JP4550141B2 - 放射性フッ素標識有機化合物の製造方法 - Google Patents
放射性フッ素標識有機化合物の製造方法 Download PDFInfo
- Publication number
- JP4550141B2 JP4550141B2 JP2008515492A JP2008515492A JP4550141B2 JP 4550141 B2 JP4550141 B2 JP 4550141B2 JP 2008515492 A JP2008515492 A JP 2008515492A JP 2008515492 A JP2008515492 A JP 2008515492A JP 4550141 B2 JP4550141 B2 JP 4550141B2
- Authority
- JP
- Japan
- Prior art keywords
- column
- solution
- reaction
- radioactive fluorine
- deesterification
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *C(C1)(CC1F)C(O*)=O Chemical compound *C(C1)(CC1F)C(O*)=O 0.000 description 2
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/14—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof
- C07C227/18—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions involving amino or carboxyl groups, e.g. hydrolysis of esters or amides, by formation of halides, salts or esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/001—Acyclic or carbocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/14—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof
- C07C227/18—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions involving amino or carboxyl groups, e.g. hydrolysis of esters or amides, by formation of halides, salts or esters
- C07C227/20—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton from compounds containing already amino and carboxyl groups or derivatives thereof by reactions involving amino or carboxyl groups, e.g. hydrolysis of esters or amides, by formation of halides, salts or esters by hydrolysis of N-acylated amino-acids or derivatives thereof, e.g. hydrolysis of carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/46—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino or carboxyl groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C229/48—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino or carboxyl groups bound to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups and carboxyl groups bound to carbon atoms of the same non-condensed ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Description
syn−1−(N−(t−ブトキシカルボニル)アミノ)−3−[((トリフルオロメチル)スルフォニル)オキシ]−シクロブタン−1−カルボン酸エチルエステルの合成
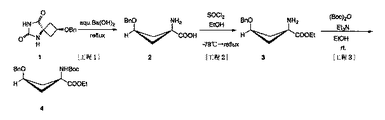
syn−5−(3−ベンジルオキシシクロブタン)ヒダントイン6.15g(25mmol相当)に、飽和水酸化バリウム水溶液250 mLを加え、114℃の油浴にて24時間以上加熱還流した。クロロホルム:メタノール=5:1(syn−ヒダントインのRf値=0.6付近)及びクロロホルム:メタノール=95:1(syn−ヒダントインのRf値=0.3付近)の2種類の系を展開溶媒として使用したTLC分析を行い、反応終了の確認を行った(UVとリンモリブデン酸による呈色によって確認)。
充分に乾燥させ水分を取り除いたsyn−1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸5.67gを、エタノール200mLに溶解させた。この液に、トリエチルアミン 9.5mL(75mmol相当)を加え、‐78℃にて20分間冷却し、塩化チオニル 4.6mL(62.5mmol相当)を加えた。反応液を、0℃で1時間、室温で1時間それぞれ攪拌した後、95℃の油浴にて、1晩加熱還流した。クロロホルム:メタノール=95:1(目的物のRf値=0.6付近)を展開溶媒として使用したTLC分析(UVとリンモリブデン酸による呈色にて確認)により、反応終了の確認を行った。反応終了確認後、反応液を減圧濃縮してsyn−1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸エチルエステル7.64gを白色結晶として得た。
syn−1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸エチルエステル7.64gを、エタノール:トリエチルアミン=9:1混液250mLに溶解させた。この溶液を氷浴で15分冷却した後、二炭酸ジ−t−ブチル 8.6mL(37.5mmol相当)を加え、室温下1晩攪拌した。ヘキサン:酢酸エチル=1:1(反応目的物のRf値=0.6付近)を展開溶媒として使用したTLC(UV及びモリブデン酸による呈色にて確認)にて、反応終了を確認した。反応終了確認後、反応液を減圧濃縮し、残渣として白色結晶を得た。この残渣に、冷酢酸エチル 150mLと0.5mol/Lの冷塩酸150mLを加え、室温で5分攪拌し、次いで静置分離した。有機層を水150mL×2、飽和炭酸水素ナトリウム水溶液150mL、水150mL×2,飽和食塩水150mL×2の順で抽出・洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮し、黄色油状物を得た。別に、水層を酢酸エチル150mL×2、水150mL×2、飽和食塩水150mLの順で抽出・洗浄し、無水硫酸ナトリウムで乾燥後減圧濃縮することにより、少量の黄色油状物を回収した。一連の操作により、淡黄色油状物8.82gを得た。残渣をシリカゲルカラムクロマトグラフィーにより分離精製(ヘキサン:酢酸エチル=1:1)することにより、白色結晶のsyn−1−(N−(t−ブトキシカルボニル)アミノ)−3−ベンジルオキシ−シクロブタン−1−カルボン酸エチルエステル8.04g(23mmol相当)を得た。
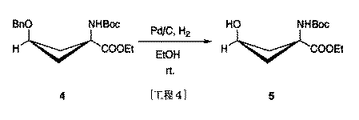
syn−1−(N−(t−ブトキシカルボニル)アミノ)−3−ベンジルオキシ−シクロブタン−1−カルボン酸エチルエステル 8.04g(23mmol相当)にエタノール150mLを加えた後、パラジウム−活性炭素(パラジウム10%)960mgを加え、水素置換、室温下で一晩攪拌した。反応後、セライトを用いた濾過によりパラジウム−活性炭素を濾去して濾液を減圧濃縮し、残渣として白色結晶5.74gを得た。なお、ヘキサン:酢酸エチル=1:1(反応目的物のRf値=0.2付近)を展開溶媒として使用したTLC分析(UVとニンヒドリンによる呈色にて確認)にて反応追跡を行い、反応終了を確認した。次いで残渣をシリカゲルカラムクロマトグラフィー (ヘキサン:酢酸エチル=1:1,ヘキサン:酢酸エチル=4:1) により分離精製し、白色結晶としてsyn−1−(N−(t−ブトキシカルボニル)アミノ)−3−ヒドロキシ−シクロブタン−1−カルボン酸エチルエステル5.36g(20.7mmol相当)を得た。
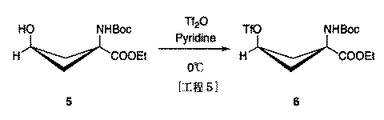
syn−1−(N−(t−ブトキシカルボニル)アミノ)−3−ヒドロキシ−シクロブタン−1−カルボン酸エチルエステル2.07g(8mmol)をピリジン26mLに溶解させ、氷浴下20分間攪拌した。無水トリフルオロメタンスルホン酸2.0mL(12mmol相当)を加え、そのまま30分間攪拌した。ヘキサン:ジエチルエーテル=1:1を展開溶媒(反応目的物のRf値=0.6付近)としたTLC分析(ニンヒドリンの呈色にて確認)を用いて反応を追跡し、反応終了を確認した。反応終了を確認後、反応液に水100mLとエーテル100mLを加え、1mol/L塩酸100mL×2、水100mL×2、飽和食塩水100mL×2の順で抽出洗浄した。無水硫酸ナトリウムで乾燥後、減圧濃縮することにより、淡黄色結晶2.78gを得た。この反応混合物をシリカゲルカラムクロマトグラフィーにより分離精製(ヘキサン:ジエチルエーテル=3:1)することにより得られた白色結晶につき、さらにペンタン:ジエチルエーテルを用いて再結晶を行うことにより、syn−1−(N−(t−ブトキシカルボニル)アミノ)−3−[((トリフルオロメチル)スルフォニル)オキシ]−シクロブタン−1−カルボン酸エチルエステル1.84g(4.7mmol相当)を得た。
[18F]フッ化物イオン含有H2 18O(13〜182GBq)を、陰イオン交換カラムに通液し、[18F]フッ化物イオンを吸着捕集した。次いで、該カラムに炭酸カリウム溶液を通液して[18F]フッ化物イオンを溶出し、さらに水でフラッシングを行って溶出液と合わせた。この液に、4、7、13、16、21、24−ヘキサオキサ−1、10−ジアザビシクロ[8.8.8]ヘキサコサン(商品名:クリプトフィックス222、メルク社製)のアセトニトリル溶液を加え、加熱蒸散を行って乾固させた。
展開相:アセトニトリル/メタノール/水/酢酸=20/5/5/1
TLCプレート:Silica Gel 60F254(商品名、膜厚:0.25mm、メルク社製)
展開長: 10cm
TLCスキャナー: Rita Star(Raytest社製)
カラム:CAPCELLPAK C18 MG(製品名、株式会社資生堂製、サイズ:5μm、4.6mmI.D.×250mm)
カラム温度:室温(約25℃)
移動相:5mmol/Lオクタンスルホン酸ナトリウム含有リン酸緩衝液 (pH2.1)をA液、アセトニトリルをB液とし、A液とB液の混合比を表1のように変えて濃度勾配制御を行った。
試料注入量:10μL
ポストカラム誘導体化条件:
反応液:0.3mol/Lホウ酸緩衝液(pH10.4)、6mmol/L o−フタルアルデヒド、6mmol/L N−アセチル−L−システイン
反応液流量:1.0mL/分
反応温度:50℃
検出器:蛍光検出器(形式:Waters2475型(日本ウォーターズ株式会社製)励起波長:330nm、蛍光波長:430nm)
[18F]フッ化物イオン含有H2 18O(7〜36GBq)を、陰イオン交換カラムに通液し、[18F]フッ化物イオンを、吸着捕集した。次いで、該カラムに炭酸カリウム溶液を通液して[18F]フッ化物イオンを溶出し、さらに水でフラッシングを行い溶出液と合わせた。この液に、4、7、13、16、21、24−ヘキサオキサ−1、10−ジアザビシクロ[8.8.8]ヘキサコサン(商品名:クリプトフィックス222、メルク社製)のアセトニトリル溶液を加え、加熱蒸散を行って乾固させた。
Claims (5)
- 下記式(1):
で表される化合物を得る脱エステル化工程と、
脱エステル化工程にて得られた前記化合物につきアミノ保護基の脱保護を行って下記式(3):
を含むことを特徴とする、放射性フッ素標識有機化合物の製造方法。 - 式(2)中、Xはナトリウム又はカリウムである請求項1に記載の放射性フッ素標識有機化合物の製造方法。
- 脱エステル化工程で用いるアルカリ溶液が、水酸化ナトリウム溶液又は水酸化カリウム溶液である請求項2に記載の放射性フッ素標識有機化合物の製造方法。
- 脱エステル化工程で用いるアルカリ溶液が、水酸化ナトリウム水溶液である請求項3に記載の放射性フッ素標識有機化合物の製造方法。
- 脱エステル化工程にて用いる逆相カラムにおける充填剤が、炭素数1〜18のアルキル鎖がケイ素を介して担体と結合した構造を有するものである、請求項1乃至4の何れか1項に記載の放射性フッ素標識有機化合物の製造方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006132089 | 2006-05-11 | ||
| JP2006132089 | 2006-05-11 | ||
| PCT/JP2007/059459 WO2007132689A1 (ja) | 2006-05-11 | 2007-05-07 | 放射性フッ素標識有機化合物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2007132689A1 JPWO2007132689A1 (ja) | 2009-09-24 |
| JP4550141B2 true JP4550141B2 (ja) | 2010-09-22 |
Family
ID=38693787
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008515492A Active JP4550141B2 (ja) | 2006-05-11 | 2007-05-07 | 放射性フッ素標識有機化合物の製造方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7897811B2 (ja) |
| EP (1) | EP2017258B1 (ja) |
| JP (1) | JP4550141B2 (ja) |
| KR (1) | KR101317258B1 (ja) |
| CN (1) | CN101443307B (ja) |
| AT (1) | ATE549312T1 (ja) |
| AU (1) | AU2007251015B2 (ja) |
| BR (1) | BRPI0711144B1 (ja) |
| CA (1) | CA2651786C (ja) |
| DK (1) | DK2017258T3 (ja) |
| ES (1) | ES2380372T3 (ja) |
| IL (1) | IL195187A (ja) |
| NO (1) | NO341173B1 (ja) |
| NZ (1) | NZ572936A (ja) |
| PT (1) | PT2017258E (ja) |
| RU (1) | RU2434846C2 (ja) |
| TW (1) | TWI389872B (ja) |
| WO (1) | WO2007132689A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10023525B2 (en) | 2012-08-09 | 2018-07-17 | Ge Healthcare Limited | Preparation of 18F-fluciclovine |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL1978015T3 (pl) | 2005-11-29 | 2018-07-31 | Nihon Medi-Physics Co., Ltd. | Związek prekursorowy związku organicznego znakowanego radioaktywnym halogenem |
| KR101408727B1 (ko) * | 2006-12-27 | 2014-06-17 | 니혼 메디피직스 가부시키가이샤 | 방사성 할로겐 표식 유기 화합물의 전구체 화합물의 제조 방법 |
| TWI406674B (zh) | 2007-02-13 | 2013-09-01 | 日本藥學物理公司 | Method for manufacturing diagnostic radiographic diagnostic agents |
| KR101583544B1 (ko) * | 2007-12-19 | 2016-01-08 | 니혼 메디피직스 가부시키가이샤 | 방사성 불소 표식 유기 화합물의 제조방법 |
| GB0812923D0 (en) * | 2008-07-15 | 2008-08-20 | Isis Innovation | Preparation of flourine-labelled compounds |
| DK2646411T3 (en) * | 2010-11-29 | 2017-10-16 | Blue Earth Diagnostics Ltd | PREPARATION OF PET PRECURSOR |
| US9061977B2 (en) * | 2010-12-20 | 2015-06-23 | Ge Healthcare Limited | Purification of precursor compound by crystallisation |
| GB201021523D0 (en) * | 2010-12-20 | 2011-02-02 | Ge Healthcare Ltd | Process simplification for precursor compound |
