JP7419302B2 - 11-メチレンステロイドの製造のための方法および新規な中間体 - Google Patents
11-メチレンステロイドの製造のための方法および新規な中間体 Download PDFInfo
- Publication number
- JP7419302B2 JP7419302B2 JP2021131984A JP2021131984A JP7419302B2 JP 7419302 B2 JP7419302 B2 JP 7419302B2 JP 2021131984 A JP2021131984 A JP 2021131984A JP 2021131984 A JP2021131984 A JP 2021131984A JP 7419302 B2 JP7419302 B2 JP 7419302B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- formula
- alkyl
- solvate
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J11/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0059—Estrane derivatives substituted in position 17 by a keto group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0081—Substituted in position 17 alfa and 17 beta
- C07J1/0088—Substituted in position 17 alfa and 17 beta the substituent in position 17 alfa being an unsaturated hydrocarbon group
- C07J1/0096—Alkynyl derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J21/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having an oxygen-containing hetero ring spiro-condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J21/005—Ketals
- C07J21/008—Ketals at position 17
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J33/00—Normal steroids having a sulfur-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J33/005—Normal steroids having a sulfur-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton spiro-condensed
- C07J33/007—Cyclic thioketals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J51/00—Normal steroids with unmodified cyclopenta(a)hydrophenanthrene skeleton not provided for in groups C07J1/00 - C07J43/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0066—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa
- C07J1/007—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa the substituent being an OH group free esterified or etherified
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0066—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa
- C07J1/007—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa the substituent being an OH group free esterified or etherified
- C07J1/0077—Ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0081—Substituted in position 17 alfa and 17 beta
- C07J1/0085—Substituted in position 17 alfa and 17 beta the substituent in position 17 alfa being a saturated hydrocarbon group
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
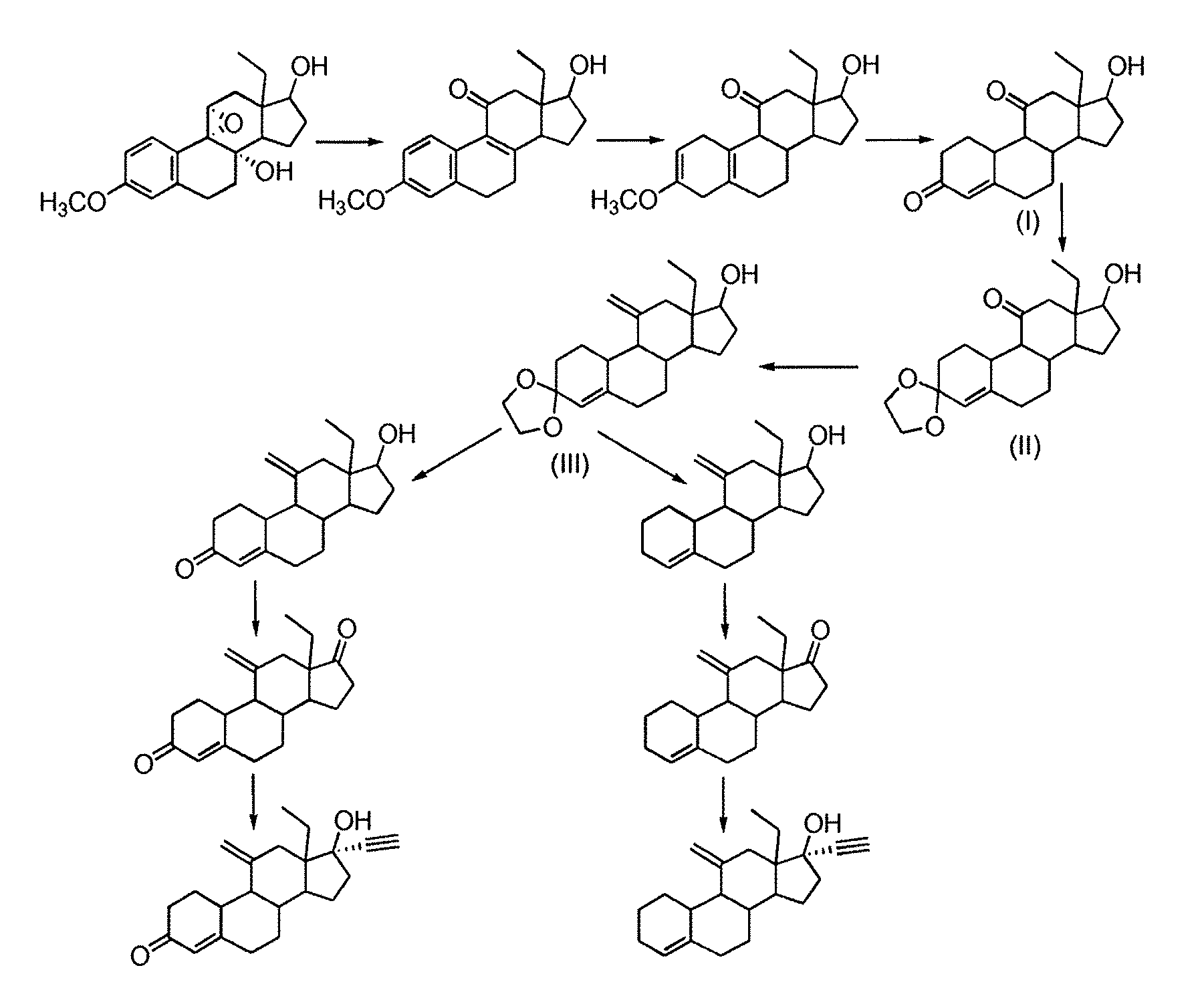
ルなどのいくつかの薬学上活性な薬剤の製造における有用な中間体である、対応する11
-メチレン-17-ケトステロイドへ選択的オレフィン化する方法に関する。
テロイドである。それらは第3世代の避妊処方物で使用される。

れ、一方、エトノゲストレルは、膣リング送達システムNuvaRing(登録商標)お
よびインプラントImplanon(登録商標)において合成プロゲスチンとして使用さ
れている。
る。
特許第2361120号(米国特許第3,927,046号としても公開)で初めて記載
された。米国特許第3,927,046号、ならびにHeuvel, M.J. et al. Recueil des
Travaux Chimiques des Pays-Bas 1988, vol.107, no.4, p.331-334に開示されているデ
ソゲストレルの合成は、重要中間体として式(IV)の化合物を使用する。この化合物は
、化合物(II)の11位におけるケトンのオレフィン化(この場合、3位と17位のケ
トン基はケタールとして保護されている)とその後の化合物(III)における保護基の
切断によって得られる。

11α-ヒドロキシ-18-メチル-エストラ-4-エン-3,17-ジオンからのデソ
ゲストレルの合成が開示されている。11位においてエキソメチレン官能基を作出する前
に、17位のケトン基が保護される。

されている方法に従って合成することもできる。11-オキソ官能基はエポキシド転位お
よびバーチ還元により得られ、式(I)の化合物が得られる。中間体(II)の11位に
おけるオレフィン化により式(III)の化合物が得られ、これはデソゲストレルおよび
エトノゲストレルのいずれの製造においても重要中間体として使用される。
398-402に開示されているエトノゲストレルの合成において、17位においてヒドロキシ
ル保護基で置換された中間化合物に対して11-ケト基へのメチルリチウムの付加が行わ
れた。

は、13β-エチル-11-ヒドロキシ-ゴン-4-エン-3,17-ジオンからのデソ
ゲストレルの合成を記載している。この場合にもまた、11位におけるケトンのオレフィ
ン化は、3位と17位におけるジエチレンケタールとしてのケトン基の保護の後に行われ
る。

の3-ケトンのジチオケタールとしての選択的保護とその後の(III)の17-カルボ
ニル基のケタールとしての保護により中間体(V)および(XIII)を得た。前記保護
中間体の11位におけるオレフィン化、ジオキソランの脱保護、17位におけるエチニル
化よびチオケタール脱保護によりエトノゲストレルが得られた。

-ケト基のエチレングリコールによる保護もまた、WO2013/135744で使用さ
れた。11-メチレン誘導体(VII)は、ウィッティヒ反応またはピーターソンオレフ
ィン化により得られた。続いてのアルキニル化および脱保護によりエトノゲストレルが得
られた。

体である化合物9の合成が記載されている。スワン条件下でのアルコール5の酸化により
ケトン6が得られ、これがピーターソンのオレフィン化条件下で処理された。トリエン7
のバーチ還元によりジエン8が得られ、これが加水分解されて11-メチレンジケトン誘
導体9が得られた。この場合にもまた、17ケトンの保護は11位におけるオレフィン化
の前に行われた。

