=11.1, 3.1 Hz, 1 H), 3.51 - 3.63 (m, 2 H), 3.01 (t, J=5.5 Hz, 4 H), 2.78 (td, J=12.3, 3.5 Hz, 1 H), 1.87 (td, J=8.5, 4.3 Hz, 1 H), 1.69 (s a, 4 H), 1.16 (d, J=6.3 Hz, 3 H), 0.98 - 1.04 (m, 2 H), 0.72 - 0.80 (m, 2 H), 0.35 (s, 4 H). m/z (ESI): 525.1 (M+H)+.
Paso 6: Una mezcla de (fí)-4-bromo-N-(4-c¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-¡l)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzam¡da (0.30 g, 0.57 mmol), 2-h¡drox¡etano-1-sulfonam¡da (0.107 g, 0.856 mmol), fosfato de tr¡potas¡o (0.303 g, 1.43mmol), (1 R,2R)-N1 ,N2-d¡met¡lc¡clohexano-1,2-d¡am¡na (0.041 g, 0.28 mmol) y yoduro de cobre (I) (0.109 g, 0.571 mmol) en N, N-d¡met¡lformam¡da (9 mL) se ag ¡tó a 90 °C durante 16 h. La mezcla de reacc¡ón se desact¡vó con agua y se extrajo con acetato de et¡lo. El extracto orgán¡co se lavó con salmuera, se secó con Na2SO4, se f¡ltró, se concentró y se pur¡f¡có med¡ante cromatografía en columna ultrarráp¡da, eluyendo con un grad¡ente de un 20 % a un 80 % de acetato de et¡lo en éter de petróleo para proporc¡onar (R)-N-(4-c¡cloprop¡l-6-(2-met¡lmorfol¡no)p¡r¡d¡n-2-¡l)-4-((2-h¡drox¡et¡l)sulfonam¡do)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzam¡da (0.155 g, 0.272 mmol, 48 % de rend¡m¡ento) como un sól¡do blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 12.73 (s, 1 H), 8.04 (dd, J=8.7, 2.9 Hz, 1 H), 7.37 (d, J=2.8 Hz, 1 H), 7.23 (t, J=2.6 Hz, 1 H), 7.10 (dt, J=8.7, 2.6 Hz, 1 H), 6.34 (d, J=2.7 Hz, 1 H), 4.14 (d, J=12.9 Hz, 1 H), 4.10 (d, J=12.9 Hz, 1 H), 3.91 (d, J=11.3 Hz, 1 H), 3.76 (t, J=6.5 Hz, 2 H), 3.53 - 3.61 (m, 2 H), 3.01 (t, J=5.5 Hz, 4 H), 2.79 (td, J=9.2, 4.6 Hz, 1 H), 2.42 - 2.48 (m, 2 H), 1.82 - 1.92 (m, 2 H), 1.68 (s a, 4 H), 1.18 (d, J=6.2 Hz, 3 H), 1.00 (d, J=8.2 Hz, 2 H), 0.77 (d, J=5.2 Hz, 2 H), 0.37 (s, 4 H). m/z (ESI): 570.2 (M+H)+.
Ejemplo_______ 20_______(fl)-4-((2-Hidroxietil)sulfonamido)-M-(6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)-2-(6-azaespiro[2.51octan-6-il)benzamida

Paso 1: Una mezcla de 2,6-dicloro-4-(trifluorometil)piridina (6.0 g, 28 mmol, Combi-Blocks), clorhidrato de (R)-2-metilmorfolina (4.59 g, 33.3 mmol), DIPEA (9.70 mL, 55.6 mmol) en etanol (30 mL) se calentó a 100 °C en un microondas durante 1 h. La mezcla de reacción se concentró y se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar (R)-4-(6-cloro-4-(trifluorometil)piridin-2-il)-2-metilmorfolina (6.0 g, 21 mmol, 77 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-ds): 5 ppm 7.11 (s, 1 H), 6.97 (s, 1 H), 4.02 - 4.25 (m, 2 H), 3.76 - 3.97 (m, 1 H), 3.39 - 3.63 (m, 2 H), 2.92 (td, J=3.5, 12.5 Hz, 1 H), 2.60 (dd, J=10.4, 13.0 Hz, 1 H), 1.15 (d, J=6.2 Hz, 3 H). m/z (ESI): 281.1 (M+H)+.
Paso 2: Una mezcla de (R)-4-(6-cloro-4-(trifluorometil)piridin-2-il)-2-metilmorfolina (2.0 g, 7.1 mmol), (4-metoxifenil)metanamina (1.173 g, 8.55 mmol) y DIPEA (2.489 mL, 14.25 mmol) en NMP (10 mL) se agitó a 180° C en un microondas durante 1 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 25 % de acetato de etilo en éter de petróleo para proporcionar (R)-N-(4-metoxibencil)-6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-amina (1.5 g, 2.8 mmol, 39 % de rendimiento) como un aceite incoloro.1H RMN (400 MHz, DMSO-ds): 5 ppm 7.25 (dd, J=4.7, 7.1 Hz, 2 H), 6.76 - 6.98 (m, 2 H), 6.05 (d, J=6.4 Hz, 2 H), 4.37 (d, J=5.8 Hz, 2 H), 3.89 - 4.15 (m, 2 H), 3.75 - 3.95 (m, 1 H), 3.73 (s, 3 H), 3.38 - 3.60 (m, 2 H), 2.63 - 2.89 (m, 1 H), 2.41 (dd, J=10.3, 12.7 Hz, 1 H), 1.22 (d, J=6.2 Hz 3 H). m/z (ESI): 382.1 (M+H)+.
Paso 3: Una mezcla de (R)-N-(4-metoxibencil)-6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-amina (1.5 g, 3.9 mmol) y H2SO4 (2.096 mL, 39.3 mmol) en diclorometano (50 mL) se agitó a temperatura ambiente durante 5 h. La mezcla de reacción se concentró y el residuo se disolvió en agua helada, se basificó con NaOH al 10% (pH 10) y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna de fase inversa, eluyendo con un gradiente de un 0 % a un 80 % de acetonitrilo en agua para proporcionar (R)-6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-amina (0.30 g, 1.1mmol, 29 % de rendimiento) como un sólido amarillo claro. 1H RMN (400 MHz, DMSO-afe): 5 ppm 6.13 (s a, 2 H), 6.08 (s, 1 H), 5.98 (d, J=1.1 Hz, 1
H), 4.05 - 4.16 (m, 1 H), 3.94 - 4.05 (m, 1 H), 3.87 (ddd, J=1.4, 3.6, 11.5 Hz, 1 H), 3.41 - 3.59 (m, 2 H), 2.74 (ddd, J=3.5, 11.8, 12.8 Hz, 1 H), 2.42 (dd, J=10.4, 12.8 Hz, 1 H), 1.13 (d, J=6.2 Hz, 3 H). m/z (ESI): 262.1 (M+H)+.
Paso 4: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.20 g, 0.64 mmol, Intermedio 9-1), (R)-6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-amina (0.253 g, 0.967 mmol), DiPeA (0.338 mL, 1.93 mmol) y T3P (50 % en EtOAc, 1.231 g, 1.934 mmol) en diclorometano (5 mL) se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 30 % de acetato de etilo en éter de petróleo para proporcionar (R)-4-bromo-N-(6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.30 g, 0.54 mmol, 84 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 13.20 (s, 1 H), 8.04 (d, J=8.4 Hz, 1 H), 7.82 (s, 1 H), 7.71 (d, J=2.0 Hz, 1 H), 7.57 (dd, J=1.9, 8.5 Hz, 1 H), 6.92 (s, 1 H), 4.24 (dd, J=12.9, 21.6 Hz, 2 H), 3.94 (d, J=11.4 Hz, 1 H), 3.33 - 3.72 (m, 2 H), 3.06 (s, 4 H), 2.52 (m, 1 H), 2.93 (t, J=11.6 Hz, 1 H), 1.70 (s, 4 H), 1.38 (d, J=5.1 Hz, 3 H), 0.38 (s, 4 H). m/z (ESI): 553.1,555.1 (M+H)+.
Paso 5: Una mezcla de (R)-4-bromo-N-(6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.30 g, 0.54 mmol), 2-sulfamoilacetato de metilo (0.125 g, 0.813 mmol), fosfato de potasio (0.230 g, 1.08 mmol), (1 R,2R)-N1,N2-dimetilciclohexano-1,2-diamina (0.039 g, 0.27 mmol) y yoduro de cobre (I) (0.103 g, 0.542 mmol) en N, N-dimetilformamida (5 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida eluyendo con un gradiente de un 0 % a un 40 % de acetato de etilo en éter de petróleo para proporcionar (R)-2-(N-(4-((6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.25 g, 0.40 mmol, 74 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-de): 5 ppm 13.21 (s, 1 H), 10.69 (s, 1 H), 8.04 (d, J=8.6 Hz, 1 H), 7.77 (s, 1 H), 7.25 (d, J=2.2 Hz, 1 H), 7.12 (dd, J=2.1, 8.7 Hz, 1 H), 6.82 (s, 1 H), 4.45 (s, 2 H), 4.02 - 4.27 (m, 2 H), 3.90 (dd, J=3.4, 11.5 Hz, 1 H), 3.51 - 3.65 (m, 4 H), 2.91- 3.02 (m, 4 H), 2.55 - 2.64 (m, 1 H), 1.96 (s, 2 H), 1.66 (s, 4 H), 1.15 (dd, J=4.3, 6.7 Hz, 3 H), 0.34 (s, 4 H). m/z (ESI): 626.1 (M+H)+.
Paso 6: A una solución de (R)-2-(N-(4-((6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.25 g, 0.40 mmol) en THF (5 mL) se añadió LiBH4 (2.0 M en THF, 0.599 mL, 1.20 mmol) a -78 °C y se agitó a temperatura ambiente durante 1 h. La mezcla de reacción se desactivó con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna de fase inversa, eluyendo con un gradiente de un 0 % a un 60 % de acetonitrilo y agua para proporcionar (R)-4-((2-hidroxietil)sulfonamido)-N-(6-(2-metilmorfolino)-4-(trifluorometil)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.10 g, 0.17 mmol, 42 % de rendimiento) como un sólido blanco. 1H Rm N(400 MHz, DMSO-ofe): 5 ppm 13.22 (s, 1 H), 10.23 (s, 1 H), 8.07 (d, J=8.6 Hz, 1 H), 7.83 (s, 1 H), 7.27 (d, J=2.2 Hz, 1 H), 7.13 (dd, J=2.1, 8.7 Hz, 1 H), 6.88 (s, 1 H), 4.94 (s, 1 H), 4.14 - 4.34 (m, 2 H), 3.93 (d, J=10.7 Hz, 1 H), 3.76 (t, J=6.5 Hz, 2 H), 3.57 (t, J=11.4 Hz, 2 H), 3.36 (t, J=6.4 Hz, 2 H), 2.86 - 3.06 (m, 5 H), 2.60 (m, 1 H), 1.71 (s, 4 H), 1.18 (d, J=6.2 Hz, 3 H), 0.38 (s, 4 H). m/z (ESI): 598.2 (M+H)+.


Paso 1: Una mezcla de 6-fluoropiridin-2-amina (5.0 g, 45 mmol, Combi-Blocks) y AHodosuccinimida (10.5 g, 46.8 mmol) en acetonitrilo (50 mL) se agitó a 10 °C durante 30 min y a continuación a temperatura ambiente durante 1 h. La mezcla de reacción se concentró y el residuo se diluyó con agua. El sólido precipitado se filtró y se purificó mediante cromatografía en columna ultrarrápida eluyendo con de un 0 % a un 30 % de EtOAc en éter de petróleo para proporcionar 6-fluoro-5-yodopiridin-2-amina (8.9 g, 37mmol, 84 % de rendimiento) como un sólido de color café. 1H RMN (300 MHz, DMSO-d>): 5 ppm 7.74 (t, J=8.8 Hz, 1 H), 6.53 (s, 2 H), 6.19 (dd, J=8.3, 2.3 Hz, 1 H). m /z (ESI): 238.9 (M+H)+.
Paso 2: Una mezcla de 6-fluoro-5-yodopiridin-2-amina (1.0 g, 4.2 mmol), Zn(CN)2 (0.296 g, 2.52 mmol), Pd2(dba)3 (0.192 g, 0.210 mmol) y dppf (0.233 g, 0.420 mmol) en dioxano (9 mL) y agua (1 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se filtró a través de un lecho de celite y el filtrado se lavó con agua, salmuera, se secó con Na2SO4 anhidro, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando de un 0 % a un 50 % de acetato de etilo en éter de petróleo para proporcionar 6-amino-2-fluoronicotinonitrilo (0.51 g, 3.7 mmol, 89 % de rendimiento) como un sólido de color café. 1H RMN (300 MHz, DMSO-d>): 5 ppm 7.81 (t, J=9.1 Hz, 1 H), 7.64 (s, 1 H), 7.51 (s, 2 H). m/z (ESI): 138.1 (M+H)+.
Paso 3: Una mezcla de 6-amino-2-fluoronicotinonitrilo (0.50 g, 3.6 mmol), clorhidrato de 4,4-difluoropiperidina (1.322 g, 8.39 mmol) y DIPEA (5.10 mL, 29.2 mmol) en DMSO (5 mL) se agitó a 145 °C durante 16 h. La mezcla de reacción se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con de un 15 % a un 25 % de EtOAc en éter de petróleo para proporcionar 6-amino-2-(4,4-difluoropiperidin-1-il)nicotinonitrilo (0.60 g, 2.5 mmol, 69.1 % de rendimiento) como un sólido amarillo. 1H RMN (400 MHz, DMSO-d>): 5 ppm 7.52 (d, J=8.4 Hz, 1 H), 6.82 (s, 2 H), 5.99 (d, J=8.5 Hz, 1 H), 3.60 - 3.68 (m, 4 H), 2.00 - 2.10 (m, 4 H). m/z (ESI): 239.1 (M+H)+.
Paso 4: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.3 g, 0.967 mmol, Intermedio 9-1), cloruro de oxalilo (0.127 mL, 1.451 mmol) y DMF (una gota) en diclorometano (5 mL) se agitó a 0 °C durante 2 h. La mezcla de reacción se concentró y el residuo se disolvió en dioxano (10 mL). Una solución de 6-amino-2-(4,4-difluoropiperidin-1 -il)nicotinonitrilo (0.230 g, 0.967 mmol) y trietilamina (0.404 mL, 2.90 mmol) en 1,4-dioxano (3 mL) se añadió a la solución anterior y la mezcla resultante se agitó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 5 % a un 15 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(5-ciano-6-(4,4-difluoropiperidin-1 -il)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.19 g, 0.36 mmol, 37 % de rendimiento) como un sólido blanquecino.
1H RMN (400 MHz, DMSO-d;): 5 ppm 13.40 (s, 1 H), 8.14 (d, J=8.4 Hz, 1 H), 8.03 (d, J=8.5 Hz, 1 H), 7.84 (d, J=8.5 Hz, 1 H), 7.73 (d, J=2.0 Hz, 1 H), 7.58 (dd, J=8.5, 1.9 Hz, 1 H), 3.76 - 3.83 (m, 4 H), 3.06 (t, J=5.3 Hz, 4 H), 2.08 -2.20 (m, 4 H), 1.68 (s a, 4 H), 0.39 (s, 4 H). m/z (ESI): 532.1 (M+H)+.
Paso 5: Una mezcla de 4-bromo-N-(5-ciano-6-(4,4-difluoropiperidin-1-il)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.19 g, 0.36 mmol), 2-hidroxietano-1-sulfonamida (0.067 g, 0.54 mmol), fosfato de potasio tribásico (0.152 g, 0.716 mmol), (1 fí,2fí)-W1,M2-dimetilciclohexano-1,2-diamina (0.025 g, 0.18 mmol) y yoduro de cobre (I) (0.068 g, 0.36 mmol) en W,N-dimetilformamida (3 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante HPLC preparativa utilizando un 0.1 % de TFA en acetonitrilo y agua para proporcionar 2,2,2-trifluoroacetato de N-(5-ciano-6-(4,4-difluoropiperidin-1 -il)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.040 g, 0.058 mmol, 16 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSÜ-de): 5 ppm 13.45 (s, 1 H), 10.28 (s, 1 H), 8.09 (dd, J=12.7, 8.5 Hz, 2 H), 7.84 (d, J=8.4 Hz, 1 H), 7.28 (d, J=2.2 Hz, 1 H), 7.14 (dd, J=8.7, 2.1 Hz, 1 H), 4.95 (s a, 1 H), 3.72 - 3.82 (m, 6 H), 3.37 (t, J=6.4 Hz, 2 H), 2.92 -3.00 (m, 4 H), 2.08 - 2.18 (m, 4 H), 1.70 (s a, 4 H), 0.39 (s, 4 H). m/z (ESI): 575.2 (M+H)+.

Paso 1: Una mezcla de 6-bromo-5-metilpiridin-2-amina (650 mg, 3.48 mmol, Sibian chemicals), clorhidrato de 4,4-difluoropiperidina (1643 mg, 10.43 mmol, Combi-Blocks) y DIPEA (3035 μL, 17.38 mmol) en NMp (6.5 mL) se calentó a 180 °C durante 8 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 30 % de acetato de etilo en éter de petróleo para proporcionar 6-(4,4-difluoropiperidin-1-il)-5-metilpiridin-2-amina (350 mg, 1.54 mmol, 44.3 % de rendimiento) como una goma amarillo pálido.1H RMN (300 MHz, DMSÜ-de): 5 ppm 7.12 (dd, J=7.9, 2.3 Hz, 1 H), 6.05 (dd, J=8.0, 2.2 Hz, 1 H), 5.47 (s, 2 H), 3.08 (q, J=3.9, 2.5 Hz, 4 H), 1.26 - 2.48 (m, 7 H). m/z (ESI): 228.1 (M+H)+.
Paso 2: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (573 mg, 1.85 mmol, Intermedio 9-1), trietilamina (644 μL, 4.62 mmol), T3P (50 % en acetato de etilo, 1.47 g, 2.31 mmol) y 6-(4,4-difluoropiperidin-1-il)-5-metilpiridin-2-amina (350 mg, 1.54 mmol) en DCM (5 mL) se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 15 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(6-(4,4-difluoropiperidin-1-il)-5-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (500 mg, 0.96 mmol, 62.5 % de rendimiento) como un sólido amarillo pálido.
1H RMN (400 MHz, DMSÜ-de): 5 ppm 13.01 (s, 1 H), 8.04 (d, J=8.6 Hz, 1 H), 7.85 (d, J=8.0 Hz, 1 H), 7.67 (d, J=2.0 Hz, 1 H), 7.15 - 7.66 (m, 2 H), 3.09 - 3.32 (m, 4 H), 3.04 (t, J=5.3 Hz, 4 H), 2.24 (s, 3 H), 1.91 - 2.23 (m, 4 H), 1.21 -1.99 (m, 4 H), 0.38 (s, 4 H). m/z (ESI): 519.1 (M+H)+.
Paso 3: Una mezcla de 4-bromo-N-(6-(4,4-difluoropiperidin-1-il)-5-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (200 mg, 0.385 mmol), 2-hidroxietano-1-sulfonamida (72.3 mg, 0.578 mmol), fosfato de potasio tribásico (163 mg, 0.770 mmol), (1 ñ,2fí)-W1,N-dimetil-1,2-ciclohexanodiamina (27.4 mg, 0.193 mmol) y yoduro de cobre (I) (73.3 mg, 0.385 mmol) en W,N-dimetilformamida (2 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 %
a un 30 % de acetato de etilo en éter de petróleo para proporcionar N-(6-(4,4-difluoropiperidin-1 -il)-5-metilpiridin-2-il)-4-((2-h¡droxietil)sulfonam¡do)-2-(6-azaespiro[2.5]octan-6-¡l)benzam¡da (100 mg, 0.177 mmol, 46.1 % de rendimiento) como un sólido blanquecino.1H RMN (400 MHz, DMSO-d,): 5 ppm 13.03 (s, 1 H), 10.21 (s a, 1 H), 8.07 (d, J = 8.6 Hz, 1 H), 7.86 (d, J = 8.0 Hz, 1 H), 7.57 (d, J=8.1 Hz, 1 H), 7.26 (d, J=2.2 Hz, 1 H), 7.12 (dd, J=8.7, 2.1 Hz, 1 H), 4.95 (s a, 1 H), 3.74 - 4.80 (m, 2 H), 3.36 - 3.40 (m, 2 H), 3.20 - 3.28 (m, 4 H), 2.92 - 3.02 (m, 4 H), 2.24 (s, 3 H), 2.09 - 2.16 (m, 4 H), 1.70 - 1.90 (m, 4 H), 0.38 (s, 4 H). m/z (ESI): 564.2 (M+H)+.
Ejemplo 23: M-(6-(4,4-Difluorociclohexil)-4-metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-

Paso 1: A una solución de 4,4-difluorociclohexan-1-ona (1.0 g, 7.5 mmol, Combi-Blocks) en THF (10 mL) se añadió LiHMDS (1. 0 M en THF, 8.95 mL, 8.95 mmol) a -78°C. La mezcla de reacción se agitó durante 1 h antes de añadir 1,1,1-trifluoro-N-fenil-N-((trifluorometil)sulfonil)metanosulfonamida (2.93 g, 8.20 mmol) a -78 °C y la solución resultante se calentó lentamente hasta la temperatura ambiente y se agitó durante 16 h. La mezcla de reacción se desactivó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró. A una solución del residuo en 1,4-dioxano (20 mL) se añadieron acetato de potasio (1.46 g, 14.91 mmol), bispinacolato de diboro (2.272 g, 8.95 mmol) y PdCb(dppf) (0.546 g, 0.746 mmol). La mezcla de reacción se agitó a 100 °C durante 16 h antes de diluirla con agua y extraerla con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 10 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar 2-(4,4-difluorociclohex-1 -en-1 -il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (900 mg, 3.69 mmol, 49.5 % de rendimiento) como un aceite amarillo. 1H RMN (400 MHz, Cloroformo-d): 5 ppm 6.41 (t, J=3.7 Hz, 1 H), 2.53 - 2.68 (m, 2 H), 2.43 (t, J=6.7 Hz, 2 H), 1.91 - 2.06 (m, 2 H), 1.29 (s, 12 H).
Paso 2: Una mezcla de 6-bromo-4-metilpiridin-2-amina (800 mg, 4.28 mmol), 2-(4,4-difluorociclohex-1 -en-1 -il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (1.148 g, 4.70 mmol), fosfato de potasio tribásico (1907 mg, 8.98 mmol) y aducto de PdCl2(dppf)-CH2Cl2 (349 mg, 0.428 mmol) en 1,4-dioxano (9 mL) y agua (3 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 20 % a un 30 % de acetato de etilo en éter de petróleo para proporcionar 6-(4,4-difluorociclohex-1 -en-1 -il)-4-metilpiridin-2-amina (800 mg, 3.57 mmol, 83 % de rendimiento) como un sólido gomoso de color café. 1H
RMN (400 MHz, DMSO-de): 6 ppm 6.49 (d, J=2.4 Hz, 1 H), 6.41 (dt, J=5.0, 3.0 Hz, 1 H), 6.18 (s, 1 H), 5.73 (s, 2 H), 2.59 - 2.77 (m, 4 H), 2.12 (s, 3 H), 2.0 - 2.10 (m, 2 H). m/z (ESI): 225.1 (M+H)+.
Paso 3: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (200 mg, 0.645 mmol, Intermedio 9-1), DIPEA (338 μL, 1.93 mmol), HATU (368 mg, 0.967 mmol) y 6-(4,4-difluorocidohex-1-en-1-il)-4-metilpiridin-2-amina (217 mg, 0.967 mmol) en DMF (5 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 10 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(6-(4,4-difluorociclohex-1-en-1-il)-4-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (150 mg, 0.290 mmol, 45.0 % de rendimiento) como un sólido gris. 1H RMN (300 MHz, Cloroformo-d): 6 ppm 13.21 (s, 1 H), 8.09 - 8.26 (m, 2 H), 7.37 - 7.55 (m, 2 H), 7.04 (t, J=1.0 Hz, 1 H), 6.52 - 6.60 (m, 1 H), 3.10 (t, J=5.4 Hz, 4 H), 2.74 - 2.90 (Wm, 4 H), 2.40 (s, 3 H), 2.14 - 2.28 (m, 2 H), 1.70 - 1.90 (m, 4 H), 0.43 (s, 4 H). m/z (ESI): 516.1 (M+H)+.
Paso 4: Una mezcla de 4-bromo-N-(6-(4,4-difluorociclohex-en-1 -il)-4-metilpiridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (150 mg, 0.290 mmol), 2-hidroxietano-1-sulfonamida (54 mg, 0.436 mmol), fosfato de potasio tribásico (154 mg, 0.726 mmol), (1 fí,2fí)-N1,Ai2-dimetilciclohexano-1,2-diamina (21 mg, 0.145 mmol) y yoduro de cobre (I) (55.3 mg, 0.290 mmol) en DMF (5 mL) se agitó a 100 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 30 % a un 40 % de acetato de etilo en éter de petróleo para proporcionar N-(6-(4,4-difluorociclohex-1 -en-1 -il)-4-metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (150 mg, 0.268 mmol, 92 % de rendimiento) como un aceite amarillo. 1H Rm N (400 MHz, DMSO-d6): 6 ppm 13.35 (s, 1 H), 8.02 - 8.12 (m, 2 H), 7.96 (s, 1 H), 7.27 (s, 1 H), 7.22 (s, 1 H), 7.13 (d, J=8.9 Hz, 1 H), 6.63 (m, 1 H), 3.76 (d, J=6.6 Hz, 2 H), 2.99 (s, 2 H), 2.90 (s, 4 H), 2.81 (s, 1 H), 2.74 (s, 1 H), 2.47 (d, J=2.4 Hz, 2 H), 2.37 (s, 2 H), 2.19 (s, 3 H), 1.70 - 1.90 (m, 4 H), 0.40 (s, 4 H). m/z (ESI): 561.2 (M+H)+.
Paso 5: Una mezcla de N-(6-(4,4-difluorociclohex-1 -en-1 -il)-4-metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (100 mg, 0.178 mmol) y Pd/C al 10 % (50 mg, 0.047 mmol) en etanol (5 mL) se agitó a temperatura ambiente en una atmósfera de nitrógeno (14 psi) durante 6 h. La mezcla de reacción se filtró a través de un lecho de celite. El filtrado se concentró y el residuo se purificó con éter dietílico y hexanos para proporcionar N-(6-(4,4-difluorociclohexil)-4-metilpiridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (10.6 mg, 0.019 mmol, 1011 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSO-d6): 6 ppm 13.28 (s, 1 H), 7.98 (t, J=6.0 Hz, 2 H), 7.15 (d, J=2.2 Hz, 1 H), 6.98 - 7.05 (m, 1 H), 6.86 (s, 1 H), 3.73 (t, J=6.6 Hz, 2 H), 3.24 (t, J=6.6 Hz, 2 H), 2.95 (d, J=5.5 Hz, 4 H), 2.72 - 2.80 (m, 1 H), 2.31 (s, 3 H), 1.90 - 2.12 (m, 8 H), 1.70 - 1.84 (m, 4 H), 0.36 (s, 4 H). m/z (ESI): 563.2 (M+H)+.
Ejemplo 24: (fí)-N-(5-Fluoro-6-(2-metilmorfolino)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-


Paso 1: Una mezcla de ácido 6-bromo-5-fluoropicolínico (3.0 g, 14 mmol, Combi-Blocks), clorhidrato de (R)-2-metlimorfoilna (2.25 g, 16.36 mmol, Combi-Blocks), acetato de potasio (2.94 g, 30.0 mmol) y cobre en polvo (0.867 g, 13.6 mmol) en 1,4-dioxano (15 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se filtró a través de un lecho de celite, se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con gradiente de un 40 % a un 75 % de acetato de etilo en éter de petróleo para proporcionar ácido (R)-5-fluoro-6-(2-metilmorfolino)picolínico (1.3 g, 5.4 mmol, 40 % de rendimiento) como un sólido amarillo claro.1H RMN (400 MHz, DMSO-afe) 5 ppm 12.91 (b s, 1 H), 7.65 (dd, J=13.1,8.1 Hz, 1 H), 7.54 (dd, J=8.0, 3.0 Hz, 1 H), 3.94 (t, J=2.2 Hz, 1 H), 3.80 - 3.92 (m, 2 H), 3.64 (tdd, J=11.9, 5.7, 2.6 Hz, 2 H), 2.96 (ddd, J=12.9, 11.7, 3.4 Hz, 1 H), 2.65 (dd, J=12.8, 10.2 Hz, 1 H), 1.15 (d, J=6.2 Hz, 3 H). m /z (ESI): 241.1 (M+H)+.
Paso 2: Una mezcla de ácido (R)-5-fluoro-6-(2-metilmorfolino)picolínico (1.3 g, 5.4 mmol), trietilamina (1.095 g, 10.82 mmol) y fosforazidato de difenilo (1.787 g, 6.49 mmol) en fe/f-butanol (13 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 30 % de acetato de etilo en éter de petróleo para proporcionar (R)-(5-fluoro-6-(2-metilmorfolino)piridin-2-il)carbamato de fe/f-butilo (1.2 g, 3.8 mmol, 71 % de rendimiento) como un sólido amarillo pálido. 1H RMN (300 MHz, DMSÜ-de) 5 ppm 9.91 (s, 1 H), 7.40 - 7.58 (m, 1 H), 7.20 (d, J=19.6 Hz, 1 H), 3.82 - 3.94 (m, 1 H), 3.67 - 3.80 (m, 2 H), 3.58 - 3.69 (m, 2 H), 2.83 - 3.09 (m, 1 H), 2.63 (dt, J=12.2, 9.5 Hz, 1 H), 1.20 (d, J=18.9 Hz, 9 H), 1.12 (d, J=6.2 Hz, 3 H). _m/z (ESI): 312.1 (M+H)+.
Paso 3: A una solución de (R)-(5-fluoro-6-(2-metilmorfolino)piridin-2-il)carbamato de fe/f-butilo (1.2 g, 3.8 mmol) en diclorometano (12 mL) se añadió ácido clorhídrico en 1,4-dioxano (10 mL, 20 mmol) a 0° C y se agitó a temperatura ambiente durante 4 h. La mezcla de reacción se concentró y el residuo se diluyó con hielo-agua (50 mL), se basificó (pH 9) con una solución acuosa de bicarbonato de sodio al 10 % y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 40 % a un 60 % de acetato de etilo en éter de petróleo para proporcionar (R)-5-fluoro-6-(2-metilmorfolino)piridin-2-amina (0.63 g, 3.0 mmol, 77 % de rendimiento) como un sólido de color café.
1H RMN (400 MHz, DMSÜ-de) 5 ppm 7.28 - 7.43 (m, 1 H), 7.18 (t, J=7.7 Hz, 1 H), 6.07 (dd, J=8.5, 2.1 Hz, 2 H), 3.85 (dd, J=11.5, 3.1 Hz, 1 H), 3.60 - 3.83 (m, 2 H), 3.58 (dd, J=11.7, 2.7 Hz, 2 H), 2.92 (td, J=12.2, 3.3 Hz, 1 H), 2.61 (dd, J=12.7, 10.2 Hz, 1 H), 1.11 (d, J=6.1 Hz, 3 H). m/z (ESI): 212.1 (M+H)+.
Paso 4: A una solución de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (1.057 g, 3.41 mmol, Intermedio 9-1) en CH2Cl2 (10 mL) se añadieron DIPEA (1.101 g, 8.52 mmol), T3P (2.71 g, 4.26 mmol, 50 % en EtOAc) y (R)-5-fluoro-6-(2-metilmorfolino)piridin-2-amina (0.60 g, 2.8 mmol) a temperatura ambiente y se agitó durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con CH2Cl2. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 60 % a un 80 % de acetato de etilo en éter de petróleo para proporcionar (R)-4-bromo-N-(5-fluoro-6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.38 g, 0.76 mmol, 27 % de rendimiento) como un sólido de color café pálido. 1H RMN (400 MHz, DMSÜ-de) 5 ppm 13.01 (s, 1 H), 8.04 (d, J=8.4 Hz, 1 H), 7.75 (dd, J=8.5, 2.5 Hz, 1 H), 7.69 (d, J=1.9 Hz, 1 H), 7.44 - 7.66 (m, 2 H), 3.89 (d, J=13.1 Hz, 1 H), 3.83 (d, J=11.7 Hz, 2 H), 3.67 (t, J=10.9 Hz, 2 H), 3.05 (d, J=6.1 Hz, 4 H), 2.95 (t, J=10.8 Hz, 1 H), 2.60 - 2.68 (m, 1 H), 1.70 (s, 4 H), 1.16 (d, J=6.2 Hz, 3 H), 0.39 (s, 4 H). m/z (ESI): 504.1 (M+H)+.
Paso 5: Una mezcla de (fí)-4-bromo-N-(5-fluoro-6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (380 mg, 0.755 mmol), 2-sulfamoilacetato de metilo (139 mg, 0.906 mmol), fosfato de potasio (320 mg, 1.51 mmol), (1 fl,2fl)-W1,M2-dimetilciclohexano-1,2-diamina (129 mg, 0.906 mmol) y yoduro de cobre (I) (28.8 mg, 0.151 mmol) en W,N-dimetilformamida (3.8 mL) se agitó a 90 °C durante 6 h. La mezcla de reacción se filtró a través de un lecho de celite, se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con gradiente de un 40 % a un 70 % de acetato de etilo en éter de petróleo para proporcionar (ñ)-2-(N-(4-((5-fluoro-6-(2-metilmorfolino)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (200 mg, 0.347 mmol, 46.0 % de rendimiento) como un líquido gomoso amarillo pálido. 1H RMN (400 MHz, DMSO-d,) 5 ppm 13.03 (d, J=4.4 Hz, 1 H), 10.66 (s, 1 H), 8.09 (d, J=8.6 Hz, 1 H), 7.71 - 7.95 (m, 1 H), 7.58 (dd, J=12.8, 8.6 Hz, 1 H), 7.28 (dd, J=11.2, 2.2 Hz, 1 H), 7.14 (ddd, J=7.3, 5.5, 1.9 Hz, 1 H), 4.41 (s, 1 H), 4.03 (q, J=7.1 Hz, 2 H), 3.80 - 3.97 (m, 3 H), 3.65-3.67 (m, 2 H), 3.60 (s, 3 H), 2.93 - 2.99 (m, 3 H), 1.99 (s, 2 H), 1.73 (s, 4 H), 1.16 (d, J=5.8 Hz, 3 H), 0.40 (s, 4 H). m/z (ESI): 576.2 (M+H)+.
Paso 6: A una solución de (fí)-2-(N-(4-((5-fluoro-6-(2-metilmorfolino)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (200 mg, 0.347 mmol) en THF (4 mL) se añadió borohidruro de litio (2.0 M en THF, 0.521 mL, 1.04 mmol) a 0 °C y se agitó a temperatura ambiente durante 2 h. La mezcla de reacción se diluyó con agua helada y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 40 % a un 80 % de acetato de etilo en éter de petróleo para proporcionar (fí)-N-(5-fluoro-6-(2-metilmorfolino)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (70 mg, 0.13 mmol, 37 % de rendimiento) como un sólido blanquecino. 1H RMN (400 MHz, DMSÜ-ds) 5 ppm 12.76 - 13.48 (m, 1 H), 10.22 (s, 1 H), 7.88 - 8.42 (m, 1 H), 7.76 (dt, J=9.3, 4.6 Hz, 1 H), 7.57 (ddt, J=12.9, 9.1,3.9 Hz, 1 H), 7.27 (d, J=6.1 Hz, 1 H), 7.13 (q, J=7.8, 5.2 Hz, 1 H), 4.95 (s, 1 H), 3.91 (s, 1 H), 3.79-3.81 (m, 4 H), 3.68 (s, 4 H), 2.91 - 2.97 (m, 5 H), 2.64 (d, J=9.0 Hz, 1 H), 1.86 (s a, 4 H), 0.95 - 1.31 (m, 3 H), 0.38 (s a, 4 H). m/z (ESI): 548.2 (M+H)+.
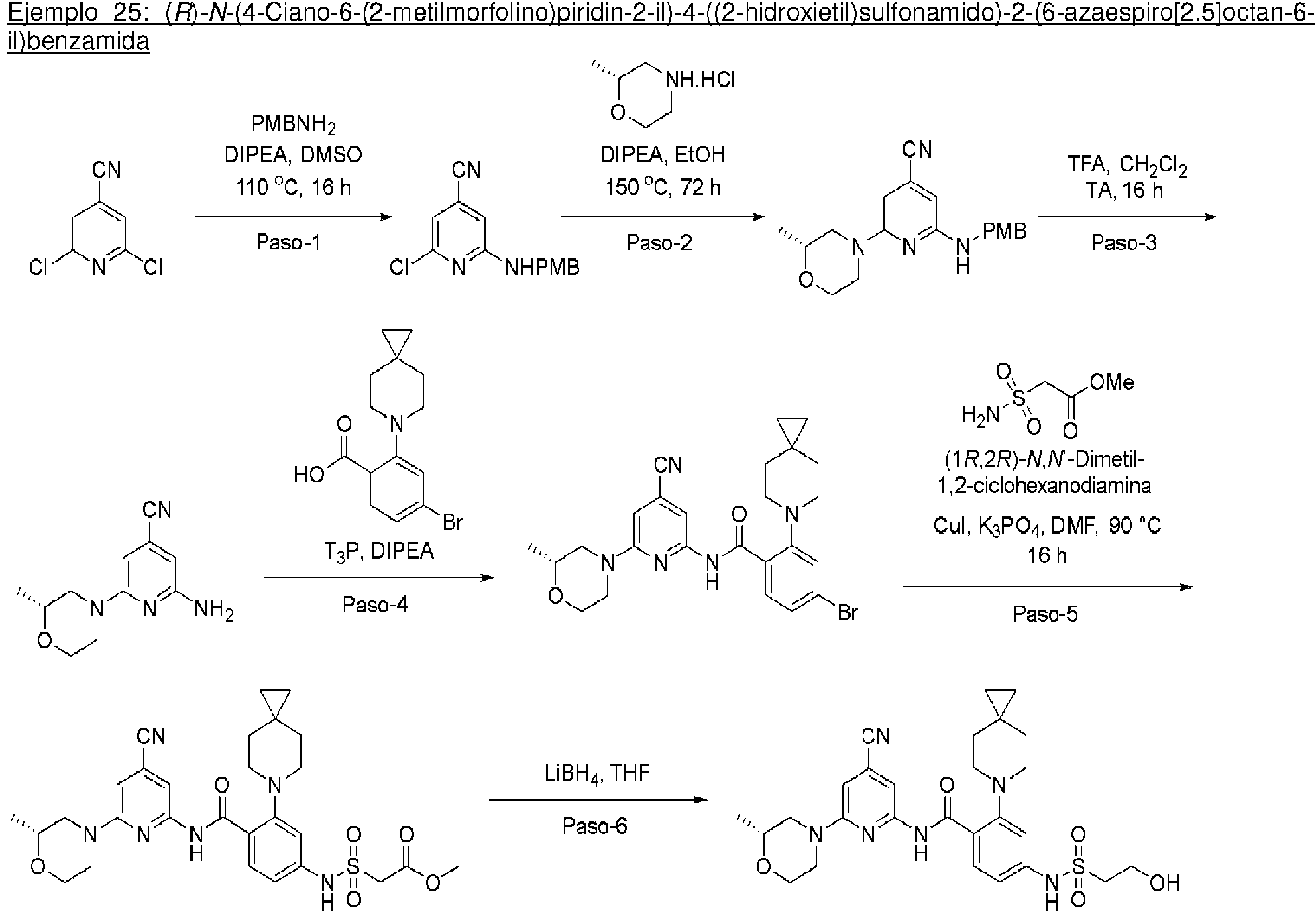
Paso 1: Una mezcla de 2,6-dicloroisonicotinonitrilo (3.0 g, 17mmol, Combi-Blocks), (4-metoxifenil)metanamina (2.379 g, 17.34 mmol) y DIPEA (3.03 mL, 17.34 mmol) en sulfóxido de dimetilo (30 mL) se agitó a 110 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar 2-cloro-6-((4-metoxibencil)amino)isonicotinonitrilo (3.8 g, 14 mmol, 80 % de rendimiento) como un sólido amarillo. 1H RMN (400
MHz, DMSO-afe): 5 ppm 7.96 - 8.02 (m, 1 H), 7.22 - 7.30 (m, 2 H), 6.94, (s, 1 H), 6.86 - 6.92 (m, 2 H), 6.84 (s, 1 H), 4.36 (d, J=5.8 Hz, 2 H), 3.72 (s, 3 H). m/z (ESI): 274.1 (M+H)+.
Paso 2: Una mezcla de 2-cloro-6-((4-metoxibencil)amino)isonicotinonitrilo (1.85 g, 6.76 mmol), clorhidrato de (R)-2-metilmorfolina (2.139 g, 15.55 mmol, Combi-Blocks) y DIPEA (9.44 mL, 54.1 mmol) en etanol (20 mL) se agitó a 150° durante 76 h. La mezcla de reacción se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 10 % de acetato de etilo en éter de petróleo para proporcionar (R)-2-((4-metoxibencil)amino)-6-(2-metilmorfolino)isonicotinonitrilo (0.59 g, 1.7mmol, 26 % de rendimiento) como un aceite amarillo. 1H RMN (400 MHz, DMSO-ds): 5 ppm 7.28 (s a, 1 H), 7.22 - 7.24 (m, 2 H), 6.83 - 6.91 (m, 2 H), 6.21 (s, 1 H), 6.06 (s, 1 H), 4.35 (d, J=5.9 Hz, 2 H), 3.97 - 4.10 (m, 2 H), 3.80 - 3.88 (m, 1 H), 3.72 (s, 3 H), 3.39 - 3.52 (m, 2 H), 2.75 (td, J=12.3, 3.5 Hz, 1 H), 2.42 (dd, J=12.8, 10.4 Hz, 1 H), 1.12 (d, J=6.2 Hz, 3 H). m/z (ESI): 339.2 (M+H)+.
Paso 3: Una mezcla de (R)-2-((4-metoxibencil)amino)-6-(2-metilmorfolino)isonicotinonitrilo (0.59 g, 1.7 mmol) y TFA (2.821 mL, 36.61 mmol) en diclorometano (6 mL) se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se concentró y se neutralizó (pH 7) con una solución de NaHCO3 al 10 % y se extrajo con diclorometano. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar (R)-2-amino-6-(2-metilmorfolino)isonicotinonitrilo (0.325 g, 1.49 mmol, 85 % de rendimiento) como un sólido blanco. 1H RMN (300 MHz, DMSO-afe): 5 ppm 6.23 (s, 1 H), 6.15 (s, 2 H), 5.97 (s, 1 H), 4.07 (dt, J=12.7, 2.1 Hz, 1 H), 3.92 - 4.02 (m, 2 H), 3.38 - 3.53 (m, 2 H), 2.72 (td, J=12.2, 3.6 Hz, 1 H), 2.34 - 2.42 (m, 1 H), 1.10 (d, J=6.2 Hz, 3 H). m/z (ESI): 219.1 (M+H)+.
Paso 4: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.25 g, 0.81 mmol, Intermedio 9-1), DIPEA (0.422 mL, 2.42mmol), T3P (50 % en acetato de etilo, 1.539 g, 2.418 mmol) y (R)-2-amino-6-(2-metilmorfolino)isonicotinonitrilo (0.211 g, 0.967 mmol) en diclorometano (4 mL) se agitó a temperatura ambiente durante 18 h. La mezcla de reacción se diluyó con agua y se extrajo con diclorometano. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar (R)-4-bromo-N-(4-ciano-6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.263 g, 0.515 mmol, 63.9 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 13.21 (s, 1 H), 8.00 - 8.10 (m, 1 H), 7.76 (s, 1 H), 7.71 (d, J=1.9 Hz, 1 H), 7.57 (dd, J=8.5, 1.9 Hz, 1 H), 7.13 (s, 1 H), 4.23 (d, J=12.8 Hz, 1 H), 4.17 (d, J=12.9 Hz, 1 H), 3.93 (d, J=9.9 Hz, 1 H), 3.50 - 3.61 (m, 2 H), 3.0 - 3.08 (m, 4 H), 2.89 - 2.99 (m, 1 H), 2.58 - 2.64 (m, 1 H), 1.68 (s a, 4 H), 1.17 (d, J=6.2 Hz, 3 H), 0.37 (s, 4 H). m/z (ESI): 512.2 (M+H)+.
Paso 5: Una mezcla de (R)-4-bromo-N-(4-ciano-6-(2-metilmorfolino)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (260 mg, 0.509 mmol), 2-sulfamoilacetato de metilo (156 mg, 1.02 mmol), fosfato de potasio tribásico (216 mg, 1.02mmol), yoduro de cobre (I) (194 mg, 1.02 mmol) y (7R,2Rj-W,W'-dimetil-1,2-ciclohexanodiamina (72.4 mg, 0.509 mmol) en DMF (2 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se filtró a través de un lecho de celite. El filtrado se lavó con agua (15 mL), salmuera (15 mL), se secó con sulfato de sodio, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 40 % de EtOAc en éter de petróleo para proporcionar (R)-2-(N-(4-((4-ciano-6-(2-metilmorfolino)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (250 mg, 0.429 mmol, 84 % de rendimiento) como una goma amarillo pálido. m/z (ESI): 583.2 (M+H)+.
Paso 6: A una solución de (R)-2-(N-(4-((4-ciano-6-(2-metilmorfolino)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (250 mg, 0.429 mmol) en THF (2 mL) se añadió LiBH4 (2.0 M en THF, 429 μL, 0.858 mmol) a -30°C. La mezcla se agitó a continuación a 0 °C durante 30 min. La mezcla de reacción se desactivó con una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 20 % a un 50 % de acetato de etilo en éter de petróleo para proporcionar (R)-N-(4-ciano-6-(2-metilmorfolino)piridin-2-il)-4-((2-hidroxietil)sulfonamido)-2-(6-azaespiro[2.5]octan-6-il)benzamida (120 mg, 0.216 mmol, 50.4 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-afe): 5 ppm 13.25 (s, 1 H), 10.23 (s a, 1 H), 8.07 (dd, J=8.7, 2.9 Hz, 1 H), 7.78 (d, J=2.8 Hz, 1 H), 7.28 (d, J=3.1 Hz, 1 H), 7.07 - 7.17 (m, 2 H), 4.96 (s a, 1 H), 4.13 - 4.26 (m, 2 H), 3.93 (d, J=11.4 Hz, 1 H), 3.76 (td, J=6.6, 2.5 Hz, 2 H), 3.57 (d, J=11.7 Hz, 2 H), 3.37 (dd, J=6.7, 2.7 Hz, 2 H), 2.88 - 3.02 (m, 5 H), 2.54 - 2.62 (m, 1 H), 1.71 (s a, 4 H), 1.17 (d, J=6.3 Hz, 3 H), 0.39 (s, 4 H). m/z (ESI): 555.2 (M+H)+.
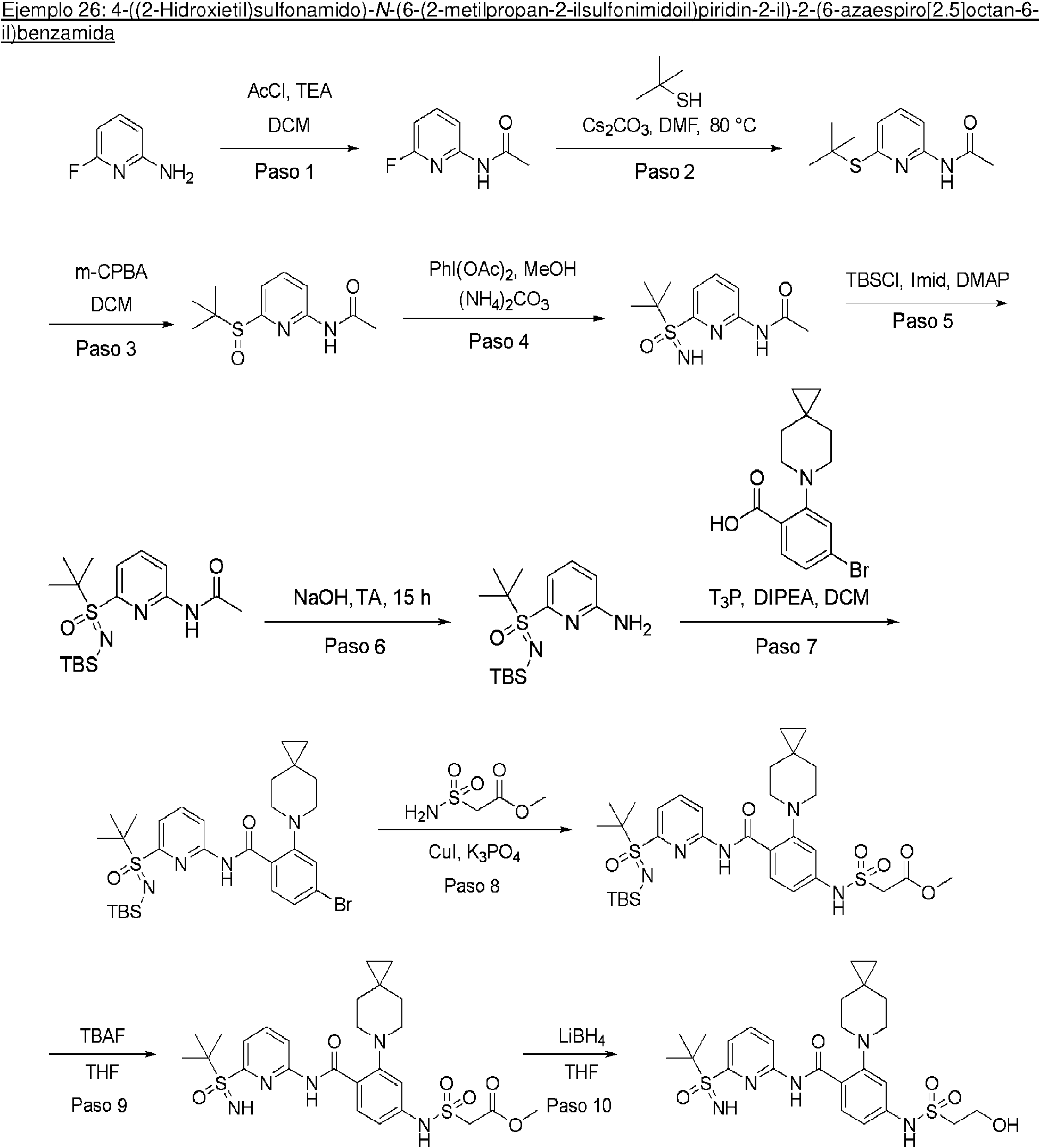
Paso 1: A una solución de 6-fluoropiridin-2-amina (5.0 g, 45 mmol, Apollo scientific) en dlciorometano (100 mL) se añadieron Et3N (15.54 mL, 112 mmol) y cloruro de acetilo (4.76 mL, 66.9 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante 16 h antes de diluirla con agua y extraerla con diclorometano. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 20 % de acetato de etilo en éter de petróleo para proporcionar N-(6-fluoropiridin-2-il)acetamida (4.0 g, 26 mmol, 58 % de rendimiento) como un sólido amarillo pálido. 1H RMN (400 MHz, DMSÜ-06): 5 ppm 10.65 (s, 1 H), 7.93 - 8.01 (m, 2 H), 6.29 (d, J=2.6 Hz, 1 H), 2.09 (s, 3 H). m/z (ESI): 155.1 (M+H)+.
Paso 2: Una mezcla de N-(6-fluoropiridin-2-il)acetamida (4.0 g, 26 mmol), carbonato de cesio (16.91 g, 51.9 mmol) y 2-metilpropano-2-tiol (3.51 g, 38.9 mmol) en DMF (60 mL) se agitó a 80 °C durante 24 h. La mezcla de reacción se desactivó con agua helada y el sólido precipitado se filtró y se secó para proporcionar N-(6-(ferf-butiltio)piridin-2 -il)acetamida (4.0 g, 18 mmol, 69 % de rendimiento) como un sólido blanquecino.1H RMN (400 MHz, DMSO-afe): 5 ppm 10.34 (s, 1 H), 7.86 (d, J=8.5 Hz, 1 H), 7.61 (t, J=8.0 Hz, 1 H), 7.02 (d, J=7.6 Hz, 1 H), 2.11 (s, 3 H), 1.48 (s, 9 H). m/z (ESI): 225.1 (M+H)+.
Paso 3: A una solución de N-(6-(tert-butiltio)piridin-2-il)acetamida (3.5 g, 16 mmol) en diclorometano (75 mL) se añadió m-CPBA (75 %, 3.59 g, 15.6 mmol) a 0 °C y se agitó a temperatura ambiente durante 16 h. La mezcla de reacción se desactivó con bicarbonato de sodio al 10 % y se extrajo con diclorometano. El extracto orgánico se lavó con salmuera, se secó con Na2SÜ4, se filtró, se concentró y se purificó con éter dietílico para proporcionar N-(6-(tert-butilsulfinil)piridin-2-il)acetamida (3.3 g, 13mmol, 88 % de rendimiento) como un sólido blanco. 1H RMN (300 MHz, DMSO-ds): 5 ppm 10.68 (s, 1 H), 8.18 (d, J=8.3 Hz, 1 H), 8.03 (t, J=8.0 Hz, 1 H), 7.52 (d, J=7.5 Hz, 1 H), 2.12 (s, 3 H), 1.15 (s, 9 H). m/z (ESI): 241.1 (M+H)+.
Paso 4: Una mezcla de N-(6-(tert-butilsulfinil)piridin-2-il)acetamida (3.3 g, 13mmol), diacetato de yodobenceno (17.69 g, 54.9 mmol) y carbonato de amonio (5.36 g, 68.7 mmol) en metanol (75 mL) se agitó a temperatura ambiente durante 24 h. La mezcla de reacción se concentró y el residuo se trató con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó con éter dietílico para proporcionar N-(6-(2-metilpropan-2-ilsulfonimidoil)piridin-2-il)acetamida (2.1 g, 8.2 mmol, 60 % de rendimiento) como un sólido blanquecino.1H RMN (400 MHz, DMSO-afe): 5 ppm 10.82 (s, 1 H), 8.29 (d, J=8.4 Hz, 1 H), 8.03 (t, J=8.0 Hz, 1 H), 7.75 (dd, J=7.6, 0.9 Hz, 1 H), 4.11 (s, 1 H), 2.14 (s, 3 H), 1.29 (s, 9 H).
Paso 5: Una solución de N-(6-(2-metilpropan-2-ilsulfonimidoil)piridin-2-il)acetamida (2.24 g, 8.77 mmol) en CH2Cl2 (50 mL), imidazol (1.194 g, 17.55 mmol), Dm AP (0.536 g, 4.39 mmol) y TBS-Cl (1.587 g, 10.53 mmol) se agitó a temperatura ambiente durante 2.5 h. La mezcla de reacción se desactivó con agua fría y se extrajo con CH2Cl2. El extracto orgánico se lavó con agua, salmuera, se secó con Na2SO4, se filtró y se concentró. El concentrado se purificó mediante cromatografía en columna ultrarrápida utilizando un gradiente de un 15 % de EtOAc en éter de petróleo para proporcionar N-(6-(N-(tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)acetamida (2.7 g, 7.3 mmol, 83 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-ak): 5 ppm 10.70 (s, 1 H), 8.27 (d, J=8.4 Hz, 1 H), 8.01 - 8.07 (m, 1 H), 7.68 (d, J=7.6 Hz, 1 H), 2.15 (s, 3 H), 1.28 (s, 9 H), 0.86 (s, 9 H), -0.07 (d, 3 H), -0.11 (d, 3 H). m/z (ESI): 256 (M-2tBu)+.
Paso 6: Una mezcla de N-(6-(N-(tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)acetamida (1.5 g, 4.1 mmol) y una solución de hidróxido de sodio acuoso (2.5 M, 32.5 mL, 81 mmol) en methanol (30 mL) se agitó a temperatura ambiente durante 15 h. La mezcla de reacción se concentró y el residuo se trató con agua. El sólido precipitado se filtró y se secó para proporcionar (6-aminopiridin-2-il)(tert-butil)((tert-butildimetilsilil)imino)- A6-sulfanona (1.2 g, 3.7 mmol, 90 % de rendimiento) como un sólido blanco. 1H r Mn (400 MHz, DMSO-afe): 5 ppm 7.58 (t, J=7.8 Hz, 1 H), 7.09 (d, J=7.3 Hz, 1 H), 6.60 (d, J=8.3 Hz, 1 H), 6.35 (s, 2 H), 1.26 (s, 9 H), 0.85 (s, 9 H), -0.07 (s, 3 H), -0.13 (s, 3 H). m/z (ESI): 214.1 (M-2tBu)+.
Paso 7: Una mezcla de ácido 4-bromo-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.50 g, 1.6 mmol, Intermedio 9-1), DIPEA (0.845 mL, 4.84 mmol), T3P (solución al 50 % en acetato de etilo, 3.08 g, 4.84 mmol) y (6-aminopiridin-2-il)(tertbutil)((tert-butildimetilsilil)imino)- A6-sulfanona (0.634 g, 1.93 mmol) en diclorometano (10 mL) se agitó a temperatura ambiente durante 48 h. La mezcla de reacción se diluyó con agua y se extrajo con diclorometano. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante cromatografía en columna ultrarrápida, eluyendo con un gradiente de un 0 % a un 15 % de acetato de etilo en éter de petróleo para proporcionar 4-bromo-N-(6-(N-(tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.32 mmol, 20 % de rendimiento) como una goma amarillo pálido. 1H RMN (400 MHz, DMSO-ds): 5 ppm 12.86 (s, 1 H), 8.51 (d, J=8.4 Hz, 1 H), 8.13 (t, J=8.0 Hz, 1 H), 8.00 (d, J=8.4 Hz, 1 H), 7.77 (d, J=7.6 Hz, 1 H), 7.68 (d, J=1.8 Hz, 1 H), 7.55 (dd, J=8.5, 1.8 Hz, 1 H), 2.97 - 3.10 (m, 4 H), 1.60 (s a, 4 H), 1.33 (s, 9 H), 0.82 (s, 9 H), 0.33 (s, 4 H), -0.12 (s, 3 H). -0.14 (s, 3 H). m/z (ESI): 507.0 (M-2tBu)+.
Paso 8: Una mezcla de 4-bromo-N-(6-(N-('tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)-2-(6-azaespiro[2.5]octan-6-il)benzamida (0.20 g, 0.32 mmol), 2-sulfamoilacetato de metilo (0.074 g, 0.48 mmol), fosfato de potasio tribásico (0.137 g, 0.645 mmol), yoduro de cobre (I) (0.061 g, 0.32 mmol) y (1 R,2R)-N,N-dimetil-1,2-ciclohexanodiamina (0.023 g, 0.16 mmol) en DMF (4 mL) se agitó a 90 °C durante 16 h. La mezcla de reacción se diluyó con agua y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró para proporcionar 2-(N-(4-((6-(N-(tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.18 g, 0.26 mmol, 81 % de rendimiento) como una goma amarillo pálido. m/z (ESI): 692.2 (M+H)+.
Paso 9 : Una mezcla de 2-(N-(4-((6-(N-(tert-butildimetilsilil)-2-metilpropan-2-ilsulfonimidoil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.18 g, 0.260 mmol) y fluoruro de tetrabutilamonio (solución 1 M en THF, 0.390 mL, 0.390 mmol) en THF (4 mL) se agitó a temperatura ambiente durante 3 h. La mezcla de reacción se desactivó con una solución acuosa saturada de cloruro de amonio y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró y se concentró para proporcionar 2-(N-(4-((6-(2-metilpropan-2-ilsulfonimidoil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.10 g, 0.17 mmol, 66 % de rendimiento) como una goma amarillo pálido. m/z (ESI): 578.1 (M+H)+.
Paso 10: A una solución de 2-(N-(4-((6-(2-metilpropan-2-ilsulfonimidoil)piridin-2-il)carbamoil)-3-(6-azaespiro[2.5]octan-6-il)fenil)sulfamoil)acetato de metilo (0.10 g, 0.17 mmol) en THF (3 mL) se añadió L¡BH4 (solución 2.0 M en Th F, 0.173 mL, 0.346 mmol) a 0 °C y se agitó a temperatura ambiente durante 1.5 h. La mezcla de reacción se desactivó con una
solución acuosa saturada de cloruro de amonio y se extrajo con acetato de etilo. El extracto orgánico se lavó con salmuera, se secó con Na2SO4, se filtró, se concentró y se purificó mediante HPLC para proporcionar 4-((2hidroxietil)sulfonamido)-N-(6-(2-metilpropan-2-ilsulfonimidoil)piridin-2-il)-2-(6-azaespiro[2.51octan-6-il)benzamida (0.016 g, 0.029 mmol, 16 % de rendimiento) como un sólido blanco. 1H RMN (400 MHz, DMSO-d>): S ppm 13.16 (s, 1 H), 10.26 (s, 1 H), 8.53 (d, J = 8.4 Hz, 1 H), 8.17 - 8.05 (m, 2 H), 7.82 (dd, J = 7.5, 0.9 Hz, 1 H), 7.29 (d, J = 2.2 Hz, 1 H), 7.14 (dd, J = 8.6, 2.1 Hz, 1 H), 4.95 (s a, 1 H), 4.04 (s, 1 H), 3.76 - 3.80 (m, 2 H), 3.37 (t, J = 6.5 Hz, 2 H), 2.99 (t, J = 5.4 Hz, 4 H), 1.60 - 1.70 (m, 3 H), 1.35 (s, 9 H), 0.36 (s, 4 H). m/z (ESI): 550.2 (M+H)+.
Ejemplos 27-1 y 27-2: 2-(6-Azaespiro[2.51octan-6-il)-4-(S-ciclopropilsulfonimidoil)-N-(6-(3,3,3-trifluoropropoxi)-2-piridinil)benzamida y 2-(6-azaespiro[2.51octan-6-il)-4-(fí-ciclopropilsulfonimidoil)-N-(6-(3,3,3-trifluoropropoxi)-2-