| GB201021530D0 (en) * | 2010-12-20 | 2011-02-02 | Ge Healthcare Ltd | Purification of precursor compound by crystallisation |
| US11534494B2 (en) | 2011-12-21 | 2022-12-27 | Ge Healthcare Limited | Formulation and method of synthesis |
| GB201411569D0 (en) | 2014-06-30 | 2014-08-13 | Ge Healthcare Ltd | Novel formulation and method of synthesis |
| KR20140102700A (ko) * | 2011-12-21 | 2014-08-22 | 지이 헬쓰케어 리미티드 | 시트레이트 완충제 내 18f―플루시클로빈 조성물 |
| GB2561122B (en) * | 2012-12-21 | 2019-07-17 | Ge Healthcare Ltd | Composition comprising [18F]-Fluciclovine |
| GB201305687D0 (en) | 2013-03-28 | 2013-05-15 | Ge Healthcare Ltd | Radiolabelling process |
| JP6755862B2 (ja) * | 2015-06-05 | 2020-09-16 | 日本メジフィジックス株式会社 | 放射性標識化合物の製造装置及び製造方法 |
| JP6485914B2 (ja) * | 2015-10-28 | 2019-03-20 | 日本メジフィジックス株式会社 | フルテメタモルの製造方法 |
| CN106770883B (zh) * | 2017-01-03 | 2019-01-29 | 原子高科股份有限公司 | 一种氟[18f]化钠注射液放化纯的薄层色谱分析方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005030677A1 (ja) * | 2003-09-30 | 2005-04-07 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素化合物の製造方法 |
| JP2006510706A (ja) * | 2002-12-20 | 2006-03-30 | ジーイー・ヘルスケア・リミテッド | 18f−標識アミノ酸の固相製造 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5808146A (en) | 1995-11-09 | 1998-09-15 | Emory University | Amino acid analogs for tumor imaging |
| BE1010280A3 (fr) | 1996-05-02 | 1998-05-05 | Coincidence S A | Procede et dispositif de synthese de 2-[18f] fluoro-2-deoxy-d-glucose. |
| RU2165266C1 (ru) * | 2000-06-28 | 2001-04-20 | Центральный научно-исследовательский рентгенорадиологический институт | Способ получения [2-18f]-2-дезоксиглюкозы |
-
2007
- 2007-05-07 NZ NZ572936A patent/NZ572936A/en unknown
- 2007-05-07 JP JP2008515492A patent/JP4550141B2/ja active Active
- 2007-05-07 US US12/227,240 patent/US7897811B2/en active Active
- 2007-05-07 WO PCT/JP2007/059459 patent/WO2007132689A1/ja not_active Ceased
- 2007-05-07 DK DK07742894.4T patent/DK2017258T3/da active
- 2007-05-07 CN CN2007800171253A patent/CN101443307B/zh active Active
- 2007-05-07 BR BRPI0711144A patent/BRPI0711144B1/pt active IP Right Grant
- 2007-05-07 AU AU2007251015A patent/AU2007251015B2/en active Active
- 2007-05-07 CA CA2651786A patent/CA2651786C/en active Active
- 2007-05-07 KR KR1020087027346A patent/KR101317258B1/ko active Active
- 2007-05-07 EP EP07742894A patent/EP2017258B1/en active Active
- 2007-05-07 PT PT07742894T patent/PT2017258E/pt unknown
- 2007-05-07 AT AT07742894T patent/ATE549312T1/de active
- 2007-05-07 RU RU2008148851/04A patent/RU2434846C2/ru active
- 2007-05-07 ES ES07742894T patent/ES2380372T3/es active Active
- 2007-05-08 TW TW096116347A patent/TWI389872B/zh active
-
2008