7873でも製造された。この場合、11位でケトンのオレフィン化を行う前に17-ケ
ト基を保護する代わりに、それが対応するヒドロキシル化合物(V)に還元され、その後
、11-メチレン基が導入された後にケトンへ再酸化された。

121(4), 710-714に開示されており、この場合には、ステロイド骨格が構築された。この
戦略は産業上利用可能であるとは思われず、17α-ヒドロキシ-11-メチレン-18
-メチルエストル-4-エン-3-オンを得るためには少なくとも13の合成工程が必要
とされる。
ている方法は長すぎ、かつ/または産業上利用可能でない。一般に、開示されているほと
んどの合成は、11位のケト基のオレフィン化を含んでなる。しかしながら、これらの総
ての方法では、17-ケト基の保護/脱保護または還元/酸化という付加的工程が必要と
され、合成の工程数が増える。
ゲストレルまたはエトノゲストレルなどのステロイドの合成における重要中間体を得るた
めの、新規な方法を開発することが必要である。
方法を提供するという課題に取り組む。特に、本発明者らは、驚くことに、11,17-
ジ-ケトステロイドが本明細書に定義される式(III)の化合物との反応によって選択
的にオレフィン化され得ることを見出した。このオレフィン化反応は、得られる11-メ
チレン-17-ケトステロイドが例えばエトノゲストレルおよびデソゲストレルなどの治
療上有価な化合物の製造における中間体であることから商業上重要である。
、大規模生産に好適な反応条件を適用し、効率的な様式で11-メチレンステロイドを製
造することを可能とする。

Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン保護基を形成し
;
Yは、それが結合している炭素原子とともにC=CH2またはC(OH)CH2Zを表
し、ここで、Zは、HおよびSiR’3から選択され、各R’は、C1-C6アルキルお
よびC6-C10アリールから独立に選択され;
R6は、H、C1-C6アルキルおよびハロゲンから選択され;
R10は、H、C1-C6アルキルおよびハロゲンから選択されるか、またはC1とC
10の間に二重結合が存在する場合には存在せず;
R13は、HおよびC1-C6アルキルから選択され;
R16は、H、C1-C6アルキルおよびハロゲンから選択され;かつ
---は、単結合または二重結合である]
の製造方法であって、
式(II)の化合物またはその溶媒和物

り得る]
を式(III)の化合物

およびMgIから選択される]
と反応させることを含んでなる方法を対象とする。
Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン保護基を形成し
;
Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよ
びC6-C10アリールから独立に選択され;
R6は、H、C1-C6アルキルおよびハロゲンから選択され;
R10は、H、C1-C6アルキルおよびハロゲンから選択されるか、またはC1とC
10の間に二重結合が存在する場合には存在せず;
R13は、HおよびC1-C6アルキルから選択され;
R16は、H、C1-C6アルキルおよびハロゲンから選択され;かつ
---は、単結合または二重結合である]
を対象とする。

Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン基またはケトン
保護基を形成し;
Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよ
びC6-C10アリールから独立に選択され;
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキ
ル、C6-C10アリールおよびハロゲンから独立に選択され;
R6は、H、C1-C6アルキルおよびハロゲンから選択され;
R10は、H、C1-C6アルキルおよびハロゲンから選択されるか、またはC1とC
10の間に二重結合が存在する場合には存在せず;
R13は、HおよびC1-C6アルキルから選択され;
R16は、H、C1-C6アルキルおよびハロゲンから選択され;かつ
---は、単結合または二重結合である]
を対象とする。
C1-C3アルキル」)の炭素原子を含み、単結合を介して分子の残りの部分に結合され
た直鎖または分岐アルカン誘導体を意味する。アルキル基の実例としては、メチル、エチ
ル、n-プロピル、i-プロピル、n-ブチル、i-ブチル、t-ブチル、ペンチル、ヘ
キシルが挙げられる。
炭素-炭素結合を介して結合されたまたは互いに縮合した1または2個の芳香核を含んで
なる芳香族基を意味する。アリール基の実例としては、フェニル、ナフチル、ジフェニル
、インデニル、フェナントリルなどが挙げられる。
3~6個(「C3-C6シクロアルキル」)の炭素原子を含むシクロアルカンに由来する
ラジカルを意味する。シクロアルキル基の実例としては、シクロプロピル、シクロブチル
、シクロペンチル、シクロヘキシルなどが挙げられる。
好ましくは1~3個のヘテロ原子からなり、完全にもしくは部分的に飽和していてもまた
は芳香族(「ヘテロアリール」)であってもよい3~10員の安定な環式基、好ましくは
、5または6員の環を意味する。本発明では、ヘテロシクリルは、一環式、二環式または
三環式系であり得、縮合環系を含んでもよい。ヘテロシクリル基の実例としては、例えば
、ピロリジン、ピペリジン、ピペラジン、モルホリン、テトラヒドロフラン、ベンズイミ
ダゾール、ベンゾチアゾール、フラン、ピロール、ピリジン、ピリミジン、チアゾール、
チオフェン、イミダゾール、インドールなどが挙げられる。
続いての反応に関してケトン官能基を遮断する基を意味する。ケトン保護基の使用は、合
成手順中の望ましくない反応に対する保護基に関して当技術分野で周知であり、このよう
な保護基は既知である(例えば、T. H. Greene and P. G. M Wuts, Protective Groups i
n Organic Synthesis, 第4版, John Wiley & Sons, 2007)。事実上、いずれのケトン保
護基も、本発明を実施するために使用可能である。ケトン保護基の限定されない実例とし
ては、以下が挙げられる:

ら独立に選択され得る。非環式ケタールおよびジチオケタールの例としては、ジメチルケ
タール、ジエチルケタール、ジイソプロピルケタール、ジブチルケタール、ジベンジルケ
タール、ジメチルチオケタール、ジエチルチオケタール、ジイソプロピルチオケタール、
ジブチルチオケタール、ジベンジルチオケタールが挙げられる。);

よびC1-C6アルキルから独立に選択され得る。環式ケタール、ジチオケタールおよび
ヘミチオケタールの例としては、1,3-ジオキソラン、4-メチル-1,3-ジオキソ
ラン、4,5-ジメチル-1,3-ジオキソラン、4,4,5,5-テトラメチル-1,
3-ジオキソラン、1,3-ジオキサン、4-メチル-1,3-ジオキサン、5-メチル
-1,3-ジオキサン、4,4-ジメチル-1,3-ジオキサン、5,5-ジメチル-1
,3-ジオキサン、4,5-ジメチル-1,3-ジオキサン、4,6-ジメチル-1,3
-ジオキサン、1,3-ジオキサパン、1,3-ジチオラン、1,3-ジチアン、1,3
-オキサチオランが挙げられる。);
られる。R’’’は、C1-C6アルキルおよびベンジルから選択され得る。エノールエ
ーテルの例としては、メチルエノールエーテル、エチルエノールエーテル、プロピルエノ
ールエーテル、ブチルエノールエーテル、ベンジルエノールエーテルが挙げられる。);

られる。各R’’’は、C1-C6アルキルおよびベンジルから独立に選択され得るか、
または2つのR’’’基はそれらが結合している窒素原子とともに5員もしくは6員複素
環式環を形成する。エナミンの例としては、ジメチルエナミン、ジエチルエナミン、ジプ
ロピルエナミン、ジブチルエナミン、ジアリルエナミン、ピロリジンエナミン、ピペリジ
ンエナミン、モルホリンエナミンが挙げられる。);

としては、オキシム、O-メチルオキシム、O-ベンジルオキシム、O-フェニルチオメ
チルオキシムが挙げられる);および

立に選択され得る。ヒドラゾンの例としては、ヒドラゾン、N,N-ジメチルヒドラゾン
、フェニルヒドラゾン、2,4-ジニトロフェニルヒドラゾン、トシルヒドラゾンが挙げ
られる)。
も可能性が高いのは極性溶媒)を有する化合物のいずれの形態も意味するものと理解され
るできである。溶媒和物の例としては、水和物およびアルコレート、例えば、メタノレー
トが挙げられる。
r2O、MeOtBu、1,4-ジオキサン、テトラヒドロフラン、メチルテトラヒドロ
フラン)、炭化水素溶媒(例えば、ペンタン、ヘキサン、ヘプタン)、ハロゲン化溶媒(
例えば、ジクロロメタン、クロロホルム)、芳香族溶媒(例えば、トルエン)、エステル
(例えば、EtOAc)、ニトリル(例えば、アセトニトリル)、アミド(例えば、DM
F)、アルコール(例えば、メタノール、エタノール、プロパノール)、スルホキシド(
DMSO)およびそれらの混合物が含まれる。

の製造方法であって、
式(II)の化合物またはその溶媒和物


Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよ
びC6-C10アリールから独立に選択され;かつ
Mは、Li、MgBr、MgClおよびMgIから選択される]
と反応させることを含んでなる方法を対象とする。
好ましくは、式(I)または(II)の化合物は、式(Ia)もしくは(IIa)の化合
物またはその溶媒和物である。

ともにケトン保護基を形成する。
オケタール、環式または非環式ヘミチオケタール、エノールエーテル、エナミン、オキシ
ムおよびヒドラゾンから選択される。好ましくは、ケトン保護基は、環式ケタール、環式
ジチオケタール、環式ヘミチオケタール、エノールエーテルおよびエナミンから選択され
る。1つの態様では、Xは、それが結合している炭素原子とともに
i)

各R’’’は、HおよびC1-C6アルキルから独立に選択される];
ii)C-O-R’’’
’’’は、C1-C6アルキルおよびベンジルから選択される);および
iii)

れ、式中、各R’’’は、C1-C6アルキルおよびベンジルから独立に選択されるか、
または2つのR’’’基は、それらが結合している窒素原子とともに5員もしくは6員複
素環式環を形成する)
から選択され基を形成する。
i)

およびC1-C6アルキルから独立に選択される];
ii)C-O-R’’’
(この場合、加えてC2とC3の間またはC3とC4の間に二重結合が存在し、式中、各
R’’’は、C1-C6アルキルから選択される);および
iii)

れ、式中、2つのR’’’基は、それらが結合している窒素原子とともに5員または6員
複素環式環を形成する)
から選択される基を形成する。
1,3-ジチオラン、メチルエノールエーテル、エチルエノールエーテルおよびピロリジ
ンエナミンから選択される基を形成する。
基を形成し、かつ、C5とC6の間に二重結合が存在する。1つの態様では、Xは、それ
が結合している炭素原子とともに1,3-ジチオラン基を形成し、かつ、C4とC5の間
に二重結合が存在する。1つの態様では、Xは、それが結合している炭素原子とともにメ
チルエノールエーテルを形成し、かつ、C2とC3の間およびC5とC10の間に二重結
合が存在する。1つの態様では、Xは、それが結合している炭素原子とともにエチルエノ
ールエーテルを形成し、かつ、C3とC4の間およびC5とC6の間に二重結合が存在す
る。1つの態様では、Xは、それが結合している炭素原子とともにピロリジンエナミンを
形成し、かつ、C3とC4の間およびC5とC10の間に二重結合が存在する。
。別の態様では、Yは、それが結合している炭素原子とともにC(OH)CH2Z基を形
成する。
C6-C10アリールから独立に選択される(ピーターソンオレフィン化反応)。
t-ブチル、n-ヘキシルおよびフェニルから独立に選択される。1つの態様では、Zは
、Me3Si-、Et3Si-、iPr3Si-、nPr3Si-、nHex3Si-、
tBu3Si-、Ph3Si-、MeEt2Si-、tBuMe2Si-、tBuPh2
Si-、MePh2Si-、EtMe2Si-およびPhMe2Si-から選択される。
より好ましくは、ZはMe3Si-である。
はLiである。特定の態様では、式(III)の化合物は、Me3Si-CH2-Liで
ある。
は、無水有機溶媒、例えば、環式または非環式エーテル(例えば、Et2O、iPr2O
、tBuOMe、1,4-ジオキサン、テトラヒドロフラン、メチルテトラヒドロフラン
)、炭化水素溶媒(例えば、ペンタン、ヘキサン、ヘプタン)、ハロゲン化溶媒(例えば
、ジクロロメタン、クロロホルム)、芳香族溶媒(例えば、トルエン)またはそれらの混
合物の存在下で行われる。好ましくは、有機溶媒は、環式または非環式エーテル、例えば
、Et2O、iPr2O、tBuOMe、1,4-ジオキサン、テトラヒドロフラン、メ
チルテトラヒドロフランまたはそれらの混合物である。特定の態様では、有機溶媒はテト
ラヒドロフランである。本明細書において、無水溶媒という用語は、含有する水が500
ppm未満の溶媒を意味する。
る。1つの態様では、この反応は、-60℃~40℃の間、好ましくは-60℃~25℃
の間の温度で行われる。
0モル当量、好ましくは2.0~4.0モル当量の量で存在する。
択的付加を可能とする。好ましくは、式(II)の化合物またはその溶媒和物と式(II
I)の化合物の反応により式(I)の化合物またはその溶媒和物が、全付加物の70%(
モル)を超える、好ましくは80%を超える、好ましくは90%を超える、より好ましく
は95%を超える、さらにより好ましくは98%を超える選択性で得られる。
れが結合している炭素原子とともにC(OH)CH2Z基を形成する式(I)の化合物が
得られる。前記化合物は単離して続いての合成工程(例えば、エチニル化、ピーターソン
脱離、脱水)に使用することができ、またはワンポット法でそのまま酸または塩基で処理
して、Yがそれが結合している炭素原子とともにC=CH2を表す式(I)の化合物を得
ることもできる。よって、特定の態様では、本発明の方法は、
(a)式(II)の化合物またはその溶媒和物を式(III)の化合物と反応させて、
Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表し、ZがHおよびS
iR’3から選択され、各R’がC1-C6アルキルおよびC6-C10アリールから独
立に選択される式(I)の化合物またはその溶媒和物を得ること;並びに
(b)Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表し、ZがH
およびSiR’3から選択され、各R’がC1-C6アルキルおよびC6-C10アリー
ルから独立に選択される式(I)の化合物またはその溶媒和物を、酸または塩基で処理し
て、Yがそれが結合している炭素原子とともにC=CH2を表す式(I)の化合物または
その溶媒和物を得ること
を含んでなる。
式(I)の化合物またはその溶媒和物をさらにエチニル化して式(IV)の化合物また
はその溶媒和物

Y、R6、R10、R13、R16および---は、上記で定義される通りであり;
Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン基もしくはケト
ン保護基を形成し;かつ
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキ
ル、C6-C10アリールおよびハロゲンから独立に選択される]
を得ることができる。
とができる。よって、特定の態様では、本発明の方法は、
(a)式(II)の化合物またはその溶媒和物を式(III)の化合物と反応させて、
Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表す式(I)の化合
物またはその溶媒和物を得ること;
(b)Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表す式(I)
の化合物またはその溶媒和物を酸または塩基で処理して、Yがそれが結合している炭素原
子とともにC=CH2を表す式(I)の化合物またはその溶媒和物を得ること;並びに
(c)Yがそれが結合している炭素原子とともにC=CH2を表す式(I)の化合物ま
たはその溶媒和物をエチニル化して、Yがそれが結合している炭素原子とともにC=CH
2を表す式(IV)の化合物またはその溶媒和物を得ること
を含んでなる。
(a)式(II)の化合物またはその溶媒和物を式(III)の化合物と反応させて、
Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表す式(I)の化合物
またはその溶媒和物を得ること;
(b)Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表す式(I)
の化合物またはその溶媒和物をエチニル化して、Yがそれが結合している炭素原子ととも
にC(OH)CH2Z基を表す式(IV)の化合物またはその溶媒和物を得ること;並び
に
(c)Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表す式(IV
)の化合物またはその溶媒和物を酸または塩基で処理して、Yがそれが結合している炭素
原子とともにC=CH2を表す式(IV)の化合物またはその溶媒和物を得ること
を含んでなる。
条件下で行うことができる。特定の態様では、エチニル化反応は、式(I)の化合物また
はその溶媒和物を式(V)の化合物

M’は、Li、Na、K、MgBr、MgClおよびMgIから選択され;かつ
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキ
ル、C6-C10アリールおよびハロゲンから独立に選択される]
で処理することによって行われる。
択される。さらなる態様では、各R’’は、メチル、エチル、n-プロピル、i-プロピ
ル、n-ブチル、t-ブチル、n-ヘキシル、PhおよびClから独立に選択される。好
ましくは、-SiR’’3は、Et3Si-、Me3Si-、iPr3Si-、nPr3
Si-、nHex3Si-、tBu3Si-、Ph3Si-、Cl3Si-、MeEt2
Si-、tBuMe2Si-、tBuPh2Si-、CliPr2Si-、ClMe2S
i-、MePh2Si-、EtMe2Si-、EtCl2Si-、MeCl2Si-、P
hMe2Si-およびPhMeClSi-から選択される。より好ましくは、-SiR’
’3は、Me3Si-、Et3Si-、iPr3Si-、PhMe2Si-、tBuMe
2Si-およびtBuPh2Si-から選択される。いっそうより好ましくは、-SiR
’’3はMe3Si-である。
であり、ここで、各R’’は、C1-C6アルキルから独立に選択され、例えば、SiM
e3である。
iR’’3基である。
、M’は、MgBr、MgClおよびMgIから選択され、かつ、R1はHである。
たは非環式エーテル(例えば、Et2O、iPr2O、1,4-ジオキサン、テトラヒド
ロフラン、メチルテトラヒドロフラン)、炭化水素溶媒(例えば、ペンタン、ヘキサン、
ヘプタン)、ハロゲン化溶媒(例えば、ジクロロメタン、クロロホルム)、芳香族溶媒(
例えば、トルエン)またはそれらの混合物の存在下で行われる。好ましくは、有機溶媒は
、環式または非環式エーテル、例えば、Et2O、iPr2O、1,4-ジオキサン、テ
トラヒドロフラン、メチルテトラヒドロフラン;炭化水素溶媒、例えば、ペンタン、ヘキ
サン、ヘプタン;またはそれらの混合物の存在下で行われる。
る。1つの態様では、この反応は、-60℃~50℃の間、好ましくは-30℃~30℃
の間の温度で行われる。
当量、好ましくは1.1~3.0モル当量の量で存在する。
基および/または酸もしくは塩基に応じて、式(I)の化合物またはその溶媒和物のXが
、それが結合している炭素原子とともにケトン保護基を形成する場合、Xがそれが結合し
ている炭素原子とともにケトン保護基またはケトン基を形成する式(IV)の化合物また
はその溶媒和物を得ることができる。
では、式(IV)の化合物またはその溶媒和物は、エチニル化反応後に維持される。
V)の化合物またはその溶媒和物は、式(IV)の化合物またはその溶媒和物を、C=C
H2基を作出するために酸または塩基で処理した後に得られる。
和物を得るためには脱シリル化を行うことができる。
ts, Protective Groups in Organic Synthesis, 第4版, John Wiley & Sons, 2007)によ
って行うことができる。特定の態様では、脱シリル化は、水、有機溶媒またはそれらの混
合物の存在下でフッ素塩または塩基を用いて行われる。フッ化ピリジニウム、フッ化カリ
ウムまたはフッ化アンモニウムなどのフッ素塩;または水酸化ナトリウム、水酸化リチウ
ム、水酸化カリウムまたは炭酸カリウムなどの無機塩基が使用できる。特定の態様では、
脱シリル化反応は、無機塩基および有機溶媒の存在下で行われる。
態様では、前記反応は-10~+60℃の間、好ましくは10~35℃の間の温度で行わ
れる。
護基の切断の前または後のいずれかで行うことができる。
1つの態様では、Yがそれが結合している炭素原子とともにC(OH)CH2Z基を表
す式(I)の化合物もしくは式(IV)の化合物またはその溶媒和物を、酸または塩基で
処理すると、Yがそれが結合している炭素原子とともにC=CH2を表す式(I)の化合
物もしくは式(IV)の化合物またはその溶媒和物が得られる。
素原子とともにC=CH2を表す化合物が得られる(ピーターソン脱離反応)。
H2を表す化合物が得られる(脱水反応)。
適な酸の例としては、酢酸、トリフルオロ酢酸、クロロ酢酸、メタンスルホン酸、トリフ
ルオロメタンスルホン酸、ギ酸、プロピオン酸、酪酸、リンゴ酸、クエン酸、安息香酸、
p-トルエンスルホン酸、シュウ酸、コハク酸、塩酸、臭化水素酸、フッ化水素酸、過塩
素酸、塩素酸、硫酸、硝酸、リン酸、ZnCl2、AlCl3およびBF3が挙げられる
。特定の態様では、酸は、酢酸、トリフルオロ酢酸、メタンスルホン酸、トリフルオロメ
タンスルホン酸、塩酸、臭化水素酸、過塩素酸、硫酸およびそれらの混合物から選択され
る。
化アルカリ金属、例えば、水素化ナトリウム、水素化カリウム、ナトリウムメトキシド、
ナトリウムエトキシド、ナトリウムt-ブトキシド、カリウムメトキシド、カリウムエト
キシド、カリウムt-ブトキシド、水酸化ナトリウムおよび水酸化カリウムが挙げられる
。
す式(I)の化合物またはその溶媒和物を、エチニル化反応前に酸または塩基で処理する
と、Yがそれが結合している炭素原子とともにC=CH2を表す式(I)の化合物または
その溶媒和物が得られる。
C(OH)CH2Z基を表す式(IV)の化合物またはその溶媒和物が、Yがそれが結合
している炭素原子とともにC=CH2を表す式(IV)の化合物またはその溶媒和物に変
換されるように、エチニル化反応の後に行われる。この場合、使用する酸または塩基、反
応条件および/またはケトン保護基に応じて、Xがそれが結合している炭素原子とともに
ケトン保護基またはケトン基を形成する式(IV)の化合物またはその溶媒和物を得るこ
とができる。
塩基はまた、Yがそれが結合している炭素原子とともにC=CH2を表し、かつ、Xがそ
れが結合している炭素原子とともにケトン基を表す式(IV)の化合物が得られるように
、3位におけるケトン保護基の切断も可能とする。
Xが水素であるか、またはそれが結合している炭素原子とともにケトン基を形成する式
(IV)の化合物を得るためには、ケトン保護基の脱保護工程が必要とされ得る。ケトン
保護基の切断は、当技術分野で公知のいずれの従来の手段によって行うこともできる(例
えば、T. H. Greene and P. G. M Wuts, Protective Groups in Organic Synthesis, 第4
版, John Wiley & Sons, 2007)。
酸性媒体中でデ3-ケト基を再生するためにそれを切断することができる。
酸性または塩基性媒体中での加水分解によって切断され得る。
で切断され得る。加えて、ケトン保護基がジチオケタールである場合、それを還元条件下
で除去してXがHである化合物を得ることができる。
H=CH2基を作出するために使用される反応条件下で切断される。
ること、および本発明を実施するためにさらなる合成工程が必要とされる場合があること
を認識するであろう。
子とともにケトン基を形成する式(IV)の化合物を得るためには、下記の工程:
(i)XがHであるか、またはそれが結合している炭素原子とともにケトン基を形成す
る式(IV)の化合物またはその溶媒和物を得るためのケトン保護基の切断、
(ii)R1がHである式(IV)の化合物またはその溶媒和物を得るためのSiR’
’3基の除去
の一方または両方を行うことが必要となる場合がある。
工程(i)は工程(ii)の前または後のいずれかに行うことができる。
またはその溶媒和物を、R1がHであり、Xがそれが結合している炭素原子とともにケト
ン基を形成し、かつ、Yがそれが結合している炭素原子とともにCH=CH2を表す式(
IV)の化合物またはその溶媒和物へ変換するために、下記の工程順序のいずれかを経る
ことができる:
・エチニル化/CH=CH2基の作出/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の切断;または
・エチニル化/CH=CH2基の作出/必要であれば、ケトン保護基の切断/必要であれ
ば、アルキンの脱シリル化;または
・エチニル化/必要であれば、アルキンの脱シリル化/CH=CH2基の作出/必要であ
れば、ケトン保護基の切断;または
・エチニル化/CH=CH2基の作出/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の切断;または
・CH=CH2基の作出/エチニル化/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の切断;または
・CH=CH2基の作出/エチニル化/必要であれば、ケトン保護基の切断/必要であれ
ば、アルキンの脱シリル化;または
・エチニル化/必要であれば、ケトン保護基の切断/CH=CH2基の作出/必要であれ
ば、アルキンの脱シリル化;または
・エチニル化/必要であれば、ケトン保護基の切断/必要であれば、アルキンの脱シリル
化/CH=CH2基の作出。
またはその溶媒和物を、R1がHであり、Xが水素であり、かつ、Yがそれが結合してい
る炭素原子とともにCH=CH2を表す式(IV)の化合物またはその溶媒和物に変換す
るために、下記の工程順序のいずれかを経ることができる:
・エチニル化/CH=CH2基の作出/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の還元的脱離;または
・エチニル化/CH=CH2基の作出/必要であれば、ケトン保護基の還元的脱離/必要
であれば、アルキンの脱シリル化;または
・エチニル化/必要であれば、アルキンの脱シリル化/CH=CH2基の作出/必要であ
れば、ケトン保護基の還元的脱離;または
・エチニル化/CH=CH2基の作出/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の還元的脱離;または
・CH=CH2基の作出/エチニル化/必要であれば、アルキンの脱シリル化/必要であ
れば、ケトン保護基の還元的脱離;または
・CH=CH2基の作出/エチニル化/必要であれば、ケトン保護基の還元的脱離/必要
であれば、アルキンの脱シリル化;または
・エチニル化/必要であれば、ケトン保護基の還元的脱離/CH=CH2基の作出/必要
であれば、アルキンの脱シリル化;または
・エチニル化/必要であれば、ケトン保護基の還元的脱離/必要であれば、アルキンの脱
シリル化/CH=CH2基の作出。
本発明の方法により得られる式(I)の化合物は、いくつかの薬学上活性な薬剤、例え
ば、エトノゲストレルおよびデソゲストレルの製造における有用な中間体である。
媒和物を本明細書で定義される式(III)の化合物と反応させることを含んでなる、エ
トノゲストレル、またはデソゲストレルまたはその溶媒和物の製造方法を対象とする。
(a)式(IIa)の化合物またはその溶媒和物

を式(III)の化合物

と反応させて式(Ia-1)の化合物またはその溶媒和物

(b)式(Ia-1)の化合物またはその溶媒和物を酸または塩基で処理して、式(I
a-2)の化合物またはその溶媒和物
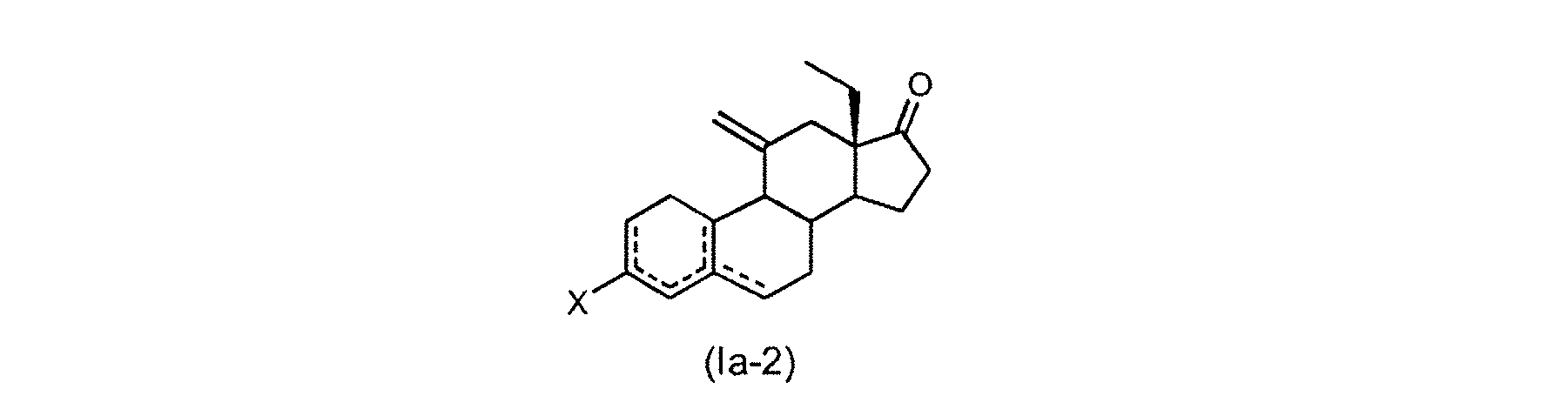
(c)式(Ia-2)の化合物またはその溶媒和物をエチニル化して、式(IVa-1
)の化合物またはその溶媒和物

Xは、それが結合している炭素原子とともにケトン基またはケトン保護基を形成し、
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキ
ル、C6-C10アリールおよびハロゲンから独立に選択される]
を得ること;並びに
(d)必要に応じて、下記の工程:
(i)Xがそれが結合している炭素原子とともにケトン保護基を形成する場合には、X
がそれが結合している炭素原子とともにケトン基を形成する式(IVa-1)の化合物ま
たはその溶媒和物を得るためのケトン保護基の切断、
(ii)R1がSiR’’3基である場合には、R1がHである式(IVa-1)の化
合物またはその溶媒和物を得るためのSiR’’3基の除去
の一方または両方を任意の順序で行う工程
を含んでなる方法により得ることができる。
(a)式(IIa)の化合物またはその溶媒和物

を式(III)の化合物

と反応させて、式(Ia-1)の化合物またはその溶媒和物

(b)式(Ia-1)の化合物またはその溶媒和物をエチニル化して、式(IVa-2
)の化合物またはその溶媒和物

アルキル、C6-C10アリールおよびハロゲンから独立に選択される]
を得ること;
(c)式(IVa-2)の化合物またはその溶媒和物を酸または塩基で処理して、式(
IVa-1)の化合物またはその溶媒和物

Xは、それが結合している炭素原子とともにケトン基またはケトン保護基を形成し、
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキ
ル、C6-C10アリールおよびハロゲンから独立に選択される]
を得ること;並びに
(d)必要に応じて、下記の工程:
(i)Xがそれが結合している炭素原子とともにケトン保護基を形成する場合には、X
がそれが結合している炭素原子とともにケトン基を形成する式(IVa-1)の化合物ま
たはその溶媒和物を得るためのケトン保護基の切断、
(ii)R1がSiR’’3基である場合には、R1がHである式(IVa-1)の化
合物またはその溶媒和物を得るためのSiR’’3基の除去
の一方または両方を任意の順序で行う工程
を含んでなる方法により得ることができる。
(a)上記に定義される方法のいずれかにより、Xがそれが結合している炭素原子とと
もに環式ジチオケタール基を形成する式(IVa-1)の化合物またはその溶媒和物を得
ること;
(b)式(IVb-1)の化合物またはその溶媒和物を得るための、還元条件下での環
式ジチオケタール基の切断;並びに
(c)R1がSiR’’3基である場合には、R1がHである式(IVb-1)の化合
物またはその溶媒和物を得るための、工程(b)の前または後いずれかでのSiR’’3
基の除去
を含んでなる方法により得ることができる。
(a)化合物(12)またはその溶媒和物


と反応させて、式(Ib-1)の化合物またはその溶媒和物

b-2)の化合物またはその溶媒和物

)の化合物またはその溶媒和物

アルキル、C6-C10アリールおよびハロゲンから独立に選択される]
を得ること;並びに
るためのSiR’’3基の除去
を含んでなる方法により得ることができる。
別の側面では、本発明は、式(Ic)の化合物またはその溶媒和物

り]
を対象とする。
る通りである。
、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択される。R
’の好ましい態様は、上記で定義される通りである。

1)の化合物またはその溶媒和物である。


れる通り]
を対象とする。
義される通りである。
ここで、各R’’は、C1-C6アルキル、C6-C10アリールおよびハロゲンから独
立に選択される。R’’の好ましい態様は、上記で定義される通りである。
Vb-2)の化合物またはその溶媒和物である。


べきである。
スワーン酸化)

した後、450mLのDCMに希釈したDMSO(63mL)を、温度を-35℃未満に
維持しつつ滴下した。添加が完了した後、反応混合物を-40℃で20分間撹拌した。次
に、450mLのDCMに溶かした45gの化合物1を、温度を-35℃未満に維持しつ
つ加えた。反応混合物を-40℃で30分間維持した後、DIPEA(235mL)を手
早く加え、冷却浴を外し、室温まで温めた(1.5時間)。675mLの3.3%酢酸溶
液を加え、水相を分離した。有機相を315mLの7%NaHCO3溶液で洗浄し、分離
し、150mLの容量まで真空濃縮した。150mLのIPAを加え、容量を150mL
に減らした。この操作を最終容量が150mLとなるまでさらに2回繰り返した。得られ
た懸濁液を氷浴中で30分間撹拌した後、濾過し、固体を45mLの冷IPAで洗浄し、
真空下、40℃で乾燥させた。39.8gの化合物2を白色固体として得た(収率=89
.5%)。
パリック・デーリング酸化)
15gの化合物1を40mLのDMSOに溶かした後、71mLのTEAを加えた。こ
の溶液を30℃で加熱し、70mLのDMSO中、SO3Py(79g)の溶液を加えた
。反応混合物を30℃で3時間撹拌した後、234mLの水中、117mLの氷酢酸の溶
液に注いで沈澱を形成させた。この懸濁液を氷浴中で1時間冷却し、濾過した。固体を8
0mLのIPAに懸濁させ、加熱して完全に溶解させた後、0℃に冷却した。生じた固体
を濾過し、真空下、40℃で乾燥させて13gの化合物2(81%)を得た。

0mgのpTsOHを加えた。反応混合物を25℃で3時間撹拌した。次に、2mLのT
EAおよび90mLの7%NaHCO3溶液を加えた。水相を50mL EtOAcで抽
出した。合わせた有機相を湿潤固体が得られるまで濃縮し、90mLのエタノールを加え
、40mLの容量まで濃縮し、氷浴中で冷却し、濾過した。固体を90mLの冷エタノー
ルで洗浄し、真空下、40℃で乾燥させ、14.7gの化合物3(75%)を得た。

35℃未満に維持しつつ、88mLのトリメチルシリルメチルリチウムをゆっくり加え、
添加が完了した後、この混合物をさらに1時間撹拌した。次に、200mLの7%NaH
CO3溶液を加え、分離し、水相を100mL EtOAcで抽出した。合わせた有機相
を30mLに濃縮し、30mLのエタノールを加え、溶媒を最終容量30mLまで蒸発さ
せた。この操作をさらに2回繰り返した後、懸濁液を氷浴中で1時間冷却した。固体を濾
過し、10mLの冷エタノールで洗浄し、真空下、40℃で乾燥させ、10.4gの化合
物4を得た(収率83%)。
1H NMR (400 MHz, CDCl3): δ 0.09 (s, 9H); 0.82 (t, 3H); 1.17-1.39 (m, 10H); 1.63
-1.66 (m, 1H); 1.68-1.72 (m, 1H); 1.84-1.87 (m, 2H); 1.92-2.12 (m, 3H); 2.21-2.3
5 (d, 3H); 2.39-2.51 (m, 4H); 3.69-3.79(m, 2H); 5.19 (s, 1H) 5.31 (d, 1H).
13C NMR (100 MHz, CDCl3): δ 1.1; 8.4; 14.7; 18.5; 21.3; 29.2; 30.7; 31.2; 35.1;
35.6; 37.1; 38.4; 43.4; 50.7; 51.5; 53.3; 62.4; 100.1; 117.3; 137.8; 156.2; 218
.8.

100mlのTHF/ヘプタン 1/3混合物中、トリメチルシリルアセチレン(30m
L)の溶液をゆっくり加えた。反応混合物を0℃で30分間撹拌した後、100mLのT
HF中、化合物4(10g)の溶液を加えた後、この混合物をさらに1時間撹拌した。水
(200mL)を加えて余分なリチウム試薬を急冷し、有機相を真空濃縮した。残渣(8
5%の化合物5と15%の化合物4を含有する、最大レベルの変換が得られた)をシリカ
ゲルにてEtOAc/ヘプタン 1/9で精製し、純粋な化合物5を油状物として得た。
1H NMR (400 MHz, CDCl3): δ 0.07 (s, 9H); 0.12 (s, 9H) 0.83-0.85 (m, 1H); 1.12-1
.18 (m, 5H); 1.22-1.28 (m, 6H); 1.33-1.38 (m, 3H); 1.58-1.66 (m, 4H); 1.91 (d, 1
H); 1.98-2.09 (m, 3H); 2.17 (d, 1H); 2.27-2.36 (m, 4H); 3.68-3.77(m, 2H); 5.18
(s, 1H) 5.29 (d, 1H).
13C (100 MHz, CDCl3): δ 1.1; 8.4; 14.7; 18.5; 21.3; 29.2; 30.7; 31.2; 35.1; 35.
6; 37.1; 38.4; 43.4; 50.7; 51.5; 53.3; 62.4; 100.1; 117.3; 137.8; 156.2; 218.8.

3 *2LiClを加えた後、温度を10℃未満に維持しつつ、塩化エチニルマグネシウム
(120mL)をゆっくり加えた。15時間後、2.5mLのTEAおよび250mLの
7%NaHCO3溶液を加えた。水相をEtOAc 50mL×2で抽出し、合わせた有
機相をブラインで洗浄した。溶媒を減圧下で20mLの容量まで蒸発せ、40mLのメタ
ノールを加えた。これを20mLまで濃縮し、2回繰り返した。最終的なメタノール溶液
を2mLのHClで処理し、25℃で1時間撹拌し、2mLの7%NaHCO3溶液を加
えた。1mLの水を加えて固体の沈殿を促した。これを濾過し、4mLの水で洗浄し、真
空下、40℃で乾燥させ、2gの粗エトノゲストレルを褐色固体として得た(hplc純
度94%)。

、100mlのTHF/ヘプタン 1/3混合物中、トリメチルシリルアセチレン(26
mL)の溶液をゆっくり加えた。反応混合物を-5℃で30分間撹拌した後、100mL
のTHF/ヘプタン 1:1中、化合物4(10g)の溶液を加え、この混合物をさらに
15時間撹拌した。水(200mL)を加えて余分なリチウム試薬を急冷し、有機相を真
空濃縮した。残渣(90%の化合物5と10%の化合物4を含有する)を50mLのメタ
ノールに溶かし、0.5mLのHClを加え、1時間20℃で撹拌した後、3mLの50
%NaOHを加え、さらに1時間撹拌した。溶媒を減圧下で蒸発させ、残渣を100mL
のDCMに溶かした。これをまず3%氷酢酸溶液で、次いで7%NaHCO3溶液で洗浄
した。得られた粗物質を30mLのアセトンに溶かし、12mLの容量まで濃縮し、12
molのIPAを加え、さらに50%の容量に減らした。この操作を2回繰り返した。こ
の懸濁液を氷浴中で冷却し、固体を濾過し、4mLの冷IPAで洗浄し、真空下、40℃
で乾燥させ、2.1gのエトノゲストレルを得た。

を加えた。反応混合物を25℃で1時間撹拌した後、溶媒を蒸発させ、50mLの水を加
え、この混合物を50mLのEtOAcで抽出した。粗生成物をシリカゲルにてEtOA
c/ヘプタン 1/9で精製し、純粋な化合物6を白色固体として得た。

OH溶液を加えた。反応混合物を25℃で1時間撹拌した後、溶媒を蒸発させ、50mL
の水を加え、この混合物を50mLのEtOAcで抽出した。粗生成物をシリカゲルにて
EtOAc/ヘプタン 1/9で精製し、純粋な化合物7を橙色の油状物として得た。

LのIPAを加え、4mLの容量まで濃縮した。この操作を3回繰り返した。次に、この
溶液を0℃で冷却し、濾過し、2mLのIPAで洗浄した。固体を真空下で乾燥させ、純
粋なエトノゲストレルを得た。

ジチオールおよび1.4gのpTsOHを加えた。反応混合物を、毎時50mLのDCM
を蒸溜しつつ(また新たな溶媒を加えつつ)5時間還流させた。140mLの7%NaH
CO3溶液を加え、水相を50mLのDCMで抽出した。合わせた有機相を最終容量15
0mLまで真空濃縮した。150mLのメタノールを加え、最終容量100mLまで減圧
下で濃縮した。得られた懸濁液を氷浴中で1時間冷却した。得られた固体を濾過し、25
mLの冷メタノールで洗浄し、真空下、40℃で乾燥させ、30gの化合物8を得た。

10℃未満に維持しつつ、640mLのトリメチルシリルメチルリチウムをゆっくり加え
、添加が完了した後、反応混合物をさらに1時間撹拌した。次に、300mLの12%N
aH4Cl溶液を加え、分離し、水相を100mLのEtOAcで抽出した。合わせた有
機相を100mLの容量まで濃縮し、懸濁液を、氷浴を用いて1時間冷却した。生じた固
体を濾過し、30mLの冷EtOAcで洗浄し、真空下、40℃で乾燥させ、11gの化
合物9を得た(43%)。

応混合物を25℃で1時間撹拌した。pHを6に調整し、TEAを加えた後、この懸濁液
を氷浴中で冷却した。生じた沈澱を濾過し、10mLのメタノールで洗浄し、真空下、4
0℃で乾燥させ、7.8gの化合物11(88%)を得た。

Cl3 *2LiClを加えた後、温度を15℃未満に維持しつつ、塩化エチニルマグネシ
ウム(159mL)をゆっくり加えた。添加が完了した後、反応混合物を30℃で2時間
加熱した。次に、5℃で冷却し、150mLの10%HCl溶液を加えた。水相を50m
LのEtOAcで抽出し、合わせた有機相をブラインで洗浄した。溶媒を40mLの容量
まで減圧下で蒸発させ、40mLのヘプタンを加えた。これを40mLまで濃縮し、2回
繰り返した。得られた懸濁液を氷浴中で1時間冷却した。固体を濾過し、10mLの冷ヘ
プタンで洗浄し、真空下、40℃で乾燥させ、収率5.8gの化合物11(77%)を得
た。

過ヨウ素酸を用いて、またはWO2013/135744の実施例8Cに記載されている
ようにSIBXを用いて除去した。

10℃未満に維持しつつ、330mLのトリメチルシリルメチルリチウムをゆっくり加え
、添加が完了した後、この混合物をさらに1.5時間撹拌した。次に、150mLの12
%NaH4Cl溶液を加え、分離し、水相を50mL EtOAcで抽出した。合わせた
有機相を20mLの容量まで濃縮し、この懸濁液を、氷浴を用いて1時間冷却した。生じ
た固体を濾過し、20mLのメタノールに溶かし、0.25mlのHClを加えた。この
反応混合物を25℃で1.5時間撹拌した。pHを6に調整し、TEAを加えた後、この
懸濁液を氷浴中で冷却した。沈澱を濾過し、5mLのメタノールで洗浄し、真空下、40
℃で乾燥させ、7.6gの化合物14(76%)を得た。

.3M)の溶液に、6.8mLのTHF/ヘキサン 1/7混合物中、トリメチルシリル
アセチレン(1.32g)の溶液をゆっくり加えた。反応混合物を-5℃で30分間撹拌
した後、8mLのヘキサン中、化合物14(1.0g)の溶液を加え、この混合物を1時
間、0/5℃で撹拌した。NaCl水溶液(8.5mL)を加え、相を分離した。有機相
を5mLのメタノールと混合した後、1.5mLの30%NaOH水溶液を加え、さらに
4時間撹拌した。10mLの3%酢酸水溶液を加えた。相を分離し、有機相を水(5.0
mL)で洗浄した。溶媒を減圧下で蒸発させ、残渣を1mLのMeOHに溶かした。溶媒
を減圧下で蒸発させ、残渣を2mLのヘキサンに溶かした。得られた粗物質を60℃で加
熱することにより4mLのヘキサンに溶かした。この溶液を氷浴中でゆっくり冷却し、生
じた固体を濾過し、1mLの冷ヘキサンで洗浄し、真空下、40℃で乾燥させ、0.89
gのデソゲストレル(89%)を得た。

ルマグネシウム(22%)を、その混合物を3時間還流下で加熱しつつゆっくり加えた。
反応をTEAで急冷した。次に、7%NaHCO3溶液を加え、水相をEtOAcで抽出
した。粗生成物をシリカゲルで精製し、化合物15(75%)およびジメチル化化合物1
6(24%)を得た。
13C NMR (100 MHz, CDCl3): δ 8.3; 14.6; 18.5; 21.1; 29.2; 30.4; 33.8; 34.5; 35.5
; 37.2; 43.2; 50.7; 51.8; 52.9; 62.3; 73.5; 100.1; 117.0; 137.7; 156.1; 219.2.

mLのメチルリチウム(3%)をゆっくり加え、この混合物を3時間0℃で撹拌した。反
応をTEAで急冷した。次に、7%NaHCO3溶液を加え、水相をEtOAcで抽出し
た。粗生成物をシリカゲルで精製し、化合物15(76%)およびジメチル化化合物16
(23%)を得た。
Claims (4)
- 式(Ic)の化合物またはその溶媒和物

Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン保護基を形成し、前記ケトン保護基が、非環式ケタール、非環式ジチオケタール、非環式ヘミチオケタール、環式ケタール、環式ジチオケタール、環式ヘミチオケタール、エノールエーテル、エナミン、オキシム、およびヒドラゾンから選択され;
Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択され;
R6は、H、C1-C6アルキルおよびハロゲンから選択され;
R10は、H、C1-C6アルキルおよびハロゲンから選択されるか、またはC 5 とC10の間に二重結合が存在する場合には存在せず;
R13は、HおよびC1-C6アルキルから選択され;
R16は、H、C1-C6アルキルおよびハロゲンから選択され;かつ
---は、単結合または二重結合である]であり、
前記化合物が、以下の条件(a)~(c)のいずれかを満たし、
(a)ZがSiR’3であり、各R’がC1-C6アルキルおよびC6-C10アリールから独立に選択され、
(b)ただし、式(Ic)の化合物は、

(c)式(Ia-1)の化合物またはその溶媒和物

Xは、それが結合している炭素原子とともにケトン保護基を形成し、前記ケトン保護基が、非環式ケタール、非環式ジチオケタール、非環式ヘミチオケタール、環式ケタール、環式ジチオケタール、環式ヘミチオケタール、エノールエーテル、エナミン、オキシム、およびヒドラゾンから選択され;
Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択され;かつ
---は、単結合または二重結合である]
並びに
式(Ib-1)の化合物またはその溶媒和物

Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択される]
から選択される、化合物またはその溶媒和物。 -

- 式(IVc)の化合物またはその溶媒和物

Xは、Hを表すか、またはそれが結合している炭素原子とともにケトン基またはケトン保護基を形成し、前記ケトン保護基が、非環式ケタール、非環式ジチオケタール、非環式ヘミチオケタール、環式ケタール、環式ジチオケタール、環式ヘミチオケタール、エノールエーテル、エナミン、オキシム、およびヒドラゾンから選択され;
Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択され;
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキル、C6-C10アリールおよびハロゲンから独立に選択され;
R6は、H、C1-C6アルキルおよびハロゲンから選択され;
R10は、H、C1-C6アルキルおよびハロゲンから選択されるか、またはC 5 とC10の間に二重結合が存在する場合には存在せず;
R13は、HおよびC1-C6アルキルから選択され;
R16は、H、C1-C6アルキルおよびハロゲンから選択され;かつ
---は、単結合または二重結合である]であり、
前記化合物が、以下の条件(a)および(b)のいずれかを満たし、
(a)R1がSiR’’3であり、ここで、各R’’は、C1-C6アルキル、C6-C10アリールおよびハロゲンから独立に選択され、
(b)式(IVa-2)の化合物またはその溶媒和物

Zは、SiR’3であり、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択され;
Xは、それが結合している炭素原子とともにケトン基またはケトン保護基を形成し、前記ケトン保護基が、非環式ケタール、非環式ジチオケタール、非環式ヘミチオケタール、環式ケタール、環式ジチオケタール、環式ヘミチオケタール、エノールエーテル、エナミン、オキシム、およびヒドラゾンから選択され;
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキル、C6-C10アリールおよびハロゲンから独立に選択され;かつ
---は、単結合または二重結合である]
並びに
式(IVb-2)の化合物またはその溶媒和物

Zは、HおよびSiR’3から選択され、ここで、各R’は、C1-C6アルキルおよびC6-C10アリールから独立に選択され;かつ
R1は、HおよびSiR’’3から選択され、ここで、各R’’は、C1-C6アルキル、C6-C10アリールおよびハロゲンから独立に選択される]
から選択される、化合物またはその溶媒和物。 -

Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16382092.1 | 2016-03-03 | ||
| EP16382092 | 2016-03-03 | ||
| JP2018546617A JP7264569B2 (ja) | 2016-03-03 | 2017-03-02 | 11-メチレンステロイドの製造のための方法および新規な中間体 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018546617A Division JP7264569B2 (ja) | 2016-03-03 | 2017-03-02 | 11-メチレンステロイドの製造のための方法および新規な中間体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021183627A JP2021183627A (ja) | 2021-12-02 |
| JP7419302B2 true JP7419302B2 (ja) | 2024-01-22 |
Family
ID=55628968
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018546617A Active JP7264569B2 (ja) | 2016-03-03 | 2017-03-02 | 11-メチレンステロイドの製造のための方法および新規な中間体 |
| JP2021131984A Active JP7419302B2 (ja) | 2016-03-03 | 2021-08-13 | 11-メチレンステロイドの製造のための方法および新規な中間体 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018546617A Active JP7264569B2 (ja) | 2016-03-03 | 2017-03-02 | 11-メチレンステロイドの製造のための方法および新規な中間体 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US11034716B2 (ja) |
| EP (2) | EP3423464B1 (ja) |
| JP (2) | JP7264569B2 (ja) |
| ES (2) | ES2881336T3 (ja) |
| HU (2) | HUE071989T2 (ja) |
| MX (1) | MX386828B (ja) |
| WO (1) | WO2017149091A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108840895B (zh) * | 2018-04-24 | 2020-09-04 | 上海共拓医药化工有限公司 | 依托孕烯以及去氧孕烯中间体的制备方法 |
| CN115260265A (zh) * | 2022-08-19 | 2022-11-01 | 上海共拓医药化工有限公司 | 一种制备11-亚甲基药物中间体的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105906680A (zh) | 2016-04-27 | 2016-08-31 | 华润紫竹药业有限公司 | 去氧孕烯的制备方法和中间体化合物 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FI54128C (fi) | 1972-12-09 | 1978-10-10 | Akzo Nv | Foerfarande foer framstaellning av nya 11,11-alkylidensteroider ur oestran- och 19-norpregnanserien med foerbaettrad hormonell verkan |
| ZA94715B (en) * | 1993-02-08 | 1994-10-24 | Akzo Nv | Steroids for treating menopausal complaints |
| JP3377856B2 (ja) * | 1993-03-25 | 2003-02-17 | イスクラ産業株式会社 | 骨吸収抑制・骨形成促進化合物 |
| CZ2002831A3 (cs) | 1999-09-06 | 2002-05-15 | Akzo Nobel N. V. | Nearomatické estrogenní steroidy |
| ITMI20021755A1 (it) | 2002-08-02 | 2002-11-01 | Poli Ind Chimica Spa | Processo e nuovi intermedi per la preparazione di steroidi ad attivita' progestinica. |
| CN1865276A (zh) | 2005-05-20 | 2006-11-22 | 上海迪赛诺化学制药有限公司 | 合成甾体孕激素的方法 |
| ITMI20060474A1 (it) * | 2006-03-16 | 2007-09-17 | Ind Chimica Srl | Professo ed intermedi per la preparazione di 11-metilen-18-metilestr-4-en-3,17-dione |
| CN101440112B (zh) * | 2008-12-19 | 2011-04-20 | 北京紫竹药业有限公司 | 甾体化合物及其用途 |
| CN102964418B (zh) | 2010-06-25 | 2015-04-15 | 华润紫竹药业有限公司 | 去氧孕烯的制备工艺及其中间体化合物 |
| US20130123523A1 (en) | 2011-11-10 | 2013-05-16 | Klaus Nickisch | Methods for the preparation of etonogestrel and desogestrel |
| ES2748597T3 (es) | 2012-03-15 | 2020-03-17 | Merck Sharp & Dohme | Vía de síntesis combinada para etonogestrel y desogestrel |
| ITMI20121472A1 (it) | 2012-09-04 | 2014-03-05 | Ind Chimica Srl | Processo per la preparazione di 11-metilen-18-metilestr-4-en-3,17-dione, utile come intermedio per la sintesi di molecole ad attivita' farmacologica |
-
2017
- 2017-03-02 US US16/081,835 patent/US11034716B2/en active Active
- 2017-03-02 HU HUE21167186A patent/HUE071989T2/hu unknown
- 2017-03-02 JP JP2018546617A patent/JP7264569B2/ja active Active
- 2017-03-02 EP EP17707356.6A patent/EP3423464B1/en active Active
- 2017-03-02 EP EP21167186.2A patent/EP3865501B1/en active Active
- 2017-03-02 ES ES17707356T patent/ES2881336T3/es active Active
- 2017-03-02 ES ES21167186T patent/ES3037073T3/es active Active
- 2017-03-02 HU HUE17707356A patent/HUE055367T2/hu unknown
- 2017-03-02 WO PCT/EP2017/054952 patent/WO2017149091A1/en not_active Ceased
- 2017-03-02 MX MX2018010559A patent/MX386828B/es unknown
-
2021
- 2021-05-13 US US17/319,545 patent/US12227537B2/en active Active
- 2021-08-13 JP JP2021131984A patent/JP7419302B2/ja active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105906680A (zh) | 2016-04-27 | 2016-08-31 | 华润紫竹药业有限公司 | 去氧孕烯的制备方法和中间体化合物 |
Non-Patent Citations (3)
| Title |
|---|
| BROEK VAN DEN A J,11-ALKYLIDENE STEROIDS IN THE 19-NOR SERIES,RECUEIL DES TRAVAUX CHIMIQUES DES PAYS-BAS / RECUEIL, 以下備考,NL,ELSEVIER SCIENCE PUBLISHERS,1975年02月,VOL:94, NR:2,PAGE(S):35 - 39,JOURNAL OF THE ROYAL NETHERLANDS CHEMICAL SOCIETY |
| JOHN A ZDERIC,STEROIDS. CXL. 1 11-METHYL STEROIDS,JOURNAL OF THE AMERICAN CHEMICAL SOCIETY,米国,1960年07月,VOL:82, NR:13,PAGE(S):3404 - 3409,http://dx.doi.org/10.1021/ja01498a042 |
| RINGOLD H J,STEROIDS―XC 11α-METHYL-11β-HYDROXY STEROIDS,TETRAHEDRON,NL,1958年,VOL:2, NR:1-2,PAGE(S):164 - 165,http://dx.doi.org/10.1016/0040-4020(58)88034-5 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210269474A1 (en) | 2021-09-02 |
| ES2881336T3 (es) | 2021-11-29 |
| US20190092807A1 (en) | 2019-03-28 |
| MX2018010559A (es) | 2019-03-28 |
| MX386828B (es) | 2025-03-19 |
| JP2019512481A (ja) | 2019-05-16 |
| HUE055367T2 (hu) | 2021-11-29 |
| EP3865501B1 (en) | 2025-04-30 |
| EP3423464A1 (en) | 2019-01-09 |
| US11034716B2 (en) | 2021-06-15 |
| HUE071989T2 (hu) | 2025-10-28 |
| ES3037073T3 (en) | 2025-09-26 |
| JP7264569B2 (ja) | 2023-04-25 |
| WO2017149091A1 (en) | 2017-09-08 |
| US12227537B2 (en) | 2025-02-18 |
| JP2021183627A (ja) | 2021-12-02 |
| EP3865501A1 (en) | 2021-08-18 |
| EP3423464B1 (en) | 2021-05-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7419302B2 (ja) | 11-メチレンステロイドの製造のための方法および新規な中間体 | |
| JP5703295B2 (ja) | 17−スピロラクトンを得る方法 | |
| JP2005528371A (ja) | ステロイドのc−17スピロラクトニゼーションおよび6,7−酸化 | |
| JP6100798B2 (ja) | 16−置換型−17−ケトステロイド類のアルキニル化方法 | |
| JPH021160B2 (ja) | ||
| JPH05509287A (ja) | 11β―置換16α,17α―メチレン―エストラ―4,9―ジエン―3―オン | |
| JP4755083B2 (ja) | 19−ノルステロイド化合物の新製造方法及び中間体 | |
| HU218484B (hu) | Új eljárás 10béta-H-szteroidok előállítására | |
| JP4629030B2 (ja) | 17−ハロゲン化19−ノルステロイド化合物の新製造方法及び中間体 | |
| MX2012009251A (es) | Un proceso para introducir un enlace doble en la posicion 15, 16 de un esteroide. | |
| JP2002533334A (ja) | 抗ヒスタミン活性を有する三環式化合物を調製するためのプロセス | |
| US20060111332A1 (en) | Process and intermediates to prepare17beta-hydroxy-7alpha-methyl-19-nor-17alpha-pregn -5(10)-en-20-yn-3-one | |
| FR2826004A1 (fr) | Procede de preparation de derives estrogenes | |
| CN106414475B (zh) | 生产21‑甲氧基‑11‑β‑苯基‑19‑去甲‑孕甾‑4,9‑二烯‑3,20‑二酮衍生物的方法 | |
| FR2645864A1 (fr) | Nouveaux derives alcoyles en 17/21 de la 19-nor progesterone, leurs procedes d'obtention et les compositions pharmaceutiques en renfermant | |
| RU2663484C1 (ru) | СПОСОБ ПОЛУЧЕНИЯ 6α-МЕТИЛГИДРОКОРТИЗОНА ИЛИ ЕГО ЭФИРОВ ИЗ 21-АЦЕТАТА ГИДРОКОРТИЗОНА | |
| NZ519802A (en) | Process for preparing and isolating 9-deoxo-9(Z)-hydroxyiminoerythromycin A | |
| SE426834B (sv) | Sett att framstella 15alfa, 16alfa-metylen-4-ostren-17beta-oler | |
| WO2019015914A1 (en) | METHODS AND INTERMEDIATES FOR THE SYNTHESIS OF OBETICHOLIC ACID AND DERIVATIVES THEREOF | |
| HUP0301765A2 (hu) | Eljárás 4-(17alfa-szubtituált-3-oxoösztra-4,9-dién-11béta-il)-benzaldehid-(1E vagy 1Z)-oximok elżállítására | |
| JPH0153680B2 (ja) | ||
| WO2005113577A1 (en) | Method for the preparation of trilostane | |
| WO2018185783A1 (en) | Novel process for preparation of 19-norsteroids | |
| HU196433B (en) | Process for producing new 20-amino-steroides | |
| AU4918697A (en) | Method of preparing 21-alkylated pregna-1,4,16-trien-3,20-diones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210820 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210831 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20211126 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220913 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220913 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20221213 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20230516 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230915 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20231012 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20231031 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20231212 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240110 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7419302 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |