WO2001062743A2 - Verfahren zur einfachen herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -quinazolin-4-yl]-amin - Google Patents
Verfahren zur einfachen herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -quinazolin-4-yl]-amin Download PDFInfo
- Publication number
- WO2001062743A2 WO2001062743A2 PCT/EP2001/000695 EP0100695W WO0162743A2 WO 2001062743 A2 WO2001062743 A2 WO 2001062743A2 EP 0100695 W EP0100695 W EP 0100695W WO 0162743 A2 WO0162743 A2 WO 0162743A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- chloro
- fluoro
- propoxy
- amine
- morpholino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- HBUGOFLVIGUDIA-UHFFFAOYSA-N Nc1cc2c(Nc(cc3Cl)ccc3F)ncnc2cc1OCCCN1CCOCC1 Chemical compound Nc1cc2c(Nc(cc3Cl)ccc3F)ncnc2cc1OCCCN1CCOCC1 HBUGOFLVIGUDIA-UHFFFAOYSA-N 0.000 description 1
- QVZKEQDJKMFEDU-UHFFFAOYSA-N [O-][N+](c1cc2c(Nc(cc3Cl)ccc3F)ncnc2cc1OCCCN1CCOCC1)=O Chemical compound [O-][N+](c1cc2c(Nc(cc3Cl)ccc3F)ncnc2cc1OCCCN1CCOCC1)=O QVZKEQDJKMFEDU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/94—Nitrogen atoms
Definitions
- the object of the invention was therefore to provide an economical and technically feasible method for producing the above.
- the invention therefore relates to the combination of the individual reaction steps into a one-pot reaction.
- the product (I) obtained in the reaction mixture was of such a high purity that the reaction solution could be used directly for the subsequent hydrogenation without isolation of (I).
- the reaction mixture is allowed to come slowly to room temperature and then stirred at room temperature for at least 24 hours.
- a solution of 324 g of potassium tert-butoxide in 1.86 L of tetrahydrofuran is added dropwise to the yellow-orange suspension with thorough stirring and gentle cooling for about 20 minutes in such a way that the temperature in the reactor is between 15 ° C. and 20 ° C. ° C remains. After adding about 1/3 of the potassium tert-butoxide THF solution, the entire reaction mixture turns dark red.
- Reactor remains between 10 ° C and 15 ° C.
- the initially yellowish suspension becomes thinner in the course of the dropping and turns orange.
- the reaction mixture is allowed to come slowly to room temperature and then stirred at room temperature for at least 24 hours.
- a solution of 43.2 g of potassium tert-butoxide in 250 ml of tetrahydrofuran is added dropwise to the yellow-orange suspension with vigorous stirring and gentle cooling for about 20 minutes in such a way that the temperature in the reactor is between 15 ° C. and stays at 20 ° C. After adding about 1/3 of the potassium tert-butoxide / THF solution, the entire reaction mixture turns dark red.
- reaction mixture After stirring for about 30 minutes, the reaction mixture is mixed at 0 ° C - 5 ° C with a mixture of 20 mL hydrochloric acid and 30 mL water and diluted with another 200 mL THF. After stirring for 20 minutes in an ice bath, the
- the reaction mixture was filtered through 50 g of Celite.
- the filter cake is washed out with 100 mL THF.
- the filtrate is mixed with 31 g of Raney nickel and hydrogenated at room temperature for 3 hours at 3.5 bar with hydrogen.
- the filtrate is concentrated to dryness and the residue verlickt with 80 mL of ethanol at about 2 C C.
- the precipitated product is filtered off and washed with a little cold ethanol. After drying in a forced-air drying cabinet at 60 ° C., 32.1 g (77.7%) of product are obtained.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description

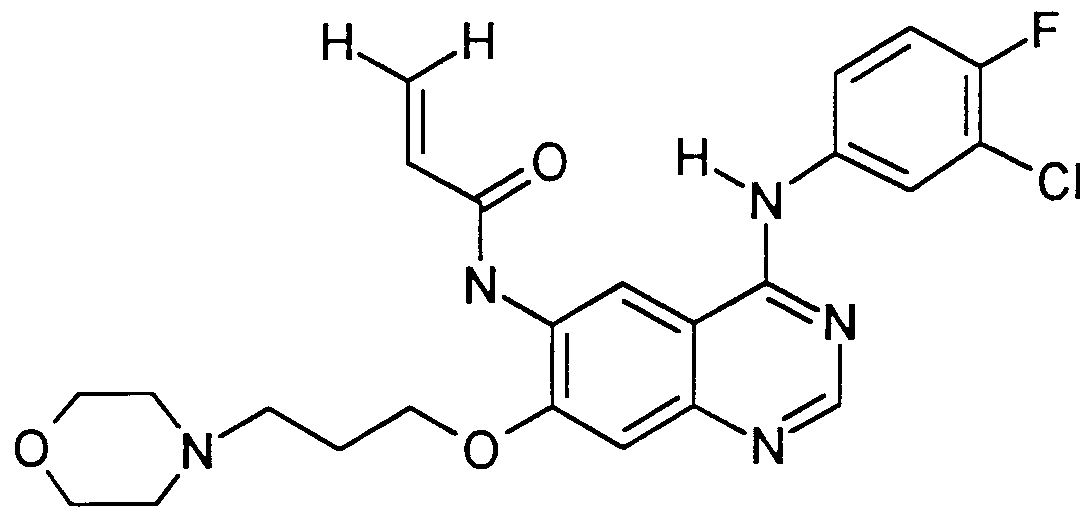








Claims





Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE50106391T DE50106391D1 (de) | 2000-02-26 | 2001-01-23 | Verfahren zur herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-chinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -chinazolin-4-yl]-amin |
| PL01357774A PL357774A1 (pl) | 2000-02-26 | 2001-01-23 | Sposób uproszczonego wytwarzania (3-chloro-4-fluorofenylo)-[7-(3-morfolinylo-4-propoksy)-6-nitro-chinazolinylo-4] aminy albo (3-chloro-4-fluorofenylo)-[7-(3-morfolinylo-4-propoksy-6-aminochinazolinylo-4] aminy |
| AU2001228478A AU2001228478A1 (en) | 2000-02-26 | 2001-01-23 | Method for the simplified production of (3-chloro-4-fluoro-phenyl)-7-(3-morpholino-4-yl-propoxy)-6-nitro-quinazoline-4-yl)-amine or (3-chloro-4-fluoro-phenyl)-(7-(3-morpholino-4-yl-propoxy)-6-amino-quinazoline-4-yl)-amine. |
| IL15120601A IL151206A0 (en) | 2000-02-26 | 2001-01-23 | Process for the simple preparation of (3-chloro-4-fluorophenyl)-7-(3-morpholin-4-yl-propoxy)-6-nitroquinazolin-4-yl]-amine or (3-chloro-4-yl-propoxy)-6-nitroquinazolin-4-yl]-amine or (3-chloro-4-fluorophenyl)-[7(3-morpholin-4-yl-propoxy)-6-aminoquinazolin-4-yl]-amine |
| HK03106705.0A HK1054551B (zh) | 2000-02-26 | 2001-01-23 | 製備(3-氯-4-氟苯基)-胺化合物的方法 |
| EA200200885A EA005213B1 (ru) | 2000-02-26 | 2001-01-23 | Способ простого получения (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-нитрохиназолин-4 -ил]амина или (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6 -аминохиназолин-4-ил]амина |
| HU0204510A HUP0204510A3 (en) | 2000-02-26 | 2001-01-23 | Method for the simplified production of (3-chloro-4-fluoro-phenyl)-[7-(3-morpholino-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluoro-phenyl)[7-(3-morpholino-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
| MXPA02008301A MXPA02008301A (es) | 2000-02-26 | 2001-01-23 | Procedimiento para la produccion simplificada de (3-cloro -4-fluoro-fenil) -[7-(3-morofolino -4-il-propoxi) -6-nitro-quinazolina -4-il] -amina o (3-cloro -4-fluoro- fenil)- [7-(3-morfolino -4-il-propoxi) -6-amino-quinazolina -4-il] -amina. |
| EP01953631A EP1265874B1 (de) | 2000-02-26 | 2001-01-23 | Verfahren zur herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-chinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -chinazolin-4-yl]-amin |
| DK01953631T DK1265874T3 (da) | 2000-02-26 | 2001-01-23 | Fremgangsmåde til fremstilling af (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin eller (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino-quinazolin-4-yl]-amin |
| CA002401171A CA2401171A1 (en) | 2000-02-26 | 2001-01-23 | Process for the simple preparation of(3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitroquinazolin-4-yl]-amine or (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-aminoquinazoline-4-yl]-amine |
| JP2001562525A JP2003524006A (ja) | 2000-02-26 | 2001-01-23 | (3−クロロ−4−フルオロフェニル)−〔7−(3−モルホリン−4−イル−プロポキシ)−6−ニトロキナゾリン−4−イル〕−アミン又は(3−クロロ−4−フルオロフェニル)−〔7−(3−モルホリン−4−イル−プロポキシ)−6−アミノキナゾリン−4−イル〕−アミンの簡単な製造方法 |
| BR0108695-2A BR0108695A (pt) | 2000-02-26 | 2001-01-23 | Processo para a preparação simples de (3-cloro-4-fluorofenil)-[7(3-morfolin-4-il-propóxi)-6-ni troquinazolin-4-il]amina ou (3-cloro-4-fluorofenil)-[7-(3-morfolin-4-il-propóxi)-6-a minoquinazolin-4-il]-amina |
| AT01953631T ATE296810T1 (de) | 2000-02-26 | 2001-01-23 | Verfahren zur herstellung von (3-chlor-4-fluor- phenyl)-(7-(3-morpholin-4-yl-propoxy)-6-nitro- chinazolin-4-yl)-amin bzw. (3-chlor-4-fluor- |
| US10/357,814 US6664390B2 (en) | 2000-02-02 | 2003-02-04 | Method for the simplified production of (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10009267.5 | 2000-02-26 | ||
| DE10009267A DE10009267A1 (de) | 2000-02-26 | 2000-02-26 | Verfahren zur einfachen Herstellung von (3-Chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin bzw. (3-Chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino-quinazolin-4-yl]-amin |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/204,911 A-371-Of-International US20030050313A1 (en) | 2000-02-02 | 2001-01-23 | Method for the simplified production of (3-chloro-4-flourophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazoline-4yl]-amine or (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
| US10/357,814 Continuation-In-Part US6664390B2 (en) | 2000-02-02 | 2003-02-04 | Method for the simplified production of (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazoline-4-yl]-amine or (3-chloro-4-fluorophenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino-quinazoline-4-yl]-amine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001062743A2 true WO2001062743A2 (de) | 2001-08-30 |
| WO2001062743A3 WO2001062743A3 (de) | 2002-03-14 |
Family
ID=7632648
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/000695 Ceased WO2001062743A2 (de) | 2000-02-02 | 2001-01-23 | Verfahren zur einfachen herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -quinazolin-4-yl]-amin |
Country Status (32)
| Country | Link |
|---|---|
| US (1) | US20030050313A1 (de) |
| EP (1) | EP1265874B1 (de) |
| JP (1) | JP2003524006A (de) |
| KR (1) | KR20030011773A (de) |
| CN (1) | CN1171877C (de) |
| AR (1) | AR027377A1 (de) |
| AT (1) | ATE296810T1 (de) |
| AU (1) | AU2001228478A1 (de) |
| BR (1) | BR0108695A (de) |
| CA (1) | CA2401171A1 (de) |
| CO (1) | CO5280058A1 (de) |
| DE (2) | DE10009267A1 (de) |
| EA (1) | EA005213B1 (de) |
| EG (1) | EG23221A (de) |
| ES (1) | ES2240483T3 (de) |
| GT (1) | GT200100025A (de) |
| HK (1) | HK1054551B (de) |
| HU (1) | HUP0204510A3 (de) |
| IL (1) | IL151206A0 (de) |
| MX (1) | MXPA02008301A (de) |
| MY (1) | MY133846A (de) |
| PA (1) | PA8512701A1 (de) |
| PE (1) | PE20011073A1 (de) |
| PL (1) | PL357774A1 (de) |
| PT (1) | PT1265874E (de) |
| PY (1) | PY0102711A (de) |
| SV (1) | SV2002000320A (de) |
| TN (1) | TNSN01031A1 (de) |
| UY (1) | UY26594A1 (de) |
| WO (1) | WO2001062743A2 (de) |
| YU (1) | YU62502A (de) |
| ZA (1) | ZA200206107B (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004069791A3 (en) * | 2003-02-05 | 2004-12-16 | Warner Lambert Co | Preparation of substituted quinazolines |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006081741A1 (en) * | 2005-02-05 | 2006-08-10 | Piaoyang Sun | Quinazoline compounds or their medical salts and preparation and medical usage thereof |
| JP5250901B2 (ja) * | 2007-02-23 | 2013-07-31 | 学校法人慶應義塾 | アニリノキナゾリン系化合物及びその用途 |
| US7829574B2 (en) | 2008-05-09 | 2010-11-09 | Hutchison Medipharma Enterprises Limited | Substituted quinazoline compounds and their use in treating angiogenesis-related diseases |
| CN105399688A (zh) * | 2015-12-02 | 2016-03-16 | 西南科技大学 | 一种吉非替尼的制备方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HU228446B1 (en) * | 1996-04-12 | 2013-03-28 | Warner Lambert Co | Kinazoline derivatives as irreversible inhibitors of protein-kinase, pharmaceutical compositions containing these compounds and use thereof |
-
2000
- 2000-02-26 DE DE10009267A patent/DE10009267A1/de not_active Withdrawn
-
2001
- 2001-01-23 US US10/204,911 patent/US20030050313A1/en not_active Abandoned
- 2001-01-23 HK HK03106705.0A patent/HK1054551B/zh not_active IP Right Cessation
- 2001-01-23 JP JP2001562525A patent/JP2003524006A/ja active Pending
- 2001-01-23 EA EA200200885A patent/EA005213B1/ru not_active IP Right Cessation
- 2001-01-23 CA CA002401171A patent/CA2401171A1/en not_active Abandoned
- 2001-01-23 WO PCT/EP2001/000695 patent/WO2001062743A2/de not_active Ceased
- 2001-01-23 AT AT01953631T patent/ATE296810T1/de not_active IP Right Cessation
- 2001-01-23 MX MXPA02008301A patent/MXPA02008301A/es active IP Right Grant
- 2001-01-23 ES ES01953631T patent/ES2240483T3/es not_active Expired - Lifetime
- 2001-01-23 EP EP01953631A patent/EP1265874B1/de not_active Expired - Lifetime
- 2001-01-23 BR BR0108695-2A patent/BR0108695A/pt not_active IP Right Cessation
- 2001-01-23 KR KR1020027010713A patent/KR20030011773A/ko not_active Withdrawn
- 2001-01-23 IL IL15120601A patent/IL151206A0/xx unknown
- 2001-01-23 HU HU0204510A patent/HUP0204510A3/hu unknown
- 2001-01-23 DE DE50106391T patent/DE50106391D1/de not_active Expired - Fee Related
- 2001-01-23 PT PT01953631T patent/PT1265874E/pt unknown
- 2001-01-23 CN CNB018056288A patent/CN1171877C/zh not_active Expired - Fee Related
- 2001-01-23 YU YU62502A patent/YU62502A/sh unknown
- 2001-01-23 AU AU2001228478A patent/AU2001228478A1/en not_active Abandoned
- 2001-01-23 PL PL01357774A patent/PL357774A1/xx not_active Application Discontinuation
- 2001-02-07 AR ARP010100547A patent/AR027377A1/es unknown
- 2001-02-08 PY PY200100102711A patent/PY0102711A/es unknown
- 2001-02-08 PE PE2001000137A patent/PE20011073A1/es not_active Application Discontinuation
- 2001-02-08 GT GT200100025A patent/GT200100025A/es unknown
- 2001-02-22 PA PA20018512701A patent/PA8512701A1/es unknown
- 2001-02-23 SV SV2001000320A patent/SV2002000320A/es not_active Application Discontinuation
- 2001-02-23 UY UY26594A patent/UY26594A1/es not_active Application Discontinuation
- 2001-02-23 TN TNTNSN01031A patent/TNSN01031A1/en unknown
- 2001-02-23 CO CO01014681A patent/CO5280058A1/es unknown
- 2001-02-23 MY MYPI20010826A patent/MY133846A/en unknown
- 2001-02-24 EG EG20010176A patent/EG23221A/xx active
-
2002
- 2002-07-31 ZA ZA200206107A patent/ZA200206107B/en unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004069791A3 (en) * | 2003-02-05 | 2004-12-16 | Warner Lambert Co | Preparation of substituted quinazolines |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69918528T2 (de) | Bicyclische heteroaromatische verbindungen als protein tyrosine kinase inhibitoren | |
| DE69922823T2 (de) | 6-SUBSTITUIERTE PYRAZOLO(3,4-d)PYRIMIDIN-4-ONE VERWENDBAR ALS CYCLIN-ABHÄNGIGE KINASEHEMMER | |
| DE69415554T2 (de) | Diaminocyclobuten-3,4-dione mit einer entspannenden wirkung auf glatte muskeln | |
| DE69804273T2 (de) | 5h-Thiazolo[3,2-a]pyrimidinderivate | |
| CH642361A5 (de) | 4-anilinochinazolinderivate, ihre herstellung und diese enthaltende pharmazeutische praeparate. | |
| EP0005205A1 (de) | Substituierte 5,6-Dimethylpyrrolo(2,3-d)pyrimidine, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel | |
| DE3418270A1 (de) | Neue aminotetralinderivate, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung | |
| JP3276371B2 (ja) | 置換イミダゾール誘導体ならびにそれらの製造法および用途 | |
| DE60004671T2 (de) | Neue piperazinylalkylthiopyrimidine derivate, diese enthaltende pharmazeutische zusammenstellungen und verfahren zu deren herstellung | |
| WO2001062743A2 (de) | Verfahren zur einfachen herstellung von (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-nitro-quinazolin-4-yl]-amin bzw. (3-chlor-4-fluor-phenyl)-[7-(3-morpholin-4-yl-propoxy)-6-amino -quinazolin-4-yl]-amin | |
| DE2052719A1 (de) | Verfahren zur Herstellung von neuen 5 Nitrofuryldenvaten | |
| DE1518373B2 (de) | 2 Amino halogen benzyiamine und Verfahren zu deren Herstellung | |
| DE2044172A1 (de) | Neue Pyrroldenvate, ein Verfahren zu ihrer Herstellung und ihre Verwendung in Arzneimittelzubereitungen | |
| EP0146642B1 (de) | Verfahren zur Herstellung von 4-Amino-6,7-dimethoxy-2-(4-(furo-2-yl)-piperazin-1-yl)-chinazolin und dessen physiologisch annehmbarer Salze | |
| DE2264374C3 (de) | 4-Hydroxy-5-phenoxy-pyrimidine, Verfahren zu ihrer Herstellung und diese enthaltendes pharmazeutisches Präparat | |
| EP0331093A1 (de) | Neue Benzimidazo[1,2-c] chinazoline, ihre Herstellung und Verwendung | |
| EP0377437B1 (de) | Verfahren zur Herstellung von 2,4-Diamino-6-piperidinyl-pyrimidin-3-N-oxid | |
| EP0167045A1 (de) | Benzo (c)(1,8)naphthyridine, Verfahren zu ihrer Herstellung und ihre Verwendung sowie diese Verbindungen enthaltende Zubereitungen | |
| DD296930A5 (de) | Dihydro-pyrimido-thiazin-derivate und verfahren zu ihrer herstellung | |
| DE1203269B (de) | Verfahren zur Herstellung von Imidazolon-2-derivaten und ihren Salzen | |
| EP0039037B1 (de) | Pyrimidylthioharnstoffe, diese enthaltende Arzneimittel und deren Verwendung | |
| DE951991C (de) | Verfahren zur Herstellung von neuen chlorierten Derivaten des Pyrimidins | |
| AT319955B (de) | Verfahren zur Herstellung von neuen 2,4-Diamino-5-benzylpyrimidinen | |
| DE1620171A1 (de) | Verfahren zur Herstellung von heterocyclischen Verbindungen | |
| DE2229359A1 (de) | Neue 1-benzoyloxy-2-niedere-alkylaminobenzocycloalkanderivate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: P-625/02 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AU BA BB BG BR BZ CA CN CR CU CZ DM DZ EE GD GE HR HU ID IL IN IS JP KP KR LC LK LR LT LV MA MG MK MN MX NO NZ PL RO SG SI SK SL TT UA US UZ VN YU ZA |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AU BA BB BG BR BZ CA CN CR CU CZ DM DZ EE GD GE HR HU ID IL IN IS JP KP KR LC LK LR LT LV MA MG MK MN MX NO NZ PL RO SG SI SK SL TT UA US UZ VN YU ZA |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/06107 Country of ref document: ZA Ref document number: 200206107 Country of ref document: ZA Ref document number: IN/PCT/2002/01036/MU Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001953631 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 151206 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027010713 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2001 562525 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2401171 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10204911 Country of ref document: US Ref document number: 018056288 Country of ref document: CN Ref document number: PA/a/2002/008301 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200200885 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001228478 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001953631 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027010713 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001953631 Country of ref document: EP |