WO2002066464A1 - Composes de beta-lactame, leur procede de production et agents d'abaissement du taux de cholesterol contenant ces composes - Google Patents
Composes de beta-lactame, leur procede de production et agents d'abaissement du taux de cholesterol contenant ces composes Download PDFInfo
- Publication number
- WO2002066464A1 WO2002066464A1 PCT/JP2002/001481 JP0201481W WO02066464A1 WO 2002066464 A1 WO2002066464 A1 WO 2002066464A1 JP 0201481 W JP0201481 W JP 0201481W WO 02066464 A1 WO02066464 A1 WO 02066464A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- general formula
- group
- solution
- compound represented
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/10—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to a novel /?-Lactam compound and a method for producing the same, and a serum cholesterol lowering agent containing the compound.
- Hypercholesterolemia is known to be a major risk factor for arteriosclerosis, and its association with heart disease, which is a leading cause of modern death, has also been reported (eg, Lipid Research Clinics) Progra ui., J. Am. Med. Assoc., 1984, 251, 351 and 365).
- HMG-CoA reductase inhibitors have been clinically used as serum cholesterol reducing agents.
- HMG-CoA reductase inhibitors have potent serum cholesterol-lowering effects, they are also considered to have safety problems (for example, Mevacor in Physician "s Desk Reference, 49th ED, Medical Economics Date Production Company, 1995, 1584. Therefore, there is a need for a highly active and safer serum cholesterol lowering agent.
- ⁇ -lactam compound exhibits cholesterol absorption inhibitory action in the small intestine by glucuronidation, it is necessary to form a single 0- bond between the 5-lactam structure and several sugars in the same molecule beforehand.
- the cholesterol-lowering effect of the compound is also reported (eg, WDVaccaro et al., Bioorg. Med. Chem. Lett. 3 1998, 8, 313).
- this compound is easily hydrolyzed at its 10-glycosidic bond by glycosidases present in the small intestine, and the inhibitory effect on cholesterol absorption in the small intestine is diminished. .
- the present invention has been made in view of the circumstances described in the above, and has a serum cholesterol-lowering agent having a lactam structure and a C monoglycoside moiety that is stable to hydrolysis by glucosidase, hydrolysis by acid or base in the same molecule. It is an object of the present invention to provide, that is, to provide a hybrid molecule of / C-glucoside which is useful as a serum cholesterol lowering agent. Disclosure of the invention
- C-glycosides which are useful as sugar derivatives stable to lactam compounds by metabolism by glycosidase, hydrolysis by acid or base (for example, RJ Linhardt et al., Tetra H., 1998, 54, 9913, DE Levy, The Chemistry of C-Glycosides; E. lsevier Science; Oxford, 1995., MHD Postema, C-Glycoside Synth esis.
- AA 3 and A 4 each represents a hydrogen atom, a halogen, a C 1 to C 5 alkyl group, a C i Cs alkoxy group, —COOR formula (b):
- R 2 is —CH 2 OH group, — CH 2 OC (0) group or — CO
- R 3 is —OH group or — OC (0) one group
- R 4 is — (CH 2 ) k R 5 (CH 2 ) i ⁇ (where k and 1 are integers of 0 or 1 or more) Yes, k + 1 is an integer less than 10)
- R 5 is a bond, which is a single bond, one CH CH CH ⁇ , one OCH 2 ⁇ , a carbonyl group or-CH (0 H) — Ru.
- any one of A ′ ′ A 3 and A 4 is a group represented by the above-mentioned formula (a).
- a 2 is a CCS alkyl chain, C i to C 5 alkoxy chain, d ⁇ C
- alkenyl chains C 1 to C 5 hydroxyalkyl chains or C 1 to C 5 It is a carbonyl alkyl chain.
- n, p, q and r> represent an integer of 0, 1 or 2.
- the present invention is also a process for producing a compound represented by the general formula (I) or a pharmaceutically acceptable salt.
- the present invention is also a serum cholesterol lowering agent containing a compound represented by the general formula (I) or a pharmaceutically acceptable salt as an active ingredient.
- the present invention is a serum cholesterol lowering agent by the combined use of a compound represented by the general formula (I) and / or a protease inhibitor.
- Examples of pharmacologically acceptable salts of the compounds represented by the general formula (I) of the present invention include sodium salts and potassium salts as salts of inorganic bases, and succinic acid, maleic acid and carbyl as organic acid salts. Acids, tartaric acid etc. may be mentioned.
- the compounds of general formula (I) can be administered orally, as such or, according to known formulation techniques, formulated into powders, granules, tablets or capsules. In addition, administration directly to the intestine and parenteral administration in the form of suppositories, injections and the like are possible. Although the dose varies depending on the condition, age, body weight, etc.
- a compound represented by the general formula (I) and a? -Lactamase inhibitor enhances the serum cholesterol lowering action.
- proteinase inhibitor is a drug that inhibits the decomposition of bacterial //-lacquer ring by clavulanic acid etc.
- the compounds of the present invention are exemplified below, but the present invention is limited thereto W is not a thing.
- the following compounds may be mentioned as specific compounds included in the present invention.
- the compounds of the present invention are illustrated by the structural formulas in Tables 1 to 12 below.
- the specific optical rotation was measured as a compound having a specific optical rotation, which was synthesized as an optically active substance or by optical resolution.
- the compound (18) is added to the diamine compound (17), heated under reflux in the presence of a base, and subjected to a Swinger reaction to obtain a complex.
- a cis / ⁇ -lactam compound is obtained by using trans 5-lactam form and LDA (lithium diisopropyamide).
- an asymmetric /?-Lactam can be obtained by adding an asymmetric ligand or the like into the system (for example, Hafez, AM et al., Org. Lett., 2000, 2 (25) 3963- 3965).
- a Grignard reagent (1-12) was reacted with the compound (1-11) to obtain a compound (1-13) (for example, MFWong et al., J. Carbohyd. Chem., 1996, 15 (6), 763, CD. Hurd et al., J. Am. Chem. Soc, 1945, 67: 1972, H. Togo et al Synthesis, 1998, 09) o With respect to the compound (1-1), Similarly, after reacting Grignard reagent (1 1 2), the resulting hydroxyl group is removed with triethylamine, or treated with a base as a leaving group such as tosyl group or halogen, The compound (1 1 1 3) was obtained by catalytic reduction, etc.
- the compound (1-13) is treated with M g to give a Grignard reagent, and when DMF (dimethyl formamide) is allowed to act, the compound (1-14) is reacted with M g, and after it is allowed to act.
- Compound (1-15) can be obtained by reacting with CO 2 (CO 2).
- Compound (3-4) is a synthetic intermediate for obtaining the general formula (I) according to Production Example 1-(1)-(), (c), (d).
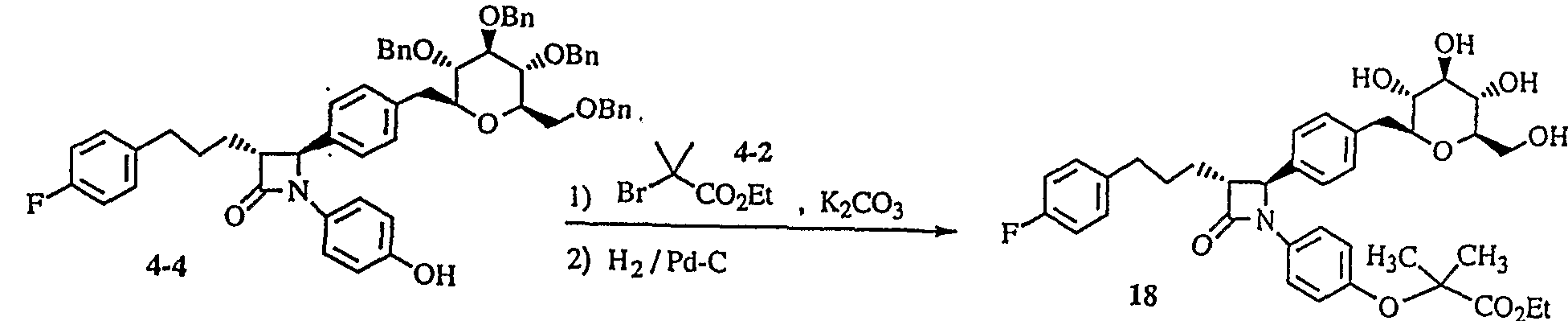
- 2-bromoisobutyric acid alkyl ester (for compound (4-1) 4-2) is allowed to act in the presence of potassium carbonate and then catalytically reduced to obtain a compound, or subsequently the ester moiety is hydrolyzed with lithium hydroxide to obtain a compound represented by compound (4-3) .
- the compound (43) is deprotected to obtain a general formula (I).
- the compounds (6-1) and (6-2) were thioglycosylated to give a compound (6-3).
- a Ramberg-Backlund reaction for example, PSBelica et al., Te trahedron Lett., 1998, 39, 8225, and FK / Griffin et al. , Tetrahedron Lett., 1998, 39, 8179
- Compound ( After catalytic reduction of 6-4), 1 was allowed to act to obtain compound (1-4).
- the compound (1-4) is a synthetic material for obtaining the general formula (I) according to Production Example 1. -
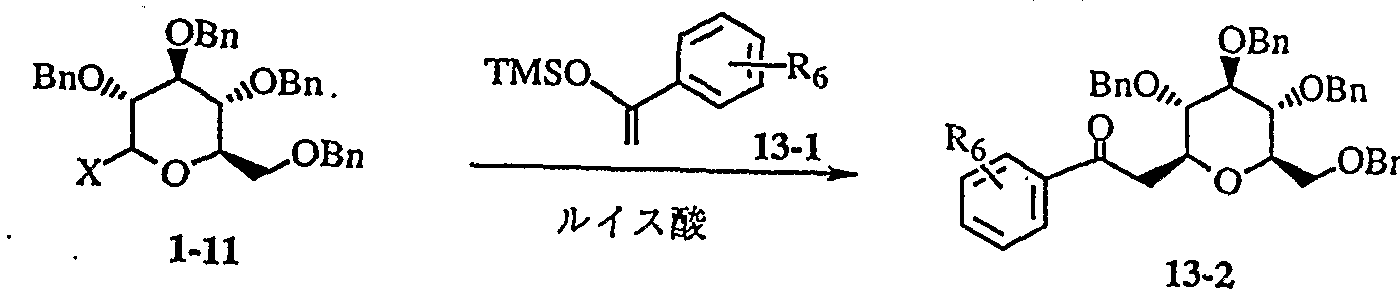
- a compound (1-11) and a compound (7-1) in the presence of a Lewis acid eg, BF a E E t 2 O, S n C 14, Ag OT f — C p 2 H f C l 2 etc.
- a Lewis acid eg, BF a E E t 2 O, S n C 14, Ag OT f — C p 2 H f C l 2 etc.
- 0- dalcosylation proceeds, followed by C 1 -darcosylation to obtain a compound (7-3) (for example, RR Schmidt et al . : Synthesis, 1993, 325).
- the compound (7-3) can be further converted to the compound (7-4) by esterifying the phenolic hydroxyl moiety.
- Compounds (7-3) and (7-4) become synthetic raw materials of the general formula (I) according to Production Examples 1 and 3.
- X is the same as the above-mentioned.
- the compound (7-6) obtained in the same manner as in the above-mentioned Preparation Example 1- (1) was deprotected to give a compound (7-7).
- carbonation reaction is carried out in the presence of carbon monoxide (for example, REDolle et al., Chem. Commun., 1987, 904), the compound Compound (7-3) is obtained according to Production Example 7- (1), Production Examples 1 and 3 as a raw material for synthesis of the general formula (I) to obtain (7-3).
- the compound (7-11) is subjected to the same force coupling as the compound (1-11), and then the asytyl group (A c) is reacted with a haloform (eg, S. Kajigaeshi et al. 3 Synthesis, 1985) , 674) as a compound (7-3).
- a haloform eg, S. Kajigaeshi et al. 3 Synthesis, 1985
- the compound (7-9) is subjected to aryl C-glucosylation reaction as shown in Production Example 1 (1) to give a compound (7-10).
- the compound (7-10) becomes a synthetic material of the general formula (I) according to Preparation Example 8.
- the compound (8-5) can also be obtained as an optically active substance as follows. That is, the compound (8-7) is reacted with an optically active amino derivative (8-8) in the presence of an acid catalyst to obtain a compound (8-9).
- Compound (8-9) directly catalytic reduction, and Compound (8-1 1) reducing the Orefu fin portion at the beginning c to (e.g. N aHB (OA c), N a BH 4 , etc.), then a strong acid (eg HC 0 2 H, E t 3 S i H , etc.) the effect is not of compound (8-1 1) may be (e.g. C.Cimarell et al., J.Org.Ch em. , 1996, 61 , 5557).
- the compound (8-11) is reacted with B ⁇ H under acidic conditions to cause transesterification to give a compound (8-5).
- Compound (8-5) can be converted to compound (8-6) in the same manner as described above.
