JP4987894B2 - ε−カプロラクタムの製造方法 - Google Patents
ε−カプロラクタムの製造方法 Download PDFInfo
- Publication number
- JP4987894B2 JP4987894B2 JP2009051764A JP2009051764A JP4987894B2 JP 4987894 B2 JP4987894 B2 JP 4987894B2 JP 2009051764 A JP2009051764 A JP 2009051764A JP 2009051764 A JP2009051764 A JP 2009051764A JP 4987894 B2 JP4987894 B2 JP 4987894B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- reaction
- zeolite
- weight
- cyclohexanone oxime
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D201/00—Preparation, separation, purification or stabilisation of unsubstituted lactams
- C07D201/02—Preparation of lactams
- C07D201/04—Preparation of lactams from or via oximes by Beckmann rearrangement
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/16—Clays or other mineral silicates
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/03—Catalysts comprising molecular sieves not having base-exchange properties
- B01J29/035—Microporous crystalline materials not having base exchange properties, such as silica polymorphs, e.g. silicalites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/40—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of the pentasil type, e.g. types ZSM-5, ZSM-8 or ZSM-11, as exemplified by patent documents US3702886, GB1334243 and US3709979, respectively
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/584—Recycling of catalysts
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
Description
特許文献1には、シリカ対アルミナの比が少なくとも12で、かつ、拘束指数(constraintindex)が1〜12であるゼオライト(ZSM−5を中心とする群)を固体酸触媒として使用する方法が記載されている。
しかし、これらの方法に共通する欠点として、活性種であるゼオライトが単独で用いられていることを挙げることができる。
例えば、特許文献5には、固体酸触媒を用いて、気相反応により、シクロヘキサノンオキシムからε−カプロラクタムを製造するにあたり、式R1−O−R2(R1はフッ素で置換されていても良い低級アルキル基、そして、R2は水素原子、低級アルキル基、または、フェニル基)で表される化合物の存在下に反応を行う方法が記載されている。(すなわち、エーテル化合物、低級アルコールを共存させる方法。)
しかし、これらに記載されているいずれの触媒も、ゼオライトを単独で用いており、適切なバインダーで成形された実用的な形態の触媒を用いた記載はない。
しかし、エーテル化合物や、低級アルコールでは、一般に沸点が低いために、キャリアーガスとの分離が困難となる欠点がある。
また、高級アルコールでは、一般に沸点が高すぎるために、シクロヘキサノンオキシムの沸点と近づき、シクロヘキサノンオキシムとの分離が困難となる欠点がある。
R1−O−R2−OH
(ただし、R1は炭素数1〜5のアルキル基、又はフェニル基であり、R2は炭素数2〜5のアルキレン基である。)
本発明の上記及び他の諸目的、諸特徴並びに諸利益は、添付の図面を参照しながら行う以下の詳細な説明及び請求範囲の記載から明らかになる。
R1−O−R2−OH
(ただし、R1は炭素数1〜5のアルキル基又はフェニル基であり、R2は炭素数2〜5のアルキレン基である。)
1.気相反応条件の下に、シクロヘキサノンオキシムを固体酸触媒と接触させて、シクロヘキサノンオキシムの転位反応を行って、ε−カプロラクタムを製造する方法において、該転位反応を、次式で表される多価アルコール誘導体の共存の下に行うことを特徴とする方法。
R1−O−R2−OH
(ただし、R1は炭素数1〜5のアルキル基又はフェニル基であり、R2は炭素数2〜5のアルキレン基である。)
2.該固体酸触媒が、ゼオライト又はゼオライトを含有する触媒であることを特徴とする、前項1に記載の方法。
3.該ゼオライトが、Si/Al原子比が10以上のアルミノシリケート、Si/金属原子比が10以上のメタロシリケート及びシリカライトよりなる群から選ばれる少なくとも1種であることを特徴とする、前項2に記載の方法。
4.該ゼオライトが、MFI型ゼオライトであることを特徴とする、前項2又は3に記載の方法。
5.該多価アルコール誘導体が、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル及びプロピレングリコールモノエチルエーテルよりなる群から選ばれる少なくとも1種であることを特徴とする、前項1〜4のいずれかに記載の方法。
6.該多価アルコール誘導体が、エチレングリコールモノメチルエーテルであることを特徴とする、前項1〜4のいずれかに記載の方法。
7.該シクロヘキサノンオキシムの該転位反応を、反応温度が200〜500℃、反応圧力が0.01〜1MPa及びシクロヘキサノンオキシムの重量空間速度が0.01〜100hr−1の条件下で行うことを特徴とする、前項1〜6のいずれかに記載の方法。
8.該転位反応を、流動床型反応方式で行うことを特徴とする、前項1〜7のいずれかに記載の方法。
9.該転位反応に使用した触媒の一部を、該転位反応のための反応器より連続的又は断続的に抜き出し、酸素含有ガス、又は、不活性ガス雰囲気下で触媒を再生し、得られる再生触媒を該反応器に戻すことを特徴とする、前項1〜8のいずれかに記載の方法。
本発明においては、固体酸触媒には、シリカアルミナ、シリカマグネシア、アルミナボリアなどの無定形の非晶質固体酸、MCM−41(米国特許第5,098,684号、J.Am.Chem.Soc、114、P10834〜P10843、1992)に代表される規則的なメソ細孔構造をもつ非晶質固体酸、及び、ゼオライトなどの結晶質固体酸などを用いることができる。
より好ましくは、Si/Al原子比が10以上のアルミノシリケート、Si/金属原子比が10以上のメタロシリケート、及びシリカライトである。更に好ましくは、Si/Al原子比が100以上のアルミノシリケート、Si/金属原子比が100以上のメタロシリケート、及びシリカライトである。特に好ましくは、Si/Al原子比が250以上のアルミノシリケート、Si/金属原子比が250以上のメタロシリケート、及びシリカライトである。更に特に好ましくは、Si/Al原子比が500以上のアルミノシリケート、Si/金属原子比が500以上のメタロシリケート、及びシリカライトであり、最も好ましくは、Si/Al原子比が1000以上のアルミノシリケート、Si/金属原子比が1000以上のメタロシリケート、及びシリカライトである。
例えば、アルミノシリケートには、A型、X型、Y型、L型、オフレタイト、モルデナイト、フェリエライト、ZSM−5(日本国特公昭46−10064号公報(カナダ国公開特許第902,334号に対応))、ZSM−11(日本国特公昭53−23280号公報(米国特許第3,709,979号に対応))、ZSM−12(日本国特公昭52−16079号公報(米国特許第3,832,449号に対応))、ZSM−23(A.C.RohrmanJr.ら著,”Zeolites”,5,P352〜P354,1985)、β型(米国特許第3,308,069号)、MCM−22(米国特許第4,954,325号)などが挙げられる。