Una mezcla de ácido 4-(c¡clopropanosulfon¡mido¡l)-2-(6-azaesp¡ro[2.5]octan-6-¡l)benzo¡co (0.200 g, 0.598 mmol, Intermedio 16), 6-(3,3,3-trifluoropropoxi)piridin-2-amina (0.160 g, 0.78 mmol, Intermedio 6), TATU (0.250 g, 0.776 mmol, Combi-Blocks), DMF (6 mL) y DIPEA (0.309 g, 0.426 mL, 2.39 mmol, Aldrich) se agitó a temperatura ambiente durante 18 h. La mezcla se diluyó con Na2CO3 saturado y EtOAc. La fase orgánica se lavó con Na2CO3, agua y salmuera, se secó con Na2SO4 y se concentró al vacío. El producto crudo se purificó mediante cromatografía en gel de sílice: 0-100 % de EtOAc en heptano. Se obtuvo el producto como un sólido blanquecino. m/z (ESI): 523.1 (M+H)+. La mezcla racémica se purificó mediante SFC quiral utilizando una Regis (S,S) Whelk-01 (250 X 21 mm, 5 mm) con una fase móvil de un 65 % de CO2 líquido y un 35 % de MeOH utilizando una tasa de flujo de 80 mL/min para generar:
Ejemplo______ 27-1: 2-(6-Azaespiro[2.51octan-6-il)-4-(S-ciclopropilsulfonimidoil)-N-(6-(3,3,3-trifluoropropoxi)-2-piridinil)benzamida. Primer pico en eluir (68 mg, ee>99 %). 1H RMN (400 MHz, CLOROFORMO-d) S 12.92 (s, 1H), 8.45 (d, J=8.29 Hz, 1H), 8.00 (d, J=7.67 Hz, 1H), 7.95 (d, J=1.66 Hz, 1H), 7.85 (dd, J=1.76, 8.19 Hz, 1H), 7.66 (t, J=7.88 Hz, 1H), 6.55 (d, J=8.09 Hz, 1 H), 4.54 (t, J=6.63 Hz, 2H), 3.15 (t, J=5.29 Hz, 4H), 2.55-2.72 (m, 3H), 1.66-1.98 (m, 5H), 1.46 (tdd, J=5.05, 6.87, 10.18 Hz, 1H), 1.24 (tdd, J=4.90, 6.95, 10.21 Hz, 1H), 1.13 (dq, J=5.18, 7.95 Hz, 1H), 0.94-1.04 (m, 1H), 0.42 (s, 4H). 19F RMN (377 MHz, CLOROFORMO-d) S -64.71 (s, 3F).
Ejemplo 27-2: 2-(6-Azaespiro[2.51octan-6-il)-4-(fí-ciclopropilsulfonimidoil)-N-(6-(3,3,3-trifluoropropoxi)-2-piridinilo). Segundo pico en eluir (67 mg, ee 99.3 %). 1H RMN (400 MHz, CLOROFORMO-d) S 12.92 (s, 1H), 8.45 (d, J=8.29 Hz, 1H), 8.00 (d, J=7.88 Hz, 1H), 7.95 (d, J=1.66 Hz, 1H), 7.85 (dd, J=1.66, 8.29 Hz, 1H), 7.66 (t, J=7.98 Hz, 1H), 6.55 (d, J=8.09 Hz, 1H), 4.54 (t, J=6.74 Hz, 2H), 3.15 (t, J=5.29 Hz, 4H), 2.54-2.73 (m, 3H), 1.69-1.80 (m, 4H), 1.41-1.49 (m, 1H), 1.19-1.29 (m, 2H), 1.08-1.17 (m, 1H), 0.98 (dq, J=5.29, 7.98 Hz, 1H), 0.42 (s, 4H). 19F RMN (376 MHz, CLOROFORMO-d) S -64.70 (s, 3F).
La estereoquímica se asignó arbitrariamente.
Tabla 16: Los Ejemplos de 18-1 a 18-3 se prepararon siguiendo un procedimiento similar al descrito para los Ejemplos 27-1 y 27-2.