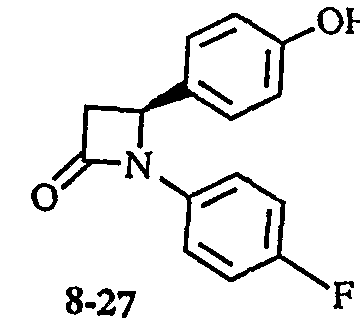
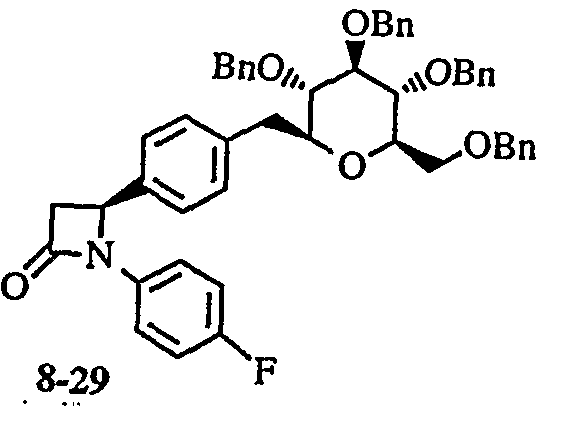
- the 5-lactam compound (8-6) is ⁇ ⁇ ⁇ ⁇ -alkylated by the method of Dominic M.T. Chan et al. (Tetra hedron Lett., 1998, 39, 2933) and then depenzylated by catalytic reduction to give a compound (8 — 1 2)
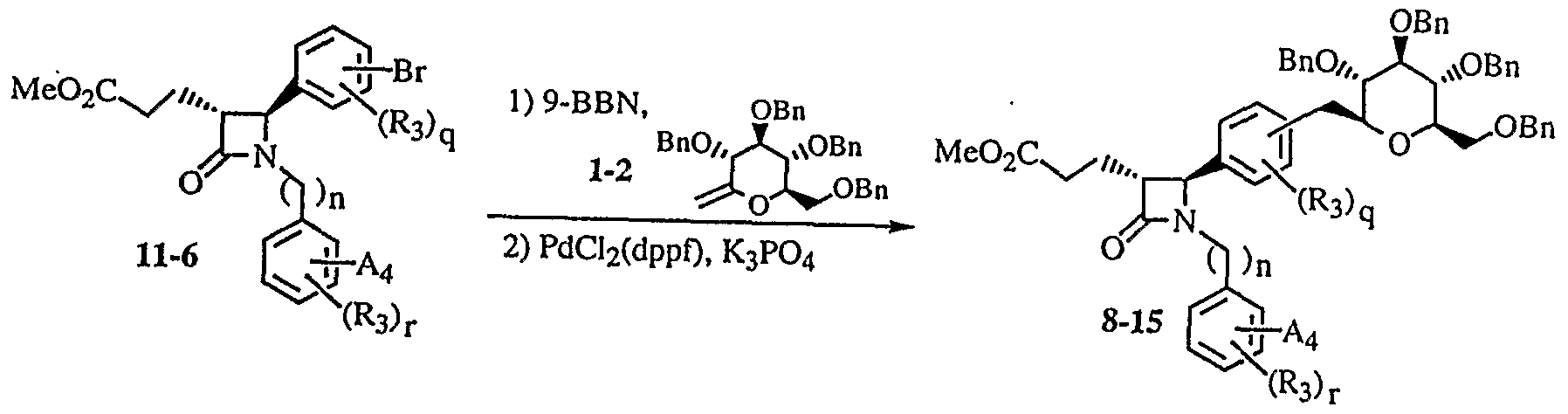
- the compound (8-12) was subjected to Suzuki reaction with a glucose derivative (112) according to the method of R. Johnson et al. (Synlett, 1997, 1406) to give a compound (8-13).
- Ai is the following formula (a) A compound of the following formula, for example compound 39, corresponds to compound (8-15) in accordance with Preparation Example 8:
- a 4 is the following formula (a):
- a compound of the following formula (8-24), for example, the compound 38 corresponds to the compound (8-12) according to Production Example 8:
- Composite (8-2 5) is a raw material of the general formula (I) in the same manner as described above by the Suzuki coupling reaction.
- the compound (11-15) is allowed to react with BSA and then with TBAF (n-tetratrapylammonium fluoride) to obtain a /?-Lactam compound (11-16).
- (1 1-6) is a synthetic raw material of the general formula (I).
- the compound (11-7) is used instead of the compound (111-14), the compound (11-8) corresponding to the compound (11-6) is obtained in the same manner.
- the compound (1.19) can be obtained in the same manner as in the preparation example ⁇ ⁇ ⁇ for the compound (1 1-8).
- the obtained compound (1 1-9) becomes a synthetic raw material of the general formula (I) according to Production Example 8.
- the obtained compound (12-3) is a synthesis raw material of the general formula (I) according to Production Example 8.
- L Carbohydr. Res., 1992, 224, 301 is an organometallic reagent (Grignard reagent) After conversion to organic zinc reagent etc.), compound (15- 3) is coupled with compound (15- 3) in the presence of a catalyst such as palladium, nickel complex and the like, followed by cyclization reaction to obtain compound (15- 4).
- a catalyst such as palladium, nickel complex and the like
- the compound (12-1) and the compound (15-3) can be force-pulled by the Heck reaction in the same manner as in Production Example 2 to obtain the compound (16-1), Compound (16-) 1) can be converted to the general formula (I) according to Production Example 1-7.
- oxychloride is not used.
- the reaction is carried out in a solvent or a solvent such as methylene chloride or dichloroethane, or DCC (1, 3-dicyclohexyl carpidine), DEPC (jetyl phosphoryl).
- a condensation agent such as lucanidide is reacted in the presence of a base in a solvent such as methylene chloride or DMF to cyclize to obtain a general formula (I).
- Mitsunobu reagent such as D EAD (jetiazo dicarboxylate), DI AD (diisopropyl azodicarboxylate) or (P y S) 2
- a base such as Na H, etc.
- the compound can be cyclized by treatment with a base such as aqueous solution of rhium to obtain the general formula (I).
- the compound (18-1) is oxidized using selenium dioxide or the like, or the compound (18-4) is oxidized by a method such as P d (0 A c) 2 -benzoquinone-perchloric acid or the like.
- the asymmetric reduction of the ketone moiety is carried out in the same manner as in Production Example 8 to obtain a compound (18-3).
- the compound (18-4) can be subjected to hydroboration to obtain the compound (18-3-), and the reaction can be performed stereoselectively with an asymmetric borane reducing agent or the like. it can.
- Compound (19-1) is subjected to asymmetric reduction (for example, a method using a transition metal complex: R. Noyori et al., J. Am. Chem. Soc., 1987, 109, 5856) to give compound (19) — Get 2).
- the hydroxyl group of the compound (19-2) is converted to a leaving group, and then the ring closure reaction is performed or the hydroxyl group is directly subjected to the Mitsunobu reaction to obtain a compound (19-3).
- the resulting double bond is catalytically reduced to obtain compound (19-4), or compound (1 9) 5) and Negishi reaction (eg, T. Hayashi et al 1., J. Am. Chem. Soc.
- Amine (20-1) is asymmetrically reduced according to Production Example 1 9 to give compound (20-2).
- the ester part of the compound (20-2) is hydrolyzed to give the corresponding carboxylic acid, and then it is lactamized (eg, DCC etc.) using a condensing agent to obtain the compound (19-3).
- the compound (19-3) can also be obtained by the /?-Lactamization (e.g., EtMgBr etc.) of the compound (20-2).
- the compound (19-3) is a raw material of the general formula (I) according to Production Example 19. Production example 2 1
- the compound (21-1) is added to obtain a compound (21-2).
- the compound (21-2) is subjected to asymmetric reduction to give a compound (21-4), or the compound (21-3-) is allowed to act on the compound (21-2-) to obtain a compound (21-5). ).
- Compound (21-3) is allowed to act on compound (21-4) to obtain compound (21-6).
- the compound (2 1-6) and the rib portion (1 2-1 or 1 9-5) are force-pulled to form a compound (2 1-8), and Get).
- compound (21-5) is asymmetrically reduced to form compound (21-7), and then the sugar portion and the compound (21-9) are cut.
- the compound (2 1-9) is also ⁇ -lactamized to obtain a compound (2 1-10).
- the compound (2 0-10) thus obtained is a raw material of the general formula (I).
- Hams tuna were divided into groups of 3 and fed with a diet containing 0.5% cholesterol (CE-2; CLEA Japan) for 4 days.
- the test compound was orally administered orally once a day to the animals at the start of cholesterol feeding.
- the administration was carried out with 0.2 mL of corn oil alone (control group) or a solution of the test compound in tocopherol oil per 100 g of body weight.
- Twenty hours after the final administration blood was collected from the abdominal aorta under light ether anesthesia to separate the serum. Serum total cholesterol was measured using cholesterol E-test (Kako Junyaku).
- the effect of the test compound was shown as the inhibition rate (%) with respect to the increase of blood cholesterol concentration by high cholesterol diet.
- glycosidase with C-aryl ( ⁇ ) and 0-aryl ( ⁇ ), ie, biological stability against mono-acetyl-1-D-galactosaminidase Were compared and tested according to the method of Mark von Itzstein et al. (Org. Lett., 1999, 1, 443-446).
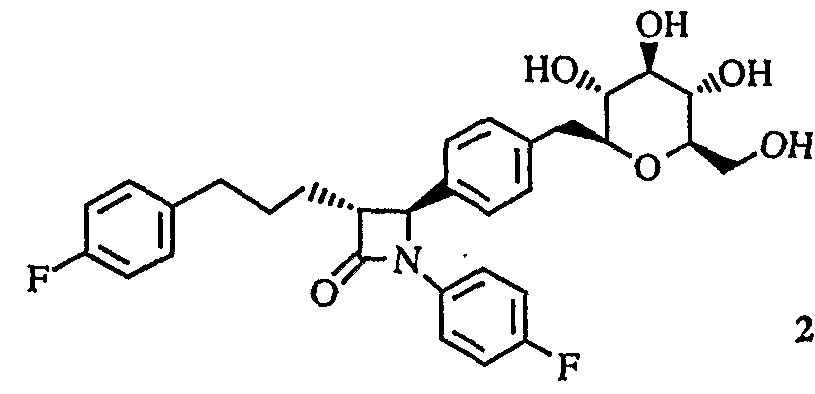
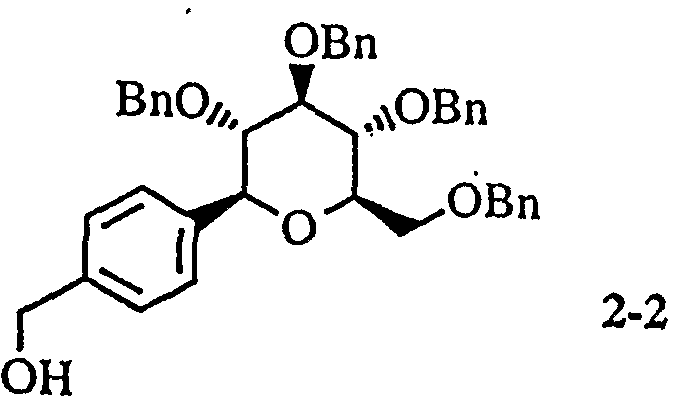
- Example 1 4-(4- ⁇ [(5 S, 2 R, 3 R, 4 R, 6 R) 1 3, 4, 5-trihydroxyloxy 6-(hydroxylmethyl) 1 perhydro drawn 2 H-blank 2-2 ⁇ ⁇ ⁇ ⁇ ⁇ ⁇ 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 4 1
- reaction solution was cooled to room temperature, K 3 PO 4 (10 mL, 3 M aqueous solution) was added, and the mixture was stirred for 15 minutes. Then add DMF solution (10 OmL) of 4-one (t-butyldimethylsilyloxy) bromobenzene (3.0 1 g), P d C 12 (dppf) (0. 7 3 g) and add 1 After stirring for 8 hours, the organic layer was washed with brine and dried over sodium sulfate. After evaporation of the organic solvent, TBAF (15 mL, 1.0 M THF solution) was added, and the mixture was stirred for 3 hours.
- MnO 2 (9.65 g) was added to a solution of diethylene compound (1-4) (3.6 g) in 2 mL of a solution in 2 ml of M 2 O, and the mixture was heated under reflux for 2 hours. The reaction solution was allowed to cool to room temperature and filtered using celite. The mixture was concentrated under reduced pressure to give the compound (1-5) as a colorless crystal (3.46 g, yield 97%).
- D-p-hydroxycarboxylic phenylglycine 8- 1) 1 6. 7 g of 2 N - N a 0 H solution 5 0 mL solution Cu S 0 4 - 5 H 2 0 in (1 2. 5 g) Add 100 mL aqueous solution of water and stir at 60 ° C for 1 hour. The reaction solution is cooled to room temperature, 50 mL of 2 N-NaOH aqueous solution, 50 mL of methanol and 3.0 mL of benzyl bromide 1 are added, and the mixture is stirred at room temperature for 20 hours.
- reaction solution is diluted with ether (100 mL), 10% aqueous HC1 solution (50 mL x 2), water (100 mL x 4), saturated brine (50 mL x 1) Wash with and dry over anhydrous sodium sulfate. It distilled off the solvent, the residue ⁇ Se Toni preparative drill (8 0 mL) solution and the after: DBU 7. 0 mL, Penjiruburomai de 5.
- the combined organic layer is washed with water (50 mL X 1), saturated aqueous sodium bicarbonate (50 mL X 1), and saturated brine (50 mL X 1), and dried over anhydrous sodium sulfate.
- the aqueous layer is further extracted with ethyl acetate (50 mL ⁇ 3), and the combined ethyl acetate layer is extracted with water (50 mL ⁇ 1), 10% aqueous HC1 solution (50 mL), saturated aqueous sodium bicarbonate (50 m Wash with LX 1) and saturated brine (50 mL ⁇ 1), and dry over anhydrous sodium sulfate.
- reaction solution is cooled to room temperature, methanol (1 mL) is added, and the mixture is stirred for 5 minutes, and then 10% aqueous hydrochloric acid solution (15 mL) is added to extract acetic acid ethyl ester (50 mL ⁇ 2).
- the extract is washed with water (50 mL 1), saturated aqueous sodium bicarbonate solution (50 mL 1) and saturated brine (50 mL 1), and dried over anhydrous sodium sulfate.
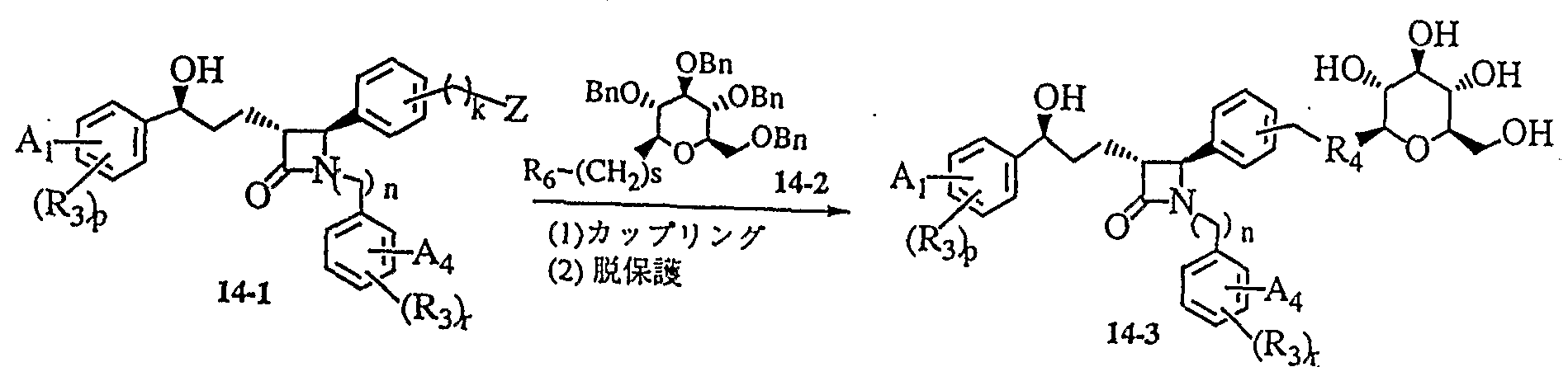
- the resulting compound is a synthetic intermediate for obtaining the general formula (I) according to Reference Example 4 (1), (j), (k) and Examples 5, 67 and 8.
- reaction solution is poured into ice water (20 ml) and extracted with ethyl acetate (30 ml ⁇ 2).
- the extract is washed with water (30 ml x 2) and saturated brine (40 mL xl), and dried over anhydrous magnesium sulfate.
- the solvent is distilled off, the residue is made into a solution of T HF (5 mL) and 1 Me OH (5 mL), 50 mg of 5% palladium-carbon is added, and the mixture is stirred at room temperature under H 2 gas atmosphere for 9 hours.
- the compound (19-8) (1.0 g) is added to a solution of Z n (C u) (106 mg) in THF-HMPA solution (3: 1, 4 mL), and the mixture is heated under reflux for 3 hours. After adding palladium acetate (1.7 mg) and 2- (di-tert-peptylphosphino) biphenyl (4.4 mg) to the reaction solution at 0 ° C. or less and stirring for 5 minutes, compound (19-9) (22) Add 3 mg). reaction After cooling the solution to room temperature, 10% aqueous hydrochloric acid solution (50 mL) and acetic acid ester (3 O mL) are added, and the insoluble matter is filtered.
- the filtrate is extracted with acetic acid ester (50 mL ⁇ 2), and the extract is washed with water (50 mL) and saturated brine (50 mL), and dried over anhydrous sodium sulfate.
- novel 5-lactam compound having in its molecule a C-glycoside which is stable to metabolism by glucosidase by the glucosidase of the present invention and to hydrolysis by acid or base has a strong serum cholesterol lowering action, and as a serum cholesterol lowering agent It is useful.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Obesity (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Hematology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Cephalosporin Compounds (AREA)
Description


































































































Claims










Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2438961A CA2438961C (en) | 2001-02-23 | 2002-02-20 | Beta-lactam compounds, manufacturing methods of the compounds and serum hypocholesterolemic agents containing the compounds |
| DK02703861T DK1362855T3 (da) | 2001-02-23 | 2002-02-20 | beta-lactam-forbindelser, en fremgangsmåde til fremstilling af disse forbindelser og serumkolesterolsænkende midler indeholdende disse forbindelser |
| DE60222742T DE60222742T2 (de) | 2001-02-23 | 2002-02-20 | Beta-lactam-verbindungen,herstellungsverfahren für diese verbindungen und serumhypocholesterinämiemittel, die diese verbindungen enthalten |
| HK04103348.9A HK1060357B (en) | 2001-02-23 | 2002-02-20 | Beta-lactam compounds, process for repoducing the same and serum cholesterol-lowering agents containing the same |
| KR1020037010927A KR100721639B1 (ko) | 2001-02-23 | 2002-02-20 | β-락탐 화합물과 그 제조방법 및 이것을 함유하는 혈청 콜레스테롤 저하제 |
| EP02703861A EP1362855B1 (en) | 2001-02-23 | 2002-02-20 | Beta-lactam compounds, process for repoducing the same and serum cholesterol-lowering agents containing the same |
| BRPI0206193A BRPI0206193B1 (pt) | 2001-02-23 | 2002-02-20 | compostos, método de preparação dos compostos, e, agentes hipocolesterolêmicos no soro |
| US10/450,171 US7045515B2 (en) | 2001-02-23 | 2002-02-20 | β-lactam compounds process for reproducing the same and serum cholesterol-lowering agents containing the same |
| JP2002565979A JP4229701B2 (ja) | 2001-02-23 | 2002-02-20 | β−ラクタム化合物及びその製造方法並びにこれを含有する血清コレステロール低下剤 |
| AU2002237522A AU2002237522B2 (en) | 2001-02-23 | 2002-02-20 | Beta-lactam compounds, process for reproducing the same and serum cholesterol-lowering agents containing the same |
| MXPA03005073A MXPA03005073A (es) | 2001-02-23 | 2002-02-20 | Compuestos de beta-lactama, proceso para producir los mismos y agentes reductores de colesterol en suero que los contienen. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001048202 | 2001-02-23 | ||
| JP2001-48202 | 2001-02-23 | ||
| JP2001-128031 | 2001-04-25 | ||
| JP2001128031 | 2001-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002066464A1 true WO2002066464A1 (fr) | 2002-08-29 |
Family
ID=26609978
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2002/001481 Ceased WO2002066464A1 (fr) | 2001-02-23 | 2002-02-20 | Composes de beta-lactame, leur procede de production et agents d'abaissement du taux de cholesterol contenant ces composes |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US7045515B2 (ja) |
| EP (1) | EP1362855B1 (ja) |
| JP (1) | JP4229701B2 (ja) |
| KR (1) | KR100721639B1 (ja) |
| CN (1) | CN1273467C (ja) |
| AT (1) | ATE374769T1 (ja) |
| AU (1) | AU2002237522B2 (ja) |
| BR (1) | BRPI0206193B1 (ja) |
| CA (1) | CA2438961C (ja) |
| DE (1) | DE60222742T2 (ja) |
| DK (1) | DK1362855T3 (ja) |
| ES (1) | ES2294101T3 (ja) |
| MX (1) | MXPA03005073A (ja) |
| PT (1) | PT1362855E (ja) |
| RU (1) | RU2301799C2 (ja) |
| TW (1) | TWI291957B (ja) |
| WO (1) | WO2002066464A1 (ja) |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6627757B2 (en) | 2001-03-28 | 2003-09-30 | Schering Corporation | Enantioselective synthesis of azetidinone intermediate compounds |
| WO2004005247A1 (en) * | 2002-07-05 | 2004-01-15 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| WO2005000353A1 (ja) * | 2003-06-27 | 2005-01-06 | Kotobuki Pharmaceutical Co., Ltd. | 血清コレステロール低下剤或はアテローム性動脈硬化症の予防又は治療剤 |
| WO2005044256A1 (en) * | 2003-10-30 | 2005-05-19 | Merck & Co., Inc. | 2-azetidinones as anti-hypercholesterolemic agents |
| WO2005047248A1 (en) * | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-ones |
| WO2005062824A2 (en) | 2003-12-23 | 2005-07-14 | Merck & Co., Inc. | Anti-hypercholesterolemic compounds |
| WO2006122020A2 (en) | 2005-05-06 | 2006-11-16 | Microbia, Inc. | Process for production of 4-biphenylyazetidin-2-ones |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| EP1832576A1 (en) * | 2003-11-10 | 2007-09-12 | Microbia Inc. | 4-Biarylyl-1-phenolyzetidin-2-ones |
| JP2007291004A (ja) * | 2006-04-25 | 2007-11-08 | Kotobuki Seiyaku Kk | アズレン誘導体及びそれを有効成分とする血清コレステロール低下剤 |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| EP1918000A2 (en) | 2003-11-05 | 2008-05-07 | Schering Corporation | Combinations of lipid modulating agents and substituted azetidinones and treatments for vascular conditions |
| WO2008060476A2 (en) | 2006-11-15 | 2008-05-22 | Schering Corporation | Nitrogen-containing heterocyclic compounds and methods of use thereof |
| DE102007005045A1 (de) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| JP2008540557A (ja) * | 2005-05-11 | 2008-11-20 | マイクロビア インコーポレーテッド | フェノール型4−ビフェニリルアゼチジン−2−オンの製造方法 |
| JP2008545700A (ja) * | 2005-05-25 | 2008-12-18 | マイクロビア インコーポレーテッド | 4−(ビフェニリル)アゼチジン−2−オンホスホン酸類の製造方法 |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| DE102007063671A1 (de) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | Neue kristalline Diphenylazetidinonhydrate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| US7635705B2 (en) | 2005-06-20 | 2009-12-22 | Schering Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| US7638526B2 (en) | 2006-09-15 | 2009-12-29 | Schering Corporation | Azetidine derivatives useful in treating pain, diabetes and disorders of lipid metabolism |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US7700597B2 (en) | 2004-12-03 | 2010-04-20 | Schering Corporation | Substituted piperazines as CB1 antagonists |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010075068A1 (en) | 2008-12-16 | 2010-07-01 | Schering Corporation | Pyridopyrimidine derivatives and methods of use thereof |
| WO2010075069A1 (en) | 2008-12-16 | 2010-07-01 | Schering Corporation | Bicyclic pyranone derivatives as nicotinic acid receptor agonists |
| WO2010100255A1 (en) | 2009-03-06 | 2010-09-10 | Lipideon Biotechnology Ag | Pharmaceutical hypocholesterolemic compositions |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| US7884080B2 (en) | 2006-09-15 | 2011-02-08 | Schering Plough Corporation | Azetidinone derivatives and methods of use thereof |
| WO2011017907A1 (zh) | 2009-08-11 | 2011-02-17 | 浙江海正药业股份有限公司 | 氮杂环丁酮类化合物及医药应用 |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US7902157B2 (en) | 2006-09-15 | 2011-03-08 | Schering Corporation | Azetidine and azetidone derivatives useful in treating pain and disorders of lipid metabolism |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012120058A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit benzyl- oder heteromethylengruppen substituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120057A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120051A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit adamantan- oder noradamantan substituierte benzyl-oxathiazinderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120050A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
| WO2020191141A1 (en) | 2019-03-20 | 2020-09-24 | Regeneron Pharmaceuticals, Inc. | Treatment of increased lipid levels with sterol regulatory element binding transcription factor 1 (srebf1) inhibitors |
| WO2020191163A1 (en) | 2019-03-20 | 2020-09-24 | Regeneron Pharmaceuticals, Inc. | Treatment of increased lipid levels with sterol regulatory element binding protein cleavage-activating protein (scap) inhibitors |
| WO2025049558A1 (en) | 2023-08-29 | 2025-03-06 | Regeneron Pharmaceuticals, Inc. | Treatment of liver disease or metabolic disorder with folliculin interacting protein 1 (fnip1) inhibitors and/or folliculin (flcn) inhibitors |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW200611896A (en) | 2004-05-21 | 2006-04-16 | Aventis Pharma Gmbh | Process for preparing 1, 4-diphenylazetidinone derivatives |
| WO2006039334A1 (en) * | 2004-09-29 | 2006-04-13 | Schering Corporation | Combinations of substituted azetidonones and cb1 antagonists |
| EP1893222A4 (en) * | 2005-06-15 | 2010-07-07 | Merck Sharp & Dohme | ANTI-HYPERCHOLESTERINAMIC COMPOUNDS |
| UY29607A1 (es) | 2005-06-20 | 2007-01-31 | Astrazeneca Ab | Compuestos quimicos |
| MY148538A (en) | 2005-06-22 | 2013-04-30 | Astrazeneca Ab | Novel 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| TW200806623A (en) * | 2005-10-05 | 2008-02-01 | Merck & Co Inc | Anti-hypercholesterolemic compounds |
| AR059021A1 (es) * | 2006-01-18 | 2008-03-05 | Schering Corp | Moduladores de receptores cannabinoides |
| US20080070890A1 (en) * | 2006-09-15 | 2008-03-20 | Burnett Duane A | Spirocyclic Azetidinone Compounds and Methods of Use Thereof |
| CA2672221A1 (en) * | 2006-12-20 | 2008-07-17 | Merck & Co., Inc. | Anti-hypercholesterolemic compounds |
| US20100197564A1 (en) * | 2007-04-19 | 2010-08-05 | Schering Corporation | Diaryl morpholines as cb1 modulators |
| US8623873B2 (en) * | 2007-06-28 | 2014-01-07 | Intervet Inc. | Substituted piperazines as CB1 antagonists |
| US20100286160A1 (en) * | 2007-06-28 | 2010-11-11 | Intervet Inc. | Substituted piperazines as cb1 antagonists |
| CN104860980B (zh) * | 2015-04-23 | 2018-06-19 | 上海弈柯莱生物医药科技有限公司 | 一种用于合成依折麦布的中间体及其制备方法和应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0076621A2 (en) * | 1981-10-01 | 1983-04-13 | Ajinomoto Co., Inc. | Azetidinone derivatives |
| WO1995008532A1 (en) * | 1993-09-21 | 1995-03-30 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| US5412092A (en) * | 1993-04-23 | 1995-05-02 | Bristol-Myers Squibb Company | N-substituted 2-azetidinones |
| WO1997016424A1 (en) * | 1995-11-02 | 1997-05-09 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(r)-(3(s)-hydroxy-3-([phenyl or 4-fluorophenyl])-propyl)-4(s)-(4-hydroxyphenyl)-2-azetidinone |
| WO1997016455A1 (en) * | 1995-10-31 | 1997-05-09 | Schering Corporation | Sugar-substituted 2-azetidinones useful as hypocholesterolemic a gents |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2107548T3 (es) | 1991-07-23 | 1997-12-01 | Schering Corp | Compuestos de beta-lactama sustituidos utiles como agentes hipocolesterolemicos y procedimientos para su preparacion. |
| SK282164B6 (sk) * | 1992-09-18 | 2001-11-06 | Merck & Co., Inc. | Spôsob výroby beta-metylkarbapenémových medziproduktov, medziprodukty na vykonávanie tohto postupu a spôsob výroby medziproduktov |
| AU2002231688A1 (en) * | 2000-12-21 | 2002-07-01 | Sanofi-Aventis Deutschland Gmbh | Diphenyl azetidinone derivatives, method for the production thereof, medicaments containing these compounds, and their use |
-
2002
- 2002-02-08 TW TW091102310A patent/TWI291957B/zh not_active IP Right Cessation
- 2002-02-20 JP JP2002565979A patent/JP4229701B2/ja not_active Expired - Lifetime