シリカライトには、米国特許第4,061,724号に記載されたシリカライトが挙げられる。
ゼオライトと類似構造を持つリン酸塩系多孔質結晶には、SAPO−5(日本国特開昭59−35018号公報(米国特許第4,440,871号に対応))、SAPO−11(日本国特開昭59−35018号公報(米国特許第4,440,871号に対応))などが挙げられる。
また、A型、X型、Y型、L型、オフレタイト、モルデナイト、フェリエライト、ZSM−5、ZSM−11については、A.Dyer著”An Introductionto Zeolite Molecular Sieves”1988、英国 P12〜P37に、組成及び構造の記載がある。
これらのうち、より好ましくは、シリカライト、ZSM−5、チタノシリケート、ホウ素シリケート、β型、フェリエライトであり、更に好ましくは、MFI型シリカライト、ZSM−5、MFI型チタノシリケートなどの、MFI型構造を有するゼオライトである。
最も好ましいゼオライトは、Si/Al原子比が1000以上のMFI型アルミノシリケート、Si/金属原子比が1000以上のMFI型メタロシリケート、及びMFI型シリカライトである。
本反応に悪影響をおよぼさない範囲であれば、ゼオライトにイオン交換させたり、又は担持させる金属種、金属量に、特に制限はない。本発明者らが行ったところによると、周期律表の11族に属する金属(Cu、Ag、Au)を、上記のいずれかの方法で交換させたり、又は担持させた場合に、触媒寿命を延長できる好ましい効果が得られており、特に好ましくはAgであった。
また、この場合には、ゼオライトのみからなる触媒であってもよいし、公知のバインダー(結合剤)で成形された触媒であってもよい。
すなわち、第1成分としてのゼオライト及び第2成分としての結晶質粘土鉱物を含み、更に、無機酸化物と焼成により該無機酸化物を形成することができる化合物とからなる群より選ばれる少なくとも1つの物質である第3成分を含んでなる乾燥触媒前駆体を焼成して得られる触媒であって、該無機酸化物は、周期律表の4族、13族及び14族から選ばれる少なくとも1つの元素の酸化物であり、且つゼオライト及び結晶質粘土鉱物中に結晶構造物の形で含まれる酸化物類以外の無機酸化物である、上記触媒を該固体酸触媒として用いることが好ましい。ここでいう「周期律表とは、1989年に、国際純粋及び応用化学連合(IUPAC)で定められた周期律表を意味する。
粘土鉱物の基本単位は、Si4+に4つのO2−が配位したSiO4四面体と、Al3+に6つのOH−またはO2−が配位したAl(OH)6八面体(Mg2+、Fe2+も同様の八面体構造をとる)の2つが存在する。そして、SiO4四面体が平面的につながったものを四面体シート、Al(OH)6八面体が平面的につながったものを八面体シートという。
これらの四面体シート及び八面体シートが積み重なることにより、層を形成している。
シートの積み重なり方により、1:1型粘土鉱物、2:1型粘土鉱物、混合層鉱物の3種が存在する。
具体的には、1:1型粘土鉱物としては、Al2Si2O5(OH)4の理論化学組成をもつカオリン鉱物、及びMg3Si2O5(OH)4の理論化学組成をもつ蛇紋石鉱物が挙げられる。カオリン鉱物には、カオリナイト、デイッカイト、ナクライト、層間に水分子をもつハロイサイトなどが含まれる。蛇紋石鉱物には、クリソタイル、リザルダイト、アンチゴライトなどが含まれる。
混合層鉱物としては、雲母とスメクタイトによる混合層鉱物、カオリン鉱物とモンモリロナイトによる混合層鉱物などが挙げられる。
ここで、本発明の好ましい態様の方法に用いられる触媒に含まれる第2成分としての結晶質粘土鉱物に含有されるSiO2、Al2O3などの酸化物は、結晶構造を構成する成分であるため、それらが結晶中に規則的に配置された構造を持つ。これに対し、後で説明する第3成分としての無機酸化物であるSiO2、Al2O3などは、それらが規則的に配置された構造を持っていない。このような点から、第2成分に含有される酸化物と第3成分である無機酸化物とを区別することができる。即ち、第3成分の無機酸化物は、ゼオライト及び結晶質粘土鉱物中に規則的に配置された結晶構造の形の結晶質酸化物以外の無機酸化物である。
上記した結晶質粘土鉱物類は、1種又は2種以上を組み合わせて用いてもよいし、天然物だけではなく人工的な合成物であってもよい。
また、これらは焼成、イオン交換、酸処理などの前処理を行った後、触媒に用いることができる。
これらのうちより好ましくは、カオリン鉱物、パイロフィライト、タルク、モンモリロナイトであり、特に好ましくは、カオリン鉱物である。更に好ましくはカオリナイトである。
特に、本反応にとって、アルミニウムの存在は副成物の生成を促進し、不利な効果を与える。それにも係わらず、例えば、アルミナ約40重量%から構成されるカオリン鉱物を存在させた方が良好な効果をもたらすことは、予想し得ない意外な事実である。
本発明の好ましい態様の方法に用いられる触媒を得るための、焼成前の乾燥前駆体の第3成分の無機酸化物と焼成により該無機酸化物を形成することができる化合物とからなる群より選ばれる少なくとも1つの物質(以下、「無機酸化物又は/及び酸化物形成化合物」と称す)は、バインダーとして本発明の方法に用いる触媒成形体の強度を向上する目的で添加される。該酸化物は、周期律表の4族、13族及び14族から選ばれる少なくとも1つの元素の酸化物である。ここで、4族に属する元素は、Ti、Zr、Hhであり、13族に属する元素はB、Al、Ga、In、Tlであり、14族に属する元素はC、Si、Ge、Sn、Pbである。これらの元素の酸化物のうち、好ましくは、Si、Al及びTiの酸化物である。具体的には、シリカ、シリカアルミナ、アルミナ、シリカチタニア、チタニア、シリカジルコニア、ジルコニアであり、より好ましくはシリカ、シリカアルミナ、アルミナであり、特に好ましくはシリカである。これらは1種又は2種以上を混合して用いることもできる。前述したように、第3成分の無機酸化物は、ゼオライト及び結晶質粘土鉱物中に規則的に配置された結晶構造の形の結晶質酸化物以外の無機酸化物である。
又、本発明の好ましい態様に使用される触媒の製造方法が、前述した日本国特開2000−202296号公報の、シリコンアルコシキドを酸水解することによって得たケイ質リガンドを、ゼオライトのバインダーとして使用する方法と異なる点は、日本国特開2000−202296号公報の方法が、シリコンアルコキシドを酸水解することを必須としているのに対し、本発明の方法に使用される無機酸化物やその原料は、塩基性であっても全く問題なく実施できる点にある。そのため、原料の選択肢が広く、より安価な原料を選択でき、また、pHの厳密なコントロールを必要としないため、調製法も簡便となる。
(1)触媒原料混合物を提供する工程
触媒原料混合物の基本的な成分は、第1成分であるゼオライト、第2成分である結晶質粘土鉱物、及び第3成分である無機酸化物又は/及び酸化物形成化合物である。
ゼオライトは、ヨーロッパ特許公開公報EP 0 380 364などに記載の公知の方法で水熱合成したゼオライト又は市販のゼオライトなどを用いることができる。
水熱合成したゼオライトが、有機テンプレート(ゼオライトの骨格を形成するために用いたテトラプロピルアンモニウムヒドロキシドなどの有機化合物)を含有する場合には、電気炉、管状炉などの炉中で、400〜700℃で1〜24時間、酸素含有雰囲気下、または、窒素雰囲気下で加熱処理することにより、有機テンプレートを焼成、分解したゼオライトを用いることが好ましい。
結晶質粘土鉱物は、市販の結晶質粘土鉱物をそのまま用いても良いし、必要に応じて500〜1200℃での焼成を施した後、用いても良い。
また、微量金属は、焼成前の乾燥前駆体を、酸で処理することにより、触媒から除去することもできる。使用できる酸に特に制限はないが、硝酸、硫酸、塩酸などが好ましい。
触媒原料混合物は、これらを含有し、均一に分散させた形態にすることが好ましい。
ケイ酸ソーダ水溶液の場合には、予め10〜15重量%の硫酸水溶液に所定量のケイ酸ソーダ水溶液を添加し、SiO2をゲル化させ、得られた溶液に上記のゼオライトと結晶性粘土鉱物の混合液を添加する方法が好ましい。
いずれにしても、ゼオライト、結晶質粘土鉱物、無機酸化物又は焼成により該酸化物を形成することができる化合物を均一に分散させることが重要である。
上記工程で得られた原料混合物を成形、乾燥、焼成することによって、本発明に用いる触媒を得ることができる。その際には、本発明の転位反応を実施する様式により、適切な成形方法、乾燥方法、及び、焼成方法を選択できる。次に代表的な好ましい例を示す。