Ejemplo 31: Trifluoroacetato de 4-(azetidin-3-ilsulfonil)-2-(6-azaespiro[2.51octan-6-il)-N-(6-(3.3.3-trifluoropropoxi)piridin-2-il)benzamida

Paso 1: Una mezcla de ácido 4-((1-(ferf-butoxicarbonil)azetidin-3-il)sulfonil)-2-(6-azaespiro[2.5]octan-6-il)benzoico (0.200 g. 0.444 mmol. Intermedio l5), 6-(3.3.3-trifluoropropoxi)piridin-2-amina (0.119 g. 0.577 mmol. Intermedio 6). DMF (4 mL). base de Hunig (0.229 g. 0.316 mL. 1.78 mmol. Aldrich) y TATU (0.250 g. 0.776 mmol. Combi-Blocks) se agitó a temperatura ambiente durante la noche. La mezcla se diluyó con Na2CÜ3 saturado y EtOAc. Se tomó la fase orgánica y se lavó con Na2CÜ3. agua y salmuera. se secó con Na2SÜ4 y se concentró al vacío. La purificación mediante cromatografía en gel de sílice (0-100 % EtOAc-heptano) proporcionó 3-((3-(6-azaespiro[2.5]octan-6-il)-4-((6-(3.3.3-trifluoropropoxi)piridin-2-il)carbamoil)fenil)sulfonil)azetidino-1 -carboxilato de ferf-butilo como un sólido blanquecino. m/z (ESI): 639.2 (M+H)+.
Paso______ 2: Se disolvió 3-((3-(6-azaespiro[2.5]octan-6-il)-4-((6-(3.3.3-trifluoropropoxi)piridin-2-il)carbamoil)fenil)sulfonil)azetidino-1-carboxilato de ferf-butilo (0.118 g. 0.184 mmol) se disolvió en DCM (8 mL) y TFA (4 mL). La mezcla de reacción se agitó a TA durante 30 min y se concentró al vacío. Se añadió EtOAc a la sal de TFA y se evaporó. Se obtuvo el producto como un sólido blanquecino. 1H RMN (400 MHz. METANOL<U) 58.38 (d. J=8.09 Hz. 1H). 7.90-7.96 (m. 2H). 7.85 (dd. J=1.66. 8.29 Hz. 1H). 7.74 (t. J=7.98 Hz. 1H). 6.60 (d. J=8.09 Hz. 1H). 4.57 (t. J=6.32 Hz. 2H). 4.42 (d. J=7.46 Hz. 4H). 3.14-3.23 (m. 4H). 2.66-2.80 (m. 2H). 1.81 (s a. 4H). 0.44 (s. 4H). 19F RMN (376 MHz. METANOL-d4) 5 -66.16 (s. 3F). -76.96 (s. 3F). m/z (ESI): 539.2 (M+H)+.
Ejemplos biológicos
Se utilizaron los siguientes ensayos en el estudio de los compuestos ejemplares de la invención. Los datos para estos ejemplos estudiados de acuerdo con los procedimientos descritos a continuación se presentan en la Tabla A más adelante.
Ensayo de enzima KIF18A: Se utilizó la actividad ATPasa estimulada por microtúbulos para medir la actividad de la enzima KIF18A tras el tratamiento con el compuesto. Los compuestos se diluyeron 2 veces en serie en DMSO (Sigma Inc) en un intervalo de concentración de 22 puntos. La proteína KIF18A humana recombinante (etiquetada con histidina 1 -467) se expresó utilizando un sistema de baculovirus y fue purificada mediante cromatografía de afinidad por Amgen Inc. Las concentraciones de la proteína KIF18A. los microtúbulos (MT). y el ATP en la reacción se optimizaron para el ensayo de la enzima homogénea estandarizado utilizando el kit de ensayo de cinasa/ATPasa ADP-GloTM (Promega Inc.). El ensayo mide el ADP formado a partir de la reacción de la ATPasa. Preparar el tampón de reacción [(Tris 15 mM. pH 7.5 (Teknova Inc). MgCl2 10 mM (JT Baker Inc). 0.01 % de Pluronic F-68 (Life Technologies Inc). Taxol 1 gM (Cytoskeleton Inc). y 30 gg/mL de microtúbulos de cerdo (Cytoskeleton Inc)]. Añadir el compuesto y la proteína KIF18A (30 nM) al tampón de reacción preparado e incubar durante 15 minutos a temperatura ambiente. a continuación añadir At o (a Km. 75 gM) a la mezcla de reacción e incubar durante 15 minutos más a temperatura ambiente. Mezclar 5 gL de reactivo ADP-GloTM y 2.5 gL de la mezcla de reacción e incubar durante 40 minutos a temperatura ambiente. Añadir 10 gL de Reactivo de detección ADP-GloTM e incubar durante 40 minutos a temperatura ambiente. Leer la luminiscencia utilizando un lector de microplacas EnVision con un módulo de ultraluminiscencia (Perkin Elmer Inc). Se realizó la determinación del ajuste de la curva de respuesta a la concentración y la CI50 utilizando el software Genedata Screener (Standard 15.0.1. Genedata Inc) con un modelo de ajuste de regresión logística de cuatro parámetros.
La Tabla A proporciona datos de los compuestos ejemplificados en la presente solicitud y documentos prioritarios de esta. como compuestos representativos de la presente invención. de la siguiente manera: nombre del compuesto y datos biológicos. (CI50 en uM. cuando esté disponible. Ej. N.° se refiere al N.° de ejemplo)
TABLA A: DATOS BIOLÓGICOS











Se ha descrito en detalle la invención anterior, a modo ilustrativo y como ejemplo, a efectos de una mejor comprensión y claridad. El experto en la técnica comprende que es posible llevar a la práctica ciertos cambios y modificaciones dentro del alcance de las reivindicaciones adjuntas. Por lo tanto, se entenderá que se pretende que la descripción anterior es ilustrativa y no restrictiva. Por lo tanto, el alcance de la invención deberá ser determinado no con referencia a la descripción anterior sino, por el contrario, deberá ser determinado con referencia a las reivindicaciones adjuntas a continuación.
Claims (21)
1. Un compuesto de fórmula I:

o cualquier sal farmacéuticamente aceptable de este, donde:
X1 es N o -CR6;
R1 es un grupo -Z-R12 donde Z es -alq C0-4-, -NR11-, -NR11SO2-alq C0-4-, -SO2NR11-alq C0-4-, -NR11SO2NR11-, -NR11SO2NR11-C(=O)-O-, -alq C0-4-S(=O)(=NH)-, alq C0-4-NR11-S(=O)(=NH), -alq C0-4-S-, -alq C0-4-S(=O)-, -alq C0-4-SO2-, alq C0-4-O-, -P-, -P(=O), -P(=O)2, -(C=O)-, -(C=O)NR11-, -C=N(OH)-, o -NR11(C=O); o el grupo -Z-R12 es -N=S(=O)-(R12)2 , donde el par de los dos R12 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1,2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S;
R2 es halo o un grupo -Y -R 13, donde Y es -alq C0-4-, -N(alq C0-1)-alq C0-4-, -C(=O)NRa(alq C1-4)-, -O-alq C0-4-, -S-, -S=O, -S(=O)2-, -SO2N(alq C0-1)-alq C0-4-, -N(alq C0-1)-SO2-alq C0-4-, -alq C0-4-S(=O)(=NH)-, -(C=O)-, -alq C0-4-(C=O)-O-; o
el grupo -Y-R13 es -N=S(=O)-(R13)2 , donde el par de los dos R13 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1,2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S;
R3 es H, metilo o etilo;
R4 es H, halo, CN, alq C1-4 o haloalq C1-4;
R5 es H, halo, alq C1-8 o haloalq C1-4;
R6 es H, halo, CN, alq C1-8, haloalq C1-4, -O-alq C0-6-, o R6a;
R7 es H, halo, alq C1-8 o haloalq C1-4;
R8 es H, halo, alq C1-8 o haloalq C1-4;
R9 es H, halo, alq C1-8 o haloalq C1-4;
Rx se selecciona del grupo que consiste en
cada uno de R10a, R10b, R10c, R10d, R10e, R10f, R10g, R10h, R10i, y R10i es H, halo, R10k, o R1
o como alternativa, cada uno del par R10a y R10b, par R10c y R10d, par R10e y R10f, par R10g y R10h o par R10i y R10j, se puede combinar independientemente con el átomo de carbono unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado espiro con el anillo Rx; donde dicho anillo monocíclico de 3, 4, 5 o 6 miembros contiene 0, 1, 2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S, y además donde dicho anillo monocíclico de 3, 4, 5 o 6 miembros está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -NRaRa, u oxo;
R11 es H o alq C1-8;
R12 es H, R12a o R12b;
R13 es R13a o R13b;
R6a, R10k, R12a y R13a se seleccionan independientemente en cada caso del grupo que consiste en un anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 4, 5, 6, 7, 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1,2 o 3 átomos de N y 0, 1, o 2 átomos seleccionados entre O y S, que está sustituido con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -C(=O)Rb, -C(=O)ORa, -C(=O)NRaRa,
-C(=NRa)NRaRa, -OC(=O)Rb, -OC(=O)NRaRa, -Oalq C2-6NRaRa, -Oalq C2-6ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRaRa, -NRaRa, -N(Ra)C(=O)Rb, -N(Ra)C(=O)ORb,-N(Ra)C(=O)NRaRa, -N(Ra)C(=NRa)NRaRa, -N(Ra)S(=O)2Rb, -N(Ra)S(=O)2NRaRa, -NRaalq C2-6NRaRa, -NRaalq C2-6ORa, -alq C1-6NRaRa, -alq C1-6ORa, -alq C1-6N(Ra)C(=O)Rb, -alq C1-6OC(=O)Rb, -alq C1-6C(=O)NRaRa, -alq C1-6C(=O)ORa, R14, y oxo;
R101, R12b y R13b se seleccionan independientemente en cada caso del grupo que consiste en alq C1-6 sustituido con 0, 1, 2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, -C(=O)ORa, -ORa, -haloalq C1-2, -Ohaloalq C1-4, CN, NH2, NH(CH3) o N(CH3)2;
R14 se selecciona independientemente en cada caso del grupo que consiste en anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 4, 5, 6, 7, 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1, 2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S, que está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -ORa, -Ohaloalq C1-4, CN, -C(=O)Rb, -C(=O)ORa, -C(=O)NRaRa, -C(=NRa)NRaRa, -OC(=O)Rb, -OC(=O)NRaRa, -Oalq C2-6NRaRa, -Oalq C2-6ORa, -SRa, -S(=O)Rb, -S(=O)2Rb, -S(=O)2NRaRa, -NRaRa, -N(Ra)C(=O)Rb, -N(Ra)C(=O)ORb,-N(Ra)C(=O)NRaRa, -N(Ra)C(=NRa)NRaRa, -N(Ra)S(=O)2Rb, -N(Ra)S(=O)2NRaRa, -NRaalq C2-6NRaRa, -NRaalq C2-6ORa, -alq C1-6NRaRa, -alq C1-6ORa, -alq C1-6N(Ra)C(=O)Rb, -alq C1-6OC(=O)Rb, -alq C1-6C(=O)NRaRa, -alq C1-6C(=O)ORa, y oxo;
Ra es independientemente, en cada caso, H o Rb; y
Rb es independientemente, en cada caso, alq C1-6, fenilo o bencilo, donde el alq C1-6 está sustituido con 0, 1, 2 o 3 sustituyentes seleccionados entre halo, -OH, -Oalq C1-4, -NH2, -NHalq C1-4, -OC(=O)alq C1-4 o -N(alq C1-4)alq C1-4; y el fenilo o bencilo está sustituido con 0, 1,2 o 3 sustituyentes seleccionados entre halo, alq C1-4, halolaq C1-3, -OH, -Oalq C1-4, -NH2, -NHalq C1-4, -OC(=O)alq C1-4 o -N(alq C1-4)alq C1-4.
2. El compuesto de la Reivindicación 1, donde RX es
3. El compuesto de una cualquiera de las reivindicaciones 1-2 donde:
X1 es -CR6; que tiene la fórmula (Ia):

X1 es N; que tiene la fórmula (Ib):

4. El compuesto de una cualquiera de las reivindicaciones 1 -3 donde R3 es H o metilo.
5. El compuesto de una cualquiera de las reivindicaciones 1 -4 donde cada uno de R10c, R10d, R10e, R10f, R10g, R10h, R10i y R10j es H, halo, alq C1-6 o haloalq C1-4; y cada uno del par R10a y R10b se combina con el átomo de carbono unido a cada uno de ellos para formar un anillo monocíclico de 3, 4 o 5 miembros saturado espiro con el anillo Rx; donde dicho anillo contiene 0, 1,2 o 3 átomos de N y 0 o 1 átomos seleccionados entre O y S; o
donde cada uno de R10c, R10d, R10e, R10f, R10g, R10h, R10i y R10j es H, metilo o etilo; y cada uno del par R10a y R10b se combina con el átomo de carbono unido a cada uno de ellos para formar un anillo ciclopropilo, ciclobutilo o ciclopentilo espiro con el anillo Rx.
6. El compuesto de una cualquiera de las reivindicaciones 1-5 donde Rx se selecciona entre:

7. El compuesto de una cualquiera de las reivindicaciones 1 -6 donde R
8. El compuesto de una cualquiera de las reivindicaciones 1-7 donde Z está ausente, es NH-, -NHSO2-(CH2)0-4-, -N(CH3)-SO2-(CH2)0-4-, -NCH3SO2NH, -NHSO2NH-C(=O)-O-, -SO2NH-(CH2)0-4-, -(CH2)0-2-S(=O)(=NH)-, -(CH2)0-2-S-,-(CH2)0-2-S(=O)-, (CH3CH)-S(=O)-, -(CH2)0-2-SO2- -O-, -P(=O), -(C=O)- o -NH(C=O).
9. El compuesto de una cualquiera de las reivindicaciones 1 -8 donde
R1 es el grupo -Z-R12 que es -N=S(=O)-(R12)2 , donde el par de los dos R12 se puede combinar como alternativa con el átomo de azufre unido a cada uno de ellos para formar un anillo monocíclico de 3, 4, 5 o 6 miembros saturado o parcialmente saturado que contiene 0, 1,2 o 3 átomos de N y 0, 1 o 2 átomos seleccionados entre O y S; que se selecciona entre:

R1 es -Z-R12, donde Z está ausente, es NH-, -NHSO2-(CH2)0-4-, -N(CH3)-SO2-(CH2)0-4-, -NCH3SO2NH, -NHSO2NH-C(=O)-O-, -SO2NH-(CH2)0-4-, -(CH2)0-2-S(=O)(=NH)-, -(CH2)0-2-S-, -(CH2)0-2-S(=O)-, (CH3CH)-S(=O)-, -(CH2)0-2-SO2-, -O-, -P(=O), -(C=O)-, o -NH(C=O); y R12 se selecciona entre:
(a) H;
(b) ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, oxiranilo, oxetanilo, tetrahidrofuranilo, azetidinilo, imidazolilo, morfolinilo, pirrolidinilo, piperazinilo,
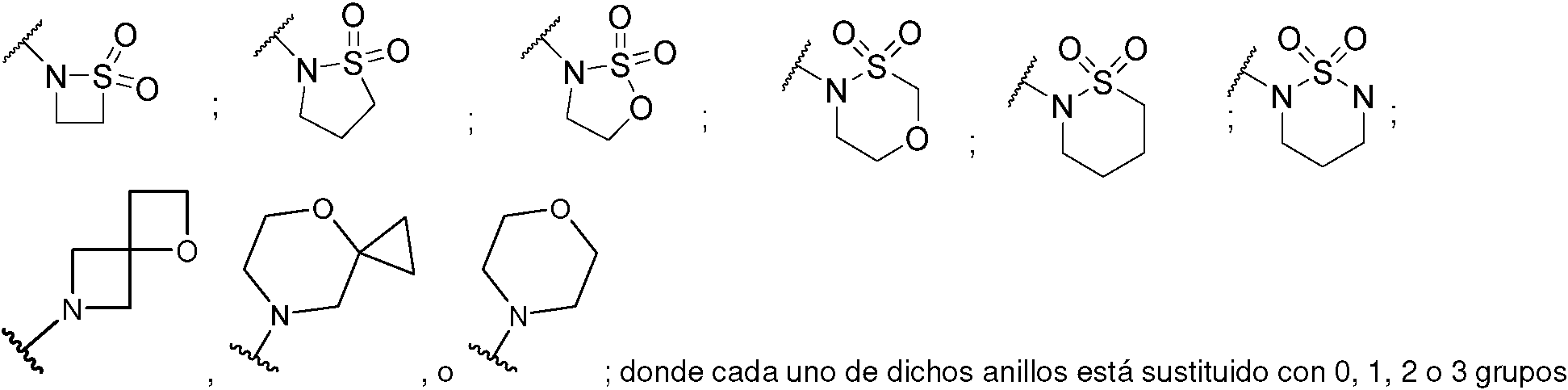
seleccionados entre donde cada anillo está sustituido con 0, 1, 2 o 3 OH, F, metilo, -CH2OH, -C(=O)OCH3, -C(=O)OC(CH3)3, NH2, CN, y oxo; o
(c) alq C1-6 sustituido con 0, 1, 2 o 3 OH, F, -C(=O)OCH3, -NH2, -NH(CH3), o -N(CH3)2; o
R1 es un grupo -Z-R12, donde Z es -NHSO2- o -SO2NH-; y R12 es oxetanilo, ciclopropilo o R12 es alq C1-6 sustituido con 0, 1, 2 o 3 grupos OH; o
R1 es un grupo -Z-R12, donde Z es -NHSO2- y R12 es -C H 2-CH2-OH.
10. El compuesto de una cualquiera de las reivindicaciones 1 -3, 4-5 o 6-9
donde R2 es halo o un grupo -Y-R 13, donde Y está ausente, es -SO2NH-(CH2)ü-4-, NH-, -NH-S02-(CH)2)ü-4-, -0-(CH2)ü-4, -O-(CH(CH3))-, -(CH2)0-4-S(=O)(=NH)-, -(C=O)-, -(CH2)0-4-(C=O)-O- o -(CH2)1-4;
R13 es un anillo monocíclico de 3, 4, 5, 6 o 7 miembros o bicíclico de 8, 9, 10, 11 o 12 miembros saturado, parcialmente saturado o insaturado que contiene 0, 1, 2 o 3 átomos de N y 0 o 1 átomos seleccionados entre O y S, que está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -OH, -Ohaloalq C1-4, CN, R14 y oxo; o;
R13 es alq C1-6 sustituido con 0, 1,2, 3, 4, o 5 grupos seleccionados entre F, Cl, Br, -OH, -Ohaloalq C1-4, o CN; o donde R2 es un anillo monocíclico de 5 o 6 miembros saturado donde cada uno de dichos anillos contiene 0, 1 o 2 átomos de N y 0 o 1 átomos de O, y donde cada uno de dichos anillos está sustituido con 0, 1, 2 o 3 grupos seleccionados entre F, Cl, Br, alq C1-6, haloalq C1-4, -OH, -OCH3, -Ohaloalq C1-4, CN, R14 y oxo; o
donde R2 es :
(a) F, Br;
(b) un grupo -Y -R 13, donde Y está ausente; y R13 es morfolinilo, piperidinilo, azetidinilo, pirrolidinilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, piperazinilo, tetrahidrofuranilo, tetrahidropiranilo, piridinilo, pirimidinilo, 3,6-dihidro-2H-piranilo,
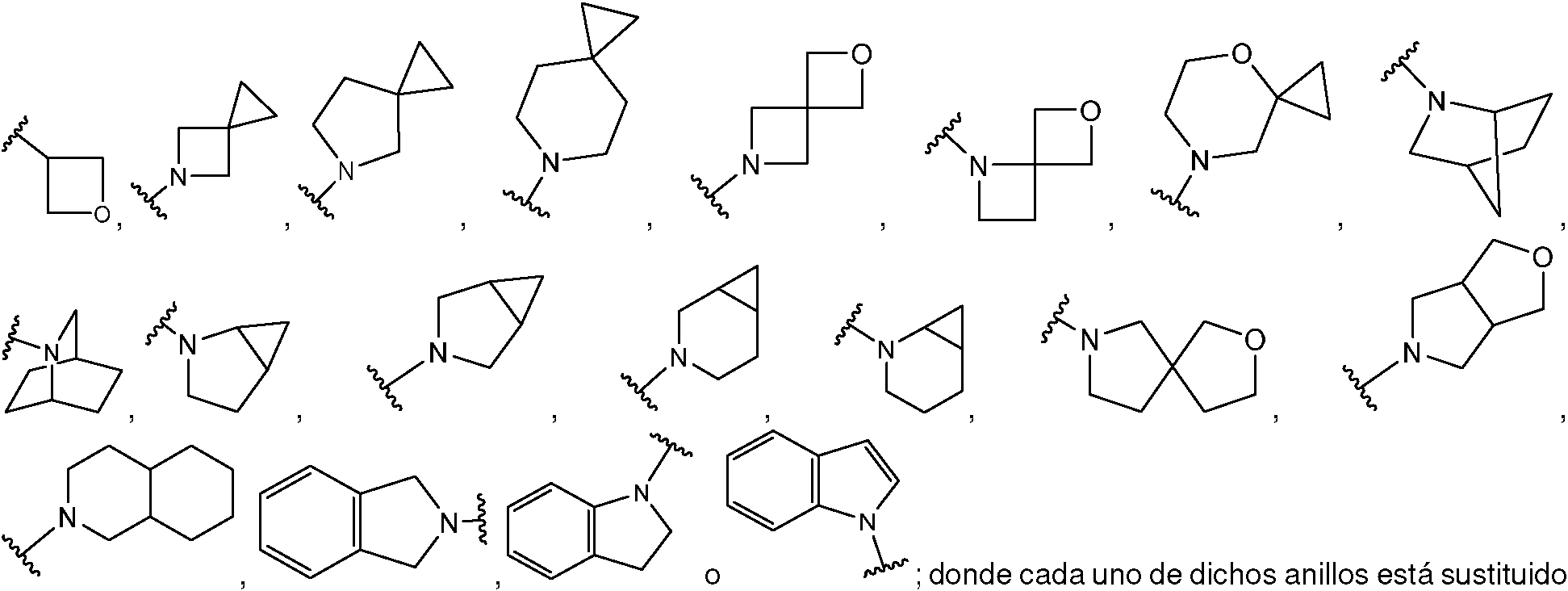
con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, metilo, CF3, CH2OH, CH(CH3)OH, C(CH3)2OH, -OH, -OCHF2, CN, oxo o ciclopropilo; o
(c) un grupo -Y -R 13, donde Y está ausente, es -SO2NH-, NH -O- S(=O)(=NH - -O-(CH2 -O-(CH(CH3))- C(=O)-,
C(=O)-O-, -CH2C(=O)-O-, o -CH2-; y donde R13 es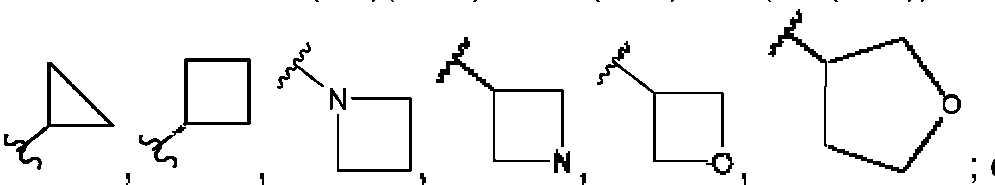 donde cada anillo está sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN;
donde cada anillo está sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN;
o R13 es H o alq C1-6 sustituido con 0, 1,2, 3, 4, o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN; o donde R2 es (a) halo; (b) un grupo -Y R 13, donde Y está ausente; y R13 es morfolinilo, piperidinilo, azetidinilo, pirrolidinilo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, piperazinilo, tetrahidrofuranilo,

uno de dichos anillos está sustituido con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH, -OCHF2, CN y oxo; o (c) un grupo -Y -R 13, donde Y es NH, -O-, -0-(CH2)-, -0-(CH2)-(CH2)- o
-0-(CH2)-(CH2)-(CH2)-, y donde R13 es o R13 es alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN; o
o R13 es alq C1-6 sustituido con 0, 1,2, 3, 4 o 5 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH o CN; o

donde R2 es morfolinilo o piperidinilo sustituido con 0, 1,2 o 3 grupos seleccionados entre F, Cl, Br, metilo, CF3, -OH, -OCHF2, CN, u oxo; o
R2 es morfolinilo sustituido con 1,2 o 3 grupos metilo.
11. El compuesto de una cualquiera de las reivindicaciones 1 -10
donde R4 se selecciona entre H, F, metilo, CN o Br; o
donde R4 es H.
12. El compuesto de una cualquiera de las reivindicaciones 1 -11
donde R5 es H.
13. El compuesto de una cualquiera de las reivindicaciones 1 -12
donde R6 es H, metilo, ciclopropilo, CN, CF3 o azetidinilo.
14. El compuesto de una cualquiera de las reivindicaciones 1-13
donde R7 es H.
15. El compuesto de una cualquiera de las reivindicaciones 1-14
donde R8 es H o F.
16. El compuesto de una cualquiera de las reivindicaciones 1 -15
donde R9 es H o F.
17. El compuesto de la reivindicación 1, seleccionado del grupo que consiste en:






o cualquier sal farmacéuticamente aceptable de estas.
18. El compuesto de una cualquiera de las reivindicaciones 1 -17 para su uso como un medicamento.
19. Una composición farmacéutica que comprende el compuesto de acuerdo con una cualquiera de las reivindicaciones 1-17 o la sal farmacéuticamente aceptable de este, y un diluyente o portador farmacéuticamente aceptable.
20. El compuesto de una cualquiera de las reivindicaciones 1-17 o la composición que comprende el compuesto de la reivindicación 19 para su uso en un método para tratar una afección que se puede tratar con inhibidores de KIF18a, comprendiendo el método administrar a un paciente que lo necesita una cantidad terapéuticamente eficaz del compuesto;
donde dicha afección es cáncer seleccionado del grupo que consiste en (a) un tumor sólido o derivado hematológicamente seleccionado entre cáncer del cáncer de vejiga, de endometrio, escamocelular de pulmón, mama, colon, riñón, hígado, pulmón, cáncer pulmonar microcítico, de esófago, vesícula biliar, cerebro, cabeza y cuello, ovario, páncreas, estómago, cuello uterino, tiroides, próstata y piel, (b) un tumor hematopoyético de linaje linfoide seleccionado entre leucemia, leucemia linfocítica aguda, leucemia linfoblástica aguda, linfoma de linfocitos B, linfoma de linfocitos T, linfoma de Hodgkin, linfoma no hodgkiniano, linfoma de tricoleucocitos y linfoma de Burkitt, (c) un tumor hematopoyético de linaje mieloide seleccionado entre leucemias mielógenas aguda y crónica, síndrome mielodisplásico y leucemia promielocítica (d) un tumor de origen mesenquimatoso seleccionado entre fibrosarcoma y rabdomiosarcoma, (e) un tumor del sistema nervioso central y periférico seleccionado entre astrocitoma, neuroblastoma, glioma y schwannoma, o (f) un melanoma, seminoma, teratocarcinoma, osteosarcoma, xenoderomia pigmentosa, queratoctantoma, cáncer folicular tiroideo o sarcoma de Kaposi.
21. El compuesto de una cualquiera de las reivindicaciones 1-17 o la composición que comprende el compuesto de la reivindicación 19 para su uso en un método para reducir el tamaño de un tumor sólido en un sujeto; o
en un método para tratar un trastorno de proliferación celular en un sujeto; o
en un método para inhibir KIF18A en una célula.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862783069P | 2018-12-20 | 2018-12-20 | |
| PCT/US2019/068172 WO2020132651A1 (en) | 2018-12-20 | 2019-12-20 | Kif18a inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2953821T3 true ES2953821T3 (es) | 2023-11-16 |
Family
ID=69411509
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19845855T Active ES2953821T3 (es) | 2018-12-20 | 2019-12-20 | Inhibidores de KIF18A |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US12459932B2 (es) |
| EP (1) | EP3897855B1 (es) |
| JP (1) | JP7686559B2 (es) |
| AU (1) | AU2019403488B2 (es) |
| CA (1) | CA3123042A1 (es) |
| ES (1) | ES2953821T3 (es) |
| MA (1) | MA54550A (es) |
| MX (1) | MX2021007104A (es) |
| WO (1) | WO2020132651A1 (es) |
Families Citing this family (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MA54543A (fr) | 2018-12-20 | 2022-03-30 | Amgen Inc | Inhibiteurs de kif18a |
| JP7686559B2 (ja) | 2018-12-20 | 2025-06-02 | アムジエン・インコーポレーテツド | Kif18a阻害剤 |
| JP2022513967A (ja) | 2018-12-20 | 2022-02-09 | アムジエン・インコーポレーテツド | Kif18a阻害剤として有用なヘテロアリールアミド |
| AU2019401495B2 (en) * | 2018-12-20 | 2025-06-26 | Amgen Inc. | Heteroaryl amides useful as KIF18A inhibitors |
| EP4007638A1 (en) | 2019-08-02 | 2022-06-08 | Amgen Inc. | Pyridine derivatives as kif18a inhibitors |
| EP4007752B1 (en) * | 2019-08-02 | 2025-09-24 | Amgen Inc. | Kif18a inhibitors |
| EP4007753B1 (en) | 2019-08-02 | 2025-09-24 | Amgen Inc. | Kif18a inhibitors |
| US20230233565A1 (en) * | 2020-05-11 | 2023-07-27 | University Of Vermont | A treatment approach involving kif18a inhibition for chromosomally unstable tumors |
| US20240059703A1 (en) | 2020-10-20 | 2024-02-22 | Amgen Inc. | Heterocyclic spiro compounds and methods of use |
| WO2022093856A1 (en) | 2020-10-27 | 2022-05-05 | Amgen Inc. | Heterocyclic spiro compounds and methods of use |
| MX2023012725A (es) | 2021-04-29 | 2024-09-23 | Amgen Inc | Compuestos de 2-aminobenzotiazol y metodos de uso de los mismos. |
| US20250214965A1 (en) * | 2021-06-25 | 2025-07-03 | Hangzhou Innogate Pharma Co., Ltd. | Compound as kif18a inhibitor |
| IL310863A (en) | 2021-08-26 | 2024-04-01 | Volastra Therapeutics Inc | Spiro indoline inhibitors of kif18a |
| WO2023041055A1 (zh) * | 2021-09-16 | 2023-03-23 | 微境生物医药科技(上海)有限公司 | Kif18a抑制剂 |
| AU2022389558A1 (en) * | 2021-11-19 | 2024-05-23 | Wigen Biomedicine Technology (shanghai) Co., Ltd. | Kif18a inhibitor |
| EP4479398A1 (en) | 2022-02-16 | 2024-12-25 | Amgen Inc. | Quinazoline compounds and use thereof as inhibtors of mutant kras proteins |
| MX2024010045A (es) | 2022-02-16 | 2024-08-26 | Amgen Inc | Compuestos de quinazolina y uso de los mismos como inhibidores de proteinas mutantes kras. |
| JP2025508192A (ja) * | 2022-03-17 | 2025-03-21 | ウィゲン・バイオメディシン・テクノロジー・(シャンハイ)・カンパニー・リミテッド | Kif18a阻害剤 |
| TW202400582A (zh) * | 2022-05-13 | 2024-01-01 | 大陸商上海湃隆生物科技有限公司 | 驅動蛋白kif18a抑制劑及其應用 |
| CN116789637B (zh) * | 2022-05-13 | 2024-08-02 | 上海湃隆生物科技有限公司 | 驱动蛋白kif18a抑制剂及其应用 |
| KR20250027560A (ko) * | 2022-06-30 | 2025-02-26 | 쑤저우 젠하우스 바이오 컴퍼니 리미티드 | 질소 함유 화합물 및 이의 용도 |
| WO2024022508A1 (zh) * | 2022-07-29 | 2024-02-01 | 武汉人福创新药物研发中心有限公司 | Kif18a抑制剂及用途 |
| CN117510463A (zh) * | 2022-08-05 | 2024-02-06 | 长春金赛药业有限责任公司 | Kif18a抑制剂化合物、药物组合物及其制备方法和应用 |
| WO2024035950A1 (en) * | 2022-08-12 | 2024-02-15 | Accent Therapeutics, Inc. | Inhibitors of kif18a and uses thereof |
| TW202416973A (zh) | 2022-08-18 | 2024-05-01 | 美商雅客森醫療公司 | Kif18a抑制劑及其用途 |
| KR20250044780A (ko) * | 2022-09-08 | 2025-04-01 | 장춘 진사이언스 파마슈티컬 씨오., 엘티디. | Kif18a 억제제 화합물, 약제학적 조성물 및 이의 제조 방법과 적용 |
| CN119907796A (zh) * | 2022-09-09 | 2025-04-29 | 海南先声再明医药股份有限公司 | 酰胺类化合物、其组合物及用途 |
| CN119072473A (zh) * | 2022-09-30 | 2024-12-03 | 长春金赛药业有限责任公司 | 稠环类kif18a抑制剂化合物、药物组合物及其制备方法和应用 |
| CN119894878A (zh) * | 2022-10-13 | 2025-04-25 | 浙江海正药业股份有限公司 | 芳香酰胺类衍生物及其制备方法和用途 |
| WO2024083208A1 (zh) * | 2022-10-21 | 2024-04-25 | 杭州邦顺制药有限公司 | Kif18a蛋白抑制剂 |
| WO2024099398A1 (zh) * | 2022-11-10 | 2024-05-16 | 南京明德新药研发有限公司 | 一类含并杂环的磺酰胺类化合物及其应用 |
| AU2023382606A1 (en) | 2022-11-14 | 2025-05-08 | Amgen Inc. | Macrocyclic kras inhibitors and methods of use |
| JP2025538601A (ja) * | 2022-11-24 | 2025-11-28 | インベンティスバイオ カンパニー リミテッド | 複素環化合物、その製造方法および用途 |
| WO2024114730A1 (zh) * | 2022-11-30 | 2024-06-06 | 微境生物医药科技(上海)有限公司 | 一种用于癌症治疗的药物组合物 |
| EP4628486A1 (en) * | 2022-12-02 | 2025-10-08 | Shenzhen Zhongge Biological Technology Co., Ltd. | Amide or urea compound |
| CN120265626A (zh) * | 2022-12-14 | 2025-07-04 | 浙江海正药业股份有限公司 | 芳香酰胺类衍生物、其制备方法和其在医药上的应用 |
| WO2024140799A1 (en) * | 2022-12-28 | 2024-07-04 | Insilico Medicine Ip Limited | Inhibitors of kif18a and uses thereof |
| CN120569373A (zh) * | 2023-01-05 | 2025-08-29 | 浙江海正药业股份有限公司 | 芳香酰胺类衍生物及其制备方法和医药上的用途 |
| CN120548308A (zh) * | 2023-01-20 | 2025-08-26 | 英矽智能科技知识产权有限公司 | Kif18a的抑制剂及其用途 |
| WO2024156288A1 (en) * | 2023-01-29 | 2024-08-02 | Shanghai Kygent Pharmaceutical Co., Ltd | Dnmt1 inhibitors, pharmaceutical compositions, and therapeutic applications |
| WO2024183710A1 (zh) * | 2023-03-06 | 2024-09-12 | 武汉人福创新药物研发中心有限公司 | 抑制kif18a的化合物及其用途 |
| MX2025012140A (es) * | 2023-04-13 | 2026-02-03 | Ideaya Biosciences Inc | Terapia combinada que comprende un inhibidor de mat2a y un inhibidor de la topoisomerasa, un inhibidor del empalme sulfonamida, o un inhibidor de kif18 |
| WO2025067239A1 (zh) * | 2023-09-26 | 2025-04-03 | 上海湃隆生物科技有限公司 | 驱动蛋白kif18a抑制剂及其应用 |
| WO2025194054A1 (en) | 2024-03-14 | 2025-09-18 | Amgen Inc. | Spirocyclic compounds as modulators of kras and uses thereof |
| TW202602882A (zh) | 2024-03-14 | 2026-01-16 | 美商安進公司 | 作為kras調節劑的大環化合物及其用途 |
| WO2025217247A1 (en) | 2024-04-10 | 2025-10-16 | Amgen Inc. | Tethered spiro-heterocyclic inhibitors of kras g12c mutant proteins and uses thereof |
| WO2025230862A1 (en) | 2024-04-29 | 2025-11-06 | Amgen Inc. | Macrocyclic amino compounds as modulators of kras and uses therof |
| WO2025230878A1 (en) | 2024-04-29 | 2025-11-06 | Amgen Inc. | Macrocyclic compounds as modulators of kras and uses thereof |
| WO2025240582A1 (en) | 2024-05-14 | 2025-11-20 | Amgen Inc. | Macrocyclic compounds as modulators of kras and uses thereof |
| WO2025242166A1 (en) * | 2024-05-22 | 2025-11-27 | InventisBio Co., Ltd. | Heterocyclic compounds, preparation methods and uses thereof |
| WO2026055477A1 (en) | 2024-09-06 | 2026-03-12 | Amgen Inc. | Macrocyclic compounds as modulators of kras and uses thereof |
| CN120665073B (zh) * | 2025-08-18 | 2025-10-28 | 山东绿叶制药有限公司 | 作为kif18a抑制剂的化合物及其用途 |
Family Cites Families (173)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT72878B (en) | 1980-04-24 | 1983-03-29 | Merck & Co Inc | Process for preparing mannich-base hydroxamic acid pro-drugs for the improved delivery of non-steroidal anti-inflammatory agents |
| EP0090505B1 (en) | 1982-03-03 | 1990-08-08 | Genentech, Inc. | Human antithrombin iii, dna sequences therefor, expression vehicles and cloning vectors containing such sequences and cell cultures transformed thereby, a process for expressing human antithrombin iii, and pharmaceutical compositions comprising it |
| GB8827305D0 (en) | 1988-11-23 | 1988-12-29 | British Bio Technology | Compounds |
| PT98990A (pt) | 1990-09-19 | 1992-08-31 | American Home Prod | Processo para a preparacao de esteres de acidos carboxilicos de rapamicina |
| US5892112A (en) | 1990-11-21 | 1999-04-06 | Glycomed Incorporated | Process for preparing synthetic matrix metalloprotease inhibitors |
| US5120842A (en) | 1991-04-01 | 1992-06-09 | American Home Products Corporation | Silyl ethers of rapamycin |
| US5100883A (en) | 1991-04-08 | 1992-03-31 | American Home Products Corporation | Fluorinated esters of rapamycin |
| US5118678A (en) | 1991-04-17 | 1992-06-02 | American Home Products Corporation | Carbamates of rapamycin |
| SG64322A1 (en) | 1991-05-10 | 1999-04-27 | Rhone Poulenc Rorer Int | Bis mono and bicyclic aryl and heteroaryl compounds which inhibit egf and/or pdgf receptor tyrosine kinase |
| US5118677A (en) | 1991-05-20 | 1992-06-02 | American Home Products Corporation | Amide esters of rapamycin |
| NZ243082A (en) | 1991-06-28 | 1995-02-24 | Ici Plc | 4-anilino-quinazoline derivatives; pharmaceutical compositions, preparatory processes, and use thereof |
| US5151413A (en) | 1991-11-06 | 1992-09-29 | American Home Products Corporation | Rapamycin acetals as immunosuppressant and antifungal agents |
| GB9125660D0 (en) | 1991-12-03 | 1992-01-29 | Smithkline Beecham Plc | Novel compound |
| GB9300059D0 (en) | 1992-01-20 | 1993-03-03 | Zeneca Ltd | Quinazoline derivatives |
| US5521184A (en) | 1992-04-03 | 1996-05-28 | Ciba-Geigy Corporation | Pyrimidine derivatives and processes for the preparation thereof |
| ZA935112B (en) | 1992-07-17 | 1994-02-08 | Smithkline Beecham Corp | Rapamycin derivatives |
| ZA935111B (en) | 1992-07-17 | 1994-02-04 | Smithkline Beecham Corp | Rapamycin derivatives |
| US5256790A (en) | 1992-08-13 | 1993-10-26 | American Home Products Corporation | 27-hydroxyrapamycin and derivatives thereof |
| GB9221220D0 (en) | 1992-10-09 | 1992-11-25 | Sandoz Ag | Organic componds |
| US5258389A (en) | 1992-11-09 | 1993-11-02 | Merck & Co., Inc. | O-aryl, O-alkyl, O-alkenyl and O-alkynylrapamycin derivatives |
| WO1994011384A1 (en) | 1992-11-13 | 1994-05-26 | Immunex Corporation | Novel cytokine designated elk ligand |
| US5455258A (en) | 1993-01-06 | 1995-10-03 | Ciba-Geigy Corporation | Arylsulfonamido-substituted hydroxamic acids |
| US5629327A (en) | 1993-03-01 | 1997-05-13 | Childrens Hospital Medical Center Corp. | Methods and compositions for inhibition of angiogenesis |
| US5516658A (en) | 1993-08-20 | 1996-05-14 | Immunex Corporation | DNA encoding cytokines that bind the cell surface receptor hek |
| WO1995009847A1 (en) | 1993-10-01 | 1995-04-13 | Ciba-Geigy Ag | Pyrimidineamine derivatives and processes for the preparation thereof |
| US5656643A (en) | 1993-11-08 | 1997-08-12 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Bis mono-and bicyclic aryl and heteroaryl compounds which inhibit EGF and/or PDGF receptor tyrosine kinase |
| CA2175215C (en) | 1993-11-19 | 2008-06-03 | Yat Sun Or | Semisynthetic analogs of rapamycin (macrolides) being immunomodulators |
| SK78196A3 (en) | 1993-12-17 | 1997-02-05 | Sandoz Ag | Rapamycin demethoxy-derivatives, preparation method thereof and pharmaceutical agent containing them |
| US5700823A (en) | 1994-01-07 | 1997-12-23 | Sugen, Inc. | Treatment of platelet derived growth factor related disorders such as cancers |
| IL112249A (en) | 1994-01-25 | 2001-11-25 | Warner Lambert Co | Pharmaceutical compositions containing di and tricyclic pyrimidine derivatives for inhibiting tyrosine kinases of the epidermal growth factor receptor family and some new such compounds |
| IL112248A0 (en) | 1994-01-25 | 1995-03-30 | Warner Lambert Co | Tricyclic heteroaromatic compounds and pharmaceutical compositions containing them |
| WO1995024190A2 (en) | 1994-03-07 | 1995-09-14 | Sugen, Inc. | Receptor tyrosine kinase inhibitors for inhibiting cell proliferative disorders and compositions thereof |
| EP0756627A1 (en) | 1994-04-15 | 1997-02-05 | Amgen Inc. | Hek5, hek7, hek8, hek11, new eph-like receptor protein tyrosine kinases |
| DE59500788D1 (de) | 1994-05-03 | 1997-11-20 | Ciba Geigy Ag | Pyrrolopyrimidinderivate mit antiproliferativer Wirkung |
| US6303769B1 (en) | 1994-07-08 | 2001-10-16 | Immunex Corporation | Lerk-5 dna |
| US5919905A (en) | 1994-10-05 | 1999-07-06 | Immunex Corporation | Cytokine designated LERK-6 |
| US6057124A (en) | 1995-01-27 | 2000-05-02 | Amgen Inc. | Nucleic acids encoding ligands for HEK4 receptors |
| US5863949A (en) | 1995-03-08 | 1999-01-26 | Pfizer Inc | Arylsulfonylamino hydroxamic acid derivatives |
| DE69536015D1 (de) | 1995-03-30 | 2009-12-10 | Pfizer Prod Inc | Chinazolinone Derivate |
| CA2214086C (en) | 1995-04-03 | 2008-07-29 | Novartis Ag | Pyrazole derivatives and processes for the preparation thereof |
| ES2153031T4 (es) | 1995-04-20 | 2001-05-16 | Pfizer | Derivados del acido arilsulfonil hidroxamico como inhibidores de mmp y tnf. |
| GB9508538D0 (en) | 1995-04-27 | 1995-06-14 | Zeneca Ltd | Quinazoline derivatives |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5880141A (en) | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| US5650415A (en) | 1995-06-07 | 1997-07-22 | Sugen, Inc. | Quinoline compounds |
| US5985890A (en) | 1995-06-09 | 1999-11-16 | Novartis Ag | Rapamycin derivatives |
| SI9620103A (sl) | 1995-07-06 | 1998-10-31 | Novartis Ag | Pirolopirimidini in postopki za njihovo pripravo |
| DE19534177A1 (de) | 1995-09-15 | 1997-03-20 | Merck Patent Gmbh | Cyclische Adhäsionsinhibitoren |
| AR004010A1 (es) | 1995-10-11 | 1998-09-30 | Glaxo Group Ltd | Compuestos heterociclicos |
| GB9523675D0 (en) | 1995-11-20 | 1996-01-24 | Celltech Therapeutics Ltd | Chemical compounds |
| ATE225343T1 (de) | 1995-12-20 | 2002-10-15 | Hoffmann La Roche | Matrix-metalloprotease inhibitoren |
| AU1441497A (en) | 1996-01-23 | 1997-08-20 | Novartis Ag | Pyrrolopyrimidines and processes for their preparation |
| JP3406763B2 (ja) | 1996-01-30 | 2003-05-12 | 東レ・ダウコーニング・シリコーン株式会社 | シリコーンゴム組成物 |
| GB9603097D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline compounds |
| GB9603095D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline derivatives |
| DE19608588A1 (de) | 1996-03-06 | 1997-09-11 | Thomae Gmbh Dr K | Pyrimido [5,4-d]pyrimidine, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| DE19629652A1 (de) | 1996-03-06 | 1998-01-29 | Thomae Gmbh Dr K | 4-Amino-pyrimidin-Derivate, diese Verbindungen enthaltende Arzneimittel, deren Verwendung und Verfahren zu ihrer Herstellung |
| WO1997034895A1 (de) | 1996-03-15 | 1997-09-25 | Novartis Ag | NEUE N-7 HETEROCYCLYL-PYRROLO[2,3-d]PYRIMIDINE UND IHRE VERWENDUNG |
| EA001595B1 (ru) | 1996-04-12 | 2001-06-25 | Варнер-Ламберт Компани | Необратимые ингибиторы тирозинкиназ |
| GB9607729D0 (en) | 1996-04-13 | 1996-06-19 | Zeneca Ltd | Quinazoline derivatives |
| CA2258548C (en) | 1996-06-24 | 2005-07-26 | Pfizer Inc. | Phenylamino-substituted tricyclic derivatives for treatment of hyperproliferative diseases |
| AU735648B2 (en) | 1996-07-12 | 2001-07-12 | Ariad Pharmaceuticals, Inc. | Materials and method for treating or preventing pathogenic fungal infection |
| EP0818442A3 (en) | 1996-07-12 | 1998-12-30 | Pfizer Inc. | Cyclic sulphone derivatives as inhibitors of metalloproteinases and of the production of tumour necrosis factor |
| AR007857A1 (es) | 1996-07-13 | 1999-11-24 | Glaxo Group Ltd | Compuestos heterociclicos fusionados como inhibidores de proteina tirosina quinasa, sus metodos de preparacion, intermediarios uso en medicina ycomposiciones farmaceuticas que los contienen. |
| HRP970371A2 (en) | 1996-07-13 | 1998-08-31 | Kathryn Jane Smith | Heterocyclic compounds |
| EA199900021A1 (ru) | 1996-07-13 | 1999-08-26 | Глаксо, Груп Лимитед | Бициклические гетероароматические соединения в качестве ингибиторов протеинтирозинкиназы |
| KR20000067904A (ko) | 1996-07-18 | 2000-11-25 | 디. 제이. 우드, 스피겔 알렌 제이 | 매트릭스 메탈로프로테아제의 포스피네이트계 억제제 |
| ES2308787T3 (es) | 1996-08-16 | 2008-12-01 | Schering Corporation | Antigenos de superficie de celular de mamiferos; reactivos relacionados. |
| US6111090A (en) | 1996-08-16 | 2000-08-29 | Schering Corporation | Mammalian cell surface antigens; related reagents |
| KR20000068248A (ko) | 1996-08-23 | 2000-11-25 | 디. 제이. 우드, 스피겔 알렌 제이 | 아릴설포닐아미노 하이드록삼산 유도체 |
| WO1998007726A1 (en) | 1996-08-23 | 1998-02-26 | Novartis Ag | Substituted pyrrolopyrimidines and processes for their preparation |
| US6251911B1 (en) | 1996-10-02 | 2001-06-26 | Novartis Ag | Pyrimidine derivatives and processes for the preparation thereof |
| ID18494A (id) | 1996-10-02 | 1998-04-16 | Novartis Ag | Turunan pirazola leburan dan proses pembuatannya |
| AU4779897A (en) | 1996-10-02 | 1998-04-24 | Novartis Ag | Fused pyrazole derivatives and processes for their preparation |
| EP0837063A1 (en) | 1996-10-17 | 1998-04-22 | Pfizer Inc. | 4-Aminoquinazoline derivatives |
| GB9621757D0 (en) | 1996-10-18 | 1996-12-11 | Ciba Geigy Ag | Phenyl-substituted bicyclic heterocyclyl derivatives and their use |
| JP3338064B2 (ja) | 1997-01-06 | 2002-10-28 | ファイザー・インク | 環状スルホン誘導体 |
| NZ336840A (en) | 1997-02-03 | 2001-01-26 | Pfizer Prod Inc | Arylsulfonylamino hydroxamic acid derivatives useful in the treatment of tumor necrosis factor and matrix metalloproteinase mediated diseases |
| WO1998033798A2 (en) | 1997-02-05 | 1998-08-06 | Warner Lambert Company | Pyrido[2,3-d]pyrimidines and 4-amino-pyrimidines as inhibitors of cell proliferation |
| JP2000507975A (ja) | 1997-02-07 | 2000-06-27 | ファイザー・インク | N−ヒドロキシ−β−スルホニルプロピオンアミド誘導体類及びそれらのマトリックスメタロプロテイナーゼ阻害薬としての使用 |
| EP0960098A1 (en) | 1997-02-11 | 1999-12-01 | Pfizer Inc. | Arylsulfonyl hydroxamic acid derivatives |
| CO4950519A1 (es) | 1997-02-13 | 2000-09-01 | Novartis Ag | Ftalazinas, preparaciones farmaceuticas que las comprenden y proceso para su preparacion |
| US6150395A (en) | 1997-05-30 | 2000-11-21 | The Regents Of The University Of California | Indole-3-carbinol (I3C) derivatives and methods |
| AU8689298A (en) | 1997-08-05 | 1999-03-01 | Sugen, Inc. | Tricyclic quinoxaline derivatives as protein tyrosine kinase inhibitors |
| CA2299355C (en) | 1997-08-08 | 2005-09-27 | Pfizer Products Inc. | Aryloxyarylsulfonylamino hydroxamic acid derivatives |
| JP2001520039A (ja) | 1997-10-21 | 2001-10-30 | ヒューマン ジノーム サイエンシーズ, インコーポレイテッド | ヒト腫瘍壊死因子レセプター様タンパク質、tr11,tr11sv1およびtr11sv2 |
| GB9725782D0 (en) | 1997-12-05 | 1998-02-04 | Pfizer Ltd | Therapeutic agents |
| RS49779B (sr) | 1998-01-12 | 2008-06-05 | Glaxo Group Limited, | Biciklična heteroaromatična jedinjenja kao inhibitori protein tirozin kinaze |
| GB9800575D0 (en) | 1998-01-12 | 1998-03-11 | Glaxo Group Ltd | Heterocyclic compounds |
| GB9801690D0 (en) | 1998-01-27 | 1998-03-25 | Pfizer Ltd | Therapeutic agents |
| AU2591599A (en) | 1998-02-09 | 1999-08-23 | Genentech Inc. | Novel tumor necrosis factor receptor homolog and nucleic acids encoding the same |
| CA2322311C (en) | 1998-03-04 | 2009-10-13 | Bristol-Myers Squibb Company | Heterocyclo-substituted imidazopyrazine protein tyrosine kinase inhibitors |
| PA8469401A1 (es) | 1998-04-10 | 2000-05-24 | Pfizer Prod Inc | Derivados biciclicos del acido hidroxamico |
| PA8469501A1 (es) | 1998-04-10 | 2000-09-29 | Pfizer Prod Inc | Hidroxamidas del acido (4-arilsulfonilamino)-tetrahidropiran-4-carboxilico |
| SK287132B6 (sk) | 1998-05-29 | 2009-12-07 | Sugen, Inc. | Farmaceutická kompozícia obsahujúca pyrolom substituovaný 2-indolinón, súprava obsahujúca uvedenú kompozíciu a použitie pyrolom substituovaného 2-indolinónu |
| UA60365C2 (uk) | 1998-06-04 | 2003-10-15 | Пфайзер Продактс Інк. | Похідні ізотіазолу, спосіб їх одержання, фармацевтична композиція та спосіб лікування гіперпроліферативного захворювання у ссавця |
| CA2336848A1 (en) | 1998-07-10 | 2000-01-20 | Merck & Co., Inc. | Novel angiogenesis inhibitors |
| CA2341409A1 (en) | 1998-08-31 | 2000-03-09 | Merck And Co., Inc. | Novel angiogenesis inhibitors |
| EP1004578B1 (en) | 1998-11-05 | 2004-02-25 | Pfizer Products Inc. | 5-oxo-pyrrolidine-2-carboxylic acid hydroxamide derivatives |
| EP1158985B1 (en) | 1999-01-13 | 2011-12-28 | Bayer HealthCare LLC | OMEGA-CARBOXY ARYL SUBSTITUTED DIPHENYL UREAS AS p38 KINASE INHIBITORS |
| JP2000256358A (ja) | 1999-03-10 | 2000-09-19 | Yamanouchi Pharmaceut Co Ltd | ピラゾール誘導体 |
| CN1660840A (zh) | 1999-03-30 | 2005-08-31 | 诺瓦提斯公司 | 治疗炎性疾病的酞嗪衍生物 |
| GB9912961D0 (en) | 1999-06-03 | 1999-08-04 | Pfizer Ltd | Metalloprotease inhibitors |
| ATE324444T1 (de) | 1999-06-07 | 2006-05-15 | Immunex Corp | Tek-antagonisten |
| US6521424B2 (en) | 1999-06-07 | 2003-02-18 | Immunex Corporation | Recombinant expression of Tek antagonists |
| WO2001003720A2 (en) | 1999-07-12 | 2001-01-18 | Genentech, Inc. | Promotion or inhibition of angiogenesis and cardiovascularization by tumor necrosis factor ligand/receptor homologs |
| ATE264863T1 (de) | 1999-08-24 | 2004-05-15 | Ariad Gene Therapeutics Inc | 28-epirapaloge |
| KR100881105B1 (ko) | 1999-11-05 | 2009-02-02 | 아스트라제네카 아베 | Vegf 억제제로서의 퀴나졸린 유도체 |
| WO2001037820A2 (en) | 1999-11-24 | 2001-05-31 | Sugen, Inc. | Ionizable indolinone derivatives and their use as ptk ligands |
| US6515004B1 (en) | 1999-12-15 | 2003-02-04 | Bristol-Myers Squibb Company | N-[5-[[[5-alkyl-2-oxazolyl]methyl]thio]-2-thiazolyl]-carboxamide inhibitors of cyclin dependent kinases |
| US6727225B2 (en) | 1999-12-20 | 2004-04-27 | Immunex Corporation | TWEAK receptor |
| NZ521437A (en) | 2000-02-25 | 2004-04-30 | Immunex Corp | Integrin antagonists suitable as inhibitors of angiogenesis |
| US6630500B2 (en) | 2000-08-25 | 2003-10-07 | Cephalon, Inc. | Selected fused pyrrolocarbazoles |
| BR0116452A (pt) | 2000-12-21 | 2003-09-30 | Glaxo Group Ltd | Composto, composição farmacêutica, uso de um composto |
| US7102009B2 (en) | 2001-01-12 | 2006-09-05 | Amgen Inc. | Substituted amine derivatives and methods of use |
| US6995162B2 (en) | 2001-01-12 | 2006-02-07 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| US7105682B2 (en) | 2001-01-12 | 2006-09-12 | Amgen Inc. | Substituted amine derivatives and methods of use |
| US6878714B2 (en) | 2001-01-12 | 2005-04-12 | Amgen Inc. | Substituted alkylamine derivatives and methods of use |
| US20020147198A1 (en) | 2001-01-12 | 2002-10-10 | Guoqing Chen | Substituted arylamine derivatives and methods of use |
| GB0215676D0 (en) | 2002-07-05 | 2002-08-14 | Novartis Ag | Organic compounds |
| US7307088B2 (en) | 2002-07-09 | 2007-12-11 | Amgen Inc. | Substituted anthranilic amide derivatives and methods of use |
| TWI329112B (en) | 2002-07-19 | 2010-08-21 | Bristol Myers Squibb Co | Novel inhibitors of kinases |
| WO2004039795A2 (en) | 2002-10-29 | 2004-05-13 | Fujisawa Pharmaceutical Co., Ltd. | Amide compounds for the treatment of hyperlipidemia |
| US7618632B2 (en) | 2003-05-23 | 2009-11-17 | Wyeth | Method of treating or ameliorating an immune cell associated pathology using GITR ligand antibodies |
| US20060173033A1 (en) | 2003-07-08 | 2006-08-03 | Michaela Kneissel | Use of rapamycin and rapamycin derivatives for the treatment of bone loss |
| WO2005007190A1 (en) | 2003-07-11 | 2005-01-27 | Schering Corporation | Agonists or antagonists of the clucocorticoid-induced tumour necrosis factor receptor (gitr) or its ligand for the treatment of immune disorders, infections and cancer |
| WO2005016252A2 (en) | 2003-07-11 | 2005-02-24 | Ariad Gene Therapeutics, Inc. | Phosphorus-containing macrocycles |
| AR045134A1 (es) | 2003-07-29 | 2005-10-19 | Smithkline Beecham Plc | Compuesto de 1h - imidazo [4,5-c] piridin-ilo, composicion farmaceutica que lo comprende, proceso para prepararla, su uso para preparar dicha composicion farmaceutica, combinacion farmaceutica, uso de la combinacion farmaceutica para la preparacion de un medicamento, procedimientos para preparar dic |
| WO2005055808A2 (en) | 2003-12-02 | 2005-06-23 | Genzyme Corporation | Compositions and methods to diagnose and treat lung cancer |
| GB0409799D0 (en) | 2004-04-30 | 2004-06-09 | Isis Innovation | Method of generating improved immune response |
| US20060002932A1 (en) | 2004-06-04 | 2006-01-05 | Duke University | Methods and compositions for enhancement of immunity by in vivo depletion of immunosuppressive cell activity |
| DE602005020465D1 (de) | 2004-08-26 | 2010-05-20 | Pfizer | Enantiomerenreine aminoheteroaryl-verbindungen als proteinkinasehemmer |
| EP1799699A1 (en) | 2004-10-13 | 2007-06-27 | Wyeth | Analogs of 17-hydroxywortmannin as pi3k inhibitors |
| US7776869B2 (en) | 2004-10-18 | 2010-08-17 | Amgen Inc. | Heteroaryl-substituted alkyne compounds and method of use |
| PT2343320T (pt) | 2005-03-25 | 2018-01-23 | Gitr Inc | Anticorpos anti-gitr e as suas utilizações |
| DK2439273T3 (da) | 2005-05-09 | 2019-06-03 | Ono Pharmaceutical Co | Humane monoklonale antistoffer til programmeret død-1(pd-1) og fremgangsmåder til behandling af cancer ved anvendelse af anti-pd-1- antistoffer alene eller i kombination med andre immunterapeutika |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| WO2007011760A2 (en) | 2005-07-15 | 2007-01-25 | Kalypsys, Inc. | Inhibitors of mitotic kinesin |
| KR20080040007A (ko) | 2005-09-20 | 2008-05-07 | 화이자 프로덕츠 인크. | 티로신 키나제 억제제를 이용한 치료의 투여형 및 방법 |
| US20070135435A1 (en) | 2005-11-02 | 2007-06-14 | Xiangping Qian | Certain chemical entities, compositions, and methods |
| WO2007133822A1 (en) | 2006-01-19 | 2007-11-22 | Genzyme Corporation | Gitr antibodies for the treatment of cancer |
| MX2009005950A (es) | 2006-12-07 | 2009-10-12 | Genentech Inc | Compuestos inhibidores de fosfoinositido 3-quinasas y metodos de uso. |
| CA2679659C (en) | 2007-03-01 | 2016-01-19 | Novartis Ag | Pim kinase inhibitors and methods of their use |
| JP5383484B2 (ja) | 2007-04-24 | 2014-01-08 | 塩野義製薬株式会社 | 環式基で置換されたアミノジヒドロチアジン誘導体 |
| US8591886B2 (en) | 2007-07-12 | 2013-11-26 | Gitr, Inc. | Combination therapies employing GITR binding molecules |
| EP2205242B1 (en) | 2007-09-12 | 2015-04-15 | Genentech, Inc. | Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents, and methods of use |
| JP5348725B2 (ja) | 2007-10-25 | 2013-11-20 | ジェネンテック, インコーポレイテッド | チエノピリミジン化合物の製造方法 |
| US20100168084A1 (en) | 2008-05-08 | 2010-07-01 | Huber L Julie | Therapeutic compounds and related methods of use |
| NZ590667A (en) | 2008-07-02 | 2013-01-25 | Emergent Product Dev Seattle | Tgf-b antagonist multi-target binding proteins |
| CN102149820B (zh) | 2008-09-12 | 2014-07-23 | 国立大学法人三重大学 | 能够表达外源gitr配体的细胞 |
| KR101790802B1 (ko) | 2009-09-03 | 2017-10-27 | 머크 샤프 앤드 돔 코포레이션 | 항-gitr 항체 |
| GB0919054D0 (en) | 2009-10-30 | 2009-12-16 | Isis Innovation | Treatment of obesity |
| CA2785907A1 (en) | 2009-12-29 | 2011-07-28 | Emergent Product Development Seattle, Llc | Ron binding constructs and methods of use thereof |
| WO2011084985A1 (en) | 2010-01-07 | 2011-07-14 | Boehringer Ingelheim International Gmbh | Cxcr3 receptor antagonists |
| WO2011085261A1 (en) * | 2010-01-08 | 2011-07-14 | Selexagen Therapeutics, Inc. | Hedgehog inhibitors |
| WO2013039954A1 (en) | 2011-09-14 | 2013-03-21 | Sanofi | Anti-gitr antibodies |
| WO2014007228A1 (ja) | 2012-07-03 | 2014-01-09 | 小野薬品工業株式会社 | ソマトスタチン受容体作動活性を有する化合物およびその医薬用途 |
| EP3033091B1 (en) | 2013-08-16 | 2022-09-07 | Versitech Limited | Probiotic composition and use thereof in the prevention and treatment of hepatocellular carcinoma |
| AU2014347027A1 (en) | 2013-11-06 | 2016-06-23 | Bristol-Myers Squibb Company | GSK -3 inhibitors |
| WO2015143654A1 (en) | 2014-03-26 | 2015-10-01 | Merck Sharp & Dohme Corp. | TrkA KINASE INHIBITORS,COMPOSITIONS AND METHODS THEREOF |
| JP6759514B2 (ja) | 2014-08-01 | 2020-09-23 | ヌエヴォリューション・アクティーゼルスカブNuevolution A/S | ブロモドメインに対して活性な化合物 |
| ES2878078T3 (es) | 2016-11-28 | 2021-11-18 | Bristol Myers Squibb Co | Inhibidores de GSK-3 |
| CA3068305A1 (en) | 2017-06-29 | 2019-01-03 | Rigel Pharmaceuticals, Inc. | Kinase inhibitors and methods for making and using |
| US20200339591A1 (en) | 2017-12-21 | 2020-10-29 | Gliapharm Sa | Compositions and methods of treatment for neurological disorders comprising a dementia |
| EP3755703B1 (en) | 2018-02-20 | 2022-05-04 | Incyte Corporation | N-(phenyl)-2-(phenyl)pyrimidine-4-carboxamide derivatives and related compounds as hpk1 inhibitors for treating cancer |
| WO2020006075A1 (en) | 2018-06-26 | 2020-01-02 | The Johns Hopkins University | Positron emission tomography (pet) radiotracers for imaging macrophage colony-stimulating factor 1 receptor (csf1r) in neuroinflammation |
| US12344595B2 (en) | 2018-11-02 | 2025-07-01 | Merck Sharp & Dohme Llc | 2-amino-n-phenyl-nicotinamides as NAV1.8 inhibitors |
| JP7288051B2 (ja) | 2018-11-02 | 2023-06-06 | メルク・シャープ・アンド・ドーム・エルエルシー | Nav1.8阻害薬としての2-アミノ-N-ヘテロアリール-ニコチンアミド類 |
| AU2019401495B2 (en) | 2018-12-20 | 2025-06-26 | Amgen Inc. | Heteroaryl amides useful as KIF18A inhibitors |
| JP2022513967A (ja) | 2018-12-20 | 2022-02-09 | アムジエン・インコーポレーテツド | Kif18a阻害剤として有用なヘテロアリールアミド |
| MA54543A (fr) | 2018-12-20 | 2022-03-30 | Amgen Inc | Inhibiteurs de kif18a |
| JP7686559B2 (ja) | 2018-12-20 | 2025-06-02 | アムジエン・インコーポレーテツド | Kif18a阻害剤 |
| MX2022012992A (es) * | 2020-04-14 | 2022-11-08 | Amgen Inc | Inhibidores de kif18a para el tratamiento de enfermedades neoplasicas. |
-
2019
- 2019-12-20 JP JP2021534697A patent/JP7686559B2/ja active Active
- 2019-12-20 AU AU2019403488A patent/AU2019403488B2/en active Active
- 2019-12-20 MX MX2021007104A patent/MX2021007104A/es unknown
- 2019-12-20 WO PCT/US2019/068172 patent/WO2020132651A1/en not_active Ceased
- 2019-12-20 US US17/416,411 patent/US12459932B2/en active Active
- 2019-12-20 EP EP19845855.6A patent/EP3897855B1/en active Active
- 2019-12-20 ES ES19845855T patent/ES2953821T3/es active Active
- 2019-12-20 MA MA054550A patent/MA54550A/fr unknown
- 2019-12-20 CA CA3123042A patent/CA3123042A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| AU2019403488A1 (en) | 2021-06-24 |
| MA54550A (fr) | 2022-03-30 |
| US12459932B2 (en) | 2025-11-04 |
| AU2019403488B2 (en) | 2025-07-24 |
| MX2021007104A (es) | 2021-08-11 |
| CA3123042A1 (en) | 2020-06-25 |
| US20220073504A1 (en) | 2022-03-10 |
| JP7686559B2 (ja) | 2025-06-02 |
| JP2022514268A (ja) | 2022-02-10 |
| EP3897855A1 (en) | 2021-10-27 |
| EP3897855B1 (en) | 2023-06-07 |
| WO2020132651A1 (en) | 2020-06-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2953821T3 (es) | Inhibidores de KIF18A | |
| ES2996960T3 (en) | Heteroaryl amides useful as kif18a inhibitors | |
| EP4007752B1 (en) | Kif18a inhibitors | |
| ES2997190T3 (en) | Heteroaryl amides useful as kif18a inhibitors | |
| EP4007753B1 (en) | Kif18a inhibitors | |
| KR102875569B1 (ko) | Kif18a 억제제 | |
| ES3060733T3 (en) | Kif18a inhibitors | |
| EP4007638A1 (en) | Pyridine derivatives as kif18a inhibitors |