- 2002-02-20 BR BRPI0206193A patent/BRPI0206193B1/pt not_active IP Right Cessation
- 2002-02-20 DK DK02703861T patent/DK1362855T3/da active
- 2002-02-20 CA CA2438961A patent/CA2438961C/en not_active Expired - Fee Related
- 2002-02-20 WO PCT/JP2002/001481 patent/WO2002066464A1/ja not_active Ceased
- 2002-02-20 AT AT02703861T patent/ATE374769T1/de active
- 2002-02-20 PT PT02703861T patent/PT1362855E/pt unknown
- 2002-02-20 ES ES02703861T patent/ES2294101T3/es not_active Expired - Lifetime
- 2002-02-20 MX MXPA03005073A patent/MXPA03005073A/es active IP Right Grant
- 2002-02-20 KR KR1020037010927A patent/KR100721639B1/ko not_active Expired - Fee Related
- 2002-02-20 AU AU2002237522A patent/AU2002237522B2/en not_active Ceased
- 2002-02-20 RU RU2003128424/04A patent/RU2301799C2/ru not_active IP Right Cessation
- 2002-02-20 CN CNB028052013A patent/CN1273467C/zh not_active Expired - Fee Related
- 2002-02-20 EP EP02703861A patent/EP1362855B1/en not_active Expired - Lifetime
- 2002-02-20 DE DE60222742T patent/DE60222742T2/de not_active Expired - Lifetime
- 2002-02-20 US US10/450,171 patent/US7045515B2/en not_active Expired - Lifetime
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0076621A2 (en) * | 1981-10-01 | 1983-04-13 | Ajinomoto Co., Inc. | Azetidinone derivatives |
| US5412092A (en) * | 1993-04-23 | 1995-05-02 | Bristol-Myers Squibb Company | N-substituted 2-azetidinones |
| WO1995008532A1 (en) * | 1993-09-21 | 1995-03-30 | Schering Corporation | Hydroxy-substituted azetidinone compounds useful as hypocholesterolemic agents |
| WO1997016455A1 (en) * | 1995-10-31 | 1997-05-09 | Schering Corporation | Sugar-substituted 2-azetidinones useful as hypocholesterolemic a gents |
| WO1997016424A1 (en) * | 1995-11-02 | 1997-05-09 | Schering Corporation | Process for preparing 1-(4-fluorophenyl)-3(r)-(3(s)-hydroxy-3-([phenyl or 4-fluorophenyl])-propyl)-4(s)-(4-hydroxyphenyl)-2-azetidinone |
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6627757B2 (en) | 2001-03-28 | 2003-09-30 | Schering Corporation | Enantioselective synthesis of azetidinone intermediate compounds |
| JP2006501184A (ja) * | 2002-07-05 | 2006-01-12 | アストラゼネカ アクチボラグ | 脂質代謝の異常を処置するためのジフェニルアゼチジノン誘導体 |
| WO2004005247A1 (en) * | 2002-07-05 | 2004-01-15 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| RU2333199C2 (ru) * | 2002-07-05 | 2008-09-10 | Астразенека Аб | Производные дифенилазетидинона, способы их получения, содержащие их фармацевтические композиции и комбинация и их применение для ингибирования всасывания холестерина |
| US7470678B2 (en) | 2002-07-05 | 2008-12-30 | Astrazeneca Ab | Diphenylazetidinone derivatives for treating disorders of the lipid metabolism |
| RU2328307C2 (ru) * | 2003-06-27 | 2008-07-10 | Котобуки Фармасьютикал Ко., Лтд. | Средство, снижающее содержание холестерина в сыворотке крови, или средство для предотвращения или терапии атеросклероза |
| WO2005000353A1 (ja) * | 2003-06-27 | 2005-01-06 | Kotobuki Pharmaceutical Co., Ltd. | 血清コレステロール低下剤或はアテローム性動脈硬化症の予防又は治療剤 |
| JP2005015434A (ja) * | 2003-06-27 | 2005-01-20 | Kotobuki Seiyaku Kk | 血清コレステロール低下剤或はアテローム性硬化症の予防又は治療剤 |
| CN1812812B (zh) * | 2003-06-27 | 2010-11-03 | 寿制药株式会社 | 降血清胆固醇药或者动脉粥样硬化的预防或治疗药 |
| US7795229B2 (en) | 2003-06-27 | 2010-09-14 | Kotobuki Pharmaceutical Co., Ltd. | Serum cholesterol lowering agent or preventative or therapeutic agent for atherosclerosis |
| AU2004251536B2 (en) * | 2003-06-27 | 2007-11-15 | Kotobuki Pharmaceutical Co., Ltd. | Serum cholesterol lowering agent or preventive or therapeutic agent for atherosclerosis |
| KR100792098B1 (ko) * | 2003-06-27 | 2008-01-04 | 고토부키 세이야쿠 가부시키가이샤 | 혈청 콜레스테롤 저하제 또는 아테롬성 동맥경화증의 예방또는 치료제 |
| WO2005044256A1 (en) * | 2003-10-30 | 2005-05-19 | Merck & Co., Inc. | 2-azetidinones as anti-hypercholesterolemic agents |
| EP1918000A2 (en) | 2003-11-05 | 2008-05-07 | Schering Corporation | Combinations of lipid modulating agents and substituted azetidinones and treatments for vascular conditions |
| EP1832576A1 (en) * | 2003-11-10 | 2007-09-12 | Microbia Inc. | 4-Biarylyl-1-phenolyzetidin-2-ones |
| EA010026B1 (ru) * | 2003-11-10 | 2008-06-30 | Майкробиа, Инк. | 4-биарилил-1-фенилазетидин-2-оны |
| WO2005047248A1 (en) * | 2003-11-10 | 2005-05-26 | Microbia, Inc. | 4-biarylyl-1-phenylazetidin-2-ones |
| US7871998B2 (en) | 2003-12-23 | 2011-01-18 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitory activity |
| WO2005062824A2 (en) | 2003-12-23 | 2005-07-14 | Merck & Co., Inc. | Anti-hypercholesterolemic compounds |
| US7700597B2 (en) | 2004-12-03 | 2010-04-20 | Schering Corporation | Substituted piperazines as CB1 antagonists |
| WO2006122020A2 (en) | 2005-05-06 | 2006-11-16 | Microbia, Inc. | Process for production of 4-biphenylyazetidin-2-ones |
| JP2008542205A (ja) * | 2005-05-06 | 2008-11-27 | マイクロビア インコーポレーテッド | 4−ビフェニリルアゼチジン−2−オンの製造方法 |
| JP2008540557A (ja) * | 2005-05-11 | 2008-11-20 | マイクロビア インコーポレーテッド | フェノール型4−ビフェニリルアゼチジン−2−オンの製造方法 |
| JP2008545700A (ja) * | 2005-05-25 | 2008-12-18 | マイクロビア インコーポレーテッド | 4−(ビフェニリル)アゼチジン−2−オンホスホン酸類の製造方法 |
| US7635705B2 (en) | 2005-06-20 | 2009-12-22 | Schering Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| US7846946B2 (en) | 2005-06-20 | 2010-12-07 | Schering Plough Corporation | Heteroatom-linked substituted piperidines and derivatives thereof useful as histamine H3 antagonists |
| US7893048B2 (en) | 2005-06-22 | 2011-02-22 | Astrazeneca Ab | 2-azetidinone derivatives as cholesterol absorption inhibitors for the treatment of hyperlipidaemic conditions |
| WO2007039177A2 (en) | 2005-09-29 | 2007-04-12 | Sanofi-Aventis | Phenyl- and pyridinyl- 1, 2 , 4 - oxadiazolone derivatives, processes for their preparation and their use as pharmaceuticals |
| JP2007291004A (ja) * | 2006-04-25 | 2007-11-08 | Kotobuki Seiyaku Kk | アズレン誘導体及びそれを有効成分とする血清コレステロール低下剤 |
| US7842684B2 (en) | 2006-04-27 | 2010-11-30 | Astrazeneca Ab | Diphenylazetidinone derivatives possessing cholesterol absorption inhibitor activity |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| US7638526B2 (en) | 2006-09-15 | 2009-12-29 | Schering Corporation | Azetidine derivatives useful in treating pain, diabetes and disorders of lipid metabolism |
| US7902157B2 (en) | 2006-09-15 | 2011-03-08 | Schering Corporation | Azetidine and azetidone derivatives useful in treating pain and disorders of lipid metabolism |
| US7884080B2 (en) | 2006-09-15 | 2011-02-08 | Schering Plough Corporation | Azetidinone derivatives and methods of use thereof |
| WO2008060476A2 (en) | 2006-11-15 | 2008-05-22 | Schering Corporation | Nitrogen-containing heterocyclic compounds and methods of use thereof |
| DE102007005045A1 (de) | 2007-01-26 | 2008-08-07 | Sanofi-Aventis | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| DE102007063671A1 (de) | 2007-11-13 | 2009-06-25 | Sanofi-Aventis Deutschland Gmbh | Neue kristalline Diphenylazetidinonhydrate, diese Verbindungen enthaltende Arzneimittel und deren Verwendung |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010075069A1 (en) | 2008-12-16 | 2010-07-01 | Schering Corporation | Bicyclic pyranone derivatives as nicotinic acid receptor agonists |
| WO2010075068A1 (en) | 2008-12-16 | 2010-07-01 | Schering Corporation | Pyridopyrimidine derivatives and methods of use thereof |
| WO2010100255A1 (en) | 2009-03-06 | 2010-09-10 | Lipideon Biotechnology Ag | Pharmaceutical hypocholesterolemic compositions |
| US9212175B2 (en) | 2009-03-06 | 2015-12-15 | Lipideon Biotechnology Ag | Pharmaceutical hypocholesterolemic compositions |
| WO2011017907A1 (zh) | 2009-08-11 | 2011-02-17 | 浙江海正药业股份有限公司 | 氮杂环丁酮类化合物及医药应用 |
| US8623855B2 (en) | 2009-08-11 | 2014-01-07 | Zhejiang Hisun Pharmaceutical Co., Ltd. | Azetidinone compounds and medical use thereof |
| JP2013501734A (ja) * | 2009-08-11 | 2013-01-17 | チュージャン・ヒスン・ファーマシューティカル・カンパニー・リミテッド | アゼチジノン化合物およびその医薬用法 |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2012120051A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit adamantan- oder noradamantan substituierte benzyl-oxathiazinderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120050A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120057A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120058A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit benzyl- oder heteromethylengruppen substituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
| WO2020191141A1 (en) | 2019-03-20 | 2020-09-24 | Regeneron Pharmaceuticals, Inc. | Treatment of increased lipid levels with sterol regulatory element binding transcription factor 1 (srebf1) inhibitors |
| WO2020191163A1 (en) | 2019-03-20 | 2020-09-24 | Regeneron Pharmaceuticals, Inc. | Treatment of increased lipid levels with sterol regulatory element binding protein cleavage-activating protein (scap) inhibitors |
| WO2025049558A1 (en) | 2023-08-29 | 2025-03-06 | Regeneron Pharmaceuticals, Inc. | Treatment of liver disease or metabolic disorder with folliculin interacting protein 1 (fnip1) inhibitors and/or folliculin (flcn) inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| TWI291957B (en) | 2008-01-01 |
| CA2438961A1 (en) | 2002-08-29 |
| ES2294101T3 (es) | 2008-04-01 |
| ATE374769T1 (de) | 2007-10-15 |
| MXPA03005073A (es) | 2003-09-05 |
| RU2301799C2 (ru) | 2007-06-27 |
| AU2002237522B2 (en) | 2007-08-02 |
| HK1060357A1 (en) | 2004-08-06 |
| DE60222742T2 (de) | 2008-07-17 |
| DE60222742D1 (de) | 2007-11-15 |
| KR100721639B1 (ko) | 2007-05-23 |
| JP4229701B2 (ja) | 2009-02-25 |
| US7045515B2 (en) | 2006-05-16 |
| BR0206193A (pt) | 2004-02-03 |
| CN1273467C (zh) | 2006-09-06 |
| RU2003128424A (ru) | 2005-01-27 |
| PT1362855E (pt) | 2007-12-04 |
| JPWO2002066464A1 (ja) | 2004-06-17 |
| DK1362855T3 (da) | 2008-01-28 |
| KR20030076690A (ko) | 2003-09-26 |
| US20040063929A1 (en) | 2004-04-01 |
| EP1362855A1 (en) | 2003-11-19 |
| EP1362855A4 (en) | 2005-02-23 |
| CA2438961C (en) | 2010-02-16 |
| CN1492865A (zh) | 2004-04-28 |
| BRPI0206193B1 (pt) | 2016-05-24 |
| EP1362855B1 (en) | 2007-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2002066464A1 (fr) | Composes de beta-lactame, leur procede de production et agents d'abaissement du taux de cholesterol contenant ces composes | |
| JP5037333B2 (ja) | 1−(2h)−イソキノロン誘導体 | |
| CN105001213B (zh) | C-芳基糖苷衍生物、其药物组合物、制备方法及应用 | |
| US7232812B2 (en) | Substituted azetidine compounds, their preparation and use as medicaments | |
| JP3887396B2 (ja) | 抗バクテリア・インドロン・オキサゾリジノン、その製造用中間体、及びそれを含有する医薬組成物 | |
| JP2789190B2 (ja) | 新規なβ−ラクタム誘導体およびその製造法 | |
| CN105017236A (zh) | C-芳基糖苷衍生物、其药物组合物、制备方法及应用 | |
| EP1727793B1 (en) | Substituted azetidine compounds as cyclooxygenase-1 - cyclooxygenase-2 inhibitors, and their preparation and use as medicaments | |
| JPS63188662A (ja) | 新規なβ−ラクタム誘導体の製造方法 | |
| JP3037965B2 (ja) | 2―アルコキシカルボニル―2―メチル―1―オキソ―1,2,3,6,7,8―ヘキサヒドロ―ベンゾ〔1,2―b;4,3―b´〕ジピロール誘導体 | |
| JP4328874B2 (ja) | 7−置換−8−ニトロキサンチン誘導体 | |
| HK1060357B (en) | Beta-lactam compounds, process for repoducing the same and serum cholesterol-lowering agents containing the same | |
| JPH0657683B2 (ja) | 光学活性アミノ酸誘導体 | |
| JPH05504337A (ja) | 新規な化合物 | |
| JPH0672875A (ja) | ペネム誘導体、その製造法および用途 | |
| JP2005232000A (ja) | ピリドンカルボン酸誘導体又はその塩 | |
| MXPA06009332A (en) | Substituted azetidine compounds as cyclooxygenase-1-cyclooxygenase-2 inhibitors, and their preparation and use as medicaments | |
| HU211665A9 (hu) | Az átmeneti oltalom az 1-19. igénypontokra vonatkozik. | |
| JPH0220637B2 (ja) | ||
| JP2000290275A (ja) | クマリン誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BR CA CN ID IN JP KR MX RU US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002237522 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2003/005073 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10450171 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002703861 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 849/KOLNP/2003 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002565979 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037010927 Country of ref document: KR Ref document number: 028052013 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2438961 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037010927 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002703861 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002703861 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002237522 Country of ref document: AU |