ES2886182T3 - C21-N-pirazolil-esteroide 19-nor C3,3-disustituido para su uso en terapia - Google Patents
C21-N-pirazolil-esteroide 19-nor C3,3-disustituido para su uso en terapia Download PDFInfo
- Publication number
- ES2886182T3 ES2886182T3 ES18203320T ES18203320T ES2886182T3 ES 2886182 T3 ES2886182 T3 ES 2886182T3 ES 18203320 T ES18203320 T ES 18203320T ES 18203320 T ES18203320 T ES 18203320T ES 2886182 T3 ES2886182 T3 ES 2886182T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- compound
- solution
- added
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC(*)(C[C@@]([C@](CC1)[C@]2C3)[C@]1C(CBr)=O)[C@@]2[C@](CC(*)[C@](*)C1)[C@@]1C3(*)*=C Chemical compound CC(*)(C[C@@]([C@](CC1)[C@]2C3)[C@]1C(CBr)=O)[C@@]2[C@](CC(*)[C@](*)C1)[C@@]1C3(*)*=C 0.000 description 15
- GFTUILSHVDSPMF-MHXSDRORSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(CBr)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(CBr)=O GFTUILSHVDSPMF-MHXSDRORSA-N 0.000 description 2
- HARRKNSQXBRBGZ-GVKWWOCJSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(C#N)c1)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(C#N)c1)=O HARRKNSQXBRBGZ-GVKWWOCJSA-N 0.000 description 2
- WVGCPEDBFHEHEZ-UHFFFAOYSA-N Brc1c[nH]nc1 Chemical compound Brc1c[nH]nc1 WVGCPEDBFHEHEZ-UHFFFAOYSA-N 0.000 description 1
- UXZQDYUBSQMFIE-UHFFFAOYSA-N C#Cc1c[nH]nc1 Chemical compound C#Cc1c[nH]nc1 UXZQDYUBSQMFIE-UHFFFAOYSA-N 0.000 description 1
- VUEWYZJJYGPJDC-UHFFFAOYSA-N C1OC11CCCCC1 Chemical compound C1OC11CCCCC1 VUEWYZJJYGPJDC-UHFFFAOYSA-N 0.000 description 1
- ADMTUGZFHLWYAU-UHFFFAOYSA-N CC(C)(C)CNN Chemical compound CC(C)(C)CNN ADMTUGZFHLWYAU-UHFFFAOYSA-N 0.000 description 1
- NWAZLPHVXXGFKE-LGPWGBJKSA-N CC(CC1)([C@@H](CC2)[C@H](C3)[C@H]1C(CCC(C1)=O)[C@H]1[C@@H]3F)/C2=C\C Chemical compound CC(CC1)([C@@H](CC2)[C@H](C3)[C@H]1C(CCC(C1)=O)[C@H]1[C@@H]3F)/C2=C\C NWAZLPHVXXGFKE-LGPWGBJKSA-N 0.000 description 1
- YWVWXJMRROJTKK-VVBLMLNTSA-N CC(CC1)([C@@H](CC2)[C@H](C[C@H]3F)[C@H]1[C@@H](CC1)C3=CC1=O)C2=O Chemical compound CC(CC1)([C@@H](CC2)[C@H](C[C@H]3F)[C@H]1[C@@H](CC1)C3=CC1=O)C2=O YWVWXJMRROJTKK-VVBLMLNTSA-N 0.000 description 1
- XDZXENYZOKSEOV-IMEXOKINSA-N CCC([C@H](CC(C)O)C(C1)F)[C@@H](CC2)C1C(CC1)[C@@]2(C)[C@H]1C(C)C Chemical compound CCC([C@H](CC(C)O)C(C1)F)[C@@H](CC2)C1C(CC1)[C@@]2(C)[C@H]1C(C)C XDZXENYZOKSEOV-IMEXOKINSA-N 0.000 description 1
- DLDUIVXFOKHKNP-FUDSMBTHSA-N CCOC[C@@](CC1)(C[C@H](CC2)C1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)C2C(C)CO)O Chemical compound CCOC[C@@](CC1)(C[C@H](CC2)C1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)C2C(C)CO)O DLDUIVXFOKHKNP-FUDSMBTHSA-N 0.000 description 1
- QMFPXNHDMZESKP-NEOIFOSFSA-N CCOC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)[C@@H]2[C@H](CC2)[C@@]1(C)[C@H]2C(C)=O)O Chemical compound CCOC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)[C@@H]2[C@H](CC2)[C@@]1(C)[C@H]2C(C)=O)O QMFPXNHDMZESKP-NEOIFOSFSA-N 0.000 description 1
- XQGMGCWFGJRIMC-YRTSKKSESA-N CCOC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)[C@@H]2[C@H](CC2)[C@@]1(C)[C@H]2C(C[Br]=C)=O)O Chemical compound CCOC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)[C@@H]2[C@H](CC2)[C@@]1(C)[C@H]2C(C[Br]=C)=O)O XQGMGCWFGJRIMC-YRTSKKSESA-N 0.000 description 1
- QLONDGWJCYMLPF-LYWOIHLFSA-N CC[C@@H](CC1)[C@@](C)(CC2)[C@@H]1[C@H](CC1)[C@H]2[C@@H](CC2)[C@H]1C[C@]2(CO)O Chemical compound CC[C@@H](CC1)[C@@](C)(CC2)[C@@H]1[C@H](CC1)[C@H]2[C@@H](CC2)[C@H]1C[C@]2(CO)O QLONDGWJCYMLPF-LYWOIHLFSA-N 0.000 description 1
- NCYKANFSYDMWBR-JTIZSSSKSA-N CC[C@@](CC1)(C[C@H](CC2)C1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)[C@H]2C(C)=O)O Chemical compound CC[C@@](CC1)(C[C@H](CC2)C1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)[C@H]2C(C)=O)O NCYKANFSYDMWBR-JTIZSSSKSA-N 0.000 description 1
- OOEJJUGCJMSQJN-OMJMBDCTSA-N CC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)[C@H]2C(CBr)=O)O Chemical compound CC[C@@](CC1)(C[C@H](CC2)[C@H]1[C@@H](CC1)C2[C@H](CC2)[C@@]1(C)[C@H]2C(CBr)=O)O OOEJJUGCJMSQJN-OMJMBDCTSA-N 0.000 description 1
- IHNXHUNMFYXQCG-UHFFFAOYSA-N CCc1c[nH]nc1 Chemical compound CCc1c[nH]nc1 IHNXHUNMFYXQCG-UHFFFAOYSA-N 0.000 description 1
- SZJSEMWMLTYQGH-MWBSRRISSA-N C[C@](CC1)(C(CC2)[C@H](C3)[C@H]1C(CC[C@](C)(C1)O)[C@H]1C3F)[C@H]2C(CBr)=O Chemical compound C[C@](CC1)(C(CC2)[C@H](C3)[C@H]1C(CC[C@](C)(C1)O)[C@H]1C3F)[C@H]2C(CBr)=O SZJSEMWMLTYQGH-MWBSRRISSA-N 0.000 description 1
- OGCDDNYKEPHMCA-HKDZEBQCSA-N C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1C(C)(CCC(C1)=O)[C@@]1(C)[C@@H]3F)C2=O Chemical compound C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1C(C)(CCC(C1)=O)[C@@]1(C)[C@@H]3F)C2=O OGCDDNYKEPHMCA-HKDZEBQCSA-N 0.000 description 1
- IOTWSBQSHDBSGV-WZEMHRCGSA-N C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1C(CC[C@](C)(C1)O)[C@H]1C3=[F])/C2=C\C Chemical compound C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1C(CC[C@](C)(C1)O)[C@H]1C3=[F])/C2=C\C IOTWSBQSHDBSGV-WZEMHRCGSA-N 0.000 description 1
- RTAUFUIZBAUEME-ZCISKLIESA-N C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1[C@@H](CCC(C1)(OC)OC)[C@H]1[C@@H]3F)C2=O Chemical compound C[C@](CC1)([C@@H](CC2)C(C3)[C@H]1[C@@H](CCC(C1)(OC)OC)[C@H]1[C@@H]3F)C2=O RTAUFUIZBAUEME-ZCISKLIESA-N 0.000 description 1
- XAVWFOOPIKBVTO-QHGSJHJHSA-N C[C@](CC1)([C@@H](CC2)C(CC3)[C@H]1[C@@H](CC1)[C@@H]3CC1=O)C2=C Chemical compound C[C@](CC1)([C@@H](CC2)C(CC3)[C@H]1[C@@H](CC1)[C@@H]3CC1=O)C2=C XAVWFOOPIKBVTO-QHGSJHJHSA-N 0.000 description 1
- MJPHSAZIPXVEMA-YWLWCBGDSA-N C[C@](CC1)([C@@H](CC2)C(CC[C@@H]3C4)[C@H]1[C@H]3C=C[C@@]4(CO)O)[C@H]2C(C)=O Chemical compound C[C@](CC1)([C@@H](CC2)C(CC[C@@H]3C4)[C@H]1[C@H]3C=C[C@@]4(CO)O)[C@H]2C(C)=O MJPHSAZIPXVEMA-YWLWCBGDSA-N 0.000 description 1
- NZDQHZGQNHSGRU-PVSAOXIISA-N C[C@](CC1)([C@@H](CC2)C3[C@H]1C(CCC(OC(C)=O)=C1)C1=CC3)C2=O Chemical compound C[C@](CC1)([C@@H](CC2)C3[C@H]1C(CCC(OC(C)=O)=C1)C1=CC3)C2=O NZDQHZGQNHSGRU-PVSAOXIISA-N 0.000 description 1
- AILRKGHFNNPTII-DIAOGVGQSA-N C[C@](CC1)([C@@H](CC2)[C@H](C3)[C@H]1[C@@H](CCC(C1)(OC)OC)[C@H]1[C@@H]3F)/C2=C\C Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](C3)[C@H]1[C@@H](CCC(C1)(OC)OC)[C@H]1[C@@H]3F)/C2=C\C AILRKGHFNNPTII-DIAOGVGQSA-N 0.000 description 1
- GDVNEVRDAKDCGD-FCEJBBPCSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@@H]3C[C@]1(COC)O)[C@H]2C(CBr)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@@H]3C[C@]1(COC)O)[C@H]2C(CBr)=O GDVNEVRDAKDCGD-FCEJBBPCSA-N 0.000 description 1
- QMEKUHVFZNKVKW-KTOOMANLSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@@H]3C[C@]1(COC)O)[C@H]2C(C[n]1nccc1)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@@H]3C[C@]1(COC)O)[C@H]2C(C[n]1nccc1)=O QMEKUHVFZNKVKW-KTOOMANLSA-N 0.000 description 1
- JIWJWZQRCFJBIU-NPXBQTALSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@@]1(COC)O)[C@H]2C(C)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@@]1(COC)O)[C@H]2C(C)=O JIWJWZQRCFJBIU-NPXBQTALSA-N 0.000 description 1
- BVGXHMPOXIJBII-VUYLISRKSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1nc(C)cc1C)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1nc(C)cc1C)=O BVGXHMPOXIJBII-VUYLISRKSA-N 0.000 description 1
- AKGYKJYYRHKTJQ-UZNJFETESA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(Br)c1)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(Br)c1)=O AKGYKJYYRHKTJQ-UZNJFETESA-N 0.000 description 1
- SPHRFVINFAOUKX-NYBDTYKHSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(S(C)(=O)=O)c1)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(S(C)(=O)=O)c1)=O SPHRFVINFAOUKX-NYBDTYKHSA-N 0.000 description 1
- RXPNKLJEOOUJCB-NYBDTYKHSA-N C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(SC)c1)=O Chemical compound C[C@](CC1)([C@@H](CC2)[C@H](CC3)[C@H]1[C@@H](CC1)[C@H]3C[C@]1(C)O)[C@H]2C(C[n]1ncc(SC)c1)=O RXPNKLJEOOUJCB-NYBDTYKHSA-N 0.000 description 1
- LLNQWPTUJJYTTE-UHFFFAOYSA-N Ic1c[nH]nc1 Chemical compound Ic1c[nH]nc1 LLNQWPTUJJYTTE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/58—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids containing heterocyclic rings, e.g. danazol, stanozolol, pancuronium or digitogenin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
- A61K31/57—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone
- A61K31/573—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane or progesterone substituted in position 21, e.g. cortisone, dexamethasone, prednisone or aldosterone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J13/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having a carbon-to-carbon double bond from or to position 17
- C07J13/007—Normal steroids containing carbon, hydrogen, halogen or oxygen having a carbon-to-carbon double bond from or to position 17 with double bond in position 17 (20)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J15/00—Stereochemically pure steroids containing carbon, hydrogen, halogen or oxygen having a partially or totally inverted skeleton, e.g. retrosteroids, L-isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J3/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by one carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
- C07J5/0046—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa
- C07J5/0053—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/001—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group
- C07J7/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa
- C07J7/002—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by a keto group not substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0059—Estrane derivatives substituted in position 17 by a keto group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0066—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa
- C07J1/007—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa the substituent being an OH group free esterified or etherified
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J1/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, not substituted in position 17 beta by a carbon atom, e.g. estrane, androstane
- C07J1/0051—Estrane derivatives
- C07J1/0066—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa
- C07J1/007—Estrane derivatives substituted in position 17 beta not substituted in position 17 alfa the substituent being an OH group free esterified or etherified
- C07J1/0074—Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J21/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having an oxygen-containing hetero ring spiro-condensed with the cyclopenta(a)hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J31/00—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring
- C07J31/006—Normal steroids containing one or more sulfur atoms not belonging to a hetero ring not covered by C07J31/003
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
- C07J5/0007—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa
- C07J5/0015—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond not substituted in position 17 alfa not substituted in position 16
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/0005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21
- C07J7/0065—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by an OH group free esterified or etherified
- C07J7/007—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms not substituted in position 21 substituted in position 20 by an OH group free esterified or etherified not substituted in position 17 alfa
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J7/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms
- C07J7/008—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms substituted in position 21
- C07J7/0085—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of two carbon atoms substituted in position 21 by an halogen atom
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Epidemiology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Addiction (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Anesthesiology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de fórmula **(Ver fórmula)** o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de un trastorno del estado de ánimo en un sujeto humano.
Description
DESCRIPCIÓN
C21-N-pirazolil-esteroide 19-nor C3,3-disustituido para su uso en terapia
Antecedentes de la invención
La excitabilidad del cerebro se define como el nivel de excitación de un animal, un continuo que abarca desde el coma hasta las convulsiones y que está regulado por diversos neurotransmisores. En general, los neurotransmisores son responsables de regular la conductancia de los iones a través de las membranas neuronales. En reposo, la membrana neuronal tiene un potencial (o voltaje de membrana) de aproximadamente -70 mV, siendo el interior de la célula negativo con respecto al exterior de la célula. El potencial (voltaje) es el resultado del equilibrio iónico (K+, Na+, Cl-, aniones orgánicos) a través de la membrana semipermeable neuronal. Los neurotransmisores se almacenan en las vesículas presinápticas y son liberados bajo la influencia de potenciales de acción neuronales. Cuando se liberan en la hendidura sináptica, un transmisor químico excitador, tal como la acetilcolina, causará la despolarización de la membrana (cambio de potencial de -70 mV a -50 mV). Este efecto está mediado por receptores nicotínicos postsinápticos que son estimulados por la acetilcolina para aumentar la permeabilidad de la membrana a los iones Na+. El potencial de membrana reducido estimula la excitabilidad neuronal en forma de un potencial de acción postsináptico.
En el caso del complejo receptor de GABA (GRC), el efecto sobre la excitabilidad del cerebro está mediado por GABA, un neurotransmisor. GABA tiene una profunda influencia sobre la excitabilidad del cerebro debido a que hasta el 40 % de las neuronas en el cerebro utilizan GABA como neurotransmisor. GABA regula la excitabilidad de las neuronas individuales regulando la conductancia de los iones de cloruro a través de la membrana neuronal. GABA interactúa con su sitio de reconocimiento en el GRC para facilitar el flujo de iones de cloruro a través de un gradiente electroquímico del GRC dentro de la célula. Un incremento intracelular en los niveles de este anión causa hiperpolarización del potencial transmembranario, lo que hace que la neurona sea menos susceptible a los estímulos excitadores (es decir, reduce la excitabilidad de las neuronas). En otras palabras, cuanto mayor sea la concentración de iones cloruro en la neurona, menor será la excitabilidad del cerebro (el nivel de excitación).
Está bien documentado que el GRC es responsable de la mediación en la ansiedad, la actividad convulsiva y la sedación. Por lo tanto, GABA y los fármacos que actúan como GABA o facilitan los efectos de GABA (por ejemplo, los barbitúricos y las benzodiacepinas (BZ) terapéuticamente útiles, tales como Valium®) producen sus efectos terapéuticos útiles interactuando con sitios reguladores específicos en el GRC.
La evidencia acumulada ha indicado ahora que, además del sitio de unión a las benzodiacepinas y los barbitúricos, el GRC contiene un sitio distinto para esteroides neuroactivos (Lan, N. C. et al., Neurochem. Res. 16:347-356 (1991)).
Los esteroides neuroactivos pueden existir de forma endógena. Los esteroides neuroactivos endógenos más potentes son pregnan-20-ona 3a-hidroxi-5-reducida y pregnan-20-ona 3a-21-dihidroxi-5-reducida, metabolitos de los esteroides hormonales progesterona y desoxicorticosterona, respectivamente. La habilidad de estos metabolitos esteroides para alterar la excitabilidad del cerebro se reconoció en 1986 (Majewska, M. D. et al., Science 232:1004-1007 (1986); Harrison, N. L. et al., J Pharmacol. Exp. Ther. 241:346-353 (1987)).
La hormona ovárica progesterona y sus metabolitos han mostrado tener profundos efectos sobre la excitabilidad del cerebro (Backstrom, T. et al., Acta Obstet. Gynecol. Scand. Suppl. 130:19-24 (1985); Pfaff, D. W y McEwen, B. S., Science 219:808-814 (1983); Gyermek et al., J Med Chem. 11:117 (1968); Lambert, J. et al., Trends Pharmacol. Sci.
8:224-227 (1987)). Los niveles de progesterona y sus metabolitos varían con las fases del ciclo menstrual. Se ha documentado bien que los niveles de la progesterona y sus metabolitos disminuyen antes del inicio de la menstruación. La reaparición mensual de ciertos síntomas físicos antes del inicio de la menstruación también ha sido bien documentada. Estos síntomas, que se han asociado con el síndrome premenstrual (SPM), incluyen estrés, ansiedad y migrañas (Dalton, K., Premenstrual Syndrome and Progesterone Therapy, 2a edición, Chicago Yearbook, Chicago (1984)). Los sujetos con SPM tienen una reaparición mensual de los síntomas que están presentes antes de la menstruación y desaparecen después de la menstruación.
De un modo similar, una reducción en la progesterona también se ha relacionado temporalmente con un incremento en la frecuencia de convulsiones en mujeres epilépticas, es decir, epilepsia catamenial (Laidlaw, J., Lancet, 1235-1237 (1956)). Se ha observado una correlación más directa con una reducción en los metabolitos de progesterona (Rosciszewska et al., J. Neurol. Neurosurg. Psych. 49:47-51 (1986)). Además, para sujetos con epilepsia de pequeño mal primaria generalizada, la incidencia temporal de las convulsiones se ha correlacionado con la incidencia de los síntomas del síndrome premenstrual (Backstrom, T. et al., J. Psychosom. Obstet. Gynaecol. 2:8-20 (1983)). Se ha encontrado que el esteroide desoxicorticosterona es eficaz en el tratamiento de sujetos con ausencias epilépticas relacionadas con sus ciclos menstruales (Aird, R. B. y Gordan, G., J. Amer. Med. Soc. 145:715-719 (1951)).
Un síndrome relacionado también con niveles bajos de progesterona es la depresión posparto (DPP). Inmediatamente después del nacimiento, los niveles de progesterona disminuyen espectacularmente, lo que conduce al inicio de la DPP. Los síntomas de la DPP varían desde depresión leve hasta psicosis que requiere hospitalización. La DPP también está asociada con ansiedad intensa e irritabilidad. La depresión asociada a DPP no es susceptible a
tratamiento con los antidepresivos clásicos y las mujeres que sufren DPP muestran una mayor incidencia de SPM (Dalton, K., Premenstrual Syndrome and Progesterone Therapy, 2a edición, Chicago Yearbook, Chicago (1984)).
En conjunto, estas observaciones implican una función crucial de la progesterona y la desoxicorticosterona y más específicamente sus metabolitos en la regulación homeostática de la excitabilidad del cerebro, lo que se manifiesta como un aumento en la actividad de las convulsiones o los síntomas asociados con la epilepsia catamenial, el SPM y la DPP. La correlación entre los niveles reducidos de progesterona y los síntomas asociados con el SPM, la DPP y la epilepsia catamenial (Backstrom, T. et al., J Psychosom. Obstet. Gynaecol. 2: 8-20 (1983)); Dalton, K., Premenstrual Syndrome and Progesterone Therapy, 2a edición, Chicago Yearbook, Chicago (1984)) ha promovido el uso de la progesterona en su tratamiento (Mattson et al., "Medroxiprogesterone therapy of catamenial epilepsy", en Advances in Epileptology: XV Epilepsy International Symposium, Raven Press, Nueva York (1984), pp. 279-282 y Dalton, K., Premenstrual Syndrome and Progesterone Therapy, 2a edición, Chicago Yearbook, Chicago (1984)). Sin embargo, la progesterona no es eficaz de forma sistemática en el tratamiento de los síndromes mencionados anteriormente. Por ejemplo, no existe relación dosis-respuesta para la progesterona en el tratamiento del SPM (Maddocks et al., Obstet. Gynecol. 154:573-581 (1986); Dennerstein et al., Brit. Med J 290:16-17 (1986)).
El documento de patente US-A-5.939.545 desvela derivados de 3.alfa.-hidroxi-pregnan-20-ona con un grupo 21-pirazolilo que modulan los receptores de GABA.
Se necesitan esteroides neuroactivos nuevos y mejorados que actúen como agentes moduladores de la excitabilidad del cerebro, así como agentes para la prevención y el tratamiento de las enfermedades relacionadas con el SNC. Los compuestos, las composiciones y los métodos descritos en el presente documento se dirigen a este fin.
Sumario de la invención
La presente invención se basa, en parte, en el deseo de proporcionar novedosos compuestos 19-nor (es decir, desmetilo C19), por ejemplo, relacionados con la progesterona, la desoxicorticosterona y sus metabolitos, con buena potencia, propiedades farmacocinéticas (FC), biodisponibilidad oral, formulabilidad, estabilidad, seguridad, depuración y/o metabolismo. Una característica clave de los compuestos como se describe en el presente documento es la disustitución en la posición C3 (por ejemplo, con un sustituyente que es un resto hidroxi 3a. Los inventores prevén que la disustitución en C-3 eliminará el potencial de oxidación del resto hidroxi a la cetona, evitará el metabolismo adicional y reducirá la posibilidad de rutas de eliminación secundarias, tales como la glucuronidación. Los inventores prevén además que el efecto total de la disustitución en C3 debería mejorar los parámetros FC totales y reducir posibles toxicidades y efectos secundarios, lo que puede permitir, en ciertas realizaciones, la administración por vía oral y/o de manera crónica. Otra característica clave de los compuestos como se describen en el presente documento es la presencia de un hidrógeno en la posición C19 ("19-nor") en vez de un grupo metilo. Los inventores prevén que los compuestos 19-nor, en comparación con sus homólogos de metilo C19, tendrán propiedades físicas mejoradas, tales como solubilidad mejorada. Los inventores prevén un mayor incremento de la solubilidad, por ejemplo, cuando el sistema de anillo AB está en la configuración cis.
Según la invención, se proporciona el compuesto SA-4 o una sal farmacéuticamente aceptable del mismo, para su uso en el tratamiento de trastornos del estado de ánimo, SPM o DPP en un sujeto humano, o para su uso en la modulación positiva o negativa de un receptor de GABAAen un sujeto humano.

El compuesto para su uso según la presente invención como se describe en el presente documento actúa, en ciertas realizaciones, como un modulador de GABA a , por ejemplo, efectuando el receptor de GAB>A,en o un modo positivo o negativo. Como moduladores de la excitabilidad del sistema nervioso central (SNC), como está medida por su capacidad para modular el receptor de GABA a , se espera que dichos compuestos tengan actividad de SNC.
Por lo tanto, se proporciona SA-4 o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento de un trastorno relacionado con el SNC en un sujeto en necesidad del mismo, que comprende administrar al sujeto una cantidad eficaz de un compuesto de la presente invención. El trastorno relacionado con el SNC se selecciona del grupo que consiste en un trastorno del estado de ánimo, SPM, DPP o la modulación del receptor de GABAA. En ciertas realizaciones, el compuesto se administra por vía oral, por vía subcutánea, por vía intravenosa o por vía intramuscular. En ciertas realizaciones, el compuesto se administra de manera crónica.
Otros objetos y ventajas serán evidentes para los expertos en la técnica a partir de una consideración de la resultante Descripción detallada, Ejemplos y Reivindicaciones.
Definiciones
El término "sal farmacéuticamente aceptable" se refiere a las sales que son, dentro del alcance de criterio médico sensato, adecuadas para su uso en contacto con los tejidos de seres humanos y animales inferiores sin excesiva toxicidad, irritación, respuesta alérgica y similares, y son proporcionales a una relación beneficio/riesgo razonable. Las sales farmacéuticamente aceptables se conocen bien en la técnica. Por ejemplo, Berge et al., describen sales farmacéuticamente aceptables en detalle en J. Pharmaceutical Sciences (1977) 66:1-19. Las sales farmacéuticamente aceptables de los compuestos de la presente invención incluyen las derivadas de ácidos y bases inorgánicos y orgánicos adecuados. Los ejemplos de sales de adición de ácido no tóxicas farmacéuticamente aceptables son las sales de un grupo amino formado con ácidos inorgánicos, tales como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico, o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico, o usando otros métodos usados en la técnica, tal como intercambio iónico. Otras sales farmacéuticamente aceptables incluyen las sales adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, canforato, canforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, yodhidrato, 2-hidroxi-etanosulfonato, lactobionato, lactato, laurato, laurilsulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato, y similares. Las sales farmacéuticamente aceptables derivadas de bases apropiadas incluyen sales de metal alcalino, metal alcalinotérreo, amonio y N+(alquilo C1-4K Sales de metales alcalinos o alcalinotérreos representivas incluyen sodio, litio, potasio, calcio, magnesio, y similares. Sales farmacéuticamente aceptables adicionales incluyen, cuando convenga, cationes
amonio, amonio cuaternario y amina no tóxicos formados usando contraiones, tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, alquil inferior-sulfonato y arilsulfonato.
El sujeto humano para el que se contempla la administración incluye, pero no se limita a, hombres y mujeres de cualquier grupo de edad, por ejemplo, un sujeto pediátrico (por ejemplo, lactante, niño, adolescente) o adulto (por ejemplo, adulto joven, adulto de mediana edad o adulto mayor)). Los términos "humano", "paciente" y "sujeto" se usan indistintamente en el presente documento.
Enfermedad, trastorno y afección se usan indistintamente en el presente documento.
Como se usa en el presente documento, y a menos que se especifique de otro modo, los términos "tratan", "tratar" y "tratamiento" contemplan una acción que ocurre mientras que un sujeto está padeciendo la enfermedad, trastorno o afección especificada, que reduce la intensidad de la enfermedad, trastorno o afección, o retrasa o ralentiza la progresión de la enfermedad, trastorno o afección ("tratamiento terapéutico"), y también contempla una acción que ocurre antes de que un sujeto empiece a padecer la enfermedad, trastorno o afección especificada ("tratamiento profiláctico").
En general, la "cantidad eficaz" de un compuesto se refiere a una cantidad suficiente para provocar la respuesta biológica deseada. Como será apreciado por los expertos habituales en esta técnica, la cantidad eficaz de un compuesto de la invención puede variar dependiendo de factores tales como el criterio de valoración biológico deseado, la farmacocinética del compuesto, la enfermedad que está tratándose, el modo de administración y la edad, salud y afección del sujeto. Una cantidad eficaz engloba tratamiento terapéutico y profiláctico.
Como se usa en el presente documento, y a menos que se especifique de otro modo, una "cantidad terapéuticamente eficaz" de un compuesto es una cantidad suficiente proporcionando un beneficio terapéutico en el tratamiento de una enfermedad, trastorno o afección, o para retardar o minimizar uno o más síntomas asociados a la enfermedad, trastorno o afección. Una cantidad terapéuticamente eficaz de un compuesto significa una cantidad de agente terapéutico, solo o en combinación con otras terapias, que proporciona un beneficio terapéutico en el tratamiento de la enfermedad, trastorno o afección. El término "cantidad terapéuticamente eficaz" puede englobar una cantidad que mejora la terapia en general, reduce o previene síntomas o causas de la enfermedad o afección, o potencia la eficacia terapéutica de otro agente terapéutico.
Como se usa en el presente documento, y a menos que se especifique de otro modo, una "cantidad profilácticamente eficaz" de un compuesto es una cantidad suficiente para prevenir una enfermedad, trastorno o afección, o uno o más síntomas asociados a la enfermedad, trastorno o afección, o prevenir su reaparición. Una cantidad profilácticamente eficaz de un compuesto significa una cantidad de un agente terapéutico, solo o en combinación con otros agentes, que proporciona un beneficio profiláctico en la prevención de la enfermedad, trastorno o afección. El término "cantidad profilácticamente eficaz" puede englobar una cantidad que mejora la profilaxis en general o potencia la eficacia profiláctica de otro agente profiláctico.
Breve descripción de los dibujos
Las FIG. 1-52 representan espectros de RMN 1H representativos de compuestos a modo de ejemplo descritos en el presente documento. El Compuesto SA-4 y las sales farmacéuticamente aceptables del mismo son los compuestos para su uso según la presente invención, siendo otros compuestos presentados aquí simplemente a efectos de comparación.
Composiciones farmacéuticas
Las composiciones farmacéuticas descritas en el presente documento forman parte de la presente invención solo en tanto que contienen el compuesto SA-4 y son para el tratamiento o la prevención de las afecciones como se especifican en las reivindicaciones.
En otro aspecto, la invención proporciona una composición farmacéutica que comprende un compuesto de la presente invención (también denominado el "principio activo") y un excipiente farmacéuticamente aceptable. En ciertas realizaciones, la composición farmacéutica comprende una cantidad eficaz del principio activo. En ciertas realizaciones, la composición farmacéutica comprende una cantidad terapéuticamente eficaz del principio activo. En ciertas realizaciones, la composición farmacéutica comprende una cantidad profilácticamente eficaz del principio activo.
Las composiciones farmacéuticas proporcionadas en el presente documento se pueden administrar mediante una variedad de vías que incluyen, pero no se limitan a, administración oral (enteral), administración parenteral (por inyección), administración rectal, administración transdérmica, administración intradérmica, administración intratecal, administración subcutánea (SC), administración intravenosa (IV), administración intramuscular (IM) y administración intranasal.
Generalmente, los compuestos proporcionados en el presente documento se administran en una cantidad eficaz. La cantidad del compuesto administrado en realidad normalmente será determinada por un médico, en vista de las
circunstancias relevantes, que incluyen la afección que se va a tratar, la vía de administración elegida, el compuesto administrado propiamente dicho, la edad, el peso y la respuesta del paciente individual, la intensidad de los síntomas del paciente, y similares.
Cuando se usa para prevenir la aparición de un trastorno del SNC, los compuestos proporcionados en el presente documento se administrarán a un sujeto en riesgo de desarrollar la afección, normalmente bajo el consejo y la supervisión de un médico, en los niveles de dosis descritos anteriormente. Los sujetos en riesgo de desarrollar una afección particular incluyen, en general, aquellos que tienen antecedentes familiares de la afección, o los que se ha identificado por pruebas genéticas o cribado genético que son particularmente susceptibles de desarrollar la afección.
Las composiciones farmacéuticas proporcionadas en el presente documento también se pueden administrar de manera crónica ("administración crónica"). La administración crónica se refiere a la administración de un compuesto o composición farmacéutica del mismo durante un periodo de tiempo prolongado, por ejemplo, por ejemplo, durante 3 meses, 6 meses, 1 año, 2 años, 3 años, 5 años, etc., o puede continuar indefinidamente, por ejemplo, durante el resto de la vida del sujeto. En ciertas realizaciones, la administración crónica pretende proporcionar un nivel constante del compuesto en la sangre, por ejemplo, dentro de la ventana terapéutica durante el periodo de tiempo prolongado.
Las composiciones farmacéuticas de la presente invención se pueden administrar además usando una variedad de métodos de administración. Por ejemplo, en ciertas realizaciones, la composición farmacéutica se puede administrar como un bolo, por ejemplo, para aumentar la concentración del compuesto en la sangre hasta un nivel eficaz. La colocación de la dosis de bolo depende de los niveles sistémicos del principio activo deseado en todo el cuerpo, por ejemplo, una dosis en embolada intramuscular o subcutánea permite una liberación lenta del principio activo, mientras que un bolo administrado directamente a las venas (por ejemplo, mediante un goteo IV) permite una administración mucho más rápida que aumenta rápidamente la concentración del principio activo en la sangre hasta un nivel eficaz. En otras realizaciones, la composición farmacéutica se puede administrar como una infusión continua, por ejemplo, por goteo IV, proporcionando el mantenimiento de una concentración en estado estacionario del principio activo en el cuerpo del sujeto. Además, en todavía otras realizaciones, la composición farmacéutica se puede administrar primero como una dosis en embolada, seguido por infusión continua.
Las composiciones para administración por vía oral pueden tomar la forma de disoluciones o suspensiones líquidas a granel, o polvos a granel. Más comúnmente, sin embargo, las composiciones se presentan en formas farmacéuticas unitarias para facilitar la administración exacta. El término "formas farmacéuticas unitarias" se refiere a unidades físicamente discretas adecuadas como administraciones unitarias para sujetos humanos y otros mamíferos, conteniendo cada unidad una cantidad predeterminada de material activo calculada para producir el efecto terapéutico deseado, en asociación con un excipiente farmacéutico adecuado. Las formas farmacéuticas unitarias típicas incluyen ampollas o jeringas precargadas y premedidas de las composiciones líquidas o píldoras, comprimidos, cápsulas o similares en el caso de composiciones sólidas. En dichas composiciones, el compuesto es normalmente un componente minoritario (desde aproximadamente 0,1 hasta aproximadamente 50 % en peso o preferentemente desde aproximadamente 1 hasta aproximadamente 40 % en peso), siendo el resto diversos vehículos o excipientes y adyuvantes de procesamiento útiles para formar la forma de administración deseada.
Con la administración oral, una a cinco y especialmente dos a cuatro y normalmente tres dosis orales por día son regímenes representativos. Usando estos patrones de administración, cada dosis proporciona desde aproximadamente 0,01 hasta aproximadamente 20 mg/kg del compuesto proporcionado en el presente documento, proporcionando cada dosis preferida desde aproximadamente 0,1 hasta aproximadamente 10 mg/kg, y especialmente aproximadamente 1 a aproximadamente 5 mg/kg.
Las dosis transdérmicas se seleccionan, en general, proporcionando niveles en sangre similares o más bajos que los logrados usando las dosis para inyección, en general, en una cantidad que varía desde aproximadamente 0,01 hasta aproximadamente 20 % en peso, preferentemente desde aproximadamente 0,1 hasta aproximadamente 20 % en peso, preferentemente desde aproximadamente 0,1 hasta aproximadamente 10 % en peso, y más preferentemente desde aproximadamente 0,5 hasta aproximadamente 15 % en peso.
Los niveles de dosis para inyección varían desde aproximadamente 0,1 mg/kg/hora hasta al menos 10 mg/kg/hora, todo ello desde aproximadamente 1 hasta aproximadamente 120 horas y especialmente 24 hasta 96 horas. También se puede administrar un bolo de precarga de desde aproximadamente 0,1 mg/kg hasta aproximadamente 10 mg/kg o más para lograr niveles en estado estacionario adecuados. No se espera que la dosis total máxima supere a aproximadamente 2 g/día para un paciente humano de 40 a 80 kg.
Las formas líquidas adecuadas para administración por vía oral pueden incluir un vehículo acuoso o no acuoso adecuado con tampones, agentes de suspensión y dispersión, colorantes, aromas y similares. Las formas sólidas pueden incluir, por ejemplo, cualquiera de los siguientes componentes, o compuestos de una naturaleza similar: un aglutinante, tal como celulosa microcristalina, goma tragacanto o gelatina; un excipiente, tal como almidón o lactosa, un agente disgregante, tal como ácido algínico, Primogel, o almidón de maíz; un lubricante, tal como estearato de magnesio; un deslizante, tal como dióxido de silicio coloidal; un edulcorante, tal como sacarosa o sacarina; o un aromatizante, tal como menta, salicilato de metilo o aromatizante de naranja.
Las composiciones inyectables se basan normalmente en solución salina estéril inyectable o solución salina tamponada con fosfato u otros excipientes inyectables conocidos en la técnica. Como antes, el compuesto activo en dichas composiciones es normalmente un componente minoritario, que frecuentemente es desde aproximadamente el 0,05 hasta el 10 % en peso, siendo el resto el excipiente inyectable y similares.
Las composiciones transdérmicas se formulan normalmente como una pomada tópica o crema que contiene el (los) principio(s) activo(s). Cuando se formulan como una pomada, los principios activos normalmente se combinarán con o una base de pomada parafínica o una miscible en agua. Alternativamente, los principios activos se pueden formular en una crema con, por ejemplo, una base de crema de aceite en agua. Dichas formulaciones transdérmicas se conocen bien en la técnica e incluyen, en general, componentes adicionales para potenciar la penetración dérmica de estabilidad de los principios activos o la formulación. Todas aquellas formulaciones transdérmicas conocidas y componentes están incluidos dentro del alcance proporcionado en el presente documento. Los compuestos proporcionados en el presente documento también se pueden administrar por un dispositivo transdérmico. Por consiguiente, la administración transdérmica se puede llevar a cabo usando un parche o del tipo depósito o de membrana porosa, o de una variedad de matrices sólidas.
Los componentes anteriormente descritos para composiciones administrables por vía oral, inyectables o administrables por vía tópica son simplemente representativos. Otros materiales, así como técnicas de procesamiento y similares, se exponen en la Parte 8 de Remington's Pharmaceutical Sciences, 17a edición, 1985, Mack Publishing Company, Easton, Pennsilvania.
Los compuestos de la presente invención también se pueden administrar en formas de liberación sostenida o de sistemas de administración de fármacos de liberación sostenida. Una descripción de materiales representativos de liberación sostenida se puede encontrar en Remington's Pharmaceutical Sciences.
La presente invención también se refiere a las formulaciones farmacéuticamente aceptables de un compuesto de la presente invención. En una realización, la formulación comprende agua. En otra realización, la formulación comprende un derivado de ciclodextrina. Las ciclodextrinas más comunes son a-, p- y Y-ciclodextrinas que consisten en 6, 7 u 8 unidades de glucosa unidas en a-1,4, respectivamente, que comprenden opcionalmente uno o más sustituyentes en los restos de azúcar unidos, que incluyen, pero no se limitan a, sustitución metilada, hidroxialquilada, acilada y de sulfoalquil éter. En ciertas realizaciones, la ciclodextrina es una p-ciclodextrina de sulfoalquil éter, por ejemplo, por ejemplo, p-ciclodextrina de sulfobutil éter, también conocida como Captisol®. Véase, por ejemplo, el documento de patente U.S. 5.376.645. En ciertas realizaciones, la formulación comprende hexapropil-p-ciclodextrina (por ejemplo, 10-50 % en agua).
La presente invención también se refiere a la sal de adición de ácido farmacéuticamente aceptable de un compuesto de la presente invención. El ácido que se puede usar para preparar la sal farmacéuticamente aceptable es la que forma una sal de adición de ácido no tóxica, es decir, una sal que contiene aniones farmacológicamente aceptables, tales como clorhidrato, yodhidrato, bromhidrato, nitrato, sulfato, bisulfato, fosfato, acetato, lactato, citrato, tartrato, succinato, maleato, fumarato, benzoato, para-toluenosulfonato, y similares.
Los siguientes ejemplos de formulación ilustran composiciones farmacéuticas representativas que se pueden preparar según la presente invención. La presente invención, sin embargo, no se limita a las siguientes composiciones farmacéuticas.
Formulación 1 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 240-270 mg (80-90 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Formulación 2 a modo de ejemplo - Cápsulas: Se puede mezclar un compuesto de la presente invención como un polvo seco con un diluyente de almidón en una relación ponderal aproximada de 1:1. La mezcla se llena en cápsulas de 250 mg (125 mg de compuesto activo por cápsula).
Formulación 3 a modo de ejemplo - Líquido: Se puede mezclar un compuesto de la presente invención (125 mg) con sacarosa (1,75 g) y goma xantana (4 mg) y la mezcla resultante se pueden combinar, pasar a través de un tamiz estadounidense de malla N.° 10 y luego se mezcla con una disolución previamente preparada de celulosa microcristalina y carboximetilcelulosa de sodio (11:89, 50 mg) en agua. Se diluyen benzoato de sodio (10 mg), aroma y color con agua y se añaden con agitación. Entonces se puede añadir agua suficiente para producir un volumen total de 5 mL.
Formulación 4 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 450-900 mg (150-300 mg de compuesto activo) en una prensa para comprimidos.
Formulación 5 a modo de ejemplo - Inyección: Se puede disolver o suspender un compuesto de la presente invención en una medio acuoso de solución salina estéril inyectable tamponada hasta una concentración de aproximadamente 5 mg/mL.
Formulación 6 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 90-150 mg (30-50 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Formulación 7 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 30-90 mg (10-30 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Formulación 8 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 0,3-30 mg (0,1-10 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Formulación 9 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 150-240 mg (50-80 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Formulación 10 a modo de ejemplo - Comprimidos: Se puede mezclar un compuesto de la presente invención como un polvo seco con un aglutinante de gelatina seca en una relación ponderal aproximada de 1:2. Se añade una cantidad menor de estearato de magnesio como un lubricante. La mezcla se conforma en comprimidos de 270-450 mg (90-150 mg de compuesto activo por comprimido) en una prensa para comprimidos.
Indicaciones médicas. Como se describe en general en el presente documento, la presente invención se refiere al compuesto SA-4 o una sal farmacéuticamente aceptable del mismo para su uso en un sujeto humano como un modulador del receptor de GABAa. En ciertas realizaciones, se prevé que dichos compuestos (compuesto SA-4 y sales farmacéuticamente aceptables del mismo) sean útiles como agentes terapéuticos para tratar un trastorno del estado de ánimo. En ciertas realizaciones, el compuesto se administra por administración intravenosa.
Estudios previos (véase, por ejemplo, Gee et al., European Journal of Pharmacology, 136:419-423 (1987)) mostraron que ciertos esteroides 3a-hidroxilados son órdenes de magnitud más potentes que los moduladores del complejo receptor de GABA (GRC) que lo que otros habían informado (véase, por ejemplo, Majewska et al., Science 232:1004-1007 (1986); Harrison et al., J Pharmacol. Exp. Ther. 241:346-353 (1987)). Majewska et al. y Harrison et al. enseñaron que los esteroides 3a-hidroxilados-5-reducidos son solo capaces de niveles muchos más bajos de eficacia. Datos experimentales in vitro e in vivo han mostrado ahora que la alta potencia de estos esteroides les permite ser terapéuticamente útiles en la modulación de la excitabilidad del cerebro por el GRC (véase, por ejemplo, Gee et al., European Journal of Pharmacology, 136:419-423 (1987); Wieland et al., Psychopharmacology 118(1):65-71 (1995)). También se han preparado diversos esteroides sintéticos como esteroides neuroactivos. Véase, por ejemplo, la patente de EE. u U. 5.232.917, que desvela compuestos esteroides neuroactivos útiles en el tratamiento de estrés, ansiedad, insomnio, trastornos convulsivos y trastornos del estado de ánimo, que son susceptibles a agentes activos del GRC, tales como depresión, en un modo terapéuticamente beneficioso. Además, se ha mostrado previamente que estos esteroides interactúan en un sitio único en el GRC que es distinto de otros sitios de interacción conocidos (por ejemplo, barbitúricos, benzodiacepinas y GABA) donde se han provocado previamente efectos terapéuticamente beneficiosos sobre el estrés, la ansiedad, el sueño, los trastornos del estado de ánimo y los trastornos convulsivos (véase, por ejemplo, Gee, K.W. y Yamamura, H.I., "Benzodiazepines and Barbiturates: Drugs for the Treatment of Anxiety, Insomnia and Seizure Disorders", en Central Nervous System Disorders, Horvell, ed., Marcel-Dekker, Nueva York (1985), pp. 123-147; Lloyd, K.G. y Morselli, P.L., "Psychopharmacology of GABAergic Drugs," en Psychopharmacology: The Third Generation of Progress, H.Y. Meltzer, ed., Raven Press, N.Y. (1987), pp. 183-195; y Gee et al., European Journal of Pharmacology, 136:419-423 (1987). Estos compuestos son deseables por su duración, potencia y actividad oral (junto con otras formas de administración).
Los compuestos de la presente invención, como se describen en el presente documento se diseñan, en general, para modular la función de GABA y, por lo tanto, para actuar de esteroides neuroactivos para el tratamiento y la prevención de afecciones relacionadas con el SNC en un sujeto. La modulación, como se usa en el presente documento, se refiere a la inhibición o potenciación de la función de receptores GABA. Por consiguiente, los compuestos y las composiciones farmacéuticas proporcionadas en el presente documento encuentran uso como terapéuticos para prevenir y/o tratar afecciones del SNC en mamíferos que incluyen seres humanos y mamíferos no humanos. Así, y como se estableció antes, la presente invención incluye dentro de su alcance, y se extiende a, los métodos de tratamiento citados, así como a los compuestos para dichos métodos, y al uso de dichos compuestos para la preparación de medicamentos útiles para dichos métodos.
Las afecciones del SNC a modo de ejemplo relacionadas con la modulación de GABA incluyen trastornos del estado de ánimo [por ejemplo, depresión, trastorno distímico (por ejemplo, depresión leve), trastorno bipolar (por ejemplo, I y/o II), trastornos de ansiedad (por ejemplo, trastorno de ansiedad generalizada (TAG), trastorno de ansiedad social), estrés, trastorno de estrés postraumático (TEPT), trastornos compulsivos (por ejemplo, trastorno obsesivo-compulsivo (TOC))].
En otro aspecto más, se proporciona una combinación de un compuesto para su uso según la presente invención y otro agente farmacológicamente activo. El compuesto SA-4 o la sal farmacéuticamente aceptable del mismo proporcionada en el presente documento se pueden administrar como el único agente activo o se pueden administrar en combinación con otros agentes. La administración en combinación puede proceder por cualquier técnica evidente para los expertos en la técnica que incluye, por ejemplo, administración separada, secuencial, concurrente y alterna.
En otro aspecto más, se proporciona el compuesto SA-4 o una sal farmacéuticamente aceptable del mismo para su uso en un método de tratamiento o prevención de estrés o ansiedad en un sujeto, que comprende administrar al sujeto en necesidad de tal tratamiento una cantidad eficaz de dicho compuesto, o una composición el mismo.
En otro aspecto más, se proporciona el compuesto SA-4 o una sal farmacéuticamente aceptable del mismo para su uso en un método de alivio o prevención de SPM o DPP en un sujeto, que comprende administrar al sujeto en necesidad de tal tratamiento una cantidad eficaz de dicho compuesto.
En otro aspecto más, se proporciona el compuesto SA-4 o una sal farmacéuticamente aceptable del mismo para su uso en un método de tratamiento o prevención de trastornos del estado de ánimo en un sujeto, que comprende administrar al sujeto en necesidad de tal tratamiento una cantidad eficaz de dicho compuesto. En ciertas realizaciones, el trastorno del estado de ánimo es depresión.
En ciertas realizaciones, el compuesto se administra al sujeto crónicamente. En ciertas realizaciones, el compuesto se administra al sujeto por vía oral, por vía subcutánea, por vía intramuscular o por vía intravenosa.
Trastornos de ansiedad
El trastorno de ansiedad es un término general que cubre varias formas diferentes de miedo anormal y patológico y ansiedad. Los actuales criterios de diagnóstico psiquiátrico reconocen una amplia variedad de trastornos de ansiedad.
El trastorno de ansiedad generalizada es un trastorno crónico común caracterizado por ansiedad de larga duración que no se concreta en ningún objeto o situación. Los que sufren de ansiedad generalizada experimentan miedo y preocupación persistentes no específicos y se preocupan de manera excesiva con los asuntos cotidianos. El trastorno de ansiedad generalizada es el trastorno de ansiedad más común que afecta a los ancianos.
En el trastorno de pánico, una persona padece breves ataque de terror intenso y aprensión, frecuentemente marcados por temblores, sacudidas, confusión, mareos, náuseas, dificultad para respirar. Estos ataques de pánico, definidos por la APA como el miedo o el malestar que aparece bruscamente y alcanza su punto máximo en menos de diez minutos, pueden durar varias horas y pueden desencadenarse por estrés, miedo o incluso ejercicio; aunque la causa específica no siempre es evidente. Además de los ataques de pánico inesperados recurrentes, a un diagnóstico de trastorno de pánico también requiere que dichos ataques tengan consecuencias crónicas: ya sean preocupación por las posibles implicaciones de los ataques, miedo persistente a ataques futuros, o cambios significativos en el comportamiento relacionados con los ataques. Por consiguiente, los que sufren de trastorno de pánico experimentan síntomas incluso fuera de los episodios específicos de pánico. Frecuentemente, los que sufren de pánico notan cambios normales en los latidos del corazón, lo que los lleva a pensar que algo está mal con su corazón o que están a punto de tener otro ataque de pánico. En algunos casos, se produce una mayor conciencia (hipervigilancia) del funcionamiento corporal durante los ataques de pánico, en donde cualquier cambio fisiológico percibido se interpreta como una posible enfermedad mortal (es decir, hipocondriasis extrema).
El trastorno obsesivo compulsivo es un tipo de trastorno de ansiedad caracterizado principalmente por obsesiones repetitivas (pensamientos o imágenes perturbadoras, persistentes y compulsivos) y compulsiones (impulsos por realizar actos o rituales específicos). El patrón de pensamiento del TOC puede compararse con las supersticiones en tanto que una creencia en una relación causal donde, en realidad, no existe. Frecuentemente, el proceso es completamente ilógico; por ejemplo, la compulsión de caminar con un cierto patrón se puede emplear para aliviar la obsesión de un daño inminente. Y en muchos casos, la compulsión es completamente inexplicable, simplemente un impulso por completar un ritual desencadenado por el nerviosismo. En una minoría de casos, los que sufren TOC solo pueden sufrir obsesiones, sin compulsiones abiertas; un número mucho menor de los que lo sufren, experimentan solamente compulsiones.
La categoría individual más grande de trastornos de ansiedad es la de la fobia, que incluye todos los casos en los que el miedo y la ansiedad se desencadenan por un estímulo o situación específicos. Los que la sufren normalmente anticipan consecuencias terribles al encontrarse con el objeto de su miedo, que puede ser cualquier cosa, desde un animal hasta una ubicación o un líquido corporal.
Es trastorno de estrés postraumático o TEPT es un trastorno de ansiedad que resulta de una experiencia traumática. El estrés postraumático puede resultar de una situación extrema, tal como combate, violación, situaciones hostiles o incluso un accidente grave. También puede resultar de una exposición a largo plazo (crónica) a un factor estresante intenso, por ejemplo, los soldados que soportan batallas individuales pero no pueden hacer frente al combate continuo. Los síntomas comunes incluyen analepsia, conductas de evitación y depresión.
Ejemplos
De los siguientes ejemplos, el Ejemplo 20 desvela la síntesis del compuesto SA-4 (indicado para su uso en los tratamientos que se definen en las reivindicaciones según la presente invención) y los métodos de ensayo que aparecen al final de los ejemplos ilustran la invención reivindicada en la medida en que apoyen las indicaciones terapéuticas de las reivindicaciones para el compuesto SA-4 y el sales farmacéuticamente aceptables del mismo.
Todos los otros ejemplos no se refieren a la presente invención y están presentes a efectos de comparación solo.
Materiales y métodos
Los compuestos proporcionados en el presente documento se pueden preparar a partir de materiales de partida fácilmente disponibles usando los siguientes métodos generales y procedimientos. Se apreciará que donde se dan condiciones de proceso típicas o preferidas (es decir, temperaturas de reacción, tiempos, relaciones molares de reactantes, disolventes, presiones, etc.), también se pueden usar otras condiciones de proceso, a menos que se establezca lo contrario. Las condiciones de reacción óptimas pueden variar con los reactantes particulares o el disolvente usado, pero dichas condiciones pueden ser determinadas por un experto en la técnica por optimización habitual.
Además, como será evidente por los expertos en la técnica, pueden ser necesarios grupos protectores convencionales se para prevenir que ciertos grupos funcionales se sometan a reacciones no deseadas. Se conocen bien en la técnica la elección de un grupo protector adecuado para un grupo funcional particular, así como condiciones adecuadas para la protección y desprotección. Por ejemplo, se describen numerosos grupos protectores, y su introducción y retirada, en T. W. Greene y P. G. M. Wuts, Protecting Groups in Organic Synthesis, segunda edición, Wiley, Nueva York, 1991, y referencias citadas en su interior.
Los compuestos proporcionados en el presente documento se pueden aislar y purificar por procedimientos convencionales conocidos. Dichos procedimientos incluyen (pero no se limitan a) recristalización, cromatografía en columna, HPLC o cromatografía de fluidos supercríticos (SFC). Los siguientes esquemas se presentan con detalles en cuanto a la preparación de pirazoles representativos que se han enumerado en el presente documento. Los compuestos proporcionados en el presente documento pueden ser preparados por un experto en la técnica de la síntesis orgánica a partir de materiales de partida y reactivos conocidos o disponibles comercialmente. Las columnas quirales a modo de ejemplo disponibles para su uso en la separación/purificación de los enantiómeros/diaestereómeros proporcionados en el presente documento incluyen, pero no se limitan a, CHIRALPAK® AD-10, CHIRALCEL® OB, CHIRALCEL® OB-H, CHIRALCEL® OD, CHIRALCEL® OD-H, CHIRALCEL® OF, CHIRALCEL® OG, CHIRALCEL® OJ y CHIRALCEL® OK.
Los RMN 1H informados en el presente documento (por ejemplo, para productos intermedios) pueden ser una representación parcial del espectro de RMN completo de un compuesto, por ejemplo, un compuesto descrito en el presente documento. Por ejemplo, los RMN 1H informados pueden excluir la región entre 8 (ppm) de aproximadamente 1 a aproximadamente 2,5 ppm. Las copias del espectro de RMN 1H completo para ejemplos representativos se proporcionan en las figuras.
Método general a modo de ejemplo para HPLC preparativa: Columna: Waters RBridge prep 10 pm C18, 19*250 mm. Fase móvil: acetonitrilo, agua (NH4HCO3) (30 L de agua, 24 g de NH4HCO3, 30 mL de NH3.H2O). Caudal: 25 mL/min
Método general a modo de ejemplo para HPLC analítica: Fase móvil: A: agua (NH4HCO310 mM), B: gradiente de acetonitrilo: 5 %-95 % de B en 1,6 o 2 min; caudal: 1,8 o 2 mL/min; columna: XBridge C18, 4,6*50mm, 3,5 pm a 45 °C.
Procedimientos de síntesis
Los compuestos de los ejemplos, que incluyen el compuesto SA-4 que aparece en las reivindicaciones y otros compuestos de los ejemplos incluidos a efectos de comparación, se pueden preparar según métodos descritos en la técnica (Upasani et al., J. Med. Chem. 1997, 40:73-84; y Hogenkamp et al., J. Med. Chem. 1997, 40:61-72) y usando los reactivos apropiados, materiales de partida y métodos de purificación conocidos por los expertos en la técnica. Los compuestos descritos en el presente documento, que incluyen el compuesto SA-4 que aparece en las reivindicaciones y otros compuestos que aparecen a efectos de comparación, se pueden preparar usando los métodos mostrados en los Esquemas generales 1-3, que comprenden una sustitución nucleófila de bromuro de 19-nor-pregnano con un nucleófilo. En estos procesos, puede ser que el nucleófilo reaccione con el bromuro de 19-nor-pregnano en presencia de K2CO3 en THF.
Esquema 1

Esquema 2

Esquema 3

Ejemplo 1. Síntesis de SA y productos intermedios de SA

Síntesis del compuesto SA-B. Se hidrogenó el compuesto SA (50 g, 184 mimóles) y negro de paladio (2,5 g) en tetrahidrofurano (300 mL) y ácido bromhídrico concentrado (1,0 mL) con 10 atm de hidrógeno. Después de agitar a temperatura ambiente durante 24 h, la mezcla se filtró a través de una almohadilla de celite y el filtrado se concentró a vacío proporcionando el compuesto en bruto. La recristalización en acetona dio compuesto SA-B (42,0 g, rendimiento: 83,4%) como un polvo blanco.
RMN 1H: (400 MHz, CDCh) 82,45-2,41 (m, 1H), 2,11-3,44 (m, 2H), 3,24 (s, 3H), 2,18-2,15 (m, 1H), 2,01-1,95 (m, 1H), 1,81-1,57 (m, 7H), 1,53-1,37 (m, 7H), 1,29-1,13 (m, 3H), 1,13-0,90 (m, 2H), 0,89 (s, 3H).
Síntesis del compuesto SA-C. Se añadió gota a gota una disolución de SA-B (42,0 g, 153,06 mmoles) en 600 mL de tolueno anhidro a la de disolución de bis(2,6-di-terc-butil-4-metilfenóxido de metilaluminio (MAD) (459,19 mmoles, 3,0 eq, recién preparada) bajo N2 a -78 °C. Después de completarse la adición, la mezcla de reacción se agitó durante 1 h a -78 °C. Entonces se añadió lentamente gota a gota MeMgBr 3,0 M (153,06 mL, 459,19 mmoles) a la mezcla anterior bajo N2 a -78 °C. Entonces la mezcla de reacción se agitó durante 3 h a esta temperatura. La CCF (éter de petróleo/acetato de etilo = 3:1) mostró que la reacción estaba completa. Entonces se añadió lentamente gota a gota NH4Cl acuoso saturado a la mezcla anterior a -78 °C. Después de completarse la adición, la mezcla se filtró, la torta de filtración se lavó con EtOAc, la fase orgánica se lavó con agua y salmuera, se secó sobre Na2SO4 anhidro, se filtró y se concentró, se purificó por cromatografía ultrarrápida sobre gel de sílice (éter de petróleo/acetato de etilo 20:1 a 3:1) proporcionando el compuesto SA-C (40,2 g, rendimiento: 90,4%) como un polvo blanco. RMN 1H: (400 MHz, CDCla) 82,47-2,41 (m, 1H), 2,13-2,03 (m, 1H), 1,96-1,74 (m, 6H), 1,70-1,62 (m, 1H), 1,54-1,47 (m, 3H), 1,45-1,37 (m, 4H), 1,35-1,23 (m, 8H), 1,22-1,10 (m, 2H), 1,10-1,01 (m, 1H), 0,87 (s, 3H).
Síntesis del compuesto SA-D. A una disolución de PPhbEtBr (204,52 g, 550,89 mmoles) en THF (500 mL) se añadió una disolución de t-BuOK (61,82 g, 550,89 mmoles) en THF (300 mL) a 0 °C. Después de completarse la adición, la mezcla de reacción se agitó durante 1 h 60 °C, entonces se añadió gota a gota SA-C (40,0 g, 137,72 mmoles) disuelto en THF (300 mL) a 60 °C. La mezcla de reacción se calentó a 60 °C durante 18 h. La mezcla de reacción se enfrió hasta temperatura ambiente y se extinguió con NH4Cl sat., se extrajo con EtOAc (3*500 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron y se concentraron dando el producto en bruto, que se purificó por una cromatografía ultrarrápida en columna (éter de petróleo/acetato de etilo 50:1 a 10:1) proporcionando el compuesto SA-D (38,4 g, rendimiento: 92 %) como un polvo blanco. RMN 1H: (400 MHz, CDCh) 85,17-5,06 (m, 1H), 2,42-2,30 (m, 1H), 2,27-2,13 (m, 2H), 1,89-1,80 (m, 3H), 1,76-1,61 (m, 6H), 1,55-1,43 (m, 4H), 1,42-1,34 (m, 3H), 1,33-1,26 (m, 6H), 1,22-1,05 (m, 5H), 0,87 (s, 3H).
Síntesis del compuesto SA-E. A una disolución de SA-D (38,0 g, 125,62 mmoles) en THF seco (800 mL) se añadió gota a gota una disolución de BH3.Me2S (126 mL, 1,26 moles) bajo baño de hielo. Después de completarse la adición,
la mezcla de reacción se agitó durante 3 h a temperatura ambiente (14-20 °C). La CCF (éter de petróleo/acetato de etilo 3:1) mostró que la reacción estaba completa. La mezcla se enfrió hasta 0 °C y se añadió disolución acuosa 3,0 M de NaOH (400 mL), seguido por 30 % de H2O2 acuosa (30 %, 300 mL). La mezcla se agitó durante 2 h a temperatura ambiente (14-20 °C), y luego se filtró, se extrajo con EtOAc (3*500 mL). Las fases orgánicas combinadas se lavaron con Na2S2O3 acuoso saturado, salmuera, se secaron sobre Na2SO4 y se concentraron a vacío dando el producto en bruto (43 g, en bruto) como un aceite incoloro. El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SA-F. A una disolución de SA-E (43,0 g, 134,16 mmoles) en diclorometano (800 mL) se añadió en porciones a 0 °C y PCC (53,8 g, 268,32 mmoles). Entonces, la mezcla de reacción se agitó a temperatura ambiente (16-22 °C) durante 3 h. La CCF (éter de petróleo/acetato de etilo 3:1) mostró que la reacción estaba completa, entonces la mezcla de reacción se filtró, se lavó con DCM. La fase orgánica se lavó con Na2S2O3 acuoso saturado, salmuera, se secó sobre Na2SO4 y se concentró a vacío dando el producto en bruto. El producto en bruto se purificó por una cromatografía ultrarrápida en columna (éter de petróleo/acetato de etilo 50:1 a 8:1) proporcionando el compuesto SA-F (25,0 g, rendimiento: 62,5 %, en dos etapas) como un polvo blanco. RMN 1H (sA-F): (400 MHz, CDCh) 52,57-2,50 (m, 1H), 2,19-2,11 (m, 4H), 2,03-1,97 (m, 1H), 1,89-1,80 (m, 3H), 1,76-1,58 (m, 5H), 1,47-1,42 (m, 3H), 1,35-1,19 (m, 10H), 1,13-1,04 (m, 3H), 0,88-0,84 (m, 1H), 0,61 (s, 3H).
Síntesis del compuesto SA. A una disolución de SA-F (10 g, 31,4 mmoles) y HBr ac. (5 gotas, 48 % en agua) en 200 mL de MeOH se añadió gota a gota bromo (5,52 g, 34,54 mmoles). La mezcla de reacción se agitó a 17 °C durante 1,5 h. La disolución resultante se inactivó con NaHCO3 acuoso saturado a 0 °C y se extrajo con EtOAc (150 mL x 2). Las fases orgánicas combinadas se secaron y se concentraron. El residuo se purificó por cromatografía en columna sobre gel de sílice eluida con (EP:AE = 15:1 a 6:1) proporcionando el compuesto SA (9,5 g, rendimiento: 76,14 %) como un sólido blanco. EM/CL: tr 5,4 min; m/z 379,0, 381,1, 396,1.
Ejemplo 2. Síntesis de SB y productos intermedios de SB
Síntesis de los compuestos SB-B y SB-C. Se añadieron pequeños trozos de litio (7,63 g, 1,1 moles) a 2,7 L de amoniaco condensado en un matraz de tres bocas a -70 °C. Tan pronto como se disolvió todo el litio, la disolución azul se calentó hasta - 50 °C. Se añadió gota a gota una disolución de 19-norandrost-4-eno-3,17-diona SB-A (1,30 g, 110 mmoles) y terc-BuOH (8,14 g, 110 mmoles) en 800 mL de tetrahidrofurano anhidro y se agitó durante 90 min hasta que la mezcla de reacción viró a amarillo claro. Se añadió cloruro de amonio (70 g) y se dejó que se evaporara el exceso de amoniaco. El residuo se extrajo con HCl 0,5 N (500 mL) y diclorometano (500 mL * 2). Las fases orgánicas combinadas se lavaron con disolución saturada de NaHCO3, se secaron sobre Na2SO4, se filtraron y se concentraron dando una mezcla de SB-B y SB-C (21 g, 70 %) que se usó directamente en la siguiente etapa sin más purificación. Se añadió una disolución de SB-B y SB-C (21 g, 76 mmoles) en 50 mL de anhidro diclorometano a una suspensión de clorocromato de piridinio (PCC) (32,8 g, 152 mmoles) en 450 mL de diclorometano. Después de agitar a temperatura ambiente durante 2 h, se añadió disolución 2 N de NaOH (500 mL) a la mezcla de reacción marrón oscura y se agitó durante otros 10 min. La disolución resultante se extrajo con diclorometano, las fases orgánicas combinadas se lavaron con HCl 2 N, salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 20:1 a 10 :1 ) proporcionando el compuesto del título SB-C (16,8 g, 80 %) como un sólido blanco. RMN 1H de SB-B (400 MHz, CDCh), 8 (ppm), 3,65 (t, 1H, 1H), 0,77 (s, 3H). RMN 1H de SB-C (400 MHz, CDCh), 8 (ppm), 0,88 (s, 3H).
Síntesis del compuesto SB-D. A una disolución del compuesto SB-C (16,8 g, 61,3 mmoles) en metanol (250 mL) se añadió yodo (1,54 g, 6,1 mmoles). Después de agitar a 60 °C durante 12 h, el disolvente se retiró a vacío. El producto en bruto se disolvió en diclorometano (200 mL) y se lavó con NaHCO3 saturado (150 mL), salmuera, se secó sobre Na2SO4, se filtró y se concentró. El residuo se purificó por cromatografía sobre alúmina básica (éter de petróleo/acetato de etilo = 100:1) dando el compuesto SB-D (14 g, 43,8 mmoles, 71 %). RMN 1H (400 MHz, CDCh), 8 (ppm), 3,18 (s, 3H), 3,12 (s, 3H), 0,85 (s, 3H).
Síntesis del compuesto SB-E. A una suspensión de t-BuOK (7,36 g, 65,7 mmoles) en THF (100 mL) a 0 °C se añadió bromuro de etiltrifenilfosfonio (26 g, 70 mmoles) lentamente. Después de agitar a 60 °C durante 3 h, se añadió el compuesto SB-D (7 g, 21,9 mmoles) y la mezcla se agitó a 60 °C durante otras 2 h. Después del enfriamiento hasta temperatura ambiente, la mezcla de reacción se vertió en cloruro de amonio saturado y se extrajo con EtOAc (2 * 500 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron proporcionando el compuesto en bruto SB-E (7,36 g, 100%). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SB-F. Se acidificó una disolución del compuesto en bruto SB-E (7,36 g, 21,9 mmoles) en THF (50 mL) hasta pH = 3 por HCl acuoso 1 N. Después de agitar a temperatura ambiente durante 12 h, la mezcla de reacción se extrajo con acetato de etilo (250 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna (éter de petróleo/acetato de etilo = 30:1 a 20:1) proporcionando el compuesto SB-F (4,8 g, 16,7 mmoles, 76 % para dos etapas). RMN 1H (400 MHz, CDCh), 8 (ppm), 5,12-5,10 (m, 1H), 0,77 (s, 3H).
Síntesis del compuesto SB-G. A una disolución de MeMgBr (28 mmoles, 1 M en THF) en THF (50 mL) a 0°C se añadió una disolución del compuesto SB-F (4,8 g, 16,8 mmoles) en THF seco (10 mL) por una bomba de jeringa durante 30 min. Después de agitar a 0 °C durante 5 h, se dejó que la mezcla de reacción se calentara y se agitó a temperatura ambiente durante la noche. La mezcla de reacción se inactivó con agua fría helada y se extrajo con acetato de etilo (150 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo blanco se purificó por cromatografía ultrarrápida en columna (éter de petróleo/acetato de etilo = 20:1 a 10:1) dando el compuesto SB-G (2,5 g, 8,28 mmoles, 49 %; Rf = 0,35, éter de petróleo/acetato de etilo = 10:1). RMN 1H (400 MHz, CDCh), 8 (ppm), 5,05-5,03 (m, 1H), 1,21 (s, 3H), 0,90 (s, 3H).
Síntesis del compuesto SB-H. A una disolución del compuesto SB-G (2 g, 6,62 mmoles) en THF seco (50 mL) se añadió el complejo de borano-tetrahidrofurano (20 mL; disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (10 mL), seguido por 30 % de disolución acuosa de H2O2 (12 mL). Después de agitar a temperatura ambiente durante una hora, la mezcla se extrajo con EtOAc (3 * 100 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secaron sobre MgSO4, se filtraron y se concentraron proporcionando el compuesto en bruto SB-H (2 g, 100 %). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SB-I. A una disolución del compuesto en bruto SB-H (2 g, 6,62 mmoles) en 60 mL de diclorometano húmedo (el diclorometano se había agitado con varios mililitros de H2O, luego se separó de la fase acuosa) se añadió peryodinato de Dess-Martin (5,5 g, 13 mmoles). Después de agitar a temperatura ambiente durante 24 h, la mezcla de reacción se extrajo con diclorometano (3 * 100 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1) proporcionando el compuesto SB-I (1 g, 3,14 mmoles, 47% para dos etapas) como un sólido blanco. RMN 1H (400 MHz, CDCh), 8 (ppm), 2,56 (t, 1H), 2,11 (s y m, 4H), 2,0 (dt, 1H), 1,8 (dm, 2H), 1,54 (m, 6 H) 1,43 (m, 1H), 1,34 (m, 2H), 1,20 (m, 12H), 0,7 (m, 2H), 0,62 (s, 3H).
Síntesis del compuesto SB. A una disolución del compuesto SB-I (600 mg, 1,89mmoles) en MeOH (20 mL) se añadió 5 gotas de HBr (48 %), seguido por bromo (302 mg, 1,89 mmoles). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se vertió en agua con hielo, luego se extrajo con acetato de etilo (100 mL x 3). Las fases orgánicas combinadas se lavaron con salmuera (200 mL), se secaron sobre MgSO4, se filtraron y se concentraron dando el compuesto en bruto SB (600 mg).
Síntesis del compuesto SB-J. Una disolución del compuesto SB (600 mg, 1,5 mmoles) en acetona 10 mL se trató con CF3COOH (6,8 mL) y EtaN (9,5 mL). Después de someterse a reflujo durante 30 min, se añadió sal de CFsCOONa (4,49 g, 33 mmoles) en partes durante un periodo de 10 h. La mezcla de reacción se dejó enfriar hasta temperatura ambiente y el disolvente se retiró a vacío. El residuo se extrajo con acetato de etilo, se secó sobre MgSO4, se filtró y se concentró. La mezcla se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 3:1) proporcionando SB-J (300 mg, rendimiento: 50 % para dos etapas). RMN 1H (400 MHz, CDCl3), 5 (ppm), 4,23 4,13 (m, 2H), 2,48-2,44 (m), 0,64 (s, 3H).
Ejemplo 3. Síntesis del compuesto SA-V
Síntesis del compuesto SA-K. Se añadió el compuesto SA-J (10 g, 36,7 mimóles) a 50 mL de cloruro de acetilo y 50 mL de anhídrido acético. La mezcla de reacción se calentó hasta 120 °C durante 5 h, se evaporó a vacío proporcionando SA-K como un sólido blanco (10 g, 87 % de rendimiento). RMN 1H (400 MHz, CDCh), 8 (ppm), 5,78 (s, 1H), 5,55 (s, 1H), 2,4 (2H,dd) , 2,13 (s, 3H), 0,90 (s, 3H).
Síntesis del compuesto SA-L. A una disolución del reactante SA-K (10 g, 31,8 mmoles) en 200 mL de THF y 20 mL de H2O, se añadió mCPBA (11 g, 63,6 mmoles) a 0 °C, se agitó a ta durante 15 h, la mezcla de reacción se extrajo con 500 mL de EtOAc, se lavó con 100 mL de Na2SO3 saturado, 100 mL de NaHCO3 saturado y 100 mL de salmuera y se evaporó a vacío, luego se purificó por cromatografía ultrarrápida en gel de sílice sobre gel de sílice (éter de petróleo/acetato de etilo = 5:1) proporcionando SA-L-1 como un sólido blanco (2,2 g, 24% de rendimiento) (eluyó primero) y SA-L como el sólido blanco (1,1 g, 12% de rendimiento) (eluyó segundo). SA-L-1: RMN 1H (400 MHz, CDCh), 8 (ppm), 5,92 (s, 1H), 4,44 (s, 1H), 0,95 (s, 3H). SA-L: RMN 1H (400 MHz, CDCh), 8 (ppm), 6,25 (s, 1H), 4,28 4,25 (m, 1H), 0,93 (s, 3H).
Síntesis del compuesto SA-M. A una disolución de SA-L (2 g, 6,94 mmoles) en 50 mL de EtOAc, se añadió 200 mg de Pd/C. La mezcla de reacción se hidrogenó en 1 atm de H2 durante 15 h. La mezcla de reacción se evaporó a vacío, luego se purificó por cromatografía (éter de petróleo/acetato de etilo = 1:2) proporcionando SA-M como un sólido blanco (1,5 g, 75 % de rendimiento). RMN 1H (400 MHz, CDCh), 8 (ppm), 3,97 (td, 1H), 0,88 (s, 3H).
Síntesis del compuesto SA-N. A una disolución de SA-M (1 g, 3,4 mmoles) en 100 mL de MeOH, se añadió 50 mg de TsOH, se calentó hasta 60 °C durante 2 h. La mezcla de reacción se extrajo con 500 mL de EtOAc, se lavó con 100 mL de NaHCO3 sat., 100 mL de disolución de salmuera y se evaporó a vacío proporcionando SA-N como un sólido blanco (1 g, 91 % de rendimiento).
Síntesis del compuesto SA-O. A una disolución de bromuro de etiltrifenilfosfonio (10,67 g, 28,84 mmoles) en 30 mL de THF, se añadió KOt-Bu (3,23 g, 28,80 mmoles). La reacción se calentó hasta 60 °C durante 1 h. Se añadió SA-N (3,23 g, 9,6 mmoles) a la mezcla, se agitó a 60 °C durante 15 h. La mezcla de reacción se extrajo con 500 mL de EtOAc, se lavó con disolución de salmuera y se evaporó a vacío, luego se purificó por cromatografía (éter de petróleo/acetato de etilo = 3:1) proporcionando SA-O como un sólido blanco (2 g, 62% de rendimiento). RMN 1H (400 MHz, MeOD), 8 (ppm) 5,15-5,12 (m, 1H), 3,80-3,78 (m, 1H), 3,21 (s, 3H), 3,15 (s, 3H), 1,67 (d, 3H), 0,95 (s, 3H).
Síntesis del compuesto SA-P. A una disolución de SA-O (0,5 g, 1,43 mmoles) en 10 mL de DCM, se añadió DAST 0,5 mL a -78 °C. La mezcla de reacción se agitó a -78 °C durante 30 min, luego se inactivó con 5 mL de NaHCO3 sat., se extrajo con 50 mL de DCM, se lavó con salmuera, se secó y se concentró a vacío, se purificó por cromatografía (éter de petróleo/acetato de etilo = 30:1) proporcionando SA-P como un sólido blanco 175 mg, 35 % de rendimiento.
Síntesis del compuesto SA-Q. A una disolución de SA-P (350 mg, 1 mmol) en 20 mL de THF, se añadió 2 mL de HCl 2 M, se agitó a ta durante 1 h. La mezcla de reacción se inactivó con 5 mL de H2O y se extrajo con 100 mL de EtOAc, se lavó con salmuera y se evaporó a vacío, luego se purificó por cromatografía (éter de petróleo/acetato de etilo = 10:1) proporcionando s A-Q como un sólido blanco (210 mg, 60 % de rendimiento). RMN 1H (400 MHz, CDCh), 8 (ppm) 5,17-5,14 (m, 1H), 4,80-4,66 (m, 1H), 2,61-2,57 (m, 1H), 1,79 (d, 3H), 0,93 (s, 3H).
Síntesis del compuesto SA-R. A una disolución con agitación de yoduro de trimetilsulfonio (3,2 g, 16 mmoles) en 10 mL de DMSO se añadió NaH (60 %; 400 mg, 16 mmoles). Después de agitar a temperatura ambiente durante 1 h, se añadió gota a gota una suspensión de SA-Q (486 mg, 1,6 mmoles) en 5 mL de DMSO. Después de 15 h, la mezcla de reacción se vertió en agua fría con hielo (100 mL) y se extrajo con 300 mL de EtOAc, se lavó con 100 mL de disolución de salmuera y se evaporó a vacío, luego se purificó por cromatografía (éter de petróleo/acetato de etilo = 10 :1 ) proporcionando s A-R y su isómero como un sólido blanco (290 mg, 58 % de rendimiento).
Síntesis del compuesto SA-S. A una disolución de SA-R y su isómero (300 mg, 0,94 mmoles) en 10 mL de THF, se añadió LiAH4 (100 mg, 2,7 mmoles), se agitó a ta durante 1 h. La mezcla de reacción se inactivó con 5 mL de H2O y se extrajo con 100 mL de EtOAc, se lavó con salmuera y se evaporó a vacío, luego se purificó por cromatografía (éter de petróleo/acetato de etilo = 3:1) proporcionando SA-S como un sólido blanco (140 mg, 48% de rendimiento).
RMN 1H (400 MHz, CDCh), 8 (ppm) 5,15-5,12 (m, 1H), 4,72-4,60 (m, 1H), 1,70 (d aparente dentro de m), 1,27 (s aparente dentro de m), 0,92 (s, 3H).
Síntesis del compuesto SA-T. A una disolución de SA-S (100 mg, 0,3 mmoles) en THF seco (5 mL) se añadió el complejo de borano-tetrahidrofurano (1 mL; disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (1 mL), seguido por 30 % de disolución acuosa de H2O2 (1 mL). Después de agitar a temperatura ambiente durante una hora, la mezcla se extrajo con EtOAc (3 * 100 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secaron sobre MgSO4, se filtraron y se concentraron proporcionando SA-T como un sólido blanco (100 mg, 91 %). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SA-U. A una disolución de SA-T (100 mg, 0,29 mmoles) en 20 mL de DCM se añadió PCC (190 mg, 0,87 mmoles), se agitó a ta durante 2 h. La mezcla de reacción se inactivó con 5 mL de H2O y se extrajo con 100 mL de EtOAc, se lavó con salmuera y se evaporó a vacío, luego se purificó por cromatografía (éter de
petróleo/acetato de etilo = 3:1) proporcionando SA-U como un sólido blanco (53 mg, 53 % de rendimiento). RMN 1H (400 MHz, CDCls), 5 (ppm) 4,71-4,57 (m, 1H), 2,54 (1H, t), 1,28 (s aparente dentro de m), 0,58 (s, 3H).
Síntesis del compuesto SA-V. A una disolución de SA-U (40 mg, 0,11 mmoles) en MeOH (5 mL) se añadió 2 gotas de HBr (48 %), seguido por bromo (150 mg, 0,33 mmoles). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se vertió en agua con hielo, luego se extrajo con EtOAc (10 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera (20 mL), se secaron sobre MgSO4, se filtraron y se concentraron, dando el compuesto en bruto SA-V como un sólido blanco (40 mg, 80 % de rendimiento). El producto en bruto se usó en la siguiente etapa sin más purificación.
Ejemplo 4. Síntesis del compuesto SB-W

A una disolución con agitación de yoduro de trimetilsulfonio (8,1 g, 36,9 mmoles) en 100 mL de DMSO se añadió NaH (60%; 1,26 g, 31,5 mmoles). Después de agitar a temperatura ambiente durante 1 h, se añadió gota a gota una suspensión del compuesto SB-F (2,2 g, 7,2 mmoles) en DMSO (20 mL). La mezcla se agitó durante otras 2,5 h, luego se vertió en agua fría con hielo y se extrajo con éter (100 mL * 3). Las fases de éter combinadas se lavaron entonces con salmuera (100 mLx 3), se secaron sobre MgSO4, se filtraron y se concentraron dando el producto en bruto SB-S (2,2 g). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SB-T. Se disolvió el compuesto SB-S (2,2 g, 7,3 mmoles) en etanol seco (250 mL) y se añadió Na (672 mg, 29,2 mmoles). La disolución se agitó a reflujo durante 6 h. Se separó por evaporación el etanol y el residuo se disolvió en diclorometano y se lavó con H2O (3 * 50 mL) y salmuera (100 mL), se secó sobre MgSO4, se filtró y se concentró. El compuesto objetivo en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1) y se concentró dando SB-T (1,8 g, 82 %) como un sólido blanco. RMN 1H (500 MHz, CDCls), 5 (ppm), 5,03-5,01 (m, 1H), 3,43 (q, 2H), 3,13 (s, 2H), 0,80 (s, 3H).
Síntesis del compuesto SB-U. A una disolución del compuesto SB-T (1,8 g, 5,2 mmoles) en THF seco (50 mL) se añadió el complejo de borano-tetrahidrofurano (20 mL de disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (10 mL), seguido por 30 % de disolución acuosa de H2O2 (12 mL). La mezcla se dejó con agitación a temperatura ambiente durante 1 hora, luego se extrajo con EtOAc (3 * 100 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secó sobre MgSO4, se filtró y se concentró proporcionando el compuesto en bruto SB-U (1,8 g, 100 %). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SB-V. A una disolución del compuesto en bruto SB-U (1,8 g, 5,2 mmoles) disuelto en 60 mL de diclorometano saturado con H2O (el diclorometano se había agitado con varios mililitros de H2O, luego se separó de la fase acuosa) se añadió peryodinato de Dess-Martin (4,4 g, 10,4 mmoles). Después de agitar a temperatura ambiente durante 24 h, la mezcla de reacción se extrajo con diclorometano (3 * 100 mL). Las fases orgánicas combinadas se lavaron con 10% de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1) proporcionando SB-V (1 g, 2,8 mmoles, 56% para dos etapas) como un sólido blanco. RM N1H (400 MHz, CDCh), 5 (ppm), 3,52 (q, 2H), 3,21 (s, 2H), 2,54 (t, 2H), 2,11 (s, 3H), 1,20 (t, 3H), 0,61 (s, 3H). EM/CL: Tr = 7,25 min. m/z = 345,1 [M-17]+.
Síntesis del compuesto SB-W. A una disolución del compuesto SB-V (600 mg, 1,65 mmoles) en MeOH (20 mL) se añadió 5 gotas de HBr (48 %), seguido por bromo (264 mg, 1,65 mmoles). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se vertió en agua con hielo, luego se extrajo con acetato de etilo (100 mL * 3). Las
fases orgánicas combinadas se lavaron con salmuera (200 mL), se secaron sobre MgSO4, se filtraron y se concentraron dando el compuesto en bruto SB-W (600 mg, 100 %). El producto en bruto se usó en la siguiente etapa sin más purificación. EM/c L: Tr = 7,25 min. m/z = 463,1 [M+Na]+.
Ejemplo 5. Síntesis del compuesto SA-AA

Síntesis del compuesto SA-X. A una disolución de EtMgBr (5 mmoles, 1 M en THF) en THF (20 mL) a 0 °Cse añadió una disolución del compuesto SA-W (858 mg, 3 mmoles) en THF seco (5 mL) por una bomba de jeringa durante 30 min. Después de agitar a 0 °C durante 5 h, se dejó que la mezcla de reacción se calentara y se agitó a temperatura ambiente durante la noche. La mezcla de reacción se inactivó con agua fría con hielo y se extrajo con EtOAc (15 mL x 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo blanco se purificó por cromatografía ultrarrápida en columna (éter de petróleo/acetato de etilo = 20:1 a 10:1) dando el compuesto SA-X (900 mg).
Síntesis del compuesto SA-Y. A una disolución del compuesto SA-X (200 mg, 0,66 mmoles) en THF seco (5 mL) se añadió el complejo de borano-tetrahidrofurano (2 mL de disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (1 mL), seguido por 30 % de disolución acuosa de H2O2 (1,2 mL). La mezcla se dejó con agitación a temperatura ambiente durante 1 hora, luego se extrajo con EtOAc (3 x 10 mL). Las fases orgánicas combinadas se lavaron con 10% de Na2S2O3 acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron proporcionando el compuesto SA-Y (260 mg, en bruto). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SA-Z. A una disolución del compuesto SA-Y (260 mg, en bruto) disuelta en 10 mL de diclorometano se añadió PCC (449 mg,). Después de agitar a temperatura ambiente durante 24 h, la mezcla de reacción se extrajo con diclorometano (3 x 10 mL). Las fases orgánicas combinadas se lavaron con 10% de NaCl acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 4:1 a 2:1) proporcionando SA-Z del título (15 mg) como un sólido blanco. RMN 1H (500 MHz, CDCla), 8 (ppm), 2,49 (1H, t), 0,84(,t 3H), 0,59 (s, 3H).
Síntesis del compuesto SA-AA. A una disolución del compuesto SA-Z (30 mg, 0,09 mmoles) en MeOH (5 mL) se añadió 2 gotas de HBr (48 %), seguido por bromo (100 mg, 0,62 mmoles). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se vertió en agua con hielo, luego se extrajo con acetato de etilo (15 mL x 3), Las fases orgánicas combinadas se lavaron con salmuera (20 mL), se secaron sobre MgSO4, se filtraron y se concentraron dando el compuesto SA-AA (36 mg en bruto). El producto en bruto se usó en la siguiente etapa sin más purificación.
Ejemplo 6. Síntesis del compuesto SA-JJ

Síntesis del compuesto SA-DD y SA-EE. Se disolvió la mezcla de compuestos SA-BB y SA-CC (5,0 g, 16,7 mimóles) en metanol seco (250 mL), y se añadió metal Na (1,2 g, 50,0 mimóles) y la disolución se sometió a reflujo durante 16 h. Entonces se separó por evaporación el metanol y el residuo se disolvió en diclorometano y se lavó con H2O (3 * 50 mL) y salmuera (100 mL), se secó sobre MgSO4, se filtró y se concentró. El compuesto objetivo en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1), y se concentró dando la mezcla de productos SA-DD y SA-EE (4,6 g, 83 %) como un sólido blanco.
Síntesis del compuesto SA-FF y SA-GG. A una disolución de la mezcla de reactantes SA-DD y SA-EE (4,6 g, 13.9 mmoles) en THF anhidro (30 mL) se añadió BH3.THF (1,0 M, 27,7 mL, 27,7 mmoles), la disolución se agitó a 25 °C durante la noche, luego la reacción se inactivó mediante la adición de agua (5 mL). Se añadió disolución 2 M de NaOH (30 mL), seguido por 30 % de H2O2 (30 mL). La mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla se diluyó con acetato de etilo (200 mL) y la disolución resultante se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. La mezcla de productos en bruto se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SA-HH y SA-II. A una disolución de la mezcla de reactantes en bruto SA-FF y SA-GG (4,9 g, 13.9 mmoles, cantidad teórica) en diclorometano (40 mL) se añadió clorocromato de piridinio (PCC) en porciones (6,0 g, 27,8 mmoles). La disolución se agitó a 25 °C durante la noche, entonces la mezcla se filtró a través de una almohadilla corta de gel de sílice y el gel de sílice se lavó con diclorometano (3 * 50 mL). Todos los filtrados se combinaron y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo = 15:1) proporcionando el producto SA-HH (2,1 g, 6,03 mmoles, rendimiento=43 % (2 etapas)) como un sólido blanco y el producto SA-II (2,2 g, 6,32 mmoles, rendimiento=45 % (2 etapas)) como un sólido blanco. Compuesto SA-HH: RMN 1H (500 MHz, CDCla) 6 (ppm): 3,40 (s, 3H), 3,20 (s, 2H), 2,62-2,51 (m, 2H), 2,11 (s, 3H), 2,02-1,99 (m, 2H), 0,62 (s, 3H). Compuesto SA-II: RMN 1H (500 MHz, CDCla) 6 (ppm): 3,42 (AB, 1H), 3,38 (AB, 1H), 3,40 (s, 3H), 2,65 (s, 1H), 2,54 (t, 1H), 2,16-2,14 (m, 1H), 2,11 (s, 3H), 2,02-1,98 (m, 1H), 0,61 (s, 3H).
Síntesis del compuesto SA-JJ. A una disolución del reactante SA-II (100 mg, 0,301 mmoles) en metanol (10 mL) se añadió 48 % de ácido bromhídrico (152 mg, 0,903 mmoles), seguido por bromo (241 mg, 0,077 mL, 1,51 mmoles). La disolución se calentó a 25 °C durante 1,5 horas, luego la mezcla se vertió en agua fría (50 mL) y el sólido resultante se extrajo con acetato de etilo (2 * 50 mL). Los extractos orgánicos combinados se lavaron con salmuera (50 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SA-JJ se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 8. Síntesis del compuesto SB-R

Síntesis del compuesto SB-K. A una disolución del compuesto SB-E (5 g, 15 mimóles) en THF seco (20 mL) se añadió el complejo de borano-tetrahidrofurano (30 mL de disolución 1,0 M en THF) y la mezcla de reacción se agitó a temperatura ambiente durante 1 hora, luego se añadió lentamente 10 % de NaOH acuoso (56 mL). La mezcla se enfrió en hielo y se añadió lentamente 30 % de disolución acuosa de H2O2 (67 mL). La mezcla se agitó a temperatura ambiente durante 1 hora y luego se extrajo con EtOAc (3 * 100 mL). Los extractos de EtOAc combinados se lavaron con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secaron sobre MgSO4. La filtración y la retirada del disolvente dieron 3,2 g del producto en bruto para la reacción de la siguiente etapa.
Síntesis del compuesto SB-L. A una disolución del compuesto SB-K (3,2 g, 9 mmoles) en THF (40 mL) se añadió HCl 2 M (3 mL). La disolución de reacción se agitó a TA durante 12 h, luego se retiró el disolvente a presión reducida. El compuesto objetivo en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1) dando 2,2 g del producto como un sólido blanco, rendimiento: 81,40 %.
Síntesis del compuesto SB-M. A una disolución con agitación de yoduro de trimetilsufonio (6,43 g, 31,5 mmoles) en 100 mL de DMSO se añadió 60% en peso NaH (1,26 g, 31,5 mmoles). Después de agitar a temperatura ambiente (15 °C) durante 1 h, se añadió gota a gota una disolución del compuesto SB-L (2,2 g, 7,2 mmoles) en 20 mL de DMSO. Después de 2,5 h, la mezcla de reacción se vertió en agua fría con hielo y se extrajo con éter (100 mL * 3). Las capas de éter combinadas se lavaron entonces con salmuera (100 mL * 3), se secaron (MgSO4), se filtraron y se concentraron dando el producto en bruto 1,6 g para la reacción de la siguiente etapa.
Síntesis del compuesto SB-N. Se disolvió el compuesto SB-M (1,6 g, 5 mmoles) en 60 mL de CH2Ch saturado con H2O (usando un embudo de decantación, el CH2Ch se había agitado con varios mililitros de H2O y luego se separó de la fase acuosa). Se añadió DMP (4,2 g, 10 mmoles), y la mezcla de reacción resultante se agitó vigorosamente durante 24 h. La disolución de reacción se diluyó con DCM (100 mL), se lavó con 10 % de Na2S2O3 acuoso (100 mL), salmuera (100 mL), se secó sobre MgSO4, se filtró y se concentró. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 20:1 a 10:1) proporcionando el compuesto del título (1,2 g, 3,79 mmoles, 75 %) como un sólido blanco. RMN 1H (400 MHz, CDCh) 8 (ppm): 2,63 (s, 1H), 2,59 (s, 1H), 2,12 (s, 3H), 0,63 (s, 3H).
Síntesis de SB-P y SB-Q. Se disolvió el compuesto SB-N (1,2 g, 3,8 mmoles) en metanol seco (250 mL) y se añadió Na (262 mg, 11,4 mmoles). La disolución se sometió a reflujo durante 16 h. Se separó por evaporación metanol y el residuo se disolvió en diclorometano y se lavó con H2O (3 * 50 mL) y salmuera (100 mL), se secó sobre MgSO4, se filtró y se concentró. El compuesto objetivo en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10:1 a 5:1) dando SB-P (300 mg, 25 %), SB-Q (300 mg, 25 %) como un sólido blanco. SB-P: RMN 1H (400 MHz, CDCla) 8 (ppm): 3,39 (s, 3H), 3,19 (s, 2H), 2,54 (t, 1H), 0,61 (s, 3H). SB-Q: RMN 1H (400 MHz, CDCl3) 8 (ppm): 3,39 (s, 5H), 3,37 (s, 2H), 2,52 (t, 1H), 0,62 (s, 3H).
Síntesis del compuesto SB-R. A una disolución del reactante SB-P (190 mg, 0,545 mmoles) en metanol (15 mL) se añadió 48 % de ácido bromhídrico (275 mg, 1,635 mmoles), seguido por bromo (435 mg, 0,139 mL, 2,725 mmoles). La disolución se calentó a 25 °C durante 1,5 horas. Entonces, la mezcla se vertió en agua enfriada (50 mL). La disolución resultante se extrajo con acetato de etilo (2 * 100 mL). Los extractos orgánicos combinados se lavaron con
salmuera (100 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 9. Síntesis del compuesto SB-FF

Síntesis del compuesto SB-KK. A una disolución de SA-K (68 g, 216,27 mmoles) en 600 mL de CH3CN, se añadió selectfluor (90,22 g, 324,4 mmoles) en porciones a -4 °C. La mezcla de reacción resultante se agitó a -4 °C durante 3 h. Después de que la CCF mostrara que la reacción estaba completa, entonces la mezcla se filtró y se concentró. El producto se purificó por cromatografía en columna sobre gel de sílice eluida con (éter de petróleo/acetato de etilo 20:1 -15:1-10:1-8:1-6:1-5:1) proporcionando SB-KK (26,3 g, 41,8 % de rendimiento) como un sólido blanco. RMN 1H (SB-KK) (400 MHz, CDCla), 8 (ppm), 6,02-5,94 (m, 1H,), 5,20-5,01 (m, 1H), 2,55-2,26 (m, 6H), 2,16-2,05 (m, 1H), 2,01 1,83 (m, 4H), 1,48-1,22 (m, 5H), 0,98-0,78 (m, 6H).
Síntesis del compuesto SB-X. A una disolución de SB-KK (27 g, 92,98 mmoles) en EtOAc (350 mL) a 20 °C, entonces se añadió Pd/C (2,7 g, 5 %) en la mezcla. La disolución se agitó a 20 °C, 1 atm durante 10 h bajo hidrógeno. Después de que la EM/CL mostrara que la reacción estaba completa, entonces la mezcla se filtró y se concentró. El producto se purificó por cromatografía en columna sobre gel de sílice eluida con (éter de petróleo/acetato de etilo 40:1-35:1-30:1-25:1-20:1-15:1-10:1-6:1) dando SB-X (15,6 g, 56,38 %) como un sólido blanco. RMN 1H (SB-X) (400 MHz, CDCh), 8 (ppm)=4,68-4,56 (m, 1H), 2,64-2,51 (m, 1H), 2,53-2,03 (m, 8H), 1,97-1,80 (m, 4H), 1,49-1,20 (m, 6H), 0,96 0,92 (m, 2H), 0,88-0,78 (m, 1H).
Síntesis del compuesto SB-Y. A una disolución de SB-X (47 g, 160,75 mmoles) en MeOH (600 mL) a 23 °C, entonces se añadió 2,35 g de TsOH en la mezcla. La disolución se agitó a 60 °C durante 1,5 h. Después de que la CCF mostrara que la reacción estaba completa, entonces la mezcla se filtró y se concentró dando SB-Y (35 g, 64,33 %) como un sólido blanco. RMN 1H (SB-Y) (400 MHz, CDCla), 8 (ppm)=4,74-4,57 (m, 1H), 3,16 (s, 3H), 3,10 (s, 3H), 2,47 2,35 (m, 1H), 2,15-2,09 (m, 1H), 2,06-1,82 (m, 6H), 1,77-1,15 (m, 11H), 1,05-0,96 (m, 1H), 0,89 (s, 3H), 0,83-0,77 (m, 1H).
Síntesis del compuesto SB-Z. A una disolución de bromuro de etiltrifenilfosfonio (115,17 g, 310,23 mimóles) en 150 mL de THF, se añadió KOt-Bu (34,81 g, 310,23 mmoles). La mezcla de reacción se calentó hasta 60 °C durante 1 h y se añadió SB-Y (35 g, 103,41 mmoles) a la mezcla que se agitó a 60 °C durante 15 h adicionales. La mezcla de reacción se enfrió y se extrajo con 1500 mL de EtOAc, se lavó con salmuera y se concentró proporcionando SB-Z como el sólido blanco (120 g, en bruto). RMN 1H (SB-Z) (400 MHz, CDCk), 8 (ppm)=5,13-5,07 (m, 1H), 4,67-4,54 (m, 1H), 3,14 (s, 3H), 3,09 (s, 3H), 2,42-2,15 (m, 3H), 1,92-1,79 (m, 3H), 1,67-1,61 (m, 4H) , 1,57-1,50 (m, 2H), 1,45-1,15 (m, 10H) , 1,01-0,94 (m, 1H), 0,92 (s, 3H), 0,90-0,84 (m, 1H).
Síntesis del compuesto SB-AA. A una disolución de SB-Z (120 g, en bruto) en 600 mL de THF, se añadió 90 mL de HCl acuoso 2M, La mezcla de reacción se agitó a 22 °C durante 1 h. Después de que la CCF mostrara que la reacción estaba completa, entonces la reacción se inactivó con NaHCO3 ac. La reacción se extrajo con 500 mL de EtOAc, se lavó con salmuera y se evaporó a vacío. El residuo resultante se purificó por cromatografía (éter de petróleo/acetato de etilo =150:1-125:1-100:1-80:1-60:1-50:1) proporcionando s B-AA como el sólido blanco (24 g, 76,23% de rendimiento). RMN 1H (SB-AA) (400 MHz, CDCla), 8 (ppm)=5,13 (m, 1H), 4,65-4,48 (m, 1H), 2,62-2,42 (m, 1H), 2,44 2,07 (m, 8H), 1,92-1,80 (m, 1H), 1,72-1,55 (m, 8H), 1,36-1,08 (m, 6H), 0,92 (s, 3H), 0,83-0,73 (m, 1H).
Síntesis del compuesto SB-BB. A una disolución de Me3SOI (78,07 g, 354,75 mmoles) en 50 mL de THF, se añadió una disolución de t-BuOK (39,81 g, 354,75 mmoles) en 50 mL de THF. La mezcla de reacción se agitó a 60 °C durante 1,5 h. Entonces se añadió una disolución de SB-AA (24 g, 78,83 mmoles) en THF (300 mL) a la reacción. La reacción se agitó durante 2,5 h a 23 °C. Después de que la CCF mostrara que la reacción estaba completa, entonces la reacción se inactivó con agua con hielo. La reacción se extrajo con 500 mL de EtOAc, se lavó con salmuera y se evaporó a vacío proporcionando SB-BB como el producto en bruto (50 g). RMN 1H (SB-BB) (400 MHz, CDCk), 8 (ppm)=5,20-5,11 (m, 1H), 4,65-4,52 (m, 1H), 2,74-2,68 (m, 2H), 2,48-1,81 (m, 9H), 1,72-1,64 (m, 4H), 1,55-1,06 (m, 10H), 0,97 0,89 (m, 3H), 0,85-0,77 (m, 1H).
Síntesis del compuesto SB-CC. A una disolución de SB-BB (50 g, en bruto) en 300 mL de THF, se añadió LiAlH4 (8,99 g, 236,49 mmoles) a 0 °C. La mezcla de reacción se agitó a 23 °C durante 1,5 h. Después de que la CCF mostrara que la reacción estaba completa, entonces la reacción se inactivó con agua. La reacción se extrajo con 1000 mL de EtOAc, se lavó con salmuera y se evaporó a vacío. El residuo resultante se purificó por cromatografía (éter de petróleo/acetato de etilo =100:1-80:1-60:1-50:1-40:1-30:1) proporcionando SB-CC como el sólido blanco (19 g, 75,19 % de rendimiento). RMN 1H (SB-CC) (400 MHz, CDCk), 8 (ppm)=5,17-5,07 (m, 1H), 4,66-4,48 (m, 1H), 2,41-2,32 (m, 1H), 2,28-2,15 (m, 2H), 2,09-2,05 (m, 1H), 1,88-1,75 (m, 2H), 1,68-1,64 (m, 3H), 1,40-1,31 (m, 1H), 1,25 1,13 (m, 9H), 0,89 (s, 3H), 0,81-0,72 (m, 1H).
Síntesis del compuesto SB-DD. A una disolución de SB-CC (19 g, 59,29 mmoles) en THF seco (500 mL) se añadió C2H9BS (59,29 mL; disolución 10 M en THF) a 0 °C. Después de agitar a temperatura ambiente durante 2 horas, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con NaOH acuoso 3 M (160 mL), seguido por 30 % de disolución acuosa de H2O2 (100 mL). Después de agitar a 20 °C durante 1,5 h, la mezcla se filtró y se extrajo con EtOAc (300 mL). Las fases orgánicas combinadas se trataron con Na2S2O3 ac., se extrajeron, se secaron y se concentraron proporcionando SB-DD en bruto (21 g, en bruto). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SB-EE. A una disolución de SB-DD (21 g, 59,29 mmoles) en 200 mL e CH2Ch, se añadió PCC (25,56 g, 118,58 mmoles) a 0 °C, se agitó a 22 °C durante 2 h. La mezcla de reacción se filtró y se extrajo con 20 mL de CH2Ch, se lavó con NaHCO3 ac., Na2S2O3 ac., salmuera y se evaporó a vacío. El residuo se purificó por cromatografía (éter de petróleo/acetato de etilo = 15:1-10:1-6:1) proporcionando SB-EE como el sólido blanco (12 g, 60,15 % de rendimiento). RMN 1H (SB-EE) (400 MHz, CDCk), 8 (ppm)=4,65-4,46 (m, 1H), 2,55-2,51 (m, 1H), 2,22 2,09 (m, 4H), 2,06-1,97 (m, 32H), 1,88-1,77 (m, 2H), 1,69-1,54 (m, 5H), 1,48-1,30 (m, 3H), 1,28-1,05 (m, 11H), 0,83 0,72 (m, 1H), 0,63 (s, 3H).
Síntesis del compuesto SB-FF. A una disolución de SB-EE (12 g, 35,66 mmoles) en 1500 mL de MeOH, se añadió HBr (5 gotas) y Br2 (2,01 mL, 39,23 mmoles) a 0 °C. La reacción se agitó a 16 °C durante 2 h. La mezcla de reacción se inactivó con NaHCO3 ac. y se concentró. Entonces, la mezcla se extrajo con 1000 mL de EtOAc, se lavó con salmuera y se evaporó a vacío. El producto se purificó por cromatógrafo de columna sobre gel de sílice eluido con (éter de petróleo/acetato de etilo = 12:1-10:1-8:1-6:1-3:1) proporcionando SB-FF como el sólido blanco (12,3 g, 83,03 % de rendimiento). RMN 1H (SB-FF) (400 MHz, CDCk), 8 (ppm)=4,64-4,47 (m, 1H), 3,95-3,86 (m, 2H), 2,89 2,80 (m, 1H), 2,23-2,16 (m, 1H), 2,07-1,64 (m, 8H) 1,46-1,06 (m, 14H), 0,83-0,74 (m, 1H), 0,67 (s, 3H).
Ejemplo 12. Síntesis del compuesto SC-O

Síntesis del compuesto SC-B. A una disolución del reactante SC-A (10,0 g, 36,44 mimóles) en piridina (30 mL) se añadió anhídrido acético (5,0 mL, 52,89 mimóles). La mezcla se agitó a 60 °C durante la noche. Entonces la disolución se vertió en agua con hielo (200 mL). El precipitado blanco se filtró y se disolvió en acetato de etilo (300 mL). La disolución resultante se lavó con disolución sat. de CuSO4.5H2O (2*200 mL) para retirar la piridina residual. La fase orgánica se lavó adicionalmente con salmuera (200 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo = 4:1) proporcionando el producto SC-B (11,125 g, 35,16 mmoles, rendimiento = 96 %) como un sólido blanco. RMN 1H (500 MHz, CDCh) 8 (ppm): 5,83 (1H, s), 4,62 (1H, dd), 2,05 (3H, s), 0,86 (3H, s).
Síntesis del compuesto SC-C. A una disolución del reactante SC-B (4,68 g, 14,79 mmoles) en THF (150 mL) se añadió LiHMDS (1,0 M en disolución de THF, 17,74 mL, 17,74 mmoles) a -78 °C. La disolución se agitó a -78 °C
durante 30 minutos. Entonces se añadió HMPA (3,09 mL, 17,74 mimóles). La disolución se agitó a -78 °C durante otros 30 minutos. Entonces se añadió yodometano (2,76 mL, 44,37 mmoles). La disolución se agitó adicionalmente a -78 °C durante 2 horas y se calentó hasta temperatura ambiente y se agitó durante 1 hora. La reacción se inactivó mediante la adición de agua (10 mL). La mayoría del disolvente de THF se retiró a vacío. Entonces, el residuo se diluyó con acetato de etilo (300 mL) y la disolución resultante se lavó con salmuera (2 * 200 mL), se secó sobre sulfato de magnesio. La retirada de disolvente a vacío proporcionó el producto en bruto SC-C (4,50 g, 13,62 mmoles, rendimiento=92 %) como un aceite denso. El producto en bruto se usó en la siguiente etapa sin más purificación. RMN 1H (500 MHz, CDCla) 8 (ppm): 5,75 (1H, s), 4,62 (1H, t), 2,05 (3H, s), 1,10 (3H, d), 0,86 (3H, s).
Síntesis del compuesto SC-D & SC-E. A una disolución del reactante en bruto SC-C (11,62 g, 35,16 mmoles, cantidad teórica) en metanol (100 mL) y agua (20 mL) se añadió hidróxido sódico (2,81 g, 70,32 mmoles). La disolución se calentó a 60 °C durante 1 hora. Entonces se retiró a vacío la mayoría del disolvente de metanol. La disolución residual se acidificó por HCl 2 M hasta pH 5-6. La fase acuosa se extrajo con acetato de etilo (3*100 mL). Los extractos orgánicos combinados se lavaron con salmuera (200 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =5:1) proporcionando el producto puro SC-D (2,354 g, 8,162 mmoles, rendimiento=23 %) y el producto puro SC-E (5,306 g, 18,40 mmoles, rendimiento=50 %) como un sólido blanco. Compuesto SC-D: r MN 1H (500 MHz, CDCla) 8 (ppm): 5,81 (1 H, s), 3,67 (1H, t), 1,11 (3H, d), 0,81 (3H, s).
Compuesto SC-E: RMN 1H (500 MHz, CDCh) 8 (ppm): 5,74 (1H, s), 3,67 (1H, t, J=8,5 Hz), 1,11 (3H, d), 0,81 (3H, s).
Síntesis del compuesto SC-F. A amoniaco líquido (200 mL) se añadió litio (1,80 g, 260 mmoles) a -78 °C. El líquido viró entonces a azul oscuro. Entonces se añadió una disolución del reactante SC-D (3,0 g, 10,40 mmoles) en t-BuOH (1,0 mL, 10,40 mmoles) y THF (100 mL) a disolución de Li-amoniaco. La mezcla se agitó a -78 °C durante 4 horas. Entonces se añadió NH4Cl sólido (20 g) para extinguir la reacción. La mezcla viró de azul oscuro a blanco. Se dejó que la mezcla se calentara hasta temperatura ambiente y el amoniaco se evaporó en una campana durante la noche. Al residuo se añadió agua (300 mL). La mezcla se acidificó por HCl conc. hasta pH 6-7. Entonces se añadió acetato de etilo (300 mL). La fase acuosa separada se extrajo adicionalmente con acetato de etilo (2*100 mL). Los extractos orgánicos combinados se lavaron con salmuera (300 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SC-F se usó directamente sin más purificación en la siguiente etapa.
Síntesis del compuesto SC-G. A una disolución del reactante en bruto SC-F (1,749 g, 6,022 mmoles) en diclorometano (60 mL) se añadió dicromato de piridinio (PDC) (3,398 g, 9,033 mmoles). La mezcla se agitó a temperatura ambiente durante la noche. La disolución se filtró a través de una almohadilla corta de celite. El Celite se lavó con CH2Cl2 (3*50 mL). La disolución de CH2Ch combinada se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =5:1) proporcionando el producto SC-G (1,298 g, 4,50 mmoles, rendimiento=75 %) como un sólido blanco. Compuesto SC-G: RMN 1H (400 Mhz, CDCh) 8 (ppm): 1,02 (3H, d), 0,91 (3H, s).
Síntesis del compuesto SC-H. A una disolución del reactante SC-G (1,948 g, 6,754 mmoles) en metanol anhidro (50 mL) se añadió monohidrato de ácido p-toluenosulfónico (128 mg, 0,6754 mmoles). La disolución se calentó a 70 °C durante 3 horas. La reacción se inactivó mediante la adición de disolución sat. De Na2CO3 (10 mL). La mayoría del disolvente de metanol se retiró a vacío. Entonces, el residuo se diluyó con acetato de etilo (200 mL). La disolución resultante se lavó con disolución sat. de Na2CO3 (2* 100 mL). Las fases acuosas combinadas se extrajeron con acetato de etilo (50 mL). Los extractos orgánicos combinados se lavaron con salmuera (100 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo = 10:1, se añadió 0,1 % de NEt3) proporcionando el producto SC-H (652 mg, 1,949 mmoles, rendimiento=29 %) como un sólido blanco. Además, también se recuperó material de partida (1,338 g). Así, el rendimiento basado en material de partida recuperado es 92 %. RMN 1H (500 MHz, d6-acetona) 8 (ppm): 3,079 (3H, s), 3,075 (3H, s), 2,38 (1H, dd), 1,98 (1H, dd), 0,91 (3H, d), 0,85 (3H, s).
Síntesis del compuesto SC-I. A una disolución de bromuro de etiltrifenilfosfonio (8,795 g, 23,69 mmoles) en THF anhidro (20 mL) se añadió t-BuOK (2,658 g, 23,69 mmoles). Entonces, la disolución se volvió rojiza en color y se calentó a 70 °C durante 2 horas. Entonces, el reactante SC-H (1,642 g, 4,909 mmoles) se añadió en una porción. La disolución se calentó a 70 °C durante la noche. La reacción se inactivó mediante la adición de agua (10 mL). La mezcla se diluyó con acetato de etilo (200 mL) y la disolución resultante se lavó con salmuera (2 *100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto SC-I se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-J. Al producto en bruto SC-I (1,702 g, 4,909 mmoles, cantidad teórica) en THF (30 mL) se añadió HCl 2 M (3 mL). La disolución se agitó a temperatura ambiente durante 1 hora. La mezcla se diluyó con acetato de etilo (300 mL) y la disolución resultante se lavó con disolución sat. de Na2CO3 (2*100 mL). Las fases acuosas combinadas se extrajeron con acetato de etilo (100 mL). Los extractos orgánicos combinados se lavaron con salmuera (100 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =100:3) proporcionando el producto en bruto SC-J (1,746 g) como un sólido blanco que estaba contaminando con algo de PPh3 no separada. Evaluado por la integración del espectro de RMN 1H, la relación entre el producto deseado y PPh3 es 3:1, por lo que la cantidad de producto
deseado SC-J es 1,354 g (4,506 mimóles), el rendimiento es 92 %. RMN 1H (500 MHz, CDCI3) 8 (ppm): 5,13 (1H, qt), 1,66 (3H, dt), 1,02 (3H, d), 0,91 (3H, s).
Síntesis del compuesto SC-K. A una disolución de yoduro de trimetilsulfoxonio (5,213 g, 23,69 mimóles) en DMSO anhidro (30 mL) se añadió hidruro de sodio (60 % en peso, 948 mg, 23,69 mmoles). La mezcla se agitó a 25 °C durante 1 hora. Entonces se añadió una disolución del reactante en bruto (1,746 g, contaminado con algo de PPh3 residual, cantidad teórica, 1,354 g, 4,506 mmoles) en THF anhidro (10 mL). La mezcla se agitó a 25 °C durante la noche. La reacción se inactivó mediante la adición de agua (5 mL). La mezcla se diluyó con acetato de etilo (300 mL) y la disolución resultante se lavó con agua (2 * 100 mL), seguido por salmuera (100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto SC-K se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-L. A una disolución del reactante en bruto SC-K (cantidad teórica, 1,417 g, 4,506 mmoles) en THF anhidro (30 mL) se añadió hidruro de litio y aluminio (342 mg, 9,012 mmoles) en porciones. La suspensión se agitó a 25 °C durante 1 hora. Entonces la reacción se inactivó mediante la adición de acetato de etilo (5 mL), seguido por agua (5 mL). Se filtró un sólido blanco y se lavó minuciosamente con acetato de etilo (5 * 100 mL). El filtrado combinado se lavó con salmuera (200 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =20:1 ) proporcionando el producto SC-L (458 mg, 1,447 mmoles, rendimiento total de 2 etapas=32 %) como un sólido blanco.
Síntesis del compuesto SC-M. A una disolución del reactante SC-L (458 mg, 1,447 mmoles) en THF anhidro (15 mL) se añadió BH3.THF (1,0 M, 7,23 mL, 7,23 mmoles), La disolución se agitó a 25 °C durante la noche. Entonces la reacción se inactivó mediante la adición de agua (5 mL). Se añadió disolución 2 M de NaOH (10 mL), seguido por 30 % de H2O2 (10 mL). La mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla se diluyó con acetato de etilo (200 mL) y la disolución resultante se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-N. A una disolución del reactante en bruto SC-M (484 mg, 1,447 mmoles, cantidad teórica) en diclorometano (40 mL) se añadió dicromato de piridinio (PDC) en porciones (1633 mg, 4,341 mmoles). La disolución se agitó a 25 °C durante la noche. Entonces la mezcla se filtró a través de una almohadilla corta de gel de sílice y el gel de sílice se lavó con diclorometano (3*50 mL). Todo el filtrado se combinó y se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =8:1 ) proporcionando el producto SC-N (305 mg, 0,917 mmoles, rendimiento=63 % (2 etapas)) como un sólido blanco. Rm N 1H (500 MHz, CDCh) 8 (ppm): 2,54 (1H, t,), 2,12-2,19 (1H, m), 2,12 (3H, s 0,92 (3H, d), 0,61 (3H, s). RMN 13C (100 MHz, CDCh) 8 (ppm): 209,75, 71,09, 63,96, 55,89, 47,96, 47,80, 47,00, 44,35, 41,19, 40,22, 39,05, 37,95, 34,49, 33,14, 31,54, 30,92, 28,46, 25,82, 24,22, 22,76, 15,14, 13,45.
Síntesis del compuesto SC-O. A una disolución del reactante SC-N (100 mg, 0,301 mmoles) en metanol (10 mL) se añadió 48 % de ácido bromhídrico (152 mg, 0,903 mmoles), seguido por bromo (241 mg, 0,077 mL, 1,505 mmoles). La disolución se calentó a 25 °C durante 1,5 horas. Entonces la mezcla se vertió en agua enfriada (50 mL). El sólido resultante se extrajo con acetato de etilo (2 * 50 mL). Los extractos orgánicos combinados se lavaron con salmuera (50 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SC-O se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 13. Síntesis del compuesto SC-Y

Síntesis del compuesto SC-P. A NH3 (líquido, 2,0 L) se añadió litio (7,0 g, 1 moles) a -78 °C. Después de que el líquido virara a azul oscuro, se añadió gota a gota una disolución del compuesto SC-A (27,0 g, 100 mmoles) en t-BuOH (7,4 g, 100 mmoles) y THF (20 mL). La mezcla se agitó a -78 °C durante 4 horas. Entonces se añadió NH4Cl sólido (50 g) para extinguir la reacción. La mezcla viró de azul oscuro a blanco. Se dejó que la mezcla se calentara hasta temperatura ambiente y se evaporó el amoniaco durante la noche. El residuo se disolvió en HCl 0,5 N acuoso (50 mL) y se extrajo con diclorometano (200 mL*3). Las fases orgánicas combinadas se lavaron con NaHCO saturado (200 mL) y salmuera (200 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo = 4:1) para conseguir el producto SC-P (18,98 g, 68,7 %) como un sólido blanco. RMN 1H (500 MHz, CDCla) 8 (ppm): 3,66 (1H, t), 2,29-2,27 (2H, m), 2,12 2,07 (2H, m), 1,83-1,81 (2H, m), 1,50 (1H, s), 0,77 (3H, s).
Síntesis del compuesto SC-Q. Se disolvió una muestra de 19,0 g de compuesto SC-P (68,84 mmoles) en 50 mL de THF a 0 °C. Entonces se añadió gota a gota 70 mL de MeMgBr en THF (3m ) en 30 min. La reacción se mantuvo a 0 °C durante 8 h. La mezcla de reacción se inactivó con agua fría en hielo y se extrajo con EtOAc (200 mL*3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo blanco se purificó por cromatografía ultrarrápida en columna (éter de petróleo/acetato de etilo = 5:1) dando el producto SC-Q (19,0 g, 94 %) como un sólido blanco. RMN 1H (500 MHz, CDCla) 8 (ppm): 5,78 (1H, a), 5,36 (1H, t), 3,67 (1H, t), 1,73 (3H, s), 0,77 (3H, s).
Síntesis del compuesto SC-R. A una disolución del compuesto SC-Q (19,0 g, 65,07 mmoles) en diclorometano (100 mL) se añadió dicromato de piridinio (PDC) (48,9 g, 130,14 mmoles). La mezcla se agitó a temperatura ambiente durante la noche. La disolución se filtró a través de una almohadilla corta de celite. El celite se lavó con CH2Ch (3 * 100 mL). La disolución de CH2Ch combinada se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =5:1) proporcionando el producto SC-R (10,0 g, 53%) como un sólido blanco.
RMN 1H (500 MHz, CDCla) 8 (ppm): 2,44 (1H, dd), 2,07 (1H, m), 1,21 (3H, s), 0,87 (3H, s).
Síntesis del compuesto SC-S. A una disolución del compuesto SC-R (5,0 g, 17,2 mimóles) en tolueno anhidro (100 mL) se añadió al ácido p-toluenosulfónico sobre gel de sílice (80 g), la mezcla se agitó bajo 45 °C durante 1 hora. Se retiraron los bi-productos insolubles de gel de sílice por elución con éter de petróleo/acetato de etilo (10 / 1). Se usó el producto en bruto SC-S (3,20 g, 11,75 mmoles) en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-T. A una disolución del compuesto SC-S (3,20 g, 11,75 mmoles) en 10 mL de diclorometano anhidro se añadió mCPBA (4,04 g, 23,50 mmoles), y la mezcla de reacción se agitó durante la noche a temperatura ambiente. La mezcla de reacción se extrajo entonces con CH2Ch, la fase orgánica combinada se lavó dos veces con NaHCO3 (100 mL) y salmuera, se secó sobre Na2SO4 y se concentró. El producto en bruto SC-T se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-U. A una disolución del compuesto SC-T (11,75 mmoles) en metanol se añadió H2SO4 (0,5 mL), y la mezcla de reacción se agitó durante 2 h a temperatura ambiente. La disolución de reacción se extrajo entonces con CH2Ch (200 mL *3), la fase orgánica combinada se lavó con NaHCO3 (100 mL) y salmuera, se secó sobre Na2SO4 y se concentró. El residuo se purificó por cromatografía (éter de petróleo/acetato de etilo = 10:1) proporcionando el compuesto SC-U (3,30 g, 10,30 mmoles, rendimiento = 87% para dos etapas) como un sólido blanco.
Síntesis del compuesto SC-V. A una disolución de bromuro de etiltrifenilfosfonio (11,52 g, 31,0 mmoles) en THF anhidro (20 mL) se añadió t-BuOK (3,48 g, 31,0 mmoles). La disolución viró a rojizo y se calentó a 70 °C durante 3 horas. Entonces se añadió el compuesto SC-U (3,30 g, 10,30 mmoles) en una porción. La disolución de reacción se calentó a 70 °C durante la noche, entonces se inactivó mediante la adición de agua (10 mL). La mezcla se diluyó con EtOAc (200 mL) y la disolución resultante se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto SC-V (1,90 g) se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-W. A una disolución del compuesto SC-V (1,90 g, 5,72 mmoles) en THF seco (20 mL) se añadió BH3-THF (18 mL de disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (12 mL), seguido por 30 % de H2O2 (20 mL). La mezcla se dejó con agitación a temperatura ambiente durante 1 h, entonces se extrajo con AE (100 mL*3). La fase orgánica combinada se lavó con 10 % de Na2S2O3 acuoso (50 mL), salmuera, se secó sobre Na2SO4, se filtró y se concentró proporcionando el compuesto en bruto SC-W (1,86 g, 5,31 mmoles). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-X. A una disolución del compuesto en bruto SC-W (1,86 g, 5,31 mmoles) en diclorometano (50 mL) se añadió dicromato de piridinio (PDC) en porciones (3,98 g, 10,62 mmoles). La disolución se agitó a 25 °C durante la noche. Entonces la mezcla se filtró a través de una almohadilla corta de gel de sílice y el gel de sílice se lavó con diclorometano (3 * 50 mL). Todo el filtrado se combinó y se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =10:1) proporcionando el producto SC-X (1,20 g, 3,45 mmoles, 65 %) como un sólido blanco. RMN 1H (500 MHz, CDCh) 8 (ppm): 3,33 (3H, s), 3,04 (1H, s), 2,53 (1H, t), 2,12 (3H, s dentro de m), 1,26 (3H, s dentro de m), 0,62 (3H, s)
Síntesis del compuesto SC-Y. A una disolución del reactante SC-X (100 mg, 0,287 mmoles) en metanol (10 mL) se añadió 48 % de HBr (152 mg, 0,903 mmoles), seguido por bromo (0,08 mL, 1,505 mmoles). La disolución se calentó a 25 °C durante 1,5 horas. Entonces la mezcla se vertió en agua enfriada (50 mL). El sólido resultante se extrajo con acetato de etilo (2*50 mL). Los extractos orgánicos combinados se lavaron con salmuera (50 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SC-Y se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 14. Síntesis del compuesto SC-EE

Síntesis del compuesto SC-Z y SC-AA. A una disolución del compuesto SA-E (800 mg, 2,79 mimóles) y PhSO2CF2H (540 mg, 2,79 mmoles) en THF (25 mL) y HMPA (0,5 mL) a -78 °C bajo N2 se añadió LHMDS (4 mL, 1 M en THF) gota a gota. Después de agitar a -78 °C durante 2 h, la mezcla de reacción se inactivó con disolución acuosa saturada de NH4Cl (10 mL) y se dejó que se calentara hasta temperatura ambiente, entonces se extrajo con Et2O (20 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna de gel de sílice (éter de petróleo/acetato de etilo = 10/ 1) dando la mezcla de compuestos SC-Z y SC-AA (700 mg). La mezcla se purificó más por HPLC quiral proporcionando el compuesto SC-Z (200 mg, t= 4,31 min). RMN 1H (400 MHz, CDCh), 8 (ppm), 7,99-7,97 (d, 2H), 7,77-7,75 (m, 1H), 7,64-7,60 (m, 2H), 5,14-5,08 (m, 1H), 0,88 (s, 3H); compuesto SC-AA (260 mg, t= 5,66 min). RMN 1H (400 MHz, CDCla), 8 (ppm), 8,00--7,98 (d, 2H), 7,77-7,75 (m, 1H), 7,64-7,60 (m, 2H), 5,14-5,09 (m, 1H), 0,88 (s, 3H).
Síntesis del compuesto SC-BB. A una disolución del compuesto SC-AA (100 mg, 0,209 mmoles) y Na2HPO4 anhidro (100 mg) en metanol anhidro (5 mL) a -20 °C bajo N2 se añadió amalgama de Na/Hg (500 mg). Después de agitar a -20 °C a 0 °C durante 1 h, se separó por decantación la disolución de metanol y el residuo sólido se lavó con Et2O (5 * 3 mL). Las fases orgánicas combinadas se lavaron con salmuera (20 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10/ 1) dando el compuesto SC-BB (36 mg, 0,106 mmoles, 51 %). RMN 1H (400 MHz, CDCla), 8 (ppm), 6,02-5,88 (t, 1H), 5,17 5,15 (m, 1H), 0,88 (s, 3H).
Síntesis del compuestoSC-CC. A una disolución del compuesto SC-BB (150 mg, 0,443 mmoles) en THF seco (5 mL) se añadió el complejo de borano-tetrahidrofurano (1,34 mL de disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10% de NaOH acuoso (1 mL), seguido por disolución acuosa al 30 % de H2O2(1,2 mL). La mezcla se dejó con agitación a temperatura ambiente durante 1 hora, luego se extrajo con EtOAc (3 * 10 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron proporcionando el compuesto en bruto SC-CC (210 mg). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-DD. A una disolución del compuesto en bruto SC-CC (210 mg) disuelto en 10 mL de diclorometano saturado con H2O (el diclorometano se había agitado con varios mililitros de H2O, luego se separó de la fase acuosa) se añadió peryodinato de Dess-Martin (380 mg, 0,896 mmoles). Después de agitar a temperatura ambiente durante 24 h, la mezcla de reacción se extrajo con diclorometano (3 * 10 mL). Las fases orgánicas combinadas se lavaron con 10 % de Na2S2O3 acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 5: 1)
proporcionando el compuesto SC-DD (90 mg, 0,254 mmoles, 57 %) como un sólido blanco. RMN 1H (400 MHz, CDCla), 5 (ppm), 6,01-5,73 (t, 1H), 2,55-2,54 (m, 1H), 2,12 (s, 3H), 0,62 (S, 3H).
Síntesis del compuesto SC-EE. A una disolución del compuesto SC-DD (80 mg, 0,226 mmoles) en MeOH (5 mL) se añadió 2 gotas de HBr (48 %), seguido por bromo (100 mg, 0,63 mmoles). Después de agitar a temperatura ambiente durante 1 h, la mezcla de reacción se vertió en agua con hielo, luego se extrajo con acetato de etilo (15 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera (20 mL), se secaron sobre MgSO4, se filtraron y se concentraron dando el compuesto en bruto SC-EE (95 mg). El producto en bruto se usó en la siguiente etapa sin más purificación.
Ejemplo 15. Síntesis del compuesto SC-II

Síntesis del compuesto SC-FF. A una disolución del reactante SB-F (4,4 g, 15,38 mmoles) en THF seco (50 mL) se añadió bromuro de etilmagnesio (3 M en THF, 51,28 mL) gota a gota a 0 °C. Entonces se calentó lentamente la disolución y se agitó a temperatura ambiente durante 15 h. Se añadió disolución sat. de NH4Cl (20 mL) para extinguir la reacción y la disolución resultante se extrajo con acetato de etilo (3*100 mL). Los extractos se lavaron con salmuera, se secaron sobre Na2SO4 y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo:acetato de etilo =10:1) proporcionando el producto SC-FF (3,15 g, 10,00 mmoles, 64,8%) como un sólido blanco.
Síntesis del compuesto SC-GG. A una disolución del reactante SC-FF (500 mg, 1,58 mmoles) en THF anhidro (10 mL) se añadió BH3.THF (1,0 M, 7,23 mL, 7,23 mmoles) a temperatura ambiente, y la disolución se agitó a 25 °C durante la noche. Entonces la reacción se inactivó mediante la adición de agua (5 mL), se añadió disolución 2 M de NaOH (10 mL), seguido por 30 % de H2O2 (10 mL). La mezcla resultante se agitó a temperatura ambiente durante 1 hora. Entonces, la mezcla se diluyó con acetato de etilo (200 mL) y la disolución resultante se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto SC-GG se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-HH. A una disolución del reactante SC-GG (6,53 g, 19,67 mmoles) en DCM anhidro (100 mL) enfriado en un baño de refrigeración de agua con hielo se añadió clorocromato de piridinio (8,48 g, 39,34 moles) en porciones. La mezcla se agitó a temperatura ambiente durante la noche. Entonces se diluyó la disolución con DCM (50 mL) y se filtró. Las disoluciones orgánicas combinadas se lavaron con salmuera (100 mL), se secaron sobre Na2SO4 y se concentraron a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo:acetato de etilo=10:1) proporcionando el producto SC-HH (2,5 g, 7,53 mmoles, rendimiento 39 %) como un sólido blanco. RMN 1H (500 MHz, CDCla) 5 (ppm): 2,54 (1H, t), 2,11 (3H, s), 1,42-1,45 (2H, q), 0,91 (3H, t), 0,62 (3H, s).
Síntesis del compuesto SC-II. A una disolución del reactante SC-HH (80 mg, 0,24 mmoles) en metanol (5 mL) se añadió 48 % de ácido bromhídrico (148 mg, 0,884 mmoles), seguido por bromo (241 mg, 0,077 mL, 1,505 mmoles). La disolución se calentó a 25 °C durante 1,5 horas, entonces la mezcla se vertió en agua enfriada (50 mL). El sólido resultante se extrajo con acetato de etilo (2 * 50 mL). Los extractos orgánicos combinados se lavaron con salmuera (20 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SC-II se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 16. Síntesis del compuesto SC-SS

Síntesis del compuesto SC-MM y SC-NN. A una mezcla de la mezcla de reactantes SA-KK y SA-LL (3,0 g, 10,0 mmoles, 1:1) se añadió (Bu)4NF seco, entonces la mezcla se calentó 100 °C durante la noche. La mezcla residual se vertió en 50 mL de H2O y se extrajo con EtOAc (2 * 50 mL). Las fases orgánicas combinadas se lavaron con disolución de salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo =20:1) proporcionando la mezcla de productos SC-MM y SC-NN (2,1 g, 6,5 mmoles, 65 %) como un sólido blanco.
Síntesis del compuesto SC-OO y SC-PP. A una disolución de la mezcla de reactantes SC-MM y SC-NN (2,1 g, 6,5 mmoles) en THF anhidro (30 mL) se añadió BH3.THF (1,0 M, 13,0 mL, 13,0 mmoles), la disolución se agitó a 25 °C durante la noche. Entonces la reacción se inactivó mediante la adición de agua (5 mL). Se añadió disolución 2 M de NaOH (20 mL), seguido por 30 % de H2O2 (20 mL). La mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla se diluyó con acetato de etilo (200 mL) y la disolución resultante se lavó con salmuera (2 * 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. La mezcla de producto en bruto se usó directamente en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-QQ y SC-RR. A una disolución de la mezcla de reactantes en bruto SC-OO y SC-PP (2,2 g, 6,5 mmoles, cantidad teórica) en diclorometano (40 mL) se añadió clorocromato de piridinio (Pcc) en porciones (2,8 g, 13,0 mmoles). La disolución se agitó a 25 °C durante la noche. Entonces la mezcla se filtró a través de una almohadilla corta de gel de sílice y el gel de sílice se lavó con diclorometano (3 * 50 mL). Todo el filtrado se combinó y se concentró a vacío. El residuo se purificó por cromatografía ultrarrápida (éter de petróleo/acetato de etilo = 15:1) proporcionando el producto SC-QQ (910 mg, 2,7 mmoles, rendimiento=41 % (2 etapas)) como un sólido blanco y el producto SC-RR (850 mg, 2,5 mmoles, rendimiento=39 % (2 etapas)) como un sólido blanco. Compuesto SC-QQ: RMN 1H (500 MHz, CDCla) 8 (ppm): 4,17 (d, 2H), 2,53 (t, 1H), 2,17-2,13 (m, 2H), 2,11 (s, 3H), 2,03-2,00 (m, 1H), 0,62 (s, 3H). Compuesto SC-RR: RMN 1H (500 MHz, CDCla) 8 (ppm): 4,45 (AB*d, 1H), 4,39 (AB*d, 1H), 2,54 (t, 1H), 0,62 (s, 3H).
Síntesis del compuesto SC-SS. A una disolución del reactante SC-RR (100 mg, 0,301 mmoles) en metanol (10 mL) se añadió 48 % de ácido bromhídrico (152 mg, 0,903 mmoles), seguido por bromo (241 mg, 0,077 mL, 1,505 mmoles). La disolución se calentó a 25 °C durante 1,5 horas. Entonces la mezcla se vertió en agua enfriada (50 mL). El sólido resultante se extrajo con acetato de etilo (2 * 50 mL). Los extractos orgánicos combinados se lavaron con salmuera (50 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto SC-SS se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 17. Síntesis del compuesto SA-ZZ

Síntesis del compuesto SC-TT y SC-UU. A una disolución del compuesto SB-F (1,3 g, 4,5 mmoles) y PhSO2CH2F (790 mg, 4,5 mmoles) en THF (25 mL) y HMPA (0,5 mL) a -78 °C bajo N2 se añadió LHMDS (5,5 mL, 1 M en THF) gota a gota. Después de agitar a -78 °C durante 2 h, la mezcla de reacción se inactivó con disolución acuosa saturada de NH4Cl (10 mL) y se dejó que se calentara hasta temperatura ambiente, luego se extrajo con Et2O (20 mL * 3). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por cromatografía en columna de gel de sílice (éter de petróleo/acetato de etilo =10/1) dando la mezcla de compuestos SC-TT y SC-UU (1,53 g). La mezcla se purificó aún más por HPLC quiral proporcionando el compuesto SC-TT-1 (220 mg, t= 3,41 min). RMN 1H (500 MHz, CDCls), 5 (ppm), 7,99-7,97 (m, 2H), 7,75-7,74 (m, 1H), 7,62-7,55 (m, 2H), 5,13-5,09 (m, 1H), 4,86-4,78 (d, 1H,), 0,88 (s, 3H); SC-TT-2 (200 mg, t= 3,66 min); RMN 1H (500 MHz, CDCls), 5 (ppm), 7,96-7,95 (m, 1H), 7,71-7,69 (m, 1H), 7,62-7,58 (m, 2H), 5,13-5,09 (m, 1H), 4,87-4,77 (d, 1H), 0,88 (s, 3H); SC-UU-1 (235 mg, t= 4,9 min). RMN 1H (500 MHz, CDCh), 5 (ppm), 7,99-7,97 (m, 1H), 7,72-7,70 (m, 1H), 7,62-7,59 (m, 2H), 5,29-5,20 (d, 1H), 4,88-4,78 (m, 1H), 0,88 (s, 3H); SC-UU-2 (220 mg, t= 5,2 min). RMN 1H (500 MHz, CDCh), 5 (ppm), 7,99-7,97 (m, 2H), 7,72 (m, 1H), 7,62-7,59 (m, 2H), 5,30-5,20 (d, 1H), 5,09-5,08 (m, 1H), 0,88 (s, 3H).
Síntesis del compuesto SC-WW. A una disolución del compuesto SC-TT-1 (200 mg, 0,434 mmoles) y Na2HPO4 anhidro (100 mg) en metanol anhidro (15 mL) a -20 °C bajo N2 se añadió amalgama de Na/Hg (400 mg). Después de agitar a -20 °C a 0 °C durante 1 h, se separó por decantación la disolución de metanol y el residuo sólido se lavó con Et2O (5 * 3 mL). El disolvente de fase orgánica combinada se retiró a vacío, y se añadieron 20 mL de salmuera, seguido por extracción con Et2O. La fase de éter combinada se secó con MgSO4, y el éter se retiró dando el producto en bruto, que se purificó aún más por cromatografía en gel de sílice (éter de petróleo/acetato de etilo = 10/ 1) dando el producto 99 mg, 69 %. RMN 1H (500 MHz, CDCh), 5 (ppm), 5,12-5,10 (m, 1H,), 4,21-24,11 (d, 2H), 0,88 (s, 3H).
Síntesis del compuesto SC-XX. A una disolución del compuesto SC-WW (95 mg, 0,296 mmoles) en THF seco (5 mL) se añadió el complejo de borano-tetrahidrofurano (1 mL de disolución 1,0 M en THF). Después de agitar a temperatura ambiente durante 1 hora, la mezcla de reacción se enfrió en un baño de hielo, luego se extinguió lentamente con 10 % de NaOH acuoso (1 mL), seguido por 30 % de disolución acuosa de H2O2 (1,2 mL). La mezcla se dejó con agitación a temperatura ambiente durante 1 hora, luego se extrajo con EtOAc (3 * 10 mL). Las fases orgánicas combinadas se lavaron con 10% de Na2S2O3 acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron proporcionando el compuesto SC-XX (120 mg en bruto). El producto en bruto se usó en la siguiente etapa sin más purificación.
Síntesis del compuesto SC-YY. A una disolución del compuesto SC-XX (120 mg en bruto) disuelta en 10 mL de diclorometano húmedo (el diclorometano se había agitado con varios mililitros de H2O, luego se separó de la fase acuosa) se añadió peryodinato de Dess-Martin (300 mg, 707 mmoles). Después de agitar a temperatura ambiente durante 24 h, la mezcla de reacción se extrajo con diclorometano (3 * 10 mL). Las fases orgánicas combinadas se
lavaron con 10% de Na2S2O3 acuoso (10 mL), salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por cromatografía sobre gel de sílice (éter de petróleo/acetato de etilo = 1: 5) proporcionando el compuesto SC-y Y (70 mg, 70 % para dos etapas) como un sólido blanco. RMN 1H (500 MHz, CDCh), 5 (ppm), 4,21-4,11 (d, 2H), 2,19 (s, 3H), 0,62 (s, 3H).
Síntesis del compuesto SC-ZZ. A una disolución del reactante (200 mg, 0,594 mmoles) en metanol (5 mL) se añadió 48% de ácido bromhídrico (300 mg, 1,782 mmoles), seguido por bromo (475 mg, 0,152 mL, 2,97 mmoles). La disolución se calentó a 25 °C durante 2 horas. Entonces, la mezcla se vertió en agua enfriada (50 mL). El sólido resultante se extrajo con acetato de etilo (2 * 100 mL). Los extractos orgánicos combinados se lavaron con salmuera (100 mL), se secaron sobre sulfato de magnesio y se concentraron a vacío. El producto en bruto se usó directamente sin más purificación en la siguiente etapa.
Ejemplo 18. Síntesis de los compuestos SA-1 y SA-2

A una suspensión de K2CO3 (50 mg, 0,36 mmoles) en THF (5 mL) se añadió 5-(trifluorometil)-1H-pirazol (80 mg, 0,59 mmoles) y SA (100 mg, 0,25 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo mezcla se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco SA-1 (15 mg, 13,2 %). SA-2 (5 mg, 4,4 %).
SA-1: RMN 1H (500 MHz, CDCh), 5 (ppm), 7,47 (d, 1H),6,59 (d, 1H), 4,99 (1H, AB), 4,95(1H, AB), 2,58 (1H, t), 1,00 2,20 (m, 24H),0,68 (s, 3H). SA-2: RMN 1H (500 MHz, CDCh), 5 (ppm), 7,57 (d, 1H), 6,66 (d, 1H), 5,03 (1H, AB), 4,93(1H, AB), 2,77 (1H, t), 1,00-2,2 (m, 24H), 0,9 (s, 3H).
Ejemplo 19. Síntesis del compuesto SA-3

A una suspensión de K2CO3 (50 mg, 0,36 mmoles) en THF (5 mL) se añadió 1H-pirazol-4-carboxilato de etilo (100 mg, 0,71 mmoles) y SA (72 mg, 0,18 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo mezcla se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (18 mg, 21,6%). RMN1H (500 MHz, CDCl3), 5 (ppm) 7,93 (s, 1H), 7,91 (s, 1H), 4,97 (1H, AB), 4,86 (1H, AB), 4,28 (q, 2H), 2,60 (1H, t), 1,34 (t, 3H), 1,00 2,25 (m, 24H), 0,67 (s,3H).
Ejemplo 20. Síntesis del compuesto SA-4

A una suspensión de K2CO3 (50 mg, 0,36 mmoles) en THF (5 mL) se añadió etil-1H-pirazol-4-carbonitrilo (100 mg, 0,97 mmoles) y SA (50 mg, 0,12 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo mezcla se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (9 mg, 17,4 %). RMN 1H (500 MHz, CDCl3), 5 (ppm) 7,87 (1H, s), 7,82 (1H, s), 5,02 (1H, AB), 4,92 (1H, AB), 2,61 (1H, t), 2,16-2,24 (1H, m), 2,05 (1H, dxt), 1,70 1,88 (6H, m), 1,61-1,69 (2H, m), 1,38-1,52 (6H, m), 1,23-1,38 (5H, m), 1,28 (3H, s), 1,06-1,17 (3H, m), 0,67 (3H, s). EM/CL: tr = 2,24 min, m/z = 410,1 [M+H]+.
Ejemplo 21. Síntesis del compuesto SA-5

A una suspensión de K2CO3 (50 mg, 0,36 mmoles) en THF (5 mL) se añadió etil-1-(1H-pirazol-5-il)etanona (100 mg, 0,91 mmoles) y SA (50 mg, 0,12 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El mezcla de residuos se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (37 mg, 65 %): RMN 1H (500 MHz, CDCl3), 5 (ppm) 7,41 (d, 1H), 6,85 (d, 1H), 4,98 (1H, AB), 4,86 (1H, AB), 2,59 (t, 1H), 2,55 (s,3H), 1,00-2,25 (m, 24H), 0,69 (s,3H).
Ejemplo 22. Síntesis del compuesto SA-6
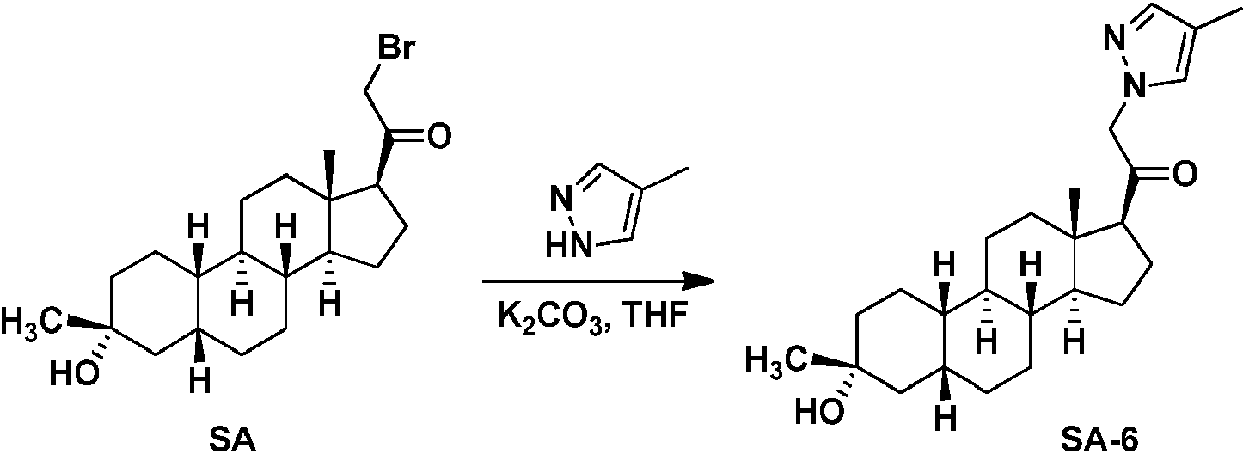
A una disolución de SA (350 mg, 0,88 mmoles) y K2CO3 (243,5 mg, 1,76 mmoles) en 10 mL de DMF seca se añadió 4-metil-1H-pirazol (144,6 mg, 1,76 mmoles) bajo N2 a temperatura ambiente. La mezcla de reacción se agitó durante 18 h a esta temperatura. La mezcla de reacción se vertió en agua, se extrajo con EtOAc (2*50 mL), las fases orgánicas se lavaron con salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron, se purificaron por cromatografía ultrarrápida en columna de sílice (éter de petróleo/acetato de etilo 10:1 a 2:1) proporcionando sA-6 (230 mg, rendimiento: 65,5 %) como un polvo blanco. RMN 1H (400 MHz, CDCl3), 5 (ppm), 7,35 (s, 1H), 7,18 (s, 1H),
4,92-4,79 (m, 2H), 2,59-2,55 (m, 1H), 2,23-2,15 (m, 1H), 2,10 (s, 3H), 2,07-2,03 (m, 1H), 1,88-1,80 (m, 3H), 1,76-1,61 (m, 6H), 1,49-1,22 (m, 16H), 1,13-1,05 (m, 3H), 0,68 (s, 3H). EM/CL: tr = 1,29 min, m/z = 399,2 [M+H]+.
Ejemplo 23. Síntesis del compuesto SA-7
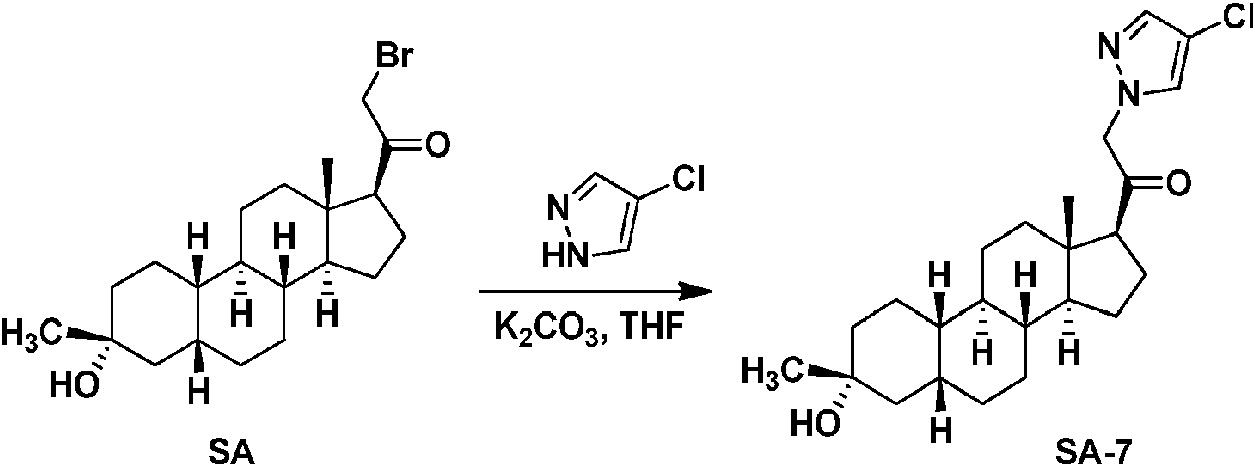
A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 4-cloro-4H-pirazol (21 mg, 0,21 mmoles) y SA (36 mg, 0,09 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (8 mg, 21 %): RMN 1H (500 MHz, CDCh), 5 (ppm), 7,45 (s, 1H), 7,41 (s, 1H), 4,90 (AB, 1H), 4,81 (AB, 1H), 2,57 (t, 1H), 2,22-2,16 (m, 1H), 2,05-2,01 (m, 1H), 1,00-1,90 (m, 22H), 0,67 (s, 3H). EM/CL: tr=2,52 min, m/z=419,1 [M+H]+
Ejemplo 24. Síntesis del compuesto SA-8

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 4-nitro-4H-pirazol (20 mg, 0,18 mmoles) y SA (36 mg, 0,09 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (12 mg, 31 %): RMN 1H (500 MHz, CDCl3), 5 (ppm) 8,11 (s, 1H), 8,01 (s, 1H), 4,93 (AB, 1H), 4,83 (AB, 1H), 2,55 (t, 1H), 2,15-2,10 (m, 1H), 1,99-1,96 (m, 1H), 1,00-1,80 (m, 22H), 0,68 (s, 3H).
Ejemplo 25. Síntesis del compuesto SA-9

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 4-bromo-4H-pirazol (26 mg, 0,18 mmoles) y SA (36 mg, 0,09 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando SA-9 como un sólido blanco (9 mg, 22 %): RMN 1H (500 MHz, CDCl3), 5 (ppm), 7,41 (s, 1H), 7,37 (s, 1H), 4,85 (AB, 1H), 4,77 (AB, 1H), 2,59 (t, 1H), 2,22-2,18 (m, 1H), 2,06-2,01 (m, 1H), 0,90-1,80 (m, 22H), 0,68 (s, 3H), 0,90-1,80 (m, 22H).
Ejemplo 26. Síntesis de los compuestos SA-10 y SA-11

A una suspensión de K2CO3 (55 mg, 0,4 mmoles) en THF (5 mL) se añadió 3-metil-4H-pirazol (33 mg, 0,4 mmoles) y SA (79 mg, 0,2 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando SA-10 como un sólido blanco (9 mg, 11 %) y SA-11 como un sólido blanco (11 mg, 14 %). Compuesto SA-10: RMN 1H (400 MHz, CDCl3), 5 (ppm), 7,41 (d, 1H), 6,07 (s, 1H), 4,85 (s, 2H), 2,84-2,83 (m, 1H), 2,59 (t, 1H), 2,17 (s, 3H), 2,07-2,04 (m, 1H), 1,00-1,90 (m, 22H), 0,69 (s, 3H). Compuesto SA-11: RMN 1H (400 MHz, CDCl3), 5 (ppm), 7,28 (s, 1H), 6,09 (d, 1H), 4,84 (AB, 1H), 4,83 (AB, 1H), 2,56 (t, 1H), 2,27 (s, 3H), 2,22-2,14 (m, 1H), 2,05-2,02 (m, 1H), 1,00-1,90 (m, 22H), 0,67 (s, 3H), 1,00-1,90 (m, 22H).
Ejemplo 27. Síntesis del compuesto SA-12
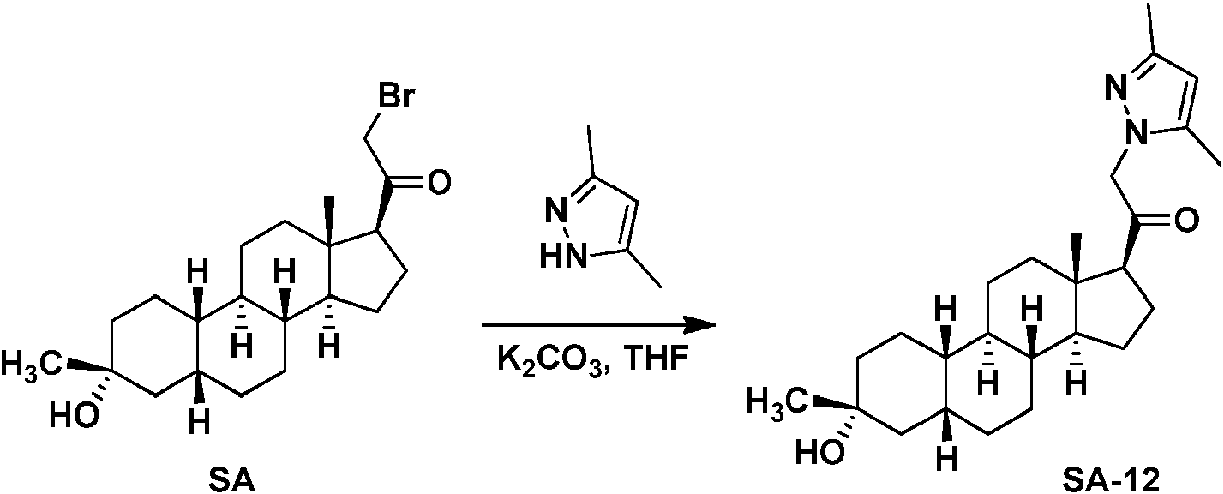
A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 3,5-dimetil-4H-pirazol (17 mg, 0,18 mmoles) y SA (36 mg, 0,09 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (11 mg, 30 %): RMN 1H (500 MHz, CDCl3), 5 (ppm), 5,86 (s, 1H), 4,79 (AB, 1H), 4,74 (AB, 1H), 2,57 (t, 1H), 2,21(s, 3H), 2,18-2,16 (m, 1H), 2,11(s, 3H), 2,05-2,02 (m, 1H), 0,90-1,80 (m, 22H), 0,68 (s, 3H).
Ejemplo 28. Síntesis del compuesto SA-13

A una suspensión de K2CO3 (50 mg, 0,36 mmoles) en THF (6 mL) se añadió 3H-pirazol (16 mg, 0,23 mmoles) y SA (36 mg, 0,09 mmoles). La mezcla se agitó a TA durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (11 mg, 31,3 %). RMN 1H (400 MHz, CDCl3), 5 (ppm), 7,56 (d, 1H), 7,44 (d, 1H), 6,35 (s, 1H), 4,95 (AB, 1H), 4,92 (AB, 1H), 2,60 (1H, t), 1,00-2,25 (m, 24 H), 0,68 (s, 3H).
Ejemplo 29. Síntesis del compuesto SA-14

A una disolución del reactante en bruto (124,8 mg, 0,315 mmoles, cantidad teórica) en THF anhidro (2,5 mL) se añadió 4-(trifluorometil)-1H-pirazol (85,5 mg, 0,628 mmoles), seguido por carbonato de potasio (86,8 mg, 0,628 mmoles). La disolución se calentó a temperatura ambiente durante la noche, entonces la disolución se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =1:1) proporcionando el producto (69 mg, 0,152 mmoles, rendimiento=48 % (2 etapas)) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,72 (2H, s), 4,99 (1H, AB), 4,89 (1H, AB), 2,61 (1H, t), 2,2 (bq, 1H), 1,00-2,10 (23H, m), 0,69 (3H, s). 1,00-2,10 (24H, m). RMN 19F (376 MHz, CDCls) 5 (ppm): -56,46. EM/CL: tr = 2,52 min, m/z = 453,2 [M+H]+
Ejemplo 30. Síntesis del compuesto SA-15

A una disolución del reactante en bruto (249,5 mg, 0,629 mmoles, cantidad teórica) en THF anhidro (5 mL) se añadió 3,4-dimetil-1H-pirazol (120,7 mg, 1,256 mmoles), seguido por carbonato de potasio (173,6 mg, 1,256 mmoles). La disolución se agitó a 25 °C durante la noche, luego la disolución se diluyó con acetato de etilo (200 mL). La disolución resultante se lavó con salmuera (2 x 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =1:3) proporcionando el producto (56 mg, 0,136 mmoles, rendimiento=22 % (2 etapas)) como un sólido blanco. RMN 1H (400 MHz, CDCl3) 5 (ppm): 7,08 (1H, s), 4,77 (1H, AB), 4,76 (1H, AB), 2,55 (1H, t), 2,18 (3H, s), 1,00-2,20 (24H, m), 0,67 (3H, s).
EM/CL: tr = 2,41 min, m/z = 413,2 [M+H]+
Síntesis de 4-etil-1H-pirazol

Síntesis de 4-etiniltrimetilsilano-1H-pirazol. A una disolución del reactante (3,88 g, 20 mmoles), Pd(dppf)Cl2.CH2Cl2 (2,45 g, 3 mmoles), Cul (0,571 g, 3 mmoles) en Et2NH (30 mL) y THF (30 mL) se añadió etiniltrimetilsilano (9,823 g, 14,1 mL, 100 mmoles) bajo N2 atmósfera y la mezcla se agitó a temperatura ambiente durante la noche. Entonces se diluyó la disolución negra con acetato de etilo (300 mL) y la disolución resultante se lavó con salmuera (2 x 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =7,5:1) proporcionando el producto (1,90 g, 11,57 mmoles, rendimiento=58 %) como un sólido parduzco. RMN 1H (500 MHz, DMSO-d6) 5 (ppm): 13,12 (1H, a), 8,07 (1H, s), 7,65 (1H, s), 0,19 (9H, s).
Síntesis de 4-etiniM H-pirazol. A una disolución del reactante (1,90 g, 11,57 mmoles) en THF (20 mL) y agua (4 mL) se añadió hidrato de hidróxido de litio (970 mg, 23,14 mmoles). La disolución se agitó a temperatura ambiente durante la noche, luego se retiró la mayor parte del disolvente de THF a vacío. La disolución se neutralizó mediante la adición de ácido acético y la mezcla resultante se diluyó con diclorometano (200 mL) y salmuera (50 mL). La fase orgánica se separó, se secó sobre sulfato de magnesio y se concentró a vacío. El residuo se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =4:1) proporcionando el producto (828 mg, 8,99 moles, rendimiento=78 %) como un sólido parduzco pálido. RMN 1H (500 MHz, DMSO-d6) 5 (ppm): 13,11 (1H, a), 8,05 (1H, s), 7,65 (1H, s), 3,95 (1H, s).
Síntesis de 4-etil-1H-pirazol. A una disolución del reactante (828 mg, 8,99 mmoles) en etanol (50 mL) se añadió 10 % en peso de Pd/C sobre carbono (165,6 mg, 0,16 mmoles). La mezcla de reacción se hidrogenó con un balón de hidrógeno durante la noche. Se filtró una disolución de muestra pequeña, se concentró a vacío y se caracterizó por RMN 1H para determinar que la reacción estaba completa. Toda la mezcla de reacción se filtró por celite y el celite se lavó con etanol (20 mL). El filtrado combinado se concentró a vacío. El residuo se purificó por una almohadilla corta de gel de sílice (éter de petróleo/acetato de etilo =3:1) proporcionando el producto (643 mg, 6,69 mmoles, rendimiento=74 %) como un líquido amarillo pálido. RMN 1H (500 MHz, DMSO-d6) 5 (ppm): 12,48 (1H, s), 7,39 (2H, s), 2,43 (2H, q, J=7,6 Hz), 1,13 (3H, t, J=7,6 Hz).
Ejemplo 31. Síntesis del compuesto SA-16

A una disolución del reactante en bruto (249,5 mg, 0,629 mmoles, cantidad teórica) en THF anhidro (5 mL) se añadió 4-etil-1H-pirazol (120,7 mg, 1,256 mmoles), seguido por carbonato de potasio (173,6 mg, 1,256 mmoles). La disolución se agitó a 25 °C durante la noche y entonces la disolución se diluyó con acetato de etilo (200 mL). La disolución resultante se lavó con salmuera (2 x 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (éter de petróleo / acetato de etilo =2:3) proporcionando el producto (29,5 mg, 0,0714 mmoles, rendimiento=11 % (2 etapas)) como un sólido blanco. RMN 1H (400 MHz, CDCls) 5 (ppm): 7,38 (1H, s), 7,18 (1H, s), 4,89 (1H, AB), 4,82 (1H, AB), 2,57 (1H, t), 2,51 (2H, q), 0,80 2,20 (24H, m), 0,68 (3H, s). EM/CL: tr = 2,34 min, m/z = 413,1 [M+H]+
Síntesis de 4-metilsulfonil-1H-pirazol

Síntesis de 4-metiltio-1H-pirazol. A una disolución de 4-bromo-1H-pirazol (200 mg, 1,361 mmoles) en THF anhidro (5 mL) se añadió n-BuLi (2,5 M, 1,8 mL, 4,5 mmoles) a 0 °C. La disolución se agitó a temperatura ambiente durante 1 hora. Se añadió MeSSMe (128 mg, 0,12 mL, 1,361 mmoles) a 0 °C y disolución de reacción se agitó a temperatura ambiente durante 2 horas. La reacción se vertió en acetato de etilo (50 mL) y agua (50 mL). La fase orgánica separada se lavó con salmuera (50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. Debido a su olor, el producto en bruto se usó en la siguiente reaccionó de oxidación sin más purificación.
Síntesis de 4-metilsulfonil-1H-pirazol. A una disolución del reactante en bruto (155,4 mg, 1,361 mmoles, cantidad teórica) en diclorometano (2,7 mL) se añadió ácido trifluoroacético (0,1 mL) a 0 °C. Entonces se añadió ácido 3-cloroperbenzoico (m-CPBA, 85 % en peso, 863 mg, 4,25 mmoles) en porciones y la disolución se agitó a temperatura ambiente durante la noche. La disolución se diluyó con acetato de etilo (100 mL) y la disolución resultante se lavó con disolución sat. de Na2CO3 (3 x 50 mL), seguido por salmuera (50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (acetato de etilo con respecto a acetato de etilo:metanol =10:1) proporcionando el producto (51 mg, 0,349 mmoles, rendimiento=26 % (2 etapas)) como un aceite denso amarillo pálido. RMN 1H (500 MHz, CDCh) 5 (ppm): 8,04 (2H, s), 3,14 (3H, s).
Ejemplo 32. Síntesis del compuesto SA-17
(a)

A una disolución del reactante en bruto (124,8 mg, 0,315 mmoles, cantidad teórica) en THF anhidro (2,5 mL) se añadió 4-(metilsulfonil)-1H-pirazol (51 mg, 0,349 mmoles), seguido por carbonato de potasio (48 mg, 0,349 mmoles). La disolución se calentó a 40 °C durante 2 horas, entonces la disolución se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SA-17 (4 mg, 0,00865 mmoles, rendimiento=2,8 % (2 etapas) como un sólido blanco. RMN 1H (400 MHz, CDCls) 5 (ppm): 7,93 (1H, s), 7,87 (1H, s), 5,02 (1H, AB), 4,92 (1H, AB), 3,14 (3H, s), 2,63 (1H, t), 2,17-2,26 (1H, s), 2,04 (1H, dxt), 1,70-1,89 (6H, m), 1,56-1,69 (1H, m), 1,20-1,54 (12H, m), 1,27 (3H, s), 1,04-1,18 (3H, m), 0,68 (3H, s). EM/CL: tr = 2,35 min, m/z = 463,1 [M+H]+.
(b)

A una disolución de SA (200 mg, 0,46 mmoles) en 30 mL de DCM se añadió m-CPBA (236 mg, 1,16 mmoles) a temperatura ambiente (15-19 °C). La mezcla de reacción se agitó durante 6 h a la misma temperatura. La CCF mostró que la reacción estaba completa. La mezcla de reacción se vertió en Na2S2O3 acuoso saturado y se extrajo con DCM (50 mL x 2). Las fases orgánicas se lavaron con Na2S2O3 acuoso saturado (10 mL), salmuera (10 mL), se secaron sobre Na2SO4 anhidro y se concentraron a vacío. El residuo se purificó por columna de gel de sílice (éter de petróleo/acetato de etilo 5/1-1/2) dando SA-17 (140,5 mg, rendimiento: 65 %) como un sólido blanco. RMN 1H: (400 MHz, CDCl3) 57,92 (s, 1H), 7,86 (s, 1H), 5,04-4,89 (m, 2H), 3,13 (s, 3H), 2,64-2,59 (m, 1H), 2,24-2,16 (m, 1H), 2,06-2,03 (m, 1H), 1,87-1,75 (m, 6H), 1,64-1,42 (m, 11H), 1,35 1,27 (m, 7H), 1,16-1,06 (m, 3H), 0,67 (s, 3H).
Ejemplo 33. Síntesis del compuesto SA-18

A una mezcla de SA (200 mg, 0,50 mmoles) y K2CO3 (138,2 mg, 1,00 mmoles) en 5 mL DMF seca se añadió 4-(metiltio)-1H-pirazol (114,2 mg, 1,00 mmoles) bajo N2 a temperatura ambiente (25 °C). La mezcla de reacción se agitó a la misma temperatura durante 18 h. La mezcla de reacción se vertió en agua y se extrajo con EtOAc (50 mL x 2). Las fases orgánicas se lavaron con salmuera, se secaron sobre Na2SO4, se filtraron y se concentraron a vacío. El residuo se purificó por columna de gel de sílice (éter de petróleo/acetato de etilo 10/1 a 2/1) dando el compuesto SA-
18 (165 mg, rendimiento: 76 %) como un polvo blanco. RMN 1H: (400 MHz, CDCI3) 57,53 (s, 1H), 7,42 (s, 1H), 4,94 4,80 (m, 2H), 2,60-2,56 (m, 1H), 2,34 (s, 3H), 2,23-2,16 (m, 1H), 2,06-2,02 (m, 1H), 1,87-1,58 (m, 12H, contuvo H2O), 1,49-1,27 (m, 14H), 1,15-1,07 (m, 2H), 0,67 (s, 3H). EM/CL: tr = 1,32 min, m/z = 431,2 [M+H]+
Ejemplo 35. Síntesis del compuesto SA-20

A una disolución de SA (100,0 mg, 0,23 mmoles) en 10 mL de DCM se añadió m-CPBA (51,86 mg, 0,26 mmoles) a -78 °C. Entonces la mezcla de reacción se agitó a -78 °C durante 3 h. La EM/CL indicó que la reacción estaba completa. A continuación se añadió Na2S2O3 acuoso saturado a la mezcla a -78 °C. Entonces se dejó que la reacción se calentara hasta temperatura ambiente (16-22 °C). La mezcla resultante se extrajo con EtOAc (50 mL * 2), se lavó con agua (10 mL), salmuera (10 mL), se secó sobre Na2SO4 anhidro y se concentró a vacío. El residuo se purificó por columna de gel de sílice (éter de petróleo/acetato de etilo = 1/1 a EtOAc) dando SA-20 (90 mg, rendimiento: 72,3 %) como un sólido blanco. RMN 1H: (400 MHz, CDCls) 57,82 (s, 1H), 7,81 (s, 1H), 5,05-4,88 (m, 2H), 2,89 (d, 3H), 2,64 2,59 (m, 1H), 2,25-2,17 (m, 1H), 2,06-2,03 (m, 1H), 1,87-1,74 (m, 6H), 1,65-1,58 (m, 2H, contuvo H2O), 1,48-1,40 (m, 7H), 1,33-1,28 (m, 8H), 1,15-1,07 (m, 3H), 0,68 (s, 3H). EM/CL: tr = 1,14 min, m/z = 429,2 [M-H2O], 469,2 [M+Na]. Ejemplo 36. Síntesis del compuesto SA-21

A una suspensión del compuesto SA (100 mg, 0,25 mmoles) en THF (25 mL) se añadió 4-fluoro-1H-pirazol (64,5 mg, 0,75 mmoles) y K2CO3 (103 mg, 0,75 mmoles). La mezcla se agitó a 35 °C durante 15 h. Entonces la mezcla de reacción se extrajo con 50 mL de EtOAc, se lavó con 100 mL de H2O y 100 mL de salmuera y se evaporó a vacío. El residuo se purificó por HPLC preparativa de fase inversa proporcionando SA-21 como un sólido blanco (19 mg, 0,05 mmoles, 20 % de rendimiento). RMN 1H (500 MHz, CDCls), 5 (ppm), 7,37 (1H, d), 7,30 (1H, d), 4,85 (1H, AB), 4,77 (1H, AB), 2,57 (t, 1H), 2,2 (bq, 1H), 2,1 (bd, 1H), 1,00-1,9 (22H, m), 0,67 (s, 3H). EM/CL: tr = 2,31 min, EM (ESI) m/z: 403,4 [M+H]+.
Ejemplo 37. Síntesis del compuesto SA-22
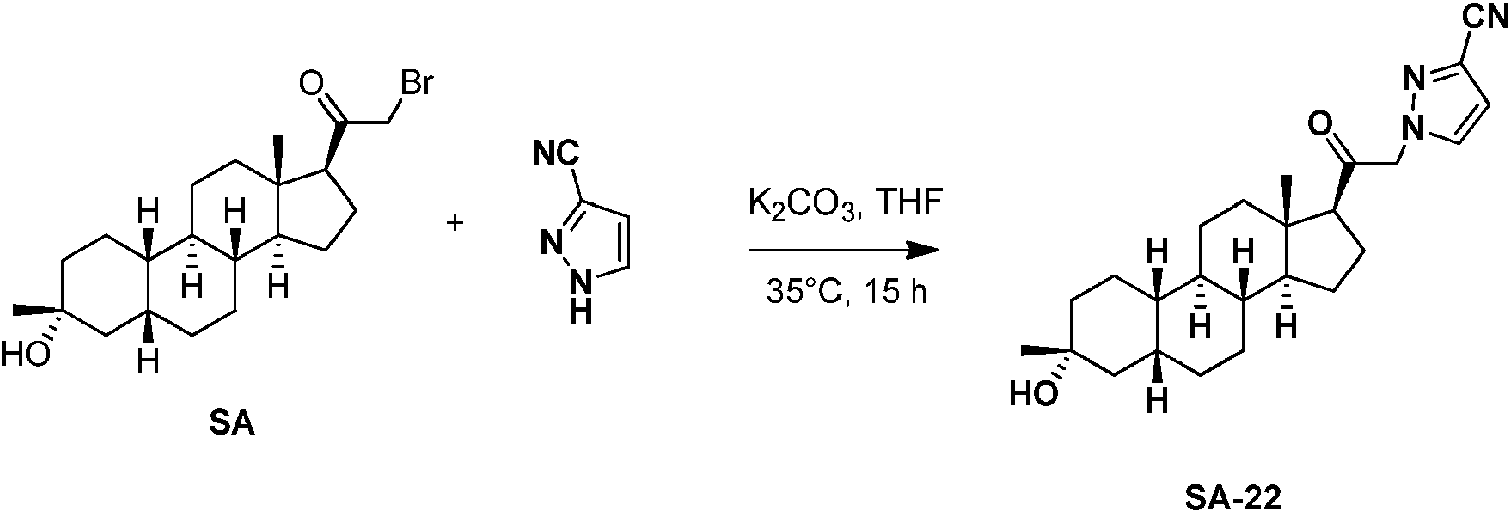
A una suspensión del compuesto SA (100 mg, 0,25 mmoles) en THF (25 mL) se añadió 1H-pirazol-3-carbonitrilo (70 mg, 0,75 mmoles) y K2CO3 (103 mg, 0,75 mmoles). La mezcla se agitó a 35 °C durante 15 h. Entonces la mezcla de reacción se extrajo con 50 mL de EtOAc, se lavó con 100 mL de H2O y 100 mL de salmuera y se evaporó a vacío. El residuo resultante se purificó por HPLC preparativa de fase inversa proporcionando SA-22 como un sólido blanco
(23 mg, 0,056 mmoles, 24 % de rendimiento). RMN 1H (500 MHz, CDCI3), 5 (ppm), 7,48 (d, 1H), 6,73 (d, 1H), 5,03 (1H, AB), 4,93 (1H, AB), 2,60 (t, 1H), 1,00-2,25 (24H, m), 0,68 (s, 3H). EM/CL: tr = 2,38 min, EM (ESI) m/z: 410,2 [M+H]+.
Ejemplo 38. Síntesis del compuesto SA-23

A una suspensión de K2CO3 (55 mg, 0,4 mmoles) en THF (5mL) se añadió IH-pirazol (28 mg, 0,4 mmoles) y el compuesto SA-JJ (85 mg, 0,2 mmoles). La mezcla se agitó a TA durante 15 h, entonces la mezcla residual se vertió en 5 mL de H2O y se extrajo con EtoAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando SA-23 como un sólido blanco (29 mg, 35 %). RMN 1H (500 m Hz , CDCl3) 5 (ppm): 7,55 (d, 1H), 7,41 (d, 1H), 6,33 (t, 1H), 4,97 (AB, 1H), 4,88 (AB, 1H), 3,42-3,37 (m, 5H), 2,58 (t, 1H), 2,22-2,16 (m, 1H), 2,06 2,03 (m, 1H), 1,00-1,90 (m, 22H), 0,68 (s, 3H). EM-CL: tr = 2,27 min, m/z = 415,3 [M+H]+
Ejemplo 39. Síntesis del compuesto SA-24

A una disolución del compuesto SA-JJ (120 mg, 0,28 mmoles) en THF (3 mL) se añadió K2CO3 (190 mg, 1,4 mmoles) y 1H-pirazol-4-carbonitrilo (130 mg, 1,4 mmoles). La disolución resultante se agitó a temperatura ambiente durante la noche, entonces la reacción se diluyó con EtoAc (20 mL). La disolución resultante se lavó con salmuera (10 mL), se secó sobre Na2SO4 y se concentró a vacío. El residuo se purificó por HPLC preparativa dando SA-24 (30 mg, 24 %) como un sólido blanco. RMN 1H: (500 MHz, CDCh), 5 (ppm), 7,86 (1H, s), 7,81 (1H, s), 5,0 (1H, AB), 4,88 (1H, AB), 3,39 (3H, s), 3,19 (2H, s), 2,59 (1H, t), 2,2 (m, 1H), 0,69 (3H, s), 0,60-2,1 (23H, m). EM-CL: tr=2,25 min; m/z=440,4 (M+H)+
Ejemplo 40. Síntesis del compuesto SA-25
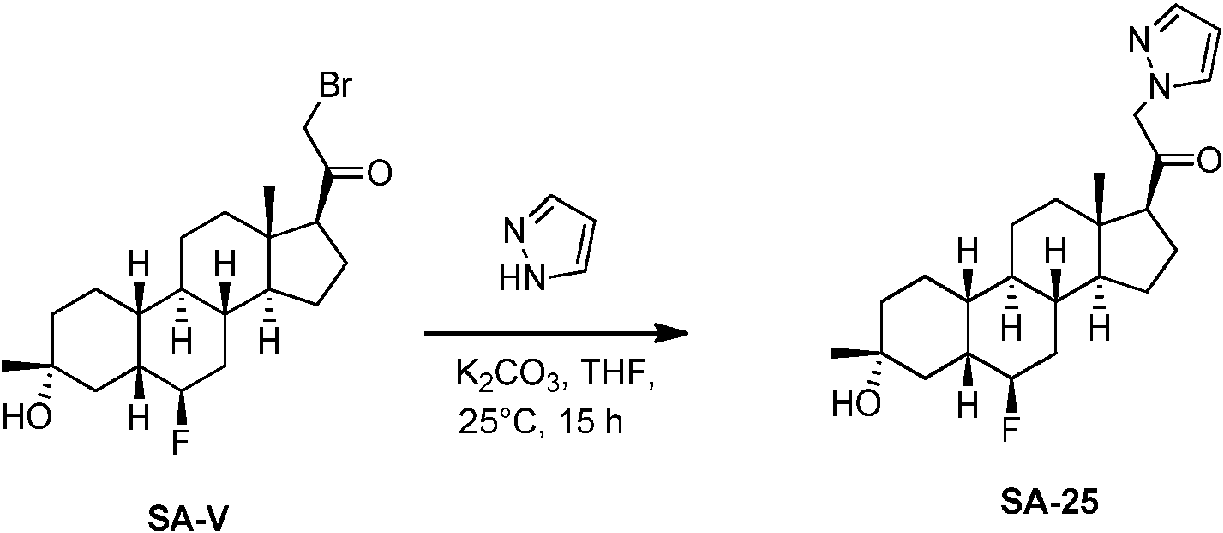
A una suspensión de SA-V (20 mg, 0,04 mmoles) en THF (5 mL) se añadió pirazol (30 mg, 0,45 mmoles) y K2CO3 (60 mg, 0,45 mmoles). La mezcla se agitó a 25 °C durante 15 h. Entonces la mezcla de reacción se purificó por HPLC preparativa de fase inversa proporcionando SA-25 como un sólido blanco (11 mg, 57% de rendimiento). RMN 1H (500 MHz, CDCh), 5 (ppm), 7,56 (s, 1H), 7,42 (s, 1H), 6,33 (s, 1H), 4,97 (1H, AB), 4,89 (1H, AB), 4,86-4,69 (m, 1H), 2,60 (1H, t), 1,00-2,20 (22H, m), 0,72 (s, 3H).
Ejemplo 42. Síntesis del compuesto SA-27

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 3H-pirazol (16 mg, 0,23 mmoles) y SC-EE (36 mg, 0,08 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (12 mg, 34,3 %). RMN 1H (500 MHz, CDCl3) 5 (ppm), 7,55 (d, 1H), 7,42-7,41 (d, 1H), 6,34 (t, 1H), 5,87 (t, 1H), 4,97 (1H, AB), 4,88 (1H, AB), 2,55(t, 1H), 0,69 (s, 3H), 1,10 2,25 (m, 24H), 0,69 (s, 3H).
Ejemplo 43. Síntesis del compuesto SA-28

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió1H-pirazol-4-carbonitrilo (20 mg, 0,23 mmoles) y SC-EE (36 mg, 0,09 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (22 mg, 61,6 %). RMN 1H (400 MHz, CDCl3), 5 (ppm): 7,86 (s, 1H), 7,81(s, 1H), 5,87 (t, 1H), 5,02 (AB, 1H), 4,90 (AB, 1H), 2,61 (t, 1H), 1,00-2,25 (m, 24H), 0,68 (s, 3H). EM-CL: tr=2,30 min, m/z = 446,2 (M+ 1).
Ejemplo 44. Síntesis del compuesto SA-29

A una suspensión de K2CO3 (127 mg, 0,92 mmoles) en THF (5 mL) se añadió 4-(metilsulfonil)-1H-pirazol (67 mg, 0,46 mmoles) y el reactante (200 mg, 0,46 mmoles) y la mezcla resultante se agitó a temperatura ambiente durante 15 h. Entonces la mezcla se vertió en 20 mL de H2O y se extrajo con EtOAc (2 x 50 mL). Las fases orgánicas
combinadas se lavaron con salmuera (50 mL), se secaron sobre sulfato de sodio, se filtraron y se concentraron a vacío. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título SA-29 como un sólido blanco (46 mg, 0,0923 mmoles, rendimiento=20 %). RMN 1H (500 MHz, CDCl3) 5 (ppm): 7,93 (s, 1H), 7,87 (s, 1H), 5,87 (t, 1H), 5,02 (AB, 1H), 4,92 (AB, 1H), 3,14 (s, 3H), 2,63 (t, 1H), 2,25-2,17 (m, 1H), 2,08-2,04 (m, 1H), 1,00-2,00 (m, 22H), 0,69 (s, 3H). EM-CL: tr = 2,10 min, m/z = 499,3 [M+H]+
Ejemplo 61. Síntesis del compuesto SA-30

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió 1H-pirazol-4-carbonitrilo (20 mg, 0,21 mmoles) y SA-AA (36 mg, 0,087 mmoles). La mezcla se agitó a ta durante 15 h. Entonces la mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (10 mg, 27,0 %). RMN 1H (400 MHz, CDCl3), 5 (ppm): 7,86(s, 1H), 7,81(s, 1H), 5,99 (AB, 1H), 5,85 (AB, 1H), 2,65 (t, 1H), 1,59 (q, 2H), 0,88 (t, 3H), 1,00 2,25 (m, 24H), 0,89 (t, 3H), 0,68 (s, 3H). EM-CL: tr=2,45 min, m/z = 424,3 (M+ 1).
Ejemplo 45. Síntesis del compuesto SA-31

A una suspensión de K2CO3 (55 mg, 0,4 mmoles) en THF (5 mL) se añadió 1H-pirazol (28 mg, 0,4 mmoles) y el compuesto SC-SS (83 mg, 0,2 mmoles). La mezcla se agitó a TA durante 15 h, entonces la mezcla residual se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando SA-31 como un sólido blanco (7 mg, 9%). Compuesto SA-31: RMN 1H (500 MHz, CDCl3) 5 (ppm): 7,55 (d, 1H), 7,41 (d, 1H), 6,33 (t, 1H), 4,97 (AB, 1H), 4,88 (AB, 1H), 4,48 (ABxd, 1H), 4,38 (ABxd, 1H), 2,59 (t, 1H), 2,23-2,16 (m, 1H), 2,09-2,05 (m, 1H), 1,00-1,90 (22H, m), 0,68 (s, 3H). EM-CL: tr = 2,15 min, m/z = 403,3 [M+H]+
Ejemplo 46. Síntesis del compuesto SA-32
A una suspensión de K2CO3 (55 mg, 0,4 mmoles) en THF (5 mL) se añadió 1H-pirazol-4-carbonitrilo (37 mg, 0,4 mmoles) y el compuesto SC-SS (83 mg, 0,2 mmoles). La mezcla se agitó a TA durante 15 h, entonces la mezcla residual se vertió en 5 mL de H2O y se extrajo con EtoAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando SA-32 como un sólido blanco (20 mg, 23 %). Compuesto SA-32: RMN 1H (500 MHz, CDCl3) 5 (ppm): 7,86 (s, 1H), 7,81 (s, 1H), 5,02 (AB, 1H), 4,91 (AB, 1H), 4,48 (ABxd, 1H), 4,38 (ABx d, 1H), 2,61 (t, 1H), 2,23 (s, 1H), 2,21-2,17 (m, 1H), 2,07-2,03 (m, 1H), 1,00-1,90 (m, 21H), 0,67 (s, 3H). EM-CL: tr = 2,22 min, m/z = 428,3 [M+H]+
Ejemplo 47. Síntesis del compuesto SA-33

A una suspensión de K2CO3 (119 mg, 0,86 mmoles) en THF (5 mL) se añadió 4-(metilsulfonil)-1H-pirazol (63 mg, 0,43 mmoles) y el reactante SC-SS (180 mg, 0,43 mmoles) y la mezcla se agitó a TA durante 15 h. La mezcla residual se vertió en 20 mL de H2O y se extrajo con EtOAc (2 x 50 mL). Las fases orgánicas combinadas se lavaron con salmuera (50 mL), se secaron sobre sulfato de sodio, se filtraron y se concentraron a vacío. La mezcla residual se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título SA-33 como un sólido blanco (53 mg, 0,110 mmoles, rendimiento=25,6 %). RMN 1H (500 MHz, CDCls) 5 (ppm): 7,93 (s, 1H), 7,87 (s, 1H), 5,02 (AB, 1H), 4,92 (AB, 1H,), 4,48 (ABxd), 4,39 (ABxd, 1H), 3,14 (s, 1H), 2,63 (t, 1H), 2,24-2,17 (m, 1H), 2,07-2,04 (m, 1H), 1,00-1,90 (m, 24H), 0,68 (s, 3H). EM-CL: tr = 2,06 min, m/z = 481,2 [M+H]+
Ejemplo 49. Síntesis del compuesto SA-35

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió1H-pirazol (20 mg, 0,23 mmoles) y SA-AA (36 mg, 0,09 mmoles). La mezcla se agitó a ta durante 15 h. La mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. El residuo se purificó por HPLC preparativa de fase inversa proporcionando SA-35 como un sólido blanco (8 mg, 21,6 %). RMN 1H (400 MHz, CDCls), 5 (ppm), 7,53 (1H, s), 7,41 (1H, s) 6,33 (s, 1H), 4,97 (AB, 1H), 4,88 (AB, 1H), 2,58 (1H, t), 1,00-2,25 (24H, m), 0,88 (3H, t), 0,68 (s, 3H). EM-CL: tr=2,39 min, m/z = 399,4 (M++1).
Ejemplo 50. Síntesis del compuesto SB-1

A una suspensión de K2CO3 (25 mg, 0,18mmoles) en THF (5mL) se añadió pirazol (13 mg, 0,18mmoles) y el compuesto SB (36 mg, 0,09 mmoles). Después de agitar a temperatura ambiente durante 15 h, la mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla de reacción se purificó por HPLC preparativa de fase inversa dando 7,54 (d, 1H), 7,41 (d, 1H), 6,33 (t, 1H), 4,97 (AB, 1H), 4,87 (AB, 1H), 2,58 (t, 1H), 0,90-2,25 (m, 21 H), 0,69 (s, 3H).
Ejemplo 51. Síntesis del compuesto SB-2

A una disolución de SB en bruto (124,8 mg, 0,314 mmoles, cantidad teórica) en THF anhidro (3 mL) se añadió 4-cianopirazol (58,5 mg, 0,628 mmoles), seguido por carbonato de potasio (86,8 mg, 0,628 mmoles). La disolución se calentó a 50 °C durante 2 horas. Entonces la disolución se diluyó con acetato de etilo (200 mL). La disolución resultante se lavó con salmuera (2 x 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto deseado (34,6 mg, 0,0845 mmoles, rendimiento global de dos etapas =27 %) como un sólido blanco. RMN 1H (400 MHz, Cd C|3) 5 (ppm): 7,86 (1H, s), 7,82 (1H, s), 5,01 (1H, AB), 4,91 (1H, AB), 2,61 (1H, t), 2,16-2,26 (2H, m), 2,04 (1H, m), 1,00-1,90 (21H, m), 0,68 (3H, s). EM/CL: tr = 2,26 min, m/z = 410,2 [M+H]+
Ejemplo 52. Síntesis del compuesto SB-3
A una disolución del reactante en bruto (374,3 mg, 0,942 mmoles, cantidad teórica) en THF anhidro (7,5 mL) se añadió 4-metilsulfonil-1H-pirazol (110 mg, 0,754 mmoles), seguido por carbonato de potasio (130 mg, 0,942 mmoles). La disolución se calentó a 25 °C durante la noche y entonces la disolución se diluyó con diclorometano (200 mL). La disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =1:3) proporcionando el producto en bruto que se contaminó con 4-metilsulfonil-1H-pirazol. El producto en bruto se recristalizó entonces en acetato de etilo proporcionando el producto puro (38,4 mg, 0,083 mmoles, rendimiento global de dos etapas =8,8 %) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,92 (1H, s), 7,87 (1H, s), 5,02 (1H, AB), 4,91 (1H, AB), 3,14 (3H, s), 2,63 (1H, t), 0,9-2,25 (21H, m), 0,68 (3H, s). EM/CL: tr = 2,15 min, m/z = 463,3 [M+H]+
Ejemplo 53. Síntesis del compuesto SB-4

A una disolución del reactante en bruto (61,1 mg, 0,143 mmoles, cantidad teórica) en THF anhidro (5 mL) se añadió 1H-pirazol (97 mg, 1,43 mmoles), seguido por carbonato de potasio (198 mg, 1,43 mmoles). La disolución se calentó a 50 °C durante la noche. Entonces la disolución se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SB-4 (7 mg, 0,0169 mmoles, rendimiento global de dos etapas =12 %) como un sólido blanco. RMN 1H (400 MHz, CDCls) 5 (ppm) 7,55 (1H, d), 7,42 (1H, d), 6,33 (1H, t), 4,97 (1H, AB), 4,88 (1H, AB), 3,39 (3H, s), 3,19 (2H, s), 2,59 (1H, t, J=8,9 Hz), 0,69 (3H, s), 0,60-2,25 (24H, m). EM-CL: tr = 2,31 min, m/z =415,3 [M+H]+
Ejemplo 54. Síntesis de los compuestos SB-5

A una disolución del reactante en bruto (122,6 mg, 0,287 mmoles, cantidad teórica) en THF anhidro (3 mL) se añadió 4-cianopirazol (134 mg, 1,435 mmoles), seguido por carbonato de potasio (198 mg, 1,435 mmoles). La disolución se calentó a 60 °C durante la noche. Entonces, la disolución se diluyó con acetato de etilo (200 mL). La disolución resultante se lavó con salmuera (2 x 100 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto deseado SB-5 (12,4 mg, 0,0282 mmoles, rendimiento global de dos etapas=9,8 %) y un subproducto (4,2 mg, 0,00955 mmoles, rendimiento global de dos etapas=3,3 %) como un sólido blanco. Compuesto s B-5: RMN 1H (400 MHz, CDCl3) 5 (ppm): 7,86 (s, 1H), 7,81 (s, 1H), 5,02 (AB, 1H), 4,90 (AB, 1H), 3,42 (AB, 1H), 3,40 (S, 3H), 3,39 (AB, 1H), 2,64 (s, 1H), 2,61 (t, 1H), 1,00-2,25 (m, 23H), 0,67 (s, 3H). EM-CL: tr = 2,32 min, m/z =440,2 [M+H]+
Ejemplo 55. Síntesis del compuesto SB-7

A una disolución del reactante en bruto (368 mg, 0,861 mmoles, cantidad teórica) en THF anhidro (7,5 mL) se añadió 4-metilsulfonil-1H-pirazol (126 mg, 0,861 mmoles), seguido por carbonato de potasio (119 mg, 0,861 mmoles). La disolución se calentó a 25 °C durante la noche, luego la disolución se diluyó con diclorometano (200 mL) y la disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por cromatografía en gel de sílice (éter de petróleo/acetato de etilo =1:3) proporcionando el producto en bruto que se contaminó con 4-metilsulfonil-1H-pirazol. El producto en bruto se recristalizó entonces en acetato de etilo proporcionando el producto puro (50 mg, 0,101 mmoles, rendimiento global de dos etapas=12 %) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,92 (1H, s), 7,87 (1H, s), 5,02 (1H, AB), 4,91 (1H, AB), 3,39 (3H, s), 3,19 (2H, s), 3,14 (3H, s), 2,63 (1H, t), 0,9-2,25 (21H, m), 0,68 (3H, s). EM/CL: tr = 2,13 min, m/z = 493,0 [M+H]+
Ejemplo 56. Síntesis del compuesto SB-8

A una suspensión de K2CO3 (25 mg, 0,18 mmoles) en THF (5 mL) se añadió pirazol (13 mg, 0,18 mmoles) y el compuesto SB-W (36 mg, 0,09 mmoles). Después de agitar a temperatura ambiente durante 15 h, la mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de sodio, se filtraron y se concentraron. La mezcla de reacción se purificó por HPLC preparativa de fase inversa proporcionando el compuesto del título como un sólido blanco (15,6 mg, 0,073 mmoles, 40,4 %). RMN 1H (500 MHz, CDCls) 5 (ppm): 7,54 (d, 1H), 7,41 (d, 1H), 6,33 (t, 1H), 4,97 (AB, 1H), 4,87 (AB, 1H), 3,52 (q, 2H), 3,21 (s, 2H), 2,59 (t, 1H), 0,69 (s, 3H), 0,69-2,25 (m, 24H). EM/CL: tr = 2,35 min. m/z = 429,4 [M+H]+.
Ejemplo 57. Síntesis del compuesto SB-9
A una suspensión de K2CO3 (63 mg, 0,46 mmoles) en THF (10 mL) se añadió 4-cianopirazol (43 mg, 0,46 mmoles) y el compuesto SB-W (100 mg, 0,23 mmoles). Después de agitar a temperatura ambiente durante 15 h, la mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 x 10 mL). Las fases orgánicas combinadas se lavaron con salmuera (2 x 10 mL), se secaron sobre sulfato de sodio, se filtraron y se concentraron a vacío. El residuo se purificó por HPLC preparativa de fase inversa proporcionando SB-9 como un sólido blanco (43,5 mg, 0,095 mmoles, 41,7 %). RMN 1H (500 MHz, CDCl3) 5 (ppm) 7,86 (1H, s), 7,82 (1H, s), 5,01 (1H, AB), 4,91 (1H, AB), 3,53 (2H, q), 3,22 (2H, s), 2,61 (1H, t), 0,67 (3H, s), 0,67-2,25 (24H, m). EM/CL: tr = 2,37 min. m/z = 454,4 [M+H]+.
Ejemplo 58. Síntesis del compuesto SB-10

A una suspensión de SB-FF (40 mg, 0,09 mmoles) en THF (5 mL) se añadió 1H-pirazol (30 mg, 0,45 mmoles) y K2CO3 (60 mg, 0,45 mmoles). La mezcla se agitó a 25 °C durante 15 h. La disolución se diluyó entonces con acetato de etilo (100 mL) y la disolución resultante se lavó con salmuera (100 mL), se secó sobre sulfato de sodio y se concentró a vacío. La mezcla de reacción se purificó por HPLC preparativa de fase inversa proporcionando SB-10 como un sólido blanco (15 mg, 38 % de rendimiento). RMN 1H (400 MHz, CDCls), 5 (ppm), 7,55 (s, 1H), 7,41 (s, 1H), 6,33 (s, 1H), 4,99-4,95 (AB, 1H), 4,90-4,87 (AB, 1H), 4,55 (1H, d, 1H), 2,60 (t, 1H), 0,70-2,25 (m, 22H), 0,71 (s, 3H).
Ejemplo 59. Síntesis del compuesto SB-11

A una disolución del reactante en bruto SB-FF (50,7 mg, 0,122 mmoles, cantidad teórica) en THF anhidro (1,5 mL) se añadió 4-cianopirazol (22,7 mg, 0,244 mmoles), seguido por carbonato de potasio (33,7 mg, 0,244 mmoles). La disolución se agitó a 25 °C durante la noche. Entonces, la disolución se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 x 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto deseado (14,2 mg, 0,0332 mmoles, rendimiento global de dos etapas=27 %) como un sólido blanco. RMN 1H (400 MHz, CDCl3) 5 (ppm): 7,85 (s, 1H), 7,81 (s, 1H), 5,03-4,87 (m, 2H), 4,62-4,50 (m, 1H), 2,63-2,62 (m, 1H), 2,30-2,20 (m, 1H), 2,05-1,95 (m, 2H), 1,90-1,60 (m, 6H), 1,50-1,20 (m, 15H), 0,70 (s, 3H). RMN 19F (376 MHz, CDCl3) 5 (ppm): -193,13. EM/CL: tr = 2,13 min, m/z = 428,0 [M+H]+
Ejemplo 60. Síntesis del compuesto SB-12
A una disolución de SB-FF (85 mg, 0,20 mmoles) en 2m L de DMF se añadió 4-metiMH-pirazol (33,6 mg, 0,41 mmoles) y K2CO3 (84,84 mg, 0,61 mmoles). La mezcla de reacción se agitó a 28 °C durante 1 h. La disolución resultante se inactivó con agua (10 mL) y se extrajo con EtOAc (15 mL x 2). Las fases orgánicas combinadas se secaron y se concentraron a vacío. El residuo se purificó por cromatografía en columna sobre gel de sílice eluida con (éter de petróleo/acetato de etilo = 12/1 a 2/1) dando SB-12 (23,1 mg, rendimiento: 31,6 %) como un sólido blanco.
RMN 1H (SB-12): (400 MHz, CDCl3) 57,34 (s, 1 H), 7,17 (s, 1H), 4,92-4,75 (m, 2H), 4,66-4,47 (m, 1H), 2,60-2,56 (m, 1H), 2,25-1,99 (m, 6H), 1,91-1,61 (m, 6H), 1,54-1,03 (m, 15H), 0,84-0,74 (m, 1H), 0,70 (s, 3H). EM/CL: tr = 1,23 min, m/z = 417,2 [M+H]+.
Ejemplo 61. Síntesis del compuesto SB-13

Se agitó una mezcla de SB-FF (100 mg, 0,241 mmoles), 1H-pirazol-3-carbonitrilo (45 mg, 0,48 mmoles), K2CO3 (66 mg, 0,48 mmoles) y DMF (3 mL) a temperatura ambiente durante 2 h. La CCF mostró que la reacción se había terminado. La mezcla de reacción se vertió en salmuera (10 mL) y se extrajo con EtOAc (10 mL x 2). Se combinaron las fases orgánicas y se secaron sobre Na2SO4, se concentraron dando el producto en bruto, que se purificó por columna de gel de sílice dando SB-13 (30 mg, rendimiento: 28 %) como un sólido blanco. RMN 1H: (400 MHz, CDCl3) 5 7,48 (s, 1H), 6,73 (s, 1H), 4,79-4,97 (m, 2H), 4,47-4,65 (m, 1H), 2,56-2,63 (m, 1H), 2,30-2,20 (m, 1H), 2,10-2,00 (m, 1H), 1,90-1,60 (m, 6H), 1,50-1,20 (m, 15H), 0,85-0,75 (m, 1H), 0,70 (s, 3H). EM/CL: tr = 1,23 min, m/z = 428,2 [M+H]+.
Ejemplo 62. Síntesis de SB-14

A una disolución de SB-FF (100 mg, 0,24 mmoles) en DMF (2 mL) se añadió A1 (55 mg, 0,48 mmoles) y K2CO3 (100 mg, 0,72 mmoles) a 19 °C. La reacción se agitó a 19 °C durante 16 h. La mezcla resultante se vertió en agua (3 mL). La mezcla se extrajo con EtOAc (2 mL x 3). Las fases orgánicas combinadas se lavaron con salmuera (5 mL), se secaron sobre Na2SO4 y se concentraron a vacío. El residuo se purificó por columna de gel de sílice (éter de petróleo/acetato de etilo = 10/1 a 3/1) dando SB-14 (80 mg, rendimiento: 74%) como un sólido rosa. RM N1H:
(400 MHz, CDCl3) 57,53 (s, 1H), 7,43 (s, 1H), 4,79-4,97 (m, 2H), 4,47-4,65 (m, 1H), 2,56-2,63 (m, 1H), 2,35 (s, 3H), 2,19-2,26 (m, 1H), 2,00-2,08 (m, 2H), 1,63-1,92 (m, 5H), 1,35-1,57 (m, 5H), 1,20-1,1,32 (m, 5H), 1,07-1,18 (m, 5H), 0,75-0,91 (m, 1H), 0,71 (s, 3H). EM/CL: tr = 1,25 min, m/z = 449,2 [M+H]+.
Ejemplo 63. Síntesis de SB-15

A una disolución de SB-14 (80 mg, 0,19 mmoles) en DCM (5 mL) se añadió m-CPBA (90 mg, 0,45 mmoles) a 0 °C. La mezcla de reacción se agitó a 20 °C durante 2 h. Se añadió disolución acuosa saturada de NaS2O3 (5 mL). La mezcla resultante se agitó a 20 °C durante 30 min, y se extrajo con EtOAc (5 mL x 3). Las fases orgánicas combinadas se lavaron con salmuera (10 mL), se secaron sobre Na2sO4 y se concentraron a vacío. El residuo se purificó por columna de gel de sílice (éter de petróleo/acetato de etilo = 1/2) dando SB-15 (30 mg, 47 %) como un sólido blanco.
RMN 1H: (400 MHz, CDCl3) 57,93 (s, 1H), 7,87 (s, 1H), 4,87-5,07 (m, 2H), 4,48-4,66 (m, 1H), 3,14 (s, 3H), 2,58-2,68
(m, 1H), 2,17-2,30 (m, 1H), 1,97-2,12 (m, 2H), 1,65-1,90 (m, 6H), 1,45-1,55 (m, 3H), 1,05-1,40 (m, 12H), 0,80-0,91 (m, 1H), 0,71 (s, 3H). EM/CL: tr = 0,85 min, m/z = 481,2 [M+H]+.
Ejemplo 66. Síntesis del compuesto SB-18

A una disolución del reactante en bruto SC-O (62 mg, 0,150 mmoles) en THF anhidro (5 mL) se añadió 1H-pirazol (20,4 mg, 0,30 mmoles), seguido por carbonato de potasio (41,5 mg, 0,30 mmoles). La disolución se calentó a 50 °C durante la noche. Entonces la disolución se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 * 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SB-18 (10 mg, 0,0251 mmoles, rendimiento=17 %) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,55 (1H, s), 7,41 (1H, s), 6,33 (1H, s), 4,97 (1H, AB), 4,89 (1H, AB), 2,59 (1H, t), 2,20 (1H, dd), 0,60-2,05 (22H, m), 0,69 (3H, s).
Ejemplo 67. Síntesis de SB-19

A una disolución del compuesto SC-Y (60 mg, en bruto) en THF seco (2 mL) se añadió carbonato de potasio (100 mg) y 1H-pirazol (60 mg, 0,09 mmoles). La mezcla de reacción se agitó a temperatura ambiente durante 16 horas, y entonces se extrajo con EtOAc (3 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera (10 mL), se secaron sobre MgSO4, se filtraron y se concentraron. El residuo se purificó por HPLC preparativa proporcionando el compuesto del título SB-19 (7 mg, 12 %) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,54 (1H, d), 7,41 (1H, d), 6,33 (1H, t), 4,96 (1H, AB), 4,88 (1H, AB), 3,33 (3H, s), 3,04 (1H, s), 2,58 (1H, t), 0,60-2,20 (22H, m), 0,68 (3H, s).
Ejemplo 68. Síntesis del compuesto SB-20

A una disolución del reactante en bruto SC-II (100 mg, 0,241 mmoles) en THF anhidro (5 mL) se añadió 3H-pirazol (82 mg, 1,2 mmoles), seguido por carbonato de potasio (170 mg, 1,2 mmoles) y la disolución se calentó a 60 °C durante 2 h. Entonces, la mezcla de reacción se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 * 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SB-20 (24 mg, 0,06 mmoles, rendimiento=25 %)
como un sólido blanco. RMN 1H (500 MHz, CDCI3) 5 (ppm): 7,55 (1H, d), 7,41 (1H, d), 6,33 (1H, t), 4,95 (1H, AB), 4,89 (1H, AB), 2,59 (1H, t), 0,69 (3H, s), 0,69-2,25 (24H, m). EM/CL: tr=2,46 min, m/z=399,2 [M+H]+
Ejemplo 69. Síntesis del compuesto SB-21

A una disolución del reactante en bruto SC-II (100 mg, 0,241 mmoles) en THF anhidro (5 mL) se añadió 1H-pirazol-4-carbonitrilo (112 mg, 1,2 mmoles), seguido por carbonato de potasio (170 mg, 1,2 mmoles) y la disolución se calentó a 60 °C durante 2 h. Entonces la mezcla de reacción se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 * 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SB-21 (46 mg, 0,109 mmoles, rendimiento=45 %) como un sólido blanco. RMN 1H (500 MHz, CDCls) 5 (ppm): 7,86 (1H, s), 7,81 (1H, s), 5,00 (1H, AB), 4,92 (1H, AB), 2,61 (1H, t), 0,67 (3H, s), 0,67-2,25 (24H, m). EM/CL: tr=2,47 min, m/z=424,2 [M+H]+ Ejemplo 70. Síntesis del compuesto SB-22

A una disolución del reactante en bruto SC-ZZ (100 mg, 0,241 mmoles) en THF anhidro (5 mL) se añadió 1H-pirazol-4-carbonitrilo (112 mg, 1,2 mmoles), seguido por carbonato de potasio (170 g, 1,2 mmoles). La disolución se calentó a 60 °C durante 2 h, entonces la disolución se enfrió hasta temperatura ambiente y se diluyó con acetato de etilo (100 mL). La disolución resultante se lavó con salmuera (2 * 50 mL), se secó sobre sulfato de magnesio y se concentró a vacío. El producto en bruto se purificó por HPLC preparativa de fase inversa proporcionando el producto SB-22 (38 mg, 0,09 mmoles, rendimiento=38 %) como un sólido blanco. RMN 1H (500 MHz, CDCh) 5 (ppm): 7,86 (1H, s), 7,81 (1H, s), 5,87 (2H, d), 5,02 (1H, AB), 4,90 (1H, AB), 4,17 (2H, d), 2,61 (1H, t), 0,70-2,25 (22H, m), 0,68 (3H, s). EM/CL: tr=2,24 min, m/z=428 [M+H]+
Ejemplo 71. Síntesis de SB-23
A una suspensión de K2CO3 (19 mg, 0,14 mimóles) en THF (5 mL) se añadió pirazol (10 mg, 0,14 mimóles) y el compuesto SB (30 mg, 0,07 mimóles). Después de agitar a temperatura ambiente durante 15 h, la mezcla de reacción se vertió en 5 mL de H2O y se extrajo con EtOAc (2 * 10 mL). Las fases orgánicas combinadas se lavaron con salmuera (2 * 10 mL), se secaron sobre sulfato de sodio, se filtraron y se concentraron a vacío. El residuo se purificó por HPLC preparativa de fase inversa proporcionando SB-23 como un sólido blanco (19,3 mg, 66 %). RMN 1H (500 MHz, CDCh), 8 (ppm), 7,55 (d, 1H), 7,41 (d, 1H), 6,33 (t, 1H), 4,97 (AB, 1H), 4,88 (AB, 1H), 4,17 (d, 2H), 2,59 (t, J = 9,0 Hz, 1H), 0,69 (s, 3H), 0,60-2,20 (m, 24H). EM/CL: tr = 2,27 min. m/z = 403,2 [M+H]+.
Métodos de ensayo
Los compuestos proporcionados en el presente documento se pueden evaluar usando diversos ensayos; ejemplos de los cuales se describen a continuación.
Inhibición por esteroides de la unión de TBPS
Se han descrito ensayos de unión a [35S]-t-butilbiciclofosforotionato (TBPS) usando membranas corticales de cerebro de rata en presencia de GABA 5 pM (Gee et al, J. Pharmacol. Exp. Ther. 1987, 241, 346-353; Hawkinson et al, Mol. Pharmacol. 1994, 46, 977-985; Lewin, A.H et al., Mol. Pharmacol. 1989, 35, 189-194).
Brevemente, se extirpan rápidamente las cortezas después de la decapitación de ratas Sprague-Dawley (200-250 g) anestesiadas con dióxido de carbono. Las cortezas se homogeneízan en 10 volúmenes de sacarosa 0,32 M fría en hielo usando un homogeneizador de vidrio/teflón y se centrifugan a 1500 * g durante 10 min a 4 °C. Los sobrenadantes resultantes se centrifugan a 10.000 * g durante 20 min a 4 °C para obtener los sedimentos P2. Los sedimentos P2 se resuspenden en tampón NaCl 200 mM/fosfato de Na-K 50 mM a pH 7,4 y se centrifugan a 10.000 * g durante 10 min a 4 °C. Este procedimiento de lavado se repite dos veces y los sedimentos se resuspenden en 10 volúmenes de tampón. Se incuban alícuotas (100 pL) de las suspensiones de membrana con [35S]-TBPS 3 nM y se disuelven alícuotas de 5 pL de fármaco de prueba en sulfóxido de dimetilo (DMSO) (0,5 % final) en presencia de GABA 5 pM. La incubación se lleva a un volumen final de 1,0 mL con tampón. Se determina la unión no específica en presencia de TBPS 2 pM no marcado y oscila del 15 al 25 %. Tras una incubación de 90 min a temperatura ambiente, los ensayos se terminan por filtración a través de filtros de fibra de vidrio (Schleicher y Schuell N.° 32) usando un recolector de células (Brandel) y se aclaran tres veces con tampón frío en hielo. Se mide la radiactividad unida al filtro por espectrometría de centelleo líquido. Se hace el ajuste de curva no lineal de los datos globales para cada fármaco promediados para cada concentración usando Prism (GraphPad). Los datos se ajustan a un modelo de inhibición parcial en lugar de uno completo si la suma de cuadrados es significativamente inferior por la prueba de la F. Similarmente, los datos se ajustan a un modelo de inhibición de dos componentes en lugar de uno de un componente si la suma de cuadrados es significativamente inferior por la prueba de la F. Se determina la concentración de compuesto de prueba que produce el 50 % de inhibición (CI50) de unión específica y el máximo grado de inhibición (Imáx) para los experimentos individuales con el mismo modelo usado para los datos globales y entonces se calcula la media ± EEM de los experimentos individuales. La picrotoxina sirve de control positivo para estos estudios, ya que se ha demostrado que inhibe de forma consistente la unión de TBPS.
Se criban o se pueden cribar diversos compuestos para determinar su potencial como moduladores de la unión de [35S]-TBPS in vitro. Estos ensayos se realizan o se pueden realizar según los procedimientos anteriormente tratados.
Electrofisiología de pinzamiento zonal de receptores de GABAa aip2Y2 y ct4@35 recombinantes
Se usa electrofisiología celular para medir las propiedades farmacológicas de los moduladores de receptores de GABAa de los presentes inventores en sistemas celulares heterólogos. Se prueba la capacidad de cada compuesto para afectar las corrientes mediadas por GABA en dosis de agonista inferiores a las máximas (CE20 de GABA = 2 pM). Se transfectan establemente células LTK con las subunidades cnp2Y2 del receptor de GABA y se transfectan transitoriamente células CHO con las subunidades 04^35 por el método de Lipofectamine. Las células se sometieron a pases a una confluencia de aproximadamente el 50-80 % y luego se sembraron en placas de cultivo estériles de 35 mm que contenían 2 mL de medio completo de cultivo sin antibióticos ni antimicóticos. Se acoplan eléctricamente agrupaciones confluentes de células (Pritchett et al., Science, 1988, 242, 1306-1308). Debido a que las respuestas en células distantes no son suficientemente sometidas a pinzamiento de voltaje y debido a las incertidumbres sobre el grado de acoplamiento (Verdoorn et al., Neuron 1990, 4, 919-928), las células se cultivaron a una densidad que permite el registro de células individuales (sin conexiones visibles con otras células).
Se midieron las corrientes de células completas con amplificadores de HEKA EPC-10 usando el software PatchMaster o usando la plataforma QPatch de alto rendimiento (Sophion). La disolución de baño para todos los experimentos contuvo (en mM): NaCl 137 mM, KCl 4 mM, CaCh1,8 mM, MgCh 1 mM, HEPES 10 mM, D-glucosa 10 mM, pH (NaOH) 7,4. En algunos casos, también se añadió 0,005 % de Cremophor. La disolución intracelular (pipeta) contuvo: KCl 130 mM, MgCl21 mM, Mg-ATP 5 mM, HEPES 10 mM, EGTA 5mM, pH 7,2. Durante los experimentos, las células y las disoluciones se mantuvieron a temperatura ambiente (19 °C - 30 °C). Para los registros de pinzamiento zonal manual, se pusieron placas de cultivo celular sobre el portaplacas del microscopio y se perfundieron continuamente (1 mL/min) con disolución de baño. Después de la formación de un sello gigaohm entre los electrodos del parche y la célula (intervalo de resistencia de la pipeta: 2,5 MQ - 6,0 MQ; intervalo de resistencia del precinto:>1 GQ), se rompió la
membrana celular a través de la punta de la pipeta para garantizar el acceso eléctrico al interior de la célula (configuración de pinzamiento de células completas). Para experimentos usando el sistema QPatch, las células se transfirieron como suspensión al sistema QPatch en la disolución de baño y se realizaron registros automáticos de células completas.
Las células se sometieron a pinzamiento de voltaje a un potencial de mantenimiento de -80 mV. Para el análisis de los productos experimentales, los receptores de GABA se estimularon por GABA 2 pM después de la preincubación secuencial de concentraciones crecientes del producto experimental. La duración de la preincubación fue 30 s y la duración del estímulo de GABA fue 2s. Se disolvieron los productos experimentales en DMSo para formar disoluciones madre (10 mM). Los productos experimentales se diluyeron hasta 0,01, 0,1, 1 y 10 pM en disolución de baño. Todas las concentraciones de productos experimentales se probaron en cada célula. La potenciación en porcentaje relativo se definió como la amplitud del pico en respuesta a la CE20 de GABA en presencia del producto experimental dividido entre la amplitud del pico en respuesta a CE20 de GABA solo, multiplicado por 100.
Pérdida de reflejo de enderezamiento en ratas
Se obtuvieron la farmacocinética plasmática y una evaluación cualitativa de la sedación en ratas Sprague Dawley macho según el siguiente procedimiento. Se les administró a las ratas por dosis intravenosa en embolada (60 segundos) por la vena dorsal de la pata dosis que variaban desde 5 hasta 15 mg/kg en un vehículo apropiado. Para evaluar la sedación, las ratas se sujetaron suavemente con la mano hasta una posición lateral para la administración de dosis. Si se observó una disminución del tono muscular durante la administración de dosis, se redujo gradualmente la sujeción. Si el animal fue incapaz de volver a una posición enderezada, se anotó el tiempo como la aparición de pérdida de reflejo de enderezamiento (LRR). En el supuesto caso de que LRR no ocurriera durante la administración, los animales se evaluaron en intervalos de 5 minutos a partir de aquí colocándolos en decúbito supino. La flojera o el enderezamiento incompleto dos veces consecutivas en un intervalo de 30 segundos se caracterizan como una pérdida del reflejo de enderezamiento. Después de la aparición de LRR, los animales se evaluaron cada 5 minutos del mismo modo. La recuperación del reflejo de enderezamiento se define como la capacidad de una rata para enderezarse ella misma completamente en 20 segundos desde que se puso en posición supina. La duración de LRR se define como el intervalo de tiempo entre LRR y el regreso del reflejo de enderezamiento.
Método de PTZ agudo
Se evaluó el efecto anticonvulsivo de los compuestos de prueba en el ensayo de convulsiones inducidas por pentilentetrazol en ratones similares a los métodos descritos en Giardina & Gasior (2009) Curr Protoc Pharmacol., Capítulo 5. Se alojaron ratones CD-1 macho en grupos de cinco en condiciones controladas (temperatura de 22±2 °C y ciclo de luz-oscuridad de 12:12, luces encendidas a las 8:00 am) y el agua y la comida estuvieron disponibles a voluntad. Los ratones se alojaron durante 1 semana antes de la prueba conductual, momento en el que pesaron 25 35 g. Se disolvió pentilentetrazol (PTZ, Sigma) en solución salina estéril al 0,9 % en una concentración de 12 mg/mL de concentración para administración subcutánea. Los compuestos de prueba se formularon y se administraron por sonda nasogástrica oral o inyección intraperitoneal en un momento predeterminado (normalmente 30 o 60 minutos) antes de la inyección de PTZ. Todas las disoluciones se prepararon nuevas y se administraron en un volumen de 10 mL/kg de peso corporal.
Los ratones se aclimataron a la sala de pruebas durante menos de 30 min antes de la administración del compuesto. Los ratones se aleatorizaron en al menos cuatro grupos de prueba (vehículo y al menos tres dosis del compuesto de prueba) con 10 ratones por grupo. Después de la administración del compuesto, los ratones se observaron para la evaluación cualitativa de la sedación durante un momento predeterminado (30 o 60 minutos). Después del momento de pretratamiento con fármaco, los ratones se inyectaron s.c. con PTZ (120 mg/kg). Inmediatamente después de la inyección de PTZ, los ratones se pusieron individualmente en cámaras de observación (25 * 15 * 15 cm) y se inició un cronómetro de tres canales. Cada ratón se observó continuamente durante 30 min y se registraron los siguientes comportamientos por observadores no conocedores de los tratamientos: 1) latencia a convulsiones clónicas que persisten durante 3 s y seguido por una ausencia de reflejo de enderezamiento 2) latencia a convulsiones tónicas, caracterizada por la extensión rígida de las cuatro extremidades que superó un ángulo de 90 grados con el cuerpo 3) latencia a muerte 4) número de convulsiones clónicas y tónicas. Los datos se presentan como media ± EEM y se usó el análisis de la varianza unidireccional con prueba a posteriori de Dunnett o Bonferroni para detectar diferencias significativas en la latencia y el número entre el grupo de vehículo y de dosis. Se consideraron valores de de p <0,05 como estadísticamente significativos.
Tabla 1. Unión de TBPS de los compuestos a modo de ejemplo.

Para la Tabla 1: TBPS: "A" indica una CI50 <10 nM, "B" indica una CI5010 a <50 nM, "C" indica una CI5050 nM a <100 nM, "D" indica una CI50100 nM a <500 nM, y "E" indica CI50 superior o igual a 500 nM.
Tabla 2. Evaluación electrofisiológica de los compuestos a modo de ejemplo en GABAa-R.

Para la Tabla 2, CE50: "A" indica una CE50 <100 nM, "B" indica una CE50100 a inferior o igual a 500 nM, "C" indica una CE50 >500 nM a 1000 nM, "D" indica CI50 >1000 nM a 2000 nM, y "E" indica CE50 >2000 nM. Emáx: "A" indica una Emáx de 0 a 500, "B" indica una Emáx de >500 a 1000, "C" indica una Emáx de >1000.
Tabla 3. Evaluación electrofisiológica de los compuestos a modo de ejemplo en GABAa-R.


Para la Tabla 3. % de eficacia de los receptores de GABAA a1p2Y2 y a4p38: "A" 10-100, "B" > 100-500, "C" >500; D indica que los datos no están disponibles o no se han determinado.
Tabla 4. Pérdida de reflejo de enderezamiento (IV en rata, 5 mpk)

Tabla 5. Las dosis anticonvulsivas efectivas mínimas se definen como la dosis más baja que reduce significativamente la latencia a convulsiones tónicas en ratones tratados con PTZ

Claims (13)
1. Un compuesto de fórmula

o una sal farmacéuticamente aceptable del mismo,
para su uso en el tratamiento de un trastorno del estado de ánimo en un sujeto humano.
2. El compuesto para su uso según la reivindicación 1, en donde el trastorno del estado de ánimo es depresión.
3. El compuesto para su uso según las reivindicaciones 1 o 2, en donde el compuesto es para administración oral, subcutánea, intravenosa o intramuscular.
4. El compuesto para su uso según la reivindicación 3, en donde el compuesto es para administración por vía oral.
5. Un compuesto de la fórmula

o una sal farmacéuticamente aceptable del mismo,
para su uso en aliviar o prevenir SPM o DPP en un sujeto humano.
6. El compuesto para su uso según la reivindicación 5, en donde el compuesto es para administración oral, subcutánea, intravenosa o intramuscular.
7. El compuesto para su uso según la reivindicación 6, en donde el compuesto es para administración por vía oral.
8. Un compuesto de la fórmula

o una sal farmacéuticamente aceptable del mismo,
para su uso en modular positiva o negativamente un receptor de GABAa en un sujeto humano.
9. El compuesto para su uso según la reivindicación 8, en donde el compuesto afecta el receptor de GABAa de un modo positivo.
10. El compuesto para su uso según las reivindicaciones 8 o 9, en donde el sujeto humano tiene un trastorno del estado de ánimo.
11. El compuesto para su uso según la reivindicación 10, en donde el trastorno del estado de ánimo es depresión.
12. El compuesto para su uso según cualquiera de las reivindicaciones 8 a 10, en donde el compuesto es para administración oral, subcutánea, intravenosa o intramuscular.
13. El compuesto para su uso según la reivindicación 12, en donde el compuesto es para administración por vía oral.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2013074323 | 2013-04-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2886182T3 true ES2886182T3 (es) | 2021-12-16 |
Family
ID=51730809
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14785712T Active ES2709434T3 (es) | 2013-04-17 | 2014-04-17 | Esteroides C21-n-pirazolilo 19-nor C3,3-disustituidos y métodos de uso de los mismos |
| ES21178047T Active ES3044385T3 (en) | 2013-04-17 | 2014-04-17 | 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid for use in therapy |
| ES18203320T Active ES2886182T3 (es) | 2013-04-17 | 2014-04-17 | C21-N-pirazolil-esteroide 19-nor C3,3-disustituido para su uso en terapia |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14785712T Active ES2709434T3 (es) | 2013-04-17 | 2014-04-17 | Esteroides C21-n-pirazolilo 19-nor C3,3-disustituidos y métodos de uso de los mismos |
| ES21178047T Active ES3044385T3 (en) | 2013-04-17 | 2014-04-17 | 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid for use in therapy |
Country Status (29)
| Country | Link |
|---|---|
| US (6) | US9512165B2 (es) |
| EP (4) | EP4640277A3 (es) |
| JP (7) | JP6470258B2 (es) |
| KR (4) | KR102396328B1 (es) |
| CN (4) | CN113527400B (es) |
| AU (5) | AU2014256229B2 (es) |
| BR (1) | BR112015026380A8 (es) |
| CA (1) | CA2909545A1 (es) |
| CY (2) | CY1121228T1 (es) |
| DK (3) | DK3498725T3 (es) |
| ES (3) | ES2709434T3 (es) |
| FI (1) | FIC20260008I1 (es) |
| FR (1) | FR26C1007I1 (es) |
| HR (2) | HRP20190232T1 (es) |
| HU (2) | HUE041369T2 (es) |
| IL (5) | IL310348B1 (es) |
| LT (3) | LT2986623T (es) |
| MX (3) | MX395023B (es) |
| NO (1) | NO2026015I1 (es) |
| PL (2) | PL3498725T3 (es) |
| PT (2) | PT3498725T (es) |
| RS (2) | RS62312B1 (es) |
| RU (1) | RU2675855C2 (es) |
| SG (2) | SG10201802389SA (es) |
| SI (2) | SI2986623T1 (es) |
| SM (2) | SMT202100509T1 (es) |
| TR (1) | TR201901745T4 (es) |
| WO (1) | WO2014169833A1 (es) |
| ZA (1) | ZA201507740B (es) |
Families Citing this family (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7858609B2 (en) | 2005-11-28 | 2010-12-28 | Marinus Pharmaceuticals | Solid ganaxolone formulations and methods for the making and use thereof |
| EP3572417B1 (en) | 2011-10-14 | 2025-12-17 | Sage Therapeutics, LLC | 3,3 disubstituted 19-nor pregnane compounds, compositions, and uses thereof |
| EP2806877B2 (en) | 2012-01-23 | 2025-01-29 | Sage Therapeutics, Inc. | Neuroactive steroid formulations comprising a complex of allopregnanolone and sulfobutyl ether beta-cyclodextrin |
| WO2013139861A1 (en) | 2012-03-20 | 2013-09-26 | Luc Montagnier | Methods and pharmaceutical compositions of the treatment of autistic syndrome disorders |
| CA2882708A1 (en) | 2012-08-21 | 2014-02-27 | Sage Therapeutics, Inc. | Methods of treating epilepsy or status epilepticus |
| EP2986624B1 (en) | 2013-04-17 | 2020-03-25 | Sage Therapeutics, Inc. | 19-nor neuroactive steroids for methods of treatment |
| US9725481B2 (en) | 2013-04-17 | 2017-08-08 | Sage Therapeutics, Inc. | 19-nor C3, 3-disubstituted C21-C-bound heteroaryl steroids and methods of use thereof |
| KR102396328B1 (ko) | 2013-04-17 | 2022-05-10 | 세이지 테라퓨틱스, 인크. | 19-노르 c3,3-이치환된 c21-n-피라졸릴 스테로이드 및 그의 사용 방법 |
| US20160068563A1 (en) | 2013-04-17 | 2016-03-10 | Boyd L. Harrison | 19-nor neuroactive steroids and methods of use thereof |
| SI3021852T1 (sl) | 2013-07-19 | 2021-07-30 | Sage Therapeutics, Inc. | Nevroaktivni steroidi, sestavki in uporabe le-teh |
| AU2014308621C1 (en) | 2013-08-23 | 2022-01-06 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| US10246482B2 (en) | 2014-06-18 | 2019-04-02 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| JOP20200195A1 (ar) | 2014-09-08 | 2017-06-16 | Sage Therapeutics Inc | سترويدات وتركيبات نشطة عصبياً، واستخداماتها |
| CN117024501A (zh) | 2014-10-16 | 2023-11-10 | 萨奇治疗股份有限公司 | 靶向cns障碍的组合物和方法 |
| ME03809B (me) | 2014-10-16 | 2021-04-20 | Sage Therapeutics Inc | KOMPOZICIJE I POSTUPCI ZA LEČENJE POREMEĆAJA CNS-a |
| WO2016082789A1 (en) | 2014-11-27 | 2016-06-02 | Sage Therapeutics, Inc. | Compositions and methods for treating cns disorders |
| CN104558092B (zh) * | 2014-12-29 | 2017-01-18 | 北京理工大学 | 一种甾体皂苷化合物及其制备方法和用途 |
| RS61530B1 (sr) | 2015-01-26 | 2021-04-29 | Sage Therapeutics Inc | Kompozicije i postupci za lečenje poremećaja cns |
| EP4155314A1 (en) | 2015-02-20 | 2023-03-29 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| AR105044A1 (es) | 2015-06-18 | 2017-08-30 | Sage Therapeutics Inc | Soluciones de esteroides neuroactivos y sus métodos de uso, recipiente |
| CN104910236B (zh) * | 2015-06-26 | 2016-08-17 | 西北农林科技大学农药研究所 | 吡唑基甾体衍生物及其制备方法、用途 |
| AU2016338672A1 (en) | 2015-10-16 | 2018-04-12 | Marinus Pharmaceuticals, Inc. | Injectable neurosteroid formulations containing nanoparticles |
| KR20240157128A (ko) | 2016-03-08 | 2024-10-31 | 세이지 테라퓨틱스, 인크. | 신경활성 스테로이드, 조성물, 및 그의 용도 |
| MA45599A (fr) | 2016-07-11 | 2019-05-15 | Sage Therapeutics Inc | Stéroïdes neuroactifs substitués en c17, c20 et c21 et leurs procédés d'utilisation |
| WO2018013615A1 (en) | 2016-07-11 | 2018-01-18 | Sage Therapeutics, Inc. | C7, c12, and c16 substituted neuroactive steroids and their methods of use |
| EP3481387A4 (en) | 2016-08-11 | 2020-04-08 | Ovid Therapeutics Inc | METHOD AND COMPOSITIONS FOR TREATING EPILEPTIC DISEASES |
| EP3504189A1 (en) * | 2016-08-23 | 2019-07-03 | Sage Therapeutics, Inc. | A crystalline 19-nor c3, 3-disubstituted c21-n-pyrazolyl steroid |
| US10391105B2 (en) | 2016-09-09 | 2019-08-27 | Marinus Pharmaceuticals Inc. | Methods of treating certain depressive disorders and delirium tremens |
| EP3612186A1 (en) * | 2017-04-18 | 2020-02-26 | Marinus Pharmaceuticals, Inc. | Sustained release injectable neurosteroid formulations |
| KR20200011946A (ko) * | 2017-06-01 | 2020-02-04 | 수미토모 케미칼 컴퍼니 리미티드 | 복소환 화합물 및 그것을 함유하는 조성물 |
| WO2019051264A1 (en) * | 2017-09-07 | 2019-03-14 | Sage Therapeutics, Inc. | NEUROACTIVE STEROIDS AND METHODS OF USE |
| CA3075872A1 (en) * | 2017-09-14 | 2019-03-21 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid and methods of use thereof |
| US20210228596A1 (en) * | 2017-10-12 | 2021-07-29 | Sage Therapeutics, Inc. | Method of treating cns disorders with neurosteroids and gabaergic compounds |
| MA51046A (fr) | 2017-12-08 | 2021-03-17 | Sage Therapeutics Inc | Dérivés de 21-[4-cyano-pyrazol-1-yl]-19-nor-pregan-3. alpha-ol-20-one deutérés pour le traitement de troubles du snc |
| MX2020006599A (es) * | 2017-12-22 | 2020-11-06 | Sage Therapeutics Inc | Composiciones y metodos para el tratamiento de trastornos en el sistema nervioso central. |
| BR112020012761A2 (pt) | 2017-12-22 | 2021-02-17 | Sage Therapeutics, Inc. | composições e métodos para o tratamento de distúrbios do snc |
| TW201930269A (zh) * | 2018-01-12 | 2019-08-01 | 美商賽吉醫療公司 | 用於治療cns病症之組合物及方法 |
| WO2019154257A1 (zh) * | 2018-02-11 | 2019-08-15 | 江苏豪森药业集团有限公司 | 一种甾族类衍生物调节剂及其制备方法和应用 |
| EP3750909B1 (en) * | 2018-02-11 | 2025-03-05 | Jiangsu Hansoh Pharmaceutical Group Co., Ltd. | Steroid derivative regulators, method for preparing the same, and uses thereof |
| CA3103421A1 (en) * | 2018-06-12 | 2019-12-19 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21 -n-pyrazolyl steroid and methods of use thereof |
| CN111040015B (zh) * | 2018-10-15 | 2022-03-18 | 南京诺瑞特医药科技有限公司 | 19-去甲-c21取代类固醇衍生物 |
| CN109503694A (zh) * | 2018-11-21 | 2019-03-22 | 苏州闻天医药科技有限公司 | 一种新型gabaa受体调节剂及其用途 |
| KR20250174100A (ko) | 2018-12-05 | 2025-12-11 | 세이지 테라퓨틱스, 인크. | 신경활성 스테로이드 및 이의 사용 방법 |
| US11266662B2 (en) | 2018-12-07 | 2022-03-08 | Marinus Pharmaceuticals, Inc. | Ganaxolone for use in prophylaxis and treatment of postpartum depression |
| MX2021007142A (es) | 2018-12-17 | 2021-08-11 | Intra Cellular Therapies Inc | Compuestos organicos. |
| CN115651054B (zh) * | 2019-01-08 | 2024-06-21 | 成都康弘药业集团股份有限公司 | 甾体类化合物、用途及其制备方法 |
| CN113301901B (zh) * | 2019-01-14 | 2022-09-09 | 北京轩义医药科技有限公司 | 四唑啉酮取代的类固醇及其用途 |
| CN111454318A (zh) * | 2019-01-20 | 2020-07-28 | 浙江易众化工有限公司 | 抗抑郁药物sage-217的晶型及其制备方法 |
| CN111770915A (zh) * | 2019-01-31 | 2020-10-13 | 深圳仁泰医药科技有限公司 | γ-氨基丁酸调节剂的晶型X及其制备方法和应用 |
| MX2021013695A (es) | 2019-05-10 | 2022-01-26 | Brii Biosciences Inc | Composición farmacéutica que contiene brexanolona, ganaxolona y zuranolona y usos de esta. |
| WO2020242772A1 (en) | 2019-05-28 | 2020-12-03 | Teva Czech Industries S.R.O | Solid state forms of sage-217 and processes for preparation thereof |
| SG11202112391UA (en) | 2019-05-31 | 2021-12-30 | Sage Therapeutics Inc | Neuroactive steroids and compositions thereof |
| JP7754725B2 (ja) * | 2019-06-27 | 2025-10-15 | セージ セラピューティクス, インコーポレイテッド | Cns障害を治療するための組成物及び方法 |
| CN114729000A (zh) | 2019-06-27 | 2022-07-08 | 萨奇治疗股份有限公司 | 用于治疗cns病症的化合物 |
| CA3143509A1 (en) * | 2019-06-27 | 2020-12-30 | Sage Therapeutics, Inc. | Compositions and methods for treating cns disorders |
| JP7754799B2 (ja) | 2019-08-05 | 2025-10-15 | マリナス ファーマシューティカルズ, インコーポレイテッド | てんかん重積状態の治療に使用するためのガナキソロン |
| CN118027131A (zh) * | 2019-08-07 | 2024-05-14 | 上海翰森生物医药科技有限公司 | 一种甾族类衍生物调节剂的盐及其晶型 |
| JP2022552788A (ja) * | 2019-09-30 | 2022-12-20 | アテネン セラピューティクス, インコーポレイテッド | Gabaaレセプターのサブタイプを優先的に増強する組成物およびその使用方法 |
| US10857163B1 (en) | 2019-09-30 | 2020-12-08 | Athenen Therapeutics, Inc. | Compositions that preferentially potentiate subtypes of GABAA receptors and methods of use thereof |
| KR20220112803A (ko) * | 2019-12-05 | 2022-08-11 | 세이지 테라퓨틱스, 인크. | 19-nor c3,3-이치환된 c21-n-피라졸릴 스테로이드 및 그의 사용 방법 |
| WO2021113834A1 (en) | 2019-12-06 | 2021-06-10 | Marinus Pharmaceuticals, Inc. | Ganaxolone for use in treating tuberous sclerosis complex |
| EP4125921A1 (en) * | 2020-03-25 | 2023-02-08 | Sage Therapeutics, Inc. | Use of agents for treatment of respiratory conditions |
| KR20230041049A (ko) * | 2020-07-20 | 2023-03-23 | 세이지 테라퓨틱스, 인크. | 19-노르 c3,3-이치환된 c21-n-피라졸릴 스테로이드의 제형 및 이의 사용 방법 |
| KR20230136599A (ko) * | 2021-01-28 | 2023-09-26 | 세이지 테라퓨틱스, 인크. | 성 기능장애의 치료를 위한 신경활성 스테로이드의용도 |
| US20240245711A1 (en) * | 2021-03-17 | 2024-07-25 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid for the treatment of major depressive disorder |
| WO2022232504A1 (en) | 2021-04-29 | 2022-11-03 | Sage Therapeutics, Inc. | 19-nor c3,3-disubstituted c21 -n-pyrazolyl steroid for use in treating major depressive disorder and postpartum depression |
| EP4329769A1 (en) | 2021-04-29 | 2024-03-06 | Sage Therapeutics, Inc. | Neuroactive steroid for use in treating major depressive disorder and postpartum depression in a lactating female |
| IL315023A (en) | 2022-02-16 | 2024-10-01 | Sage Therapeutics Inc | Neuroactive steroids for treatment of cns-related disorders |
| WO2023163879A1 (en) | 2022-02-28 | 2023-08-31 | Sage Therapeutics, Inc. | Neuroactive steroids for treatment of gastrointestinal diseases or conditions |
| JP2025538551A (ja) | 2022-11-21 | 2025-11-28 | セージ セラピューティクス, エルエルシー | 負のnmda調節化合物及びその使用方法 |
| JP2026509303A (ja) | 2023-03-14 | 2026-03-17 | キュリア、スペイン、エセ、ア、ウ | 3アルファ-ヒドロキシ-3ベータ-アルキルステロイドを調製するための方法および中間体 |
| EP4711377A1 (en) | 2023-05-11 | 2026-03-18 | Shujing Biopharma Co., Ltd | Steroid compound, preparation method therefor, and use thereof |
| WO2026055127A2 (en) | 2024-09-04 | 2026-03-12 | Sage Therapeutics, Inc., A Wholly Owned Subsidiary Of Supernus Pharmaceuticals, Inc. | Process for producing a gaba receptor modulator |
Family Cites Families (162)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2856415A (en) | 1957-11-13 | 1958-10-14 | Searle & Co | 3,19-dihydroxy-5-androstene derivatives |
| FR1380417A (fr) | 1962-05-15 | 1964-12-04 | Roussel Uclaf | Nouveaux androstanyl-pyrazoles et procédé de préparation |
| NL297205A (es) * | 1962-08-28 | |||
| US3206459A (en) | 1962-10-19 | 1965-09-14 | Syntex Corp | 10alpha-pregnan-19-ol derivatives |
| US3169134A (en) | 1963-03-21 | 1965-02-09 | Searle & Co | 2, 3-oxygenated-17alpha-methyl-5alpha-androstan-17beta-ols |
| US3511860A (en) * | 1966-09-26 | 1970-05-12 | Syntex Corp | Synthesis of 19-nor and ring a aromatic steroids and intermediates therefor |
| NL6801449A (es) * | 1968-02-01 | 1969-08-05 | ||
| BE754111A (fr) | 1969-07-29 | 1971-01-29 | Upjohn Co | Nouveaux 7alpha- et 7beta-methyl-3alpha,5alpha- cycloandrostanes et composes analogues 19-nor et leur procede de preparation |
| GB1434919A (en) | 1972-06-15 | 1976-05-12 | Glaxo Lab Ltd | 3alpha-hydroxy androstanes |
| GB1380246A (en) | 1970-12-17 | 1975-01-08 | Glaxo Lab Ltd | 3alpha-hydroxy-androstanes and esters thereof |
| US3943124A (en) | 1970-12-17 | 1976-03-09 | Gordon Hanley Phillipps | Chemical compounds |
| GB1430942A (en) | 1972-05-05 | 1976-04-07 | Glaxo Lab Ltd | 21-substituted 3alpha-hydroxy pregnanes |
| US3983111A (en) | 1972-05-05 | 1976-09-28 | Glaxo Laboratories Limited | Steroidal anaesthetics of the pregnane and 19-norpregnane series |
| GB1436324A (en) | 1972-05-12 | 1976-05-19 | Glaxo Lab Ltd | Anaesthetic 3alpha-hydroxy pregnanes |
| ES432106A1 (es) | 1973-11-30 | 1976-11-01 | Schering Ag | Procedimiento para la preparacion de d-homo-20-cetopregna- nos. |
| US4071625A (en) | 1974-05-13 | 1978-01-31 | Richardson-Merrell Inc. | 19-Oxygenated-5α-androstanes for the enhancement of libido |
| DE2438020A1 (de) | 1974-08-05 | 1976-02-26 | Schering Ag | 18-methyl-19-nor-20-keto-pregnane und verfahren zu ihrer herstellung |
| IL48628A0 (en) | 1974-12-23 | 1976-02-29 | Schering Ag | D-homo-20-keto-pregnanes and process for their manufactur |
| DE2526373C2 (de) | 1975-06-11 | 1983-11-10 | Schering AG, 1000 Berlin und 4709 Bergkamen | Verfahren zur Herstellung von Δ↑9↑↑(↑↑1↑↑1↑↑)↑-5α-20-Ketosteroiden |
| GB1570394A (en) | 1976-01-06 | 1980-07-02 | Glaxo Lab Ltd | 11-acyloxy-3-hydroxy steroids |
| US4192871A (en) | 1976-01-06 | 1980-03-11 | Glaxo Laboratories Limited | Chemical compounds |
| GB1581234A (en) | 1976-04-05 | 1980-12-10 | Glaxo Operations Ltd | 11a - amino - 3a - hydroxysteroids |
| DE2632677A1 (de) | 1976-07-16 | 1978-01-26 | Schering Ag | Verfahren zur herstellung von androstan-17-on-derivaten und deren verwendung |
| GB1581235A (en) | 1977-04-04 | 1980-12-10 | Glaxo Operations Ltd | 11a-amino-3a-hydroxy-steroids |
| US4389345A (en) | 1981-10-09 | 1983-06-21 | G.D. Searle & Co. | 3-Oxoestra-17-acetonitrile and unsaturated analogs |
| US4495102A (en) | 1982-09-03 | 1985-01-22 | G. D. Searle & Co. | Aminoalkyl steroids |
| US5232917A (en) | 1987-08-25 | 1993-08-03 | University Of Southern California | Methods, compositions, and compounds for allosteric modulation of the GABA receptor by members of the androstane and pregnane series |
| US5319115A (en) | 1987-08-25 | 1994-06-07 | Cocensys Inc. | Method for making 3α-hydroxy, 3β-substituted-pregnanes |
| US5120723A (en) | 1987-08-25 | 1992-06-09 | University Of Southern California | Method, compositions, and compounds for modulating brain excitability |
| US5376645A (en) | 1990-01-23 | 1994-12-27 | University Of Kansas | Derivatives of cyclodextrins exhibiting enhanced aqueous solubility and the use thereof |
| EP0603312A4 (en) | 1991-09-13 | 1995-06-07 | Cocensys Inc | NEW GABA A RECEPTOR WITH STEROID BINDING POINTS. |
| DE4232681C2 (de) | 1992-09-29 | 1994-11-24 | Sigma Tau Ind Farmaceuti | 17-Phenyl- und 17-Furyl-14beta,5alpha-androstan- und androsten- Derivate, Verfahren zu deren Herstellung und diese enthaltende pharmazeutische Zusammensetzung |
| DE69435286D1 (de) | 1993-05-24 | 2010-05-20 | Purdue Pharma Ltd | Verfahren und zusammensetzungen zum hervorrufen von schlaf |
| CZ300694A3 (en) | 1993-12-02 | 1996-05-15 | Akzo Nobel Nv | Substituted 2beta-morpholinandrostane derivatives, process of their preparation, their use for preparing pharmaceutical preparations and pharmaceutical compositions containing thereof |
| EP0656365B1 (en) | 1993-12-02 | 1997-04-09 | Akzo Nobel N.V. | Substituted 2beta-morpholinoandrostane derivatives |
| US5567830A (en) | 1994-02-14 | 1996-10-22 | Cocensys, Inc. | Process for synthesis of acetylenic carbinols |
| ES2296594T3 (es) | 1994-02-14 | 2008-05-01 | Euro-Celtique S.A. | Androstanos y pregnanos para la modulacion alosterica del receptor de gaba. |
| US5939545A (en) | 1994-02-14 | 1999-08-17 | Cocensys, Inc. | Method, compositions, and compounds for allosteric modulation of the gaba receptor by members of the androstane and pregnane series |
| AU3125695A (en) | 1994-07-21 | 1996-02-22 | Pharmacia & Upjohn Company | Neurologically active aminosteroids |
| US5792757A (en) * | 1994-08-04 | 1998-08-11 | Pherin Pharmaceuticals | 19-nor-pregnane steroids as neurochemical initiators of change in human hypothalamic function |
| NZ298567A (en) | 1994-11-23 | 2000-01-28 | Cocensys Inc | Androstane and pregnane derivatives, pharmaceutical compositions and methods of treatment |
| ATE284895T1 (de) * | 1995-06-06 | 2005-01-15 | Euro Celtique Sa | Neuroaktive steroide der androstan- und pregnanreihe |
| WO1998005337A1 (en) | 1996-08-01 | 1998-02-12 | Cocensys, Inc. | Use of gaba and nmda receptor ligands for the treatment of migraine headache |
| US5994334A (en) | 1997-02-05 | 1999-11-30 | University Of Maryland | Androgen synthesis inhibitors |
| US5935545A (en) | 1997-07-11 | 1999-08-10 | E. I. Du Pont De Nemours And Company | Process for producing an aqueous solution comprising ferric chloride |
| UA73736C2 (en) * | 1999-04-29 | 2005-09-15 | Euro Celtic S A | Method for alleviating or preventing insomnia and inducing anesthesia |
| US7018406B2 (en) | 1999-11-17 | 2006-03-28 | Corevalve Sa | Prosthetic valve for transluminal delivery |
| DE60111304T2 (de) | 2000-02-18 | 2005-11-10 | Taiho Pharmaceutical Co. Ltd. | Verfahren zur herstellung von steroid-derivaten |
| US6855836B2 (en) | 2000-10-26 | 2005-02-15 | Jenapharm Gmbh & Co. Kg | 17-Methylene steroids, process for their production and pharmaceutical compositions that contain these compounds |
| ES2287184T3 (es) | 2000-11-03 | 2007-12-16 | Washington University | Estructuras aromaticas modificadas con sustituyentes hidroxi-, que tienen actividad citoprotectora. |
| GR1003861B (el) | 2000-12-29 | 2002-04-11 | Νεα νευροστεροειδη που αλληλεπιδρουν με τον υποδοχεα gabaa. | |
| PL378577A1 (pl) | 2003-03-24 | 2006-05-02 | Sterix Limited | Pochodne estrogenu jako inhibitory sulfatazy steroidowej |
| WO2005000869A1 (en) | 2003-05-29 | 2005-01-06 | Washington University | Neuroactive 13,24-cyclo-18,21-dinorcholanes and structurally-related pentacyclic steroids |
| WO2005051972A1 (en) | 2003-11-24 | 2005-06-09 | Merck & Co., Inc. | Estrogen receptor modulators |
| WO2005105822A2 (en) | 2004-04-23 | 2005-11-10 | Euro-Celtique S.A. | 3-alpha-hydroxy 21-n- heteroaryl-pregnane derivatives for modulation of brain excitability and a process for the production thereof |
| CA2578789A1 (en) * | 2004-09-03 | 2006-03-16 | Celgene Corporation | Substituted heterocyclic compounds and uses thereof |
| US8604011B2 (en) | 2004-09-27 | 2013-12-10 | The Regents Of The University Of California | Therapy for treatment of chronic degenerative brain diseases and nervous system injury |
| EP2471536A1 (en) | 2004-09-29 | 2012-07-04 | Harbor BioSciences, Inc. | Steroid analogs and characterization and treatment methods |
| US8030298B2 (en) * | 2005-05-26 | 2011-10-04 | Abbott Products Gmbh | 17β-HSD1 and STS inhibitors |
| ATE538797T1 (de) | 2005-06-09 | 2012-01-15 | Euro Celtique Sa | Pharmazeutische zusammensetzungen mit neuroaktivem steroid und verwendungen davon |
| CN100362013C (zh) * | 2006-03-16 | 2008-01-16 | 中国科学院上海有机化学研究所 | 一类c22-羰基甾体化合物、合成方法及其用途 |
| JP2009532471A (ja) * | 2006-04-05 | 2009-09-10 | ワイス | 5−ヒドロキシトリプタミン−6リガンドとしてのスルホニル−3−ヘテロシクリルインダゾール誘導体 |
| CN101506221B (zh) * | 2006-09-19 | 2012-09-12 | 索尔瓦药物有限公司 | 雌三烯衍生物及其作为17β-羟基类固醇脱氢酶抑制剂的用途 |
| US20090203658A1 (en) | 2007-01-08 | 2009-08-13 | Duke University | Neuroactive steroid compositions and methods of use therefor |
| DE102007027636A1 (de) | 2007-06-12 | 2008-12-18 | Bayer Schering Pharma Aktiengesellschaft | 17ß-Cyano-18a-homo-19-nor-androst-4-en-Derivat, dessen Verwendung und das Derivat enthaltende Arzneimittel |
| CA2698172C (en) | 2007-06-15 | 2014-02-18 | Cook, Kevin M. | Androstane and pregnane steroids with potent allosteric gaba receptor chloride ionophore modulating properties |
| GB0711948D0 (en) | 2007-06-20 | 2007-08-01 | Bionature E A Ltd | Neurosteriod compounds |
| CN101412742B (zh) | 2007-10-19 | 2013-07-31 | 天津金耀集团有限公司 | 一种抑制血管新生的硝酸酯药物 |
| KR100958286B1 (ko) * | 2007-11-15 | 2010-05-19 | 한국과학기술원 | a1G T―타입 칼슘 채널을 억제하여 본태성 진전증을예방 및 치료하는 방법 |
| US20090264443A1 (en) | 2008-04-18 | 2009-10-22 | David Helton | Treatment of organophosphate exposure with tetrahydroindolone arylpiperazine compounds |
| CN101624414B (zh) | 2008-07-07 | 2013-02-13 | 天津金耀集团有限公司 | 一种抑制血管新生的硝酸酯药物 |
| CZ2008434A3 (cs) | 2008-07-10 | 2009-12-09 | Ústav organické chemie a biochemie Akademie ved CR, v. v. i. | Pregnanové anionické slouceniny, zpusob jejich výroby a jejich použití |
| WO2010054158A2 (en) | 2008-11-07 | 2010-05-14 | Auspex Pharmaceuticals, Inc. | Steroid modulators of glucocorticoid receptor |
| US20110152840A1 (en) | 2009-12-23 | 2011-06-23 | Drugtech Corporation | Methods for reducing the occurrence of preterm delivery and other pregnancy-related conditions |
| US20130303500A1 (en) | 2010-07-30 | 2013-11-14 | Medexis S.A. | Compounds and methods for treating neoplasia |
| CN103347525A (zh) | 2010-12-15 | 2013-10-09 | 哈博生物科学公司 | 制备3α-氧取代的甾族化合物的方法及化合物 |
| CZ201181A3 (cs) | 2011-02-15 | 2012-09-12 | Ústav organické chemie a biochemie Akademie ved CR, v.v.i. | Deriváty pregnanolonu substituované v poloze 3alfa kationickou skupinou, zpusob jejich výroby, jejich použití a prostredek je obsahující |
| US9457033B2 (en) | 2011-02-15 | 2016-10-04 | Socpra Sciences Et Genie, S.E.C. | Steroid alkaloids and uses thereof as antimicrobial agents against electron transport-deficient microbes and as potentiators for antimicrobial agents against pathogenic bacteria |
| US9388210B2 (en) | 2011-02-25 | 2016-07-12 | Washington University | Neuroactive 17(20)-Z-vinylcyano-substituted steroids, prodrugs thereof, and methods of treatment using same |
| US9851309B2 (en) | 2011-03-23 | 2017-12-26 | Satoshi Watabe | Ultra-high-sensitive assay of protein and nucleic acid and kit, and novel enzyme substrate |
| JP2014521662A (ja) | 2011-07-29 | 2014-08-28 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | GABAA受容体のモジュレーターとしての新規な17β−ヘテロアリール置換ステロイド |
| CA2848212C (en) * | 2011-09-08 | 2022-03-29 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| EP3572417B1 (en) | 2011-10-14 | 2025-12-17 | Sage Therapeutics, LLC | 3,3 disubstituted 19-nor pregnane compounds, compositions, and uses thereof |
| EP2806877B2 (en) | 2012-01-23 | 2025-01-29 | Sage Therapeutics, Inc. | Neuroactive steroid formulations comprising a complex of allopregnanolone and sulfobutyl ether beta-cyclodextrin |
| US20150175651A1 (en) | 2012-06-15 | 2015-06-25 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| NZ734451A (en) | 2012-06-19 | 2018-12-21 | Intercept Pharmaceuticals Inc | Preparation, uses and solid forms of obeticholic acid |
| US20140050789A1 (en) | 2012-08-13 | 2014-02-20 | The Regents Of The University Of California | Mitigation of epileptic seizures by combination therapy using benzodiazepines and neurosteroids |
| CA2882708A1 (en) | 2012-08-21 | 2014-02-27 | Sage Therapeutics, Inc. | Methods of treating epilepsy or status epilepticus |
| WO2014058736A1 (en) | 2012-10-08 | 2014-04-17 | Washington University | Neuroactive 19-alkoxy-17(20)-z-vinylcyano-substituted steroids, prodrugs thereof, and methods of treatment using same |
| ES3056529T3 (en) | 2012-11-09 | 2026-02-23 | Drawbridge Pharmaceuticals Pty Ltd | Neuroactive steroids and their use to facilitate neuroprotection |
| AU2013352141B2 (en) | 2012-11-30 | 2018-04-05 | The Regents Of The University Of California | Anticonvulsant activity of steroids |
| HUE039057T2 (hu) | 2012-12-18 | 2018-12-28 | Univ Washington | Kezelési eljárásokban alkalmazható neuroaktív 19-alkoxi-17-szubsztituált szteroidok |
| US8939545B2 (en) | 2012-12-20 | 2015-01-27 | Eastman Kodak Company | Inkjet printing with managed airflow for condensation control |
| WO2014108808A2 (en) | 2013-01-09 | 2014-07-17 | Henry James Lorne | Pharmaceutical formulations for the treatment and prevention of trauma-induced neuropathology and neurodegeneration |
| CA2831054C (en) | 2013-01-09 | 2016-08-30 | Sapna Life Sciences Corp. | Formulations, and methods for their use in treatment of neuropathology and neurodegeneration as a result of traumatic injury |
| GB201302368D0 (en) | 2013-02-11 | 2013-03-27 | Univ Bath | Compound |
| US9512170B2 (en) | 2013-03-01 | 2016-12-06 | Washington University | Neuroactive 13, 17-substituted steroids as modulators for GABA type-A receptors |
| US9562026B2 (en) | 2013-03-14 | 2017-02-07 | Washington University | Neuroactive substituted cyclopent[a]anthracenes as modulators for GABA type-A receptors |
| US9725481B2 (en) | 2013-04-17 | 2017-08-08 | Sage Therapeutics, Inc. | 19-nor C3, 3-disubstituted C21-C-bound heteroaryl steroids and methods of use thereof |
| EP2986624B1 (en) | 2013-04-17 | 2020-03-25 | Sage Therapeutics, Inc. | 19-nor neuroactive steroids for methods of treatment |
| US20160068563A1 (en) | 2013-04-17 | 2016-03-10 | Boyd L. Harrison | 19-nor neuroactive steroids and methods of use thereof |
| KR102396328B1 (ko) | 2013-04-17 | 2022-05-10 | 세이지 테라퓨틱스, 인크. | 19-노르 c3,3-이치환된 c21-n-피라졸릴 스테로이드 및 그의 사용 방법 |
| SI3021852T1 (sl) | 2013-07-19 | 2021-07-30 | Sage Therapeutics, Inc. | Nevroaktivni steroidi, sestavki in uporabe le-teh |
| AU2014308621C1 (en) | 2013-08-23 | 2022-01-06 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| ES2805598T3 (es) | 2014-05-29 | 2021-02-12 | Sage Therapeutics Inc | Esteroides neuroactivos, composiciones y usos de los mismos |
| US10246482B2 (en) | 2014-06-18 | 2019-04-02 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| WO2016036724A1 (en) | 2014-09-02 | 2016-03-10 | The Texas A&M University System | Method of treating organophosphate intoxication |
| JOP20200195A1 (ar) | 2014-09-08 | 2017-06-16 | Sage Therapeutics Inc | سترويدات وتركيبات نشطة عصبياً، واستخداماتها |
| CN117024501A (zh) | 2014-10-16 | 2023-11-10 | 萨奇治疗股份有限公司 | 靶向cns障碍的组合物和方法 |
| ME03809B (me) | 2014-10-16 | 2021-04-20 | Sage Therapeutics Inc | KOMPOZICIJE I POSTUPCI ZA LEČENJE POREMEĆAJA CNS-a |
| WO2016082789A1 (en) | 2014-11-27 | 2016-06-02 | Sage Therapeutics, Inc. | Compositions and methods for treating cns disorders |
| RS61530B1 (sr) | 2015-01-26 | 2021-04-29 | Sage Therapeutics Inc | Kompozicije i postupci za lečenje poremećaja cns |
| CN105985396A (zh) | 2015-02-16 | 2016-10-05 | 苏州泽璟生物制药有限公司 | 氘代鹅去氧胆酸衍生物以及包含该化合物的药物组合物 |
| EP4155314A1 (en) | 2015-02-20 | 2023-03-29 | Sage Therapeutics, Inc. | Neuroactive steroids, compositions, and uses thereof |
| EP3280420B1 (en) | 2015-04-10 | 2024-03-27 | Sage Therapeutics, Inc. | Compositions and methods for treating cns disorders |
| AR105044A1 (es) | 2015-06-18 | 2017-08-30 | Sage Therapeutics Inc | Soluciones de esteroides neuroactivos y sus métodos de uso, recipiente |
| UY36741A (es) | 2015-06-21 | 2016-12-30 | Prevacus Inc | Compuesto de esteroide c-20, sus composiciones y usos para tratar la lesión cerebral traumática (tbi), que incluye las conmociones cerebrales |
| CA2998134A1 (en) | 2015-09-08 | 2017-03-16 | Viewpoint Therapeutics, Inc. | Compounds and formulations for treating ophthalmic diseases |
| AU2016338672A1 (en) | 2015-10-16 | 2018-04-12 | Marinus Pharmaceuticals, Inc. | Injectable neurosteroid formulations containing nanoparticles |
| MA43284A (fr) | 2015-11-20 | 2018-09-26 | Sage Therapeutics Inc | Composés et leurs méthodes d'utilisation |
| KR20240157128A (ko) | 2016-03-08 | 2024-10-31 | 세이지 테라퓨틱스, 인크. | 신경활성 스테로이드, 조성물, 및 그의 용도 |
| US10226550B2 (en) | 2016-03-11 | 2019-03-12 | Brigham Young University | Cationic steroidal antimicrobial compositions for the treatment of dermal tissue |
| MA45599A (fr) | 2016-07-11 | 2019-05-15 | Sage Therapeutics Inc | Stéroïdes neuroactifs substitués en c17, c20 et c21 et leurs procédés d'utilisation |
| WO2018013615A1 (en) | 2016-07-11 | 2018-01-18 | Sage Therapeutics, Inc. | C7, c12, and c16 substituted neuroactive steroids and their methods of use |
| EP3504189A1 (en) | 2016-08-23 | 2019-07-03 | Sage Therapeutics, Inc. | A crystalline 19-nor c3, 3-disubstituted c21-n-pyrazolyl steroid |
| US10391105B2 (en) | 2016-09-09 | 2019-08-27 | Marinus Pharmaceuticals Inc. | Methods of treating certain depressive disorders and delirium tremens |
| CN108727453A (zh) | 2017-04-20 | 2018-11-02 | 华东理工大学 | 新型pd-1抑制剂及其应用 |
| WO2019018119A1 (en) | 2017-07-18 | 2019-01-24 | Pairnomix, Llc | METHODS FOR TREATING EPILEPSY AND DISORDERS ASSOCIATED WITH KCNTI |
| US11285139B2 (en) | 2017-08-31 | 2022-03-29 | Takeda Pharmaceutical Company Limited | Treatment of CNS conditions |
| WO2019051264A1 (en) | 2017-09-07 | 2019-03-14 | Sage Therapeutics, Inc. | NEUROACTIVE STEROIDS AND METHODS OF USE |
| US20200276209A1 (en) | 2017-09-11 | 2020-09-03 | Sage Therapeutics, Inc. | Methods of treating epilepsy or status epilepticus |
| CA3075872A1 (en) | 2017-09-14 | 2019-03-21 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid and methods of use thereof |
| MY204493A (en) | 2017-11-10 | 2024-08-30 | Marinus Pharmaceuticals Inc | Ganaxolone for use in treating genetic epileptic disorders |
| MA51046A (fr) | 2017-12-08 | 2021-03-17 | Sage Therapeutics Inc | Dérivés de 21-[4-cyano-pyrazol-1-yl]-19-nor-pregan-3. alpha-ol-20-one deutérés pour le traitement de troubles du snc |
| MX2020006599A (es) | 2017-12-22 | 2020-11-06 | Sage Therapeutics Inc | Composiciones y metodos para el tratamiento de trastornos en el sistema nervioso central. |
| BR112020012761A2 (pt) | 2017-12-22 | 2021-02-17 | Sage Therapeutics, Inc. | composições e métodos para o tratamento de distúrbios do snc |
| TW201930269A (zh) | 2018-01-12 | 2019-08-01 | 美商賽吉醫療公司 | 用於治療cns病症之組合物及方法 |
| AR116659A1 (es) | 2018-05-04 | 2021-06-02 | Acerus Pharmaceuticals Corp | Derivados de neuroesteroides y usos de estos |
| CA3103421A1 (en) | 2018-06-12 | 2019-12-19 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21 -n-pyrazolyl steroid and methods of use thereof |
| EP4321519B1 (en) | 2018-10-12 | 2026-04-08 | Sage Therapeutics, LLC | Neuroactive steroids substituted in position 10 with a cyclic group for use in the treatment of cns disorders |
| ES3039267T3 (en) | 2018-10-19 | 2025-10-20 | Sage Therapeutics Inc | 9(11)-unsaturated neuroactive steroids and their methods of use |
| KR20250174100A (ko) | 2018-12-05 | 2025-12-11 | 세이지 테라퓨틱스, 인크. | 신경활성 스테로이드 및 이의 사용 방법 |
| WO2020132504A1 (en) | 2018-12-21 | 2020-06-25 | Sage Therapeutics, Inc. | 3.alpha.-hydroxy-17.beta.-amide neuroactive steroids and compositions thereof |
| TW202523658A (zh) | 2019-05-24 | 2025-06-16 | 美商賽吉醫療公司 | 化合物、組合物及使用方法 |
| SG11202112391UA (en) | 2019-05-31 | 2021-12-30 | Sage Therapeutics Inc | Neuroactive steroids and compositions thereof |
| CN114729000A (zh) | 2019-06-27 | 2022-07-08 | 萨奇治疗股份有限公司 | 用于治疗cns病症的化合物 |
| CA3143509A1 (en) | 2019-06-27 | 2020-12-30 | Sage Therapeutics, Inc. | Compositions and methods for treating cns disorders |
| JP7754725B2 (ja) | 2019-06-27 | 2025-10-15 | セージ セラピューティクス, インコーポレイテッド | Cns障害を治療するための組成物及び方法 |
| KR20220112803A (ko) | 2019-12-05 | 2022-08-11 | 세이지 테라퓨틱스, 인크. | 19-nor c3,3-이치환된 c21-n-피라졸릴 스테로이드 및 그의 사용 방법 |
| WO2021188778A2 (en) | 2020-03-18 | 2021-09-23 | Sage Therapeutics, Inc. | Neuroactive steroids and their methods of use |
| EP4125921A1 (en) | 2020-03-25 | 2023-02-08 | Sage Therapeutics, Inc. | Use of agents for treatment of respiratory conditions |
| US20230250129A1 (en) | 2020-06-24 | 2023-08-10 | Sage Therapeutics, Inc. | Neuroactive steroids and compositions thereof |
| KR20230041049A (ko) | 2020-07-20 | 2023-03-23 | 세이지 테라퓨틱스, 인크. | 19-노르 c3,3-이치환된 c21-n-피라졸릴 스테로이드의 제형 및 이의 사용 방법 |
| WO2022115381A1 (en) | 2020-11-25 | 2022-06-02 | Sage Therapeutics, Onc. | Compositions and methods for treating cns disorders cross reference to related application |
| KR20230136599A (ko) | 2021-01-28 | 2023-09-26 | 세이지 테라퓨틱스, 인크. | 성 기능장애의 치료를 위한 신경활성 스테로이드의용도 |
| WO2022177718A1 (en) | 2021-02-18 | 2022-08-25 | Sage Therapeutics, Inc. | Use of neuroactive steroid for treatment of sexual dysfunction |
| US20240245711A1 (en) | 2021-03-17 | 2024-07-25 | Sage Therapeutics, Inc. | A 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid for the treatment of major depressive disorder |
| JP2024513581A (ja) | 2021-04-12 | 2024-03-26 | セージ セラピューティクス, インコーポレイテッド | 本態性振戦の治療 |
| EP4329769A1 (en) | 2021-04-29 | 2024-03-06 | Sage Therapeutics, Inc. | Neuroactive steroid for use in treating major depressive disorder and postpartum depression in a lactating female |
| WO2022232504A1 (en) | 2021-04-29 | 2022-11-03 | Sage Therapeutics, Inc. | 19-nor c3,3-disubstituted c21 -n-pyrazolyl steroid for use in treating major depressive disorder and postpartum depression |
| IL315023A (en) | 2022-02-16 | 2024-10-01 | Sage Therapeutics Inc | Neuroactive steroids for treatment of cns-related disorders |
| WO2023163879A1 (en) | 2022-02-28 | 2023-08-31 | Sage Therapeutics, Inc. | Neuroactive steroids for treatment of gastrointestinal diseases or conditions |
-
2014
- 2014-04-17 KR KR1020217010184A patent/KR102396328B1/ko active Active
- 2014-04-17 HU HUE14785712A patent/HUE041369T2/hu unknown
- 2014-04-17 PT PT182033209T patent/PT3498725T/pt unknown
- 2014-04-17 SM SM20210509T patent/SMT202100509T1/it unknown
- 2014-04-17 CN CN202110717941.7A patent/CN113527400B/zh active Active
- 2014-04-17 RU RU2015149008A patent/RU2675855C2/ru active
- 2014-04-17 CA CA2909545A patent/CA2909545A1/en active Pending
- 2014-04-17 SM SM20190066T patent/SMT201900066T1/it unknown
- 2014-04-17 SG SG10201802389SA patent/SG10201802389SA/en unknown
- 2014-04-17 LT LTEP14785712.2T patent/LT2986623T/lt unknown
- 2014-04-17 IL IL310348A patent/IL310348B1/en unknown
- 2014-04-17 CN CN201480031476.XA patent/CN105339381B/zh active Active
- 2014-04-17 RS RS20211106A patent/RS62312B1/sr unknown
- 2014-04-17 HR HRP20190232TT patent/HRP20190232T1/hr unknown
- 2014-04-17 PT PT14785712T patent/PT2986623T/pt unknown
- 2014-04-17 DK DK18203320.9T patent/DK3498725T3/da active
- 2014-04-17 EP EP25185895.7A patent/EP4640277A3/en active Pending
- 2014-04-17 IL IL326689A patent/IL326689A/en unknown
- 2014-04-17 MX MX2019000996A patent/MX395023B/es unknown
- 2014-04-17 IL IL286794A patent/IL286794B2/en unknown
- 2014-04-17 MX MX2015014600A patent/MX362543B/es active IP Right Grant
- 2014-04-17 KR KR1020227015252A patent/KR102614507B1/ko active Active
- 2014-04-17 DK DK21178047.3T patent/DK3909966T3/da active
- 2014-04-17 BR BR112015026380A patent/BR112015026380A8/pt not_active Application Discontinuation
- 2014-04-17 ES ES14785712T patent/ES2709434T3/es active Active
- 2014-04-17 CN CN201810263557.2A patent/CN108440633B/zh active Active
- 2014-04-17 LT LTEP18203320.9T patent/LT3498725T/lt unknown
- 2014-04-17 HU HUE18203320A patent/HUE055554T2/hu unknown
- 2014-04-17 WO PCT/CN2014/075594 patent/WO2014169833A1/en not_active Ceased
- 2014-04-17 US US14/785,171 patent/US9512165B2/en active Active
- 2014-04-17 EP EP18203320.9A patent/EP3498725B1/en active Active
- 2014-04-17 SI SI201431072T patent/SI2986623T1/sl unknown
- 2014-04-17 KR KR1020157032454A patent/KR102239541B1/ko active Active
- 2014-04-17 SI SI201431867T patent/SI3498725T1/sl unknown
- 2014-04-17 KR KR1020237042912A patent/KR20230172622A/ko active Pending
- 2014-04-17 RS RS20190136A patent/RS58297B1/sr unknown
- 2014-04-17 ES ES21178047T patent/ES3044385T3/es active Active
- 2014-04-17 CN CN202410741773.9A patent/CN118754924A/zh active Pending
- 2014-04-17 AU AU2014256229A patent/AU2014256229B2/en active Active
- 2014-04-17 DK DK14785712.2T patent/DK2986623T3/en active
- 2014-04-17 SG SG11201508550XA patent/SG11201508550XA/en unknown
- 2014-04-17 EP EP14785712.2A patent/EP2986623B1/en active Active
- 2014-04-17 TR TR2019/01745T patent/TR201901745T4/tr unknown
- 2014-04-17 JP JP2016507995A patent/JP6470258B2/ja active Active
- 2014-04-17 PL PL18203320T patent/PL3498725T3/pl unknown
- 2014-04-17 PL PL14785712T patent/PL2986623T3/pl unknown
- 2014-04-17 ES ES18203320T patent/ES2886182T3/es active Active
- 2014-04-17 EP EP21178047.3A patent/EP3909966B1/en active Active
-
2015
- 2015-10-15 ZA ZA2015/07740A patent/ZA201507740B/en unknown
- 2015-10-15 IL IL242089A patent/IL242089B/en active IP Right Grant
- 2015-10-16 MX MX2022007490A patent/MX2022007490A/es unknown
-
2016
- 2016-10-19 US US15/297,845 patent/US10172871B2/en active Active
-
2018
- 2018-06-27 US US16/020,641 patent/US10342810B2/en active Active
- 2018-09-04 JP JP2018164995A patent/JP6784735B2/ja active Active
- 2018-09-04 JP JP2018164996A patent/JP6710730B2/ja active Active
- 2018-12-11 AU AU2018278844A patent/AU2018278844B2/en active Active
-
2019
- 2019-02-05 CY CY20191100154T patent/CY1121228T1/el unknown
- 2019-05-31 US US16/428,386 patent/US11241446B2/en active Active
-
2020
- 2020-09-28 JP JP2020161921A patent/JP7085601B2/ja active Active
- 2020-10-15 AU AU2020256400A patent/AU2020256400B2/en active Active
- 2020-11-04 IL IL278487A patent/IL278487B/en unknown
-
2021
- 2021-08-13 HR HRP20211314TT patent/HRP20211314T1/hr unknown
- 2021-09-07 CY CY20211100787T patent/CY1125416T1/el unknown
- 2021-12-23 US US17/560,732 patent/US12201640B2/en active Active
-
2022
- 2022-01-26 JP JP2022010037A patent/JP7527318B2/ja active Active
- 2022-07-12 AU AU2022205157A patent/AU2022205157B2/en active Active
-
2023
- 2023-08-09 JP JP2023129979A patent/JP7668850B2/ja active Active
-
2024
- 2024-12-10 US US18/975,467 patent/US20250319106A1/en active Pending
- 2024-12-12 AU AU2024278325A patent/AU2024278325A1/en active Pending
- 2024-12-27 JP JP2024232815A patent/JP2025060975A/ja active Pending
-
2026
- 2026-02-25 FR FR26C1007C patent/FR26C1007I1/fr active Active
- 2026-03-05 FI FIC20260008C patent/FIC20260008I1/fi unknown
- 2026-03-10 NO NO2026015C patent/NO2026015I1/no unknown
- 2026-03-12 LT LTPA2026511C patent/LTPA2026511I1/lt unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2886182T3 (es) | C21-N-pirazolil-esteroide 19-nor C3,3-disustituido para su uso en terapia | |
| ES2927007T3 (es) | Esteroides neuroactivos, composiciones y usos de los mismos | |
| ES2680582T3 (es) | Esteroides 19-alcoxi-17-sustituidos neuroactivos, profármacos de los mismos y métodos de tratamiento con los mismos | |
| ES2864719T3 (es) | Esteroides neuroactivos, composiciones y usos de los mismos | |
| ES2935476T3 (es) | Esteroides neuroactivos, composiciones y usos de los mismos | |
| ES2706180T3 (es) | Esteroides neuroactivos, composiciones y usos de los mismos | |
| ES2807264T3 (es) | Esteroides neuroactivos 19-nor para métodos de tratamiento | |
| ES3039267T3 (en) | 9(11)-unsaturated neuroactive steroids and their methods of use | |
| WO2019113494A1 (en) | Deuterated 21 -[4-cyano-pyrazol-1 -yl]-19-nor-pregan-3. alpha-ol-20-one derivatives for treating cns disorders | |
| HK40010209B (en) | 19-nor c3,3-disubstituted c21-n-pyrazolyl steroid for use in therapy | |
| RU2812930C2 (ru) | 19-нор c3,3-дизамещенные c21-n-пиразолильные стероиды и способы их применения | |
| BR122024013785A2 (pt) | Compostos esteroides neuroativos, composição farmacêutica e seus usos | |
| NZ752693A (en) | 19-nor C3,3-disubstituted C21-N-pyrazolyl steroids and methods of use thereof | |
| BR112016027508B1 (pt) | Composto esteróide neuroativo, seu sal, composição farmacêutica, seus usos e processo para preparar o composto | |
| BR122023011040B1 (pt) | Compostos esteróides neuroativos e seus sais, composição farmacêutica e seus usos | |
| NZ752696B2 (en) | 19-nor C3,3-disubstituted C21-N-pyrazolyl steroids and methods of use thereof |