- 2008-11-10 IL IL195187A patent/IL195187A/en active IP Right Grant
- 2008-12-04 NO NO20085076A patent/NO341173B1/no unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006510706A (ja) * | 2002-12-20 | 2006-03-30 | ジーイー・ヘルスケア・リミテッド | 18f−標識アミノ酸の固相製造 |
| WO2005030677A1 (ja) * | 2003-09-30 | 2005-04-07 | Nihon Medi-Physics Co., Ltd. | 放射性フッ素化合物の製造方法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10023525B2 (en) | 2012-08-09 | 2018-07-17 | Ge Healthcare Limited | Preparation of 18F-fluciclovine |
Also Published As
| Publication number | Publication date |
|---|---|
| IL195187A0 (en) | 2009-08-03 |
| NO341173B1 (no) | 2017-09-04 |
| US7897811B2 (en) | 2011-03-01 |
| TWI389872B (zh) | 2013-03-21 |
| RU2008148851A (ru) | 2010-06-20 |
| NO20085076L (no) | 2009-02-10 |
| AU2007251015B2 (en) | 2011-09-01 |
| CN101443307B (zh) | 2012-05-16 |
| JPWO2007132689A1 (ja) | 2009-09-24 |
| RU2434846C2 (ru) | 2011-11-27 |
| EP2017258A1 (en) | 2009-01-21 |
| KR20090034805A (ko) | 2009-04-08 |
| BRPI0711144B1 (pt) | 2017-03-28 |
| TW200811084A (en) | 2008-03-01 |
| HK1129884A1 (en) | 2009-12-11 |
| IL195187A (en) | 2013-04-30 |
| US20090198085A1 (en) | 2009-08-06 |
| CA2651786A1 (en) | 2007-11-22 |
| WO2007132689A1 (ja) | 2007-11-22 |
| NZ572936A (en) | 2011-01-28 |
| EP2017258A4 (en) | 2011-05-25 |
| BRPI0711144A2 (pt) | 2011-08-23 |
| DK2017258T3 (da) | 2012-07-09 |
| KR101317258B1 (ko) | 2013-10-14 |
| PT2017258E (pt) | 2012-04-12 |
| ES2380372T3 (es) | 2012-05-11 |
| CN101443307A (zh) | 2009-05-27 |
| CA2651786C (en) | 2014-11-04 |
| EP2017258B1 (en) | 2012-03-14 |
| AU2007251015A1 (en) | 2007-11-22 |
| ATE549312T1 (de) | 2012-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4550141B2 (ja) | 放射性フッ素標識有機化合物の製造方法 | |
| JP5732198B2 (ja) | 放射性フッ素標識有機化合物の製造方法 | |
| ES2663496T3 (es) | Compuesto precursor de compuesto orgánico marcado con halógeno radiactivo | |
| HK1129884B (en) | Process for production of radioactive fluorine-labeled organic compound | |
| HK1151513B (en) | Process for production of radioactive-fluorine-labeled organic compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20091104 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20091104 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20091203 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20091215 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100105 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100217 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100615 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100707 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4550141 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130716 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |