ES2746530T3 - 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5- il)urea como un inhibidor de TrkA cinasa - Google Patents
1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5- il)urea como un inhibidor de TrkA cinasa Download PDFInfo
- Publication number
- ES2746530T3 ES2746530T3 ES15726455T ES15726455T ES2746530T3 ES 2746530 T3 ES2746530 T3 ES 2746530T3 ES 15726455 T ES15726455 T ES 15726455T ES 15726455 T ES15726455 T ES 15726455T ES 2746530 T3 ES2746530 T3 ES 2746530T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- trka
- pain
- use according
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 101150111783 NTRK1 gene Proteins 0.000 title claims description 90
- 229940043355 kinase inhibitor Drugs 0.000 title claims description 8
- 239000003757 phosphotransferase inhibitor Substances 0.000 title claims description 8
- BGKSBHPSVMJTFL-IZZNHLLZSA-N 1-[(3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl]-3-[4-methyl-5-(2-methylpyrimidin-5-yl)-2-phenylpyrazol-3-yl]urea Chemical compound FC=1C=C(C=CC1)[C@H]1[C@@H](CN(C1)CCOC)NC(=O)NC1=C(C(=NN1C1=CC=CC=C1)C=1C=NC(=NC1)C)C BGKSBHPSVMJTFL-IZZNHLLZSA-N 0.000 title claims description 7
- 150000001875 compounds Chemical class 0.000 claims abstract description 137
- 150000003839 salts Chemical class 0.000 claims abstract description 135
- 229940125904 compound 1 Drugs 0.000 claims description 197
- 208000002193 Pain Diseases 0.000 claims description 104
- 206010028980 Neoplasm Diseases 0.000 claims description 79
- 230000036407 pain Effects 0.000 claims description 79
- 238000011282 treatment Methods 0.000 claims description 72
- 201000011510 cancer Diseases 0.000 claims description 68
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 56
- 239000003814 drug Substances 0.000 claims description 53
- 238000000034 method Methods 0.000 claims description 51
- 201000010099 disease Diseases 0.000 claims description 50
- 238000002360 preparation method Methods 0.000 claims description 39
- 239000003112 inhibitor Substances 0.000 claims description 31
- 229940124597 therapeutic agent Drugs 0.000 claims description 27
- 208000027866 inflammatory disease Diseases 0.000 claims description 20
- 238000002560 therapeutic procedure Methods 0.000 claims description 20
- 230000011664 signaling Effects 0.000 claims description 17
- 208000032131 Diabetic Neuropathies Diseases 0.000 claims description 16
- 239000002246 antineoplastic agent Substances 0.000 claims description 16
- 229940127089 cytotoxic agent Drugs 0.000 claims description 16
- 229940079593 drug Drugs 0.000 claims description 16
- 206010016654 Fibrosis Diseases 0.000 claims description 15
- 206010061218 Inflammation Diseases 0.000 claims description 15
- 238000006243 chemical reaction Methods 0.000 claims description 15
- 230000004054 inflammatory process Effects 0.000 claims description 15
- 230000004770 neurodegeneration Effects 0.000 claims description 15
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 15
- 208000000094 Chronic Pain Diseases 0.000 claims description 14
- 230000001594 aberrant effect Effects 0.000 claims description 14
- 239000003795 chemical substances by application Substances 0.000 claims description 14
- 230000008482 dysregulation Effects 0.000 claims description 14
- 208000011580 syndromic disease Diseases 0.000 claims description 14
- 108010039419 Connective Tissue Growth Factor Proteins 0.000 claims description 13
- 208000005298 acute pain Diseases 0.000 claims description 12
- 230000004761 fibrosis Effects 0.000 claims description 12
- 201000009273 Endometriosis Diseases 0.000 claims description 11
- 208000000450 Pelvic Pain Diseases 0.000 claims description 11
- 208000004296 neuralgia Diseases 0.000 claims description 11
- 208000021722 neuropathic pain Diseases 0.000 claims description 11
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 201000007094 prostatitis Diseases 0.000 claims description 11
- 230000026749 regulation of bone remodeling Effects 0.000 claims description 11
- 208000010392 Bone Fractures Diseases 0.000 claims description 10
- 206010065390 Inflammatory pain Diseases 0.000 claims description 10
- 206010029260 Neuroblastoma Diseases 0.000 claims description 10
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 10
- 238000001356 surgical procedure Methods 0.000 claims description 10
- 206010009944 Colon cancer Diseases 0.000 claims description 9
- 206010060862 Prostate cancer Diseases 0.000 claims description 9
- 230000003831 deregulation Effects 0.000 claims description 9
- 208000010749 gastric carcinoma Diseases 0.000 claims description 9
- 201000001441 melanoma Diseases 0.000 claims description 9
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 8
- 206010033701 Papillary thyroid cancer Diseases 0.000 claims description 8
- 206010017758 gastric cancer Diseases 0.000 claims description 8
- 208000005017 glioblastoma Diseases 0.000 claims description 8
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 claims description 8
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 claims description 8
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 8
- 201000000498 stomach carcinoma Diseases 0.000 claims description 8
- 208000030045 thyroid gland papillary carcinoma Diseases 0.000 claims description 8
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 7
- 208000024699 Chagas disease Diseases 0.000 claims description 7
- 201000010915 Glioblastoma multiforme Diseases 0.000 claims description 7
- 208000005615 Interstitial Cystitis Diseases 0.000 claims description 7
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 7
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 claims description 7
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 7
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 7
- 206010001935 American trypanosomiasis Diseases 0.000 claims description 6
- 208000008035 Back Pain Diseases 0.000 claims description 6
- 230000003110 anti-inflammatory effect Effects 0.000 claims description 6
- 201000010989 colorectal carcinoma Diseases 0.000 claims description 6
- 208000035475 disorder Diseases 0.000 claims description 6
- 230000004927 fusion Effects 0.000 claims description 6
- 230000002757 inflammatory effect Effects 0.000 claims description 6
- 229940005483 opioid analgesics Drugs 0.000 claims description 6
- 108010008165 Etanercept Proteins 0.000 claims description 5
- 102000042838 JAK family Human genes 0.000 claims description 5
- 108091082332 JAK family Proteins 0.000 claims description 5
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 claims description 5
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 claims description 5
- 206010039491 Sarcoma Diseases 0.000 claims description 5
- 230000004913 activation Effects 0.000 claims description 5
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 5
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 5
- 230000037430 deletion Effects 0.000 claims description 5
- 238000012217 deletion Methods 0.000 claims description 5
- 230000009977 dual effect Effects 0.000 claims description 5
- 229960000485 methotrexate Drugs 0.000 claims description 5
- 230000035772 mutation Effects 0.000 claims description 5
- 230000002018 overexpression Effects 0.000 claims description 5
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 claims description 5
- 229960000215 ruxolitinib Drugs 0.000 claims description 5
- 150000003431 steroids Chemical class 0.000 claims description 5
- 208000016718 Chromosome Inversion Diseases 0.000 claims description 4
- 201000004624 Dermatitis Diseases 0.000 claims description 4
- 208000034951 Genetic Translocation Diseases 0.000 claims description 4
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 4
- 208000019693 Lung disease Diseases 0.000 claims description 4
- 239000004012 Tofacitinib Substances 0.000 claims description 4
- 238000011374 additional therapy Methods 0.000 claims description 4
- 230000003305 autocrine Effects 0.000 claims description 4
- 239000003085 diluting agent Substances 0.000 claims description 4
- 238000003780 insertion Methods 0.000 claims description 4
- 230000037431 insertion Effects 0.000 claims description 4
- 229960001350 tofacitinib Drugs 0.000 claims description 4
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 claims description 4
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 claims description 4
- 206010058029 Arthrofibrosis Diseases 0.000 claims description 3
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 3
- 102000004310 Ion Channels Human genes 0.000 claims description 3
- 208000002260 Keloid Diseases 0.000 claims description 3
- 206010023330 Keloid scar Diseases 0.000 claims description 3
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 claims description 3
- JOOXLOJCABQBSG-UHFFFAOYSA-N N-tert-butyl-3-[[5-methyl-2-[4-[2-(1-pyrrolidinyl)ethoxy]anilino]-4-pyrimidinyl]amino]benzenesulfonamide Chemical compound N1=C(NC=2C=C(C=CC=2)S(=O)(=O)NC(C)(C)C)C(C)=CN=C1NC(C=C1)=CC=C1OCCN1CCCC1 JOOXLOJCABQBSG-UHFFFAOYSA-N 0.000 claims description 3
- 208000003510 Nephrogenic Fibrosing Dermopathy Diseases 0.000 claims description 3
- 206010067467 Nephrogenic systemic fibrosis Diseases 0.000 claims description 3
- 208000003782 Raynaud disease Diseases 0.000 claims description 3
- 208000012322 Raynaud phenomenon Diseases 0.000 claims description 3
- 206010038748 Restrictive cardiomyopathy Diseases 0.000 claims description 3
- 206010039710 Scleroderma Diseases 0.000 claims description 3
- 201000009594 Systemic Scleroderma Diseases 0.000 claims description 3
- 206010042953 Systemic sclerosis Diseases 0.000 claims description 3
- 229960002964 adalimumab Drugs 0.000 claims description 3
- 230000033115 angiogenesis Effects 0.000 claims description 3
- 230000003432 anti-folate effect Effects 0.000 claims description 3
- 230000000340 anti-metabolite Effects 0.000 claims description 3
- 229940125681 anticonvulsant agent Drugs 0.000 claims description 3
- 239000001961 anticonvulsive agent Substances 0.000 claims description 3
- 229940127074 antifolate Drugs 0.000 claims description 3
- 229940100197 antimetabolite Drugs 0.000 claims description 3
- 239000002256 antimetabolite Substances 0.000 claims description 3
- 230000001746 atrial effect Effects 0.000 claims description 3
- 239000002775 capsule Substances 0.000 claims description 3
- 229960003115 certolizumab pegol Drugs 0.000 claims description 3
- 230000007882 cirrhosis Effects 0.000 claims description 3
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 3
- 231100000433 cytotoxic Toxicity 0.000 claims description 3
- 230000001472 cytotoxic effect Effects 0.000 claims description 3
- 201000010048 endomyocardial fibrosis Diseases 0.000 claims description 3
- 229960000403 etanercept Drugs 0.000 claims description 3
- 239000004052 folic acid antagonist Substances 0.000 claims description 3
- 229960001743 golimumab Drugs 0.000 claims description 3
- 230000001969 hypertrophic effect Effects 0.000 claims description 3
- 229960000598 infliximab Drugs 0.000 claims description 3
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 3
- 210000001117 keloid Anatomy 0.000 claims description 3
- 201000003445 large cell neuroendocrine carcinoma Diseases 0.000 claims description 3
- 229950001845 lestaurtinib Drugs 0.000 claims description 3
- 229950008814 momelotinib Drugs 0.000 claims description 3
- ZVHNDZWQTBEVRY-UHFFFAOYSA-N momelotinib Chemical compound C1=CC(C(NCC#N)=O)=CC=C1C1=CC=NC(NC=2C=CC(=CC=2)N2CCOCC2)=N1 ZVHNDZWQTBEVRY-UHFFFAOYSA-N 0.000 claims description 3
- 206010028537 myelofibrosis Diseases 0.000 claims description 3
- 229950011410 pacritinib Drugs 0.000 claims description 3
- HWXVIOGONBBTBY-ONEGZZNKSA-N pacritinib Chemical compound C=1C=C(C=2)NC(N=3)=NC=CC=3C(C=3)=CC=CC=3COC\C=C\COCC=2C=1OCCN1CCCC1 HWXVIOGONBBTBY-ONEGZZNKSA-N 0.000 claims description 3
- 230000000750 progressive effect Effects 0.000 claims description 3
- 238000001959 radiotherapy Methods 0.000 claims description 3
- 230000037390 scarring Effects 0.000 claims description 3
- 230000019491 signal transduction Effects 0.000 claims description 3
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 claims description 2
- 229940123445 Tricyclic antidepressant Drugs 0.000 claims description 2
- 230000000202 analgesic effect Effects 0.000 claims description 2
- 229940076005 apoptosis modulator Drugs 0.000 claims description 2
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 claims description 2
- 230000001747 exhibiting effect Effects 0.000 claims description 2
- 230000008569 process Effects 0.000 claims description 2
- 201000001514 prostate carcinoma Diseases 0.000 claims description 2
- 125000006239 protecting group Chemical group 0.000 claims description 2
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 claims description 2
- 239000003029 tricyclic antidepressant agent Substances 0.000 claims description 2
- 239000002451 tumor necrosis factor inhibitor Substances 0.000 claims description 2
- 102000015225 Connective Tissue Growth Factor Human genes 0.000 claims 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 claims 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 claims 2
- 206010006002 Bone pain Diseases 0.000 claims 1
- 108090000932 Calcitonin Gene-Related Peptide Proteins 0.000 claims 1
- 102000004414 Calcitonin Gene-Related Peptide Human genes 0.000 claims 1
- 102000015098 Tumor Suppressor Protein p53 Human genes 0.000 claims 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 claims 1
- 229940035676 analgesics Drugs 0.000 claims 1
- 239000000730 antalgic agent Substances 0.000 claims 1
- 201000003911 head and neck carcinoma Diseases 0.000 claims 1
- 230000002685 pulmonary effect Effects 0.000 claims 1
- 239000002464 receptor antagonist Substances 0.000 claims 1
- 229940044551 receptor antagonist Drugs 0.000 claims 1
- 206010041823 squamous cell carcinoma Diseases 0.000 claims 1
- 238000002626 targeted therapy Methods 0.000 claims 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 abstract description 9
- 239000004202 carbamide Substances 0.000 abstract description 5
- LNJMHEJAYSYZKK-UHFFFAOYSA-N 2-methylpyrimidine Chemical compound CC1=NC=CC=N1 LNJMHEJAYSYZKK-UHFFFAOYSA-N 0.000 abstract description 2
- 241000124008 Mammalia Species 0.000 description 67
- 239000000203 mixture Substances 0.000 description 65
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 39
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 38
- 239000000243 solution Substances 0.000 description 37
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 36
- -1 RN-624 Chemical compound 0.000 description 33
- 229940125782 compound 2 Drugs 0.000 description 32
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 30
- 125000000217 alkyl group Chemical group 0.000 description 29
- 239000011541 reaction mixture Substances 0.000 description 28
- 229940126214 compound 3 Drugs 0.000 description 25
- 239000007787 solid Substances 0.000 description 19
- 238000012360 testing method Methods 0.000 description 19
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 241000700159 Rattus Species 0.000 description 17
- 235000019439 ethyl acetate Nutrition 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 16
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 16
- 239000002585 base Substances 0.000 description 16
- 229910052736 halogen Inorganic materials 0.000 description 16
- 150000002367 halogens Chemical class 0.000 description 16
- 230000005764 inhibitory process Effects 0.000 description 16
- 239000010410 layer Substances 0.000 description 16
- 102100031168 CCN family member 2 Human genes 0.000 description 15
- 108010025020 Nerve Growth Factor Proteins 0.000 description 15
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 15
- 238000001990 intravenous administration Methods 0.000 description 15
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 14
- 108091000080 Phosphotransferase Proteins 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 14
- 102000020233 phosphotransferase Human genes 0.000 description 14
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 14
- 125000003545 alkoxy group Chemical group 0.000 description 13
- 108090000623 proteins and genes Proteins 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 239000002904 solvent Substances 0.000 description 13
- 239000011550 stock solution Substances 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 238000011534 incubation Methods 0.000 description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 12
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- 238000004458 analytical method Methods 0.000 description 10
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 10
- 239000000543 intermediate Substances 0.000 description 10
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 10
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 9
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- HCWFKVHARKTZKK-UHFFFAOYSA-N 2-methoxy-n-(trimethylsilylmethyl)ethanamine Chemical compound COCCNC[Si](C)(C)C HCWFKVHARKTZKK-UHFFFAOYSA-N 0.000 description 8
- LYMABWZJXOWOCQ-UHFFFAOYSA-N 4-methyl-5-(2-methylpyrimidin-5-yl)-2-phenylpyrazol-3-amine Chemical compound CC1=C(N)N(C=2C=CC=CC=2)N=C1C1=CN=C(C)N=C1 LYMABWZJXOWOCQ-UHFFFAOYSA-N 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- 102000015336 Nerve Growth Factor Human genes 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 210000004556 brain Anatomy 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 239000012086 standard solution Substances 0.000 description 8
- NTOIKDYVJIWVSU-WOJBJXKFSA-N (2r,3r)-2,3-dihydroxy-2,3-bis(4-methylbenzoyl)butanedioic acid Chemical class C1=CC(C)=CC=C1C(=O)[C@@](O)(C(O)=O)[C@](O)(C(O)=O)C(=O)C1=CC=C(C)C=C1 NTOIKDYVJIWVSU-WOJBJXKFSA-N 0.000 description 7
- 208000035473 Communicable disease Diseases 0.000 description 7
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 7
- 206010027452 Metastases to bone Diseases 0.000 description 7
- 102000007072 Nerve Growth Factors Human genes 0.000 description 7
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 7
- 238000013103 analytical ultracentrifugation Methods 0.000 description 7
- 210000003494 hepatocyte Anatomy 0.000 description 7
- 230000037361 pathway Effects 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- IWWQROMVQSKLDE-UHFFFAOYSA-N 2-methoxy-n-(methoxymethyl)-n-(trimethylsilylmethyl)ethanamine Chemical compound COCCN(COC)C[Si](C)(C)C IWWQROMVQSKLDE-UHFFFAOYSA-N 0.000 description 6
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 6
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 6
- 239000005695 Ammonium acetate Substances 0.000 description 6
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 229940043376 ammonium acetate Drugs 0.000 description 6
- 235000019257 ammonium acetate Nutrition 0.000 description 6
- 208000037765 diseases and disorders Diseases 0.000 description 6
- 238000005516 engineering process Methods 0.000 description 6
- 125000001153 fluoro group Chemical group F* 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 6
- 230000002440 hepatic effect Effects 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 229960005181 morphine Drugs 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 6
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 5
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 5
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 208000001132 Osteoporosis Diseases 0.000 description 5
- 229960001138 acetylsalicylic acid Drugs 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000012491 analyte Substances 0.000 description 5
- 229940043279 diisopropylamine Drugs 0.000 description 5
- 238000004821 distillation Methods 0.000 description 5
- 238000000605 extraction Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 108020001507 fusion proteins Proteins 0.000 description 5
- 102000037865 fusion proteins Human genes 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 229960001680 ibuprofen Drugs 0.000 description 5
- 229960000905 indomethacin Drugs 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 5
- 229960000991 ketoprofen Drugs 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 235000015320 potassium carbonate Nutrition 0.000 description 5
- 206010039073 rheumatoid arthritis Diseases 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 5
- 102000015533 trkA Receptor Human genes 0.000 description 5
- 108010064884 trkA Receptor Proteins 0.000 description 5
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 4
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 206010058019 Cancer Pain Diseases 0.000 description 4
- 241000282472 Canis lupus familiaris Species 0.000 description 4
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 4
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 4
- 101100291385 Drosophila melanogaster p38a gene Proteins 0.000 description 4
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 4
- UGJMXCAKCUNAIE-UHFFFAOYSA-N Gabapentin Chemical compound OC(=O)CC1(CN)CCCCC1 UGJMXCAKCUNAIE-UHFFFAOYSA-N 0.000 description 4
- 101000777550 Homo sapiens CCN family member 2 Proteins 0.000 description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 4
- 239000007832 Na2SO4 Substances 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 229960004544 cortisone Drugs 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 229960003957 dexamethasone Drugs 0.000 description 4
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 4
- 229960002714 fluticasone Drugs 0.000 description 4
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 4
- 238000000338 in vitro Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 125000000842 isoxazolyl group Chemical group 0.000 description 4
- 208000003849 large cell carcinoma Diseases 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 230000000955 neuroendocrine Effects 0.000 description 4
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical class C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 4
- 125000002971 oxazolyl group Chemical group 0.000 description 4
- 229940124641 pain reliever Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 238000000159 protein binding assay Methods 0.000 description 4
- 125000004076 pyridyl group Chemical group 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- 125000001544 thienyl group Chemical group 0.000 description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 4
- XINQFOMFQFGGCQ-UHFFFAOYSA-L (2-dodecoxy-2-oxoethyl)-[6-[(2-dodecoxy-2-oxoethyl)-dimethylazaniumyl]hexyl]-dimethylazanium;dichloride Chemical compound [Cl-].[Cl-].CCCCCCCCCCCCOC(=O)C[N+](C)(C)CCCCCC[N+](C)(C)CC(=O)OCCCCCCCCCCCC XINQFOMFQFGGCQ-UHFFFAOYSA-L 0.000 description 3
- RJIDSVJDKGCEFC-UHFFFAOYSA-N (5-amino-4-methyl-1-phenylpyrazol-3-yl) trifluoromethanesulfonate Chemical compound NC1=C(C)C(OS(=O)(=O)C(F)(F)F)=NN1C1=CC=CC=C1 RJIDSVJDKGCEFC-UHFFFAOYSA-N 0.000 description 3
- IUURCNSBVAESQR-UHFFFAOYSA-N 3-amino-4-methyl-2-phenyl-1h-pyrazol-5-one Chemical compound N1C(=O)C(C)=C(N)N1C1=CC=CC=C1 IUURCNSBVAESQR-UHFFFAOYSA-N 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 206010009900 Colitis ulcerative Diseases 0.000 description 3
- 208000011231 Crohn disease Diseases 0.000 description 3
- 206010012438 Dermatitis atopic Diseases 0.000 description 3
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 3
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 3
- 235000009827 Prunus armeniaca Nutrition 0.000 description 3
- 244000018633 Prunus armeniaca Species 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 201000006704 Ulcerative Colitis Diseases 0.000 description 3
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 208000006673 asthma Diseases 0.000 description 3
- 201000008937 atopic dermatitis Diseases 0.000 description 3
- 230000027455 binding Effects 0.000 description 3
- 230000017531 blood circulation Effects 0.000 description 3
- 210000000988 bone and bone Anatomy 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 238000011088 calibration curve Methods 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 230000037011 constitutive activity Effects 0.000 description 3
- 238000012937 correction Methods 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 238000007865 diluting Methods 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 239000002054 inoculum Substances 0.000 description 3
- 230000010354 integration Effects 0.000 description 3
- 229960002725 isoflurane Drugs 0.000 description 3
- 210000001853 liver microsome Anatomy 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 230000003228 microsomal effect Effects 0.000 description 3
- 230000005012 migration Effects 0.000 description 3
- 238000013508 migration Methods 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000012488 sample solution Substances 0.000 description 3
- 238000013207 serial dilution Methods 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 3
- 235000019798 tripotassium phosphate Nutrition 0.000 description 3
- 210000003462 vein Anatomy 0.000 description 3
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 2
- QXWMHBLZOICQSI-QWHCGFSZSA-N (3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidine-3-carboxylic acid Chemical compound FC=1C=C(C=CC1)[C@H]1[C@@H](CN(C1)CCOC)C(=O)O QXWMHBLZOICQSI-QWHCGFSZSA-N 0.000 description 2
- DCHKJXBSVRDRCN-QWHCGFSZSA-N (3s,4r)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-amine Chemical compound C1N(CCOC)C[C@@H](N)[C@@H]1C1=CC=CC(F)=C1 DCHKJXBSVRDRCN-QWHCGFSZSA-N 0.000 description 2
- ZTVHJKAWOVGJTF-CQSOCPNPSA-N (3s,4r)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-amine;dihydrochloride Chemical compound Cl.Cl.C1N(CCOC)C[C@@H](N)[C@@H]1C1=CC=CC(F)=C1 ZTVHJKAWOVGJTF-CQSOCPNPSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- ZEUITGRIYCTCEM-KRWDZBQOSA-N (S)-duloxetine Chemical compound C1([C@@H](OC=2C3=CC=CC=C3C=CC=2)CCNC)=CC=CS1 ZEUITGRIYCTCEM-KRWDZBQOSA-N 0.000 description 2
- HMZMJVUIFWVPPL-JYFHCDHNSA-N 1-[(3s,4r)-4-(3,4-difluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl]-3-[4-methyl-5-(2-methylpyrimidin-5-yl)-2-phenylpyrazol-3-yl]urea Chemical compound N([C@@H]1CN(C[C@H]1C=1C=C(F)C(F)=CC=1)CCOC)C(=O)NC1=C(C)C(C=2C=NC(C)=NC=2)=NN1C1=CC=CC=C1 HMZMJVUIFWVPPL-JYFHCDHNSA-N 0.000 description 2
- JDIIGWSSTNUWGK-UHFFFAOYSA-N 1h-imidazol-3-ium;chloride Chemical compound [Cl-].[NH2+]1C=CN=C1 JDIIGWSSTNUWGK-UHFFFAOYSA-N 0.000 description 2
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 2
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- 239000012099 Alexa Fluor family Substances 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 2
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 2
- 108091006146 Channels Proteins 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 2
- 230000004544 DNA amplification Effects 0.000 description 2
- 208000016192 Demyelinating disease Diseases 0.000 description 2
- 206010012305 Demyelination Diseases 0.000 description 2
- HCYAFALTSJYZDH-UHFFFAOYSA-N Desimpramine Chemical compound C1CC2=CC=CC=C2N(CCCNC)C2=CC=CC=C21 HCYAFALTSJYZDH-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 229910052693 Europium Inorganic materials 0.000 description 2
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 2
- 208000037767 Gallbladder pain Diseases 0.000 description 2
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 2
- 238000006842 Henry reaction Methods 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 108090000862 Ion Channels Proteins 0.000 description 2
- 239000007836 KH2PO4 Substances 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 2
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 2
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 2
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 101150117329 NTRK3 gene Proteins 0.000 description 2
- PHVGLTMQBUFIQQ-UHFFFAOYSA-N Nortryptiline Chemical compound C1CC2=CC=CC=C2C(=CCCNC)C2=CC=CC=C21 PHVGLTMQBUFIQQ-UHFFFAOYSA-N 0.000 description 2
- 101150056950 Ntrk2 gene Proteins 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 102000004257 Potassium Channel Human genes 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 239000005708 Sodium hypochlorite Substances 0.000 description 2
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 2
- 241000223109 Trypanosoma cruzi Species 0.000 description 2
- IKWTVSLWAPBBKU-UHFFFAOYSA-N a1010_sial Chemical compound O=[As]O[As]=O IKWTVSLWAPBBKU-UHFFFAOYSA-N 0.000 description 2
- 229960002833 aflibercept Drugs 0.000 description 2
- 108010081667 aflibercept Proteins 0.000 description 2
- 229960005310 aldesleukin Drugs 0.000 description 2
- 108700025316 aldesleukin Proteins 0.000 description 2
- 229940008201 allegra Drugs 0.000 description 2
- 229960000836 amitriptyline Drugs 0.000 description 2
- KRMDCWKBEZIMAB-UHFFFAOYSA-N amitriptyline Chemical compound C1CC2=CC=CC=C2C(=CCCN(C)C)C2=CC=CC=C21 KRMDCWKBEZIMAB-UHFFFAOYSA-N 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 229960002594 arsenic trioxide Drugs 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 229940077737 brain-derived neurotrophic factor Drugs 0.000 description 2
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 description 2
- 229960001573 cabazitaxel Drugs 0.000 description 2
- 239000003735 calcitonin gene related peptide receptor antagonist Substances 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 229960004117 capecitabine Drugs 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- OOCUOKHIVGWCTJ-UHFFFAOYSA-N chloromethyl(trimethyl)silane Chemical compound C[Si](C)(C)CCl OOCUOKHIVGWCTJ-UHFFFAOYSA-N 0.000 description 2
- 229940090100 cimzia Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 229960005061 crizotinib Drugs 0.000 description 2
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 229960003901 dacarbazine Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 229960003914 desipramine Drugs 0.000 description 2
- 229960003529 diazepam Drugs 0.000 description 2
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 2
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 2
- 229960004166 diltiazem Drugs 0.000 description 2
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 2
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 2
- 235000019797 dipotassium phosphate Nutrition 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 229960002866 duloxetine Drugs 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 229940073621 enbrel Drugs 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 2
- 229960005167 everolimus Drugs 0.000 description 2
- 238000013213 extrapolation Methods 0.000 description 2
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 229960002870 gabapentin Drugs 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 229960002584 gefitinib Drugs 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 229940048921 humira Drugs 0.000 description 2
- 238000005984 hydrogenation reaction Methods 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- XMBWDFGMSWQBCA-RNFDNDRNSA-M iodine-131(1-) Chemical compound [131I-] XMBWDFGMSWQBCA-RNFDNDRNSA-M 0.000 description 2
- 229960005386 ipilimumab Drugs 0.000 description 2
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- 229960004891 lapatinib Drugs 0.000 description 2
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 201000005296 lung carcinoma Diseases 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- 229960000600 milnacipran Drugs 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 2
- 235000019796 monopotassium phosphate Nutrition 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 210000000653 nervous system Anatomy 0.000 description 2
- 229960001346 nilotinib Drugs 0.000 description 2
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 229960003301 nivolumab Drugs 0.000 description 2
- 229960001158 nortriptyline Drugs 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 210000000963 osteoblast Anatomy 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 229960000639 pazopanib Drugs 0.000 description 2
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 2
- 229960002621 pembrolizumab Drugs 0.000 description 2
- 229960002087 pertuzumab Drugs 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- CPJSUEIXXCENMM-UHFFFAOYSA-N phenacetin Chemical compound CCOC1=CC=C(NC(C)=O)C=C1 CPJSUEIXXCENMM-UHFFFAOYSA-N 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 108020001213 potassium channel Proteins 0.000 description 2
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- AYXYPKUFHZROOJ-ZETCQYMHSA-N pregabalin Chemical compound CC(C)C[C@H](CN)CC(O)=O AYXYPKUFHZROOJ-ZETCQYMHSA-N 0.000 description 2
- 229960001233 pregabalin Drugs 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 239000000700 radioactive tracer Substances 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 229960004836 regorafenib Drugs 0.000 description 2
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 2
- 229940116176 remicade Drugs 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229940068638 simponi Drugs 0.000 description 2
- 229960000714 sipuleucel-t Drugs 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 229960001796 sunitinib Drugs 0.000 description 2
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 229960000235 temsirolimus Drugs 0.000 description 2
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 2
- 230000007838 tissue remodeling Effects 0.000 description 2
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 2
- 229960004066 trametinib Drugs 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- ODLHGICHYURWBS-LKONHMLTSA-N trappsol cyclo Chemical compound CC(O)COC[C@H]([C@H]([C@@H]([C@H]1O)O)O[C@H]2O[C@@H]([C@@H](O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O[C@H]3O[C@H](COCC(C)O)[C@H]([C@@H]([C@H]3O)O)O3)[C@H](O)[C@H]2O)COCC(O)C)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@@H]3O[C@@H]1COCC(C)O ODLHGICHYURWBS-LKONHMLTSA-N 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- 125000004950 trifluoroalkyl group Chemical group 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 229960001722 verapamil Drugs 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 1
- CMIBUZBMZCBCAT-HOTGVXAUSA-N (2s,3s)-2,3-bis[(4-methylbenzoyl)oxy]butanedioic acid Chemical compound C1=CC(C)=CC=C1C(=O)O[C@H](C(O)=O)[C@@H](C(O)=O)OC(=O)C1=CC=C(C)C=C1 CMIBUZBMZCBCAT-HOTGVXAUSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- ZMCGJMHVCOACEF-GXFFZTMASA-N (3s,4r)-4-(3,4-difluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-amine Chemical compound C1N(CCOC)C[C@@H](N)[C@@H]1C1=CC=C(F)C(F)=C1 ZMCGJMHVCOACEF-GXFFZTMASA-N 0.000 description 1
- RTSIUKMGSDOSTI-SNAWJCMRSA-N (e)-3-(3-fluorophenyl)prop-2-enoic acid Chemical compound OC(=O)\C=C\C1=CC=CC(F)=C1 RTSIUKMGSDOSTI-SNAWJCMRSA-N 0.000 description 1
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- JNTHQYPNOWOSDA-LOSJGSFVSA-N 1-[(3s,4r)-4-(4-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl]-3-[4-methyl-5-(1-methylpyrazol-4-yl)-2-phenylpyrazol-3-yl]urea Chemical compound N([C@@H]1CN(C[C@H]1C=1C=CC(F)=CC=1)CCOC)C(=O)NC1=C(C)C(C2=CN(C)N=C2)=NN1C1=CC=CC=C1 JNTHQYPNOWOSDA-LOSJGSFVSA-N 0.000 description 1
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- COBZMDPXIDGRHY-UHFFFAOYSA-N 2-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidine Chemical compound C1=NC(C)=NC=C1B1OC(C)(C)C(C)(C)O1 COBZMDPXIDGRHY-UHFFFAOYSA-N 0.000 description 1
- PIAOLBVUVDXHHL-UHFFFAOYSA-N 2-nitroethenylbenzene Chemical compound [O-][N+](=O)C=CC1=CC=CC=C1 PIAOLBVUVDXHHL-UHFFFAOYSA-N 0.000 description 1
- DXSUORGKJZADET-UHFFFAOYSA-N 3,3-dimethylbutan-2-amine Chemical compound CC(N)C(C)(C)C DXSUORGKJZADET-UHFFFAOYSA-N 0.000 description 1
- 108020004021 3-ketosteroid receptors Proteins 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- RLFWWDJHLFCNIJ-UHFFFAOYSA-N Aminoantipyrine Natural products CN1C(C)=C(N)C(=O)N1C1=CC=CC=C1 RLFWWDJHLFCNIJ-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- VKZVMFBJTKVCJP-JYFHCDHNSA-N Cc1c(NC(N[C@H](CN(C/C=[O]/C)C2)[C@@H]2c(cc2F)ccc2F)=O)[n](-c2ccccc2)nc1-c1cnc(C)nc1 Chemical compound Cc1c(NC(N[C@H](CN(C/C=[O]/C)C2)[C@@H]2c(cc2F)ccc2F)=O)[n](-c2ccccc2)nc1-c1cnc(C)nc1 VKZVMFBJTKVCJP-JYFHCDHNSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 206010011796 Cystitis interstitial Diseases 0.000 description 1
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 101001111439 Homo sapiens Beta-nerve growth factor Proteins 0.000 description 1
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 1
- 101001077015 Homo sapiens Rab GTPase-activating protein 1-like Proteins 0.000 description 1
- 101001077011 Homo sapiens Rab GTPase-activating protein 1-like, isoform 10 Proteins 0.000 description 1
- 208000037147 Hypercalcaemia Diseases 0.000 description 1
- 108010078049 Interferon alpha-2 Proteins 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 1
- MKXZASYAUGDDCJ-SZMVWBNQSA-N LSM-2525 Chemical compound C1CCC[C@H]2[C@@]3([H])N(C)CC[C@]21C1=CC(OC)=CC=C1C3 MKXZASYAUGDDCJ-SZMVWBNQSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 230000037364 MAPK/ERK pathway Effects 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- 108020000002 NR3 subfamily Proteins 0.000 description 1
- 229910019093 NaOCl Inorganic materials 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- 102100033857 Neurotrophin-4 Human genes 0.000 description 1
- 239000006057 Non-nutritive feed additive Substances 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 102100038277 Prostaglandin G/H synthase 1 Human genes 0.000 description 1
- 108050003243 Prostaglandin G/H synthase 1 Proteins 0.000 description 1
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 1
- 108050003267 Prostaglandin G/H synthase 2 Proteins 0.000 description 1
- 108010026552 Proteome Proteins 0.000 description 1
- 102100025165 Rab GTPase-activating protein 1-like, isoform 10 Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 1
- 206010061934 Salivary gland cancer Diseases 0.000 description 1
- 208000005250 Spontaneous Fractures Diseases 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 125000005097 aminocarbonylalkyl group Chemical group 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- VEQOALNAAJBPNY-UHFFFAOYSA-N antipyrine Chemical compound CN1C(C)=CC(=O)N1C1=CC=CC=C1 VEQOALNAAJBPNY-UHFFFAOYSA-N 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 230000005775 apoptotic pathway Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 239000012911 assay medium Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229950003054 binimetinib Drugs 0.000 description 1
- ACWZRVQXLIRSDF-UHFFFAOYSA-N binimetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F ACWZRVQXLIRSDF-UHFFFAOYSA-N 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 208000024883 bone remodeling disease Diseases 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 229960001292 cabozantinib Drugs 0.000 description 1
- ONIQOQHATWINJY-UHFFFAOYSA-N cabozantinib Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 ONIQOQHATWINJY-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 230000008473 connective tissue growth Effects 0.000 description 1
- 239000010779 crude oil Substances 0.000 description 1
- 125000004966 cyanoalkyl group Chemical group 0.000 description 1
- 239000003260 cyclooxygenase 1 inhibitor Substances 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 229960001985 dextromethorphan Drugs 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 239000002895 emetic Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- MIHRVXYXORIINI-UHFFFAOYSA-N ethyl 2-cyanopropionate Chemical compound CCOC(=O)C(C)C#N MIHRVXYXORIINI-UHFFFAOYSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000035558 fertility Effects 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000001207 fluorophenyl group Chemical group 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 102000046917 human NGF Human genes 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 description 1
- 230000000148 hypercalcaemia Effects 0.000 description 1
- 208000030915 hypercalcemia disease Diseases 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 230000004968 inflammatory condition Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229960003507 interferon alfa-2b Drugs 0.000 description 1
- 230000002262 irrigation Effects 0.000 description 1
- 238000003973 irrigation Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 210000003292 kidney cell Anatomy 0.000 description 1
- 229960001632 labetalol Drugs 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 208000004731 long QT syndrome Diseases 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 238000010907 mechanical stirring Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 125000006393 methylpyrimidinyl group Chemical group 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 230000023105 myelination Effects 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000019382 nerve compression syndrome Diseases 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000006576 neuronal survival Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 210000004248 oligodendroglia Anatomy 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 230000011164 ossification Effects 0.000 description 1
- 230000000010 osteolytic effect Effects 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- 230000003076 paracrine Effects 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229960003893 phenacetin Drugs 0.000 description 1
- 229960005222 phenazone Drugs 0.000 description 1
- HKOOXMFOFWEVGF-UHFFFAOYSA-N phenylhydrazine Chemical compound NNC1=CC=CC=C1 HKOOXMFOFWEVGF-UHFFFAOYSA-N 0.000 description 1
- 229940067157 phenylhydrazine Drugs 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- MZVRPCYUUFGMLJ-UHFFFAOYSA-N pyrrolidin-1-ylurea Chemical class NC(=O)NN1CCCC1 MZVRPCYUUFGMLJ-UHFFFAOYSA-N 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 210000003752 saphenous vein Anatomy 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 1
- 229950010746 selumetinib Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- MIXCUJKCXRNYFM-UHFFFAOYSA-M sodium;diiodomethanesulfonate;n-propyl-n-[2-(2,4,6-trichlorophenoxy)ethyl]imidazole-1-carboxamide Chemical compound [Na+].[O-]S(=O)(=O)C(I)I.C1=CN=CN1C(=O)N(CCC)CCOC1=C(Cl)C=C(Cl)C=C1Cl MIXCUJKCXRNYFM-UHFFFAOYSA-M 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000002462 tachykinin receptor antagonist Substances 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000011287 therapeutic dose Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 230000017423 tissue regeneration Effects 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- FTNWXGFYRHWUKG-UHFFFAOYSA-N triflupromazine hydrochloride Chemical compound [H+].[Cl-].C1=C(C(F)(F)F)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 FTNWXGFYRHWUKG-UHFFFAOYSA-N 0.000 description 1
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical compound C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C403/00—Derivatives of cyclohexane or of a cyclohexene or of cyclohexadiene, having a side-chain containing an acyclic unsaturated part of at least four carbon atoms, this part being directly attached to the cyclohexane or cyclohexene or cyclohexadiene rings, e.g. vitamin A, beta-carotene, beta-ionone
- C07C403/06—Derivatives of cyclohexane or of a cyclohexene or of cyclohexadiene, having a side-chain containing an acyclic unsaturated part of at least four carbon atoms, this part being directly attached to the cyclohexane or cyclohexene or cyclohexadiene rings, e.g. vitamin A, beta-carotene, beta-ionone having side-chains substituted by singly-bound oxygen atoms
- C07C403/12—Derivatives of cyclohexane or of a cyclohexene or of cyclohexadiene, having a side-chain containing an acyclic unsaturated part of at least four carbon atoms, this part being directly attached to the cyclohexane or cyclohexene or cyclohexadiene rings, e.g. vitamin A, beta-carotene, beta-ionone having side-chains substituted by singly-bound oxygen atoms by esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/50—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Dermatology (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Communicable Diseases (AREA)
- Gynecology & Obstetrics (AREA)
- Transplantation (AREA)
- Pulmonology (AREA)
- Gastroenterology & Hepatology (AREA)
Abstract
Compuesto**Fórmula** 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5- il)urea, o una sal farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea como un inhibidor de TrkA cinasa.
Antecedentes de la invención
La presente invención se refiere a nuevos compuestos, a composiciones farmacéuticas que comprenden los compuestos, a procedimientos y a productos intermedios para preparar los compuestos, y a los compuestos para la utilización en terapia. Más particularmente, se refiere a 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea, o una sal farmacéuticamente aceptable del mismo, que muestra inhibición de la TrkA cinasa, y que resulta útil en el tratamiento de dolor, cáncer, inflamación/enfermedades inflamatorias, enfermedades neurodegenerativas, determinadas enfermedades infecciosas, síndrome de Sjogren, endometriosis, neuropatía periférica diabética, prostatitis, síndrome del dolor pélvico, enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, y enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
Los regímenes de tratamiento actuales para las condiciones de dolor utilizan varias clases de compuestos. Los opioides (tales como la morfina) adolecen de varias desventajas, entre ellas, los efectos eméticos, astringentes y respiratorios negativos, así como el potencial de adicción. Los analgésicos antiinflamatorios no esteroideos (AINE, tales como el tipo COX-1 o COX-2) asimismo adolecen de desventajas, entre ellas la insuficiente eficacia en el tratamiento del dolor severo. Además, los inhibidores de COX-1 pueden causar úlceras en las mucosas. De acuerdo con lo anterior, continúa existiendo una necesidad de nuevos y más eficaces tratamientos para el alivio del dolor, especialmente el dolor crónico.
Los Trk son los receptores tirosina cinasas de alta afinidad activadas por un grupo de factores de crecimiento solubles denominados neurotrofinas (NT). La familia de receptores Trk presenta tres miembros: TrkA, TrkB y TrkC. Entre las neurotrofina se encuentran: (i) el factor de crecimiento nervioso (NFG), que activa TrkA, (ii) el factor neurotrófico derivado del cerebro (BDNF) y NT-4/5, que activan TrkB, y (iii) NT3, que activa TrkC. Las Trk se expresan ampliamente en el tejido neuronal y participan en el mantenimiento, señalización y supervivencia de las células neuronales (Patapoutian, A. et al., Current Opinion in Neurobiology 11,272-280, 2001).
Los inhibidores de la ruta de Trk/neurotrofina se ha demostrado que resultan eficaces en numerosos modelos animales preclínicos de dolor. Por ejemplo, los anticuerpos antagonistas de NGF y TrkA, tales como RN-624, se ha demostrado que resultan eficaces en modelos animales de dolor inflamatorio y neuropático (Woolf C. J. et al., Neuroscience 62, 327-331, 1994; Zahn, P. K. et al., J. Pain 5, 157-163, 2004; McMahon, S. B. et al., Nat. Med. 1, 774-780, 1995; Ma, Q. P. y Woolf, C. J., NeuroReport 8, 807-810, 1997; Shelton, D. L. et al. Pain 116, 8-16, 2005; Delafoy, L. et al., Pain 105, 489-497, 2003; Lamb, K. et al., Neurogastroenterol. Motil. 15, 355-361, 2003; Jaggar, S. I. et al., Br. J. Anaesth. 83, 442-448, 1999) y modelos animales de dolor neuropático (Ramer, M. S. y Bisby, M. A., Eur. J. Neurosci. 11, 837-846, 1999; Ro, L. S. et al., Pain 79, 265-274, 1999; Herzberg, U. et al., Neuroreport 8, 1613-1618, 1997; Theodosiou, M. et al., Pain 81, 245-255, 1999; Li, L. et al., Mol. Cell. Neurosci. 23, 232-250, 2003; Gwak, Y. S. et al., Neurosci. Lett. 336, 117-120, 2003).
Asimismo se ha demostrado que NGF secretado por células tumorales y macrófagos invasores de tumores estimulan directamente TrkA situado sobre fibras de dolor periféricas. Mediante la utilización de diversos modelos tumorales, tanto en ratones como en ratas, se ha demostrado que la neutralización de NGF con un anticuerpo monoclonal inhibe el dolor relacionado con el cáncer en un grado similar o superior a la dosis máxima tolerada de morfina. Debido a que la cinasa TrkA puede servir como mediador de las respuestas biológicas controladas por NGF, los inhibidores de TrkA y/o otras cinasas Trk podrían proporcionar un tratamiento eficaz para los estados de dolor crónico y el dolor relacionado con el cáncer.
La literatura reciente asimismo ha demostrado que la sobreexpresión, activación, amplificación y/o mutación de las cinasas Trk están asociados a muchos cánceres, incluyendo el neuroblastoma (Brodeur G. M., Nat. Rev. Cancer 3:203-216, 2003), el ovárico (Davidson, B., et al., Clin. Cancer Res. 9, 2248-2259, 2003), el cáncer colorrecta (Bardelli, A., Science 300, 949, 2003), el melanoma (Truzzi, F., et al., Dermato-Endocrinology 3(1), pp. 32-36, 2011), el cáncer de cabeza y cuellor (Yilmaz, T., et al., Cancer Biology and Therapy 10(6), pp. 644-653, 2010), el carcinoma gástrico (Du, J. et al., World Journal of Gastroenterology 9 (7), pp. 1431-1434, 2003), el carcinoma pulmonar (Ricci A., et al., American Journal of Respiratory Cell and Molecular Biology 25 (4), pp. 439-446), el cáncer de mama (Jin, W., et al., Carcinogenesis 31 (11), pp. 1939-1947, 2010), el cáncer de mama secretorio (Euthus, D.M., et al., Cancer Cell 2 (5), pp. 347-348, 2002), el glioblastoma (Wadhwa, S., et al., Journal of Biosciences 2003, 28 (2), pp. 181-188), meduloblastoma (Gruber-Olipitz, M., et al., Journal of Proteome Research 7 (5), pp. 1932-1944, 2008), cáncer de glándulas salivales (Li, Y.-G., et al., Chinese Journal of Cancer Prevention and Treatment 16 (6), pp. 428-430, 2009), carcinoma de tiroides papilar (Greco, A., et al., Molecular and Cellular Endocrinology 321 (1), pp. 44-49, 2010) y leucemia mieloide adulta (Eguchi, M., et al., Blood 93 (4), pp. 1355-1363, 1999). En modelos preclínicos de cáncer, los inhibidores no selectivos de molécula pequeña de TrkA, B y C resultaron eficaces tanto en la inhibición del crecimiento tumoral como en la detención de la metástasis tumoral
(Nakagawara, A., Cáncer Letters 169:107-114, 2001; Meyer, J., et al. Leukemia, 21(10):2171 -2180, 2007; Pierottia, M.A. y Greco A., Cancer Letters 232:90-98, 2006; Eric Adriaenssens, E., et al. Cancer Res. 68:(2) 346-351, 2008). Estos datos respaldan la utilización de inhibidores de Trk para el tratamiento del cáncer.
Además, la inhibición de la ruta de neurotrofina/Trk se ha demostrado que resulta eficaz en el tratamiento de modelos preclínicos de enfermedades inflamatorias con anticuerpos de NGF o inhibidores no selectivos de molécula pequeña de TrkA. Por ejemplo, la inhibición de la ruta de neurotrofina/Trk se ha implicado en modelos preclínicos de enfermedades pulmonares inflamatorias, incluyendo el asma (Freund-Michel, V; Frossard, N., Pharmacology & Therapeutics 117(1), 52-76, 2008), cistitis intersticial (Hu, Vivian Y; et. al. The Journal of Urology 173(3), 1016-21, 2005), síndrome doloroso vesical (Liu, H.-T., et al., BJU International, 106 (11), pp. 1681-1685, 2010), enfermedades intestinales inflamatorias, entre ellas la colitis ulcerosa y la enfermedad de Crohn (Di Mola, F. F, et. al., Gut 46(5), 670-678, 2000) y las enfermedades inflamatorias de la piel, tales como la dermatitis atópica (Dou, Y.-C., et. al., Archives of Dermatological Research 298(1), 31-37, 2006), eccema y soriasis (Raychaudhuri, S. P., et al., J. Investigative Dermatology 122(3), 812-819, 2004). Estos datos respaldan la utilización de inhibidores de Trk para el tratamiento de enfermedades inflamatorias.
Asimismo se cree que el receptor de TrkA resulta crítico para el proceso de la enfermedad de la infección parasitaria por Trypanosoma cruzi (enfermedad de Chagas) en huéspedes humanos (de Melo-Jorge, M. et al., Cel1Host & Microbe 1(4), 251-261,2007).
Los inhibidores de Trk asimismo pueden encontrar utilidad en el tratamiento de enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, tal como la osteoporosis, la artritis reumatoide y las metástasis óseas. Las metástasis óseas son una complicación frecuente del cáncer, produciéndose en hasta el 70 por ciento de los pacientes con cáncer de mama o de próstata avanzado y en aproximadamente 15 a 30 por ciento de los pacientes con carcinoma de pulmón, colon, estómago, vejiga, útero, recto, tiroides o riñón. Las metástasis osteolíticas pueden causar dolor severo, fracturas patológicas, hipercalcemia potencialmente letal, compresión de la médula espinal y otros síndromes de compresión nerviosa. Por estos motivos, la metástasis ósea es una complicación grave y costosa del cáncer. Por lo tanto, resultarían altamente ventajosos los agentes que pueden inducir apoptosis de los osteoblastos proliferantes. Se ha observado la expresión de receptores de TrkA en la zona de formación ósea en modelos de ratón de fractura ósea (K. Asaumi et al., Bone 26(6):625-633, 2000). Además, la localización de NGF se ha observado en prácticamente la totalidad de las células formadoras de hueso (K. Asaumi et al., Bone 26(6):625-633, 2000). Recientemente se ha demostrado que un inhibidor de Trk inhibe la señalización activada por la unión de las neurotrofinas a la totalidad de los tres receptores de Trk en los osteoblastos hFOB humanos (J. Pinski, et al., Cancer Research 62, 986-989, 2002). Estos datos respaldan la utilización de inhibidores de Trk para el tratamiento de enfermedades del remodelado óseo, tales como las metástasis óseas en pacientes de cáncer.
Los inhibidores de Trk asimismo podrían encontrar utilidad en el tratamiento de enfermedades y trastornos tales como el síndrome de Sjogren (Fauchais, A.L., et al., Scandinavian Journal of Rheumatology, 38(1), pp. 50-57, 2009), la endometriosis (Barcena De Arellano, M. L., et al., Reproductive Sciences, 18(12), pp. 1202-1210, 2011; Barcena De Arellano, et al., Fertility and Sterility, 95(3), pp. 1123-1126, 2011; Cattaneo, A., Current Opinion in Molecular Therapeutics, 12(1), pp. 94-106, 2010), la neuropatía periférica diabética (Kim, H.C., et al., Diabetic Medicine, 26 (12), pp. 1228-1234, 2009; Siniscalco, D., et al., Current Neuropharmacology, 9(4), pp. 523-529, 2011; Ossipov, M. H., Current Pain and Headache Reports, 15(3), pp. 185-192, 2011) y la prostatitis y síndrome doloroso pélvico (Watanabe, T., et al., BJU International, 108(2), pp. 248-251, 2011 y Miller, L. J., et al., Urology, 59(4), pp.
603-608, 2002).
Los inhibidores de TrkA asimismo pueden resultar útiles para tratar enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo (CTGF, asimismo denominado CCN2), por ejemplo enfermedades que implican el remodelado y fibrosis de tejidos. El CTGF es un mediador central del remodelado y fibrosis de los tejidos (Lipson, K. E., et al., Fibrogenesis & Tissue Repair 5(Supl. 1):S24, 2012) y las terapias que reducen la señalización de CTGF han demostrado ser eficaces como tratamientos de la fibrosis (Li, G., et al., J. Gene Med. 8:889-900, 2006). CTGF interactúa con TrkA y lo inactiva (Wahab, N. A., J. Am. Soc. Nephrol. 16:340-351,2005) y la inhibición de esta ruta por inhibidores de TrkA podría demostrar utilidad en el tratamiento de diversas enfermedades fibróticas, tales como el síndrome de Raynaud, la fibrosis pulmonar idiopática, la cicatrización (hipertrófica, queloides y otros), la cirrosis, la fibrosis endomiocárdica, la fibrosis auricular, la mielofibrosis, la fibrosis masiva progresiva (pulmón), la fibrosis sistémica nefrogénica, el escleroderma, la esclerosis sistémica, la artrofibrosis y la fibrosis ocular.
Varias clases de inhibidores de molécula pequeña de las cinasas de Trk se afirma que resultan útiles para tratar el dolor o el cáncer (Wang, T. et al., Expert Opin. Ther. Patents 19(3), 305-319, 2009; McCarthy C. y Walker E., Expert Opin. Ther. Patents 24(7):731-744, 2014).
La solicitud publicada de patente internacional n° WO 2010/032856 describe compuestos representados mediante la fórmula:

en la que el anillo B es un anillo aromático, el anillo D es un anillo aromático, y L es NR3, NR3C(R4aR4b), O o OC(R4aR4b), que se afirma que son antagonistas de receptores de taquicinina.
La solicitud publicada de patente internacional n° WO 2012/158413 da a conocer un subgrupo de compuestos de pirrolidinil urea como inhibidores de TrkA que presentan la fórmula general:

en la que:
Y es un enlace, -O- o -OCH2-,
X es O, S o NH,
R1 es (alcoxi 1-3C)alquilo (1-6C), (trifluorometoxi)alquilo(1-6C), (1-3C sulfanil)alquilo(1-6C), monofluoroalquilo(1-6C), difluoroalquilo(1-6C), trifluoroalquilo(1-6C), tetrafluoro(2-6C)alquilo, pentafluro(2-6C)alquilo, cianoalquilo(1-6C), aminocarbonilalquilo(1-6C), hidroxialquilo(1-6C), dihidroxi(2-6C)alquilo, alquilo(1-6C), (1-3Calquiloamino)alquilo(1-3C), (1-4C alcoxicarbonil)alquilo(1-6c), aminoalquilo(1-6C), hidroxi(1-3C alcoxi)alquilo(1-6c), di(1-3C alcoxi)alquilo(1-6C), (1-3c alcoxi)trifluoroalquilo(1-6c), hidroxitrifluoroalquilo(1-6C), (1-4C alcoxicarbonil)(1-3c alcoxi)alquilo(1-6C), hidroxicarbonil(1-3C alcoxi)alquilo(1-6C), hetAr5(cH2)o-1, o Ar5(CH2)o-1;
hetAr5 es un anillo heteroarilo de 5-6 elementos que presenta 1 a 3 heteroátomos anulares seleccionados independientemente de N, O o S, en el que el anillo se sustituye opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno, alquilo(1-6), alcoxi(1-6C) y CF3,
Ar5 es fenilo sustituido opcionalmente con uno o más grupos seleccionados independientemente de halógeno, alquilo(1-6C), alcoxi(1-6C), CF3O-, alcoxicarbonilo(1-4C) y aminocarbonilo,
B es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno, CF3, CF3O-, alcoxi(1-4C), hidroxi-alquilo(1-4C), alquilo(1-6C) y CN; un heteroarilo de 5-6 elementos que presenta 1-3 heteroátomos anulares seleccionados independientemente de N, S y O, y sustituidos opcionalmente con 1-2 grupos seleccionados independientemente de alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxialquilo(1-2C), alquilo 1-6C, o alcoxi(1-6C), y
el anillo C es de fórmula C-1, C-2 o C-3:

Entre los ejemplos de dichos compuestos en el documento n° WO 2012/158413 se incluyen el compuesto del Ejemplo 511 que presenta la estructura:

que asimismo es conocido como 1-((3S,4R)-4-(3,4-difluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea (en adelante, “Compuesto 2) y el compuesto del Ejemplo 441, que presenta la estructura:

que asimismo es conocido como 1-(1',4-dimetiM-feniMH,1'H-[3,4-bipirazol]-5-il)-3-((3S,4R)-4-(4-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)urea (en adelante, “Compuesto 3").
Se ha descubierto que puede obtenerse un compuesto que presenta propiedades inesperadas y particularmente deseables mediante la selección de 2-metilpirimidín-5-ilo como el grupo R4 y un 3-fluorofenilo como el grupo B.
Sumario de la invención
En la presente memoria se proporciona el compuesto 1:

o una sal farmacéuticamente aceptable del mismo, que es un inhibidor de TrkA. El compuesto asimismo puede describirse con el nombre químico 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea.
Inesperadamente se ha descubierto que el compuesto 1 presenta un lavado IV (intravenoso) predicho sustancialmente más bajo en ratas respecto a los compuestos 2 y 3. Inesperadamente se ha encontrado que el compuesto 1 consigue una AUC (superficie bajo la curva de concentración plasmática de fármaco-tiempo) oral más elevada tras una dosis oral de 10 mg/kg en ratas respecto a los compuestos 2 y 3. Inesperadamente se ha encontrado que el compuesto 1 consigue una Cmax más elevada (concentración máxima alcanzada en la curva de concentración plasmática de fármaco-tiempo) tras una dosis oral de 10 mg/kg en ratas respecto a los compuestos
2 y 3. Inesperadamente se ha encontrado que el compuesto 1 consigue una concentración valle más elevada (concentración mínima alcanzada en la curva de concentración plasmática de fármaco-tiempo) tras una dosis oral de 10 mg/kg en ratas respecto a los compuestos 2 y 3. Inesperadamente se ha encontrado que el compuesto 1 presenta una inhibición estimada más elevada de TrkA respecto a los compuestos 2 y 3. El compuesto 1 asimismo ha mejorado inesperadamente la distribución sistema nervioso periférico-a-central respecto al compuesto 2, tal como pone de manifiesto la proporción plasmática a cerebral más elevada tras la administración oral en ratas. El compuesto 1 asimismo presenta una reducción inesperada de la actividad inhibidora del canal de hERG (el gen Ether-a-go-go humano) respecto al compuesto 2.
El compuesto 1 es para la utilización en métodos de tratamiento de una enfermedad o trastorno modulado por TrkA, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
El compuesto 1 asimismo se utiliza en métodos de tratamiento de dolor crónico y agudo, incluyendo, aunque sin limitarse a ellos, dolor inflamatorio, dolor neuropático y dolor asociado a cáncer, cirugía o fractura ósea, que comprende administrar en un mamífero que requiere dicho tratamiento, una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto para la utilización en métodos de tratamiento de cáncer, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 es para la utilización en métodos de tratamiento de inflamación o enfermedades inflamatorias, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 es para la utilización en métodos de tratamiento de enfermedades neurodegenerativas, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 para la utilización en métodos de tratamiento de prostatitis o síndrome doloroso pélvico, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 para la utilización en métodos de tratamiento de enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, tal como la osteoporosis, la artritis reumatoide y las metástasis óseas, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 es para la utilización en métodos de tratamiento de enfermedades y trastornos seleccionados de determinadas enfermedades infecciosas, síndrome de Sjogren, endometriosis y neuropatía periférica diabética, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se describe además un compuesto 1 para la utilización en métodos de tratamiento de enfermedades que resulta de la señalización aberrante del factor de crecimiento del tejido conectivo, que comprende administrar en el mamífero que necesita dicho tratamiento una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En una forma de realización, cualquiera de los tratamientos anteriores comprende administrar en el mamífero que necesita dicho tratamiento, una cantidad eficaz de compuesto 1, o una sal farmacéuticamente aceptable del mismo, en combinación con un agente terapéutico adicional.
En la presente memoria se proporciona además una composición farmacéutica que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en terapia.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de dolor crónico y agudo, incluyendo, aunque sin limitación, dolor inflamatorio, dolor neuropático y dolor asociado a cáncer, cirugía o fractura ósea.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del cáncer.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de inflamación o enfermedades inflamatorias.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades neurodegenerativas.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de prostatitis o síndrome doloroso pélvico.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades relacionadas con un desequilibrio en la regulación del remodelado óseo, tal como osteoporosis, artritis reumatoide y metástasis óseas.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de determinadas enfermedades infecciosas, síndrome de Sjogren, endometriosis y neuropatía periférica diabética.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
En la presente memoria se describe además compuesto 1, o una sal farmacéuticamente aceptable del mismo, en la preparación de un medicamento para el tratamiento de enfermedades y trastornos tales como el dolor crónico y agudo, incluyendo, aunque sin limitación, dolor inflamatorio y dolor asociado a cáncer, cirugía o fractura ósea. En la presente memoria se describe además la utilización de compuesto 1, o una sal farmacéuticamente aceptable del mismo, en la preparación de un medicamento para el tratamiento de enfermedades y trastornos seleccionados de cáncer, inflamación o enfermedades inflamatorias, enfermedades neurodegenerativas, determinadas enfermedades infecciosas, síndrome de Sjogren, endometriosis, neuropatía periférica diabética, prostatitis, síndrome doloroso pélvico y enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, tal como osteoporosis, artritis reumatoide y metástasis óseas.
En la presente memoria se describe además la utilización de compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la preparación de un medicamento para el tratamiento de enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
En la presente memoria se describen además productos intermedios para preparar compuestos inhibidores de TrkA, tales como compuesto 1, y métodos de preparación de tales productos intermedios.
En la presente memoria se proporcionan además métodos de preparación, métodos de separación y métodos de purificación de compuesto 1, o una sal farmacéuticamente aceptable del mismo.
Breve descripción de las figuras
La figura 1 es un gráfico que compara la concentración plasmática de fármaco (pg/ml) frente al tiempo (horas) para el compuesto 1 (triángulos) y el compuesto 2 (cuadrados) tras una dosis oral de 10 mg/kg en ratas. La figura 2 es un gráfico que compara la concentración plasmática de fármaco (pg/ml) frente al tiempo (horas) para el compuesto 1 (triángulos) y el compuesto 3 (círculos) tras una dosis oral de 10 mg/kg en ratas.
La figura 3 es un gráfico que compara el porcentaje estimado de inhibición de TrkA frente al tiempo (horas) para el compuesto 1 (triángulos) y el compuesto 2 (cuadrados) tras una dosis oral de 10 mg/kg en ratas. La figura 4 es un gráfico que compara el porcentaje estimado de inhibición de TrkA frente al tiempo (horas) para el compuesto 1 (triángulos) y el compuesto 3 (círculos) tras una dosis oral de 10 mg/kg en ratas.
Descripción detallada de la invención
En la presente memoria se proporciona el compuesto 1:

o una sal farmacéuticamente aceptable del mismo, que es un inhibidor de TrkA. El compuesto asimismo puede describirse con el nombre químico 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea.
Se ha descubierto que el compuesto 1 presenta propiedades inesperadas y deseables. Una propiedad particularmente ventajosa del compuesto 1 es el lavado predicho de IV humano, que es sustancialmente inferior respecto al lavado predicho de IV humano para tanto el compuesto 2 como el compuesto 3, tal como se muestra en la tabla 1.
Tabla 1

En la tabla 1, los valores predichos de lavado de IV humano se calcularon a partir de los datos de concentración de compuesto-versus-tiempo en plasma recogido de especies preclínicas (ratón, rata, perro y mono) tras dosis intravenosas (IV) de 1 mg/kg. Se midieron las concentraciones de compuesto en el plasma mediante CL-EM (cromaografía líquida-espectrometría de masas). Se determinaron los valores de AUCinf (superficie bajo la curva de concentración plasmática-tiempo para un intervalo de administración entre tiempo 0 y la extrapolación a infinito) para curvas Cp/t individuales (concentración del fármaco en e plasma en cualquier tiempo t) utilizando la integración lineal-trapezoidal y la extrapolación de primer orden a partir de los puntos terminales de las curvas Cp/t y se calculó el lavado (CL) utilizando Dosis/AUCinf.
Se calculó el valor de lavado hepático predicho (CLh) escalando la semivida in vitro (ti/2) para la estabilidad del compuesto en microsomas hepáticos (1 mg/ml; 20 minutos) utilizando los factores de escalado físico y fisiológico utilizados para predecir el lavado intrínseco (CLint) y el lavado hepático (CLhep) presentados en la tabla 2 y utilizados en las ecuaciones siguientes:

En las que D es la cantidad de proteína relacionada con citocromo P450 por unidad de masa de hígado; W es la masa media de hígado presente por unidad de peso de animal; C es la concentración microsómica del hígado y Q es el flujo sanguíneo hepático según especie.
Tabla 2

A continuación, se determinó el factor de corrección in vivo/n vitro mediante la división del valor de CLh microsómico por el valor de CL de IV. Se utilizó la media aritmética de los factores de corrección preclinicos para cada compuesto para predecir el lavado de IV humano a partir de CLh microsómico humano medido (CLh/factor de corrección).
Adicionalmente, el compuesto 1 alcanza inesperadamente un AUC oral más elevado (superficie bajo la curva de concentración plasmática de fármaco-tiempo), una Cmax más elevada (concentración máxima alcanzada en la curva de concentración plasmática de fármaco-tiempo) y una concentración valle más elevada (concentración mínima alcanzada en la curva de concentración plasmática de fármaco-tiempo) tras una dosis oral (10 mg/kg), tal como se ejemplifica mediante sus curvas farmacocinéticas en la rata respecto a los compuestos 2 y 3, tal como se muestra en las figuras 1 y 2 y se resume en la tabla 3.
Tabla 3

Las figuras 3 y 4 ilustran la inhibición estimada de TrkA para el compuesto 1 tras una dosis oral de 10 mg/kg en ratas respecto al compuesto 2 y para el compuesto 1 respecto al compuesto 3, respectivamente. Las curvas en las figuras 3 y 4 se calcularon utilizando la ecuación siguiente:

En la que Imin y Imax son la inhibición mínima y máxima posible de la diana, respectivamente; IC50 es la concentración a la que la diana resulta inhibida al 50%; C es la concentración del inhibidor y n es la pendiente de Hill. Tal como se muestra en las figuras 3 y 4, la inhibición estimada de TrkA es más elevada para el compuesto 1 que para el compuesto 2 y el compuesto 3.
A menos lavado, mayor AUC, mayor Cmax y mayor concentración valle de compuesto 1, mayor inhibición del receptor de TrkA durante el día tras la administración de compuesto 1 en un paciente en comparación con los compuestos 2 y 3. Lo anterior se traduce en una mayor eficacia terapéutica del compuesto 1 respecto a los compuestos 2 y 3. La capacidad de conseguir una eficacia terapéutica mejorada resulta importante en el tratamiento de pacientes que sufren, por ejemplo, de dolor moderado a severo, determinadas condiciones inflamatorias o cáncer (incluyendo cánceres controlados por una señalización aberrante de TrkA).
Además, el menor lavado de compuesto 1 resulta en una semivida más prolongada del compuesto tras la administración en comparación con los compuestos 2 y 3, resultando en una inhibición más sostenida del receptor de TrkA tras la administración oral, intravenosa o subcutánea del compuesto. Una semivida incrementada presenta beneficios en términos de proporcionar una eficacia más sostenida y/o una frecuencia de administración reducida del fármaco. Adicionalmente, el menor lavado en el ser humano y a Uc oral más elevada del compuesto 1 resultan ventajosos en el aspecto de que una dosis más baja de compuesto 1 resulta necesaria para inhibir eficazmente el receptor de TrkA y proporcionar una eficacia terapéutica dada en comparación con los compuestos 2 y 3. La reducción de la dosis de un compuesto requerida para eficacia se traduce en una mejora de la tolerabilidad del fármaco, el coste del tratamiento y el cumplimiento del paciente con los regímenes de administración.
El compuesto 1 asimismo presenta una distribución sistema nervioso periférico-a-central mejorada en comparación con el compuesto 2, tal como pone de manifiesto la proporción plasmática a cerebral más elevada en ratas, tal como se muestra en la Tabla 4. El mantenimiento de una baja exposición cerebral presenta el beneficio de reducir el riesgo de efectos secundarios relacionados con el sistema nervioso central, tales como efectos secundarios relacionados con la cognición o el deterioro motor. La exposición reducida al sistema nervioso central resulta beneficiosa en el aspecto de que se producen menos efectos secundarios o efectos secundarios reducidos en el paciente para un efecto terapéutico dado y/o se consigue un efecto terapéutico mayor antes de que se observe ningún efecto secundario limitante de dosis.
El compuesto 1 asimismo presenta una actividad inhibidora inesperadamente reducida de hERG (el gen Ether-ágo-go humano) respecto al compuesto 2, tal como se muestra en la Tabla 4. La inhibición de la función del canal hERG puede resultar en un efecto secundario de síndrome de QT largo inducido farmacológicamente (adquirido) grave debido a la naturaleza grave de este efecto secundario, resulta altamente deseable una actividad inhibidora reducida de hERG.
Tabla 4

Las propiedades anteriormente indicadas del compuesto 1 respecto al compuesto 2 no podrían preverse razonablemente a partir de la diferencia estructural menor entre estos compuestos. La única diferencia estructural entre el compuesto 1 y el compuesto 2 es la eliminación de un único átomo de flúor. Sin embargo, se ha demostrado en la técnica que la eliminación de un único átomo de flúor de un compuesto generalmente no proporciona un compuesto que presente el cambio mejorado de lavado predicho en el ser humano que se ha observado para el compuesto 1 respecto al compuesto 2. De hecho, existen varios ejemplos en la técnica con otros motivos de fármaco que demuestran que los análogos con más átomos de flúor tienden a presentar un lavado in vivo más bajo que sus análogos que presentan menos átomos de flúor (ver, por ejemplo, Barker A.J. Et al., Bioorg. Med. Chem. Lett. 11:1911-1914, 2001; Rosenblum, S. B. et al., J. Med. Chem., 41:973-980, 1998; Brown, M. F. et al., Bioorg. Med. Chem. Lett. 14:2175-2179, 2004). En contraste, el compuesto monoflúor 1 presenta un lavado humano predicho más bajo que el compuesto diflúor 2. De manera similar, las ventajas farmacocinéticas (FC) de una Cmax más elevada, una AUC más elevada, exposición valle más elevada e inactivación de diana TrkA más elevada con el compuesto 1 que el compuesto 2 según se demuestra en los datos FC preclínicos en la rat son bastante notables e inesperados para un cambio estructural tan pequeño.
Adicionalmente, una comparación de los parámetros FC de compuesto 1 y compuesto 3 demuestra que la farmacocinética mejorada del compuesto 1 no es un fenómeno general de todos los análogos monoflúor. De hecho, el compuesto 3 muestra un lavado humano proyectado peor y valores FC inferiores en términos de Cmax, AUC, exposición valle e inactivación de diana TrkA que el compuesto 1, a pesar de presentar un grupo fenilo sustituido con monoflúor. La combinación única de fracciones metilpirimidinilo y fenilo sustituido con monoflúor en el compuesto 1 proporciona inesperadamente las propiedades superiores indicadas de manera general anteriormente.
En la presente memoria se proporcionan además sales farmacéuticamente aceptables de compuesto 1. Son sales particulares las sales hidrocloruro. En una forma de realización, en la presente memoria se proporciona una sal monohidrocloruro del compuesto 1. En una forma de realización, en la presente memoria se proporciona una sal dihidrocloruro del compuesto 1.
En una forma de realización, en la presente memoria se proporciona la forma base libre del compuesto 1.
En una forma de realización, el compuesto 1 o sus sales pueden aislarse en forma de solvatos y, por consiguiente, cualquiera de dichos solvatos se encuentra comprendido dentro del alcance de la presente invención. Por ejemplo, el compuesto 1 o sus sales pueden existir en forma no solvatada, además de formas solvatadas con solventes farmacéuticamente aceptables, tales como agua, etanol y similares.
La expresión "farmacéuticamente aceptable" indica que la sustancia o la composición debe ser compatible química y/o toxicológicamente, con los otros ingredientes que comprende una formulación y/o con los que se trata el mamífero.
En la presente memoria se proporciona además un procedimiento para preparar compuesto 1 o una sal farmacéuticamente aceptable del mismo, que comprende:
(a) hacer reaccionar un compuesto de fórmula II-A

con un compuesto que presenta la fórmula III

en presencia de carbonildiimidazol o trifosgeno y una base, o
(b) hacer reaccionar un compuesto de fórmula II-A

con un compuesto que presenta la fórmula IV

en la que L1 es un grupo saliente, en presencia de una base, o
(c) hacer reaccionar un compuesto que presenta la fórmula V

en la que L2 es un grupo saliente, con un compuesto que presenta la fórmula III

en presencia de una base, o
(d) hacer reaccionar un compuesto que presenta la fórmula VI

con azida de difenilfosforilo para formar un producto intermedio, seguido de la reacción del producto intermedio con un compuesto que presenta la fórmula III

en presencia de una base, o
(e) hacer reaccionar un compuesto de fórmula II-A

con un compuesto que presenta la fórmula VII

en presencia de una base, y eliminar grupos protectores en su caso, y opcionalmente preparar una sal farmacéuticamente aceptable del mismo.
En referencia al método (a), la base puede ser una base amina, tal como trietilamina o diisopropilamina. Entre los solventes adecuados se incluyen diclorometano, dicloroetano, THF, DMA y DMF. La reacción se lleva a cabo convenientemente a temperatura ambiente.
En referencia al método (b), el grupo saliente puede ser, por ejemplo, fenoxi o 4-nitrofenoxi. La base puede ser una base amina, tal como trietilamina o diisopropilamina. Entre los solventes adecuados se incluyen DMA, DMF y DCE. La reacción se lleva a cabo convenientemente a temperatura ambiente.
En referencia al método (c), el grupo saliente puede ser, por ejemplo, fenoxi o 4-nitrofenoxi. La base puede ser una base amina, tal como trietilamina o diisopropilamina. Entre los solventes adecuados se incluyen DCE, DMA y DMF. La reacción se lleva a cabo convenientemente a temperatura ambiente.
En referencia al método (d), la base puede ser una base amina, tal como trietilamina o diisopropilamina. Entre los solventes adecuados se incluyen tolueno y DMF. La reacción se lleva a cabo convenientemente a temperaturas elevadas, por ejemplo, la temperatura de reflujo del solvente.
En referencia al método (e), la base puede ser una base amina, tal como trietilamina o diisopropilamina. Entre los solventes adecuados se incluyen DCM, DCE, DMF y THF. La reacción se lleva a cabo convenientemente a temperaturas de entre aproximadamente 0°C y la temperatura ambiente.
En la presente memoria se describe además un procedimiento para preparar una mezcla racémica de un compuesto de fórmula II,

en la que el anillo B y la fracción NH2 se encuentran en la configuración trans. Los compuestos de fórmula II (tales como los compuestos de fórmula II-A) resultan útiles para preparar compuestos tales como los compuestos inhibidores de TrkA, tales como el compuesto 1.
De acuerdo con lo anterior, en la presente memoria se describe un procedimiento para preparar una mezcla racémica de un compuesto de fórmula II,

en la que:
el anillo B es Ar1 o hetAr1,
Ar1 es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno, CF3, CF3O-, alcoxi(1-4C), hidroxi-alquilo(1-4C), alquilo(1-6C) y CN, y
hetAr1 es un heteroarilo de 5-6 elementos que presenta 1-3 heteroátomos anulares seleccionados independientemente deN, S y O y sustituidos opcionalmente con 1-2 grupos seleccionados independientemente de entre alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxi-alquilo(1-2C), comprendiendo dicho procedimiento:
(a) hacer reaccionar un compuesto de fórmula (a):

en la que el anillo B se define mediante la fórmula II, en donde 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina presenta la fórmula:

en presencia de una cantidad catalítica de ácido, proporcionando un compuesto de fórmal (b)

en la que el anillo B es tal como se define para la fórmula II,
(b) hacer reaccionar dicho compuesto de fórmula (b) con carbonil diimidazol en presencia de una cantidad catalítica de hidrocloruro de imidazol, seguido del tratamiento con amonio, proporcionando un compuesto que presenta la fórmula (c),
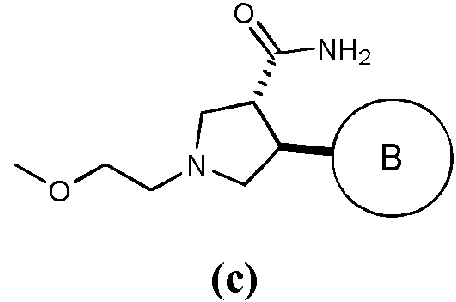
en la que el anillo B es tal como se define para la fórmula II, y
(c) hacer reaccionar dicho compuesto de fórmula (c) con hipoclorito sódico, seguido del tratamiento con KOH, seguido del tratamiento con HCl, proporcionando dicho compuesto de fórmula II en forma de mezcla racémica de fórmula II trans.
En referencia a la etapa (a), el ácido puede ser un ácido orgánico, tal como ácido trifluoroacético.
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno, CF3, CF3O-, alcoxi (1-4C), hidroxi-alquilo(1-4C), alquilo(1-6C) y CN.
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es fenilo sustituido opcionalmente con uno o más átomos de flúor.
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es 3-fluorofenilo.
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es 3,4-difluorofenilo.
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de n compuesto de fórmula II, el anillo B es un heteroarilo de 5-6 elementos que presenta 1-3 heteroátomos anulares seleccionados independientemente de N, S y O y sustituidos opcionalmente con 1-2 grupos seleccionados independientemente de alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxi-alquilo(1-2C).
En una forma de realización del procedimiento anterior, el anillo B es un anillo piridilo, tiofenilo, tiazolilo, oxazolilo o isoxazolilo sustituido opcionalmente con 1-2 grupos seleccionados independientemente de alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxi-alquilo(1-2C).
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, el anillo B es un anillo piridilo, tiofenilo, tiazolilo, oxazolilo o isoxazolilo sustituido opcionalmente con 1-2 grupos seleccionados independientemente de halógeno y alquilo(1-6C).
En una forma de realización del procedimiento anterior de preparación de una mezcla racémica de un compuesto de fórmula II, puede prepararse 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina que presenta la fórmula:

mediante un procedimiento que comprende:
(i) hacer reaccionar (dorometil)trimetilsilano con 2-metoxietanamina, proporcionando (2-metoxi-N-((trimetilsilil)metil)etanamina que presenta la estructura:

y
(ii) tratar dicha (2-metoxi-N-((trimetilsilil)metil)etanamina con formaldehído en metanol, proporcionando dicha 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina.
en la presente memoria se describe además un procedimiento para aislar el enantiómero 1 de fórmula II trans, como la base libre o como la sal de ácido di-p-toluoil-D-tartárico, que comprende:
tratar la fórmula II trans racémica con ácido di-p-toluoil-D-tartárico, proporcionando la sal de ácido di-p-toluoil-D-tartárico de II trans racémico:
recristalizar la sal de ácido di-p-toluoil-D-tartárico de II trans, proporcionando la sal de ácido di-p-toluoil-D-tartárico del enantiómero 1 de II trans, y
opcionalmente tratar la sal de ácido di-p-toluoil-D-tartárico del enantiómero 1 de II trans con una base inorgánica, proporcionando la base libre del enantiómero 1 de fórmula II trans que presenta la configuración absoluta que se ilustra:

En una forma de realización, la sal de ácido di-p-toluoil-D-tartárico del enantiómero 1 de II trans se utiliza para preparar compuestos tales como compuestos inhibidores de TrkA, tales como el compuesto 1. Por ejemplo, en una forma de realización, la sal de ácido di-p-toluoil-D-tartárico del enantiómero 1 de fórmula II trans se utiliza para preparar compuestos tales como compuestos inhibidores de TrkA, tales como el compuesto 1 bajo condiciones de Shotten-Bauman, en el que la base libre de la sal de ácido di-p-toluoil-D-tartárico del enantiómero 1 de fórmula II trans se genera bajo un sistema de solventes de dos fases, que consiste en agua y un solvente orgánico (tal como diclorometano), en presencia de una base, tal como hidróxido sódico, y se hace reaccionar con un compuesto que presenta la fórmula IV.
En una forma de realización, la base libre del enantiómero 1 de fórmula II trans se utiliza para preparar compuestos tales como compuestos inhibidores de TrkA, tales como el compuesto 1.
En una forma de realización del procedimiento anterior para aislar enantiómero 1 de fórmula II trans, el anillo B es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno, CF3, CF3O-, alcoxi (1-4C), hidroxi-alquilo(1-4C), alquilo(1-6C) y CN.
En una forma de realización del procedimiento anterior para aislar enantiómero 1 de fórmula II trans, el anillo B es fenilo sustituido opcionalmente con uno o más sustituyentes seleccionados independientemente de halógeno. En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es fenilo sustituido opcionalmente con uno o más átomos de flúor.
En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es 3-fluorofenilo.
En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es 3,4-difluorofenilo.
En una forma de realización, el procedimiento anterior para aislar el enantiómero 1 de fórmula II trans proporciona un método para preparar (3S,4R)-4-(3,4-difluorofenil)-1-(2-metoxietil)pirrolidín-3-amina.
En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es un heteroarilo de 5-6 elementos que presenta 1-3 heteroátomos anulares seleccionados independientemente de N, S y O y sustituidos opcionalmente con 1-2 grupos seleccionados independientemente de alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxi-alquilo(1-2C).
En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es un anillo piridilo, tiofenilo, tiazolilo, oxazolilo o isoxazolilo sustituido opcionalmente con 1-2 grupos seleccionados independientemente de alquilo(1-6C), halógeno, OH, CF3, NH2 e hidroxi-alquilo(1-2C).
En una forma de realización del procedimiento anterior para aislar el enantiómero 1 de fórmula II trans, el anillo B es un anillo piridilo, tiofenilo, tiazolilo, oxazolilo o isoxazolilo sustituido opcionalmente con 1-2 grupos seleccionados independientemente de halógeno y alquilo(1-6C).
El procedimiento anterior para preparar una mezcla racémica de un compuesto de fórmula II ofrece varias ventajas respecto al procedimiento descrito en la solicitud publicada de patente internacional n° WO 2012/158413. Por ejemplo, el procedimiento descrito en el documento n° WO 2012/158413 incluye una reacción de nitro-aldol entre un reactivo benzaldehído y nitrometano. El producto intermedio 2-nitrovinilbenceno resultante se reduce, a continuación, mediante hidrogenación a alta presión. Ambas etapas de reacción no permiten la síntesis a gran escala, ya que las reacciones de nitro-aldol y los productos intermedios nitro derivados de las mismas son inestables y pueden resultar peligrosos. En contraste, el procedimiento anterior para preparar una mezcla racémica de compuesto de fórmula II no utiliza ninguna reacción altamente peligrosa y evita los productos intermedios que contienen nitro y las hidrogenaciones. Además, todas las condiciones utilizadas en el procedimiento anterior para preparar una mezcla racémica de un compuesto de fórmula II se encuentran dentro de los parámetros operativos estándares típicamente presentes en las plantas piloto para las síntesis a gran escala, por ejemplo, el procedimiento no utiliza temperaturas extremas y no requiere recipientes a alta presión.
La capacidad del compuesto 1 o una sal farmacéuticamente aceptable del mismo de actuar como un inhibidor de TrkA puede demostrarse mediante los ensayos indicados en los Ejemplos A y B.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, resulta útil en el tratamiento de enfermedades y trastornos, entre ellos dolor, cáncer, inflamación/enfermedades inflamatorias, enfermedades neurodegenerativas, determinadas enfermedades infecciosas, síndrome de Sjogren, endometriosis, neuropatía periférica diabética, prostatitis, síndrome doloroso pélvico y enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, y enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
Tal como se utiliza en la presente memoria, los términos “tratar” o “tratamiento” se refieren a medidas terapéuticas o paliativas. Entre los resultados clínicos beneficiosos o deseados se incluyen, aunque sin limitarse a ellos, el alivio total o parcial de los síntomas asociados a un trastorno o condición, la reducción de la extensión de la enfermedad, un estado estabilizado (es decir, que no empeora) de enfermedad, el retraso o enlentecimiento de la progresión de la enfermedad, la mejora o mitigación del estado de la enfermedad y la remisión (parcial o total), sea detectable o indetectable. El "tratamiento" asimismo puede referirse a prolongar la supervivencia en comparación con la supervivencia esperada en caso de no recibir tratamiento.
En determinadas formas de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, resulta útil para evitar enfermedades y trastornos tal como se definen en la presente memoria. El término “evitar” tal como se utiliza en la presente memoria se refiere a la evitación de la aparición, recurrencia o extensión, total o parcialmente, de la enfermedad o condición indicada en la presente memoria, e incluye la administración de compuesto 1 o una sal farmacéuticamente aceptable del mismo, antes de la aparición de los síntomas.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, puede utilizarse en combinación con uno o más agentes terapéuticos adicionales que funcionan mediante un mecanismo de acción igual o diferente.
La expresión “combinación farmacéutica” tal como se utiliza en la presente memoria se refiere a una terapia farmacéutica que resulta de la mezcla o combinación de más de un principio activo e incluye combinaciones tanto fijas como no fijas de los principios activos. La expresión “combinación fija” se refiere a que el compuesto 1 o una sal farmacéuticamente aceptable del mismo, y por lo menos un agente terapéutico adicional, se administran en el paciente simultáneamente en forma de una única entidad o dosis. La expresión “combinación no fija” se refiere a que el compuesto 1 o una sal farmacéuticamente aceptable del mismo, y por lo menos un agente terapéutico adicional, se administran en un paciente como entidades separadas simultánea, concurrente o secuencialmente
con límites intermedios variables, en donde dicha administración proporciona niveles eficaces de los dos o más compuestos en el cuerpo del paciente. Lo anterior se aplica además a las terapias de cóctel, por ejemplo la administración de tres o más principios activos.
Tal como se utiliza en la presente memoria, el término “coadministrar” pretende comprender la administración de los agentes terapéuticos seleccionados en un único paciente, y pretende incluir los regímenes de tratamiento en los que los agentes se administran mediante la misma o diferentes vías de administración o en el mismo o diferentes tiempos. El término comprende la administración de dos o más agentes en un mamífero de manera que ambos agentes y/o sus metabolitos se encuentran presentes en el mamífero simultáneamente. Incluye la administración simultánea en composiciones separadas; la administración en tiempos diferentes en composiciones separadas, y/o la administración en una composición en la que se encuentran presentes ambos agentes. En una forma de realización, el compuesto o compuestos de la invención y el otro u otros agentes terapéuticos se administran en una única composición. En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, y el otro agente o agentes se mezclan en la composición.
En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, resulta útil para tratar el dolor, incluyendo el dolor crónico y el dolor agudo. Por ejemplo, el compuesto 1, o una sal farmacéuticamente aceptable del mismo, resulta útil en n el tratamiento de múltiples tipos de dolor, incluyendo el dolor inflamatorio, el dolor neuropático, el dolor de espalda y el dolor asociado a cáncer, cirugía o fractura ósea.
En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, resulta útil para tratar el dolor agudo. El dolor agudo, tal como define la International Association for the Study of Pain, resulta de enfermedad, inflamación o lesión de los tejidos. Este tipo de dolor generalmente aparece abruptamente, por ejemplo tras un traumatismo o cirugía, y puede verse acompañado de ansiedad o estrés, y se limita a un periodo de tiempo y a una severidad dados En algunos casos, puede volverse crónico.
En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, resulta útil para tratar el dolor crónico. El dolor crónico, tal como define la International Association for the Study of Pain, se cree en general que representa una enfermedad en sí misma. Puede verse muy agravado por factores medioambientales y psicológicos. El dolor crónico persiste durante un periodo más prolongado que el dolor agudo y es resistente a la mayoría de tratamientos médicos, generalmente durante 3 meses o más. Puede causar problemas graves para los pacientes, y ello ocurre con frecuencia.
De acuerdo con lo anterior, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, está destinado a la utilización en un método de tratamiento del dolor en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho dolor. En una forma de realización, el dolor es dolor agudo. En una forma de realización, el dolor es dolor crónico. En una forma de realización, el dolor es dolor de espalda crónico. En una forma de realización, el dolor es dolor neuropático, tal como dolor asociado a neuropatía periférica diabética o neuropatía periférica no diabética (por ejemplo, inducida por quimioterapia). En una forma de realización, el dolor es dolor inflamatorio, tal como dolor asociado a osteoartritis. En una forma de realización, el dolor es dolor asociado a cáncer. En una forma de realización, el dolor es dolor asociado a cirugía. En una forma de realización, el dolor es dolor asociado a fractura ósea.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en la prevención del dolor en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para evitar dicho dolor. En una forma de realización, el dolor es dolor agudo. En una forma de realización, el dolor es dolor crónico. En una forma de realización, el dolor es dolor de espalda crónico. En una forma de realización, el dolor es dolor neuropático, tal como dolor asociado a neuropatía periférica diabética o neuropatía periférica no diabética (por ejemplo, inducida por quimioterapia). En una forma de realización, el dolor es dolor inflamatorio, tal como dolor asociado a osteoartritis. En una forma de realización, el dolor es dolor asociado a cáncer. En una forma de realización, el dolor es dolor asociado a cirugía. En una forma de realización, el dolor es dolor asociado a fractura ósea.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de dolor en un mamífero, que comprende coadministrar en un mamífero que lo necesita, una cantidad eficaz de: (a) compuesto 1 o una sal farmacéuticamente aceptable del mismo, y (b) por lo menos un agente terapéutico adicional seleccionado de compuestos antiinflamatorios, esteroides (por ejemplo, dexametasona, cortisona y fluticasona), analgésicos, tales como AINE (por ejemplo, aspirina, ibuprofeno, indometacina y ketoprofeno), opioides (tales como morfina), antagonistas de receptores del péptido relacionado con el gen de la calcitonina, moduladores de canales iónicos selectivos de subtipo, anticonvulsivos (por ejemplo, pregabalina y gabapentina), inhibidores duales de la recaptación de serotonina-norepinefrina (por ejemplo, duloxetina, venlaxina y milnaciprán), inhibidores de cinasa de la familia de JAK (por ejemplo, ruxolitinib o tofacitinib) y antidepresivos tricíclicos (tales como amitriptilina, nortriptilina y desipramina). Estos agentes terapéuticos adicionales pueden administrarse con el compuesto 1 o una sal farmacéuticamente aceptable del mismo como
parte de las mismas o diferentes formas de administración, mediante la misma o diferentes vías de administración, y en el mismo o diferentes programas de administración según la práctica farmacéutica estándar conocida por el experto en la materia.
De acuerdo con lo anterior, en la presente memoria se proporciona además una combinación farmacéutica que comprende una cantidad eficaz de: (a) compuesto 1 o una sal farmacéuticamente aceptable del mismo y (b) por lo menos un agente terapéutico adicional seleccionado de compuestos antiinflamatorios, esteroides (por ejemplo, dexametasona, cortisona y fluticasona), analgésicos tales como AINE (por ejemplo, aspirina, ibuprofeno, indometacina y ketoprofeno) y opioides (tales como morfina), para la utilización en el tratamiento del dolor en un mamífero, en el que (a) y (b) pueden encontrarse en formas de administración separadas o en la misma forma de administración. En una forma de realización, en la presente memoria se proporciona una combinación farmacéutica que comprende: (a) una cantidad eficaz de compuesto 1 o sal farmacéuticamente aceptable del mismo, y (b) una cantidad eficaz de un analgésico, tal como un AINE (por ejemplo, aspirina, ibuprofeno, indometacina y ketoprofeno).
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, asimismo resulta útil para tratar el cáncer. Entre los ejemplos particulares se incluyen neuroblastoma, cáncer ovárico, pancreático, colorrectal y prostático.
De acuerdo con lo anterior, en la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del cáncer en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho cáncer.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, asimismo resulta útil para tratar un cáncer que presenta una desregulación de TrkA.
De acuerdo con lo anterior, en la presente memoria se describe además un compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de un paciente en el que se ha diagnosticado cáncer que presenta una desregulación de TrkA, que comprende administrar en el paciente una cantidad terapéuticamente eficaz de compuesto 1 o de una sal farmacéuticamente aceptable del mismo.
En una forma de realización, la desregulación de TrkA comprende la sobreexpresión de TrkA de tipo salvaje (activación autocrina/paracrina).
En una forma de realización, la desregulación de TrkA comprende la amplificación génica o una o más traslocaciones o inversiones cromosómicas que resultan en fusiones génicas de TrkA. En una forma de realización, la desregulación es un resultado de traslocaciones genéticas en las que la proteína expresada es una proteína de fusión que contiene residuos de proteínas no TrkA y TrkA y como mínimo el dominio de cinasa de TrkA. En una forma de realización, la proteína de fusión de TrkA es LMNA-TrkA, TFG-TrkA, TPM3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, o TPR-TrkA. En una forma de realización, la proteína de fusión de TrkA es LMNA-TrkA, TFG-TrkA, TPM3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, TP53-TrkA, RNF213-TrkA, RABGAP1L-TrkA, IRF2BP2-TrkA, SQSTM1-TrkA, SSBP2-TrkA, o TPR-TrkA, donde:

En una forma de realización, la desregulación de TrkA comprende una o más eliminaciones, inserciones o mutaciones en la proteína TrkA. En una forma de realización, la desregulación comprende una eliminación de uno o más residuos de la proteína TrkA, dando como resultado actividad constitutiva de la cinasa de TrkA. En una forma de realización, la eliminación incluye la eliminación de los residuos 303 a 377 en la isoforma 2 de TrkA.
En una forma de realización, la desregulación de TrkA comprende una variación de corte y empalme en la que la proteína expresada es una variante de corte y empalme alternativa de TrkA que presenta uno o más residuos
eliminados, dando como resultado actividad constitutiva de la cinasa de TrkA. En una forma de realización, una forma de corte y empalme alternativa de TrkA con actividad constitutiva presenta eliminaciones de los exones 8, 9 y 11, dando como resultado una proteína expresada en la que faltan los residuos 192 a 284 y 393 a 398 respecto a la isoforma 2 de TrkA.
Entre los cánceres en los que se ha identificado la desregulación de TrkA (ver las referencias de la literatura, a continuación; ver asimismo www.cancer.gov and www.nccn.org) se incluyen:
(A) Cánceres en los que la desregulación de TrkA comprende la amplificación génica o una o más traslocaciones o inversiones cromosómicas que resultan en fusiones génicas de TrkA, incluyendo:

(B) Cánceres en los que la desregulación de TrkA comprende una o más eliminaciones, inserciones o mutaciones en la proteína TrkA, incluyendo:

(C) Cánceres regulados por la sobreexpresión de TrkA de tipo salvaje (activación autocrina), incluyendo:

En una forma de realización, en la presente memoria se describe un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de un paciente en el que se ha diagnosticado cáncer que presenta una desregulación de TrkA, que comprende administrar en el paciente, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho cáncer, en el que el cáncer se selecciona de cáncer pulmonar de células no pequeñas, carcinoma papilar tiroideo, glioblastoma multiforme, leucemia mieloide agudo, carcinoma colorrectal, carcinoma neuroendocrino de células grandes, cáncer de próstata, neuroblastoma, carcinoma pancreático, melanoma, carcinoma de células escamosas de cabeza y cuello y carcinoma gástrico. En una forma de realización, en la presente memoria se describe un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de un paciente en el que se ha diagnosticado un cáncer que presenta una desregulación de TrkA, que comprende administrar en el paciente compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho cáncer, en el que el cáncer se selecciona de cáncer de pulmón de células no pequeñas, carcinoma tiroideo papilar, glioblastoma multiforme, leucemia mieloide aguda, carcinoma colorrectal, carcinoma neuroendocrino de células grandes, cáncer de próstata, neuroblastoma, carcinoma pancreático, melanoma, carcinoma de células escamosas de cabeza y cuello, carcinoma gástrico, colangiocarcinoma y sarcoma.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de cáncer en un mamífero, comprendiendo el método: (a) determinar si el cáncer está asociado a una desregulación de la cinasa de TrkA, y (b) si se determina que el cáncer está asociado a una desregulación de la cinasa de TrkA, administrar en el mamífero una cantidad terapéuticamente eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo. En una forma de realización, el compuesto 1 es una sal dihidrocloruro.
En una forma de realización, la desregulación de TrkA comprende una o más traslocaciones o inversiones cromosómicas que resultan en fusiones génicas de TrkA. En una forma de realización, la fusión génica de TrkA es LMNA-TrkA, TFG-TrkA, TPM3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, TP53-TrkA, RNF213-TrkA, RABGAP1L-TrkA, IRF2BP2-TrkA, SQSTM1-TrkA, SSBP2-TrkA, o TPR-TrkA. En una forma de realización, el cáncer es cáncer de pulmón de células no pequeñas, carcinoma papilar tiroideo, glioblastoma multiforme, carcinoma colorrectal, melanoma, colangiocarcinoma o sarcoma.
En una forma de realización, la desregulación de TrkA comprende una o más eliminaciones, inserciones o mutaciones en la proteína TrkA. En una forma de realización, el cáncer es leucemia mieloide aguda, carcinoma neuroendocrino de células grandes o neuroblastoma.
En una forma de realización, la desregulación de TrkA es sobreexpresión de TrkA de tipo salvaje (activación autocrina). En una forma de realización, el cáncer es carcinoma de próstata, neuroblastoma, carcinoma pancreático, melanoma, carcinoma de células escamosas de cabeza y cuello o carcinoma gástrico.
En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de cáncer en un mamífero que lo necesita, comprende además administrar una cantidad eficaz de compuesto 1 o una sal farmacéuticamente aceptable del mismo en combinación con una cantidad eficaz de por lo menos un agente terapéutico adicional seleccionado de una o más terapias adicionales o agentes quimioterápicos.
En una forma de realización, compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del cáncer en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en combinación con uno o más agentes quimioterápicos adicionales.
En una forma de realización, el agente o agentes quimioterápicos se seleccionan de agentes terapéuticos dirigidos a (“targeted”) receptor tirosina cinasa, incluyendo cabozantinib, crizotinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, pazopanib, pertuzumab, regorafenib, sunitinib y trastuzumab.
En una forma de realización, el agente o agentes quimioterápicos adicionales se seleccionan de inhibidores de la ruta de transducción de señales, incluyendo inhibidores de la ruta de Ras-Raf-MEK-ERK (por ejemplo, binimetinib, selumetinib, encorafinib, sorafenib, trametinib, vemurafenib), inhibidores de la ruta de PI3K-Akt-mTOR-S6K (por ejemplo, everolimus, rapamicina, perifosina, temsirolimus) y moduladores de la ruta de apoptosis (por ejemplo, obataclax).
En una forma de realización, el agente o agentes quimioterápicos adicionales se seleccionan de quimioterápicos citotóxicos, incluyendo trióxido de arsénico, bleomicina, cabazitaxel, capecitabina, carboplatino, cisplatino, ciclofosfamida, citarabina, dacarbazina, daunorrubicina, docetaxel, doxorrubicina, etopósido, fluorouracilo, gemcitabina, irinotecán, lomustina, metotrexato, mitomicina C, oxaliplatino, paclitaxel, pemetrexed, temozolomida y vincristina.
En una forma de realización, el agente o agentes quimioterápicos adicionales se seleccionan de terapias dirigidas a angiogénesis, incluyendo aflibercept y bevacizumab.
En una forma de realización, el agente o agentes quimioterápicos adicionales se seleccionan de agentes inmunodirigidos, incluyendo aldesleuquina, ipilimumab, lambrolizumab, nivolumab y sipuleucel-T.
En una forma de realización, el agente o agentes quimioterápicos adicionales se seleccionan de agentes activos contra la ruta de TrkA, incluyendo biofarmacéuticos dirigidos a NGF, tales como anticuerpos de NGF e inhibidores panTrk.
En una forma de realización, el agente terapéutico o terapia adicional es radioterapia, incluyendo terapia de radioyodo, radiación de haz externo y terapia de radio-223.
En una forma de realización, el agente o agentes terapéuticos adicionales incluyen cualquiera de las terapias o agentes terapéuticos anteriormente listados que son estándares de cuidado en cánceres en los que el cáncer presenta una desregulación de TrkA.
En una forma de realización descrita en la presente memoria, el compuesto 1 o una sal farmacéuticamente aceptable del mismo para la utilización en el tratamiento de cáncer en un paciente que lo necesita, comprende administrar en dicho paciente compuesto 1 o una sal farmacéuticamente aceptable del mismo, en combinación con por lo menos una terapia o agente quimioterápico adicional seleccionado de radioterapia (por ejemplo, terapia de radioyodo, radiación de haz externo, terapia de radio-223), quimioterápicos citotóxicos (por ejemplo, trióxido de arsénico, bleomicina, cabazitaxel, capecitabina, carboplatino, cisplatino, ciclofosfamida, citarabina, dacarbazina, daunorrubicina, docetaxel, doxorrubicina, etopósido, fluorouracilo, gemcitabina, irinotecán, lomustina, metotrexato, mitomicina C, oxaliplatino, paclitaxel, pemetrexed, temozolomida, vincristina), tratamiento dirigido a tirosina cinasa (por ejemplo, afatinib, cabozantini, cetuximab, crizotinib, dabrafenib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, pazopanib, panitumumab, pertuzumab, regorafenib, sunitinib, trastuzumab), moduladores de la apóptosis e inhibidores de la transducción de señales (por ejemplo, everolimus, perifosina, rapamicina, binimetinib, selumetinib, encorafinib, sorafenib, temsirolimus, trametinib, vemurafenib), terapias inmunodirigidas (por ejemplo, aldesleuquina, interferón alfa-2b, ipilimumab, lambrolizumab, nivolumab, prednisona, sipuleucel-T) y terapias dirigidas a la angiogénesis (por ejemplo, aflibercept, bevacizumab), en el que la cantidad de compuesto 1 o una sal farmacéuticamente aceptable del mismo es, en combinación con la terapia o agente terapéutico adicional, eficaz en el tratamiento de dicho cáncer. Estos agentes terapéuticos adicionales pueden administrarse con el compuesto 1 o una sal farmacéuticamente aceptable del mismo como parte de las mismas o diferentes formas de administración, mediante la misma o diferentes vías de administración, y en el mismo o diferentes programas de administración según la práctica farmacéutica estándar conocida por el experto en la materia.
En la presente memoria se proporciona además: (i) una combinación farmacéutica para la utilización en el tratamiento del cáncer en un paciente que lo necesita, que comprende: (a) compuesto 1 o una sal
farmacéuticamente aceptable del mismo, (b) un agente terapéutico adicional, y (c) opcionalmente por lo menos un portador farmacéuticamente aceptable para la utilización simultánea, separada o secuencial para el tratamiento del cáncer, en el que las cantidades de compuesto 1 o de una sal farmacéuticamente aceptable del mismo y del agente terapéutico adicional resultan eficaces conjuntamente en el tratamiento de dicho cáncer, (ii) una composición farmacéutica que comprende dicha combinación, (iii) la utilización de dicha combinación para la preparación de un medicamento para el tratamiento de cáncer, y (iv) un paquete o producto comercial que comprende dicha combinación en forma de una preparación combinada para la utilización simultánea, separada o secuencia, y a un método de tratamiento de cáncer en un paciente que lo necesita.
En una forma de realización, la terapia de combinación es para la utilización en el tratamiento de un cáncer seleccionado de cáncer de pulmón de células no pequeñas, carcinoma papilar tiroideo, glioblastoma multiforme, leucemia mieloide aguda, carcinoma colorrectal, carcinoma neuroendocrino de células grandes, cáncer de próstata, carcinoma pancrático, melanoma, carcinoma de células escamosas de cabeza y cuello, y carcinoma gástrico. En una forma de realización, la terapia de combinación es para el tratamiento de un cáncer seleccionado de cáncer de pulmón de células no pequeñas, carcinoma papilar tiroideo, glioblastoma multiforme, leucemia mieloide aguda, carcinoma colorrectal, carcinoma neuroendocrino de células grandes, cáncer de próstata, neuroblastoma, carcinoma pancrático, carcinoma de células escamosas de cabeza y cuello, carcinoma gástrico, colangiocarcinoma y sarcoma.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, asimismo resulta útil para tratar la inflamación o una enfermedad o trastorno inflamatorio.
De acuerdo con lo anterior, en la presente memoria se describe un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de inflamación o una enfermedad o trastorno inflamatoria en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha inflamación. En una forma de realización, la enfermedad inflamatoria se selecciona de enfermedades pulmonares inflamatorias (tales como asma), cistitis intersticial (CI), síndrome doloroso vesicular (SDV), enfermedades intestinales inflamatorias (incluyendo la colitis ulcerosa y la enfermedad de Crohn) y enfermedades inflamatorias de la piel, tales como la dermatitis atópica y la soriasis.
En una forma de realización, compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de la inflamación o una enfermedad o trastorno inflamatorio comprende administrar en un mamífero que lo necesita compuesto 1 o una sal farmacéuticamente aceptable del mismo en combinación con uno o más agentes adicionales. Entre los ejemplos de agentes adicionales se incluyen agentes anti-TNF (por ejemplo, anticuerpo monoclonal, tal como infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), y golimumab (Simponi), o una proteína de fusión de receptor circulante, tal como etanercept (Enbrel), un antimetabolito y fármacos antifolato (por ejemplo, metotrexato) o inhibidores de cinasa dirigidos (por ejemplo, inhibidores de la familia JAK, tales como ruxolitinib, tofacitinib, CYT387, lestaurtinib, pacritinib y TG101348). Estos agentes terapéuticos adicionales pueden administrarse con compuesto 1 o una sal farmacéuticamente aceptable del mismo como parte de las mismas o diferentes formas de administración, mediante la misma o diferentes vías de administración, y en el mismo o diferentes programas de administración según la práctica farmacéutica estándar conocida por el experto en la materia.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, asimismo resulta útil para tratar una enfermedad neurodegenerativa en un mamífero. En una forma de realización, el compuesto 1 o una sal farmacéuticamente aceptable del mismo, asimismo puede utilizarse para tratar la desmielinización y la desmielinización mediante la estimulación de la mielinización, supervivencia neuronal y diferenciación de los oligodendrocitos mediante el bloqueo de la interacción Sp35-TrkA.
De acuerdo con lo anterior, en la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de una enfermedad neurodegenerativa en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha enfermedad neurodegenerativa. En una forma de realización, la enfermedad neurodegenerativa es la esclerosis múltiple. En una forma de realización, la enfermedad neurodegenerativa es la enfermedad de Parkinson. En una forma de realización, la enfermedad neurodegenerativa es la enfermedad de Alzheimer.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de determinadas enfermedades infecciosas, tales como la infección por Trypanosoma cruzi en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha infección por Trypanosoma cruzi.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del síndrome de Sjogren en un mamífero, que comprende administrar en dicho
mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho síndrome.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de la endometriosis en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha endometriosis.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de la neuropatía periférica diabética en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha neuropatía periférica diabética.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de la prostatitis en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha prostatitis.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del síndrome doloroso pélvico en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicho síndrome doloroso pélvico.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de un desequilibrio de la regulación del remodelado óseo en un mamífero, que comprende administrar en dicho mamífero que lo necesita, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha enfermedad. En una forma de realización, la enfermedad es osteoporosis, artritis reumatoide o metástasis óseas.
En una forma de realización, compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades relacionadas con un desequilibrio en la regulación del remodelado óseo en un mamífero que lo necesita, que comprende administrar compuesto 1 o una sal farmacéuticamente aceptable del mismo en combinación con uno o más agentes terapéuticos o terapias adicionales. Entre los ejemplos de agentes terapéuticos adicionales se incluyen tratamientos anti-TNF (por ejemplo, anticuerpo monoclonal, tal como infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), y golimumab (Simponi), o una proteína de fusión de receptor circulante, tal como etanercept (Enbrel), un antimetabolito y fármacos antifolato (por ejemplo, metotrexato) o inhibidores de cinasa dirigidos (por ejemplo, inhibidores de la familia JAK, tales como ruxolitinib, tofacitinib, CYT387, lestaurtinib, pacritinib y TG101348). Estos agentes terapéuticos adicionales pueden administrarse con el compuesto 1 o una sal farmacéuticamente aceptable del mismo como parte de las mismas o diferentes formas de administración, mediante la misma o diferentes vías de administración, y en el mismo o diferentes programas de administración según la práctica farmacéutica estándar conocida por el experto en la materia.
En la presente memoria se describe además un compuesto 1 o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades que resulta de la señalización aberrante del factor de crecimiento del tejido conectivo en un mamífero que lo necesita, que comprende administrar en dicho mamífero, compuesto 1 o una sal farmacéuticamente aceptable del mismo, en una cantidad eficaz para tratar dicha enfermedad. En una forma de realización, la enfermedad que resulta de la señalización aberrante del factor de crecimiento del tejido conectivo es el síndrome de Raynaud, la fibrosis pulmonar idiopática, la cicatrización (hipertrófica, queloide y otros), la cirrosis, la fibrosis endomiocárdica, la fibrosis auricular, la mielofibrosis, la fibrosis masiva progresiva (pulmón), la fibrosis sistémica nefrogénica, el escleroderma, la esclerosis sistémica, la artrofibrosis y la fibrosis ocular.
Tal como se utiliza en la presente memoria, una “cantidad eficaz” se refiere a una cantidad de compuesto que, al administrarla en un mamífero que necesita dicho tratamiento, resulta suficiente para (i) tratar una enfermedad, condición o trastorno particular que puede tratarse con compuesto 1 o una sal farmacéuticamente aceptable del mismo, o (ii) atenuar, aliviar o eliminar uno o más síntomas de la enfermedad, condición o trastorno particular indicado en la presente memoria. La cantidad de compuesto 1 que se corresponderá con dicha cantidad variará dependiendo de factores tales como la condición de la enfermedad y su severidad, y la identidad (por ejemplo, el peso) de mamífero que requiere tratamiento, aunque, sin embargo, puede ser rutinariamente determinada por el experto en la materia.
Tal como se utiliza en la presente memoria, el término “mamífero” se refiere a un animal de sangre caliente que presenta o está en riesgo de desarrollar una enfermedad descrita en la presente memoria e incluye, aunque sin limitación, cobayas, perros, gatos, ratas, ratones, hámsters y primates, incluyendo seres humanos.
El compuesto 1 o una sal farmacéuticamente aceptable del mismo, puede administrarse mediante cualquier vía conveniente, por ejemplo, en el tracto gastrointestinal (por ejemplo, rectal u oralmente), la nariz, pulmones, musculatura o vasculatura, irrigación de heridas, o transdérmica o dérmicamente. El compuesto 1 o una sal farmacéuticamente aceptable del mismo, pueden administrarse en cualquier forma de administración conveniente, por ejemplo comprimidos, polvos, cápsulas, soluciones, dispersiones, suspensiones, jarabes, pulverizadores, supositorios, geles, emulsiones, parches, etc. Dichas composiciones pueden contener componentes convencionales en las preparaciones farmacéuticas, por ejemplo diluyentes, portadores, modificadores del pH, edulcorantes, agentes de carga y agentes activos adicionales. Si se desea la administración parenteral, las composiciones serán estériles y en una forma de solución o suspensión adecuada para la inyección o infusión. Dichas composiciones forman un aspecto adicional de la invención.
Puede prepararse otra formulación mediante la mezcla de compuesto 1 o una sal farmacéuticamente aceptable del mismo, y un portador o excipiente. Los portadores y excipientes adecuados son bien conocidas por el experto en la materia. Las formulaciones asimismo pueden incluir uno o más tampones, agentes estabilizadores, surfactantes, agentes humectantes, agentes lubricantes, emulsionantes, agentes de suspensión, conservantes, antioxidantes, agentes opacificadores, deslizantes, adyuvantes de procesamiento, colorantes, edulcorantes, agentes perfumantes, agentes saborizantes, diluyentes y otros aditivos conocidos, proporcionando una presentación elegante del fármaco (es decir, un compuesto de la presente memoria o composición farmacéutica del mismo) o adyuvante en la preparación del producto farmacéutico (es decir, medicamento).
De acuerdo con lo anterior, en la presente memoria se proporciona además una composición farmacéutica que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo, junto con un diluyente o portador farmacéuticamente aceptable. En una forma de realización, una composición farmacéutica que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo, se formula para la administración oral. En una forma de realización, una composición farmacéutica que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo, se formula para la administración oral en forma de un comprimido o una cápsula.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del dolor en un mamífero. En una forma de realización, el dolor es dolor crónico. En una forma de realización, el dolor es dolor agudo. En una forma de realización, el dolor es dolor de espalda crónico, dolor inflamatorio, dolor neuropático o dolor asociado a cáncer, cirugía o fractura ósea.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento del cáncer en un mamífero. En una forma de realización, el cáncer se selecciona de cáncer de pulmón de células no pequeñas, carcinoma papilar tiroideo, glioblastoma multiforme, leucemia mieloide aguda, carcinoma colorrectal, carcinoma neuroendocrino de células grandes, cáncer de próstata, neuroblastoma, carcinoma pancrático, melanoma, carcinoma de células escamosas de cabeza y cuello, carcinoma gástrico, colangiocarcinoma y sarcoma.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de inflamación o una enfermedad o trastorno inflamatorio en un mamífero. En una forma de realización, la enfermedad inflamatoria es enfermedad pulmonar inflamatoria (tal como asma), cistitis intersticial, síndrome doloroso vesicular, enfermedades intestinales inflamatorias (incluyendo colitis ulcerosa y enfermedad de Crohn) y enfermedades inflamatorias de la piel, tales como dermatitis atópica.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades infecciosas, por ejemplo la infección por Trypanosoma cruzi, en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de síndrome de Sjogren en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de endometriosis en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de neuropatía periférica diabética en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de prostatitis en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de síndrome doloroso pélvico en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedad neurodegenerativa en un mamífero.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de una enfermedad relacionada con un desequilibrio en la regulación del remodelado óseo.
En la presente memoria se proporciona además compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la utilización en el tratamiento de enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
En la presente memoria se describe además la utilización de compuesto 1, o una sal farmacéuticamente aceptable del mismo, para la preparación de un medicamento para el tratamiento de una condición seleccionada de dolor, cáncer, inflamación, enfermedades neurodegenerativas, infección por Trypanosoma cruzi, y enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo. En una forma de realización, el dolor es dolor crónico. En una forma de realización, el dolor es dolor agudo. En una forma de realización, el dolor es dolor de espalda crónico. En una forma de realización, el dolor es dolor inflamatorio, dolor neuropático o dolor asociado a cáncer, cirugía o fractura ósea. En una forma de realización, la condición es cáncer. En una forma de realización, la condición es inflamación. En una forma de realización, la condición es una enfermedad neurodegenerativa. En una forma de realización, la condición es infección por Trypanosoma cruzi. En una forma de realización, la condición es síndrome de Sjogren. En una forma de realización, la condición es endometriosis. En una forma de realización, la condición es neuropatía periférica diabética. En una forma de realización, la condición es prostatitis. En una forma de realización, la condición es síndrome doloroso pélvico. En una forma de realización, la enfermedad es una enfermedad relacionada con un desequilibrio en la regulación del remodelado óseo. En una forma de realización, la enfermedad es una enfermedad que resulta de la señalización aberrante del factor de crecimiento del tejido conectivo.
En la presente memoria se proporciona además un medicamento que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo, para el tratamiento del dolor en un mamífero, en combinación con un agente terapéutico adicional seleccionado de compuestos antiinflamatorios, esteroides (por ejemplo, dexametasona, cortisona y fluticasona), analgésicos, tales como AINE (por ejemplo, aspirina, ibuprofeno, indometacina y ketoprofeno), opioides (tales como morfina), antagonistas de receptores del péptido relacionado con el gen de la calcitonina, moduladores de canales iónicos selectivos de subtipo, anticonvulsivos (por ejemplo, pregabalina y gabapentina), inhibidores duales de la recaptación de serotonina-norepinefrina (por ejemplo, duloxetina, venlaxina y milnaciprán), inhibidores de cinasa de la familia de JAK (por ejemplo, ruxolitinib o tofacitinib) y antidepresivos tricíclicos (tales como amitriptilina, nortriptilina y desipramina). En una forma de realización, en la presente memoria se proporciona un medicamento que comprende compuesto 1 o una sal farmacéuticamente aceptable del mismo para el tratamiento del dolor en un mamífero en combinación con un AINE.
En la presente memoria se proporciona además un medicamento que comprende un agente terapéutico seleccionado de compuestos antiinflamatorios, esteroides (por ejemplo, dexametasona, cortisona y fluticasona), analgésicos tales como AINE (por ejemplo, aspirina, ibuprofeno, indometacina y ketoprofeno) y opioides (tales como morfina) para el tratamiento del dolor en un mamífero en combinación con compuesto 1 y o una sal farmacéuticamente aceptable del mismo.
Ejemplos
Preparación de productos intermedios sintéticos
Preparación A

Dihidrocloruro de (3S,4ft)-4-(3-fluorofeniD-1-(2-metoxietiDpirrolidín-3 -amina
Etapa A: preparación de (3S,4ffl-4-(3-fluorofeniD-1-(2-metoxietiDpirrolidín-3-ilcarbamato de terc-butilo: una solución de (3S,4R)-4-(3-fluorofenil)pirrolidín-3-ilcarbamato de terc-butilo (250 mg, 0.89 mmoles, disponible comercialmente), DIEA (0,48 ml, 2,68 mmoles) y 1-bromo-2-metoxietano (149 mg, 1.07 mmoles) en Dm F (3
ml) se agitó a temperatura ambiente durante 2 horas, después se calentó a 60°C durante 4 horas, después se enfrió hasta la temperatura ambiente durante 16 horas. Tras dividir entre EtOAc y NaHCO3 saturado (10 ml cada uno), la capa orgánica se lavó con agua y solución hipersalina (2 x 10 ml cada uno), se secó sobre Na2SO4, se filtró y se concentró, rindiendo (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-ilcarbamato de terc-butilo (250 mg, rendimiento: 83%) en forma de un aceite naranja viscoso. CL-e M (apci) m/z = 339,1 (M+H).
Etapa B: preparación de dihidrocloruro de (3S.4ft)-4-(3-fluorofenil)-1-(2-metoxietN)pirrolidín-3-amina: una solución de (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-ilcarbamato de terc-butilo (250 mg, 0.74 mmoles) en HCl 5-6 N en alcohol isopropílico (14.8 ml, 73.9 mmoles) se agitó a temperatura ambiente durante 1 hora. La mezcla se concentró bajo vacío y se trituró con Et2O, proporcionando el compuesto del título (230 mg, rendimiento: 100%) en forma de un sólido de color beige. CL-EM (apci) m/z = 239.1 (M+H).
Preparación B

4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-amina
Etapa A: preparación de 5-amino-4-metil-1-fenil-1H-pirazol-3(2H)-ona: una mezcla de 2-cianopropanoato de etilo (50.5 g, 397.2 mmoles) y fenilhidrazina (39 ml, 397.2 mmoles) en dioxano (100 ml) se calentó a 110°C durante 5 días. La mezcla enfriada se concentró a 1/2 volumen y después se enfrió en hielo y se trituró con Et2O frío. Se filtraron los sólidos resultantes, se lavaron extensamente con EtO y se secaron bajo vacío, proporcionando 5-amino-4-metil-1-fenil-1H-pirazol-3(2H)-ona (34.69 g, rendimiento: 46%) en forma de unos polvos blancos esponjosos. EM (apci) m/z = 190.1 (M+H).
Etapa B: preparación de sulfonato de 5-amino-4-metil-1-fenil-1H-pirazol-3-il trifluorometano: una suspensión de 5-amino-4-metil-1-fenil-1H-pirazol-3(2H)-ona (13,72 g, 72,5 mmoles) y N-fenilbis(trifluorometilsulfonamida) (27.2 g, 76.1 mmoles) en DMF (100 ml) se trató con DIEA (37.9 ml, 217.5 mmoles) y la mezcla se agitó a temperatura ambiente durante 16 horas. La mezcla se dividió entre NaHCO3 saturado (400 ml) y EtOAc (200 ml) y la capa acuosa se extrajo con EtOAc (2 x 200 ml). Las fases orgánicas agrupadas se lavaron con agua (5 x 50 ml) y solución hipersalina (50 ml), después se secaron sobre Na2SO4, se filtraron y se concentraron bajo vacío. El residuo se purificó mediante cromatografía de columna de sílice, eluyendo con hexanos/EtOAC 4:1, proporcionando sulfonato de 5-amino-4-metil-1-fenil-1H-pirazol-3-il trifluorometano (23.1 g, rendimiento: 99%) en forma de un sólido amarillo pálido. EM (apci) m/z = 322.0 (M+H).
Etapa C: preparación de 4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-amina: se agruparon en tolueno (10 ml), agua (5 ml) y EtOH (2.5 ml), sulfonato de 5-amino-4-metil-1-fenil-1H-pirazol-3-il trifluorometano (900 mg, 2.8 mmoles), 2-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)pirimidina (925 mg, 4.2 mmoles), K2CO3 (1.55 g, 11.2 mmoles) y Pd(PPh3)4 (324 mg, 0.28 mmoles) y se calentaron a 95°C en un tubo sellado durante 16 horas. La mezcla fría se filtró y el filtrado se dividió entre agua (50 ml) y EtOAc (50 ml). Se extrajo la capa acuosa con EtOAc (2 x 30 ml) y las fases orgánicas agrupadas se lavaron con solución hipersalina (30 ml), se secaron sobre Na2SO4, se filtraron y se concentraron bajo vacío. El residuo se purificó mediante cromatografía de columna de gel de sílice (eluyendo con MeOH al 2%/DCM, proporcionando el compuesto del título (533 mg, rendimiento: 72%) en forma de un sólido rosa. EM (apci) m/z = 266.1 (M+H).
Preparación C

2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina
Etapa A: preparación de 2-metoxi-N-((trimetilsilil)metil)etanamina: a una solución en DMSO (15 ml) de 2-metoxietanamina (14.2 ml, 163 mmoles) a 90°C se le añadió una solución en DMSO (10 ml) de (clorometil)trimetilsilano (11.4 ml, 81.5 mmoles) mediante embudo de adición durante 40 minutos. La mezcla se calentó a 90°C durante 3,5 horas y después se enfrió hasta la temperatura ambiente. A continuación, la mezcla se diluyó con H2O (150 ml) y se extrajo con EtOAc (2 x 150 ml). Los extractos orgánicos agrupados se
lavaron con solución hipersalina (150 ml), se secaron con MgSO4, se filtraron y se concentraron, proporcionando 2-metoxi-N-((trimetilsilil)metil)etanamina (8.14 g, rendimiento 62%) en forma de un aceite amarillo. EM (apci) m/z = 162.0 (M+H).
Etapa B: preparación de 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina: Una solución en MeOH (2.45 ml) de formaldehído (acuoso al 37%, 4.91 g, 60.6 mmoles) se enfrió a 0°C y se trató con una adición gota a gota de 2-metoxi-N-((trimetilsilil)metil)etanamina (8.14 g, 50.5 mmoles9. La mezcla bifásica se agitó a 0°C durante 3 horas, después se añadió K2CO3 (6.97 g, 50.5 mmoles) y la mezcla se agitó a 0°C durante 1 hora. El aceite amarillo se decantó sobre K2CO3 (2.00 g, 14.4 mmoles) y la mezcla se agitó a temperatura ambiente durante 2 horas. Tras decantar el aceite amarillo, el K2CO3 sólido se lavó con Et2O (2 x 10 ml) y se agruparon los lavados de Et2O con el aceite amarillo decantado y se concentraron en un evaporador rotatorio, rindiendo el compuesto del título (9,92 g, rendimiento: 96%) en forma de un aceite amarillo. RMN 1H (CDCh) 54.00 (s.
2H). 3.37-3.43 (m. 2H). 3.29 (s. 3H). 3.19 (s. 3H). 2.77-2.82 (m. 2H). 2.18 (s. 2H). 0.00 (s. 9H) ppm.
Preparación D

(3S.4ft)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina
Etapa A: preparación de ácido (trans)-4-(3-fluorofeniD-1-(2-metoxietN)pirrolidín-3-carboxílico: se trató el ácido (E)-3-(3-fluorofenil)acrílico (25.20 g, 151.7 mmoles secuencialmente con EtOAc (75 ml) y heptano (75 ml) seguido de TFA (1.17 ml, 15.17 mmoles). La mezcla se introdujo en un baño de agua fría (temperatura interna: 15°C) y se añadió gota a gota 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina [Preparación C] (69.82 g, 197.2 mmoles) desde un embudo de adición durante 20 minutos, añadiendo hielo al baño de agua para mantener la temperatura interna entre 13°C y 19°C durante la adición. Se retiró el baño de hielo y la mezcla se agitó a temperatura ambiente durante 18 horas, después se enfrió (temperatura interna: 13°C) y se añadió gota a gota 2-metoxi-N-(metoximetil)-N-((trimetilsilil)metil)etanamina adicional (Preparación C, 34 g) desde un embudo de adición durante 6 minutos. Se retiró el baño y tras agitar durante 4 horas adicionales a temperatura ambiente, la mezcla se concentró hasta la mitad del volumen. Se añadió heptano (100 ml) y la mezcla se concentró parcialmente, eliminando aproximadamente 150 ml de solvente. Se repitió lo anterior dos veces, eliminando 100 ml de solvente cada vez. La suspensión residual se enjuagó en el fondo del matraz con heptano (50 ml), después se selló y se agitó a temperatura ambiente durante 64 horas. Se filtraron los sólidos, se enjuagaron con heptano (2 x 50 ml) y se secaron al vacío, proporcionando ácido (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-carboxílico (44.99 g, rendimiento: 111%) en forma de un sólido amarillo pálido que se utilizó directamente en la etapa siguiente, asumiendo un rendimiento de 100%. EM (apci) m/z = 268.1 (M+H).
Etapa B: preparación de (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-carboxamida: a una suspensión de ácido (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-carboxílico (40.54 g, 151.7 mmoles) en THF (440 ml) se añadieron CDI (31.97 g, 197.2 mmoles) seguido de hidrocloruro de imidazol (3.171 g, 30.33 mmoles). La mezcla de reacción se agitó a temperatura ambiente durante 16 horas y a continuación se añadió amonio (135.0 ml, 2 M en iPrOH, 270.0 mmoles) mediante el embudo de adición durante 30 minutos. Tras agitar durante 3 horas adicionales, se filtraron los sólidos resultantes, se lavaron con EtOAc y se concentró el filtrado. Se disolvió el residuo en EtOAc (200 ml) y se lavó con 25/75 de agua:solución hipersalina (2 x 200 ml). La fase orgánica se secó sobre MgSo4, se filtró, se concentró y se secó al vacío, proporcionando (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-carboxamide (54.97 g, rendimiento: 136%) en forma de un sólido amarillo que se utilizó asumiendo un rendimiento de 100%. Em (apci) m/z = 267.2 (M+H).
Etapa C: preparación de (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina: a una solución de (trans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-carboxamide (29.33 g, 85.90 mmoles) en MeOH (345 ml), en un matraz de fondo redondo de 1 l de 3 cuellos, dotado de sonda de temperatura, enfriado en un baño de hielo, se añadió NaOCl (solución acuosa con 10-15% Cl disponible) (116.3 ml, 189.0 mmoles) desde un embudo de adición durante 20 minutos [la temperatura interna era de entre 7°C y 13°C]. Se retiró el baño de hielo y la mezcla heterogénea se agitó a temperatura ambiente durante 90 minutos, seguido de a 55°C durante 3 horas. Se añadió una solución de KOH (67,48 g, 1203 mmoles) en H2O (225.9 ml, 12.542 mmoles) y la solución se agitó a 75°C durante 19 horas. La mezcla se enfrió a 0°C y se añadió lentamente HCl concentrado (147.1 ml,
4.802 mmoles) durante 30 minutos. Se ajustó el pH a 6 mediante la adición de KíPO4 acuoso (al 30% en peso, 155 ml) y la mezcla se concentró parcialmente para eliminar los solventes orgánicos. Se añadió Kí PO4 acuoso (al 30% en peso, 280 ml) hasta pH 10 y la mezcla se extrajo con EtOAc (2 x 450 ml). Se concentraron los extractos orgánicos agrupados y se secaron al vacío, proporcionando (frans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (28.08 g, rendimiento: 91%) en forma de un jarabe de color ámbar oscuro. EM (apci) m/z = 239.2 (M+H).
Etapa D: preparación de (3S,4ft)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina(2S.3S)-23-bis((4-metilbenzoil)oxi)succinato: se pesó (frans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (125 mg, 0.52 mmoles) y ácido (2S,3S)-2,3-bis((4-metilbenzoil)oxi)succínico (222,9 mg, 0,58 mmoles) en un vial de 4 ml, y después se trató con MeOH (1,57 ml), seguido de agua (0,175 ml). Se tapó el vial y la mezcla de reacción se agitó a 50°C durante 5 minutos, después se dejó enfriar lentamente hasta la temperatura ambiente durante 17 horas. Se filtraron los sólidos resultantes, se lavaron con Et2O (4 x 0,2 ml) y se secaron al vacío, proporcionando -(3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina(2S,3S)-2,3-bis((4-metilbenzoil)oxi)succinato (131.6 mg, rendimiento: 40%) en forma de un sólido blanco. El análisis de SFC quiral de una muestra libre indicó >95% ee. Se utilizó este material como cristales de inóculo en la etapa siguiente:
se disolvió (frans)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (7,39 g, 18,61 mmoles) en MeOH (52.7 ml) y se trató con ácido (2S,3S)-2,3-bis((4-metilbenzoil)oxi)succínico (10,78 g, 27,91 mmoles) seguido de H2O (9.3 ml). La mezcla se agitó a 50°C durante 5 minutos. Se añadieron cristales de inóculo (90 mg) a la solución y la mezcla se dejó que se calentase lentamente hasta la temperatura ambiente durante 16 horas. Se filtraron los sólidos resultantes, se enjuagaron con Et2O (4 x 5 ml) y se secaron al vacío. El residuo se suspendió en MeOH (15 ml) y H2O (2.7 ml), se agitó a 50°C durante 15 minutos, después se dejó enfriar lentamente hasta la temperatura ambiente durante 19 horas. Se filtraron los sólidos, se lavaron con Et2O (ml) y se secaron al vacío, proporcionando (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina(2S,3S)-2,3-bis((4-metilbenzoil)oxi)succinato (3.12 g, rendimiento: 27%) en forma de un sólido blanco. El análisis quiral de una muestra libre indicó >99.7% ee.
Etapa E: preparación de (3S.4ft)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina: una suspensión de (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (2S,3S)-2,3-bis((4-metilbenzoil)oxi)succinato (2.89 g, 4.63 mmol) en DCM (25 ml) se lavó con solución acuosa 1 M de NaOH (2 x 15 ml). Las fases acuosas agrupadas se extrajeron con DCM (25 ml) y los extractos orgánicos agrupados se lavaron con solución hipersalina (30 ml), se secaron sobre Na2SO4, se filtraron, se concentraron y se secaron al vacío, proporcionando (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (998 mg, rendimiento: 90%) en forma de un aceite de color beige amarillo. EM (apci) m/z = 239.2 (M+H).
Ejemplos sintéticos
Ejemplo 1
Dihidrocloruro de 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea

Etapa A: preparación de 1 -((3SAR)-4-(3-fluorofenil)-1 -(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea: a una solución de 4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-amina (Preparación B; 61 mg, 0.16 mmoles) en DCM (2 ml) se añadió trifosgeno (24 mg, 0.08 mmoles) seguido de DieA (84 pl, 0.48 mmoles). La mezcla se agitó a temperatura ambiente durante 15 minutos y después se trató con dihidrocloruro de (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (Preparación A; 50 mg, 0.16 mmoles) y DIEA (84 pl, 0.48 mmoles). La mezcla de reacción se agitó durante 16 horas. La mezcla se dividió entre agua (10 ml) y DCM (10 ml) y la capa acuosa se extrajo con DCM (2 x 10 ml). Las fases orgánicas agrupadas se lavaron con solución hipersalina (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó mediante cromatografía en columna de sílice, eluyendo con MeOH al 2,5-5%/DCM. Las fracciones que contenían el producto se purificaron adicionalmente mediante HPLC de fase inversa (ACN al 595%/agua/TFA al 0.1%), proporcionando el compuesto del título tras el tratamiento acuoso básico final (28 mg, rendimiento: 33%) en forma de un sólido blanco. EM (apci) m/z = 530.3 (M+H).
Etapa B: preparación de dihidrocloruro de 1-((3SAR)-4-(3-fluorofenil)-1-(2-metoxiethly)pirrolidín-3-N)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea: a una solución de 4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-amina (Preparación B; 2,56 g, 9,64 mmoles) en DCM (96 ml) enfriada a 0°C se le añadió trifosgeno (1.72 g, 5.78 mmoles) en una porción, seguido de la adición gota a gota de DIEA (8.40 ml, 48.20 mmoles). La mezcla se agitó a 0°C durante 1 hora. Se añadió dihidrocloruro de (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (Preparación A; 3.0 g, 9.64 mmoles) a la mezcla en porciones pequeñas durante 10 minutos. La mezcla de reacción se agitó a temperatura ambiente durante 40 minutos y después se vertió en agua (100 ml). Tras la separación de fases, se extrajo la fase acuosa con DCM (2 x 100 ml). Las fases orgánicas agrupadas se lavaron con solución hipersalina (100 ml), se filtraron y se concentraron. El residuo se purificó mediante cromatografía de fase inversa (SNAP, 120 g C18, MeOH al 5 a 75%/agua con HCl al 0,1%) y se secó al vacío, proporcionando el compuesto del título (3.33 g, rendimiento: 57%) en forma de una espuma de color crema. Em (apci) m/z = 530.3 (M+H).
Ejemplo 2

1-((3S.4^)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea
A una suspensión de 4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-amina (Preparación B; 800 mg, 3.02 mmoles) en DCM (15 ml) se le añadió trietilamina (2.1 ml, 15.1 mmoles) seguido de CDI (587 mg, 3.62 mmoles). La mezcla se selló bajo N2 y se agitó a temperatura ambiente durante 22 horas, después se trató con (3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (Preparación D; 826 mg, 3,47 mmoles) en DCM (4 ml). Se continuó la agitación durante 100 minutos. La mezcla se dividió entre DCM (60 ml) y agua (100 ml). Tras la separación de fases, la capa de DCM se extrajo con HCl 0,5 N (50 ml) seguido de HCl 0,2 N (2 x 25 ml). Se agruparon los extractos ácidos, se decantaron a un matraz limpio, se introdujeron en un baño de agua a temperatura ambiente y se basificaron con NaOH(aq.) 6 N y después con NaOH(aq) 1 N a pH 9-10 bajo agitación. La suspensión se agitó en un baño de hielo durante 5 minutos Se filtraron los sólidos resultantes, se enjuagaron con agua y éter (3 x 20 ml, cada uno) y se secaron al vacío, proporcionando el compuesto del título (1.44 g, rendimiento: 90%) en forma de un sólido blanco. EM (apci) m/z = 530.3 (M+H).
Ejemplo 3
Preparación de (2S,3S)-2,3-bis((4-metilbenzoil)oxi)succinato de (3S,4R)-4-(3,4-difluorofenil)-1-(2-metoxietil)pirrolidín-3-amina
Etapa A: preparación de (2-metoxi-N-((trimetilsilil)metil)etanamina: un matraz se cargó con (clorometil)trimetilsilano (3330.0 g, 1,0 eq.), acetonitrilo (5,28 l) y 2-metoxietanamina (4041.0 g, 2.0 eq.). La mezcla de reacción se agitó a 74°C durante 16 horas y después se dejó que la reacción se enfriase hasta 25°C. Se añadió pentano (16 l) y la mezcla de reacción se agitó durante 1 minuto. Se separaron las capas. Se añadió de vuelta la capa del fondo (capa de acetonitrilo) al embudo de separación y se añadió pentano (16 l) al embudo de separación. Tras la agitación, se separaron las capas. Se repitió la extracción con pentano (16 l). Las capas de pentano agrupadas se añadieron al embudo de separación y se añadió agua (3.3 l). Tras la agitación, se separaron las capas. Se añadió MeOH (3.0 l) a un reactor de 22 l dotado de vapor, agitación mecánica, condensador, sonda de temperatura de J-Kem y embudo de adición. Se llevó a cabo la destilación atmosférica a 25°C-45°C. Durante la destilación, se añadió solución de pentano al matraz de destilación mediante embudo de adición a una tasa que mantuvo el volumen en aproximadamente 6.6 l hasta que toda la solución de pentano se encontraba en el matraz de destilación. Se continuó la destilación hasta que el volumen total en el matraz era de aproximadamente 6,6 l. La mezcla de reacción se enfrió a 25°C y se agitó a temperatura ambiente bajo una atmósfera de nitrógeno, proporcionando (2-metoxi-N-((trimetilsilil)metil)etanamina (rendimiento: 72%).
Etapa B: preparación de 2-metox¡-N-(metox¡met¡l)-N-((tr¡met¡ls¡l¡l)met¡l)etanam¡na: se cargó un matraz con (2-metoxi-N-((trimetilsilil)metil)etanamina (380.0 g, 1.0 eq.), metanol (332 ml) y pentano (193 ml). La mezcla de reacc¡ón se ag¡tó en un baño de h¡elo-agua a menos de 5°C. Se añad¡ó soluc¡ón acuosa al 37% de formaldehído (97.9 ml, 1.2 eq.) a la mezcla de reacc¡ón a una tasa que mantenía una temperatura ¡nterna ¡nfer¡or a 10°C y la mezcla de reacc¡ón se ag¡tó a una temperatura ¡nfer¡or a 10°C durante 2 horas. Se añad¡ó éter terc-but¡l-metíl¡co (700 ml, 5.2 eq.) y agua (300.0 ml) a la mezcla de reacc¡ón y la mezcla se ag¡tó a temperatura amb¡ente durante aprox¡madamente 1 m¡nuto. Se separaron las capas y se añad¡ó la capa orgán¡ca de vuelta al embudo de separac¡ón. Se añad¡ó soluc¡ón h¡persal¡na (300 ml) al embudo de separac¡ón. Se ag¡tó la mezcla y se separaron las capas. Se añad¡ó carbonato potás¡co, K2CO3 (10.0 g) a la capa orgán¡ca, y tras mezclar la capa orgán¡ca, se f¡ltró por un papel de f¡ltro. La torta se enjuagó con éter terc-but¡l-metíl¡co. Las capas orgán¡cas agrupadas se concentraron, proporc¡onando 2-metox¡-N-(metox¡met¡l)-N-((tr¡met¡ls¡l¡l)met¡l)etanam¡na en forma de un ace¡te (rend¡m¡ento: 75%).
Etapa C: preparac¡ón de ác¡do trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxíl¡co: se cargó un matraz con ác¡do (E)-3-(3,4-d¡fluorofen¡l)acríl¡co (57.0 g, 1.0 eq.), acetato de et¡lo (143 ml) y heptano (143 ml) y la mezcla se ag¡tó. Se añad¡ó ác¡do tr¡fluoroacét¡co (2.4 ml, 0.1 eq.) y la mezcla de reacc¡ón se enfr¡ó a una temperatura ¡nfer¡or a 20°C. Se añad¡ó 2-metox¡-N-(metox¡met¡l)-N-((tr¡met¡ls¡l¡l)etanam¡na med¡ante embudo de ad¡c¡ón a una tasa que mantenía una temperatura ¡nterna entre 20°C y 25°C y la mezcla de reacc¡ón se ag¡tó a 20°C durante 2 horas. La mezcla de reacc¡ón se concentró bajo pres¡ón reduc¡da a una temperatura de entre 10°C y 27°C. Se añad¡ó acetato de et¡lo (200 ml) al res¡duo y la mezcla se concentró bajo pres¡ón reduc¡da. El res¡duo se ag¡tó durante la noche a 20°C. Se llevó a cabo la el¡m¡nac¡ón azeotróp¡ca de ¡mpurezas med¡ante dest¡lac¡ón al vacío med¡ante la ad¡c¡ón de heptano en porc¡ones (volumen total: 1200 ml). La suspens¡ón resultante se dejó en reposo durante la noche a 20°C y después se f¡ltró sobre papel de f¡ltro. La torta se enjuagó con heptano y se secó en un horno de vacío a 45°C durante aprox¡madamente 4 horas, proporc¡onando ác¡do trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxíl¡co (rend¡m¡ento: 90%).
Etapa D: preparac¡ón de trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxam¡de: un matraz se cargó con ác¡do trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxíl¡co (98,3 g, 1,0 eq.), carbon¡l d¡¡m¡dazol (69.8 g, 1.3 eq.), ¡m¡dazol HCl (7.2 g, 0.2 eq.) y tetrah¡drofurano (1000 ml). La mezcla de reacc¡ón se ag¡tó durante 1 hora a 25°C y después durante la noche a temperatura amb¡ente (aprox¡madamente 20°C) bajo n¡trógeno. Se añad¡ó amonio (2,0 M en alcohol ¡sopropíl¡co) (307 ml, 1.8 eq.) a la mezcla de reacc¡ón durante 15 m¡nutos. La mezcla de reacc¡ón se ag¡tó a 25°C durante aprox¡madamente 30 m¡nutos. La mezcla de reacc¡ón se f¡ltró por papel de f¡ltro y los sól¡dos f¡ltrados se enjuagaron con EtOAc (200 ml). Las capas orgán¡cas agrupadas se concentraron bajo pres¡ón reduc¡da a 37°C. Se añad¡ó acetato de et¡lo (200 ml) al ace¡te en bruto resultante y la mezcla se concentró bajo pres¡ón reduc¡da a 37°C. Se añad¡ó acetato de et¡lo (200 ml), agua (500 ml) y NaCl (82.0 g, 4.1 eq.) al res¡duo y la mezcla se ag¡tó durante aprox¡madamente 2 m¡nutos a temperatura amb¡ente. Se separaron las capas y las capas orgán¡cas agrupadas se concentraron bajo pres¡ón reduc¡da a 37°C, proporc¡onando trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxam¡de (rend¡m¡ento: 99%).
Etapa E: preparac¡ón de trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-am¡na: se cargó un matraz con trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-carboxam¡de (96,8 g, 1,0 eq.) y metanol (1,450 ml) y la mezcla de reacc¡ón se enfr¡ó a 5°C. Se añad¡ó una soluc¡ón al 10% de h¡poclor¡to sód¡co (462.2 ml, 2.2 eq.) med¡ante embudo de ad¡c¡ón a una tasa que mantuv¡ese una temperatura ¡nterna ¡nfer¡or a 20°C. La mezcla de reacc¡ón se ag¡tó durante 1 hora a 20°C y después a 55°C durante 1 hora. Se añad¡ó h¡dróx¡do potás¡co (314.6 g, 14.0 eq.) en forma de una soluc¡ón en agua (100 ml) a la mezcla de reacc¡ón durante 5 m¡nutos y la mezcla de reacc¡ón se ag¡tó a 72°C-75°C durante 12 horas. La mezcla de reacc¡ón se enfr¡ó a 55°C y se añad¡ó HCl al 37% en peso (583.8 ml, 55.9 eq.) durante 10 m¡nutos para ajustar el pH a un n¡vel ¡nfer¡or a 2. Se añad¡ó K3PO4 tr¡bás¡co (al 30% en peso, 100,0 ml) para ajustar el pH de la mezcla de reacc¡ón a un valor entre 6 y 7. La mezcla de reacc¡ón se concentró bajo pres¡ón reduc¡da a 37°C. La mezcla acuosa resultante se transf¡r¡ó a un embudo de separac¡ón y se añad¡ó K3PO4 tr¡bás¡co al 30% en peso (540.0 ml) y K3PO4 tr¡bás¡co al 37% en peso para ajustar el pH a >10. Se añad¡ó EtOAc (1000 ml) y la mezcla se ag¡tó durante aprox¡madamente 2 m¡nutos a temperatura amb¡ente. Se separaron las capas y la capa acuosa se añad¡ó de vuelta al embudo de separac¡ón. Se añad¡ó EtOAc (500 ml) al embudo de separac¡ón y se ag¡tó la mezcla. Se separaron las capas y las capas orgán¡cas agrupadas se concentraron bajo pres¡ón reduc¡da a 37°C, proporc¡onando trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-am¡na (rend¡m¡ento: 99%).
Etapa F: preparac¡ón de (2S.3S)-2.3-b¡s((4-met¡lbenzo¡l)ox¡)succ¡nato de (3S,4R)-4-(3,4-d¡fluorofen¡l)-1-('2-metox¡et¡l)-p¡rrol¡dín-3-am¡na: se cargó un matraz con trans-4-(3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-am¡na (123.3 g, 1.0 eq.) y metanol (1.603 ml). La mezcla se ag¡tó a temperatura amb¡ente. Se añad¡ó ác¡do (2S,3S)-2,3-B¡s((4-met¡lbenzo¡l)ox¡)succín¡co (145.0 g, 1.5 eq.) y agua (295 ml) y la mezcla de reacc¡ón se calentó a 65°C. La mezcla de reacc¡ón se sembró con 3S,4R)-4-(2S,3S)-2,3-b¡s((4-met¡lbenzo¡l)ox¡)-succ¡nato de (3,4-d¡fluorofen¡l)-1-(2-metox¡et¡l)p¡rrol¡dín-3-am¡na (0.5 g, 0.7 eq.). (se prepararon ¡nóculos llevando a cabo la reacc¡ón a pequeña escala, lo que proporc¡onó el producto en forma de un sól¡do), y la mezcla de reacc¡ón se enfr¡ó lentamente a 25°C durante 16 horas. La suspens¡ón se f¡ltró por un papel de f¡ltro. La torta se enjuagó con éter terc-but¡l metíl¡co (700 ml) y después se secó en un horno de vacío a 45°C durante aprox¡madamente
4 horas, proporcionando (2S,3S)-2,3-bis((4-metilbenzoil)oxi)succinato de (3S,4R)-4-(3,4-difluorofenil)-1-(2-metoxietil)pirrolidín-3-amina (rendimiento de 35% en dos etapas).
Ensayos biológicos
En los ensayos biológicos, el compuesto 1 se refiere a 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3 -il)-3 -(4-metil-3 -(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea; el compuesto 2 se refiere a 1-((3S,4R)-4-(3,4-difluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5 - yl)urea (dado a conocer en el documento n° WO 2012/158413), y el compuesto 3 se refiere a 1-(1',4-dimetil-1-fenil-1H,1'H-[3,4-bipirazol]-5-il)-3-((3S,4R)-4-(4-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)urea (dado a conocer en el documento n° WO 2012/158413).
Ejemplo A
Ensayo de unión de TrkA cinasa
Se determinó la actividad de unión de TrkA en un ensayo de unión a Eu de cinasa TrkA LanthaScreen™. Se incubó TrkA humano recombinante etiquetado con His 5 nM (dominio citoplasmático etiquetado con 6HIS de Invitrogen, n° de catálogo PV3144) con trazador Alexa-Fluor® 236, 4 nM (Invitrogen, n° de catálogo PV5592), anti-His biotinilado 2 nM (Invitrogen, n° de cat. PV6090) y estreptavidina marcada con europio 2 nM (Invitrogen, n° de cat. PV5899), en tampón (MOPS 25 mM, pH 7.5, MgCh 5 mM, Triton X-100 al 0.005%). Se añadieron diluciones en serie de tres veces del compuesto 1, compuesto 2 o compuesto 3 en DMSO a un porcentaje final de 2% de DMSO. Tras 60 minutos de incubación a 22°C, se midió la reacción utilizando el lector de placas multimodo EnVision (PerkinElmer) mediante detección dual de longitudes de onda TR-FRET a 615 nM y 665 nM. Se calculó el porcentaje de control utilizando un factor de emisión ratiométrico. Se determinó el valor de IC50 mediante el ajuste de un modelo de cuatro parámetros al porcentaje de datos de control.
El compuesto 1 presentaba un valor medio de IC50 de 1.1 nM al someterlo a ensayo en el ensayo de Ejemplo A. El compuesto 2 presentaba un valor medio de IC50 de 1.5 nM al someterlo a ensayo en el ensayo del Ejemplo A. El compuesto 3 presentaba un valor medio de IC50 de 1.7 nM al someterlo a ensayo en el ensayo del Ejemplo A.
Ejemplo B
Ensayo celular de TrkA
Se sembraron células CHO-K1 transfectadas con TrkA de tipo salvaje humano en una placa de fondo plano de 96 pocillos a razón de 3x105 células/pocillo en medio DMEM completo que contenía FBS al 10% y se dejó que se adhiriesen durante 24 horas a 37°C, 5% de CO2. A continuación, se sustituyó el medio por medio de ensayo libre de suero completo y las células se sometieron a ayuno de suero durante 1 hora a 37°C, 5% de CO2. Las células se trataron con compuesto 1, 2 o 3 utilizando diluciones en serie 1:3 con concentraciones finales comprendidas entre 1 pM y 152 pM. El compuesto se incubó sobre las células durante 1 hora a 37°C, 5% de CO2. A continuación, las células se estimularon con una concentración final de 30 ng/ml de NGF humano durante 7 minutos a 37°C, 5% de CO2. Se eliminó el medio y las células se lisaron con tampón de lisis que contenía inhibidores de fosfatasa y de proteasa. Se midió fosfo-TrkA mediante ELISA (R&D, n° de catálogo DYC2578). El ELISA capturó el TrkA total y detectó el total de fosfo-tirosina. Se midió la densidad óptica en cada pocillo utilizando un lector Versamax a una longitud de onda de 450 nm. Se determinaron los valores de IC50 mediante ajuste de un modelo de cuatro parámetros al porcentaje de datos de control.
El compuesto 1 presentaba un valor medio de IC50 de 1.9 nM al someterlo a ensayo en el ensayo de Ejemplo B. El compuesto 2 presentaba un valor medio de IC50 de 1.3 nM al someterlo a ensayo en el ensayo del Ejemplo B. El compuesto 3 presentaba un valor medio de IC50 de 6.1 nM al someterlo a ensayo en el ensayo del Ejemplo B.
Ejemplo C
Lavado de microsomas humanos y de hepatocitos humanos de los compuestos 1, 2 y 3
Incubaciones de microsomas hepáticos
Se preparó una solución tampón de ensayo de fosfato potásico (KPB) 100 mM de la manera siguiente: Se disolvió tanto KH2PO4 como K2HPO4 separadamente en agua de grado reactivo, resultando en concentraciones finales de 100 mM. Se preparó una mezcla 75:25 v/v de K2HPO4:KH2PO4 y se ajustó el pH de la solución a 7.4 utilizando soluciones diluidas de HCl o NaOH. Se prepararon soluciones madre de compuesto 1, compuesto 2 y compuesto 3 a una concentración de 10 mM (compuesto activo) en DMSO. La solución madre se diluyó inmediatamente antes de la utilización a 2.5 pM utilizando la solución de KPB para crear el estándar de trabajo. Todos los compuestos de ensayo eran completamente solubles en DMSO según la inspección visual a temperatura ambiente. Se preparó solución de regeneración de NADPH (NRS) el día del análisis mediante la dilución e un volumen de NADP+ 17
mg/ml con un volumen de glucosa-6-fosfato 78 mg/ml (ambos preparados en KPB, pH 7.4) y 7.9 volúmenes de MgCl2 20 mM. Las concentraciones finales de NADP+ y glucosa-6-fosfato eran de 1.7 mg/ml y 7.8 mg/ml, respectivamente. Inmediatamente antes de la utilización, se activó el NRS mediante la adición de 10 pl de glucosa-6- fosfato deshidrogenasa (150 unidades/ml en KPB, pH 7.4) por ml de solución madre de NRS. Se diluyeron microsomas hepáticos a una concentración de 2.5 mg de proteína/ml utilizando KPB.
Para los compuestos 1,2 y 3, y cada control positivo (es decir, dextrometorfano, diazepam, diltiazem, fenacetina, tolbutamida y verapamilo), se añadieron 20 pl de solución estándar de trabajo 2.5 pM de compuesto de ensayo y 20 pl de microsomas (2.5 mg de proteína/ml) a cada pocillo de una placa de polipropileno de 96 pocillos (Costar, VWR, West Chester, PA) por duplicado. Las placas se introdujeron en un incubador a 37°C durante 5 minutos antes de añadir la solución inicial. Se añadió una alícuota de 10 pl de la solución de NRS a cada pocillo original para iniciar el metabolismo. La concentración del compuesto de ensayo al inicio de la incubación era de 1 pM. Se preparó una placa de incubación para cada punto temporal (es decir, 0 y 20 minutos). Las incubaciones se llevaron a cabo a 37°C y con 100% de humedad relativa. En cada punto temporal, se sacó la placa de incubación apropiada del incubador y se añadió a cada pocillo una solución que contenía estándar interno (150 pl, 0,25 pM marcado en acetonitrilo al 60%). La placa se centrifugó inmediatamente en una centrífuga a 2.095 x g durante 7 minutos a temperatura ambiente utilizando una centrífuga de laboratorio Allegra (Beckman Coulter, Fullerton, CA). Se transfirió una alícuota de 200 pl del sobrenadante a cada pocillo de una placa poco profunda de 96 pocillos (Costar). Las placas se sellaron utilizando alfombrillas de placas reutilizables.
Incubaciones de hepatocitos
Se prepararon soluciones madre de compuesto 1, compuesto 2 y compuesto 3 a una concentración de 10 mM (compuesto activo) en DMSO. Se evaluaron las estabilidades in vitro de los compuestos 1, 2 y 3 (1 pM) en presencia de hepatocitos de la manera siguiente. Los hepatocitos crioconservados se descongelaron, se aislaron de los medios utilizados para el transporte y se diluyeron a una densidad de 1 x 106 células viables/ml, siguiendo las directrices del proveedor, utilizando medio de Eagle modificado por Dulbecco, IX, rico en glucosa (DMEM, Invitrogen, Carlsbad, CA). Se determinó la viabilidad mediante exclusión de azul tripán utilizando un hemocitómetro (3500 Hausser, VWR, West Chester, PA). Las soluciones madre 10 mM de los compuestos 1,2 y 3 se diluyeron a 2 pM utilizando DMEM complementado para crear el estándar de trabajo. Se añadió una alícuota de 20 pE de compuesto de ensayo o de control (es decir, antipirina, diazepam, diltiazem, lorazepam, propanolol, verapamilo y 7- etil-10-hidroxicampotetcina [SN-38]) a cada pocillo de ensayo de una placa de polipropileno de 96 pocillos (Costar, VWR, West Chester, PA) inmediatamente seguida de la adición de 20 pl de la suspensión de hepatocitos. Se preparó una placa de incubación para cada punto temporal (es decir, 0, 60 y 120 minutos), preparando las muestras por duplicado. Las incubaciones se llevaron a cabo a 37°C y con 100% de humedad relativa. En cada punto temporal, se sacó la placa de incubación apropiada del incubador y se añadió a cada pocillo una solución que contenía estándar interno (200 pl, labetalol 0.25 pM en acetonitrilo al 60%). La placa se mezcló a 700 rpm durante 1 minuto en un agitador de placas (agitador de microtitulación Digital IkA MtS 2/4, VWR) y se centrifugó inmediatamente en una centrífuga a 2,095 x g durante 10 minutos a temperatura ambiente utilizando una centrífuga de laboratorio Allegra (Beckman Coulter, Fullerton, CA). Se transfirió una alícuota de 200 pl del sobrenadante a cada pocillo de una placa poco profunda de 96 pocillos (Costar). Las placas se sellaron utilizando alfombrillas de placas reutilizables.
Cálculos
Todos los cálculos se llevaron a cabo utilizando BioAssay Enterprise. Las proporciones de superficies medias de los picos se calcularon promediando las proporciones de superficies de pico (n=2) de compuesto 1, compuesto 2 y compuesto 3, y el estándar interno para cada muestra. Se calculó el porcentaje remanente mediante la determinación de la proporción de la superficie de pico en cada punto temporal a la proporción de superficie de pico de las muestras de tiempo cero. Se determinó la tasa de pérdida de compuesto de ensayo (km) mediante regresión lineal de - ln(f(t)) frente al tiempo. La regresión utilizó la forma “y = mx”; por lo tanto, el modelo forzó un corte de 100% remanente y asumía que el metabolismo seguía una cinética de primer orden. Se determinó el t1/2 dividiendo ln(2) por km. El lavado intrínseco predicho (CLint) se calculó escalando la semivida in vitro para la estabilidad de cada uno de los compuestos 1, 2 y 3 utilizando los factores de escalado físicos y fisiológicos listados en la Tabla 5 y utilizados en la ecuación siguiente:

donde D es el número de hepatocitos por unidad de masa de hígado para una especie particular. W es la masa media de hígado presente por unidad de peso de animal, y C es el número de hepatocitos presente durante las incubaciones por unidad de volumen. El lavado hepático predicho (CLh) se calculó utilizando los factores de escalado físico y fisiológico listados en la Tabla 5 y utilizados en la ecuación a continuación:

donde Q es el flujo sanguíneo hepático dependiente de especie. No se realizó ningún ajuste para la fracción no unida del compuesto de ensayo (fu). Se determinó la proporción de extracción hepática predicha (ER) mediante el cálculo de la proporción entre el CLh predicho y el flujo sanguíneo hepático.
CLh
ER
Q
Tabla 5

Ejemplo D
Farmacocinética en ratas Sprague-Dawley macho
Se formuló cada uno de los compuestos 1, 2 y 3 mediante la adición de compuesto (seco) a Trappsol® al 20% (hidroxipropil-beta-ciclodextrina, CDT, Inc.) (aq.), pH 5.0, y se agitó con vórtex hasta conseguir una suspensión (PO) o solución (IV) homogénea.
Para la administración intravenosa, los animales fueron anestesiados con isoflurano al 3%, O2 equilibrado. Se administró 1 mg/kg de compuesto 1, compuesto 2 y compuesto 3 a un volumen de dosis de 1 ml/kg en la vena lateral de la cola. Se extrajo sangre de la vena lateral opuesta de la cola mientras el animal se encontraba bajo anestesia (isoflurano al 3% equilibrado con O2, 0.2 ml/punto temporal) en tubos con Na-EDTA (al 1.5% v/v) en los puntos temporales siguientes: 1, 5, 15, 30 minutos y 1,2, 4, 8 y 24 horas, y se mezclaron a fondo.
Para la administración oral, las ratas recibieron una única dosis oral tras ayuno durante la noche. Se extrajo sangre de la vena lateral de la cola mientras el animal se encontraba bajo anestesia de isoflurano al 3% equilibrado con O2 (0.2 ml/punto temporal) en tubos con Na-EDTA (al 1.5% v/v) en los puntos temporales siguientes: 5, 15 y 30 minutos y 1, 2, 4, 8, 24 y 26 horas, y se mezclaron a fondo. Tras la recolección de sangre de las 24 horas, los animales recibieron nuevamente dosis orales y se recogió plasma y cerebro 2 horas después de la dosis para el análisis de compuestos. Los cerebros se perfundieron con solución salina antes de la extracción, se pesaron y se congelaron instantáneamente en nitrógeno líquido hasta el análisis mediante CLM/EM. Se separó el plasma mediante centrifugación a 14.000 rpm durante 10 minutos y se transfirieron las muestras de plasma a tubos de placa de 96 pocillos con tapones de goma y se almacenaron a 20 ± 5 °C hasta el análisis.
Análisis de plasma
Se preparó una única curva de calibración de 12 puntos en primer lugar diluyendo en serie (3 veces) una solución madre 40 pg/ml de compuesto 1, compuesto 2 y compuesto 3 en DMSO, proporcionando soluciones estándares. A continuación, se añadió plasma (20 pl) a la placa de extracción y se añadieron 2.5 pl de cada solución estándar al plasma no expuesto. A continuación, se añadió una solución madre de un estándar interno (10.0 pl de 2.5 pg/ml en acetonitrilo) a cada estándar y solución de muestra. El material proteico se precipitó a partir de 20 pl de plasma con la adición de 317.5 pl de acetonitrilo para un volumen total de 350 pl. Las muestras se agitaron con vórtex durante 5 minutos y se centrifugaron en una centrífuga Allegra X-12R (Beckman Coulter, Fullerton, CA) durante 15 minutos a aproximadamente 1,500 x g a 4°C. Se transfirió una alícuota de 100 pl e cada sobrenadante mediante una pipeteadora Personal de 550 pl (Apricot Designs, Monrovia, CA) a placas de 96 pocillos y se diluyó 1:1 con agua de grado HPLC. Las placas resultantes se sellaron con alfombrillas de placas y se analizó la cantidad de compuesto en el plasma mediante CL-EM/EM.
El sistema de CL-EM/EM comprendía un automuestreador HTC-PAL (Leap Technologies, Inc., Carrboro, NC), una HPLC HP1100 (Agilent Technologies Inc., Santa Clara, CA) y un espectrómetro de masas de triple cuadrupolo
API4000 (Applied Biosystems, Foster City, CA). La retención cromatografía del analito y el estándar interno se consiguió utilizando una columna Phenomenex® Synergi 4|j Hydro-RP 80A (2.1 x 30 mm, tamaño de partícula: 4 |jm; Phenomenex, Torrance, CA) junto con condiciones de gradiente utilizando las fases móviles A (acetato amónico 10 mM en ácido fórmico acuoso al 0.1% y alcohol isopropílico al 1%) y B (acetato amónico 10 mM en 89.9% de acetonitrilo, 10% de metanol y 0.1% de ácido fórmico). El tiempo de migración total, incluyendo el tiempo de reequilibrado, para una única inyección fue de 3.5 minutos y el caudal, de 0.8 ml/minuto. El solvente B se incrementó de 5% a 95% en 1.0 minuto, se mantuvo a 95% durante 1.0 minuto y después se redujo a 5% en 0.1 minutos.

Análisis de cerebro
Se preparó una única curva de calibración de 12 puntos en primer lugar diluyendo en serie (3 veces) soluciones madre de 40 jg/m l de compuesto 1, compuesto 2 y compuesto 3 en DMSO. Se preparó el homogeneizado de cerebro mediante la adición de 5,0 ml de agua a tubos cónicos de 50 ml que contenían los cerebros. El contenido de los tubos se homogeneizó utilizando un homogeneizador multimuestra Omni-Prep (Kennesaw, GA) a 22,000 rpmi durante 180 segundos. Se añadió el homogeneizado (100 j l) a la placa de extracción y se añadieron 2.5 j l de cada solución estándar al homogeneizado cerebral no expuesto. A continuación, se añadió una solución madre de un estándar interno (10.0 j l de 2.5 jg/m l en acetonitrilo) a cada estándar y solución de muestra. El material proteico se precipitó a partir de 100 j l de homogeneizado tumoral de ratón con la adición de 237.5 j l de acetonitrilo para un volumen total de 350 jl. Las muestras se agitaron con vórtex durante 5 minutos y se centrifugaron en una centrífuga Allegra X-12R (Beckman Coulter, Fullerton, CA) durante 15 minutos a aproximadamente 1,500 x g a 4°C. Se transfirió una alícuota de 100 j l e cada sobrenadante mediante una pipeteadora Personal de 550 j l (Apricot Designs, Monrovia, CA) a placas de 96 pocillos y se diluyó 1:1 con agua de grado HPLC. Las placas resultantes se sellaron con alfombrillas de placas y se analizó la cantidad de compuesto en el cerebro mediante CL-EM/EM.
El sistema de CL-EM/EM comprendía un automuestreador HTC-PAL (Leap Technologies, Inc., Carrboro, NC), una HPLC HP1100 (Agilent Technologies Inc., Santa Clara, CA) y un espectrómetro de masas de triple cuadrupolo API4000 (Applied Biosystems, Foster City, CA). La retención cromatográfica del analito y el estándar interno se consiguió utilizando una columna Phenomenex® Synergi 4 j Hydro-RP 80A (2.1 x 30 mm, tamaño de partícula: 4 jm ; Phenomenex, Torrance, CA) junto con condiciones de gradiente utilizando las fases móviles A (acetato amónico 10 mM en ácido fórmico acuoso al 0.1% y alcohol isopropílico al 1%) y B (acetato amónico 10 mM en 89.9% de acetonitrilo, 10% de metanol y 0.1% de ácido fórmico).

El tiempo de migración total, incluyendo el tiempo de reequilibrado, para una única inyección fue de 3.5 minutos y el caudal, de 0.8 ml/minuto. El solvente B se incrementó de 5% a 95% en 1.0 minuto, se mantuvo a 95% durante 1.0 minuto y después se redujo a 5% en 0,1 minutos. La detección mediante espectrometría de masas de los analitos se llevó a cabo utilizando el modo de ionización ESI+. Se optimizó la corriente iónica durante la infusión de una solución madre de compuesto 1, compuesto 2 y compuesto 3. Se midieron las respuestas de los analitos mediante la monitorización de múltiples reacciones (MRM) de las transiciones únicas a cada compuesto. Se calcularon los parámetros farmacocinéticos mediante el modelo no compartimentado establecido utilizando un software propietario del propio laboratorio (Sherlock, versión 2.1). Se calcularon las AUC utilizando integración trapezoidal lineal.
Ejemplo E
Farmacocinética en perros
Se preparó una formulación de dosis IV para la administración de una dosis de 1 mg/kg en un volumen de dosis de 1 ml/kg de la manera siguiente. El vehículo para la formulación era Trappsol® al 20% (hidroxipropil-betaciclodextrina, CDT, Inc.) (aq.) en agua. Se añadió un volumen final del vehículo a cada uno de compuesto 1, compuesto 2 y compuesto 3 (en forma de compuestos secos) y la mezcla se agitó hasta obtener una solución homogénea. El pH final de la solución final era de 7. La solución se filtró utilizando un filtro de 0.2 pm antes de la administración en el animal.
Perros Beagle macho recibieron una única dosis oral tras ayuno durante la noche. Se recogió sangre/plasma (1.5 ml/punto temporal) en tubos de recolección de sangre con K2EDTA en los puntos temporales posdosis siguientes: 0, 1, 5, 15 y 30 minutos y 1, 2, 4, 8, 12, 24 y 48 horas, y se mezclaron a fondo. Se recogieron muestras de sangre de las venas yugular, cefálica o sáfena de animales conscientes. Las muestras de sangre se mantuvieron sobre hielo hasta el procesamiento para plasma. Las muestras de sangre se centrifugaron a 3,200 rpm durante 10 minutos a aproximadamente 5°C. Las muestras de plasma se transfirieron directamente a tubos de placas de 96 pocillos (MatrixTech, 0.75 ml). Se colocaron tapas inyectables de goma en los tubos de placas de 96 pocillos. Las muestras de plasma se almacenaron a -20 ± 5°C hasta el análisis.
Análisis de plasma
Se preparó una única curva de calibración de 12 puntos en primer lugar diluyendo en serie (3 veces) una solución madre 40 pg/ml de compuesto 1, compuesto 2 y compuesto 3 en DMSO, proporcionando soluciones estándares. A continuación, se añadió plasma (20 pl) a la placa de extracción y se añadieron 2.5 pl de cada solución estándar al plasma no expuesto. A continuación, se añadió una solución madre de un estándar interno (10.0 pl de 2.5 pg/ml en acetonitrilo) a cada estándar y solución de muestra. El material proteico se precipitó a partir de 20 pl de plasma con la adición de 317.5 pl de acetonitrilo para un volumen total de 350 pl. Las muestras se agitaron con vórtex durante 5 minutos y se centrifugaron en una centrífuga Allegra X-12R (Beckman Coulter, Fullerton, CA) durante 15 minutos a aproximadamente 1,500 x g a 4°C. Se transfirió una alícuota de 100 pl e cada sobrenadante mediante una pipeteadora Personal de 550 pl (Apricot Designs, Monrovia, CA) a placas de 96 pocillos y se diluyó 1:1 con agua de grado HPLC. Las placas resultantes se sellaron con alfombrillas de placas y se analizó la cantidad de compuesto en el plasma mediante CL-EM/EM.
El sistema de CL-EM/EM comprendía un automuestreador HTC-PAL (Leap Technologies, Inc., Carrboro, NC), una HPLC HP1100 (Agilent Technologies Inc., Santa Clara, CA) y un espectrómetro de masas de triple cuadrupolo API4000 (Applied Biosystems, Foster City, CA). La retención cromatográfica del analito y el estándar interno se consiguió utilizando una columna Phenomenex® Synergi 4p Hydro-RP 80A (2.1 x 30 mm, tamaño de partícula: 4 pm; Phenomenex, Torrance, CA) junto con condiciones de gradiente utilizando las fases móviles A (acetato amónico 10 mM en ácido fórmico acuoso al 0.1% y alcohol isopropílico al 1%) y B (acetato amónico 10 mM en 89.9% de acetonitrilo, 10% de metanol y 0.1% de ácido fórmico). El tiempo de migración total, incluyendo el tiempo de reequilibrado, para una única inyección fue de 3,5 minutos y el caudal, de 0.8 ml/minuto. El solvente B se incrementó de 5% a 95% en 1.0 minuto, se mantuvo a 95% durante 1.0 minuto y después se redujo a 5% en 0.1 minutos.

La detección mediante espectrometría de masas de los analitos se llevó a cabo utilizando el modo de ionización ESI+. Se optimizó la corriente iónica durante la infusión de una solución madre de compuesto 1, compuesto 2 y compuesto 3. Se midieron las respuestas de los analitos mediante la monitorización de múltiples reacciones (MRM) de las transiciones únicas a cada compuesto. Se calcularon los parámetros farmacocinéticos mediante el modelo no compartimentado establecido utilizando un software propietario del propio laboratorio (Sherlock, versión 2.1). Se calcularon las AUC utilizando integración trapezoidal lineal.
Ejemplo F
Ensayo de hERG FASTPatch®
Se utilizó este ensayo para medir los efectos del compuesto 1, el compuesto 2 y el compuesto 3 sobre canales de potasio de hERG clonados expresados en células renales embrionarias humanas (HEK293). Se evaluaron los efectos in vitro del compuesto 1, el compuesto 2 y el compuesto 3 sobre la corriente de los canales de potasio de hERG (gen Ether-a-go-go humano) (una variable sustitutiva de IKr, el componente de activación rápida de la corriente rectificadora retardada de potasio cardiaco) expresada en células de mamífero, a temperatura ambiente utilizando un QPatch HT® (Sophion Bioscience A/S, Dinamarca), un sistema automático paralelo de pinza de parche. Se evaluó el artículo de ensayo a 0.3, 1, 3, 10, 30 y 100 pM, sometiendo a ensayo cada concentración en
tres o más células (n > 3). La duración de la exposición a cada concentración de artículo de ensayo era de 3 minutos. Un control positivo confirmó la sensibilidad del sistema de ensayo a la inhibición de hERG.
Ejemplo G
Ensayo de unión de cinasa p38
Se determinó la actividad de unión de p38a en un ensayo de unión a Eu de cinasa LanthaScreen™ de p38a. Se incubaron 5 nM de p38a humano recombinante etiquetado con GST inactivo (dominio citoplasmático etiquetado con GST de Invitrogen, n° de catálogo PV3305) con trazador 199 Alexa-Fluor® 5 nM (Invitrogen, n° de catálogo PV5830) y anticuerpo anti-GST marcado con europio 2 nM (Invitrogen, n° de cat. PV5594), en tampón ([Na+] HEPES 25 mM, pH 7.3, MgCh 10 mM, TNaVO4l0o |jM). Se añadieron diluciones en serie de tres veces del compuesto 1 en DMSO a un porcentaje final de 2% de DMSO. Tras 60 minutos de incubación a 22°C, se midió la reacción utilizando el lector de placas multimodo EnVision (PerkinElmer) mediante detección dual de longitudes de onda TR-FRET a 615 nM y 665 nM. Se calculó el porcentaje de control utilizando un factor de emisión ratiométrico. Se determinó el valor de IC50 mediante el ajuste de un modelo de cuatro parámetros al porcentaje de datos de control. Se encontró que el compuesto 1 era 1000 más potente contra TrkA que p38a.
Ejemplo H
Perfilado de cinasas fuera de diana
Se sometió a ensayo el compuesto 1 para actividad de cinasa fuera de diana a una concentración de 10 j M en Millipore, Inc., en su servicio de KinaseProfiler™ contra todas las cinasas disponibles en su panel completo de cinasas. El compuesto 1 se sometió a ensayo por duplicado a una concentración de ATP próxima a la Km de cada cinasa individual según las especificaciones de Millipore. Se muestran los resultados en la tabla, a continuación. Los datos se expresan en porcentaje del control (POC) y son medias de dos réplicas.
En el KinaseProfiler™ el compuesto 1 mostró una notable e inesperada selectividad para la inhibición de TrkA frente a otras cinasas en el panel. De hecho, el compuesto 1 era en gran parte inactivo contra cinasas fueras de diana a una concentración de 10 j M, de esta manera, no se esperaría que inhibiese las cinasas fuera de diana a dosis terapéuticas en mamíferos. La capacidad del compuesto 1 de inhibir selectivamente la ruta de Trk sin inhibir otras cinasas fuera de diana se traduciría en un perfil farmacológico esencialmente libre de efectos secundarios relacionados con la inhibición de las cinasas fuera de diana. Dicho perfil farmacológico representaría un enfoque más seguro en el tratamiento del dolor, la inflamación, el cáncer y determinadas enfermedades de la piel de lo informado anteriormente.





Claims (28)
1. Compuesto
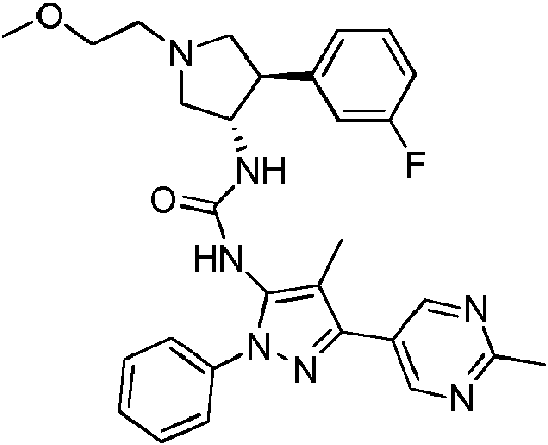
1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea, o una sal farmacéuticamente aceptable del mismo.
2. Compuesto según la reivindicación 1, en el que el compuesto es una sal dihidrocloruro de 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5-il)urea.
3. Composición farmacéutica, que comprende un compuesto según la reivindicación 1 o 2, y un diluyente o portador farmacéuticamente aceptable.
4. Composición farmacéutica según la reivindicación 3, en la que dicha composición farmacéutica se formula para la administración oral, preferentemente dicha composición farmacéutica se encuentra en la forma de un comprimido o una cápsula.
5. Compuesto según la reivindicación 1 o 2 para la utilización en terapia.
6. Compuesto según la reivindicación 1 o 2 para la utilización en el tratamiento de una enfermedad o un trastorno seleccionada/o de entre el grupo que consiste en dolor, cáncer, inflamación o enfermedades inflamatorias, enfermedades neurodegenerativas, infección por Trypanosoma cruzi, síndrome de Sjogren, endometriosis, neuropatía periférica diabética, prostatitis, síndrome de dolor pélvico, enfermedades relacionadas con un desequilibrio de la regulación del remodelado óseo, y enfermedades que resultan de la señalización aberrante del factor de crecimiento del tejido conectivo.
7. Compuesto para la utilización según la reivindicación 6, en el que la enfermedad o el trastorno tratada/o es el dolor.
8. Compuesto para la utilización según la reivindicación 7, en el que dicho dolor es el dolor crónico, preferentemente el dolor de espalda crónico.
9. Compuesto para la utilización según la reivindicación 7, en el que dicho dolor es el dolor agudo.
10. Compuesto para la utilización según la reivindicación 7, en el que dicho dolor se selecciona de entre el grupo que consiste en dolor neuropático, dolor inflamatorio, dolor asociado al cáncer, dolor asociado a fractura ósea y dolor asociado a cirugía.
11. Compuesto para la utilización según la reivindicación 10, en el que dicho dolor neuropático es un dolor asociado a una neuropatía periférica diabética.
12. Compuesto para la utilización según cualquiera de las reivindicaciones 7 a 11, en combinación con una cantidad eficaz de por lo menos un agente terapéutico adicional seleccionado de entre el grupo que consiste en compuestos antiinflamatorios, esteroides, analgésicos, opioides, antagonistas de receptor de péptido relacionado con el gen de la calcitonina, moduladores de canales iónicos selectivos de subtipo, anticonvulsivos, inhibidores de la recaptación de serotonina-norepinefrina duales, inhibidores de cinasa de la familia JAK y antidepresivos tricíclicos.
13. Compuesto para la utilización según la reivindicación 12, en el que el analgésico es un fármaco antiinflamatorio no esteroideo (AlNE).
14. Compuesto para la utilización según la reivindicación 6, en el que dicha enfermedad es una enfermedad inflamatoria, preferentemente seleccionada de entre el grupo que consiste en enfermedades pulmonares inflamatorias, cistitis intersticial, síndrome de vejiga dolorosa, enfermedades intestinales inflamatorias y enfermedades de la piel inflamatorias.
15. Compuesto para la utilización según la reivindicación 14, en combinación con una cantidad eficaz de uno o más agentes adicionales seleccionados de entre el grupo que consiste en agentes anti-TNF, fármacos antifolato y antimetabolito e inhibidores de cinasa dirigidos.
16. Compuesto para la utilización según la reivindicación 15, en combinación con una cantidad eficaz de uno o más agentes adicionales seleccionados de entre el grupo que consiste en infliximab, adalimumab, certolizumab pegol, golimumab, etanercept, metotrexato, ruxolitinib, tofacitinib, CYT387, lestaurtinib, pacritinib y TG101348.
17. Compuesto para la utilización según la reivindicación 6, en el que la enfermedad es una enfermedad que resulta de la señalización aberrante del factor de crecimiento del tejido conectivo.
18. Compuesto para la utilización según la reivindicación 17, en el que la enfermedad se selecciona de entre el grupo que consiste en síndrome de Raynaud, fibrosis pulmonar idiopática, cicatrización (hipertrófica, queloide y otras), cirrosis, fibrosis endomiocárdica, fibrosis auricular, mielofibrosis, fibrosis masiva progresiva (pulmonar), fibrosis sistémica nefrógena, escleroderma, esclerosis sistémica, artrofibrosis y fibrosis ocular.
19. Compuesto para la utilización según la reivindicación 6, en el que dicha enfermedad es el cáncer.
20. Compuesto para la utilización según la reivindicación 19, en el que dicho cáncer es un cáncer que presenta una desregulación de TrkA.
21. Compuesto para la utilización según la reivindicación 20, en el que la desregulación de TrkA comprende una o más traslocaciones o inversiones cromosómicas que dan como resultado unas fusiones de gen de TrkA.
22. Compuesto para la utilización según la reivindicación 21, en el que la fusión de gen de TrkA es LMNA-TrkA, TFG-TrkA, TPM3-TrkA, CD74-TrkA, NFASC-TrkA, MPRIP-TrkA, BCAN-TrkA, TP53-TrkA, RNF213-TrkA, RABGAP1L-TrkA, IRF2BP2-TrkA, SQSTM1-TrkA, SSBP2-TrkA, o TPR-TrkA.
23. Compuesto para la utilización según la reivindicación 22, en el que dicho cáncer es cáncer de pulmón no microcítico, carcinoma papilar tiroideo, glioblastoma multiforme, carcinoma colorrectal, melanoma, colangiocarcinoma o sarcoma.
24. Compuesto para la utilización según la reivindicación 20, en el que la desregulación de TrkA comprende una o más eliminaciones, inserciones o mutaciones en la proteína TrkA, siendo dicho cáncer preferentemente leucemia mieloide aguda, carcinoma neuroendocrino de células grandes o neuroblastoma.
25. Compuesto para la utilización según la reivindicación 20, en el que la desregulación de TrkA es la sobreexpresión de TrkA de tipo salvaje (activación autocrina), siendo dicho cáncer preferentemente carcinoma de próstata, neuroblastoma, carcinoma pancreático, melanoma, carcinoma de células escamosas de cabeza y cuello o carcinoma gástrico.
26. Compuesto para la utilización según cualquiera de las reivindicaciones 19 a 25, en combinación con una cantidad eficaz de por lo menos un agente terapéutico adicional seleccionado de entre uno o más terapias o agentes quimioterápicos adicionales, seleccionándose preferentemente dicha/o terapia o agente quimioterápico adicional de entre radioterapia, quimioterápicos citotóxicos, tratamiento dirigido a tirosina cinasa, moduladores de la apoptosis, inhibidores de la transducción de señales, terapias inmunodirigidas y terapias dirigidas a la angiogénesis.
27. Compuesto para la utilización según la reivindicación 26, en el que dicho agente quimioterápico se selecciona de entre tratamiento dirigido a tirosina cinasa.
28. Procedimiento para la preparación de un compuesto según la reivindicación 1 o 2, que comprende:
(a) hacer reaccionar un compuesto que presenta la fórmula II-A

con un compuesto que presenta la fórmula III
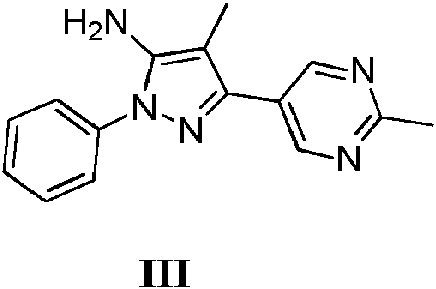
en presencia de carbonildiimidazol o trifosgeno y una base; o
(b) hacer reaccionar un compuesto que presenta la fórmula II-A

con un compuesto que presenta la fórmula IV

en la que L1 es un grupo saliente, en presencia de una base; o
(c) hacer reaccionar un compuesto que presenta la fórmula V

en la que L2 es un grupo saliente, con un compuesto que presenta la fórmula III

en presencia de una base; o
(d) hacer reaccionar un compuesto que presenta la fórmula VI
con azida de difenilfosforilo para formar un producto intermedio, seguido de la reacción del producto intermedio con un compuesto que presenta la fórmula III

en presencia de una base; o
(e) hacer reaccionar un compuesto que presenta la fórmula II-A

con un compuesto que presenta la fórmula VII

en presencia de una base; y
eliminar los grupos protectores si están presentes y opcionalmente preparar su sal farmacéuticamente aceptable.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461993426P | 2014-05-15 | 2014-05-15 | |
| PCT/US2015/030795 WO2015175788A1 (en) | 2014-05-15 | 2015-05-14 | 1-((3s,4r)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1h-pyrazol-5-yl)urea as a trka kinase inhibitor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2746530T3 true ES2746530T3 (es) | 2020-03-06 |
Family
ID=53276291
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15726455T Active ES2746530T3 (es) | 2014-05-15 | 2015-05-14 | 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5- il)urea como un inhibidor de TrkA cinasa |
Country Status (30)
| Country | Link |
|---|---|
| US (1) | US10835533B2 (es) |
| EP (2) | EP3154959B1 (es) |
| JP (2) | JP6499672B2 (es) |
| KR (2) | KR101998461B1 (es) |
| CN (2) | CN109970614B (es) |
| AU (2) | AU2015259075B2 (es) |
| CA (1) | CA2949160C (es) |
| CL (1) | CL2016002900A1 (es) |
| CR (1) | CR20160583A (es) |
| CY (1) | CY1122075T1 (es) |
| DK (1) | DK3154959T3 (es) |
| ES (1) | ES2746530T3 (es) |
| HR (1) | HRP20191593T1 (es) |
| HU (1) | HUE045340T2 (es) |
| IL (1) | IL248924B (es) |
| LT (1) | LT3154959T (es) |
| MA (1) | MA40434B1 (es) |
| MX (2) | MX2016014945A (es) |
| MY (1) | MY191956A (es) |
| NZ (1) | NZ727418A (es) |
| PH (2) | PH12016502242A1 (es) |
| PL (1) | PL3154959T3 (es) |
| PT (1) | PT3154959T (es) |
| RS (1) | RS59286B1 (es) |
| RU (1) | RU2719489C2 (es) |
| SG (2) | SG11201609550PA (es) |
| SI (1) | SI3154959T1 (es) |
| SM (1) | SMT201900522T1 (es) |
| UA (1) | UA122126C2 (es) |
| WO (1) | WO2015175788A1 (es) |
Families Citing this family (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010033941A1 (en) | 2008-09-22 | 2010-03-25 | Array Biopharma Inc. | Substituted imidazo[1,2b]pyridazine compounds as trk kinase inhibitors |
| MY169791A (en) | 2008-10-22 | 2019-05-15 | Array Biopharma Inc | Substituted pyrazolo [1,5-a] pyrimidine compounds as trk kinase inhibitors |
| AR077468A1 (es) | 2009-07-09 | 2011-08-31 | Array Biopharma Inc | Compuestos de pirazolo (1,5 -a) pirimidina sustituidos como inhibidores de trk- quinasa |
| ME03376B (me) | 2010-05-20 | 2020-01-20 | Array Biopharma Inc | Makrociklička jedinjenja kао inhibiтori trk kinaze |
| ES2752733T3 (es) | 2013-11-28 | 2020-04-06 | Kyorin Seiyaku Kk | Derivados de urea deuterados o marcados isotópicamente o sales farmacológicamente aceptables de los mismos útiles como agonistas de FPRL-1 |
| TWI672141B (zh) | 2014-02-20 | 2019-09-21 | 美商醫科泰生技 | 投予ros1突變癌細胞之分子 |
| MA40434B1 (fr) | 2014-05-15 | 2019-09-30 | Array Biopharma Inc | 1-((3s,4r)-4-(3-fluorophényl)-1-(2-méthoxyéthyl)pyrrolidin-3-yl)-3-(4-méthyl-3-(2-méthylpyrimidin-5-yl)-1-phényl-1h-pyrazol-5-yl)urée comme inhibiteur de la kinase trka |
| WO2016021629A1 (ja) | 2014-08-06 | 2016-02-11 | 塩野義製薬株式会社 | TrkA阻害活性を有する複素環および炭素環誘導体 |
| CN113354649B (zh) | 2014-11-16 | 2024-12-10 | 阵列生物制药公司 | 一种新的晶型 |
| HK1253093A1 (zh) | 2015-06-01 | 2019-06-06 | Loxo Oncology Inc. | 诊断和治疗癌症的方法 |
| JP6812059B2 (ja) * | 2015-07-07 | 2021-01-13 | 塩野義製薬株式会社 | TrkA阻害活性を有する複素環誘導体 |
| BR112018000808A2 (pt) | 2015-07-16 | 2018-09-04 | Array Biopharma Inc | compostos de pirazolo[1,5-a]piridina substituída como inibidores de ret cinase |
| EP3368039A1 (en) | 2015-10-26 | 2018-09-05 | The Regents of The University of Colorado, A Body Corporate | Point mutations in trk inhibitor-resistant cancer and methods relating to the same |
| AU2016370846B2 (en) * | 2015-12-18 | 2022-08-25 | Ignyta, Inc. | Combinations for the treatment of cancer |
| WO2017135399A1 (ja) | 2016-02-04 | 2017-08-10 | 塩野義製薬株式会社 | TrkA阻害活性を有する含窒素複素環および炭素環誘導体 |
| US10045991B2 (en) | 2016-04-04 | 2018-08-14 | Loxo Oncology, Inc. | Methods of treating pediatric cancers |
| FI3439662T3 (fi) | 2016-04-04 | 2024-09-04 | Loxo Oncology Inc | (s)-n-(5-((r)-2-(2,5-difluorifenyyli)pyrrolidin-1-yyli)pyratsolo[1,5-a]pyrimidin-3-yyli)-3-hydroksipyrrolidiini-1-karboksamidin nesteformulaatioita |
| AU2017246547B2 (en) | 2016-04-04 | 2023-02-23 | Loxo Oncology, Inc. | Methods of treating pediatric cancers |
| EP3458456B1 (en) | 2016-05-18 | 2020-11-25 | Loxo Oncology Inc. | Preparation of (s)-n-(5-((r)-2-(2,5-difluorophenyl)pyrrolidin-1-yl)pyrazolo[1,5-a]pyrimidin-3-yl)-3-hydroxypyrrolidine-1-carboxamide |
| TWI752098B (zh) | 2016-10-10 | 2022-01-11 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡唑并[1,5-a]吡啶化合物 |
| TWI704148B (zh) | 2016-10-10 | 2020-09-11 | 美商亞雷生物製藥股份有限公司 | 作為ret激酶抑制劑之經取代吡唑并[1,5-a]吡啶化合物 |
| JOP20190092A1 (ar) | 2016-10-26 | 2019-04-25 | Array Biopharma Inc | عملية لتحضير مركبات بيرازولو[1، 5-a]بيريميدين وأملاح منها |
| WO2018136663A1 (en) | 2017-01-18 | 2018-07-26 | Array Biopharma, Inc. | Ret inhibitors |
| WO2018136661A1 (en) | 2017-01-18 | 2018-07-26 | Andrews Steven W | SUBSTITUTED PYRAZOLO[1,5-a]PYRAZINE COMPOUNDS AS RET KINASE INHIBITORS |
| JOP20190213A1 (ar) | 2017-03-16 | 2019-09-16 | Array Biopharma Inc | مركبات حلقية ضخمة كمثبطات لكيناز ros1 |
| ES2992887T3 (en) | 2017-04-27 | 2024-12-19 | Mochida Pharm Co Ltd | Novel tetrahydronaphthyl urea derivatives as inhibitors of tropomyosin receptor kinase a for the treatment of pain |
| CN107245073B (zh) * | 2017-07-11 | 2020-04-17 | 中国药科大学 | 4-(芳杂环取代)氨基-1h-3-吡唑甲酰胺类flt3抑制剂及其用途 |
| KR102718538B1 (ko) | 2017-07-19 | 2024-10-21 | 이그니타, 인코포레이티드 | 엔트렉티닙을 포함하는 약학적 조성물 |
| JP2019026646A (ja) * | 2017-08-03 | 2019-02-21 | 塩野義製薬株式会社 | 含窒素複素環および炭素環誘導体を含有する、疼痛の治療および/または予防用医薬組成物 |
| FI3661935T3 (fi) * | 2017-08-11 | 2023-01-13 | Kinaasin estäjinä käyttökelpoiset pyratsolipyrimidiinit | |
| JP7134983B2 (ja) | 2017-09-15 | 2022-09-12 | 持田製薬株式会社 | テトラヒドロナフチルウレア誘導体の結晶 |
| TWI876442B (zh) | 2017-10-10 | 2025-03-11 | 美商絡速藥業公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之調配物 |
| TWI791053B (zh) | 2017-10-10 | 2023-02-01 | 美商亞雷生物製藥股份有限公司 | 6-(2-羥基-2-甲基丙氧基)-4-(6-(6-((6-甲氧基吡啶-3-基)甲基)-3,6-二氮雜雙環[3.1.1]庚-3-基)吡啶-3-基)吡唑并[1,5-a]吡啶-3-甲腈之結晶形式及其醫藥組合物 |
| CN111225662B (zh) | 2017-10-17 | 2022-11-22 | 伊尼塔公司 | 药物组合物和剂型 |
| WO2019084285A1 (en) | 2017-10-26 | 2019-05-02 | Qian Zhao | FORMULATIONS OF A MACROCYCLIC TRK KINASE INHIBITOR |
| JP7060694B2 (ja) | 2018-01-18 | 2022-04-26 | アレイ バイオファーマ インコーポレイテッド | Retキナーゼ阻害剤としての置換ピロロ[2,3-d]ピリミジン化合物 |
| EP3740490A1 (en) | 2018-01-18 | 2020-11-25 | Array Biopharma, Inc. | Substituted pyrazolo[3,4-d]pyrimidine compounds as ret kinase inhibitors |
| US11472802B2 (en) | 2018-01-18 | 2022-10-18 | Array Biopharma Inc. | Substituted pyrazolyl[4,3-c]pyridine compounds as RET kinase inhibitors |
| US10676431B2 (en) | 2018-03-05 | 2020-06-09 | Bristol-Myers Squibb Company | Phenylpyrrolidinone formyl peptide 2 receptor agonists |
| WO2019191659A1 (en) | 2018-03-29 | 2019-10-03 | Loxo Oncology, Inc. | Treatment of trk-associated cancers |
| CN108794484B (zh) * | 2018-04-28 | 2020-04-24 | 北京施安泰医药技术开发有限公司 | [1,2,4]三唑并[4,3-a]吡嗪衍生物、其药物组合物、制备方法和应用 |
| US11932652B2 (en) | 2018-07-12 | 2024-03-19 | Zhangzhou Pien Tze Huang Pharmaceutical Co., Ltd. | Pyrrolidinyl urea derivatives and application thereof in TrkA-related diseases |
| CN117285467A (zh) | 2018-07-31 | 2023-12-26 | 罗索肿瘤学公司 | 喷雾干燥的分散体和制剂 |
| TWI714231B (zh) * | 2018-09-04 | 2020-12-21 | 美商美國禮來大藥廠 | 2,6-二胺基吡啶化合物 |
| CN112996794A (zh) | 2018-09-10 | 2021-06-18 | 阿雷生物药品公司 | 作为ret激酶抑制剂的稠合杂环化合物 |
| WO2020083377A1 (zh) * | 2018-10-26 | 2020-04-30 | 南京明德新药研发有限公司 | 吡咯烷基脲衍生物及其应用 |
| US12351571B2 (en) | 2018-12-19 | 2025-07-08 | Array Biopharma Inc. | Substituted quinoxaline compounds as inhibitors of FGFR tyrosine kinases |
| EP3898626A1 (en) | 2018-12-19 | 2021-10-27 | Array Biopharma, Inc. | Substituted pyrazolo[1,5-a]pyridine compounds as inhibitors of fgfr tyrosine kinases |
| EA202192575A1 (ru) | 2019-03-21 | 2022-01-14 | Онксео | Соединения dbait в сочетании с ингибиторами киназ для лечения рака |
| CN114761006A (zh) | 2019-11-08 | 2022-07-15 | Inserm(法国国家健康医学研究院) | 对激酶抑制剂产生耐药性的癌症的治疗方法 |
| SG11202109079YA (en) | 2019-12-05 | 2021-09-29 | Illumina Inc | Rapid detection of gene fusions |
| US12441707B2 (en) | 2019-12-30 | 2025-10-14 | Tyra Biosciences, Inc. | Indazole compounds |
| JP7337279B2 (ja) | 2020-01-10 | 2023-09-01 | ▲ザン▼州片仔▲ファン▼薬業股▲フン▼有限公司 | ピロリジニル尿素誘導体の調製方法 |
| JP7289017B2 (ja) * | 2020-01-10 | 2023-06-08 | ▲ザン▼州片仔▲ファン▼薬業股▲フン▼有限公司 | ピロリジニルウレア誘導体の結晶及びその使用 |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2022218289A1 (en) * | 2021-04-12 | 2022-10-20 | Cullgen (Shanghai), Inc. | Tropomyosin receptor kinase (trk) degradation compounds and methods of use |
Family Cites Families (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU679887B2 (en) | 1995-09-11 | 1997-07-10 | Nihon Nohyaku Co., Ltd. | Azole derivatives, their use, production and usage |
| CA2206201A1 (en) | 1996-05-29 | 1997-11-29 | Yoshiaki Isobe | Pyrazole derivatives and their pharmaceutical use |
| US6083986A (en) | 1996-07-26 | 2000-07-04 | Icagen, Inc. | Potassium channel inhibitors |
| US5998424A (en) | 1997-06-19 | 1999-12-07 | Dupont Pharmaceuticals Company | Inhibitors of factor Xa with a neutral P1 specificity group |
| EP1028953A1 (en) | 1997-11-03 | 2000-08-23 | Boehringer Ingelheim Pharmaceuticals Inc. | Aromatic heterocyclic compounds as anti-inflammatory agents |
| US7329670B1 (en) | 1997-12-22 | 2008-02-12 | Bayer Pharmaceuticals Corporation | Inhibition of RAF kinase using aryl and heteroaryl substituted heterocyclic ureas |
| IL136768A0 (en) | 1997-12-22 | 2001-06-14 | Bayer Ag | INHIBITION OF p38 KINASE ACTIVITY USING ARYL AND HETEROARYL SUBSTITUTED HETEROCYCLIC UREAS |
| ATE529109T1 (de) | 1997-12-22 | 2011-11-15 | Bayer Healthcare Llc | Hemmung der p38 kinase aktivität durch substituierte heterocyclische harnstoffe |
| US6197798B1 (en) | 1998-07-21 | 2001-03-06 | Novartis Ag | Amino-benzocycloalkane derivatives |
| KR100711538B1 (ko) | 1998-12-25 | 2007-04-27 | 아스카 세이야쿠 가부시키가이샤 | 아미노피라졸 유도체 |
| UA73492C2 (en) | 1999-01-19 | 2005-08-15 | Aromatic heterocyclic compounds as antiinflammatory agents | |
| US6387900B1 (en) | 1999-08-12 | 2002-05-14 | Pharmacia & Upjohn S.P.A. | 3(5)-ureido-pyrazole derivatives process for their preparation and their use as antitumor agents |
| CA2384240A1 (en) | 1999-10-15 | 2001-04-26 | Dean A. Wacker | Benzylcycloalkyl amines as modulators of chemokine receptor activity |
| US6410533B1 (en) | 2000-02-10 | 2002-06-25 | Genzyme Corporation | Antibacterial compounds |
| JP2004517805A (ja) | 2000-06-30 | 2004-06-17 | ブリストル−マイヤーズ・スクイブ・ファーマ・カンパニー | ケモカイン受容体活性調節剤としてのn−ウレイドヘテロシクロアルキル−ピペリジン |
| EP1389194A2 (en) | 2001-04-27 | 2004-02-18 | Vertex Pharmaceuticals Incorporated | Inhibitors of bace |
| GB0110901D0 (en) | 2001-05-02 | 2001-06-27 | Smithkline Beecham Plc | Novel Compounds |
| WO2003037274A2 (en) | 2001-11-01 | 2003-05-08 | Icagen, Inc. | Pyrazole-amides and-sulfonamides |
| CA2468159A1 (en) | 2001-11-27 | 2003-06-05 | Merck & Co., Inc. | 4-aminoquinoline compounds |
| SE0104248D0 (sv) | 2001-12-14 | 2001-12-14 | Astrazeneca Ab | Method of treatment |
| EP1601666A2 (en) | 2002-07-02 | 2005-12-07 | Schering Corporation | New neuropeptide y y5 receptor antagonists |
| ZA200502612B (en) | 2002-10-08 | 2007-07-25 | Rinat Neuroscience Corp | Methods for treating post-surgical pain by administering a nerve crowth factor antagonist and compositions containing the same |
| KR101023035B1 (ko) | 2002-10-30 | 2011-03-24 | 와쿠나가 세이야쿠 가부시키 가이샤 | 세펨 화합물 |
| WO2005024755A2 (en) | 2002-12-31 | 2005-03-17 | Deciphera Pharmaceuticals, Llc. | Medicaments for the treatment of neurodegenerative disorders or diabetes |
| US7202257B2 (en) | 2003-12-24 | 2007-04-10 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US20040171075A1 (en) | 2002-12-31 | 2004-09-02 | Flynn Daniel L | Modulation of protein functionalities |
| US7144911B2 (en) | 2002-12-31 | 2006-12-05 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| KR101138216B1 (ko) | 2003-06-12 | 2012-09-05 | 아보트 러보러터리즈 | 바닐로이드 수용체 아형 1(vr1) 수용체를 억제하는융합 화합물 |
| EP1684762A4 (en) | 2003-11-13 | 2009-06-17 | Ambit Biosciences Corp | UREA DERIVATIVES AS MODULATORS OF KINASE |
| US20080220497A1 (en) | 2003-12-24 | 2008-09-11 | Flynn Daniel L | Modulation of protein functionalities |
| JP4573223B2 (ja) | 2004-01-23 | 2010-11-04 | 東レ・ファインケミカル株式会社 | 光学活性trans−4−アミノ−1−ベンジル−3−ピロリジノールの製造方法 |
| EP2295426A1 (en) * | 2004-04-30 | 2011-03-16 | Bayer HealthCare, LLC | Substituted pyrazolyl urea derivatives useful in the treatment of cancer |
| WO2006068591A1 (en) | 2004-12-20 | 2006-06-29 | Astrazeneca Ab | Novel pyrazole derivatives and their use as modulators of nicotinic acetylcholine receptors |
| AU2005325676A1 (en) | 2004-12-23 | 2006-08-03 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| CA2592118C (en) | 2004-12-23 | 2015-11-17 | Deciphera Pharmaceuticals, Llc | Urea derivatives as enzyme modulators |
| WO2006070198A1 (en) | 2004-12-30 | 2006-07-06 | Astex Therapeutics Limited | Pyrazole derivatives as that modulate the activity of cdk, gsk and aurora kinases |
| GB0510141D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds B3 |
| US20060281768A1 (en) | 2005-06-10 | 2006-12-14 | Gaul Michael D | Thienopyrimidine and thienopyridine kinase modulators |
| EP1960394A2 (en) | 2005-11-15 | 2008-08-27 | Bayer HealthCare AG | Pyrazolyl urea derivatives useful in the treatment of cancer |
| US7514435B2 (en) | 2005-11-18 | 2009-04-07 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| EP1957450B1 (en) | 2005-12-01 | 2009-06-24 | F.Hoffmann-La Roche Ag | Serotonin transporter (sert) inhibitors |
| US20110195110A1 (en) | 2005-12-01 | 2011-08-11 | Roger Smith | Urea compounds useful in the treatment of cancer |
| GB0607953D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| WO2008016811A2 (en) | 2006-07-31 | 2008-02-07 | Neurogen Corporation | Aminopiperidines and realted compounds |
| GB0615619D0 (en) | 2006-08-05 | 2006-09-13 | Astrazeneca Ab | Chemical process for preparation of intermediates |
| WO2008021859A1 (en) | 2006-08-09 | 2008-02-21 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| US7897762B2 (en) | 2006-09-14 | 2011-03-01 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of proliferative diseases |
| US8188113B2 (en) | 2006-09-14 | 2012-05-29 | Deciphera Pharmaceuticals, Inc. | Dihydropyridopyrimidinyl, dihydronaphthyidinyl and related compounds useful as kinase inhibitors for the treatment of proliferative diseases |
| US7790756B2 (en) | 2006-10-11 | 2010-09-07 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of myleoproliferative diseases and other proliferative diseases |
| WO2008131276A1 (en) | 2007-04-20 | 2008-10-30 | Deciphera Pharmaceuticals, Llc | Kinase inhibitors useful for the treatment of myleoproliferative diseases and other proliferative diseases |
| WO2008150899A1 (en) | 2007-05-29 | 2008-12-11 | Emory University | Combination therapies for treatment of cancer and inflammatory diseases |
| NZ582443A (en) | 2007-07-12 | 2011-04-29 | Novartis Ag | Oral pharmaceutical solutions containing telbivudine |
| ITMI20071731A1 (it) | 2007-09-06 | 2009-03-07 | Univ Degli Studi Genova | Nuovi derivati ureici dell'acido 1h-pirazol-4-carbossilico con attivita inibente nei confronti della chemiotassi di neutrofili |
| KR101324804B1 (ko) | 2008-05-13 | 2013-11-01 | 아이알엠 엘엘씨 | 키나제 억제제로서의 질소 함유 융합 헤테로사이클 및 그의 조성물 |
| US8592454B2 (en) | 2008-09-19 | 2013-11-26 | Takeda Pharmaceutical Company Limited | Nitrogen-containing heterocyclic compound and use of same |
| WO2010033941A1 (en) | 2008-09-22 | 2010-03-25 | Array Biopharma Inc. | Substituted imidazo[1,2b]pyridazine compounds as trk kinase inhibitors |
| BRPI0920306A2 (pt) | 2008-10-09 | 2019-09-24 | Hoffmann La Roche | derivados de n-benzil pirrolidina |
| MY169791A (en) | 2008-10-22 | 2019-05-15 | Array Biopharma Inc | Substituted pyrazolo [1,5-a] pyrimidine compounds as trk kinase inhibitors |
| JP2012509338A (ja) | 2008-11-19 | 2012-04-19 | メリアル リミテッド | 二量体1−アリールピラゾール誘導体 |
| JP5752601B2 (ja) | 2008-12-08 | 2015-07-22 | ブイエム ファーマ エルエルシー | タンパク質受容体チロシンキナーゼ阻害薬の組成物 |
| SG172352A1 (en) | 2008-12-23 | 2011-07-28 | Abbott Lab | Anti-viral compounds |
| EP2398323A4 (en) | 2009-02-19 | 2012-08-08 | Univ Connecticut | NEW HETEROPYRROLANALOGES THAT ACTIVATE ON CANNABINOID RECEPTORS |
| US20120071475A1 (en) | 2009-04-27 | 2012-03-22 | Shionogi & Co., Ltd. | Urea derivatives having pi3k-inhibiting activity |
| AR077468A1 (es) | 2009-07-09 | 2011-08-31 | Array Biopharma Inc | Compuestos de pirazolo (1,5 -a) pirimidina sustituidos como inhibidores de trk- quinasa |
| US20120220564A1 (en) | 2009-09-18 | 2012-08-30 | Zalicus Pharmaceuticals Ltd. | Selective calcium channel modulators |
| ME03376B (me) | 2010-05-20 | 2020-01-20 | Array Biopharma Inc | Makrociklička jedinjenja kао inhibiтori trk kinaze |
| CA2835835C (en) * | 2011-05-13 | 2019-04-02 | Array Biopharma Inc. | Pyrrolidinyl urea and pyrrolidinyl thiourea compounds as trka kinase inhibitors |
| EP2770987B1 (en) | 2011-10-27 | 2018-04-04 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| US8969325B2 (en) | 2011-12-19 | 2015-03-03 | Abbvie Inc. | TRPV1 antagonists |
| WO2013176970A1 (en) | 2012-05-22 | 2013-11-28 | Merck Sharp & Dohme Corp. | TrkA KINASE INHIBITORS, COMPOSITIONS AND METHODS THEREOF |
| WO2014052566A1 (en) | 2012-09-28 | 2014-04-03 | Merck Sharp & Dohme Corp. | Novel compounds that are erk inhibitors |
| RU2660429C2 (ru) | 2012-09-28 | 2018-07-06 | Мерк Шарп И Доум Корп. | Новые соединения, которые являются ингибиторами erk |
| WO2014078372A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| WO2014078378A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | Pyrrolidinyl urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| US9822118B2 (en) | 2012-11-13 | 2017-11-21 | Array Biopharma Inc. | Bicyclic heteroaryl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| WO2014078331A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | N-(arylalkyl)-n'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| WO2014078325A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | N-(monocyclic aryl),n'-pyrazolyl-urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| WO2014078417A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | Pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| MX374242B (es) | 2012-11-13 | 2025-03-05 | Array Biopharma Inc | Compuestos de n-pirrolidinil, n' -pirazolil-urea, tiourea, guanidina y cianoguanidina como inhibidores de trka cinasa. |
| WO2014078328A1 (en) | 2012-11-13 | 2014-05-22 | Array Biopharma Inc. | N-bicyclic aryl,n'-pyrazolyl urea, thiourea, guanidine and cyanoguanidine compounds as trka kinase inhibitors |
| RS55593B1 (sr) * | 2012-11-13 | 2017-06-30 | Array Biopharma Inc | Jedinjenja biciklične uree, tiouree, guanidina i cijanoguanidina korisna za lečenje bola |
| US9981959B2 (en) | 2012-11-13 | 2018-05-29 | Array Biopharma Inc. | Thiazolyl and oxazolyl urea, thiourea, guanidine and cyanoguanidine compounds as TrkA kinase inhibitors |
| WO2015039333A1 (en) | 2013-09-22 | 2015-03-26 | Merck Sharp & Dohme Corp. | TrkA KINASE INHIBITORS, COMPOSITIONS AND METHODS THEREOF |
| MA40434B1 (fr) | 2014-05-15 | 2019-09-30 | Array Biopharma Inc | 1-((3s,4r)-4-(3-fluorophényl)-1-(2-méthoxyéthyl)pyrrolidin-3-yl)-3-(4-méthyl-3-(2-méthylpyrimidin-5-yl)-1-phényl-1h-pyrazol-5-yl)urée comme inhibiteur de la kinase trka |
-
2015
- 2015-05-14 MA MA40434A patent/MA40434B1/fr unknown
- 2015-05-14 SG SG11201609550PA patent/SG11201609550PA/en unknown
- 2015-05-14 US US15/311,148 patent/US10835533B2/en active Active
- 2015-05-14 JP JP2016567819A patent/JP6499672B2/ja active Active
- 2015-05-14 KR KR1020167035164A patent/KR101998461B1/ko active Active
- 2015-05-14 PT PT15726455T patent/PT3154959T/pt unknown
- 2015-05-14 EP EP15726455.7A patent/EP3154959B1/en active Active
- 2015-05-14 CR CR20160583A patent/CR20160583A/es unknown
- 2015-05-14 AU AU2015259075A patent/AU2015259075B2/en active Active
- 2015-05-14 CN CN201811493142.0A patent/CN109970614B/zh active Active
- 2015-05-14 RU RU2016149233A patent/RU2719489C2/ru active
- 2015-05-14 SG SG10201903532XA patent/SG10201903532XA/en unknown
- 2015-05-14 PL PL15726455T patent/PL3154959T3/pl unknown
- 2015-05-14 HR HRP20191593 patent/HRP20191593T1/hr unknown
- 2015-05-14 CA CA2949160A patent/CA2949160C/en active Active
- 2015-05-14 RS RSP20191168 patent/RS59286B1/sr unknown
- 2015-05-14 ES ES15726455T patent/ES2746530T3/es active Active
- 2015-05-14 KR KR1020197019130A patent/KR102321883B1/ko active Active
- 2015-05-14 MY MYPI2016704187A patent/MY191956A/en unknown
- 2015-05-14 UA UAA201612765A patent/UA122126C2/uk unknown
- 2015-05-14 CN CN201580032260.XA patent/CN106459013B/zh active Active
- 2015-05-14 SI SI201530875T patent/SI3154959T1/sl unknown
- 2015-05-14 LT LTEP15726455.7T patent/LT3154959T/lt unknown
- 2015-05-14 DK DK15726455.7T patent/DK3154959T3/da active
- 2015-05-14 NZ NZ727418A patent/NZ727418A/en unknown
- 2015-05-14 MX MX2016014945A patent/MX2016014945A/es active IP Right Grant
- 2015-05-14 HU HUE15726455A patent/HUE045340T2/hu unknown
- 2015-05-14 WO PCT/US2015/030795 patent/WO2015175788A1/en not_active Ceased
- 2015-05-14 SM SM20190522T patent/SMT201900522T1/it unknown
-
2016
- 2016-11-11 PH PH12016502242A patent/PH12016502242A1/en unknown
- 2016-11-13 IL IL248924A patent/IL248924B/en active IP Right Grant
- 2016-11-14 MX MX2019012600A patent/MX2019012600A/es unknown
- 2016-11-15 CL CL2016002900A patent/CL2016002900A1/es unknown
-
2019
- 2019-02-27 AU AU2019201400A patent/AU2019201400A1/en not_active Abandoned
- 2019-03-15 JP JP2019048124A patent/JP2019085427A/ja active Pending
- 2019-03-19 EP EP19163878.2A patent/EP3564236A1/en not_active Withdrawn
- 2019-09-25 CY CY20191101009T patent/CY1122075T1/el unknown
-
2020
- 2020-01-23 PH PH12020500171A patent/PH12020500171A1/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2746530T3 (es) | 1-((3S,4R)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidín-3-il)-3-(4-metil-3-(2-metilpirimidín-5-il)-1-fenil-1H-pirazol-5- il)urea como un inhibidor de TrkA cinasa | |
| ES2834027T3 (es) | Compuestos de N-pirrolidinil, N'-pirazolil-urea como inhibidores de TrkA quinasa | |
| ES2747909T3 (es) | Inhibidores duales de quinasas ITK y JAK3 de 3,5-(no)sustituida-1H-pirrolo[2,3-b]piridina, 1H-pirazolo[3,4-b]piridina y 5H-pirrolo[2,3-b]pirazina | |
| JP6437559B2 (ja) | Wee−1キナーゼ阻害剤として有用なピリミドピリミジノン | |
| ES2718043T3 (es) | Compuestos macrocíclicos como inhibidores de quinasas Trk | |
| JP6706630B2 (ja) | TrkAキナーゼの阻害剤 | |
| WO2024061333A1 (zh) | 一种kras突变蛋白抑制剂、及其制备方法和应用 | |
| CN111225916B (zh) | 用于治疗炎性障碍的新化合物及其药物组合物 | |
| KR20250070059A (ko) | 신규한 테트라헤테로사이클 화합물 | |
| EA034558B1 (ru) | 4-ИМИДАЗО[1,5-a]ПИРИДАЗИН-1-ИЛ-БЕНЗАМИДЫ В КАЧЕСТВЕ Btk-ИНГИБИТОРОВ | |
| ES2734268T3 (es) | Derivados de urea y amida de aminoalquilpiperazinas y uso de los mismos | |
| TW201940473A (zh) | 作為多激酶抑制劑的胺基碳酸鹽及尿素化合物 | |
| CN118434735A (zh) | 8-氮杂喹唑啉作为脑渗透sos1抑制剂 | |
| BR112016026672B1 (pt) | Composto 1-((3s,4r)-4-(3-fluorofenil)-1-(2-metoxietil)pirrolidin-3-il)-3-(4-metil-3-(2-metilpirimidin-5-il)-1-fenil-1h-pirazol-5-il)ureia, seu uso, processos para a preparação do mesmo e de sua mistura racêmica e composição farmacêutica que o compreende | |
| EA053265B1 (ru) | Тетрагетероциклическое соединение | |
| HK1231879A1 (en) | 1-((3s,4r)-4-(3-fluorophenyl)-1-(2methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2methylpyrimidin-5-yl)-1-phenyl-1h-pyrazol-5-yl)urea as a trka kinase inhibitor | |
| HK1231879B (en) | 1-((3s,4r)-4-(3-fluorophenyl)-1-(2methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2methylpyrimidin-5-yl)-1-phenyl-1h-pyrazol-5-yl)urea as a trka kinase inhibitor | |
| HK40035135A (en) | Carbamate and urea compounds as multikinase inhibitors |