この場合の触媒の製造工程は、上記の原料混合物を噴霧乾燥法により乾燥する工程(その際に流動床反応に適した球形に成形した触媒前駆体粉末を得ることができる。)と、それを焼成する工程を包含する。
噴霧乾燥法における原料混合物の噴霧化は、遠心方式、二流体ノズル方式、高圧ノズル方式を採用することができる。乾燥熱源は、スチーム、電気ヒーターなどによって加熱された空気を用いることができる。この時の熱風の乾燥機入口温度は150〜500℃が好ましい。この熱風を向流または並流に原料混合物と接触させることにより、原料混合物中の水分を蒸発させ、20〜150μm程度の球形に成形された乾燥触媒前駆体粉末を得ることができる。得られた乾燥触媒前駆体粉末は、電気炉、管状炉などの炉中で、500〜1,000℃で1〜48時間、好ましくは600〜800℃で1〜10時間、空気雰囲気下で焼成する。
この場合の触媒の製造工程は、上記の触媒原料混合物を乾燥または半乾燥した後に、押し出し成形法、打錠成形法、圧縮成形法などにより円柱状、円筒状、粒状などに触媒を成形する工程と、成形物を焼成する工程を包含する。
R1−O−R2−OH
(ただし、R1は炭素数1〜5のアルキル基、又はフェニル基であり、R2は炭素数2〜5のアルキレン基である。)
例えば、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールイソプロピルエーテル、エチレングリコールモノブチルエーテル、エチレングリコールイソブチルエーテル、エチレングリコールイソアミルエーテル、エチレングリコールモノフェニルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールイソプロピルエーテル、プロピレングリコールモノブチルエーテル、プロピレングリコールイソブチルエーテル、1−メトキシ−2−プロパノール、1−メトキシ−2−ブタノール、3−メトキシ−1−ブタノール、3−メトキシ−3−メチルブタノールなどが挙げられる。
これらの多価アルコール誘導体を反応系に共存させることにより、触媒の活性劣化を抑制し、更に、ε−カプロラクタム選択性の向上にも大きく寄与する効果がある。
シクロヘキサノンオキシムは多価アルコール誘導体と共存させると、タールやピッチを生成する副反応を抑制し、触媒寿命を顕著に延ばし、ε−カプロラクタムを長期間にわたり、安定して高収率で得ることができる点で好ましい。
反応は、固定床、移動床、流動床などの反応器に充填した固体酸触媒に、原料気化器又は反応器内で気化させたシクロヘキサノンオキシムを、気相で適切な反応条件の下で、接触させることにより行われる。
この時、ゼオライト、結晶質粘土鉱物、及び無機酸化物又は/及び酸化物形成化合物を含む固体酸触媒を用い、多価アルコール誘導体の共存の下に反応を行うことが、最も望ましい反応様式である。
反応温度は、200〜500℃の範囲が好ましく、300〜450℃の範囲がより好ましい。更に好ましくは330〜380℃の範囲である。200℃未満では反応速度が十分ではない傾向があり、500℃を越えるとシクロヘキサノンオキシムが熱分解してしまう傾向がある。
反応圧力は0.01〜1MPaの範囲が好ましく、0.03〜0.5MPaの範囲がより好ましい。更に好ましくは0.06〜0.3MPaの範囲である。
上記の重量空間速度は以下の式で算出される。
重量空間速度=F/C(hr−1)
F=シクロヘキサノンオキシム供給量(Kg/hr)
C=触媒重量(Kg)
この再生処理は、空気、空気を不活性ガス(窒素ガス、二酸化炭素ガス、アルゴンガスなどであり、好ましくは窒素ガス。)で所望の酸素濃度(1〜10容量%程度)になる様に希釈したガス、不活性ガスを流通させながら、触媒上に蓄積したタール、ピッチ、炭素質物質が燃焼、分解、揮発するのに十分な温度と時間、保持することによって行われる。その際の温度は、400〜700℃が好ましい。また、保持する時間は、通常0.5〜48時間である。
本反応に共存させた多価アルコール誘導体は、反応生成物から分離され、再使用することができる。
尚、実施例、及び、比較例において行われた種々の測定は以下のように行った。
ゼオライト粉末を下記の装置にて、粉末X線回折解析を行い、得られた回折パターンより、ゼオライトの結晶構造を同定する。
ゼオライトの同定には、R.v.Ballmoos and J.B.Higgins著”COLLECTION OF SIMULATED XRD POWDER PATTERNSFOR ZEOLITES”(ZEOLITES vol.10、NO.5、JUNE、1990)を参照した。
装置:日本国理学電機製、粉末X線回折測定装置RAD−IIIA
測定条件:CuKα線
X線管球電圧 40kV
X線管球電流 30mA
測定角度 2θ 5〜45°
ゼオライト0.2gと5N水酸化ナトリウム水溶液50gをテフロン(登録商標)製マイクロボンベに移し、マイクロボンベを密封する。これをオイルバス中で、150℃で12〜70時間保持することにより、ゼオライトを完全に溶解させる。得られたゼオライトの溶解水溶液を、イオン交換水で希釈する。(下記、ICP装置を用いた測定に適する希釈度に関しては、ゼオライト組成などにより異なるため、約5〜100倍の内から適宜選択する。)得られた希釈液中のケイ素、アルミニウム、及び、チタン濃度を、下記のICP(プラズマ発光分光分析)装置にて下記の条件にて測定し、その結果からゼオライトのSi/Al原子比、Si/Ti原子比を計算する。
ICP装置、及び、測定条件
装置:日本国理学電機製JOBIN YVON (JY138 ULTRACE)
測定条件:ケイ素測定波長 251.60nm
アルミニウム測定波長 396.152nm
チタン測定波長 334.94nm
プラズマパワー 1.0kw
ネブライザーガス 0.28リットル/min
シースガス 0.3〜0.8リットル/min
クーラントガス 13リットル/min
反応によって生成したε−カプロラクタムを含有する反応液は、下記のガスクロマトグラフィーにて、下記の条件にて測定し、その結果から反応成績(シクロヘキサノンオキシム転化率、ε−カプロラクタム選択率)を計算する。
ガスクロマトグラフィー分析装置、及び、分析条件
装置:日本国島津製作所製GC−17A
カラム:キャピラリーカラムHR−20M
(内径0.25mm、長さ50m、膜厚さ0.25μm)
分析サンプル前処理:反応液1gに反応溶媒に用いた試薬3g、内標としてエチルベンゼン0.15gを加えたものを分析する。(各重量は精秤する。)
分析サンプル注入量:1マイクロリットル
昇温プログラム:100℃で5分保持し、次いで10℃/分で240℃まで昇温した後、240℃で42分保持する。
スプリット比:100:1
キャリアガス(窒素)全流量:200ml/min
FID検出器:エアー供給圧 50kPa(約500ml/min)、水素供給圧 60kPa(約50ml/min)
アトリッションインデックスとは、流動床反応に使用される様な、粒子状触媒の機械的強度を表す指標である。これは、ある特定の条件で粒子状触媒の摩耗テストを行ない、その際の触媒中の19μm以下の粒子の増加率で定義される。アトリッションインデックスの小さい方が、耐摩耗性が高いことを表す。
以下に、摩耗テスト及びアトリッションインデックス測定の具体的方法について述べる。
粒子状触媒25gに予め水を10重量%含有させ、円筒空状のアトリッション測定装置(円筒部長さ27.5インチ、内径1.5インチ、上部長さ22インチ、内径5インチ、塔頂に微粉末捕集容器を持つ。)に仕込み、吸湿させた空気425リットル/hrを円筒底部の1/64インチ細孔(3ヶ所)より吹き出しながら装置内で5時間触媒を循環流動させ、粒子状触媒を摩耗させた。循環前後の粒子状触媒を光学式粒度分布計で測定し、19μm以下の粒子量の変化を測定した。
アトリッションインデックス(I.D.)は以下の式で計算される。
I.D.=(A−B)/(C−B)×100
A:5時間循環後の触媒中の19μm以下の触媒量(g)
B:仕込み触媒中の19μm以下の触媒量(g)
C:仕込み触媒量(g)
以下に、下記の参考例に用いる結晶質粘土鉱物の組成、特徴を列挙する。
含水カオリンASP072(商品名、米国ENGELHARD社製)
カオリナイト含有率:約100重量%
組成:Al2O3 38.5重量%
SiO2 45.4重量%
TiO2 1.6重量%
その他の微量金属成分 0.9重量%
結晶水 13.6重量%
平均粒径:0.3μm
焼成カオリンSATINTONE SP33(商品名、米国ENGELHARD社製)
焼成温度:800℃
カオリナイト含有率:約100重量%
組成:Al2O3 44.3重量%
SiO2 52.2重量%
TiO2 1.8重量%
その他の微量金属成分 1.2重量%
結晶水 0.5重量%
平均粒径:1.4μm
5M(商品名、日本国土屋カオリン社製)
パイロフィライト含有率:約60重量%(残りは主に非晶質のシリカから成る)
組成:Al2O3 17.0重量%
SiO2 78.0重量%
その他の微量金属成分 0.4重量%
結晶水 4.6重量%
平均粒径:0.3μm
含水カオリンASP600(商品名、米国ENGELHARD社製)
カオリナイト含有率:約100重量%
組成:Al2O3 38.5重量%
SiO2 45.4重量%
TiO2 1.6重量%
その他の微量金属成分 0.9重量%
結晶水 13.6重量%
平均粒径:0.6μm
ミクロエースK−1(商品名、日本国日本タルク社製)
タルク含有率:約100重量%
組成:MgO 30.7重量%
SiO2 60.1重量%
微量金属 1.6重量%
結晶水 5.3重量%
オルト珪酸テトラエチル130gにエタノール278gを添加後、10重量%テトラプロピルアンモニウムヒドロキシド水溶液291gを添加した。その溶液をホモジナイザーで5000回転/分で30分撹拌した後、1Lオートクレーブに移し105〜110℃で150時間、500回転/分で撹拌しながら水熱合成を行った。合成されたスラリーをろ過器でろ過後、ほぼ中性まで水洗し、120℃で12時間乾燥し白色結晶を得た。この結晶を500〜550℃で6時間電気炉中、空気下で焼成し、上述の粉末X線回折法により分析した。
分析の結果、面間隔(d)の値(Å)で表して、10.99、9.87、3.83、3.79、3.73、3.69で特徴的なピークが見られた。
このd値は上記の分析結果と一致することから、得られたゼオライトはMFI型であると同定した。
このゼオライトのSi/Al原子比を上述の方法で測定したところ、このゼオライトのAl含有量は10ppm以下であり、Si/Al原子比は4.5万以上であった。(これは事実上、Alを含有していないと判断できる。)この結果より、このゼオライトはシリカライトであると同定した。
尚、触媒のこの組成は焼成前の乾燥触媒前駆体の組成であり、このことは、以下の参考例(2)〜(19)で得られた触媒の全てについて同様である。
シリカゾル(スノーテックスSTN30、日本国日産化学製)66.7g、純水113g、H型シリカライト20gを用いた以外は、参考例1と同様にして触媒(B)を得た。触媒(B)の成分は、H型シリカライト50重量%、SiO250重量%である。
不純物として含有する微量金属の少ないシリカゾル[これは、日本国特開平4−231319号公報の実施例記載の方法に従って調製したもの(以降、「高純度シリカゾル」と称す。)であって30重量%SiO2含有、SiO2当たりナトリウム120重量ppm以下、pH10強塩基性。]を用いた以外は、参考例1と同様にして触媒(C)を得た。触媒(C)の成分は、H型シリカライト50重量%、SiO225重量%、カオリナイト25重量%である。
高純度シリカゾル66.7g、純水113g、H型シリカライト20gを用いた以外は、参考例1と同様にして触媒(D)を得た。触媒(D)の成分は、H型シリカライト50重量%、SiO250重量%である。
高純度シリカゾル25.0gに、アルミナゾル(日本国日産化学製アルミナゾル200、10.5重量%Al2O3含有、pH4〜6弱酸性。)23.8g、純水121g、H型シリカライト20g、カオリナイト(商品名:ASP072)10gを用い、又、焼成温度を700℃とした以外は、参考例1と同様にして触媒(E)を得た。触媒(E)の成分は、H型シリカライト50重量%、SiO219重量%、Al2O36重量%、カオリナイト25重量%である。
高純度シリカゾル50.0g、アルミナゾル(日本国日産化学製アルミナゾル200)を47.6g、純水82g、H型シリカライト20gを用いた以外は、参考例5と同様にして触媒(F)を得た。触媒(F)の成分は、H型シリカライト50重量%、SiO238重量%、Al2O312重量%である。
アルミナゾル(日本国日産化学製アルミナゾル200)95.2g、純水275g、H型シリカライト20g、カオリナイト(商品名:ASP072)10gを用いた以外は、参考例5と同様にして触媒(G)を得た。触媒(G)の成分は、H型シリカライト50重量%、Al2O325重量%、カオリナイト25重量%である。
アルミナゾル(日本国日産化学製アルミナゾル200)を190g、純水190g、H型シリカライト20gを用いた以外は、参考例5と同様にして触媒(H)を得た。触媒(H)の成分は、H型シリカライト50重量%、Al2O350重量%である。
結晶質粘土鉱物の含有量の異なる本発明の触媒の調製例。
高純度シリカゾル53.3g、純水123g、H型シリカライト20g、カオリナイト(商品名:ASP072)4gを用いた以外は、参考例1と同様にして触媒(I)を得た。触媒(I)の成分は、H型シリカライト50重量%、SiO240重量%、カオリナイト10重量%である。
高純度シリカゾル33.3gに、純水137g、H型シリカライト20g、タルク(商品名:ミクロエースK−1、日本国日本タルク社製)10gを用いた以外は、参考例1と同様にして、触媒(J)を得た。触媒(J)の成分は、H型シリカライト50重量%、SiO225重量%、タルク25重量%である。
高純度シリカゾル33.3gに、純水137g、H型シリカライト20g、モンモリロナイト(商品名:K−10、米国アルドリッチ社製)10gを用いた以外は、参考例1と同様にして、触媒(K)を得た。触媒(K)の成分は、H型シリカライト50重量%、SiO225重量%、モンモリロナイト25重量%である。
特3号ケイ酸ソーダ(日本国富士化学社製、SiO225.2重量%含有、Al2O30.01重量%含有、SiO2/Na2O=3.3モル比、強塩基性)47.4g、純水71g、硫酸6.0gの溶液に、カオリナイト(商品名:ASP072)10g、H型シリカライト18gを添加し、ホモジナイザーで5000回転/分で十分撹拌した後、このスラリーを小型噴霧器にて200℃に加熱したホットプレート上に吹き付け、乾燥触媒前駆体粉末を得た。これを1モル濃度の硝酸水溶液中(10重量%スラリー)で25℃、1Hr処理した。これを水洗、乾燥して、最後に600℃で5時間焼成して触媒(L)を得た。触媒(L)の成分は、H型シリカライト45重量%、SiO230重量%、カオリナイト25重量%である。
カオリナイト(商品名:SATINTONE SP33)10gを用いた以外は、参考例12と同様にして、触媒(M)を得た。触媒(M)の成分は、H型シリカライト45重量%、SiO230重量%、カオリナイト25重量%である。
パイロフィライト(商品名:5M)10gを用いた以外は、参考例12と同様にして、触媒(N)を得た。触媒(N)の成分は、H型シリカライト45重量%、SiO240重量%、パイロフィライト15重量%である。
カオリナイト(商品名:ASP600)10gを用いた以外は、参考例12と同様にして、触媒(O)を得た。触媒(O)の成分は、H型シリカライト45重量%、SiO230重量%、カオリナイト25重量%である。
オルト珪酸テトラエチル130gにエタノール78gを添加後、純水65gにチタンイソプロポキシド0.0886gを添加した溶液を加え、10重量%テトラプロピルアンモニウムヒドロキシド水溶液291gを添加した。その溶液を参考例1と同様に水熱合成、後処理を行い白色結晶を得た。この結晶を参考例1と同様に焼成した後、粉末X線回折法により分析した。
分析の結果、粉末X線回折パターンから、面間隔(d)の値(Å)で表して、10.99、9.87、3.83、3.79、3.73、3.69で特徴的なピークが見られた。
このd値は上記の文献に記載されたMFI型ゼオライトの粉末X線回折パターンと一致することから、得られたゼオライトはMFI型であると同定した。
このゼオライトのSi/Ti原子比を測定したところ、Si/Ti原子比は1900であった。この結果より、このゼオライトはチタノシリケートであると同定した。このゼオライトを、参考例1と同様にイオン交換して、H型チタノシリケートを得た。
H型シリカライトの代わりにH型チタノシリケートを用いた以外は、参考例3と同様にしてカオリナイト(商品名:ASP072)、高純度シリカゾルと混合し、調製して、触媒(P)を得た。触媒(P)の成分は、H型チタノシリケート50重量%、SiO225重量%、カオリナイト25重量%である。
オルト珪酸テトラエチル130gにエタノール278gを添加後、純水13gに溶解した硫酸アルミニウム・14〜18水塩0.197gを加え、10重量%テトラプロピルアンモニウムヒドロキシド水溶液291gを添加した。その溶液を参考例1と同様に水熱合成、後処理を行い白色結晶を得た。この結晶を参考例1と同様に焼成した後、粉末X線回折法により分析した。
分析の結果、の位置に特徴的な粉末X線回折パターンから、面間隔(d)の値(Å)で表して、10.99、9.87、3.83、3.79、3.73、3.69で特徴的なピークが見られた。
このd値は上記の文献に記載されたMFI型ゼオライトの粉末X線回折パターンと一致することから、得られたゼオライトはMFI型であると同定した。
このゼオライトのSi/Al原子比を測定したところ、1000であった。
この結果より、このゼオライトがZSM−5であると同定した。このゼオライトを参考例1と同様にイオン交換して、H型ZSM−5(Si/Al原子比=1000)を得た。
H型シリカライトの代わりにH型ZSM−5(Si/Al原子比=1000)を用い、又、焼成温度を600℃とした以外は、参考例3と同様にしてカオリナイト(商品名:ASP072)、高純度シリカゾルと混合、調製し、触媒(Q)を得た。触媒(Q)の成分は、H型ZSM−5(Si/Al原子比=1000)50重量%、SiO225重量%、カオリナイト25重量%である。
β型ゼオライト(商品名:CP814B−50、米国ZEOLYST社製、Si/Al原子比=25)を粉末X線回折法により分析した結果、面間隔(d)の値(Å)で表して、11.58、6.56、4.16、3.94、3.01に特徴的なピークが見られた。
一方、米国特許第3,308,069に記載のβ型ゼオライトの特徴的な粉末X線回折パターンは、d値で表して、11.4±0.2、6.7±0.2、4.25±0.1、3.97±0.1、3.0±0.1で特徴的なピークが示されている。
このd値は上記の分析結果と一致することからこのゼオライトはβ型ゼオライトであると同定した。
このゼオライトを1モル濃度の塩化アンモニウム水溶液に添加して10重量%スラリーとし、70℃で3時間イオン交換した。ろ過水洗後、120℃で12時間乾燥し、更に500℃で4時間焼成して、H型β型ゼオライトを得た。
H型ZSM−5(Si/Al原子比=1000)の代わりにH型β型ゼオライトを用いた以外は、参考例17と同様にして、カオリナイト(商品名:ASP072)、高純度シリカゾルと混合、調製し、触媒(R)を得た。触媒(R)の成分は、H型β型ゼオライト50重量%、SiO225重量%、カオリナイト25重量%である。
フェリエライト(CP914、米国ZEOLYST社製、Si/Al原子比=28)を粉末X線回折法により分析した結果、面間隔(d)の値(Å)で表して、9.38、5.62、3.97、3.53、3.45に特徴的なピークが見られた。
一方、R.v.Ballmoos and J.B.Higgins著”COLLECTION OF SIMULATED XRD POWDER PATTERNSFOR ZEOLITES”(ZEOLITES vol.10,NO.5,JUNE,1990)P398Sに記載のフェリエライトの粉末X線回折パターンは、d値で表して、9.58±0.2、5.82±0.2、4.00±0.1、3.53±0.05、3.49±0.05で特徴的なピークが示されている。
このd値は上記の分析結果と一致することから、このゼオライトはフェリエライトであると同定した。
このゼオライトを参考例18と同様にイオン交換して、H型フェリエライトを得た。
H型ZSM−5(Si/Al原子比=1000)の代わりにH型フェリエライトを用いた以外は、参考例17と同様にして、カオリナイト(商品名:ASP072)、高純度シリカゾルと混合、調製し、触媒(S)を得た。触媒(S)の成分は、H型フェリエライト50重量%、SiO225重量%、カオリナイト25重量%である。
触媒(A)を圧縮成形後粉砕し0.5〜1.5mmに整粒した後、1.5gを固定床反応装置である石英ガラス製反応管(長さ40cm、内径12mm)に充填し、窒素ガスを200Ncc/minで流し、400℃1時間保持した[Nccとは、0℃1気圧の標準状態下での体積(cc)を示す]。次いで窒素ガスを70Ncc/min流しながら350℃に保持し、メタノールにシクロヘキサノンオキシム35.7重量%溶解させた原料溶液を8.4g/hrで供給し、常圧下で反応させた。この時の重量空間速度は、2.0hr−1(ゼオライト基準4.0hr−1)であった。この反応ガスを約3℃に保たれた冷却管を通過させ凝縮させた後、氷冷又はドライアイスエタノールで冷却したトラップに反応液を回収し、上述の方法でガスクロマトグラフィーにより反応液を分析した。この時の反応結果の一部を表1に示す。
シクロヘキサノンオキシムの転化率、ε−カプロラクタムの選択率は、以下の様に計算する。
シクロヘキサノンオキシム転化率(%)=[(O−R)/O]×100
ε−カプロラクタム選択率(%)=[L/(O−R)]×100
O=シクロヘキサノンオキシム供給量(mol/hr)
R=未反応シクロヘキサノンオキシム量(mol/hr)
L=ε−カプロラクタム生成量(mol/hr)
触媒(B)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
参考例21
触媒(C)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。参考例20と比較すると、高純度シリカゾルを用いた場合には、選択性に優れることがわかる。
比較例2
触媒(D)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
触媒(E)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
アルミナを無機酸化物の一部として含有する触媒(E)は、アルミナを含有しない触媒(C)より選択性は低下したが、触媒成形体の機械的強度は向上した。(機械的強度の測定は、各々の触媒を直径3mm、長さ4mmの円柱状に同一の圧力で圧縮成形し、木屋式硬度計を用いて硬度を測定した。)これは工業触媒として有益な特性である。
比較例3
触媒(F)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
触媒(G)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
触媒(G)は触媒(E)より選択性は低下したが、触媒(E)よりアルミナ含有量が多いため成形触媒の機械的強度は、触媒(E)より更に向上した。(参考例22と同様に硬度を測定した。)これは工業触媒として有益な特性である。
触媒(H)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
参考例24
触媒(I)を用いて参考例20と同一条件で反応した結果の一部を表1に示す。
参考例25
触媒(J)を用いて参考例20と同一条件で反応した結果の一部を表2に示す。
参考例26
触媒(K)を用いて参考例20と同一条件で反応した結果の一部を表2に示す。
触媒(L)1.50gを用いて、原料として、エチレングリコールモノメチルエーテルにシクロヘキサノンオキシム35.7重量%溶解させた原料溶液を8.4g/hrで供給し、窒素ガスを106Ncc/minに変更した以外は、参考例20と同一条件で反応させた。この時の重量空間速度は、2.0hr−1(ゼオライト基準4.5hr−1)であった。この時の反応結果の一部を表3に示す。
触媒(M)を用いて実施例1と同一条件で反応した結果の一部を表3に示す。
実施例3
触媒(N)を用いて実施例1と同一条件で反応した結果の一部を表3に示す。
実施例4
触媒(O)を用いて実施例1と同一条件で反応した結果の一部を表3に示す。
参考例27
触媒(P)を用いて参考例20と同一条件で反応した結果の一部を表4に示す。
参考例28
触媒(Q)を用いて参考例20と同一条件で反応した結果の一部を表4に示す。
触媒(R)1.2gを用いて、窒素ガスを20Ncc/min流しながら、1−ヘキサノールにシクロヘキサノンオキシムを9.0重量%溶解させた原料溶液を4.4g/hrで供給した以外は参考例20と同一条件で反応を行った。この時の重量空間速度は、0.33hr−1(ゼオライト基準0.66hr−1)であった。この時の反応結果の一部を表4に示す。
触媒(S)1.5gを用いて、窒素ガスを50Ncc/min流しながら、1−ヘキサノールにシクロヘキサノンオキシムを5.0重量%溶解させた原料溶液を5.0g/hrで供給した以外は、参考例20と同一条件で反応を行った。この時の重量空間速度は、0.17hr−1(ゼオライト基準0.33hr−1)であった。この時の反応結果の一部を表4に示す。
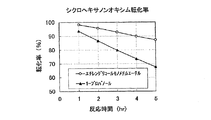
参考例1で調製したH型シリカライトを圧縮成形後粉砕し、0.5〜1.5mmに整粒した、ゼオライトのみからなる触媒0.375gを用いて、窒素ガスを53Ncc/min流しながら、エチレングリコールモノメチルエーテルにシクロヘキサノンオキシムを35.7重量%溶解させた原料溶液を4.2g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は7.0容量%であった。この時の反応結果の一部を表5、及び、図1、2に示す。
実施例5と同一の触媒0.375gを用いて、窒素ガスを49Ncc/min流しながら、1−プロパノールにシクロヘキサノンオキシムを35.7重量%溶解させた原料溶液を4.2g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は7.0容量%であった。この時の反応結果の一部を表5、及び、図1、2に示す。
尚、本比較例は、実施例5の反応条件で、本発明の多価アルコール誘導体と、同一炭素数のアルコールとの比較を行ったものである。
実施例5と同一の触媒0.375gを用いて、窒素ガスを57Ncc/min流しながら、エチレングリコールモノメチルエーテルにシクロヘキサノンオキシムを45.0重量%溶解させた原料溶液を3.3g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は6.9容量%であった。この時の反応結果の一部を表5、及び、図3、4に示す。
実施例5と同一の触媒0.375gを用いて、窒素ガスを54Ncc/min流しながら、アルコールとエーテルの1:1モル混合溶液(27重量%メタノール、73重量%メチル−tert−ブチルエーテル)に、シクロヘキサノンオキシムを45.0重量%溶解させた原料溶液を3.3g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は7.2容量%であった。この時の反応結果の一部を表5、及び、図3、4に示す。
尚、本比較例は、実施例6の反応条件で、本発明の多価アルコール誘導体と、アルコールとエーテルの等モルの物理的な混合物との比較を行ったものである。
実施例5と同一の触媒0.375gを用いて、窒素ガスを42Ncc/min流しながら、エチレングリコールモノエチルエーテルにシクロヘキサノンオキシムを20.0重量%溶解させた原料溶液を7.5g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は6.9容量%であった。この時の反応結果の一部を表5に示す。
実施例5と同一の触媒0.375gを用いて、窒素ガスを44Ncc/min流しながら、1−ヘキサノールにシクロヘキサノンオキシムを20.0重量%溶解させた原料溶液を7.5g/hrで供給し、それ以外の条件は、参考例20と同様にして反応を行った。この時のゼオライト基準の重量空間速度は、4.0hr−1であり、全原料ガス中のシクロヘキサノンオキシム濃度は7.0容量%であった。この時の反応結果の一部を表5に示す。
流動床反応に用いる触媒の調製例、及び、反応例を示す。
特3号ケイ酸ソーダ(日本国富士化学社製、SiO225.2重量%含有、Al2O30.01重量%含有、SiO2/Na2O=3.3モル比、強塩基性)2370gに、純水3570g、硫酸300gを氷冷下に十分混合し、その溶液にカオリナイト(商品名:ASP072)500g、H型シリカライト900gを添加し、ホモジナイザーで5000回転/分で1時間撹拌した。このスラリーを、スプレードライヤー(日本国ニロジャパン社製、モービルマイナ)で噴霧乾燥成形し乾燥触媒前駆体の成形粉末を得た。噴霧乾燥条件は、二流体ノズルを用いて、熱風入口温度250℃、出口温度100℃、スラリーフィード量2Kg/hrで行った。この成形粉末の一部を110℃で乾燥後、1モル濃度の硝酸水溶液中(10重量%スラリー)で25℃、1hr処理した。これを水洗、110℃で12時間乾燥して、最後に600℃で5時間焼成して触媒(T)を得た。触媒(T)の成分は、H型シリカライト45重量%、SiO230重量%、カオリナイト25重量%であった。光学顕微鏡で観察したこの粉末の粒子径は、主には50〜100μmの範囲の球状粒子であった。
この触媒(T)の上述した方法で測定したアトリッションインデックスは0.2重量%であった。すなわち、触媒は、耐摩耗性に極めて優れ、流動床反応触媒として実用的な形状、及び、機械的強度を有していることがわかった。
特定の多価アルコール誘導体を共存させる本発明の方法により、本発明の反応に好ましい反応形態である流動床反応様式を用いる場合においても、非常に優れた触媒性能を示すことがわかった。

Claims (9)
- 気相反応条件の下に、シクロヘキサノンオキシムを固体酸触媒と接触させて、シクロヘキサノンオキシムの転位反応を行って、ε−カプロラクタムを製造する方法において、該転位反応を、次式で表される多価アルコール誘導体の共存の下に行うことを特徴とする方法。
R1−O−R2−OH
(ただし、R1は炭素数1〜5のアルキル基又はフェニル基であり、R2は炭素数2〜5のアルキレン基である。) - 該固体酸触媒が、ゼオライト又はゼオライトを含有する触媒であることを特徴とする、請求項1に記載の方法。
- 該ゼオライトが、Si/Al原子比が10以上のアルミノシリケート、Si/金属原子比が10以上のメタロシリケート及びシリカライトよりなる群から選ばれる少なくとも1種であることを特徴とする、請求項2に記載の方法。
- 該ゼオライトが、MFI型ゼオライトであることを特徴とする、請求項2又は3に記載の方法。
- 該多価アルコール誘導体が、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル及びプロピレングリコールモノエチルエーテルよりなる群から選ばれる少なくとも1種であることを特徴とする、請求項1〜4のいずれかに記載の方法。
- 該多価アルコール誘導体が、エチレングリコールモノメチルエーテルであることを特徴とする、請求項1〜4のいずれかに記載の方法。
- 該シクロヘキサノンオキシムの該転位反応を、反応温度が200〜500℃、反応圧力が0.01〜1MPa及びシクロヘキサノンオキシムの重量空間速度が0.01〜100hr−1の条件下で行うことを特徴とする、請求項1〜6のいずれかに記載の方法。
- 該転位反応を、流動床型反応方式で行うことを特徴とする、請求項1〜7のいずれかに記載の方法。
- 該転位反応に使用した触媒の一部を、該転位反応のための反応器より連続的又は断続的に抜き出し、酸素含有ガス、又は、不活性ガス雰囲気下で触媒を再生し、得られる再生触媒を該反応器に戻すことを特徴とする、請求項1〜8のいずれかに記載の方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009051764A JP4987894B2 (ja) | 2001-02-14 | 2009-03-05 | ε−カプロラクタムの製造方法 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001037189 | 2001-02-14 | ||
| JP2001037189 | 2001-02-14 | ||
| JP2001065271 | 2001-03-08 | ||
| JP2001065271 | 2001-03-08 | ||
| JP2009051764A JP4987894B2 (ja) | 2001-02-14 | 2009-03-05 | ε−カプロラクタムの製造方法 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002564493A Division JP4312461B2 (ja) | 2001-02-14 | 2002-02-14 | ε−カプロラクタムの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009114219A JP2009114219A (ja) | 2009-05-28 |
| JP4987894B2 true JP4987894B2 (ja) | 2012-07-25 |
Family
ID=26609388
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002564493A Expired - Fee Related JP4312461B2 (ja) | 2001-02-14 | 2002-02-14 | ε−カプロラクタムの製造方法 |
| JP2009051764A Expired - Fee Related JP4987894B2 (ja) | 2001-02-14 | 2009-03-05 | ε−カプロラクタムの製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002564493A Expired - Fee Related JP4312461B2 (ja) | 2001-02-14 | 2002-02-14 | ε−カプロラクタムの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6894163B2 (ja) |
| EP (2) | EP1361211B1 (ja) |
| JP (2) | JP4312461B2 (ja) |
| DE (2) | DE60232433D1 (ja) |
| RU (1) | RU2240312C1 (ja) |
| TW (1) | TW570911B (ja) |
| WO (1) | WO2002064560A1 (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4312461B2 (ja) * | 2001-02-14 | 2009-08-12 | 旭化成ケミカルズ株式会社 | ε−カプロラクタムの製造方法 |
| FR2887250B1 (fr) * | 2005-06-21 | 2009-10-02 | Arkema Sa | Procede de synthese du lauryllactame (l12) par rearrangement catalytique en phase gazeuse de la cyclododecanone oxime |
| CN100374418C (zh) * | 2006-03-01 | 2008-03-12 | 湘潭大学 | 一种合成己内酰胺及其低聚物的方法 |
| KR101241777B1 (ko) * | 2008-12-19 | 2013-03-14 | 제일모직주식회사 | 무기계 인조대리석 및 무기계 인조대리석용 조성물 |
| JP5607024B2 (ja) | 2009-03-02 | 2014-10-15 | 旭化成ケミカルズ株式会社 | プロピレンの製造方法 |
| EP2697202B1 (en) | 2011-04-13 | 2015-07-22 | Rennovia, Inc. | Production of caprolactam from carbohydrate-containing materials |
| US8946411B2 (en) | 2012-02-23 | 2015-02-03 | Rennovia, Inc. | Production of caprolactam from adipic acid |
| CN104513202B (zh) * | 2013-09-29 | 2017-05-24 | 中国石油化工股份有限公司 | 一种环己酮肟转化的方法 |
| EP3527553B1 (en) * | 2016-09-14 | 2021-11-03 | Sumitomo Chemical Company, Ltd | Process for producing caprolactam from cyclohexanone oxime by beckmann-rearrangement in the gas-phase in the presence of a zeolite catalyst with a magnesium content of 3-10.000 ppm by mass |
| CN110156042A (zh) * | 2019-05-28 | 2019-08-23 | 河南师范大学 | 一种雪花状h-zsm-5分子筛的制备方法 |
| CN119059952A (zh) * | 2023-05-31 | 2024-12-03 | 中国石油化工股份有限公司 | 联产n-取代己内酰胺的方法 |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4359421A (en) * | 1981-01-15 | 1982-11-16 | Mobil Oil Corporation | Process for making epsilon-caprolactam |
| FR2498493A1 (fr) * | 1981-01-26 | 1982-07-30 | Rondolotti Ets | Tour de repoussage perfectionne |
| JPH0794428B2 (ja) * | 1985-08-28 | 1995-10-11 | 住友化学工業株式会社 | ε−カプロラクタムの製造方法 |
| JPS62281856A (ja) * | 1986-02-27 | 1987-12-07 | Sumitomo Chem Co Ltd | ε−カプロラクタムの製造法 |
| JP2676895B2 (ja) * | 1989-03-23 | 1997-11-17 | 住友化学工業株式会社 | ε―カプロラクタムの製造法 |
| US4968793A (en) * | 1989-01-26 | 1990-11-06 | Sumitomo Chemical Company, Limited | Process for producing ε-caprolactam |
| JPH0687981B2 (ja) * | 1989-03-08 | 1994-11-09 | 広栄化学工業株式会社 | 触媒の再生方法 |
| US5071802A (en) * | 1989-03-08 | 1991-12-10 | Sumitomo Chemical Co., Ltd. | Regeneration of catalysts by burning in the presence of alcohols |
| US5102643A (en) | 1990-01-25 | 1992-04-07 | Mobil Oil Corp. | Composition of synthetic porous crystalline material, its synthesis |
| JP3221021B2 (ja) * | 1991-11-27 | 2001-10-22 | 住友化学工業株式会社 | ε−カプロラクタムの製造方法 |
| TW213896B (ja) | 1991-11-27 | 1993-10-01 | Sumitomo Chemical Co | |
| KR100224333B1 (ko) | 1991-11-27 | 1999-10-15 | 고오사이 아끼오 | ε-카프로락탐의 제조방법 |
| JP3254751B2 (ja) * | 1991-11-27 | 2002-02-12 | 住友化学工業株式会社 | ε−カプロラクタムの製造法 |
| JP3254752B2 (ja) * | 1991-11-27 | 2002-02-12 | 住友化学工業株式会社 | ε−カプロラクタムの製法 |
| RU2053227C1 (ru) * | 1992-01-13 | 1996-01-27 | Гродненское производственное объединение "Азот" им.С.О.Притыцкого | Способ получения капролактама |
| RU2035453C1 (ru) * | 1992-02-03 | 1995-05-20 | Акционерное общество "Куйбышевазот" | Способ получения капролактама бекмановской перегруппировкой циклогексаноноксима |
| US5292880A (en) * | 1992-05-11 | 1994-03-08 | Mobil Oil Corporation | Synthesis of caprolactam using catalysts |
| KR100229405B1 (ko) * | 1992-06-25 | 1999-11-01 | 고오사이 아끼오 | 제올라이트성형체의 강도향상방법 |
| JP3023581B2 (ja) * | 1992-06-25 | 2000-03-21 | 住友化学工業株式会社 | ゼオライト成形体の強度向上方法 |
| JP3254753B2 (ja) * | 1992-10-01 | 2002-02-12 | 住友化学工業株式会社 | ε−カプロラクタムの製造法 |
| JPH07324070A (ja) * | 1994-04-08 | 1995-12-12 | Kazuo Sugiyama | ラクタムの製造方法 |
| BE1009465A3 (fr) * | 1995-05-04 | 1997-04-01 | Degussa | Procede de fabrication de -caprolactame. |
| JPH1087611A (ja) | 1996-09-12 | 1998-04-07 | Ube Ind Ltd | ε−カプロラクタムの製法 |
| IT1303713B1 (it) | 1998-11-06 | 2001-02-23 | Enichem Spa | Processo per la preparazione di catalizzatori a base di zeolite tipomfi. |
| JP4465731B2 (ja) * | 1999-03-03 | 2010-05-19 | 住友化学株式会社 | ε−カプロラクタムの製造方法 |
| JP4716536B2 (ja) * | 1999-12-27 | 2011-07-06 | 旭化成ケミカルズ株式会社 | ε−カプロラクタムの製造方法 |
| JP4312461B2 (ja) * | 2001-02-14 | 2009-08-12 | 旭化成ケミカルズ株式会社 | ε−カプロラクタムの製造方法 |
-
2002
- 2002-02-14 JP JP2002564493A patent/JP4312461B2/ja not_active Expired - Fee Related
- 2002-02-14 US US10/466,888 patent/US6894163B2/en not_active Expired - Fee Related
- 2002-02-14 DE DE60232433T patent/DE60232433D1/de not_active Expired - Lifetime
- 2002-02-14 EP EP02712365A patent/EP1361211B1/en not_active Expired - Lifetime
- 2002-02-14 WO PCT/JP2002/001270 patent/WO2002064560A1/ja not_active Ceased
- 2002-02-14 EP EP06006573A patent/EP1741699B1/en not_active Expired - Lifetime
- 2002-02-14 DE DE60239085T patent/DE60239085D1/de not_active Expired - Lifetime
- 2002-02-14 RU RU2003124652/04A patent/RU2240312C1/ru not_active IP Right Cessation
- 2002-02-15 TW TW091102612A patent/TW570911B/zh not_active IP Right Cessation
-
2009
- 2009-03-05 JP JP2009051764A patent/JP4987894B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| EP1361211A4 (en) | 2004-03-24 |
| EP1361211B1 (en) | 2009-05-27 |
| RU2240312C1 (ru) | 2004-11-20 |
| RU2003124652A (ru) | 2005-01-10 |
| EP1741699A2 (en) | 2007-01-10 |
| US20040054169A1 (en) | 2004-03-18 |
| JP4312461B2 (ja) | 2009-08-12 |
| JP2009114219A (ja) | 2009-05-28 |
| JPWO2002064560A1 (ja) | 2004-06-10 |
| EP1741699A3 (en) | 2008-03-19 |
| WO2002064560A1 (fr) | 2002-08-22 |
| DE60232433D1 (de) | 2009-07-09 |
| DE60239085D1 (de) | 2011-03-10 |
| EP1741699B1 (en) | 2011-01-26 |
| TW570911B (en) | 2004-01-11 |
| US6894163B2 (en) | 2005-05-17 |
| EP1361211A1 (en) | 2003-11-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4987894B2 (ja) | ε−カプロラクタムの製造方法 | |
| KR101852143B1 (ko) | NOx 환원을 위한 신규한 금속―함유 제올라이트 베타 | |
| KR102290266B1 (ko) | 인을 함유하는 aei 형 제올라이트 및 그 제조 방법 | |
| EP2592049A2 (en) | Zeolite or an analogous material thereof including mesopores arranged regularly or irregularly, and preparation method for same | |
| KR20170113573A (ko) | 분자체의 제조 방법 | |
| CN110540215A (zh) | Aei型铝硅酸盐沸石、催化剂和废气处理方法 | |
| US20140058180A1 (en) | Modified catalyst for converting oxygenates to olefins | |
| CA2581309C (en) | Method for the synthesis of zeolite beta with diethylenetriamine | |
| JPH03122009A (ja) | ベータ型ゼオライトおよびその調製方法 | |
| KR101451902B1 (ko) | 메조기공을 갖는 mre 구조의 제올라이트 또는 유사 mre 제올라이트 물질 및 그의 제조 방법 | |
| CN107848821B (zh) | 制备沸石ssz-98的方法 | |
| KR102697497B1 (ko) | 유기주형에 의해 로딩된 종정을 사용한 제올라이트 물질의 연속 합성 방법 | |
| JPS59162952A (ja) | バインダ−レスゼオライト触媒とその製造方法並びにそれを用いた触媒反応 | |
| EP1466866B1 (en) | Porous crystalline material (itq-21) and the method of obtaining same in the absence of fluoride ions | |
| JP2020523266A (ja) | モレキュラーシーブssz−111、その合成及び使用 | |
| CN101279745A (zh) | 制备mel-结构型沸石的方法 | |
| JP4243360B2 (ja) | アミンの製造方法 | |
| KR0157704B1 (ko) | 메틸아민류의 제조방법 | |
| KR102391078B1 (ko) | 분자체 ssz-107, 이의 합성 및 용도 | |
| JP6659729B2 (ja) | 分子ふるいssz−27及びその合成 | |
| JP7207154B2 (ja) | ゼオライトの製造方法 | |
| JP4716536B2 (ja) | ε−カプロラクタムの製造方法 | |
| KR20230015379A (ko) | 분자체 ssz-120, 이의 합성 및 용도 | |
| CN108640122A (zh) | 利用元素前驱体制备沸石材料的方法 | |
| KR100503512B1 (ko) | 금속 이온이 담지된 세공성 촉매, 이의 제조방법 및 이를이용한 ε-카프로락탐의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090402 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120424 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120425 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150511 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| LAPS | Cancellation because of no payment of annual fees |