ES2734432T3 - Compuestos antiamiloides que contienen benzofurazano - Google Patents
Compuestos antiamiloides que contienen benzofurazano Download PDFInfo
- Publication number
- ES2734432T3 ES2734432T3 ES15755943T ES15755943T ES2734432T3 ES 2734432 T3 ES2734432 T3 ES 2734432T3 ES 15755943 T ES15755943 T ES 15755943T ES 15755943 T ES15755943 T ES 15755943T ES 2734432 T3 ES2734432 T3 ES 2734432T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- etoac
- added
- reaction
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 106
- -1 4- (pyrrolidin-1-yl) piperidin-1-yl Chemical group 0.000 claims abstract description 126
- 125000001424 substituent group Chemical group 0.000 claims abstract description 76
- 125000004566 azetidin-1-yl group Chemical group N1(CCC1)* 0.000 claims abstract description 41
- 150000003839 salts Chemical class 0.000 claims abstract description 33
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims abstract description 28
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 28
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims abstract description 26
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical group C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims abstract description 14
- 229930192474 thiophene Natural products 0.000 claims abstract description 13
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 12
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims abstract description 11
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims abstract description 10
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims abstract description 10
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 9
- 150000001408 amides Chemical class 0.000 claims abstract description 9
- 239000001257 hydrogen Substances 0.000 claims abstract description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 9
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims abstract description 8
- 125000002971 oxazolyl group Chemical group 0.000 claims abstract description 8
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims abstract description 8
- 125000003226 pyrazolyl group Chemical group 0.000 claims abstract description 8
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims abstract description 8
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims abstract description 8
- UQFQONCQIQEYPJ-UHFFFAOYSA-N N-methylpyrazole Chemical group CN1C=CC=N1 UQFQONCQIQEYPJ-UHFFFAOYSA-N 0.000 claims abstract description 7
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical group C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims abstract description 7
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 7
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical group C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 claims abstract description 7
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 claims abstract description 7
- 239000000460 chlorine Substances 0.000 claims abstract description 6
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 6
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 6
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 claims abstract description 6
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims abstract description 4
- 206010002022 amyloidosis Diseases 0.000 claims description 22
- 238000000034 method Methods 0.000 claims description 18
- 239000008194 pharmaceutical composition Substances 0.000 claims description 10
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 7
- 208000024827 Alzheimer disease Diseases 0.000 claims description 6
- 230000037396 body weight Effects 0.000 claims description 6
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 claims description 5
- 206010033799 Paralysis Diseases 0.000 claims description 5
- 208000018737 Parkinson disease Diseases 0.000 claims description 5
- 230000000750 progressive effect Effects 0.000 claims description 5
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 5
- 208000000044 Amnesia Diseases 0.000 claims description 3
- 201000011240 Frontotemporal dementia Diseases 0.000 claims description 3
- 208000026139 Memory disease Diseases 0.000 claims description 3
- 230000019771 cognition Effects 0.000 claims description 3
- 230000006984 memory degeneration Effects 0.000 claims description 3
- 208000023060 memory loss Diseases 0.000 claims description 3
- 125000004287 oxazol-2-yl group Chemical group [H]C1=C([H])N=C(*)O1 0.000 claims description 3
- 125000004304 oxazol-5-yl group Chemical group O1C=NC=C1* 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 claims description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 620
- 235000019439 ethyl acetate Nutrition 0.000 description 308
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 305
- 239000000243 solution Substances 0.000 description 272
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 243
- 238000006243 chemical reaction Methods 0.000 description 242
- 239000000203 mixture Substances 0.000 description 236
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 206
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 201
- 239000011780 sodium chloride Substances 0.000 description 181
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 168
- 239000010410 layer Substances 0.000 description 156
- 239000013058 crude material Substances 0.000 description 155
- 239000007832 Na2SO4 Substances 0.000 description 143
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 143
- 229910052938 sodium sulfate Inorganic materials 0.000 description 143
- 235000011152 sodium sulphate Nutrition 0.000 description 143
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 116
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 113
- 238000005160 1H NMR spectroscopy Methods 0.000 description 110
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 93
- 239000000284 extract Substances 0.000 description 87
- 229910052786 argon Inorganic materials 0.000 description 84
- 238000003818 flash chromatography Methods 0.000 description 83
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 78
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 76
- 238000001816 cooling Methods 0.000 description 73
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 72
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 67
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 66
- 239000000463 material Substances 0.000 description 61
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 59
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 58
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 58
- 235000019341 magnesium sulphate Nutrition 0.000 description 58
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 56
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 55
- 239000012044 organic layer Substances 0.000 description 55
- 239000012074 organic phase Substances 0.000 description 54
- 150000001299 aldehydes Chemical class 0.000 description 49
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 48
- 239000007787 solid Substances 0.000 description 48
- HJBGZJMKTOMQRR-UHFFFAOYSA-N (3-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC(C=O)=C1 HJBGZJMKTOMQRR-UHFFFAOYSA-N 0.000 description 47
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 47
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 41
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 41
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 41
- 229910000029 sodium carbonate Inorganic materials 0.000 description 38
- 235000017550 sodium carbonate Nutrition 0.000 description 38
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 36
- 238000004587 chromatography analysis Methods 0.000 description 35
- 238000011282 treatment Methods 0.000 description 30
- 101150041968 CDC13 gene Proteins 0.000 description 29
- 235000012239 silicon dioxide Nutrition 0.000 description 29
- 238000003756 stirring Methods 0.000 description 29
- 238000010992 reflux Methods 0.000 description 28
- CWWZUAXJZKSULT-UHFFFAOYSA-N 4,6-dibromo-2,1,3-benzoxadiazole Chemical compound C1=C(Br)C=C(Br)C2=NON=C21 CWWZUAXJZKSULT-UHFFFAOYSA-N 0.000 description 27
- 229910052681 coesite Inorganic materials 0.000 description 27
- 229910052906 cristobalite Inorganic materials 0.000 description 27
- 238000007865 diluting Methods 0.000 description 27
- 239000000377 silicon dioxide Substances 0.000 description 27
- 229910052682 stishovite Inorganic materials 0.000 description 27
- 229910052905 tridymite Inorganic materials 0.000 description 27
- 239000003921 oil Substances 0.000 description 23
- 235000019198 oils Nutrition 0.000 description 23
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 22
- 239000006196 drop Substances 0.000 description 22
- 239000003153 chemical reaction reagent Substances 0.000 description 21
- 238000001914 filtration Methods 0.000 description 21
- 238000000746 purification Methods 0.000 description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 239000002253 acid Substances 0.000 description 18
- 239000010779 crude oil Substances 0.000 description 17
- 201000010099 disease Diseases 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 238000001035 drying Methods 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 238000005406 washing Methods 0.000 description 13
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 230000002776 aggregation Effects 0.000 description 12
- 238000004220 aggregation Methods 0.000 description 12
- FCNOMZNLCYJVNC-UHFFFAOYSA-N 6-bromo-4-chloro-2,1,3-benzoxadiazole Chemical compound ClC1=CC(Br)=CC2=NON=C12 FCNOMZNLCYJVNC-UHFFFAOYSA-N 0.000 description 11
- UOUHGBYZUOXVBE-UHFFFAOYSA-N 6-bromo-n-[(4-fluorophenyl)methyl]-n-methyl-2,1,3-benzoxadiazol-4-amine Chemical compound C=1C(Br)=CC2=NON=C2C=1N(C)CC1=CC=C(F)C=C1 UOUHGBYZUOXVBE-UHFFFAOYSA-N 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- SJGGDZCTGBKBCK-UHFFFAOYSA-N 3-acetylphenylboronic acid Chemical compound CC(=O)C1=CC=CC(B(O)O)=C1 SJGGDZCTGBKBCK-UHFFFAOYSA-N 0.000 description 10
- 239000007821 HATU Substances 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 235000019441 ethanol Nutrition 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- GWCHWDHVHNPCNL-UHFFFAOYSA-N 6-bromo-4-pyrrolidin-1-yl-2,1,3-benzoxadiazole Chemical compound C12=NON=C2C=C(Br)C=C1N1CCCC1 GWCHWDHVHNPCNL-UHFFFAOYSA-N 0.000 description 9
- 102000009091 Amyloidogenic Proteins Human genes 0.000 description 9
- 108010048112 Amyloidogenic Proteins Proteins 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 235000019502 Orange oil Nutrition 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 239000010502 orange oil Substances 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 102000013498 tau Proteins Human genes 0.000 description 8
- 108010026424 tau Proteins Proteins 0.000 description 8
- NJFWAMUFMSQKRO-UHFFFAOYSA-N 1-(6-bromo-2,1,3-benzoxadiazol-4-yl)azetidin-3-ol Chemical compound C1C(O)CN1C1=CC(Br)=CC2=NON=C12 NJFWAMUFMSQKRO-UHFFFAOYSA-N 0.000 description 7
- DQTDHCJLAKUOSV-UHFFFAOYSA-N 4-(azetidin-1-yl)-6-bromo-2,1,3-benzoxadiazole Chemical compound C12=NON=C2C=C(Br)C=C1N1CCC1 DQTDHCJLAKUOSV-UHFFFAOYSA-N 0.000 description 7
- 239000002552 dosage form Substances 0.000 description 7
- 239000002502 liposome Substances 0.000 description 7
- 231100000252 nontoxic Toxicity 0.000 description 7
- 230000003000 nontoxic effect Effects 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 229910052717 sulfur Inorganic materials 0.000 description 7
- DBVFWZMQJQMJCB-UHFFFAOYSA-N 3-boronobenzoic acid Chemical compound OB(O)C1=CC=CC(C(O)=O)=C1 DBVFWZMQJQMJCB-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- 235000019270 ammonium chloride Nutrition 0.000 description 6
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000010438 heat treatment Methods 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 239000000651 prodrug Substances 0.000 description 6
- 229940002612 prodrug Drugs 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 150000003509 tertiary alcohols Chemical class 0.000 description 6
- UPYXZDBCRHUYNF-UHFFFAOYSA-N 6-bromo-4-(1,3-dihydroisoindol-2-yl)-2,1,3-benzoxadiazole Chemical compound C1C2=CC=CC=C2CN1C1=CC(Br)=CC2=NON=C21 UPYXZDBCRHUYNF-UHFFFAOYSA-N 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 208000035475 disorder Diseases 0.000 description 5
- 150000002576 ketones Chemical class 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 239000012279 sodium borohydride Substances 0.000 description 5
- 229910000033 sodium borohydride Inorganic materials 0.000 description 5
- 239000011593 sulfur Substances 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 4
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- 208000023105 Huntington disease Diseases 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- WHLAXDUXKMECTM-UHFFFAOYSA-N oxadiazol-4-amine Chemical compound NC1=CON=N1 WHLAXDUXKMECTM-UHFFFAOYSA-N 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 150000003138 primary alcohols Chemical class 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 239000001993 wax Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 3
- YGZJTYCCONJJGZ-UHFFFAOYSA-N (3,5-dimethoxyphenyl)methanamine Chemical compound COC1=CC(CN)=CC(OC)=C1 YGZJTYCCONJJGZ-UHFFFAOYSA-N 0.000 description 3
- SZJIQLSCDIEJFC-UHFFFAOYSA-N 1-(4-fluorophenyl)-n-methylmethanamine Chemical compound CNCC1=CC=C(F)C=C1 SZJIQLSCDIEJFC-UHFFFAOYSA-N 0.000 description 3
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 3
- BZPGUFQHXWFCJF-UHFFFAOYSA-N 1-(6-bromo-2,1,3-benzoxadiazol-4-yl)azetidine-3-carboxylic acid Chemical compound C1C(C(=O)O)CN1C1=CC(Br)=CC2=NON=C12 BZPGUFQHXWFCJF-UHFFFAOYSA-N 0.000 description 3
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 3
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 3
- 241000416162 Astragalus gummifer Species 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 208000012902 Nervous system disease Diseases 0.000 description 3
- 208000024777 Prion disease Diseases 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 229920001615 Tragacanth Polymers 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 230000006933 amyloid-beta aggregation Effects 0.000 description 3
- UQUPQEUNHVVNKW-UHFFFAOYSA-N azetidin-1-ium-3-ol;chloride Chemical compound Cl.OC1CNC1 UQUPQEUNHVVNKW-UHFFFAOYSA-N 0.000 description 3
- HGQULGDOROIPJN-UHFFFAOYSA-N azetidin-1-ium;chloride Chemical compound Cl.C1CNC1 HGQULGDOROIPJN-UHFFFAOYSA-N 0.000 description 3
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 125000004494 ethyl ester group Chemical group 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- RIKMMFOAQPJVMX-UHFFFAOYSA-N fomepizole Chemical compound CC=1C=NNC=1 RIKMMFOAQPJVMX-UHFFFAOYSA-N 0.000 description 3
- 229960004285 fomepizole Drugs 0.000 description 3
- 229940093915 gynecological organic acid Drugs 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- CTZGPJRMFPQATC-UHFFFAOYSA-N n-benzyl-6-bromo-2,1,3-benzoxadiazol-4-amine Chemical compound C12=NON=C2C=C(Br)C=C1NCC1=CC=CC=C1 CTZGPJRMFPQATC-UHFFFAOYSA-N 0.000 description 3
- MCSAQVGDZLPTBS-UHFFFAOYSA-N n-methyl-1-pyridin-3-ylmethanamine Chemical compound CNCC1=CC=CN=C1 MCSAQVGDZLPTBS-UHFFFAOYSA-N 0.000 description 3
- XHFGWHUWQXTGAT-UHFFFAOYSA-N n-methylpropan-2-amine Chemical compound CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 238000006384 oligomerization reaction Methods 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 239000006201 parenteral dosage form Substances 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 229960004063 propylene glycol Drugs 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- 235000019345 sodium thiosulphate Nutrition 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 235000010487 tragacanth Nutrition 0.000 description 3
- 239000000196 tragacanth Substances 0.000 description 3
- 229940116362 tragacanth Drugs 0.000 description 3
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical class OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- DEQOVKFWRPOPQP-UHFFFAOYSA-N (5-formylthiophen-2-yl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)S1 DEQOVKFWRPOPQP-UHFFFAOYSA-N 0.000 description 2
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 2
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 2
- KOHBEDRJXKOYHL-UHFFFAOYSA-N 2-methoxy-n-methylethanamine Chemical compound CNCCOC KOHBEDRJXKOYHL-UHFFFAOYSA-N 0.000 description 2
- GRFNBEZIAWKNCO-UHFFFAOYSA-N 3-pyridinol Chemical compound OC1=CC=CN=C1 GRFNBEZIAWKNCO-UHFFFAOYSA-N 0.000 description 2
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N 4-methylimidazole Chemical compound CC1=CNC=N1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 2
- STWODXDTKGTVCJ-UHFFFAOYSA-N 4-pyrrolidin-1-ylpiperidine Chemical compound C1CCCN1C1CCNCC1 STWODXDTKGTVCJ-UHFFFAOYSA-N 0.000 description 2
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 2
- 241000349731 Afzelia bipindensis Species 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 2
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- POMVPZZXIWSJFU-UHFFFAOYSA-N [1-(6-bromo-2,1,3-benzoxadiazol-4-yl)azetidin-3-yl]methanol Chemical compound C1C(CO)CN1C1=CC(Br)=CC2=NON=C12 POMVPZZXIWSJFU-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 150000001499 aryl bromides Chemical class 0.000 description 2
- 150000001503 aryl iodides Chemical class 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- GFZWHAAOIVMHOI-UHFFFAOYSA-N azetidine-3-carboxylic acid Chemical compound OC(=O)C1CNC1 GFZWHAAOIVMHOI-UHFFFAOYSA-N 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- DUJGQVVONTYHLT-RGMNGODLSA-N ethyl (2s)-pyrrolidine-2-carboxylate;hydrochloride Chemical compound Cl.CCOC(=O)[C@@H]1CCCN1 DUJGQVVONTYHLT-RGMNGODLSA-N 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 229940093471 ethyl oleate Drugs 0.000 description 2
- DUJGQVVONTYHLT-UHFFFAOYSA-N ethyl pyrrolidine-2-carboxylate;hydrochloride Chemical compound Cl.CCOC(=O)C1CCCN1 DUJGQVVONTYHLT-UHFFFAOYSA-N 0.000 description 2
- LIWAQLJGPBVORC-UHFFFAOYSA-N ethylmethylamine Chemical compound CCNC LIWAQLJGPBVORC-UHFFFAOYSA-N 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical compound C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 2
- IHLVCKWPAMTVTG-UHFFFAOYSA-N lithium;carbanide Chemical compound [Li+].[CH3-] IHLVCKWPAMTVTG-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 239000002184 metal Chemical class 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- SIGOIUCRXKUEIG-UHFFFAOYSA-N methyl 2-dimethoxyphosphorylacetate Chemical compound COC(=O)CP(=O)(OC)OC SIGOIUCRXKUEIG-UHFFFAOYSA-N 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- MNHGOWYUJYMPGV-UHFFFAOYSA-N n-methyl-1-(1,3-thiazol-2-yl)methanamine Chemical compound CNCC1=NC=CS1 MNHGOWYUJYMPGV-UHFFFAOYSA-N 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 239000002304 perfume Substances 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 230000003285 pharmacodynamic effect Effects 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 235000015320 potassium carbonate Nutrition 0.000 description 2
- LJCNRYVRMXRIQR-OLXYHTOASA-L potassium sodium L-tartrate Chemical compound [Na+].[K+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O LJCNRYVRMXRIQR-OLXYHTOASA-L 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical compound OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- QPMSJEFZULFYTB-UHFFFAOYSA-N pyrrolidin-1-ium-3-ol;chloride Chemical compound Cl.OC1CCNC1 QPMSJEFZULFYTB-UHFFFAOYSA-N 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 150000003333 secondary alcohols Chemical class 0.000 description 2
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 235000011006 sodium potassium tartrate Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 239000003206 sterilizing agent Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 2
- 125000005309 thioalkoxy group Chemical group 0.000 description 2
- JADVWWSKYZXRGX-UHFFFAOYSA-M thioflavine T Chemical compound [Cl-].C1=CC(N(C)C)=CC=C1C1=[N+](C)C2=CC=C(C)C=C2S1 JADVWWSKYZXRGX-UHFFFAOYSA-M 0.000 description 2
- 125000005505 thiomorpholino group Chemical group 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 235000019798 tripotassium phosphate Nutrition 0.000 description 2
- 238000003828 vacuum filtration Methods 0.000 description 2
- 208000027121 wild type ATTR amyloidosis Diseases 0.000 description 2
- DGUWACLYDSWXRZ-UHFFFAOYSA-N (2-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=CC=C1C=O DGUWACLYDSWXRZ-UHFFFAOYSA-N 0.000 description 1
- AFSSVCNPDKKSRR-UHFFFAOYSA-N (3-bromophenyl)boronic acid Chemical compound OB(O)C1=CC=CC(Br)=C1 AFSSVCNPDKKSRR-UHFFFAOYSA-N 0.000 description 1
- VXWBQOJISHAKKM-UHFFFAOYSA-N (4-formylphenyl)boronic acid Chemical compound OB(O)C1=CC=C(C=O)C=C1 VXWBQOJISHAKKM-UHFFFAOYSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- FHUDAMLDXFJHJE-UHFFFAOYSA-N 1,1,1-trifluoropropan-2-one Chemical compound CC(=O)C(F)(F)F FHUDAMLDXFJHJE-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- ORFDPMMQDVBIMD-UHFFFAOYSA-N 1-(1H-pyrazol-5-yl)-4-pyrrolidin-1-ylpiperidine Chemical compound C1CCN(C1)C1CCN(CC1)C1=NNC=C1 ORFDPMMQDVBIMD-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- KWHWEKRYATWZFW-UHFFFAOYSA-N 1-pyrrolidin-1-ylpropan-1-amine Chemical compound CCC(N)N1CCCC1 KWHWEKRYATWZFW-UHFFFAOYSA-N 0.000 description 1
- ZPOROQKDAPEMOL-UHFFFAOYSA-N 1h-pyrrol-3-ol Chemical compound OC=1C=CNC=1 ZPOROQKDAPEMOL-UHFFFAOYSA-N 0.000 description 1
- SFFUEHODRAXXIA-UHFFFAOYSA-N 2,2,2-trifluoroacetonitrile Chemical compound FC(F)(F)C#N SFFUEHODRAXXIA-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003660 2,3-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- RGHPCLZJAFCTIK-UHFFFAOYSA-N 2-methylpyrrolidine Chemical compound CC1CCCN1 RGHPCLZJAFCTIK-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- MCSXGCZMEPXKIW-UHFFFAOYSA-N 3-hydroxy-4-[(4-methyl-2-nitrophenyl)diazenyl]-N-(3-nitrophenyl)naphthalene-2-carboxamide Chemical class Cc1ccc(N=Nc2c(O)c(cc3ccccc23)C(=O)Nc2cccc(c2)[N+]([O-])=O)c(c1)[N+]([O-])=O MCSXGCZMEPXKIW-UHFFFAOYSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- ROADCYAOHVSOLQ-UHFFFAOYSA-N 3-oxetanone Chemical compound O=C1COC1 ROADCYAOHVSOLQ-UHFFFAOYSA-N 0.000 description 1
- ZOXVJCUXZNPIGB-UHFFFAOYSA-N 4-(1-methylpiperazin-2-yl)morpholine Chemical compound CN1CCNCC1N1CCOCC1 ZOXVJCUXZNPIGB-UHFFFAOYSA-N 0.000 description 1
- 125000004176 4-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1F)C([H])([H])* 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 208000037259 Amyloid Plaque Diseases 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 230000007134 Aβ oligomerisation Effects 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- JKAKOMNYEHRYON-UHFFFAOYSA-N Cc1cnc([nH]1)N1CCOCC1 Chemical compound Cc1cnc([nH]1)N1CCOCC1 JKAKOMNYEHRYON-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000020406 Creutzfeldt Jacob disease Diseases 0.000 description 1
- 208000003407 Creutzfeldt-Jakob Syndrome Diseases 0.000 description 1
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- YIIMEMSDCNDGTB-UHFFFAOYSA-N Dimethylcarbamoyl chloride Chemical compound CN(C)C(Cl)=O YIIMEMSDCNDGTB-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 102000016252 Huntingtin Human genes 0.000 description 1
- 108050004784 Huntingtin Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 1
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 150000007945 N-acyl ureas Chemical class 0.000 description 1
- OHLUUHNLEMFGTQ-UHFFFAOYSA-N N-methylacetamide Chemical compound CNC(C)=O OHLUUHNLEMFGTQ-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 108010071690 Prealbumin Proteins 0.000 description 1
- 102000029797 Prion Human genes 0.000 description 1
- 108091000054 Prion Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- 244000000231 Sesamum indicum Species 0.000 description 1
- 235000003434 Sesamum indicum Nutrition 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102000009190 Transthyretin Human genes 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 230000006229 amino acid addition Effects 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- JXLHNMVSKXFWAO-UHFFFAOYSA-N azane;7-fluoro-2,1,3-benzoxadiazole-4-sulfonic acid Chemical class N.OS(=O)(=O)C1=CC=C(F)C2=NON=C12 JXLHNMVSKXFWAO-UHFFFAOYSA-N 0.000 description 1
- GMWFCJXSQQHBPI-UHFFFAOYSA-N azetidin-3-ol Chemical compound OC1CNC1 GMWFCJXSQQHBPI-UHFFFAOYSA-N 0.000 description 1
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000007910 chewable tablet Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 150000005827 chlorofluoro hydrocarbons Chemical class 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- IQFVPQOLBLOTPF-HKXUKFGYSA-L congo red Chemical compound [Na+].[Na+].C1=CC=CC2=C(N)C(/N=N/C3=CC=C(C=C3)C3=CC=C(C=C3)/N=N/C3=C(C4=CC=CC=C4C(=C3)S([O-])(=O)=O)N)=CC(S([O-])(=O)=O)=C21 IQFVPQOLBLOTPF-HKXUKFGYSA-L 0.000 description 1
- 239000000562 conjugate Chemical class 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- QPJDMGCKMHUXFD-UHFFFAOYSA-N cyanogen chloride Chemical compound ClC#N QPJDMGCKMHUXFD-UHFFFAOYSA-N 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Substances CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 1
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 1
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- QPNJHVDIRZNKOX-LURJTMIESA-N ethyl (2s)-pyrrolidine-2-carboxylate Chemical compound CCOC(=O)[C@@H]1CCCN1 QPNJHVDIRZNKOX-LURJTMIESA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 238000010575 fractional recrystallization Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000008274 jelly Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- IMAKHNTVDGLIRY-UHFFFAOYSA-N methyl prop-2-ynoate Chemical compound COC(=O)C#C IMAKHNTVDGLIRY-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- TUCUVZHDJRMHQX-UHFFFAOYSA-N n,4-dimethylpentan-1-amine Chemical compound CNCCCC(C)C TUCUVZHDJRMHQX-UHFFFAOYSA-N 0.000 description 1
- YFJAIURZMRJPDB-UHFFFAOYSA-N n,n-dimethylpiperidin-4-amine Chemical compound CN(C)C1CCNCC1 YFJAIURZMRJPDB-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- RIWRFSMVIUAEBX-UHFFFAOYSA-N n-methyl-1-phenylmethanamine Chemical compound CNCC1=CC=CC=C1 RIWRFSMVIUAEBX-UHFFFAOYSA-N 0.000 description 1
- HQFYIDOMCULPIW-UHFFFAOYSA-N n-methylprop-2-yn-1-amine Chemical compound CNCC#C HQFYIDOMCULPIW-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000037125 natural defense Effects 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- KSVFXAZDBKITEF-UHFFFAOYSA-N phenyl(piperazin-1-yl)methanone;hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(=O)N1CCNCC1 KSVFXAZDBKITEF-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 125000005544 phthalimido group Chemical group 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 230000004845 protein aggregation Effects 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N pyridin-3-ylboronic acid Chemical compound OB(O)C1=CC=CN=C1 ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 150000003235 pyrrolidines Chemical class 0.000 description 1
- 125000002112 pyrrolidino group Chemical group [*]N1C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 239000001476 sodium potassium tartrate Substances 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 230000009967 tasteless effect Effects 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000006211 transdermal dosage form Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 235000016804 zinc Nutrition 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/12—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/08—1,2,5-Oxadiazoles; Hydrogenated 1,2,5-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Psychology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Un compuesto de fórmula I:**Fórmula** o una de sus sales farmacéuticamente aceptables, en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan- 1-amino, N1,N1,N3 trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1-ilo, 3- (pirrolidin-1-ilmetanon)azetidin-1-ilo, 3-(morfolina)-1-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3- (morfolin-4-il)pirrolidin-1-1-ilo, 3-(morfolin-4-ilmetil)pirrolidin-1-ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-1- ilo, 4-(morfolin-4-il)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2- il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo, y 3-(metilamino-Nmetil) azetidin-1-ilo; R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más sustituyentes A, oxadiazol opcionalmente sustituido con uno o más sustituyentes A, tiofeno opcionalmente sustituido con uno o más sustituyentes A, tiazol opcionalmente sustituido con uno o más sustituyentes A, furano opcionalmente sustituido con uno o más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más de los sustituyentes A, Nmetilpirazol opcionalmente sustituido con uno o más sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con uno o más alquilo, - CH2OH, 1-ol-2,2,2-trifluoroetan-1-ilo, 2-ol-propan-2-ilo, y ciclopropilo; y R12 y R14 son cada uno independientemente hidrógeno o alquilo, con la condición de que si A es 1-ol-2,2,2- trifluoroetan-1-ilo, entonces no puede conectarse a tiofeno.
Description
DESCRIPCIÓN
Compuestos antiamiloides que contienen benzofurazano
Antecedentes
La acumulación de proteínas amiloides en el tejido vivo, una afección conocida como amiloidosis, es la causa o un factor importante en la patología de muchas enfermedades llamadas amiloides, como la enfermedad de Parkinson, la enfermedad de Huntington y la enfermedad priónica de Alzheimer, incluidas trastornos no relacionados con el SNC, como la diabetes mellitus tipo 2. Históricamente, las agregaciones de proteínas se clasificaron como amiloides si mostraban birrefringencia verde manzana bajo luz polarizada cuando se teñían con los tintes rojo Congo o Tioflavina T (ThT) (Sipe y Cohen, 2000. J. Struct. Biol. 130: 88-98). Esa definición de amiloide se ha ampliado en los últimos años para aplicarla a cualquier polipéptido que pueda polimerizarse en una conformación de hoja beta cruzada in vitro o in vivo, independientemente de la secuencia (Xu, 2007. Amyloid 14: 119-31). Ciertos tipos de amiloidosis pueden ocurrir principalmente en el sistema nervioso central, como con la agregación de la proteína beta-amiloide en la enfermedad de Alzheimer, la proteína tau en la parálisis supranuclear progresiva, la alfasinucleína en la enfermedad de Parkinson, la proteína huntingtina en la enfermedad de Huntington y la proteína priónica en la enfermedad de Creutzfeldt -Jacob y otras enfermedades priónicas. Otros tipos de amiloidosis son de naturaleza sistémica, como ocurre con la agregación de transtiretina en la amiloidosis sistémica senil.
Todas las enfermedades enumeradas anteriormente son invariablemente fatales usando la práctica médica actual. En ninguna de estas enfermedades hay una terapia o tratamiento conocido y ampliamente aceptado que pueda detener y/o revertir la agregación de depósitos de amiloide. Como tal, sigue habiendo una necesidad urgente de tratamientos.
Resumen
En el presente documento se divulga que los anti-amiloides previenen o reducen la velocidad de oligomerización o agregación de proteínas amiloides. Se espera que este efecto tenga efectos beneficiosos en las enfermedades afectadas por la amiloidosis.
En general, en un aspecto, se proporcionan compuestos de Fórmula I, o una sal farmacéuticamente aceptable de los mismos:

en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1-amino, N1,N1,N3-trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1-ilo, 3-(pirrolidin-1-ilmetanon)azetidin-1-ilo, 3-(morfolina)-1-ilmetil)azetidin-1-ilo, 3-(2 etanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-4-il) pirrolidin-1 -1 -ilo, 3-(morfolin-4-ilmetil)pirrolidin-1-ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolin-4-il)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo; R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más sustituyentes A, oxadiazol opcionalmente sustituido con uno o más sustituyentes A, tiofeno opcionalmente sustituido con uno o más sustituyentes A, tiazol opcionalmente sustituido con uno o más sustituyentes A, furano opcionalmente sustituido con uno o más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más de los sustituyentes A, N metilpirazol opcionalmente sustituido con uno o más sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4 piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con uno o más alquil, -CH2OH, 1-ol-2,2,2-trifluoroetan-1-ilo, 2-ol-propan-2-ilo, y ciclopropilo; y R12 y R14 son cada uno independientemente hidrógeno o alquilo, con la condición de que si A es 1-ol-2,2,2-trifluoroetan-1-ilo, entonces no puede conectarse a tiofeno.
En general, en un aspecto, se proporciona un compuesto de acuerdo con la fórmula anterior o una sal farmacéuticamente aceptable del mismo, en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il) piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1 -amino, N1,N1,N3-trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1 -ilo, 3-(pirrolidin-1-ilmetanon)azetidin-1 -ilo, y 3-(morfolin-4-ilmetil)azetidin-1 -ilo; y R13 se selecciona del grupo que consiste en tiofeno opcionalmente sustituido con uno o más de los sustituyentes A, tiazol opcionalmente sustituido con uno o más de los sustituyentes A, furano opcionalmente sustituido con uno o
más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más de sustituyentes A, N-metilpirazol opcionalmente sustituido con uno o más de los sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alctiolilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con uno o más alquilos, y -CH2OH, con la condición de que si A es 1-ol-2,2,2-trifluoroetan-1-ilo, entonces no puede conectarse al tiofeno.
En general, en un aspecto, se proporciona un compuesto de acuerdo con la fórmula anterior o una sal farmacéuticamente aceptable del mismo, en el que R11 se selecciona del grupo que consiste en 3-(morfolin-4-ilmetil)azetidin-1-ilo, 3-(2-metanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-4-il)pirrolidin-1 -ilo, 3-(morfolin-4-ilmetil)pirrolidina-1-ilo, 4-(etilamido)piperidin-1 -ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolin-4-ilo)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1 -ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo; y R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más de los sustituyentes A, oxadiazol opcionalmente sustituido con uno o más de los sustituyentes A, y 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en 1-ol-2,2,2-trifluoroetan-1-ilo, oxetan-3-ol-3-ilo, 2-ol-propan-2-ilo y ciclopropilo.
En general, en un aspecto, se proporciona un compuesto de acuerdo con la fórmula anterior o una sal farmacéuticamente aceptable del mismo, en el que R13 se selecciona del grupo que consiste en 5-(2-ol-propan-2-il) oxadizol-3-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-5-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)piridina-2-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il) piridin-3-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)tiazol-2-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)piridin-6-ilo, 2-(1-ol-2,2,2-trifluoroetan-1-il)-piridin-6-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-2-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-3-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)tiofen-2-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)tiofen-3-ilo, 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-5-ilo, 3-(1-ol-2,2,2-trifluoroetan-1-il)piridin-2-ilo, 3-(1-ol-2,2,2-trifluoroetan-1-il)piridin-6-ilo, y 2-(1-ol-2,2,2-trifluoroetan-1-il) piridin-3-ilo.
En general, en un aspecto, se proporciona un compuesto de acuerdo con la fórmula anterior o una sal farmacéuticamente aceptable del mismo, en el que R11 es 3-(morfolin-1-ilmetil)azetidin-1 -ilo y R13 se selecciona de la grupo que consiste en 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo y 5-(2-ol-propan-2-il)oxadizol-3-ilo.
En general, en un aspecto, se proporcionan métodos útiles en el tratamiento de la amiloidosis. Los métodos incluyen administrar a un sujeto un compuesto terapéutico de la presente invención que inhibe la agregación u oligomerización de amiloide. La amiloidosis puede ser la enfermedad de Alzheimer, la parálisis supranuclear progresiva, la demencia frontotemporal, la enfermedad de Parkinson, la enfermedad de Huntington, la enfermedad por priones, la amiloidosis sistémica senil, la diabetes mellitus tipo 2 o alguna otra amiloidosis sistémica o del sistema nervioso central.
En general, en un aspecto, se proporcionan composiciones farmacéuticas para tratar la amiloidosis. Las composiciones farmacéuticas incluyen un compuesto terapéutico de la presente invención en una cantidad eficaz para inhibir la agregación de amiloides, y un excipiente o vehículo farmacéuticamente aceptable. En algunas implementaciones, la cantidad efectiva está entre aproximadamente 0.0003 y aproximadamente 50 mg/kg de peso corporal de la persona que lo necesita.
De acuerdo con lo anterior, la presente invención también está dirigida a sales, estereoisómeros, polimorfos, metabolitos, análogos y profármacos farmacéuticamente aceptables de los compuestos, y a cualquier combinación de los mismos.
Con las anteriores y otras ventajas y características de la invención que se harán evidentes a continuación, la naturaleza de la invención puede entenderse más claramente por referencia a la siguiente descripción detallada de la invención y las reivindicaciones adjuntas.
Breve descripción de los dibujos
La fig. 1 muestra un ensayo de agregación de beta-amiloide para un compuesto descrito en el presente documento.
La fig. 2 muestra un ensayo de agregación de beta-amiloide para un compuesto descrito en el presente documento.
La fig. 3 muestra un ensayo de agregación tau para un compuesto descrito en el presente documento.
La fig. 4 muestra un ensayo de agregación tau para un compuesto descrito en el presente documento.
La fig. 5 muestra un ensayo de agregación tau para un compuesto descrito en el presente documento.
La fig. 6 muestra un ensayo de agregación tau para un compuesto descrito en el presente documento.
La fig. 7 muestra un ensayo de agregación tau para un compuesto descrito en el presente documento.
La fig. 8 muestra un ensayo de oligomerización de beta-amiloide biotinilado para un compuesto descrito en el presente documento.
Descripción
En una realización, se proporcionan compuestos de Fórmula I, o una sal farmacéuticamente aceptable de los mismos:

en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1-amino, N1,N1,N3-trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1-ilo, 3-(pirrolidin-1-ilmetanon)azetidin-1-ilo, 3-(morfolin-4-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3 (morfolin-4-il)pirrolidin-1-ilo, 3-(morfolin-1 -ilmetil)pirrolidin-1-ilo, 4-(etilamido)piperidin-1 -ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolin-4-il)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidina-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo y 3-(metilamino-N metil)azetidin-1-ilo; R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más sustituyentes A, oxadiazol opcionalmente sustituido con uno o más sustituyentes A, tiofeno opcionalmente sustituido con uno o más sustituyentes A, tiazol opcionalmente sustituido con uno o más sustituyentes A, furano opcionalmente sustituido con uno o más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más de los sustituyentes A, N metilpirazol opcionalmente sustituido con uno o más de los sustituyentes A, 2-piridina opcionalmente sustituido con uno o más sustituyentes A, 3-piridina opcionalmente sustituida con uno o más de los sustituyentes A, y 4 piridina opcionalmente sustituida con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltiolilo, isopropilo, t-butilo, trifluorometilo, cloro ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con on E o más alquilo, -CH2OH, 1-ol-2,2,2-trifluoroetan-1-ilo, oxetan-3-ol-3-ilo, 2-ol propan-2-ilo y ciclopropilo; y R12 y R14 son cada uno independientemente hidrógeno o alquilo, con la condición de que si A es 1-ol-2,2,2-trifluoroetan 1 -ilo, entonces no puede conectarse a tiofeno.
En una realización, se proporcionan compuestos de Fórmula I, o una sal farmacéuticamente aceptable de los mismos:

en la que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1-amino, N1,N1N3-trimetilpropano-1.3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1 -ilo, 3 (pirrolidin-1-ilmetanon)azetidin-1-ilo, y 3-(morfolin-4-ilmetil)azetidin-1-ilo; R13 se selecciona del grupo que consiste en tiofeno opcionalmente sustituido con uno o más sustituyentes A, tiazol opcionalmente sustituido con uno o más sustituyentes A, furano opcionalmente sustituido con uno o más sustituyentes A, pirazol opcionalmente sustituido con uno o más sustituyentes A, N-metilpirazol opcionalmente sustituido con uno o más de los sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4 piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con uno o más alquilo, y -CH2OH; y R12 y R14 son cada uno independientemente hidrógeno o alquilo, con la condición de que si A es 1 -ol-2,2,2-trifluoroetan-1 -ilo, entonces no puede conectarse al tiofeno.
En una realización, se proporcionan compuestos de Fórmula I, o una sal farmacéuticamente aceptable de los mismos:

en el que R11 se selecciona del grupo que consiste en 3-(morfolin-4-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-1-ilo)pirrolidin-1-ilo, 3-(morfolin-4-ilmetil)pirrolidin-1-ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-l-ilo, 4-(morfolin-4-il)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin-1-ilo, 3-amidoazetidin1-ilo, 3 (propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo; R12 y R14 son cada uno independientemente hidrógeno o alquilo; y R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más de los sustituyentes A, oxadiazol opcionalmente sustituido con uno o más de los sustituyentes A, y 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en 1-ol-2,2,2-trifluoroetan-1-ilo, oxetan-3-ol 3-ilo, 2-ol-propan-2-ilo y ciclopropilo.
En una realización, se proporcionan compuestos de Fórmula I, o una de sus sales farmacéuticamente aceptables:

en el que R11 se selecciona del grupo que consiste en 3-(morfolin-4-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-4-il)pirrolidin-1 -ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolinil) piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidina-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo; R12 y R14 son cada uno independientemente hidrógeno o alquilo; y R13 se selecciona del grupo que consiste en 5-(2-ol-propan 2-il)oxadizol-3-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-5-ilo, 4-(1-ol-2,2,2-trifluoroetano-1-il)piridin2-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)tiazol-2-ilo, 4-(1-ol-2.2.2 trifluoroetan-1 -il)piridin-6-ilo, 2-(1-ol-2,2,2-trifluoroetan-1-il)-piridin-6-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-2-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-3-ilo, 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-5-ilo, 3-(1-ol-2,2,2-trifluoroetan-1-il)piridin-2-ilo, 3-(1-ol-2.2.2 trifluoroetan-1-il)piridin-6-ilo, y 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo.
En una realización, se proporcionan compuestos de Fórmula I, o una de sus sales farmacéuticamente aceptables:

en la que R11 es 3-(morfolin-4-ilmetil)azetidin-1-ilo; R12 y R14 son cada uno independientemente hidrógeno o alquilo; y R13 se selecciona del grupo que consiste en 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo y 5-(2-ol-propan-2-il)oxadizol-3 -ilo.
En una realización, se divulga un método de tratamiento de una enfermedad amiloide en un sujeto, que comprende administrar una cantidad terapéuticamente eficaz de un compuesto de la presente invención al sujeto. En algunas realizaciones, la enfermedad amiloide es la enfermedad de Alzheimer. En algunas realizaciones, la enfermedad amiloide es la enfermedad de Parkinson. En algunas realizaciones, la enfermedad amiloide es la enfermedad de Huntington. En algunas realizaciones, la enfermedad amiloide es la demencia frontotemporal. En algunas realizaciones, la enfermedad amiloide es una parálisis supranuclear progresiva. En algunas realizaciones, la enfermedad amiloide es diabetes mellitus tipo 2.
13. Un método para tratar una afección que es un miembro seleccionado de pérdida de memoria, pérdida de cognición y una combinación de las mismas, dicho método comprende administrar a un sujeto que lo necesite una cantidad terapéuticamente eficaz de un compuesto de cualquiera de las reivindicaciones 1-5.
En una realización, se proporciona una composición farmacéutica que comprende un compuesto de la presente invención y un portador farmacéuticamente aceptable.
En una realización, un método para tratar una afección que es un miembro seleccionado de pérdida de memoria, pérdida de cognición y una combinación de los mismos, dicho método comprende administrar a un sujeto que lo necesite una cantidad terapéuticamente eficaz de un compuesto divulgado en el presente documento. En algunas realizaciones, dicha condición está asociada con la enfermedad de Alzheimer. En algunas realizaciones, dicha condición está asociada con parálisis supranuclear progresiva. En algunas realizaciones, la cantidad terapéuticamente eficaz es una dosis diaria total de aproximadamente 0.0003 a aproximadamente 50 mg/kg de peso corporal del sujeto.
Se considera que los compuestos de la presente invención inhiben la agregación de la proteína amiloide. Los datos que respaldan esta conclusión se pueden encontrar en los ejemplos a continuación.
Definiciones
A menos que se defina lo contrario, los términos utilizados en la especificación se refieren a las siguientes definiciones, como se detalla a continuación.
Se debe entender que los términos “administración” o “administrar” compuestos significan proporcionar un compuesto de la presente invención a un individuo en una forma que puede introducirse en el cuerpo de ese individuo en una cantidad efectiva para la profilaxis, el tratamiento o el diagnóstico, según corresponda. Tales formas pueden incluir, por ejemplo, formas de dosificación oral, formas de dosificación inyectables, formas de dosificación transdérmica, formas de dosificación por inhalación y formas de dosificación rectal.
El término “alcoxi” como se usa en el presente documento significa un grupo alquilo, como se define en el presente documento, unido al resto molecular parental a través de un átomo de oxígeno. Los ejemplos representativos de alcoxi incluyen, pero no se limitan a, metoxi, etoxi, propoxi, 2-propoxi, butoxi, tert-butoxi, pentiloxi y hexiloxi. El término “alctiolilo” como se usa en este documento significa el grupo análogo que contiene azufre en lugar de oxígeno.
El término “alquilo” como se usa en este documento significa un hidrocarburo de cadena lineal o ramificada que contiene de 1 a 20 átomos de carbono, preferiblemente de 1 a 10 átomos de carbono, más preferiblemente de 1, 2, 3, 4, 5 o 6 carbonos. Ejemplos representativos de alquilo incluyen, pero no se limitan a, metilo, etilo, n-propilo, isopropilo, n-butilo, sec-butilo, iso-butilo, tert-butilo, n-pentilo, isopentilo, neopentilo, n-hexilo, 3-metilhexilo, 2,2-dimetilpentilo, 2,3-dimetilpentilo, n-heptilo, n-octilo, n-nonilo y n-decilo.
El término “carbonilo”, como se usa en este documento, significa un grupo -C(=O)-.
El término “carboxi”, como se usa en este documento, significa un grupo -COOH, que puede estar protegido como un grupo éster: -COO-alquilo.
El término “flúor” como se usa en el presente documento significa -F.
El término “halo” o “halógeno” como se usa en el presente documento significa Cl, Br, I o F.
El término “heteroarilo”, como se usa en el presente documento, se refiere a un anillo aromático que contiene uno o más heteroátomos seleccionados independientemente de nitrógeno, oxígeno o azufre, o un tautómero de los mismos. Dichos anillos pueden ser monocíclicos o bicíclicos como se describe adicionalmente en el presente documento. Los anillos de heteroarilo están conectados a una fracción molecular parental a través de un átomo de carbono o nitrógeno.
Los términos “heteroarilo” o “anillo de heteroarilo de 5 o 6 miembros”, como se usan en este documento, se refieren a anillos aromáticos de 5 o 6 miembros que contienen 1, 2, 3 o 4 heteroátomos seleccionados independientemente de nitrógeno, oxígeno, o azufre, o un tautómero del mismo. Los ejemplos de dichos anillos incluyen, pero no se limitan a, un anillo en el que un carbono se reemplaza con un O o un átomo; uno, dos o tres átomos de N dispuestos de manera adecuada para proporcionar un anillo aromático; o un anillo en el que dos átomos de carbono en el anillo se reemplazan con un átomo de O o S y un átomo de N. Dichos anillos pueden incluir, pero no se limitan a, un anillo aromático de seis miembros en el que uno a cuatro de los átomos de carbono del anillo están reemplazados por átomos de nitrógeno, anillos de cinco miembros que contienen un azufre, oxígeno o nitrógeno en el anillo; anillos de cinco miembros que contienen de uno a cuatro átomos de nitrógeno; y anillos de cinco miembros que contienen un oxígeno o azufre y uno a tres átomos de nitrógeno. Los ejemplos representativos de los anillos de heteroarilo de 5 a 6 miembros incluyen, pero no se limitan a, furilo, imidazolilo, isoxazolilo, isotiazolilo, oxazolilo, pirazinilo, pirazolilo, piridazinilo, piridinilo, pirimidinilo, pirrolilo, tetrazolilo, [1,2,3]tiadiazolilo, [1,2,3]oxadiazolilo, tiazolilo, tienilo, [1,2,3]triazinilo, [1,2,4]triazinilo, [1,3,5]triazinilo, [1,2,3]triazolilo y [1,2,4]triazolilo.
Los grupos heteroarilo de la invención pueden estar sustituidos con hidrógeno o alquilo. Los anillos heteroarilo monocíclico o heteroarilo de 5 o 6 miembros están sustituidos con 0, 1, 2, 3, 4 o 5 sustituyentes. Los grupos heteroarilo de la presente invención pueden estar presentes como tautómeros.
El término “hidroxi” como se usa en este documento significa un grupo -OH.
A menos que se indique lo contrario, el término “profármaco” abarca ésteres, carbonatos, tiocarbonatos, derivados N-acilo, derivados N-aciloxialquilo farmacéuticamente aceptables, derivados cuaternarios de aminas terciarias, bases de N-Mannich, bases de Schiff, conjugados de aminoácidos, ésteres de fosfato, sales metálicas y ésteres de sulfonato de compuestos divulgados en el presente documento. Los ejemplos de profármacos incluyen compuestos que comprenden una fracción biohidrolizable (por ejemplo, una amida biohidrolizable, carbamato biohidrolizable, carbonato biohidrolizable, éster biohidrolizable, fosfato biohidrolizable o análogo de ureido biohidrolizable). Los profármacos de los compuestos descritos en el presente documento se prevén y preparan fácilmente por los expertos en la técnica. Véase, por ejemplo, Design of Prodrugs , Bundgaard, A. Ed., Elseview, 1985; Bundgaard, hours., "Design and Application of Prodrugs," A Textbook of Drug Design and Development, Krosgaard-Larsen and hours. Bundgaard, Ed., 1991, Chapter 5, p. 113-191; and Bundgaard, hours., Advanced Drug Delivery Review, 1992, 8, 1-38.
A menos que se indique lo contrario, el término “grupo protector” o “grupo protector”, cuando se usa para referirse a parte de una molécula sometida a una reacción química, significa una fracción química que no es reactivo en las condiciones de esa reacción química, y que puede eliminarse para proporcionar una fracción que es reactiva en esas condiciones. Los grupos protectores son bien conocidos en la técnica. Ver, por ejemplo, Greene, T. W. and Wuts, P.G.M., Protective Groups in Organic Synthesis (3 rd ed., John Wiley & Sons: 1999); Larock, R. C., Comprehensive Organic Transformations (2 nd ed., John Wiley & Sons: 1999). Algunos ejemplos incluyen bencilo, difenilmetilo, tritilo, Cbz, Boc, Fmoc, metoxicarbonilo, etoxicarbonilo y ftalimido. Los grupos protectores incluyen, por ejemplo, grupos protectores de nitrógeno y grupos protectores de hidroxi.
El término “sulfonilo” como se usa en este documento significa un grupo S(O)2-.
El término “tioalcoxi” como se usa en el presente documento significa un grupo alquilo, como se define en el presente documento, unido a la fracción molecular parental a través de un átomo de azufre. Los ejemplos representativos de tioalcoxi incluyen, pero no se limitan a, metiltio, etiltio y propiltio.
Los compuestos de la invención se pueden usar en forma de sales farmacéuticamente aceptables derivadas de ácidos inorgánicos u orgánicos. Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Para mayor claridad, el término “sales farmacéuticamente aceptables” como se usa en el presente documento generalmente se refiere a sales preparadas a partir de ácidos o bases no tóxicos farmacéuticamente aceptables que incluyen ácidos y bases inorgánicos y ácidos y bases orgánicos. Las sales de adición de base farmacéuticamente aceptables adecuadas incluyen sales metálicas hechas de aluminio, calcio, litio, magnesio, potasio, sodio y zinc o sales orgánicas hechas de lisina, N,N'-dibenciletilendiamina, cloroprocaína, colina, dietanolamina, etilendiamina, meglumina (N-metilglucamina) y procaína. Los ácidos no tóxicos adecuados incluyen ácidos inorgánicos y orgánicos tales como acético, algínico, antranílico, bencenosulfónico, benzoico, canforsulfónico, cítrico, etenosulfónico, fórmico, fumárico, furoico, galacturónico, glucónico, glutámico, glicólico, bromhídrico, clorhídrico, isetiónico, láctico, maleico, málico, mandélico, metanosulfónico, múcico, nítrico, pamoico, pantoténico, fenilacético, fosfórico, propiónico, salicílico, esteárico, succínico, sulfanílico, sulfúrico, ácido tartárico y ácido p-toluensulfónico. Los ácidos no tóxicos específicos incluyen los ácidos clorhídrico, bromhídrico, fosfórico, sulfúrico y metanosulfónico. Ejemplos de sales específicas incluyen sales de clorhidrato y mesilato. Otros son bien conocidos en la técnica. Ver, por ejemplo, Los ácidos no tóxicos específicos incluyen los ácidos clorhídrico, bromhídrico, fosfórico, sulfúrico y metanosulfónico. Ejemplos de sales específicas incluyen sales de clorhidrato y mesilato. Otros son bien conocidos en la técnica. Ver, por ejemplo, Remington's Pharmaceutical Sciences, 18a ed. (Mack Publishing, Easton Pa.: 1990) y Remington: The Science and Practice of Pharmacy, 19a ed. (Mack Publishing, Easton Pa.: 1995). La preparación y el uso de sales de adición de ácido, sales de carboxilato, sales de adición de aminoácidos y sales de zwitterión de los compuestos de la presente invención también pueden considerarse farmacéuticamente aceptables si están, dentro del alcance de un buen juicio médico, adecuadas para su uso en contacto con los tejidos de humanos y animales inferiores sin toxicidad indebida, irritación, respuesta alérgica y similares, son proporcionales con una relación razonable de riesgo/beneficio y son efectivos para el uso previsto. Dichas sales también pueden incluir diversos solvatos e hidratos del compuesto de la presente invención.
Ciertos compuestos de la presente invención pueden estar marcados isotópicamente, por ejemplo, con diversos isótopos de carbono, flúor o yodo, según sea aplicable cuando el compuesto en cuestión contiene al menos uno de dichos átomos. En realizaciones preferidas, los métodos de diagnóstico de la presente invención comprenden la administración de tal compuesto marcado isotópicamente.
Ciertos compuestos de la presente invención pueden existir como estereoisómeros en los que están presentes centros asimétricos o quirales. Estos estereoisómeros son “R” o “S” dependiendo de la configuración de los sustituyentes alrededor del átomo de carbono quiral. Los términos “R” y “S” utilizados aquí son configuraciones tal como se definen en IUPAC 1974 Recommendations for Section E, Fundamental Stereochemistry, in Pure Appl.
Chem., 1976, 45: 13-30. La invención contempla varios estereoisómeros y mezclas de los mismos y estos se incluyen específicamente dentro del alcance de esta invención. Los estereoisómeros incluyen enantiómeros y diastereómeros, y mezclas de enantiómeros o diastereómeros. Los estereoisómeros individuales de los compuestos de la invención se pueden preparar sintéticamente a partir de materiales de partida disponibles comercialmente que contienen centros asimétricos o quirales o mediante la preparación de mezclas racémicas, seguido de una resolución bien conocida por los expertos en la técnica. Estos métodos de resolución se ejemplifican mediante (1) unión de una mezcla de enantiómeros a un auxiliar quiral, separación de la mezcla resultante de diastereoisómeros por recristalización o cromatografía y liberación opcional del producto ópticamente puro del auxiliar como se describe en Furniss, Hannaford, Smith, and Tatchell, "Vogel's Textbook of Practical Organic Chemistry", 5th edition (1989), Longman Scientific & Technical, Essex CM202JE, Inglaterra, o (2) separación directa de la mezcla de enantiómeros ópticos en columnas cromatográficas quirales o (3) métodos de recristalización fraccionada.
Ciertos compuestos de la presente invención pueden existir como isómeros cis o trans, en el que los sustituyentes en un anillo pueden unirse de tal manera que estén en el mismo lado del anillo (cis) uno con relación al otro, o en lados opuestos del anillo en relación entre sí (trans). Dichos métodos son bien conocidos por los expertos en la técnica y pueden incluir la separación de isómeros por recristalización o cromatografía. Debe entenderse que los compuestos de la invención pueden poseer formas tautoméricas, así como isómeros geométricos, y que estos también constituyen un aspecto de la invención.
Debe observarse que una fracción química que forma parte de un compuesto más grande puede describirse en el presente documento usando un nombre que comúnmente se le otorga cuando existe como una molécula única o un nombre comúnmente asignado a su radical. Por ejemplo, los términos “piridina” y “piridilo” tienen el mismo significado cuando se usan para describir una fracción unida a otros restos químicos. Así, por ejemplo, las dos frases “XOH, donde X es piridilo” y “XOH, donde X es piridina” tienen el mismo significado, y abarcan los compuestos piridin-2-ol, piridin-3-ol y piridin-4-ol.
El término “excipiente farmacéuticamente aceptable”, tal como se usa en el presente documento, significa un relleno, diluyente, material de encapsulación o formulación auxiliar no sólido, inerte, sólido, inerte o líquido de cualquier tipo. Algunos ejemplos de materiales que pueden servir como portadores farmacéuticamente aceptables son azúcares tales como lactosa, glucosa y sacarosa; almidones tales como almidón de maíz y almidón de patata; celulosa y sus derivados tales como carboximetilcelulosa sódica, etilcelulosa y acetato de celulosa; tragacanto en polvo; malta; gelatina; talco; manteca de cacao y ceras para supositorios; aceites tales como aceite de cacahuete, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; glicoles; tal propilenglicol; ésteres tales como oleato de etilo y laurato de etilo; agar agentes tamponantes tales como hidróxido de magnesio e hidróxido de aluminio; ácido algínico; agua libre de pirógenos; solución salina isotónica; Solución de Ringer; alcohol etílico y soluciones tampón de fosfato, así como otros lubricantes compatibles no tóxicos, como el laurilsulfato de sodio y el estearato de magnesio, así como los agentes colorantes, agentes liberadores, agentes de recubrimiento, edulcorantes, aromatizantes y perfumantes, conservantes y antioxidantes también pueden estar presente en la composición, de acuerdo con el criterio de un experto en la técnica de formulaciones.
A menos que se indique lo contrario, los términos “previene”, “prevenir” y “prevención” contemplan una acción que se produce antes de que un paciente comience a sufrir una enfermedad o trastorno específico, que inhibe o reduce la gravedad de la enfermedad o trastorno o de uno o más de sus síntomas. Los términos abarcan la profilaxis.
A menos que se indique lo contrario, una “cantidad profilácticamente eficaz” de un compuesto es una cantidad suficiente para prevenir una enfermedad o afección, o uno o más síntomas asociados con la enfermedad o afección, o prevenir su recurrencia. Una cantidad profilácticamente eficaz de un compuesto es una cantidad de agente terapéutico, solo o en combinación con otros agentes, que proporciona un beneficio profiláctico en la prevención de la enfermedad. El término “cantidad profilácticamente eficaz” puede abarcar una cantidad que mejora la profilaxis general o aumenta la eficacia profiláctica de otro agente profiláctico.
A menos que se indique lo contrario, una “cantidad eficaz para el diagnóstico” de un compuesto es una cantidad suficiente para diagnosticar una enfermedad o afección. En general, la administración de un compuesto con fines de diagnóstico no continúa durante el uso terapéutico de un compuesto, y podría administrarse solo una vez si es suficiente para producir el diagnóstico.
A menos que se indique lo contrario, una “cantidad terapéuticamente eficaz” de un compuesto es una cantidad suficiente para tratar una enfermedad o afección, o uno o más síntomas asociados con la enfermedad o afección. La cantidad apropiada depende, entre otras cosas, de la etapa de la enfermedad o afección; la edad del paciente; el peso del paciente; la biodisponibilidad del compuesto con respecto a un tejido objetivo; la concentración de compuesto requerida in vivo para dar como resultado un efecto beneficioso en relación con el control medido por comportamiento, motor, bioquímico, anatómico u otras lecturas; o la concentración de compuesto requerida para dar como resultado un efecto farmacodinámico sobre una proteína amiloide diana en el tejido diana. En algunas realizaciones, la enfermedad es un trastorno neurológico, la proteína amiloide objetivo es la proteína beta-amiloide y el tejido diana es el cerebro. En algunas realizaciones, la enfermedad es un trastorno neurológico, la proteína amiloide diana es la proteína tau y el tejido diana es el cerebro. En algunas realizaciones, las enfermedades son un
trastorno neurológico, tanto la proteína beta amiloide como la proteína tau son proteínas amiloides objetivo y el tejido objetivo es el cerebro. En algunas realizaciones, la enfermedad es un trastorno metabólico como la diabetes tipo 2, la proteína amiloide objetivo es la amilina y el tejido diana es el páncreas. En ausencia de datos clínicos, la concentración de compuesto requerida para producir un efecto farmacodinámico puede estimarse in vitro mediante la correlación de la concentración de compuesto requerida para la reducción de la oligomerización o agregación in vitro frente a la concentración de una proteína amiloide objetivo in vitro con la concentración de la proteína amiloide objetivo que se espera encontrar en el tejido objetivo, de modo que la concentración requerida in vivo sea aproximadamente igual a (concentración de compuesto in vitro * concentración de amiloide in vivo/concentración de amiloide in vitro); a partir de la cual la cantidad terapéuticamente efectiva puede estimarse adicionalmente basándose en información farmacocinética derivada de animales, como la biodisponibilidad, la penetrancia de la barrera hematoencefálica y la unión a proteínas.
El término “sujeto” pretende incluir organismos vivos en los que puede producirse una enfermedad. Los ejemplos de sujetos incluyen humanos, monos, vacas, ovejas, cabras, perros, gatos, ratones, ratas y especies transgénicas de los mismos.
El término “sustancialmente puro” significa que el material aislado es al menos un 90% puro, preferiblemente un 95% puro, incluso más preferiblemente un 99% puro como se analiza mediante técnicas analíticas conocidas en la técnica.
Las composiciones farmacéuticas pueden formularse para administración oral en forma sólida o líquida, para inyección parenteral intravenosa, subcutánea, intramuscular, intraperitoneal, intraarterial o intradérmica, o para administración vaginal, nasal, tópica o rectal. Las composiciones farmacéuticas de la presente invención adecuadas para administración oral pueden presentarse como formas de dosificación discretas, por ejemplo, comprimidos, comprimidos masticables, comprimidos oblongos, cápsulas, líquidos y jarabes aromatizados. Dichas formas de dosificación contienen cantidades predeterminadas de ingredientes activos, y pueden prepararse por métodos farmacéuticos bien conocidos por los expertos en la técnica. Ver en general, Remington's Pharmaceutical Sciences, 18a edición, Mack Publishing, Easton Pa. (1990).
Las formas de dosificación parenteral se pueden administrar a los pacientes por diversas vías, incluidas la subcutánea, la intravenosa (incluida la inyección en bolo), la intramuscular y la intraarterial. Debido a que su administración generalmente pasa por alto las defensas naturales de los pacientes contra los contaminantes, las formas de dosificación parenteral son específicamente estériles o pueden esterilizarse antes de la administración a un paciente. Los ejemplos de formas de dosificación parenteral incluyen soluciones listas para inyección, productos secos listos para ser disueltos o suspendidos en un vehículo farmacéuticamente aceptable para inyección, suspensiones listas para inyección y emulsiones. Las composiciones farmacéuticas para inyección parenteral comprenden soluciones acuosas o no acuosas estériles farmacéuticamente aceptables, dispersiones, Suspensiones o emulsiones y polvos estériles para reconstituir en soluciones o dispersiones inyectables estériles. Los ejemplos de vehículos, diluyentes, solventes o vehículos acuosos y no acuosos adecuados incluyen agua, etanol, polioles (propilenglicol, polietilenglicol, glicerol y similares, y mezclas adecuadas de los mismos), aceites vegetales (como el aceite de oliva) y ésteres orgánicos inyectables. tales como oleato de etilo, o mezclas adecuadas de los mismos. La fluidez adecuada de la composición se puede mantener, por ejemplo, mediante el uso de un recubrimiento tal como lecitina, mediante el mantenimiento del tamaño de partícula requerido en el caso de dispersiones, y mediante el uso de tensioactivos. Estas composiciones también pueden contener adyuvantes tales como agentes conservantes, agentes humectantes, agentes emulsionantes y agentes dispersantes. La prevención de la acción de los microorganismos puede garantizarse mediante diversos agentes antibacterianos y antifúngicos, por ejemplo, parabenos, clorobutanol, fenol, ácido sórbico y similares. También puede ser deseable incluir agentes isotónicos, por ejemplo, azúcares, cloruro de sodio y similares. La absorción prolongada de la forma farmacéutica inyectable se puede lograr mediante el uso de agentes que retrasan la absorción, por ejemplo, monoestearato de aluminio y gelatina.
En algunos casos, para prolongar el efecto de un fármaco, a menudo es deseable disminuir la absorción del fármaco por inyección subcutánea o intramuscular. Esto se puede lograr mediante el uso de una suspensión líquida de material cristalino o amorfo con escasa solubilidad en agua. La velocidad de absorción del fármaco depende entonces de su velocidad de disolución que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. Alternativamente, la absorción retardada de una forma de fármaco administrada por vía parenteral se logra disolviendo o suspendiendo el fármaco en un vehículo oleoso.
Las suspensiones, además de los compuestos activos, pueden contener agentes de suspensión, por ejemplo, alcoholes isoestearílicos etoxilados, polioxietilen sorbitol y ésteres de sorbitán, celulosa microcristalina, metahidróxido de aluminio, bentonita, agar-agar, tragacanto y mezclas de los mismos. Si se desea, y para una distribución más efectiva, los compuestos de la invención pueden incorporarse en sistemas de liberación lenta o de administración dirigida, tales como matrices de polímeros, liposomas y microesferas. Pueden esterilizarse, por ejemplo, por filtración a través de un filtro que retiene las bacterias o por incorporación de agentes esterilizantes en forma de composiciones sólidas estériles, que pueden disolverse en agua estéril o en algún otro medio inyectable estéril inmediatamente antes de su uso.
Las formas de depósito inyectables se hacen formando matrices microencapsuladas del fármaco en polímeros biodegradables tales como polilactida-poliglicólido. Dependiendo de la relación de fármaco a polímero y de la naturaleza del polímero particular empleado, se puede controlar la velocidad de liberación del fármaco. Ejemplos de otros polímeros biodegradables incluyen poli (ortoésteres) y poli (anhídridos). Las formulaciones inyectables de depósito también se preparan atrapando el fármaco en liposomas o microemulsiones que son compatibles con los tejidos corporales. Las formulaciones inyectables pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro de retención bacteriana o incorporando agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse o dispersarse en agua estéril u otro medio inyectable estéril justo antes de su uso.
Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleaginosas inyectables estériles pueden formularse de acuerdo con la técnica conocida usando agentes dispersantes o humectantes adecuados y agentes de suspensión. La preparación inyectable estéril también puede ser una solución, suspensión o emulsión inyectable estéril en un diluyente o disolvente no tóxico y parenteralmente aceptable, como una solución en 1.3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse están el agua, la solución de Ringer, la U.S.P. y la solución de cloruro de sodio isotónica. Además, los aceites fijos estériles se emplean convencionalmente como un disolvente o medio de suspensión. Para este propósito, se puede emplear cualquier aceite fijo insípido, incluyendo mono o diglicéridos sintéticos. Además, los ácidos grasos como el ácido oleico se utilizan en la preparación de inyectables.
Las formas de dosificación sólidas para administración oral incluyen cápsulas, comprimidos, píldoras, polvos y gránulos. En tales formas de dosificación sólidas, uno o más compuestos de la invención se mezclan con al menos un vehículo farmacéuticamente aceptable inerte tal como citrato de sodio o fosfato dicálcico y/o a) rellenos o diluyentes tales como almidones, lactosa, sacarosa, glucosa, manitol y ácido salicílico; b) aglutinantes tales como carboximetilcelulosa, alginatos, gelatina, polivinilpirrolidinona, sacarosa y acacia; c) humectantes tales como glicerol; d) agentes desintegrantes tales como agar-agar, carbonato de calcio, almidón de patata o tapioca, ácido algínico, ciertos silicatos y carbonato de sodio; e) agentes retardantes de solución tales como parafina; f) aceleradores de absorción tales como compuestos de amonio cuaternario; g) agentes humectantes tales como alcohol cetílico y monoestearato de glicerol; h) absorbentes tales como caolín y arcilla de bentonita; y i) lubricantes tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, laurilsulfato de sodio y mezclas de los mismos. En el caso de cápsulas, comprimidos y píldoras, la forma de dosificación también puede comprender agentes tamponantes.
Las composiciones sólidas de un tipo similar también se pueden emplear como rellenos en cápsulas de gelatina blandas y rellenas que utilizan lactosa o azúcar de leche, así como polietilenglicoles de alto peso molecular. Las formas de dosificación sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y envolturas, tales como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de formulación farmacéutica. Opcionalmente, pueden contener agentes opacificantes y también pueden ser de una composición que liberen el (los) ingrediente(s) activo(s) solo, o preferentemente, en una determinada parte del tracto intestinal de manera retardada. Ejemplos de materiales que pueden ser útiles para retrasar la liberación del agente activo puede incluir sustancias poliméricas y ceras.
Las composiciones para administración rectal o vaginal son preferiblemente supositorios que pueden prepararse mezclando los compuestos de esta invención con portadores adecuados no irritantes tales como manteca de cacao, polietilenglicol o una cera para supositorios que son sólidos a temperatura ambiente, pero líquidos en el cuerpo. temperatura y, por lo tanto, se funden en el recto o la cavidad vaginal y liberan el compuesto activo.
Las formas de dosificación líquidas para administración oral incluyen emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además de los compuestos activos, las formas de dosificación líquidas pueden contener diluyentes inertes comúnmente usados en la técnica como, por ejemplo, agua u otros disolventes, agentes solubilizantes y emulsionantes como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1.3-butilenglicol, dimetilformamida, aceites (en particular, semillas de algodón, maní, maíz, germen, aceite de oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurfurílico, polietilenglicoles y ésteres de ácidos grasos de sorbitán, y mezclas de los mismos.
Además de los diluyentes inertes, las composiciones orales también pueden incluir adyuvantes tales como agentes humectantes, agentes emulsionantes y agentes de suspensión, edulcorantes, aromatizantes y perfumantes. Las formas de dosificación para administración tópica o transdérmica de un compuesto de esta invención incluyen pomadas, pastas, cremas, lociones, geles, polvos, soluciones, pulverizaciones, inhalantes o parches. Un compuesto deseado de la invención se mezcla en condiciones estériles con un vehículo farmacéuticamente aceptable y cualquier conservante o tampón necesario que pueda requerirse. La formulación oftálmica, gotas para los oídos, pomadas oculares, polvos y soluciones también se consideran dentro del alcance de esta invención. Las pomadas, pastas, cremas y geles pueden contener, además de un compuesto activo de esta invención, grasas animales y vegetales, aceites, ceras, parafinas, almidón, tragacanto, celulosa. derivados, polietilenglicoles, siliconas, bentonitas, ácido silícico, talco y óxido de zinc, o mezclas de los mismos.
Los polvos y aerosoles pueden contener, además de los compuestos de esta invención, lactosa, talco, ácido silícico, hidróxido de aluminio, silicatos de calcio y poliamida en polvo, o mezclas de estas sustancias. Los aerosoles también pueden contener propelentes habituales, como los clorofluorohidrocarburos.
Los compuestos de la invención también pueden administrarse en forma de liposomas. Como se conoce en la técnica, los liposomas se derivan generalmente de fosfolípidos u otras sustancias lipídicas. Los liposomas están formados por cristales líquidos hidratados mono o multilamelares que se dispersan en un medio acuoso. Se puede usar cualquier lípido no tóxico, fisiológicamente aceptable y metabolizable capaz de formar liposomas. Las presentes composiciones en forma de liposoma pueden contener, además de los compuestos de la invención, estabilizantes, conservantes y similares. Los lípidos preferidos son los fosfolípidos naturales y sintéticos y las fosfatidilcolinas (lecitinas) que se usan por separado o juntos. Los métodos para formar liposomas son conocidos en la técnica. Ver, por ejemplo, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, NY, (1976), p. 33 y siguientes.
Los niveles de dosificación reales de los ingredientes activos en las composiciones farmacéuticas de esta invención pueden variarse para obtener una cantidad del (de los) compuesto(s) activo(s) que sea efectivo para lograr la respuesta terapéutica deseada para un paciente particular, composiciones y modo de administración. El nivel de dosificación seleccionado dependerá de la actividad del compuesto particular, la vía de administración, la gravedad de la afección que se está tratando y la afección y el historial médico anterior del paciente que se está tratando. Sin embargo, está dentro de la experiencia de la técnica iniciar dosis del compuesto a niveles inferiores a los requeridos para lograr el efecto terapéutico deseado y aumentar gradualmente la dosis hasta que se logre el efecto deseado.
Se puede emplear una cantidad eficaz de uno de los compuestos de la invención en forma pura o, cuando existan tales formas, en forma de sal farmacéuticamente aceptable. Alternativamente, el compuesto puede administrarse como una composición farmacéutica que contiene el compuesto de interés en combinación con uno o más vehículos farmacéuticamente aceptables. Se entenderá, sin embargo, que el uso diario total de los compuestos y composiciones de la invención será decidido por el médico asistente dentro del alcance de un buen juicio médico. El nivel de dosis efectiva específica para cualquier paciente en particular dependerá de una variedad de factores, que incluyen el trastorno que se está tratando y la gravedad del trastorno; actividad del compuesto específico empleado; la composición específica empleada; la edad, peso corporal, salud general, sexo y dieta del paciente. el tiempo de administración, la vía de administración y la tasa de excreción del compuesto específico empleado; la duración del tratamiento; la relación riesgo/beneficio; fármacos utilizados en combinación o coincidentes con el compuesto específico empleado; y como factores bien conocidos en las artes médicas. Por ejemplo, está dentro de la experiencia de la técnica iniciar dosis del compuesto a niveles inferiores a los requeridos para lograr el efecto terapéutico deseado y aumentar gradualmente la dosis hasta que se logre el efecto deseado.
La dosis diaria total de los compuestos de la presente invención administrada a un animal o animal inferior puede oscilar entre aproximadamente 0.0003 y aproximadamente 50 mg/kg de peso corporal. Para fines de administración oral, las dosis más preferidas pueden estar en el intervalo de aproximadamente 0.0003 a aproximadamente 5 mg/kg de peso corporal. Si se desea, la dosis diaria efectiva se puede dividir en dosis múltiples para fines de administración; en consecuencia, las composiciones de dosis única pueden contener tales cantidades o submúltiplos de las mismas para completar la dosis diaria. Para la administración oral, las composiciones de la invención se proporcionan preferiblemente en forma de comprimidos que contienen aproximadamente 1.0. aproximadamente 5.0. aproximadamente 10.0. aproximadamente 15.0. aproximadamente 25.0. aproximadamente 50.0. aproximadamente 100 aproximadamente 250. o aproximadamente 500 miligramos del ingrediente activo.
Ejemplos
Métodos sintéticos
A menos que se indique lo contrario, las ID del compuesto denotadas por “TRV-” que contienen los centros quirales encontrados en

grupos se sintetizaron y probaron como mezclas racémicas con respecto a ese centro quiral respectivo. Los compuestos que contienen pirrolidinas sustituidas pretenden abarcar los enantiómeros tanto R como S como sustancialmente entantioméricamente puros, o como racematos.
En las siguientes partes de la descripción, ciertas realizaciones y compuestos que no se incluyen en las reivindicaciones adjuntas se consideran solo como realizaciones de referencia y compuestos.
Se usaron los siguientes esquemas sintéticos y procedimientos escritos para sintetizar los compuestos descritos en el presente documento:

NR1R2 = benzilamnna, pirrolidino, N-metil-1-fenilmetanamina, pirrolidino-2-carboxilato de etilo, morfolino, 4-metilimidazol, 1-metilpiperazina, N-isopropilmetilamina, 2-metNpirrolidino, piperidina, dietilamina, (2-metoxietN)metilamina, N-metiletanamina, tiomorfolino, 4-(pirrolidin-1-il)piperidino, pirazol, 4-metilpirazol, 4-fluoro-N-metilbenzilamina, etilamina, isoindilino, 3-hidroxiazetidina, azetidina, propargilamina, ciclopropilamina, 3-hidroxipirrolidona, N-metil-N-(3-piridinilmetil)amina, 4-metoxibenzilamina, 3,5-dimetoxibenzilamina, ácido 3-azetidino carboxílico, azetidina, (S)-etil pirrolidino-2-carboxilato, N-metil-1-(tiazol-2-il)metanamina


a) i) nBuLi, -78°C, ii) I2, THF, -78°C; b) metil propiolato, Pd(PPh3)2Cl2, Cul, K2CO3, THF, 65°C; c) i) nBuLi, -78°C; ii) DMF; d) i) trimetilfosfonoacetato, NaH, THF, 0°C; e) i) CF3TMS, TBAF; f) DIBAL, DCM, -78 a -30°C; g) CF3TM, TBAF; h) i) nBuLi, -78°C; ii) metilcloroformato, THF, -78°C;

a) i) nBuLi/-78°C; ii) acetaldehído; b) NaH/THF/Mel; c) Dess-Martin; d) MeMgBr/THF/-78°C






o
A (grupo amino) = morfolino, 1-metilpiperazina, N-isopropilmetilamina, 2-metilpirrolidino, piperidino, dietilamina, (2-metoxietilo)metilamina, N-metiletanamina, tiomorfolino, 4-(pirrolidin-1-il)piperidino, pirazol, 4-metilpirazol, 4-fluoro-N-metilbenzilamina, isoindolina, N-metil propargilamina, N-metil-N-(2-piridinilmetil)amiina, N-metil-N-(3-piridinilmetil)amina, azetidina, N-metil-1-(tiazol-2-il)metanamina, 1-(azetidin-3-ilmetil)pirrolidino; 4-azetidin-3-ilmetil)morfolino; N-metil-3- 
(pirrolidin-1-il)propan-1-amina; N,4-dimetilpentan-1-amina; N,N-dimetilpiperidin-4-amina 

TRV-1397
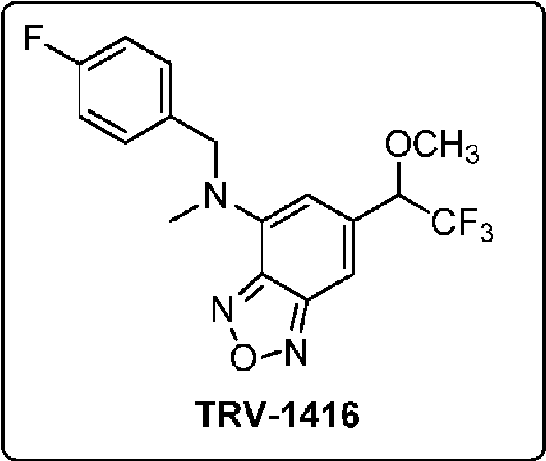

a) (COCI)? / DMSO / TEA / DCM; b) NaH / THF/ Mel



3) CF3TMS / TBAF 0 °C
NR-|= ciclopropilamina, 4-metoxibenzilamina, 
3,5-dimetoxibenzilamina
1) NaH / Mel / NMP / 0 C



21

Ċ

a) ácido 3-formilfenilborónico Pd(PPha)4/DME/100°C 2 M Na2COa; b) CF3TMS/TBAF

acido 3-aceti fem boromco
Pd(PPh3W DME / 100 °C
2 M NaoCO

acido 3-acetilfenilboronico
Pd(PPh3)4 / DME /100 °C
2 M Na2C 03



a) piperazina/HATU/DIPEA; b) ciclopropilamina/HATU/DIPEA; c) morfolino/HATU/DIPEA; d) clorhidrato de azetidina/HATU/DIPEA; e) clorhidrato de 3-hidroxiazetidina/HATU/DIPEA

a) pirrolidino/EtOAc/T3P/TEA; b) ciclopropilamina/EtOAc; c) morfolino/EtOAc/T3P/TEA



a) ácido 3-formilfenilborónico Pd(PPha)4/DME/100°C 2 M Na2COa; CF3TMS/TBAF


X, Y o Z = C, N, S, O, C=C, C=N
A, B o C = H, alquilo, Cl, Br, F, OMe, OPh, SO2Me
C=O(CH3), NHC=O(CH3), CF3CN, OCF3, B(OH)2
CH2OH, C=ON(CH3)2, CH(OH)CF3, -O(CH2)O
N Ph Boc 


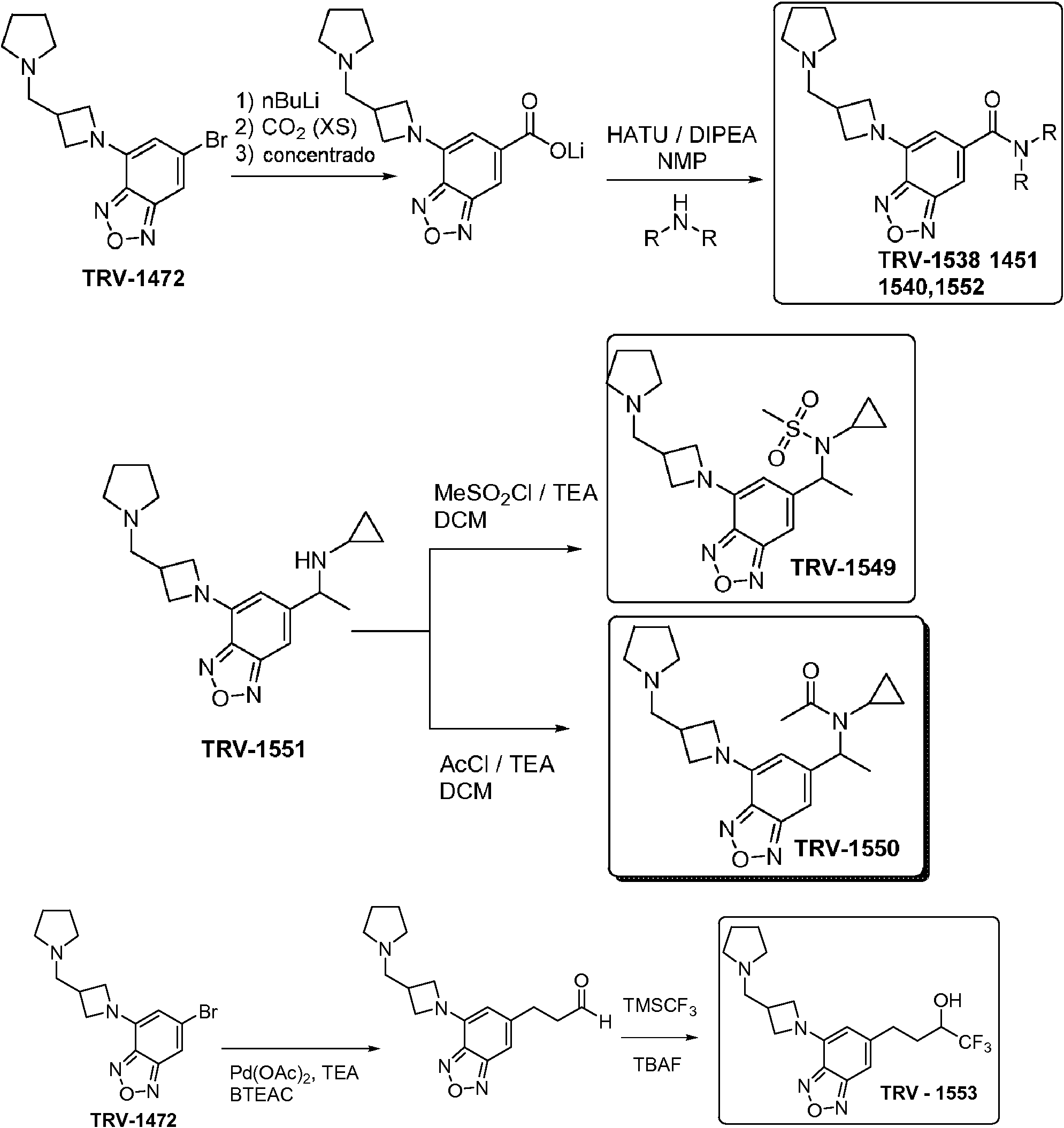



, , , , e, , , , ,
CH3CCH30H.H, Cicloalquilo
R, R"= 1-(azetidin-3-ilmetil)pirrohdino
4-(azetidin-3-ilmetil)morfohno



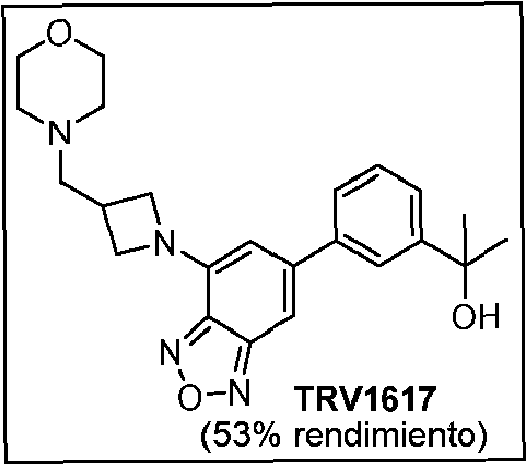



TRV1470
N a B H 4
MeOH












Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol 4 (0.9678 g, 3.48 mmol) y bencilamina (1.9 mL, 17.4 mmol) en DMSO (10.5 mL) bajo argón y se agitaron en un tubo sellado durante 3 días, después de ese tiempo, el tubo se calentó a 60°C durante 12 horas. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El sólido oscuro se recristalizó a partir de EtOH y los cristales oscuros se recolectaron mediante filtración y se secaron para producir 0.4516 g (43% de rendimiento) de N-bencil-6-bromobenzo[c][1,2,5]oxadiazol-4-amina. Este material (0.1980 g, 0.651 mmol), clorhidrato de benzoilpiperazina (0.1771 g, 0.781 mmol) y Cs2CO3 (0.6353 g, 1.95 mmol) se agregaron a un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron a tolueno (2 mL) y NMP (1.2 mL) el tubo y sus contenidos se desgasificaron durante 15 minutos, en cuyo punto se agregaron rápidamente Pd2(dba)3 (0.0119 g, 0.0130 mmol) y BINAP (0.0162 g, 0.026 mmol), el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo se purificó a través de cromatografía (40% de EtOAc/Hexanos) para dar 0.188 g (70% de rendimiento) de TRV-1256. 1H RMN (500 MHz, CDCh) 8 = 7.46-7.43 (m, 5H), 7.39-7.36 (m, 4H), 7.34-7.31 (m, 1H), 6.13 (d, J = 1.0 Hz, 1H), 5.88 (s, 1H), 5.32 (t, J = 5.5 Hz, 1H), 4.48 (d, J = 5.5 Hz, 2H), 3.90 (br s, 2H), 3.58 (br s, 2H), 3.29 (br s, 2H), 3.15 (br s, 2H).

Se agregaron N-bencil-6-bromobenzo[c][1,2,5]oxadiazol-4-amina (0.2235 g, 0.735 mmol) y ácido 3-acetilbencenoborónico (0.1566 g, 0.955 mmol) a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (1.1 mL, 2.21 mmol) y DME (1.6 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0425 g, 0.0368 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo se disolvió en metanol (15 mL) y se enfrió a 0°C. Se agregó NaBH4 (0.0556 g, 1.47 mmol) y la mezcla se agitó a 0°C durante 3 horas. La mezcla de reacción se detuvo con NaHCO3 saturado. Esta mezcla espesa se diluyó con agua y se extrajo con DCM (5X). Los extractos luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (30% de EtOAc/Hexano) para producir 0.1412 g (56% de rendimiento) de TRV-1259. 1H RMN (500 MHz, CDCh) 8 = 7.47 (s, 1H), 7.46-7.38 (m, 7H), 7.36 7.33 (m, 1H), 7.18 (s, 1H), 6.36 (s, 1H), 5.44 (t, J = 5.5 Hz, 1H), 4.99-4.95 (m, 1H), 4.57 (d, J - 5.5 Hz, 2H), 1.88 (d, J = 3.5 Hz, 1H), 1.54 (d, J = 7.0 Hz, 3H)

Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3122 g, 1.12 mmol), pirrolidina (0.10 mL, 1.12 mmol) y DIPEA (0.20 mL, 1.12 mmol) en NMP (2 mL) bajo argón y se agitaron en un tubo sellado a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar para dar el 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol crudo (0.2247 g, 75% de rendimiento). Se agregaron el material crudo (0.1964 g, 0.73 mmol) y ácido 3-acetilbencenoborónico (0.1558 g, 0.95 mmol) a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (1.1 mL, 2.21 mmol) y DME (1.6 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0422 g, 0.0365
mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo se disolvió en metanol (12 mL) y se enfrió a 0°C. Se agregó NaBH4 (0.0552 g, 1.46 mmol) y la mezcla se agitó a 0°C durante 3 horas. La mezcla de reacción se detuvo con NaHCO3 saturado. Esta mezcla espesa se diluyó con agua y se extrajo con DCM (5X). Los extractos luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (30% de EtOAc/Hexano) para producir 0.2029 g (90% de rendimiento, durante 2 etapas) de TRV-1310. 1H RMN (CDCl3) 8 = 7.65 (s, 1H), 7.55 (d, J = 7.0 Hz, 1H), 7.47-7.42 (m, 2H), 7.07 (s, 1H), 6.11 (s, 1H), 5.00 4.99 (m, 1H), 3.81 (s, 4H), 2.12-2.09 (m, 4H), 1.92 (d, J = 3.0 Hz, 1H), 1.56 (d, J = 6.5 Hz, 3H).

Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.5607 g, 2.0 mmol), N-metil-1-fenilmetanamina (0.28 mL, 2.2 mmol) y DIPEA (0.52 mL, 3.0 mmol) en NMP (3 mL) bajo argón y se agitaron en un tubo sellado a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar para dar el material crudo (N-bencil-6-bromo-N-metilbenzo[c][1,2,5]oxadiazol-4-amina) como un aceite. Se agregaron este material crudo y ácido 3-acetilbencenoborónico (0.4263 g, 2.6 mmol) a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (3.0 mL, 6.0 mmol) y DME (4.5 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.1156 g, 0.10 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo se disolvió en metanol (33 mL) y se enfrió a 0°C. Se agregó NaBH4 (0.1513 g, 4.0 mmol) y la mezcla se agitó a 0°C durante 3 horas. La mezcla de reacción se detuvo con NaHCO3 saturado. Esta mezcla espesa se diluyó con agua y se extrajo con DCM (5X). Los extractos luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (30% de EtOAc/Hexano) para producir 0.4843 g (67 % de rendimiento, durante 3 etapas) de TRV-1358 como aceite amarillo. 1H RMN (CDCla, 500 MHz) 8 = 7.60 (s, 1H), 7.51 (dt, J = 6.5, 2.0 Hz, 1H), 7.47-7.42 (m, 2H), 7.35-7.32 (m, 2H), 7.29-7.28 (m, 3H), 7.24 (s, 1H), 6.37 (s, 1H), 5.16 (s, 2H), 5.01-4.96 (m, 1H), 3.22 (s, 3H), 1.85 (d, J = 3.5 Hz, 1H), 1.55 (d, J = 6.5 Hz, 3H).

Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3765 g, 1.35 mmol), clorhidrato de pirrolidina-2-carboxilato de etilo (0.2677 g, 1.49 mmol) y D iPEa (0.59 mL, 3.38 mmol) en NMP (1.8 mL) bajo argón y se agitaron en un tubo sellado a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar para dar 0.1756 g (38 % de rendimiento) de material crudo. Este material crudo y ácido 3-acetilbencenoborónico (0.1099 g, 0.67 mmol) se agregaron a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (0.8 mL, 1.55 mmol) y DME (1.2 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0298 g, 0.0258 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo luego se disolvió en DCM (0.3 mL) y tolueno (1.6 mL) y
esta solución se enfrió a 0°C. Se agregó en forma de gotas DIBAL (1.7 mL de una solución 1.0 M en hexano) y la reacción se le permitió agitar durante la noche. Se agregaron otros 1.5 eq de DIBAL (0.8 mL) a 0°C y la reacción se agitó durante 24 horas adicionales. La mezcla se detuvo con una solución saturada de tartrato de sodio potasio y se extrajo con acetato de etilo. Los extractos combinados se lavaron con H2O y solución salina, se secaron con Na2SO4, se filtraron y se concentraron. El material crudo se purificó a través de columna de 60% de EtOAc/hexano para producir 0.1224 (70% de rendimiento, 2 etapas) de TRV-1359 como sólido naranja. 1H RMN (CDCh, 500 MHz) 8 = 7.64 (s, 1H), 7.54 (dt, J = 6.5, 2.0 Hz, 1H), 7.47-7.43 (m, 2H), 7.14 (s, 1H), 6.26 (s, 1H), 5.00-4.99 (br s, 1H), 4.74 (br s, 1H), 3.87-3.83 (m, 1H), 3.80- 3.77 (m, 1H), 3.69-3.64 (m, 1H), 3.54-3.50 (m, 1H), 2.21-2.11 (m, 4H), 1.92-1.86 (m, 2H), 1.56 (d, J = 6.5 Hz, 3H)

Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.5385 g, 1.94 mmol), morfolina (0.17 mL, 1.94 mmol) y DIPEA (0.34 mL, 1.94 mmol) en NMP (2.5 mL) bajo argón y se agitaron en un tubo sellado a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar para dar 0.5360 g (97 % de rendimiento) de sólido marrón. Este material crudo (0.5002 g, 1.76 mmol) y ácido 3-formilfenilborónico (0.3434 g, 2.29 mmol) se agregaron a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (2.6 mL, 5.3 mmol) y DME (3.9 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.1040 g, 0.09 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con DCM. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. Este material crudo luego se disolvió en THF (1.8 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.3003 g, 2.11 mmol) seguido por TBAF (0.18 mL, 0.18 mmol) a 0°C. Después de que se completa la adición, se eliminó el baño de hielo y la reacción se agitó a temperatura ambiente durante varias horas, en cuyo punto, se agregaron unos 2 eq adicionales de CF3TMS (0.52 mL) junto con 0.1 eq de TBAF (0.18 mL) a 0°C. Una vez más se calentaron a temperatura ambiente y se agitaron durante 2 horas. A esta mezcla luego se agrego TBAF (7.6 mL, 7.6 mmol) a 0°C y la reacción se agitó durante la noche. La reacción se detuvo con solución salina y se extrajo con EtOAc. Los extractos combinados se lavaron con H2O (4X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. La purificación a través de cromatografía instantánea (30% de EtOAc/hexano) produjo 0.2303 g (34 % de rendimiento, durante 3 etapas) de TRV-1360 como sólido amarillo. 1H RMN (CDCh, 500 MHz) 8 = 7.74 (s, 1H), 7.67-7.65 (m, 1H), 7.57-7.52 (m, 2H), 7.40 (s, 1H), 6.58 (s, 1H), 5.17-5.12 (m, 1H), 3.97 (t, J = 5.0 Hz, 4H), 3.65 (t, J = 5.0 Hz, 4H), 2.75 (d, J = 4.5 Hz, 1H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.4244 g, 1.58 mmol) y ácido 3-formilfenilborónico (0.3149 g, 2.1 mmol) a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (2.4 mL, 4.7 mmol) y DME (3.6 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0924 g, 0.08 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. Este material crudo luego se disolvió en THF (1.6 mL) y se enfrió a 0°C. CF3TMS (0.2702 g, 1.9 mmol) se agregó seguido por TBAF (0.16 mL, 0.16 mmol) a 0°C. Después de que se completa la adición, se eliminó el baño de hielo y la reacción se agitó a temperatura ambiente durante varias horas, en cuyo punto, se agregaron unos 2 eq adicionales de CF3TMS (0.47 mL) junto con 0.1 eq de TBAF (0.16 mL) a 0°C. Una vez más se calentaron a temperatura ambiente y se agitaron durante 2 horas. A esta mezcla luego se agrego TBAF (7.0 mL, 7.0 mmol) a 0°C y la reacción se agitó durante la noche. La reacción se detuvo con solución salina y se extrajo con EtOAc. Los extractos combinados se lavaron con H2O (4X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. La purificación a través
de cromatografía instantánea (20% de EtOAc/hexano) produjo 0.2749 g (48 % de rendimiento, durante 3 etapas) de TRV-1361 como sólido naranja. 1H RMN (CDCla, 500 MHz) 8 = 7.74 (s, 1H), 7.68 (dt, J = 7.0, 1.5 Hz, 1H), 7.55-7.50 (m, 2H), 7.07 (s, 1H), 6.09 (s, 1H), 5.13 (q, J = 6.5 Hz, 1H), 3.83-3.80 (m, 4H), 2.67 (brs, 1H), 2.14-2.09 (m, 4H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.1239 g, 0.46 mmol) y ácido 3-acetilbencenoborónico (0.0984 g, 0.60 mmol) a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO3 2 M (0.70 mL, 1.38 mmol) y DME (1.0 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0266 g, 0.023 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. El material crudo se purificó a través de cromatografía instantánea (25% de EtOAc/hexano) para producir 0.0391 g (28 % de rendimiento) de TRV-1362 como sólido naranja. 1H RMN ( CDCla, 500 MHz) 8 = 8.22 (d, J = 1.5 Hz, 1H), 8.00 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.09 (s, 1H), 6.10 (s, 1H), 3.82 (s, 4H), 2.68 (s, 3H), 2.13-2.11 (m, 4H).
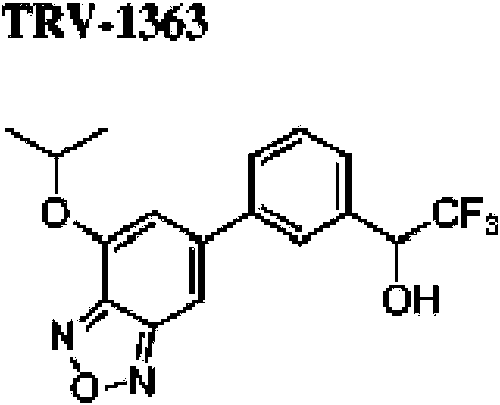
Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (1.00 g, 3.6 mmol) y isopropanol (0.31 mL, 4.0 mmol) en THF (20 mL) bajo argón y se enfrió a -78°C. Se agregó en forma de gotas NaHMDS (4.0 mL de una solución 1.0 M en THF) y la reacción se agitó durante 30 minutos a -78°C. El baño frío luego se retiró y la reacción se agitó a temperatura ambiente durante unas pocas horas. La reacción luego se enfrió a 0°C y se detuvo con solución de NH4Cl (ac.) saturada. Esta suspensión se extrajo con DCM (3X). Los extractos combinados se lavaron con H2O, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para producir 0.9407 g de aceite negro crudo. Este material crudo (0.6702 g, 2.6 mmol) y ácido 3-formilfenilborónico (0.5098 g, 3.4 mmol) se agregaron a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Na2CO32 M (3.9 mL, 7.8 mmol) y DME (5.8 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.1502 g, 0.13 mmol) y el tubo se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con H2O (5X), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar. Este material crudo se purificó a través de cromatografía instantánea (20% de EtOAc/hexano) para producir 0.4836 g (66% de rendimiento) del aldehído. Estructura aldehído (0.6702 g, 2.6 mmol) se disolvió en THF (2.0 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.23 mL, 1.56 mmol) seguido por TBAF (0.1 mL, 0.1 mmol) a 0°C. Después de que se completa la adición, se eliminó el baño de hielo y la reacción se agitó a temperatura ambiente durante varias horas. La mezcla luego se volvió a enfriar a 0°C y se agregó TBAF (2.8 mL, 2.8 mmol) a la reacción, que se agitó durante la noche. La reacción se detuvo con solución salina y se extrajo con EtOAc. Los extractos combinados se lavaron con H2O (4X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. La purificación a través de cromatografía instantánea (25% de EtOAc/hexano) produjo 0.1468 g (53% de rendimiento, durante 2 etapas) de TRV-1363 como aceite amarillo. 1H RMN (CDCh, 500 MHz) 8 = 7.74 (s, 1H), 7.66 (dt, J = 7.0, 2.0 Hz, 1H), 7.58-7.54 (m, 2H), 7.48 (s, 1H), 6.78 (s, 1H), 5.17-5.13 (m, 1H), 4.99 (sept, J = 6.0 Hz, 1H), 2.76 (d, J = 4.5 Hz, 1H), 1.52 (d, J = 6.0 Hz, 6H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.3898 g, 1.45 mmol) y ácido 5-formil-2-tienilborónico (0.2948 g, 1.89 mmol) a un tubo. El tubo se evacuó y se purgó con argón 3 veces. Luego se agregaron Na2CO32M (ac.) (2.2 mL) y DME (3.3 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0844 g, 0.073 mmol), el tubo se selló y calentó a100 °C durante la noche. Luego de enfriar a temperatura ambiente, se determinó que la reacción no se completó. Se agregó otro equivalente de ácido 5-formil-2-tienilborónico junto con 5% en mol adicional de Pd(PPh3)4 y la mezcla se calentó durante la noche de nuevo. Es necesario agregar más ácido borónico y catalizador una vez más, y se calentó durante 24 horas adicionales. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y luego la capa acuosa se extrajo de nuevo con EtOAc. Los extractos orgánicos combinados se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se tomó en THF (3 mL) y se agregó CF3TMS (0.43 mL, 2.9 mmol). Esta solución se enfrió a 0°C antes de agregar TBAF (0.15 mL, 0.145 mmol). La mezcla se agitó durante la noche y luego se agregaron 2 eq. adicionales de CF3TMS y 0.1 eq de TBAF. Después de agitar durante unas 2 horas adicionales, la reacción se enfrió a 0°C y se agregó TBAF (8 mL, 8 mmol). La mezcla se agitó durante la noche antes de diluir con agua. La capa acuosa se extrajo con EtOAc (3X). Los extractos combinados se lavaron con solución salina, se secaron (Na2SO4), filtraron y se concentraron. El material crudo se purificó a través de columna (15-20% de EtOAc/hexano) para dar 0.1043 g (19% de rendimiento, 3 etapas) de TRV-1364 como sólido naranja. 1H RMN (CDCla, 700 MHz) 8 = 7.35 (d, J = 3.2 Hz, 1H), 7.19 (d, J = 3.2 Hz, 1H), 7.13 (s, 1H), 6.10 (s, 1H), 5.31 (q, J = 3.2 Hz, 1H), 3.80 (br s, 4H), 2.90 (br s, 1H), 2.11 (s, 4H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.3948 g, 1.47 mmol) y ácido 4-formilbencenoborónico (0.2864 g, 1.91 mmol) a un tubo. El tubo se evacuó y se purgó con argón 3 veces. Luego se agregaron Na2CO32M (ac.) (2.2 mL) y DME (3.3 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0855 g, 0.074 mmol), el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y luego la capa acuosa se extrajo de nuevo con EtOAc. Los extractos orgánicos combinados se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se tomó en THF (3 mL) y se agregó CF3TMS (0.43 mL, 2.94 mmol). Esta solución se enfrió a 0°C antes de agregar TBAF (0.15 mL, 0.147 mmol). Después de agitar durante 2 horas, la reacción se enfrió a 0°C y se agregó TBAF (5.1 mL, 5.1 mmol). La mezcla se agitó durante la noche antes de diluir con agua. La capa acuosa se extrajo con EtOAc (3X). Los extractos combinados se lavaron con solución salina, se secaron (Na2SO4), filtraron y se concentraron. El material crudo se purificó a través de columna (15% de EtOAc/hexano) para dar 0.3795 g (71% de rendimiento, 3 etapas) de TRV-1365 como sólido naranja. 1H RMN (CDCla, 700 MHz) 8 = 7.68 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.07 (s, 1H), 6.10 (s, 1H), 5.11 (q, J = 6.3 Hz, 1H), 3.81 (br s, 4H), 2.72 (br s, 1H), 2.13-2.09 (m, 4H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.3856 g, 1.44 mmol) y ácido 2-formilbencenoborónico (0.2804 g, 1.87 mmol) a un tubo. El tubo se evacuó y se purgó con argón 3 veces. Luego se agregaron Na2CO32M (ac.) (2.2 mL) y DME (3.3 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.0832 g, 0.072 mmol), el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y luego la capa acuosa se extrajo de nuevo con EtOAc. Los extractos orgánicos combinados se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se tomó en THF (3 mL) y se agregó CF3TMS (0.43 mL, 2.94 mmol). Esta solución se enfrió a 0°C antes de agregar TBAF (0.14 mL, 0.144 mmol). Después de agitar durante 2 horas, la reacción se enfrió a 0°C y se agregó TBAF (5.0 mL, 5.0 mmol). La mezcla se agitó durante la noche antes de diluir con agua. La capa acuosa se extrajo con EtOAc (3X). Los extractos combinados se lavaron con solución salina, se secaron (Na2SO4), filtraron y se concentraron. El material crudo se purificó a través de columna (10% de EtOAc/hexano) para dar 0.179 g (34 % de rendimiento, 3 etapas) de TRV-1366 como sólido naranja. 1H RMN (CDCla, 500 MHz) 8 = 7.78 (d, J = 8.0 Hz, 1H), 7.52-7.45 (m, 2H), 7.34 (d, J = 7.5 Hz, 1H), 6.85 (s, 1H), 5.79 (s, 1H), 5.26 (q, J = 6.5 Hz, 1H), 3.76 (br s, 4H), 2.55 (br s, 1H), 2.11-2.08 (m, 4H).

Se disolvió 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.7754 g, 2.89 mmol) en THF y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (1.6 mL de una solución 2.0 M en ciclohexano) y la mezcla se le permitió agitar durante 30 minutos. En este punto, a la solución que se agitó a -78°C se agregó d Mf (0.25 mL, 3.18 mmol) todo de una vez a -78°C. La reacción se agitó durante 30 minutos y luego se dejó calentar a temperatura ambiente. La reacción luego se detuvo con NH4Cl saturado (ac.), se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para producir 0.4779 g de aldehido crudo. Este aldehido (0.1352 g, 0.622 mmol) se disolvió en Th F (2 mL) y se trató con CF3TMS (0.18 mL, 1.24 mmol). La solución se enfrió a 0°C y luego se agregó TBAF (0.1 mL, 0.1 mmol). Después de agitar durante 2 horas, la mezcla se volvió a enfriar a 0°C y se agregó TBAF (2.2 mL, 2.2 mmol), la reacción se le permitió agitar durante la noche. La mezcla luego se detuvo con agua y se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de columna (25% de EtOAc/hexano) para producir 0.0201 g (11% de rendimiento) de TRV-1368 como aceite rojo. 1H RMN (CDCla, 700 MHz) 8 = 7.06 (s, 1H), 5.97 (s, 1H), 5.01 (q, J = 6.3 Hz, 1H), 3.78 (br s, 4H), 2.74 (br s, 1H), 2.12 2.08 (m, 4H).

Se agregaron 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol (0.5785 g, 2.16 mmol) y ácido piridina-3-borónico (0.3442 g, 2.8 mmol) a un tubo. El tubo se evacuó y se purgó con argón 3 veces. Luego se agregaron Na2CO32M (ac.) (3.2 mL) y DME (4.8 mL) y la solución se desgasificó durante 15 minutos. Se agregó rápidamente Pd(PPh3)4 (0.1248 g, 0.108 mmol), el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y luego la capa acuosa se extrajo de nuevo con EtOAc. Los extractos orgánicos combinados se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo se purificó a través de columna (50% de EtOAc/hexano) para producir 0.1902 g de material contaminado y 0.2624 g de material puro para un rendimiento combinado de 79% de rendimiento de TRV-1369 como sólido naranja. 1H RMN (CDCh, 700 MHz) 8 = 8.90 (d, J = 1.4 Hz, 1H), 8.65 (d, J = 4.6 Hz, 1H), 7.93 (d, J = 7.7 Hz, 1H), 7.39 (dd, J = 7.7, 4.6 Hz, 1H), 7.07 (s, 1H), 6.05 (s, 1H), 3.82 (br s, 4 H), 2.14 2.10 (m, 4H).

Se disolvió 6-bromo-4-(pirrolidin-1-il)benzo[c][1,2,5]oxadiazol 1 (0.7754 g, 2.89 mmol) en THF y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (1.6 mL de una solución 2.0 M en ciclohexano) y la mezcla se le permitió agitar durante 30 minutos. En este punto, se tomó una alícuota de 2.9 mL y se agregó en forma de gotas a una solución enfriada con hielo de NH4Cl saturado (ac.). Después de calentar a temperatura ambiente, la mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo se purificó a través de columna (5% de EtOAc/hexano) para producir 0.0654 g (46% de rendimiento) de TRV-1370 como sólido naranja. 1H RMN (CDCla, 500 MHz) 8 = 7.22 (dd, J = 8.5, 8.0 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 5.87 (d, J = 8.0 Hz, 1H), 3.76-3.73 (m, 4H), 2.10-2.05 (m, 4 H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (1.0173 g, 3.66 mmol), 4-metilimidazol (0.3005 g, 3.66 mmol), NMP (5 mL) y DIPEA (0.64 mL, 3.66 mmol) se sellaron en un tubo y se calentaron a 150°C durante 3 días. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de columna de 35% de EtOAc/hexano para producir 0.5295 g (52% de rendimiento) de la anilina. La anilina (0.2988 g, 1.07 mmol) y ácido 3-formilbencenoborónico (0.2084 g, 1.39 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.6 mL, solución ac.) junto con DME (2.4 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0618 g, 0.0535 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.24 mL, 1.61 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAf (3.2 mL, 3.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (5% de MeOH/DCM) para producir 0.0616 g (15% de rendimiento, 3 etapas) de TRV-1375 como una mezcla 1:1 de regioisómeros. 1H RMN (500 MHz, CDCla) 8 = 8.353 (s, 1H), 8.350 (s, 1H), 7.89 (s, 1H), 7.88 (s, 1H), 7.85 (s, 2H), 7.71-7.69 (m, 2H), 7.65-7.63 (m, 2H), 7.61-7.56 (m, 4H), 7.48 (s, 2H), 5.20 (q, J = 7.0 Hz, 2H), 5.07 (br s, 2H), 2.332 (s, 3H), 2.331 (s, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3327 g, 1.2 mmol), 1-metilpiperazina (0.13 mL, 1.2 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.2 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2338 g, 1.56 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.7 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0693 g, 0.06 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.4 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (5% de MeOH/DCM) para producir 0.1695 g (36% de rendimiento, 4 etapas) de TRV-1376. 1H RMN (500 MHz, DMSO) 8 = 7.91 (s, 1H), 7.85-7.83 (m, 1H), 7.60 7.54 (m, 2H), 7.54 (s, 1H), 6.94 (d, J = 6.0 Hz, 1H), 6.75 (s, 1H), 5.31-5.28 (m, 1H), 3.64 (t, J = 5.0 Hz, 4H), 2.55 (t, J = 5.0 Hz, 4H), 2.25 (s, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3113 g, 1.12 mmol), N-isopropilmetilamina (0.12 mL, 1.12 mmol), NMP (2 mL) y DIPEA (0.20 mL, 1.12 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2189 g, 1.46 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.7 mL, solución ac.) junto con DME (2.5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0647 g, 0.056 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.33 mL, 2.24 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.0 mL, 4.0 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.1283 g (31% de rendimiento, 4 etapas) de TRV-1377. 1H RMN (500 MHz, CDCh) 8 = 7.74 (s, 1H), 7.68 (dt, J = 5.0, 2.0 Hz, 1H), 7.55-7.51 (m, 2H), 7.18 (d, J = 1.0 Hz, 1H), 6.30 (s, 1H), 5.24 (sept, J = 6.5 Hz, 1H), 5.13 (q, J = 6.5 Hz, 1H), 3.01 (s, 3H), 2.71 (br s, 1H), 1.30 (d, J = 6.5 Hz, 6 H).

N-bencil-6-bromo-N-metilbenzo[c][1,2,5]oxadiazol-4-amina y ácido 3-formilbencenoborónico (0.2293 g, 1.53 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.6 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0682 g, 0.059 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.36 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.2683 g (55% de rendimiento, 4 etapas) de TRV-1378. 1H RMN (500 MHz, CDCh) 8 = 7.69 (s, 1H), 7.64 (dt, J = 7.5, 1.5 Hz, 1H), 7.54-7.49 (m, 2H), 7.35-7.32 (m, 2H), 7.29 7.26 (m, 3H), 7.23 (d, J = 1.0 Hz, 1H), 6.33 (s, 1H), 5.16 (s, 2H), 5.11 (q, J = 6.5 Hz, 1H), 3.24 (s, 3H), 2.68 (s, 1H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3414 g, 1.23 mmol), 2-metilpirrolidina (0.13 mL, 1.23 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.23 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2398 g, 1.6 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.9 mL, solución ac.) junto con DME (2.8 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0716 g, 0.062 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.36 mL, 2.46 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.3 mL, 4.3 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.1319 g (28 % de rendimiento, 4 etapas) de TRV-1379, una mezcla de 1:1:1:1 de diastereómeros debido a los dos centros quirales. 1H RMN (500 MHz, CDCh) 8 = 7.74 (s, 1H), 7.67 (dt, J = 7.0, 2.0 Hz, 1H), 7.55-7.50 (m, 2H), 7.07 (s, 1H), 6.12 (s, 1H), 5.13 (q, J = 6.5 Hz, 1H), 4.75-4.71 (m, 1H), 3.87-3.83 (m, 1H), 3.65-3.60 (m, 1H), 2.71 (br s, 1H), 2.24 2.08 (m, 3H), 1.85-1.81 (m, 1H), 1.29 (d, J = 6.5 Hz, 3H).

N-bencil-6-bromobenzo[c][1,2,5]oxadiazol-4-amina y ácido 3-formilbencenoborónico (0.2488 g, 1.66 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.9 mL, solución ac.) junto con DME (2.9 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.074 g, 0.064 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (3 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.28 mL, 1.92 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (3.9 mL, 3.9 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.0655 g (13% de rendimiento, 4 etapas) de TRV-1380. 1H RMN (500 MHz, CDCh) 8 = 7.67 (s, 1H), 7.63 (dt, J = 7.5, 1.5 Hz, 1H), 7.57-7.52 (m, 2H), 7.49-7.42 (m, 4H), 7.40 7.36 (m, 1H), 7.22 (d, J = 1.0 Hz, 1H), 6.37 (s, 1H), 5.51 (t, J = 5.0 Hz, 1H), 5.14 (q, J = 6.5 Hz, 1H), 4.60 (d, J = 5.0 Hz, 2H), 2.72 (br s, 1H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3095 g, 1.11 mmol), piperidina (0.11 mL, 1.11 mmol), NMP (2 mL) y DIPEA (0.19 mL, 1.11 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2159 g, 1.44 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.7 mL, solución ac.) junto con DME (2.5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0647 g, 0.056 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.33 mL, 2.22 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (3.9 mL, 3.9 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.3097 g (74 % de rendimiento, 4 etapas) de TRV-1381. 1H RMN (500 MHz, CDCh) 8 = 7.73 (s, 1H), 7.67-7.66 (m, 1H), 7.57 7.51 (m, 2H), 7.30 (s, 1H), 6.53 (s, 1H), 5.14 (q, J = 6.5 Hz, 1H), 3.65 (t, J = 5.5 Hz, 4H), 2.70 (br s, 1H), 1.84-1.77 (m, 4H), 1.74-1.69 (m, 2H).
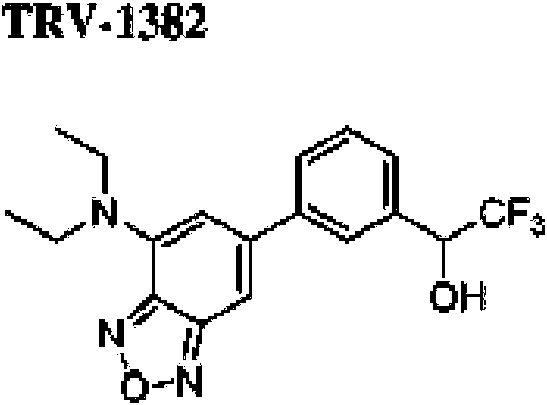
4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3340 g, 1.2 mmol), dietilamina (0.13 mL, 1.2 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.2 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2338 g, 1.56 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.7 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0693 g, 0.06 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.4 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.2184 g (50% de rendimiento, 4 etapas) de TRV-1382. 1H RMN (500 MHz, CDCh) 8 = 7.73 (s, 1H), 7.65 (dt, J = 7.0, 2.0 Hz, 1H), 7.55-7.51 (m, 2H), 7.11 (d, J = 0.5 Hz, 1H), 6.25 (s, 1H), 5.13 (q, J = 6.5 Hz, 1H), 3.81 (q, J = 7.0 Hz, 4H), 2.76 (br s, 1H), 1.31 (t, J = 7.0 Hz, 6H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3322 g, 1.2 mmol), (2-metoxietil)metilamina (0.13 mL, 1.2 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.2 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2338 g, 1.56 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.7 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0693 g, 0.06 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.4 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.2514 g (55% de rendimiento, 4 etapas) de TRV-1383. 1H RMN (500 MHz, CDCh) 8 = 7.75 (s, 1H), 7.68 (dt, J = 7.0, 2.0 Hz, 1H), 7.55-7.51 (m, 2H), 7.17 (d, J = 1.0 Hz, 1H), 6.28 (s, 1H), 5.13 (q, J = 7.0 Hz, 1H), 4.18 (t, J = 5.5 Hz, 2H), 3.68 (t, J = 5.5 Hz, 2H), 3.35 (s, 3H), 3.28 (s, 3H), 2.78 (br s, 1H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3327 g, 1.2 mmol), N-metiletanamida (0.10 mL, 1.2 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.2 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2338 g, 1.56 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.7 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0693 g, 0.06 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.4 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.2528 g (60% de rendimiento, 4 etapas) de TRV-1384. 1H RMN (500 MHz, CDCh) 8 = 7.74 (s, 1H), 7.66 (dt, J = 7.0, 2.0 Hz, 1H), 7.55-7.50 (m, 2H), 7.15 (d, J = 1.0 Hz, 1H), 6.23 (s, 1H), 5.13 (q, J = 7.0 Hz, 1H), 4.00 (q, J = 7.0 Hz, 2H), 3.22 (s, 3H), 2.78 (br s, 1H), 1.25 (t, J = 7.0 Hz, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3373 g, 1.2 mmol), tiomorfolina (0.12 mL, 1.2 mmol), NMP (2 mL) y DIPEA (0.21 mL, 1.2 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2338 g, 1.56 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución ac.) junto con DME (2.7 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0693 g, 0.06 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.35 mL, 2.4 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.2 mL, 4.2 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.1863 g (39% de rendimiento, 4 etapas) de TRV-1385. 1H RMN (500 MHz, CDCh) 8 = 7.73 (s, 1H), 7.65 (dt, J = 7.0, 2.0 Hz, 1H), 7.57-7.52 (m, 2H), 7.34 (d, J = 0.5 Hz, 1H), 6.55 (s, 1H), 5.14 (q, J = 6.5 Hz, 1H), 4.06 (m, 4H), 2.86 (m, 4H), 2.76 (br s, 1H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (8.9 g, 32.0 mmol) y ácido 3-formilbencenoborónico (5.037 g, 33.6 mmol) se cargaron a un matraz. El matraz se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (48 mL, solución ac.) junto con DME (72 mL). La solución se desgasificó durante 15 minutos y luego se agregó Pd(PPh3)4 (1.85 g, 1.6 mmol) todo de una vez. El matraz se calentó a 100°C durante 4 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron para dar 13.3 g de un sólido amarillo que fue una mezcla del producto deseado, material de partida sin reaccionar, el regioisómero incorrecto y el producto bis-acoplado. La purificación del material crudo a través de cromatografía (0, 5, 10, 15, 20% de gradiente de elución EtOAc/hexano) produjo 1.9412 g (20% de rendimiento) de 3-(7-bromobenzo[c][1,2,5]oxadiazol-5-il)benzaldehído. Este material (1.7903 g, 5.91 mmol) luego se disolvió en THF (12 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (1.75 mL, 11.8 mmol) y luego TBAF (0.6 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (22 mL, 22 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo se disolvió en THF (15 mL) y se enfrió a 0°C. Se agregó en forma de porciones NaH (0.2836 g, 7.09 mmol) y la reacción se agitó durante 10 minutos a 0°C antes de calentar a temperatura ambiente y agitar unos 30 minutos adicionales. La solución luego se volvió a enfriar y se agregó TBSCl (1.336 g, 8.87 mmol). La reacción se agitó durante la noche bajo argón. Se enfrió a 0°C y se detuvo con NH4Cl saturado (ac.) y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó mediante cromatografía (5% de EtOAc/hexano) para producir 1.2744 g (44 % de rendimiento, 3 etapas) del éter de sililo correspondiente como un sólido marrón. Este material (0.2732 g, 0.56 mmol) se disolvió en THF (5 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.31 mL, solución 2.0 M en ciclohexano, 0.62 mmol) y la solución se agitó durante 30 minutos antes de detenerla con NH4Cl saturado (ac.) y se dejó calentar a temperatura ambiente. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo se disolvió en THF (10 mL) y se enfrió a 0°C. Se agregó TBAF (1.2 mL, solución 1.0 M en THF) y la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción se detuvo con solución salina. La mezcla luego se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3832 g, 1.38 mmol), 4-(pirrolidin-1-il)piperidina (0.2127 g, 1.38 mmol), NMP (2 mL) y DIPEA (0.24 mL, 1.38 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2698 g, 1.8 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (2.1 mL, solución ac.) junto con DME (3.1 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0797 g, 0.069 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche.
Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.8 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.41 mL, 2.76 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.8 mL, 4.8 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (10% de MeOH/DCM) para producir 0.1349 g (22% de rendimiento, 4 etapas) de TRV-1387. 1H RMN (500 MHz, CDCla) 8 = 7.73 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.53-7.46 (m, 2H), 7.25 (s, 1H), 6.48 (s, 1H), 5.08 (q, J = 6.5 Hz, 1H), 4.34-4.28 (m, 2H), 3.05-3.00 (m, 2H), 2.65 (br s, 4H), 2.30-2.27 (m, 1H), 2.06 2.03 (m, 2H), 1.84 (br s, 4H), 1.77-1.66 (m, 3H); 1H RMN (DMSO, 500 MHz) 8 = 7.91 (s, 1H), 7.83 (d, J = 10 Hz, 1H), 7.59 (d, J = 10 Hz, 1H), 7.55 (t, J = 10 Hz, 1H), 7.50 (s, 1H), 6.97 (d, J = 5 Hz, 1H), 6.73 (s, 1H), 5.31-5.26 (m, 1H), 4.20 (d, J = 10 Hz, 2H), 3.16 (t, J = 10 Hz, 2H), 2.52 (s, 4H), 2.26-2.22 (m, 1H), 2.00 (d, J = 10 Hz, 2H), 1.67 (s, 4H), 1.63-1.56 (m, 2H).

A una solución de 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3106 g, 1.12 mmol) y pirazol (0.0837 g, 1.23 mmol) en THF a -78°C se agregó NaHMDS (1.2 mL, solución 1.0 M en THF) en forma de gotas. La solución se agitó durante 30 minutos luego se calentó a temperatura ambiente. La solución luego se desgasifico durante 5 minutos antes de calentar a 50°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se detuvo con NH4Cl saturado (ac.) y se diluyó con EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2189 g, 1.46 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.7 mL, solución ac.) junto con DME (2.5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0647 g, 0.056 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrío en un baño de hielo. A esta solución se agregó CF3TMS (0.33 mL, 2.24 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (3.9 mL, 3.9 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtoAc/hexano) para producir 0.0342 g (8.5% de rendimiento, 4 etapas) de TRV-1388. 1H RMN (500 MHz, CDC13) 8 = 8.91 (d, J = 2.5 Hz, 1H), 8.33 (d, J = 1.5 Hz, 1H), 7.84-7.83 (m, 3H), 7.76 (dt, J = 7.5, 1.5 Hz, 1H), 7.59-7.52 (m, 2H), 6.62-6.61 (m, 1H), 5.15-5.12 (m, 1H), 3.40 (d, J = 4.0 Hz, 1H).
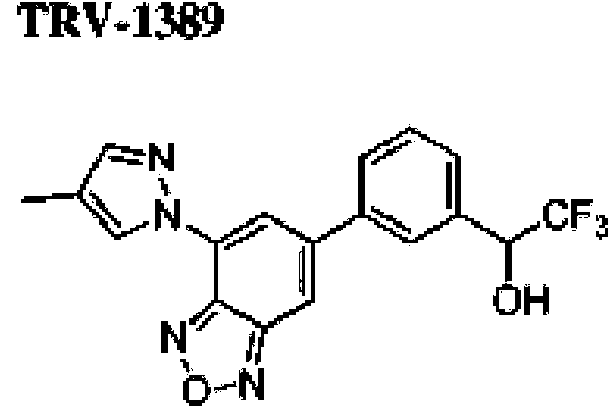
A una solución de 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3457 g, 1.24 mmol) y 4-metilpirazol (0.11 mL, 1.37 mmol) en THF a -78°C se agregó NaHMDS (1.3 mL, solución 1.0 M en THF) en forma de gotas. La solución se agitó durante 30 minutos luego se calentó a temperatura ambiente. La solución luego se desgasifico durante 5 minutos antes de calentar a 50°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se detuvo con
NH4CI saturado (ac.) y se diluyó con EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. La anilina cruda y el ácido 3-formilbencenoborónico (0.2413 g, 1.61 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Na2CO32M (1.9 mL, solución ac.) se agregó junto con DME (2.8 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0716 g, 0.062 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (2.5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.37 mL, 2.48 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.3 mL, 4.3 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (20% de EtOAc/hexano) para producir 0.0365 g (7.9% de rendimiento, 4 etapas) de TRV-1389. 1H RMN (500 MHz, CDC13) 8 = 8.68 (s, 1H), 8.28 (d, J = 1.0 Hz, 1H), 7.84 (s, 1H), 7.81 (d, J = 1.0 Hz, 1H), 7.76 (d, J = 7.5 Hz, 1H), 7.66 (s, 1H), 7.60-7.54 (m, 2H), 5.16-5.14 (m, 1H), 3.06 (d, J = 3.5 Hz, 1H), 2.23 (s, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3688 g, 1.33 mmol), 4-fluoro-N-metilbencilamina (0.18 mL, 1.39 mmol), NMP (3 mL) y DIPEA (0.26 mL, 1.5 mmol) se sellaron en un tubo y se calentaron a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron para dar 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina. La anilina cruda y el ácido 3-formilbencenoborónico (0.2593 g, 1.73 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (2.0 mL, solución ac.) junto con DME (3.0 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0768 g, 0.066 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo luego se disolvió en THF (4.0 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.39 mL, 2.66 mmol) y luego TBAF (0.1 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (4.7 mL, 4.7 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.2675 g (47 % de rendimiento, 4 etapas) de TRV-1390. 1H RMN (500 MHz, CDC13) 8 = 7.70 (s, 1H), 7.64 (dt, J = 7.0, 2.0 Hz, 1H), 7.55-7.50 (m, 2H), 7.27-7.24 (m, 3H), 7.04-7.00 (m, 2H), 6.34 (s, 1H), 5.12-5.10 (m, 3H), 3.19 (s, 3H), 2.71 (s, 1H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (8.9 g, 32.0 mmol) y ácido 3-formilbencenoborónico (5.037 g, 33.6 mmol) se cargaron a un matraz. El matraz se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (48 mL, solución ac.) junto con DME (72 mL). La solución se desgasificó durante 15 minutos y luego se agregó Pd(PPh3)4 (1.85 g, 1.6 mmol) todo de una vez. El matraz se calentó a 100°C durante 4 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron para dar 13.3 g de un sólido amarillo que fue una mezcla del producto deseado, material de partida sin reaccionar, el regioisómero incorrecto, y el producto bis-acoplado. La purificación del material crudo a través de cromatografía (0, 5, 10, 15, 20% de gradiente de elución EtOAc/hexano) produjo 1.9412 g (20% de rendimiento) de 3-(7-bromobenzo[c][1,2,5]oxadiazol-5-il)benzaldehído. Este material (1.7903 g, 5.91 mmol) luego se disolvió en THF (12 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (1.75 mL, 11.8 mmol) y luego TBAF (0.6 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (22 mL, 22 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material crudo se disolvió en THF (15 mL) y se enfrió a 0°C. Se agregó en forma de porciones NaH (0.2836 g, 7.09 mmol) y la reacción se agitó durante 10 minutos a 0°C antes de calentar a temperatura ambiente y agitar unos 30 minutos adicionales. La solución luego se volvió a enfriar y se agregó TBSCl (1.336 g, 8.87 mmol). La reacción se agitó durante la noche bajo argón. se enfrió a 0°C y se detuvo con NH4Cl saturado (ac.) y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó mediante cromatografía (5% de EtOAc/hexano) para producir 1.2744 g (44 % de rendimiento, 3 etapas) de sólido marrón. Este sólido (0.2647 g, 0.543 mmol) se disolvió en THF (5 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.30 mL, solución 2.0 M en ciclohexano, 0.60 mmol) y la solución se agitó durante 30 minutos antes de agregar DMF (0.050 mL, 0.652 mmol). La mezcla se dejó calentar lentamente a temperatura ambiente. La solución luego se volvió a enfriar a 0°C y se detuvo con NH4Cl saturado (ac.). Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el aldehído crudo. El aldehído se purificó a través de cromatografía instantánea (10% de EtOAc/hexano) para dar 0.0913 g (39% de rendimiento). Este aldehído luego se disolvió en DCM (1 mL) y se agregó pirrolidina (0.026 mL, 0.313 mmol). A esta mezcla luego se agrego NaHB(OAc)3 (0.0886 g, 0.418 mmol) con agitación vigorosa y la reacción se agitó durante la noche. La reacción se detuvo con NaHCO3 saturado (ac.) y se extrajo con DCM. Los extractos combinados se lavaron con agua, solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material luego se volvió a disolver en THF (2 mL) y se enfrió a 0°C. Se agregó TBAF (0.42 mL, solución 1.0 M en THF) y la mezcla se agitó durante la noche bajo argón. La reacción se detuvo con solución salina y se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4) se filtraron y se concentraron. El material crudo se purificó a través de cromatografía instantánea (5% de MeOH/DCM) para producir 0.0181 g (23% de rendimiento) de TRV-1391. 1H RMN (CDC13, 500 MHz) 8 = 7.84 (t, J = 0.5 Hz, 1H), 7.78 (s, 1H), 7.72-7.70 (m, 2H), 7.58-7.54 (m, 2H), 5.15 (q, J = 7.0 Hz, 1H), 4.12 (s, 2H), 2.69 (s, 4H), 1.86-1.84 (m, 4H).
Se disolvió N-bencil-6-bromo-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.3720 g, 1.17 mmol) en THF (6 mL) bajo argón, y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.65 mL, solución 2.0 M en ciclohexano) formando una solución de coloro rojo profundo. Esta mezcla se agitó durante 30 minutos a -78°C y luego se agregó rápidamente DMF (0.11 mL, 1.4 mmol). La mezcla se dejó calentar lentamente a temperatura ambiente. Luego se volvió a enfriar a 0°C y se detuvo con NH4Cl saturado (ac.). Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el aldehído crudo. El aldehído se purificó a través de cromatografía instantánea (10% de EtOAc/hexano) para dar 0.1737 g (56% de rendimiento) del aldehído 4. Este material (0.1737 g, 0.546 mmol) luego se disolvió en Th F (2.0 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.16 mL, 1.09 mmol) y luego TBAF (0.06 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (2.0 mL, 2.0 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (15% de EtOAc/hexano) para producir 0.1094 g (60% de rendimiento) de TRV-1392. 1H RMN (500 MHz, CDC13) 8 = 7.38-7.35 (m, 2H), 7.33-7.28 (m, 4H), 6.26 (s, 1H), 5.17 (s, 2H), 5.05 (q, J = 6.5 Hz, 1H), 3.23 (s, 3H), 2.79 (br s, 1 H).

Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.3753 g, 1.12 mmol) en THF (6 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.62 mL, solución 2.0 M en ciclohexano, 1.23 mmol) y la solución se agitó durante 30 minutos antes de agregar DMF (0.10 mL, 1.34 mmol). La mezcla se agitó bajo argón a -78°C. Después de 3 horas, se agregó NH4Cl saturado (ac.) y luego la mezcla se dejó calentar a temperatura ambiente. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el aldehído crudo. El aldehído se purificó a través de cromatografía instantánea (10% de EtOAc/hexano) para dar 0.1846 g (58 % de rendimiento). Este material (0.1846 g, 0.647 mmol) luego se disolvió en THF (3 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.19 mL, 1.3 mmol) y luego TBAF (0.06 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (2.3 mL, 2.26 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material se purificó a través de cromatografía (20% de EtOAc/hexano) para dar 0.0794 g (34 % de rendimiento) TRV-1397 como un aceite rojo. 1H RMN (500 MHz, CDC13) 8 = 7.26 (s, 1H), 7.24-7.22 (m, 2H), 7.03-6.99 (m, 2H), 6.23 (s, 1H), 5.09 (s, 2H), 5.02 (q, J = 7.0 Hz, 1H), 3.15 (s, 3H), 2.67 (br s, 1H).
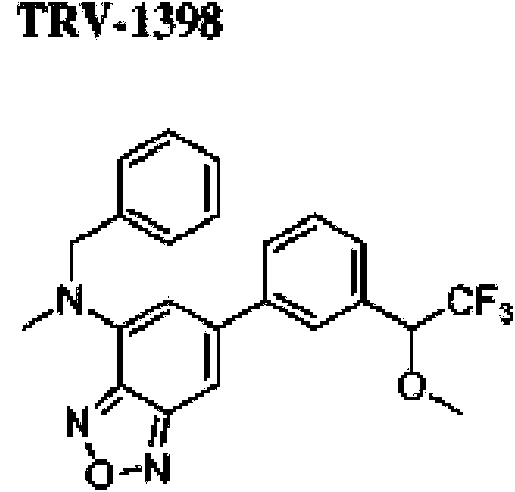
El TRV-1378 (83 mg, 0.2 mmol) se disolvió en THF (2 mL) y se agregó en forma de gotas a una suspensión de NaH (50 mg) se agitó en THF (1 mL) a 0°C. Una vez se completó la adición el baño frío se retiró y reemplazó con un baño de agua a temperatura ambiente. Después de 5 min la reacción se enfrío de nuevo a 0°C y se agregó Mel (100 pL, 0.8 mmol). El baño frío se dejó en su lugar y se le permitió a la reacción llegar a la temperatura ambiente durante la noche. Luego de un tratamiento final estándar y cromatografía instantánea (9:1 Hex/EtOAc) el producto se aisló
como un sólido naranja (54 mg, 63% de rendimiento). 1H RMN (500 MHz, CDCI3) 8 = 7.68 (m, 2H), 7.54 (m, 2H), 7.37 (m, 2H), 7.32 (m, 4H), 6.38 (s, 1H), 5.20 (s, 2H0, 4.61 (q, J = 7 Hz, 1H), 3.52 (s, 3H), 3.29 (s, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (0.9837 g, 3.53 mmol), etilamina (0.29 mL, 3.53 mmol), NMP (5 mL) y DIPEA (0.61 mL, 3.53 mmol) se sellaron en un tubo y se calentaron a 60°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O, HCl 1 N (ac.), NaHCO3 saturado (ac.), H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (3% de EtOAc/hexano) para producir 0.3952 g (46% de rendimiento). Este material (0.1298 g, 0.54 mmol) se disolvió en AC2O (5 mL) y se calentó a 140°C durante 48 horas. Después de enfriar a temperatura ambiente el material se concentró para producir 4 en 98 % de rendimiento. Este material (0.1499 g, 0.52 mmol) se agregó a un tubo junto con ácido 3-formilbenzenborónico (0.1013 g, 0.676 mmol) y el tubo se purgó y se evacuó con argón (3 veces). Se agregaron Na2CO3 (1.6 mL, 3.12 mmol, solución acuosa 2 M) y DME (2.3 mL) y la solución se desgasificó durante 10 minutos. Finalmente se agregó Pd(PPh3)4 (0.030 g, 0.026 mmol), el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la mezcla se diluyó con EtOAc y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3X). Las capas orgánicas combinadas se lavaron con H2O (4X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar a aldehido crudo 6. Este aldehido luego se disolvió en THF (3 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (0.08 mL, 0.52 mmol) y luego TBAF (0.05 mL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó durante unas 2 horas adicionales. La solución luego se volvió a enfriar a 0°C y se agregó TBAF (1.3 mL, 1.3 mmol), la mezcla se dejó calentar a temperatura ambiente durante la noche. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. Este material se purificó a través de cromatografía (40% de EtOAc/hexano) para dar 0.0327 g (17 % de rendimiento durante 3 etapas) de TRV-1399 como sólido amarillo. 1H RMN (500 MHz, CDC13) 8 = 7.98 (s, 1H), 7.78 (s, 1H), 7.68-7.67 (m, 1H), 7.61-7.57 (m, 2H), 7.52 (s, 1H), 5.19-5.14 (m, 1H), 3.97 (q, J = 10 Hz, 2H), 3.33 (d, J = 5 Hz, 1H), 2.02 (br s, 3H), 1.21 (t, J = 10 Hz, 3H).

Se agregó CsF (0.0052 g, 0.034 mmol) a una mezcla de TRV-1402 (0.1162 g, 0.34 mmol) y CF3TMS (0.10 mL, 0.68 mmol) en THF (3 mL) a temperatura ambiente. Esta solución se agitó hasta que se obtuvo el 100% de conversión del material de partida y luego se agregó TBAF (1.2 mL, solución 1.0 M en THF), este se agitó durante unas 16 horas adicionales. La reacción se detuvo con solución salina y se diluyó con EtOAc y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de columna de 10% de EtOAc/hexano y luego de nuevo con una columna de 5% de EtOAc/hexano para producir 0.020 g (13% de rendimiento) de TRV-1400 como sólido naranja. 1H RMN (500 MHz, CDC13) 8 = 7.24-7.21 (m, 2H), 7.17 (d, J = 15 Hz, 1H), 7.16 (s, 1H), 7.03-6.99 (m, 2H), 6.23 (d, J = 15 Hz, 1H), 6.18 (s, 1H), 5.11 (s, 2H), 3.30 (br s, 1H), 3.17 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (372 mg, 1.6 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó isoindolina (238 mg, 2 mmol) y trietilamina (400 j L, 2.9 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (9:1 Hex/EtOAc) para dar 208 mg (41% de rendimiento) de 6-bromo-4-(isoindolin-2-il)benzo[c][1,2,5]oxadiazol. 1H RMN (300 MHz, CDCh) 8 = 7.38 (m, 4H), 7.26 (s, 1H), 6.14 (s, 1H), 5.14 (s, 4H). A una solución del material mencionado anteriormente, (200 mg, 0.6 mmol) en d Me (4 mL)/Na2CO3 (0.9 mL) se agregó ácido 3-formil-fenilborónico (134 mg, 0.9 mmol) y Pd(P(Ph)3)4 (35 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (2 g) y se purificó mediante cromatografía de columna flash (2:1 DCM/Hex) para dar 190 mg (92% de rendimiento). 1H RMN (500 MHz, CDCl3) 8 = 10.18 (s, 1H), 8.24 (m, 1H), 7.99 (dm, J = 8 Hz, 2H), 7.70 (t, J = 7 Hz, 1H), 7.44 (m, 4H), 7.26 (s, 1H), 6.30 (s, 1H), 5.25 (s, 4 H). A una solución agitada del material mencionado anteriormente, (190 mg, 0.55 mmol) y reactivo de Rupert (150 mg, 1.1 mmol) en DCM (5 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se pasó a través de un tapón de SiO2 (DCM) y luego se purificó mediante cromatografía de columna flash (2:1 DCM/Hex). 1H RMN (500 MHz, CDCh) 8 = 7.84 (s, 1H), 7.77 (dt, J = 7 Hz, 2 Hz, 1H), 7.60 (m, 2H), 7.43 (m, 4H), 7.23 (s, 1H), 6.25 (s, 1H), 5.31 (s, 4H), 5.2 (m, 1H), 2.68 (s, 1H).
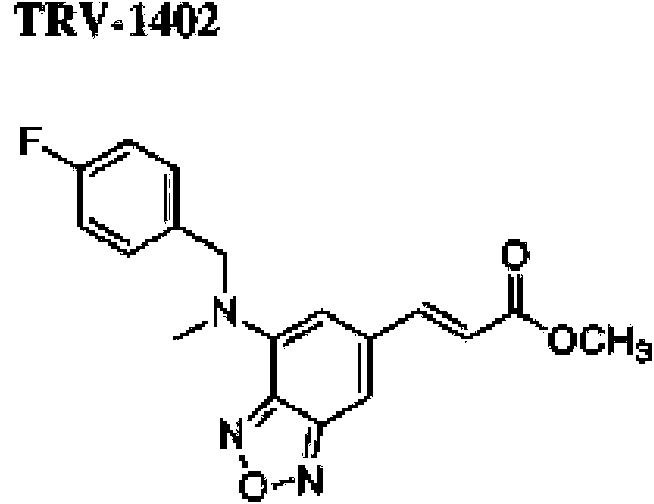
Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (1.5615 g, 4.65 mmol) en THF (45 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (2.6 mL, solución 2.0 M de ciclohexano) y la solución se agitó durante 30 minutos a -78°C. Se agregó DMF (0.54 mL, 7.0 mmol) y la reacción se agitó a -78°C durante 3 horas. Esta reacción luego se detuvo con NH4Cl saturado (ac.) y se dejó calentar lentamente a temperatura ambiente. Esta mezcla se extrajo con EtOAc y las capas orgánicas combinadas se lavaron con H2O (3X) solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el aldehído crudo. Este material se purificó a través de columna de 10% de EtOAc/hexano para producir 0.7248 g (55% de rendimiento) de aldehído. Este aldehído (0.5615 g, 1.97 mmol) en se agregó THF (5 mL) a una suspensión agitada de NaH (0.104 g, 2.6 mmol) y fosfonoacetato de trimetilo (0.31 mL, 2.17 mmol) en THF (20 mL) a 0°C. Después de que se completa la adición la mezcla se agitó durante la noche mientras que se calienta a temperatura ambiente. Después de volver a enfriar a 0°C la reacción se detuvo con NH4Cl saturado (ac.). Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de 20% de EtOAc/hexano para dar 0.5945 g (88 % de rendimiento) de TRV-1402 como el transisómero. 1H RMN (500 MHz, CDC13) 8 = 7.64 (d, J = 15 Hz, 1H), 7.25 (s, 1H), 7.23-7.21 (m, 2H), 7.03 7.00 (m, 2H), 6.46 (d, J = 15 Hz, 1H), 6.25 (s, 1H), 5.10 (s, 2H), 3.83 (s, 3H), 3.15 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (353 mg, 1.5 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó clorhidrato de 3-hidroxiazetidina (180 mg, 1.65 mmol) y trietilamina (635 j L, 4.5 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtoAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM) para dar 264 mg (65% de rendimiento) de 1-(6- bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol. 1H RMN (300 Mhz , CDCla) 8 = 7.23 (s, 1H), 5.95 (s, 1H), 4.92 (m, 1H), 4.59 (m, 2H), 4.15 (m, 2H), 2.20 (d, J = 6 Hz, 1H). A una solución agitada a 0°C de 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (480 mg, 2 mmol) en NMP (12 mL) se agregó Mel (1.4 g, 10 mmol) seguido por NaH (200 mg, 8.5 mmol). Con el baño frío en su lugar se le permitió a la reacción llegar a RT. Después de 16 horas la reacción se detuvo cuidadosamente con agua, se trató con NH4Cl saturado y se extrajo en EtOAc (3 X 20 mL). Los orgánicos combinados se lavaron con HCl (2M) seguido por solución salina, se secaron con MgSO4 y se concentraron en vacío. El material se purificó mediante cromatografía de succión en seco (1:1 DCM/Hex) para dar 46 mg (71% de rendimiento). 1H RMN (300 MHz, CDCla) 8 = 7.22 (d, J = 1 Hz, 1H), 5.92 (s, br, 1H), 4.44 (m, 3H), 4.16 (m, 2H), 3.37 (s, 3H). A una solución de TKW-I-92 (406 mg, 1.43 mmol) en DME (7 mL)/Na2CO3 (2.1 mL) se agregó ácido 3-formil-fenilborónico (322 mg, 2.1 mmol) y Pd(P(Ph)3)4 (80 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante filtración de succión en seco (DCM) para dar 411 mg de material. 1H RMN (500 MHz, CDCh) 8 = 10. 11 (s, 1H), 8.12 (m, 1H), 7.93 (dm, J = 8 Hz, 1H), 7.88 (dm, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 7.21 (m, 1H), 6.08 (m, 1H), 4.56 (dd, J = 10 Hz/5 Hz, 2H), 4.46 (m, 1H), 4.21 (dd, J = 10 Hz/4 Hz, 2H), 3.39 (s, 3H). A una solución agitada de TKW-I-93 (411 mg, 1.33 mmol) y reactivo de Rupert (378 mg, 2.7 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM) para dar 477 mg (94 % de rendimiento). 1H RMN (500 MHz, CDCl3) 8 = 7.72 (s, amplio, 1H), 7.65 (dt, J = 7 Hz/2 Hz, 2H), 7.51 (m, 2H), 7.17 (d, J = 1 Hz, 1H), 6.06 (s, 1H), 5.12 (m, 1H), 4.53 (m, 2H), 4.45 (m, 1H), 4.18 (dm, J = 10 Hz, 2H), 3.38 (s, 3H).
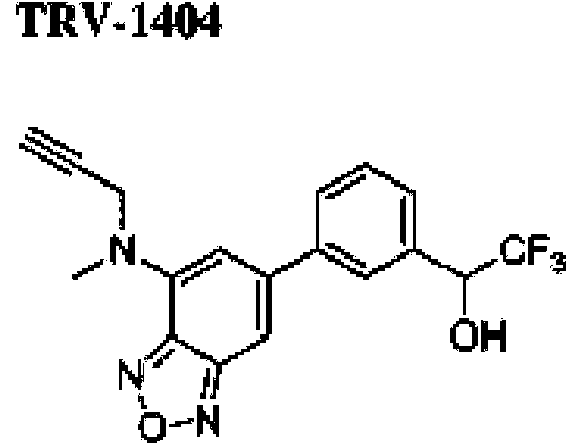
A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (355 mg, 1.52 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó N-metil propargilamina (92 mg, 1.67 mmol) y trietilamina (635 j L, 4.5 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. Debido a la naturaleza volátil del SM del material crudo solo se convirtió el 50%. La mezcla se utilizó sin purificación adicional. 1H RMN (300 MHz, CDCh) 8 = 8.03 (s, 1H, SM), 7.55 (s, 1H, SM), 7.41 (s, 1H), 6.35 (s, 1H), 4.66 (s, 2H), 3.24 (s, 3H), 2.27 (s, 1H). A una solución de TKW-I-81 (180 mg, 0.7 mmol) en DME (3 mL)/Na2CO3 (1.0 mL) se agregó ácido 3-formil-fenilborónico (152 mg, 1.0 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (9:1 Hex/EtOAc) para dar 58 mg de material. 1H RMN (500 MHz, CDCh) 8 = 10.12 (s, 1H), 8.17 (m, 1H), 7.99 (dm, J = 8 Hz, 1H), 7.96 (dm, J = 8 Hz, 1H), 7.39 (s, 1H), 6.54 (s, 1H), 4.71 (s, 2H), 3.31 (s, 3H), 2.28 (s, 1H). A una solución agitada de TkW-I-87 (277 mg, 0.94 mmol) y reactivo de Rupert (400 mg, 2.8
mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (50-80% de DCM en Hex). 1H RMN (300 MHz, CDCls) 8 = 7.76 (m, 1H), 7.69 (m, 1H), 7.55 (m, 2H), 7.34 (d, J = 1 Hz, 1H), 6.55 (d, J = 1 Hz, 1H), 5.14 (m, 1H), 4.68 (d, J = 2 Hz, 2H), 3.30 (s, 3H), 2.69 (d, J = 4 Hz, 1H), 2.29 (t, J = 2 Hz, 1H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (388 mg, 1.66 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó ciclopropilamina (104 mg, 1.83 mmol) y trietilamina (694 pL, 4.9 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM) para dar 264 mg (65% de rendimiento). 1H RMN (500 MHz, CDCla) 8 = 7.32 (m, 1H), 6.60 (m, 1H), 5.46 (s, amplio, 1H), 2.65 (m, 1H), 0.94 (m, 2H), 0.74 (m, 2H). A una solución de TKW-I-88 (214 mg, 0.8 mmol) en DME (3 mL)/Na2CO3 (1.0 mL) se agregó ácido 3-formil-fenilborónico (240 mg, 1.6 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (3 g) y se purificó mediante cromatografía de columna flash (9:1 Hex/EtOAc) para dar 100 mg (42% de rendimiento) de material. 1H RMN (300 MHz, CDCh) 8 = 10.13 (s, 1H), 8.19 (m, 1H), 7.94 (m, 2H), 7.69 (t, J = 7 Hz, 1H), 7.32 (m, 1H), 6.83 (m, 1H), 3.57 (s, 3H), 2.77 (m, 1H), 1.01 (m, 2H), 0.76 (m, 2H). A una solución agitada de TKW-I-91 (277 mg, 0.94 mmol) y reactivo de Rupert (400 mg, 2.8 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (10-15% de EtOAc en Hex) para dar 100 mg (80% de rendimiento). 1H RMN (300 MHz, CDCh) 8 = 7.77 (s, 1H), 7.68 (m, 1H), 7.53 (m, 2H), 7.28 (m, 1H), 6.82 (d, J = 1 Hz, 1H), 5.13 (m, 1H), 3.54 (s, 3H), 2.67 (m, 2H), 0.96 (m, 2H), 0.75 (m, 2H).

A una solución de 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (260 mg, 0.9 mmol) en DME (6 mL)/Na2CO3 (1.5 mL) se agregó ácido 3-formil-fenilborónico (225 mg, 0.9 mmol) y Pd(P(Ph)3)4 (45 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío para dar 277 mg. El material crudo fue un solo punto por TLC y se utilizó sin purificación adicional. 1H RMN (300 MHz, CDCh) 8 = 10.12 (s, 1H), 8.14 (s, 1H), 7.98 (dm, J = 7 Hz, 1H), 7.93 (dm, J = 8 Hz, 1H), 7.70 (t, J = 7 Hz, 1H), 7.24 (s, 1H), 6.11 (s, 1H), 4.95 (m, 1H), 4.63 (m, 2H), 4.19 (2H), 2.28 (m, 1H). A una solución agitada de aldehído (277 mg, 0.94 mmol) y reactivo de Rupert (400 mg, 2.8 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con d Cm . Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (40% de EtOAc en Hex) para dar 147 mg (42% de rendimiento) de TRV-1406. 1H RMN (500 MHz, CDCh) 8 = 7.76 (s, 1H), 7.69 (dt, J
= 7 Hz, 2 Hz, 1H), 7.57 (m, 2H), 7.23 (s, 1H), 6.12 (s, 1H), 5.16 (q, J = 6 Hz, 1H), 4.96 (m, 1H), 4.65 (dd, J = 10 Hz/6 Hz, 2H), 4.20 (dd, J = 10 Hz/6 Hz, 2H), 2.71 (s, amplio, 1H), 2.22 (s, amplio, 1H).

Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.2959 g, 0.88 mmol) en THF (5 mL) y se enfrió a -78°C. Se agregó nBuLi (0.51 mL, 1.01 mmol, solución 2.0 M en ciclohexano) en forma de gotas y la solución se agitó durante 20 minutos a -78°C. Se agregó I2 (1.3 mL, solución 1.0 M en THF) y la solución se dejó calentar lentamente a 0°C durante la noche. La reacción se detuvo con NH4Cl saturado (ac.) y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de columna de 3% de EtOAc/hexano para proporcionar 0.1381 g (41% de rendimiento, 89% c.p.) de yoduro de arilo 8. El yoduro de arilo (0.1332 g, 0.348 mmol) y propiolato de metilo (0.12 mL, 1.39 mmol) se agregaron a un tubo. El tubo se evacuó y se purgó con argón (3X). Se agregaron Pd(PPha)2Cl2 (0.0122 g, 0.0174 mmol), CuI (0.0066 g, 0.0348 mmol) y K2CO3 (0.0962 g, 0.696 mmol). El tubo se selló y calentó a 65 °C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se lavó con H2O (3X), solución salina, se secó (Na2SO4), se filtró y se concentró. El material crudo se purificó a través de columna de 10% de EtOAc/hexano para producir 0.0154 g (13% de rendimiento) de TRV-1408. 1H RMN (500 MHz, CDC13) 8 = 7.42 (s, 1H), 7.21-7.18 (m, 2H), 7.03-7.00 (m, 2H), 6.17 (s, 1H), 5.11 (s, 2H), 3.86 (s, 3H), 3.13 (s, 3H).

A una solución agitada de TRV-1402 (0.5283 g, 1.55 mmol) en DCM (10 mL) a -78°C se agregó en forma de gotas DIBAL (3.6 mL, solución 1.0 M en hexano). Una vez se completó la adición la reacción se calentó a -40°C y se agitó bajo argón hasta el consumo completo del material de partida. Se detuvo con una solución saturada de sal de Rochelle y se agitó durante 30 minutos. Esta mezcla se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El aceite crudo se purificó a través de columna de 50% de EtOAc é hexano para producir 0.3740 g (77 % de rendimiento) de TRV-1409 como un aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.23-7.21 (m, 2H), 7.03 (s, 1H), 7.02-6.98 (m, 2H), 6.64 (d, J = 15 Hz, 1H), 6.44 (dt, J = 15, 5 Hz, 1H), 6.26 (s, 1H), 5.06 (s, 2H), 4.38 (dd, J = 5 Hz, 2H), 3.11 (s, 3H).
Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.1444 g, 0.43 mmol) en THF (5 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.23 mL, 0.45 mmol, solución 2.0 M de ciclohexano) y la mezcla se agitó durante 30 minutos. Luego se agrego cloroformato de metilo (0.050 mL, 0.65 mmol) y la reacción se dejó calentar lentamente a temperatura ambiente. Después de volver a enfriar a 0°C la reacción se detuvo con NH4C saturado (ac.). Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de 15% de EtOAc/hexano para producir 0.0234 g (17 % de rendimiento) de TRV-1410. 1H RMN (500 MHz, CDC13) 8 = 7.88 (d, J = 5 Hz, 1H), 7.22-7.19 (m, 2H), 7.03-6.98 (m, 2H), 6.75 (s, 1H), 5.12 (s, 2H), 3.95 (s, 3H), 3.17 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (396 mg, 1.7 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó clorhidrato de 3-hidroxipirrolidina (230 mg, 1.83 mmol) y trietilamina (710 pL, 5.1 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtoAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. La anilina cruda se utilizó sin purificación adicional. A una solución agitada a 0°C de anilina (480 mg, 1.7 mmol) en NMP (10 mL) se agregó Mel (1.06 mL mg, 17 mmol) seguido por NaH (200 mg, 8.5 mmol). Con el baño frío en su lugar se le permitió a la reacción llegar a RT. Después de 16 horas la reacción se detuvo cuidadosamente con agua, se trató con NH4Cl saturado y se extrajo en EtOAc (3 X 20 mL). Los orgánicos combinados se lavaron con HCl (2M) seguido por solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material obtenido se utilizó sin purificación adicional. A una solución del éter mencionado anteriormente (1.7 mmol) en DME (8 mL)/Na2CO3 (2.5 mL) se agregó ácido 3-formilfenilborónico (382 mg, 2.6 mmol) y Pd(P(Ph)3)4 (80 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía instantánea (4:1 Hex/EtOAc) para dar 415 mg de material. 1H RMN (500 MHz, CDCh) 8 = 10.11 (s, 1H), 8.15 (s, 1H), 7.91 (m, 2H), 7.64 (t, J = 8 Hz, 1H), 7.14 (s, 1H), 6.14 (s, 1H), 4.94 (m, 1H), 3.95 (m, 4H), 3.41 (s, 3H), 2.17 (m, 2H). A una solución agitada de aldehído (415 mg, 1.33 mmol) y reactivo de Rupert (365 pL, 2.6 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (10-30% de EtOAc en Hex) para dar 240 mg (48 % de rendimiento) de TRV-1411, una mezcla de 1:1:1:1 de diastereómeros debido a los dos centros quirales. 1H RMN (500 MHz, d Ms O-D6) 8 = 7.90 (s, 1H), 7.82 (dm, J = 7 Hz, 1H), 7.53 (m, 2H), 7.29 (s, 1H), 6.93 (d, J = 6 Hz, 1H), 6.29 (s, 1H), 5.28 (m, 1H), 4.18 (s, 1H), 3.87 (m, 3H), 3.77 (m, 1H), 2.15 (m, 2H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (355 mg, 1.52 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó N-metil-N-(2-piridinilmetil)amina (204 mg, 1.67 mmol) y trietilamina (400 pL, 2.9 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtoAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (4:1 Hex/EtoAc) para dar 293 mg (60% de rendimiento) de anilina. A una solución de esta anilina (293 mg, 0.91 mmol)
en DME (5 mL)/Na2CO3 (1.4 mL) se agregó ácido 3-formilfenilborónico (202 mg, 1.4 mmol) y Pd(P(Ph)3)4 (50 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (3 g) y se purificó mediante cromatografía de columna flash (4:1 Hex/EtOAc) para dar 280 mg de aldehído. 1H RMN (500 MHz, CDCl3) 8 = 10.01 (s, 1H), 8.60 (dm, J = 4 Hz, 1H), 8.09 (t, J = 2 H, 1H), 7.93 (dt, J = 7 Hz/2 Hz, 1H), 7.87 (dm, J = 8 Hz, 1H), 7.64 (m, 2H), 7.28 (m, 2H), 7.20 (m, 1H), 6.39 (s, 1H), 5.28 (s, 2H), 3.37 (s, 3H). A una solución agitada de aldehído (280mg, 0.81 mmol) y reactivo de Rupert (230 mg, 1.63 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (10% de EtOAc en Hex) para dar 120 mg (35% de rendimiento) de TRV-1412. 1H RMN (500 MHz, DMSO-Ds) 8 = 8.51 (d, J = 6 Hz, 1H), 7.87 (s, 1H), 7.80 (dm, J = 8 Hz, 1H), 7.74 (td, J = 8 Hz/2 Hz, 1H), 7.57 (m, 2H), 7.36 (s, 1H), 7.32 (d, J = 8 Hz, 1H), 7.26 (m, 1H), 6.95 (d, J = 5 Hz, 1H), 5.27 (m, 3H), 3.36 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (360 mg, 1.54 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó N-metil-N-(3-piridinilmetil)amina (207 mg, 1.69 mmol) y trietilamina (400 pL, 2.9 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtoAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (5% de EtOAc en DCM) para dar 247 mg (50% de rendimiento) de anilina. 1H RMN (500 MHz, DMSO-D6) 8 = 8.50 (d, J = 2 Hz, 1H), 8.48 (dd, J = 5 Hz/1 Hz, 1H), 7.64 (dm, J = 8 Hz, 1H), 7.52 (d, J = 1 Hz, 1H), 7.35 (dd, J = 8 Hz/5 Hz, 1H), 6.35 (m, 1H), 5.17 (s, 2H), 3.21 (s, 3H). A una solución de anilina (247 mg, 0.77 mmol) en DME (4 mL)/Na2CO3 (1.1mL) se agregó ácido 3-formil-fenilborónico (173 mg, 1.2 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío para dar 312 mg de aldehído crudo. A una solución agitada de aldehído (0.77 mmol) y reactivo de Rupert (400 mg, 2.8 mmol) en DCM (8 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (2% de MeOH en DCM). 1H RMN (300 MHz, CDCh) 8 = 8.51 (m, 2H), 7.70-7.55 (m, 3H), 7.50-7.45 (m, 2H), 7.28 (m, 1H), 7.25 (m, 1H), 6.35 (s, 1H), 5.14 (m, 3H), 4.24 (s, amplio, 1H), 3.19 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (357 mg, 1.5 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó 4-metoxibencilamina (230 mg, 1.65 mmol) y trietilamina (400 pL, 2.9 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se
concentró en vacío. La anilina cruda se utilizó sin purificación adicional. A una solución agitada a 0°C de anilina (220 mg, 0.6 mmol) en NMP (5 mL) se agregó NaH (151 mg, 6.3 mmol). Después de que la evolución de gas ha cesado se agregó en forma de gotas MeI (446 mg, 3.1 mmol). Con el baño frío en su lugar se le permitió a la reacción llegar a RT. Después de 16 horas la reacción se detuvo cuidadosamente con agua, se trató con NH4Cl saturado y se extrajo en EtOAc (3 X 20 mL). Los orgánicos combinados se lavaron con solución salina, se secaron con MgSO4 y se concentraron en vacío. La anilina obtenida se utilizó sin purificación adicional. A una solución de anilina (0.6 mmol) en DME (4 mL)/Na2CO3 (1.0 mL) se agregó ácido 3-formil-fenilborónico (142 mg, 0.9 mmol) y Pd(P(Ph)3)4 (45 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío para dar 220 mg de material crudo. 1H RMN (300 MHz, CDCh) 8 = 10.1 (s, 1H), 8.1 (s, 1H), 7.9 (m, 2H), 7.7 (m, 1H), 7.2 (m, 2H), 6.9 (d, J = 6 Hz, 2H), 6.4 (m, 1H), 5.1 (s, 2H), 3.8 (s, 3H), 3.2 (s, 3H). A una solución agitada de aldehído (220 mg, 0.59 mmol) y reactivo de Rupert (168 mg, 1.2 mmol) en d Cm (6 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (20% de EtOAc en Hex). 1H RMN (300 MHz, CDCh) 8 = 7.7 (s, 1H), 7.6 (dt, J = 7 Hz, 1 Hz, 1H), 7.5 (m, 2 H), 7.19 (m, 3H), 6.8 (d, J= 8 Hz, 2 H), 6.3 (s, 1H), 5.1 (m, 1H), 5.0 (s, 2H), 3.8 (s, 3H), 3.2 (s, 3H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (345 mg, 1.5 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó 3,5-dimetoxibencilamina (272 mg, 1.6 mmol) y trietilamina (600 pL, 4.3 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se utilizó sin purificación adicional. A una solución agitada a 0°C de anilina (1.5 mmol) en NMP (5 mL) se agregó NaH (211 mg, 8.8 mmol). Después de que la evolución de gas ha cesado se agregó en forma de gotas MeI (570 mg, 4 mmol). Con el baño frío en su lugar se le permitió a la reacción llegar a RT. Después de 16 horas la reacción se detuvo cuidadosamente con agua, se trató con NH4Cl saturado y se extrajo en EtOAc (3 X 20 mL). Los orgánicos combinados se lavaron con solución salina, se secaron con MgSO4 y se concentraron en vacío. El material crudo se fusionó a SiO2 y se destiló (10% de EtOAc en Hex) para dar 163 mg de material. A una solución de anilina (165 mg, 0.44 mmol) en DME (4 mL)/Na2CO3 (0.7 mL) se agregó ácido 3-formilfenilborónico (98 mg, 0.9 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío para dar 277 mg. El material crudo se utilizó sin purificación adicional. A una solución agitada de aldehído (277 mg, 0.4 mmol) y reactivo de Rupert (125 mg, 0.8 mmol) en DCM (4 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se fusionó a SiO2 (3 g) se purificó mediante cromatografía de columna flash (10-20% de EtOAc en Hex). 1H RMN (500 MHz, CDCh) 8 = 7.7 (s, 1H), 7.6 (dt, 7 Hz, 1 Hz, 2H), 7.5 (m, 2H), 7.2 (s, 1H), 6.4 (m, 1H), 6.3 (m, 2H), 5.1 (m, 1H), 5.0 (s, 2H), 3.7 (s, 3H), 3.3 (s, 3H), 2.7 (s, amplio, 1H)

El TRV-1397 (0.0768 g, 0.22 mmol) se disolvió en THF (5 mL) y se enfrió a 0°C. NaH (0.0132 g, 0.33 mmol) se agregó y la suspensión se agitó durante 30 minutos. Luego se agrego yodometano 0.03 mL, 0.44 mmol) y la reacción se agitó durante la noche mientras que se calienta a temperatura ambiente. La mezcla luego se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó a través de una columna de 10% de EtOAc/hexano para producir 0.0259 g (32% de rendimiento) de TRV-1416 como aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.30-7.27 (m, 2H), 7.23 (s, 1H), 7.07-7.04 (m, 2H), 6.22 (s, 1H), 5.11 (s, 2H), 4.52 (q, J = 5 Hz, 1H), 3.50 (s, 3H), 3.20 (s, 3H).

A una solución de cloruro de oxalilo (0.032 mL, 0.37 mmol) en DCM (1 mL) a -78°C se agregó una solución de DMSO (0.024 mL, 0.34 mmol) en DCM (1.1 mL). La solución se agitó durante 5 minutos y luego se agregó una solución de TRV-1397 (0.1097 g, 0.31 mmol) en DCM (1.0 mL) y la mezcla se agitó unos 15 minutos adicionales. Se agregó en una porción TEA (0.22 mL, 1.55 mmol), la reacción se agitó durante 10 minutos a -78°C y luego se dejó calentar a temperatura ambiente. La mezcla luego se diluyó con agua y acetato de etilo. Las capas se separaron y la capa acuosa se extrajo de nuevo. Las capas orgánicas combinadas se lavaron con solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar la trifluorocetona cruda. Este material luego se purificó a través de cromatografía instantánea (30% de EtOAc/hexano) para dar 0.0356 g (32% de rendimiento) de TRV-1417 como aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.93 (s, 1H), 7.23-7.20 (m, 2H), 7.04-7.00 (m, 2H), 6.67 (s, 1H), 5.16 (s, 2H), 3.23 (s, 3H).

El TRV-1421 (0.3511 g, 1.17 mmol) se disolvió en DCM (50 mL) y se agregó reactivo de Dess-Martin (1.4845 g, 3.5 mmol). La reacción se agitó durante 40 minutos y luego se detuvo con NaHCO3 saturado (ac.) y exceso de Na2S2O3. La mezcla se agitó hasta que todos los sólidos se disolvieron y luego se extrajo varias veces con DCM. Los extractos orgánicos combinados se lavaron con NaHCO3 saturado, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de columna de 10% de EtOAc/hexano para producir 0.2322 g (66% de rendimiento) de TRV-1418 como sólido naranja. 1H RMN (500 MHz, CDC13) 8 = 7.74 (s, 1H), 7.22-7.19 (m, 2H), 7.02-6.97 (m, 2H), 6.74 (s, 1H), 3.17 (s, 3H), 2.65 (s, 3H).

A una solución agitada de 6-bromo-4-(isoindolin-2-il)benzo[c][1,2,5]oxadiazol (158 mg, 0.3 mmol) en THF (5 mL) a -78°C se agregó n-BuLi (0.25 mL, 2 M). Después de agitar durante 30 min a baja temperatura oxatenanona (72 mg, 1 mmol) se disolvió en THF (4 mL). El baño frío se retiró y se le permitió a la reacción llegar a RT y se monitorizó mediante TLC. La reacción se sometió a trabajo final con HCl diluido y se extrajo en EtOAc (3 X 20 mL). La fase orgánica se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (3 g) y se purificó mediante cromatografía de columna flash (DCM modificado con MeOH) para dar 40 mg de material. 1H RMN (300 MHz, DMSO-Da) 8 = 7.5 (m, 2H), 7.4 (m, 2H), 7.2 (s, 1H), 6.6 (s, 1H) 6.5 (s, 1H), 6.5 (s, 1H), 5.1 (s, 4H), 4.8 (m, 4H).

A una solución agitada de 6-bromo-4-(isoindolin-2-il)benzo[c][1,2,5]oxadiazol (98 mg, 0.3 mmol) en THF (3 mL) a -78°C se agregó n-BuLi (0.15 mL, 2 M). Después de agitar durante 30 min a baja temperatura se agregó DMF seco (0.5 mL) y la reacción se agitó durante una hora adicional antes se detuvo con MeOH seguido por HCl (4 M). Se le permitió a la reacción llegar a RT, se diluyó con agua y se extrajo en DCM (3 X 20 mL). La fase orgánica se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (3 g) y se purificó mediante cromatografía de columna flash (1:1 DCM/Hex) para dar 71 mg de material. A una solución agitada de aldehído (71 mg, 0.26 mmol) y reactivo de Rupert (74 mg, 0.52 mmol) en DCM (3 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM). 1H RMN (500 MHz, DMSO-D6) 8 = 7.50 (m, 2H), 7.37 (m, 2H), 7.27 (s, 1H),7.11 (d, J = 5 Hz, 1H), 6.32 (s, 1H), 5.29 (m, 1H), 5.09 (s. 4H).

Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.8514 g, 2.53 mmol) en THF (25 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (1.3 mL, solución 2.0 M en ciclohexano) a -78°C y la reacción luego se agitó durante 15-20 minutos. Luego se agregó acetaldehído (0.21 mL, 3.8 mmol) y la reacción se dejó calentar lentamente a temperatura ambiente. La solución se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó a través de una columna de 40% de EtOAc/hexano para producir 0.5340 g (70% de rendimiento) de TRV-1421 como aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.24-7.21 (m, 2H), 7.10 (s, 1H), 7.02-6.98 (m, 2H), 6.16 (s, 1H), 5.06 (s, 2H), 4.87 (q, J = 5 Hz, 1H), 3.11 (s, 3H), 1.90 (s, 1H), 1.51 (d, J = 5 Hz, 3H).

El TRV-1421 (0.1381 g, 0.458 mmol) se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó NaH (0.0275 g, 0.687 mmol) y la suspensión se agitó durante 30 minutos. Luego se agrego yodometano (0.06 mL, 0.916 mmol) y la reacción se dejó calentar a temperatura ambiente. La solución se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó a través de una columna de 10% de EtOAc/hexano para producir 0.1243 g (86% de rendimiento) de TRV-1422 como aceite amarillo. 1H RMN (500 MHz, CDC13) 8 = 7.25-7.23 (m, 2H), 7.02 (s, 1H), 7.02-6.98 (m, 2H), 6.14 (s, 1H), 5.07 (d, J -15 Hz, 1H), 5.03 (d, J = 15 Hz, 1H), 4.27 (q, J = 5 Hz, 1H), 3.26 (s, 3H), 3.12 (s, 3H), 1.44 (d, J = 5 Hz, 3H).

A una solución de 6-bromo-4-(isoindolin-2-il)benzo[c][1,2,5]oxadiazol (647 mg, 2.0 mmol) en DME (10 mL)/Na2CO3 (3 mL) se agregó ácido 3-acetil-fenilborónico (500 mg, 0.9 mmol) y Pd(P(Ph)3)4 (90 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se disolvió en DCM y se eluyó a través de un corto tapón de SiO2 para dar 456 mg de material que fue adecuado para reacciones adicionales. 1H RMN (300 MHz, CDCl3) 8 = 8.3 (m, 1H), 8.0 (d, J = 8 Hz, 1H), 7.8 (d, J = 8 Hz, 1H), 7.6 (t, J = 8 Hz, 1H), 7.4 (m, 4H), 7.2 (s, 1H), 6.2 (s, 1h ), 5.1 (s, 4h ), 2.7 (s, 3H). A una solución agitada de cetona (222 mg, 0.6 mmol) disuelta en THF (4 mL) a 0°C se agregó MeMgBr (0.9 mL, 0.9 mmol). Después de 30 minutos a baja temperatura la reacción se calentó a RT y se detuvo con HCl diluido y se extrajo en EtOAc (3 X 20 mL). La fase orgánica se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM). 1H RMN (300 MHz, CDCh) 8 = 7.8 (m, 1H), 7.5 (m, 2H0, 7.4-7.3 (m, 5H), 7.2 (s, 1H), 5.1 (s, 4H), 1.0 (s, 1H), 1.6 (s, 6H).

A una solución de 6-bromo-4-(isoindolin-2-il)benzo[c][1,2,5]oxadiazol (647 mg, 2.0 mmol) en DME (10 mL)/Na2CO3 (3 mL) se agregó ácido 3-acetil-fenilborónico (500 mg, 0.9 mmol) y Pd(P(Ph)3)4 (90 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se disolvió en DCM y se eluyó a través de un corto tapón de SiO2 para dar 456 mg de material que fue adecuado para reacciones adicionales. 1H RMN (300 MHz, CDCl3) 8 = 8.3 (m, 1H), 8.0 (d, J = 8 Hz, 1H), 7.8 (d, J = 8 Hz, 1H), 7.6 (t, J = 8 Hz, 1H), 7.4 (m, 4H), 7.2 (s, 1H), 6.2 (s, 1h ), 5.1 (s, 4h ), 2.7 (s, 3H). A una solución agitada de cetona (230 mg, 0.65 mmol) disuelta en MeOH/THF a 0°C
se agregó NaBH4 (80 mg, 2 mmol). Una vez que la reacción exotérmica inicial se había calmado el baño frío se retiró y se le permitió a la reacción llegar a RT. La reacción se agitó a RT durante 1 hora y luego se vertió en agua, después de 30 min la reacción se acidificó y se extrajo en EtOAc (3 X 20 mL). La fase orgánica se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM). 1H RMN (300 MHz, CDCla) 8 = 7.7 (s, 1H), 7.6 (dt, J = 10 Hz, 1 Hz), 7.5-7.3 (m, 6H), 7.1 (s, 1H), 6.2 (s, 1H), 5.2 (s, 4H), 5.0 (m, 1H), 1.9 (m, 1h), 1.5 (d, J = 6 Hz, 3H).

El TRV-1418 (0.1195 g, 0.399 mmol) se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (0.52 mL, solución 1.0 M en Bu2O) y la reacción se agitó hasta que se completó mediante TLC. La solución se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó a través de dos columnas de 30% de EtOAc/hexano consecutivas para producir 0.0626 g (50% de rendimiento) de TRV-1425 como un aceite naranja en aproximadamente 90% cp. 1H r Mn (500 MHz, CDC13) 8 = 7.23-7.20 (m, 2H), 7.19 (s, 1H), 7.02-6.95 (m, 2H), 6.33 (s, 1H), 5.04 (s, 2H), 3.12 (s, 3H), 1.59 (s, 6H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (347 mg, 1.49 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó bencenotiol (104 mg, 1.83 mmol) y trietilamina (400 pL, 2.8 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtOAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM) para dar 264 mg (65% de rendimiento). 1H RMN (500 MHz, CDCla) 8 = 7.8 (m, 1H), 7.6 (m, 2H), 7.5 (m, 3H), 6.7 (m, 1H). A una solución de tioéter (220 mg, 0.7 mmol) en DME (6 mL)/Na2CO3 (1.5 mL) se agregó ácido 3-acetil-fenilborónico (185 mg, 1.1 mmol) y Pd(P(Ph)3)4 (75 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a tratamiento final al diluir con NaOH 1 M (40 mL) y extraer con EtOAc (3 X 20 mL). La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se purificó al empujar a través de un tapón de SiO2 con DCM. 1H RMN (500 MHz, CDCh) 8 = 8.0 (s, 1H), 7.9 (m, 1H), 7.7 (s, 1H), 7.6 (m, 3H), 7.5 (m, 1H), 7.5 (m, 3H), 7.1 (m, 1H), 2.6 (s, 3H). A una solución agitada de cetona se disolvió en MeOH/THF a 0°C se agregó NaBH4 (80 mg, 2 mmol). Una vez que la reacción exotérmica inicial se había calmado el baño frío se retiró y se le permitió a la reacción llegar a RT. La reacción se agitó a RT durante 1 hora y luego se vertió en agua, después de 30 min la reacción se acidificó y se extrajo en EtOAc (3 X 20 mL). La fase orgánica se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM). 1H RMN (500 MHz, CDCh) 8 = 7.7 (d, J = 1 Hz, 1H), 7.6 (m, 2H), 7.5-7.3 (m, 7H), 7.1 (d, J = 1 Hz, 1H), 4.9 (m, 1H), 1.8 (d, J = 3 Hz, 1H), 1.5 (d, J= 6 Hz, 3H).

El TRV-1409 (0.2077 g, 0.663 mmol) se disolvió en DCM (37 mL) y luego se agregó DMP (0.8436 g, 1.99 mmol) en una porción. La reacción se agitó durante 3 horas y luego se detuvo con NaHCO3 saturado (ac.) y exceso de Na2S2O3. Esta mezcla se agitó hasta que todos los sólidos se disolvieron y luego se extrajo con DCM. Las capas orgánicas combinadas se secaron (Na2SO4), se filtraron y se concentraron para producir el aldehído crudo. La purificación a través de cromatografía instantánea (20% de EtOAc/hexano) produjo 0.0556 g (27 % de rendimiento) del aldehído 4. Este aldehído (0.0532 g, 0.171 mmol) se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (0.19 mL, solución 1.0 M en Et2O) y la reacción se agitó hasta que se completó mediante TLC. La reacción luego se detuvo con NH4Cl y se extrajo con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó con cromatografía instantánea para producir 0.0262 g (47 % de rendimiento) de TRV-1427 como aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.23-7.21 (m, 2H), 7.03-6.99 (m, 3H), 6.59 (d, J = 15 Hz, 1H), 6.34 (dd, J = 15, 5 Hz, 1H), 6.26 (s, 1H), 4.54 (m, 1H), 3.10 (s, 3H), 1.62 (d, J = 5 Hz, 1H), 1.40 (d, J = 10 Hz, 3H).

En un frasco sellado 4,6-dibromobenzo[c][1,2,5]oxadiazol (2.2 g, 8 mmol) se combinó con, (888 mg, 8.8 mmol), Et3N (3.3 mL, 24 mmol), y NMP (13 mL). La mezcla se calentó a 85°C durante 2 días. En ese momento la reacción se diluyó con NaOH 1 M (150 mL) y el material insoluble se retiró mediante filtración. El compuesto deseado se precipitó mediante la adición de HCl (conc.) a la capa acuosa y se aíslo mediante filtración al vacío para dar ácido 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidina-3-carboxílico (1.7 g), que se utilizó sin purificación adicional. El ácido (1.5 g, 5 mmol) se disolvió en THF (50 mL) y se enfrió a 0°C. A este se agregó BH3-THF (10 mL, 10 mmol). Se le permitió a la reacción llegar a la temperatura ambiente durante la noche. Al siguiente día la reacción se detuvo con AcOH y se extrajo en EtOAc. La capa orgánica se lavó con NaOH 1 M hasta que los lavados quedaron azul tornasol y luego se concentraron en vacío. El material crudo se fusionó a SO2 y se purificó mediante cromatografía de columna flash (3:2 Hex:EtOAc) para dar 800 mg de (1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)metanol (56% de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.05 (s, 1H), 6.11 (s, 1H), 5.30 (s, 1H), 4.11 (t, J = 8 Hz, 2H), 3.85 (m, 4H), 3.00 (m, 1H). A una solución del alcohol mencionado anteriormente (400 mg, 1.4 mmol) en DME (7 mL)/Na2CO3 (2.1 mL) se agregó ácido 3-formil-fenilborónico (315 mg, 2.1 mmol) y Pd(P(Ph)3)4 (50 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar los sólidos resultantes mediante filtración al vacío. El material crudo se purificó al tapar a través de SiO2 (50% de EtOAc en Hex) para dar 350 mg de material que se utilizó como estaba. A una solución agitada de aldehído (400 mg, 1.4 mmol) y reactivo de Rupert (483 mg, 3.3 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de agitar durante la noche se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (30% de EtOAc en Hex) para dar 150 mg (34 % de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.72 7.66 (m, 2H), 7.5 (m, 2H), 7.14 (s, 1H0, 6.04 (s, 1H), 5.11 (m, 1H), 4.41 (t, J = 8 Hz, 2H), 4.15 (dd, 2H), 3.93 (d, J = 5 Hz, 2H), 3.06 1H), 2.82 (s, amplio), 1.63 (s, amplio).

Se disolvió 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (574 mg, 2.1 mmol) en DCM (25 mL) y se enfrió a -78°C y DAST (421 uL, 3.2 mmol) se agregó en forma de gotas y el baño frío se retiró. Después de 2 h a temperatura ambiente la reacción se enfrió a 0°C y se detuvo con MeOH. La mezcla de reacción luego se diluyó con agua y se extrajo con DCM. Los extractos combinados se secaron (MgSO4) y se concentraron en vacío. El residuo se purificó mediante cromatografía de columna flash (50% de DCM en Hex) para dar el producto como un sólido amarillo (160 mg, 27% de rendimiento). 1H RMN (500 MHz, CDCls) 8 = 7.27 (s, 1H), 5.96 (s, 1H), 5.51 (dm, 2JHF = 57 Hz, 1H), 4.56 (m, 2H), 4.42 (dm, 3j HF = 23 Hz, 2H). A una solución de anilina (330 mg, 1.2 mmol) en DME (7 mL)/Na2CO3 (1.8 mL) se agregó ácido 3-formil-fenilborónico (270 mg, 1.9 mmol) y Pd(P(Ph)3)4 (50 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (20% de EtOAc en Hex) y se utilizó sin purificación adicional. A una solución agitada de aldehído (aproximadamente 1.0 mmol) y reactivo de Rupert (348 mg, 2.0 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de agitar durante la noche se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (25% de EtOAc en Hex) para dar 300 mg (80% de rendimiento). 1H RMN (500 MHz, CDCla) 8 = 7.72 (s, 1H), 7.66 (dm, J = 10 Hz, 1H), 7.51 (m, 2H), 7.23 (s, 1H), 6.12 (s, 1H), 5.53 (dm, 2JHF = 56 Hz, 1H), 5.14 (m, 1H), 4.64 (m, 2H), 4.42 (ddm, J = 23 Hz/10 Hz, 2H), 2.73 (s, 1H).

El TRV-1402 (0.1992 g, 0.58 mmol) se disolvió en THF (10 mL) y se enfrió a - 78°C. MeLi (0.80 mL, solución 1.6 M en Et2O) se agregó en forma de gotas y la reacción se dejó calentar a temperatura ambiente durante la noche. La mezcla luego se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. Esta mezcla se extrajo tres veces con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el alcohol crudo. Este material luego se purificó a través de cromatografía (30% de EtOAc/hexano) para producir 0.0843 g (43% de rendimiento) de aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.23-7.21 (m, 2H), 7.03 (s, 1H), 7.02-6.98 (m, 2H), 6.61 (d, J = 15 Hz, 1H), 6.42 (d, J = 15 Hz, 1H), 6.25 (s, 1H), 5.08 (s, 2H), 3.10 (s, 3H), 1.45 (s, 6H).

Se disolvió 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.2771 g, 0.82 mmol) en THF (10 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.43 mL, solución 2.0 M en ciclohexano) y la mezcla se
agitó durante 30 minutos antes de agregar cloruro de N,N-dimetilcarbamilo (0.10 mL, 1.1 mmol) en forma de gotas. La mezcla luego se dejó calentar a temperatura ambiente durante la noche. La mezcla luego se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. Esta mezcla se extrajo tres veces con EtOAc. Los extractos orgánicos combinados se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar la amida cruda. La purificación final de este material fue con una columna 50% de EtOAc/hexano para producir 16.6 mg (6.2% de rendimiento) de aceite naranja. 1H RMN (500 MHz, CDC13) 8 = 7.23-7.20 (m, 2H), 7.08 (s, 1H), 7.03-6.99 (m, 2H), 6.12 (s, 1H), 5.11 (s, 2H), 3.14 (s, 3H), 3.12 (s, 3H), 3.01 (s, 3H).

En un frasco sellado 4,6-dibromobenzo[c][1,2,5]oxadiazol (834 mg, 3 mmol) se combinó con clorhidrato de azetidina (309 mg, 3.3 mmol), Et3N (1.25 mL, 9 mmol), y NMP (6 mL). La mezcla se calentó a 85°C durante 2 días. El material crudo se precipitó al vertir la mezcla de reacción en agua (150 mL). El material crudo se tapó a través de SiO2 (10% de EtOAc en Hex) para dar (540 mg) un sólido naranja que la RMN mostró ser una mezcla 2:1 de material de partida y 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol. 1H RMN (500 MHz, CDCla) 8 = 7.05 (s, 1H), 5.85 (s, 1H), 4.33 (m, 4h ), 2.53 (m, 2H). A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (322 mg, 1.27 mmol) en d Me (7 mL)/Na2CO3 (2.0 mL) se agregó ácido 3-formil-fenilborónico (286 mg, 1.9 mmol) y Pd(P(Ph)3)4 (50 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 115°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (DCM) para dar 280 mg de material que se utilizó como estaba. A una solución agitada del aldehido (411 mg, 1.33 mmol) y reactivo de Rupert (378 mg, 2.7 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (DCM) para dar 477 mg (94 % de rendimiento). 1H RMN (500 MHz, CDCla) 8 = 7.72 (s, 1h), 7.66 (m, 1H), 7.52 (m, 2H), 7.13 (s, 1H), 6.01 (s, 1H), 5.12 (q, J = 7 Hz, 1H), 4.35 (t, J = 8 Hz, 4H), 2.74 (s, br, 1H), 2.53 (p, J = 7 Hz, 2H).

Se disolvió ácido 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidina-3-carboxílico (“ 600 mg, 2 mmol) en MeOH (100 mL) y se agregó H2SO4 (2 gotas). Se equipó con un condensador de reflujo y la reacción se sometió a reflujo gentilmente durante 24 h. En ese momento la reacción se diluyó con EtOAc (200 mL) y se extrajo con agua (3 X 100 mL). El material crudo se fusionó a SiO2 y se purificó mediante cromatografía de columna flash (DCM) para dar esencialmente un rendimiento cuantitativo (2.08 mmol). El éster (650 mg, 2.08 mmol) se disolvió en THF (20 mL) y se enfrió a 0°C. A este se agregó MeMgBr (6 mL, 6 mmol). Después de 30 min a baja temperatura la reacción se llevó a RT y seguido por TLC. Cuando la reacción se consideró completa la mezcla se enfrío de nuevo a 0°C y se detuvo cuidadosamente con NH4Cl acuoso. La mezcla luego se extrajo en EtOAc (3 X 100 mL), se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 y se purificó mediante cromatografía de columna flash (20% de acetona en Hex) para dar 474 mg de 2-(1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)propan-2-ol (73% de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.20 (s, 1H), 5.89 (s, 1H), 4.26 (m, 4H), 2.87 (m, 1H), 1.38 (s, 1H), 1.26 (s, 6H). A una solución agitada del alcohol terciario (223 mg, 0.71 mmol) se disolvió en THF (10 mL) y se enfrió a 0°C se agregó NaH (600 mg, 20 mmol). Una vez ha cesado el burbujeo inicial se agregó en forma de gotas Mel (1.14 g, 8 mmol) y la mezcla de reacción se dejó ir a RT. Después de 18 h la reacción se enfrío de nuevo a 0°C y se agregó cuidadosamente NH4CL La mezcla de reacción se extrajo con EtOAc (3 X 50 mL), se lavó con solución salina, (1 X 50 mL) y se concentró en vacío. El material crudo luego se purificó mediante cromatografía de columna flash (10% de Acetona en Hex). A una solución del éter (193 mg, 0.6 mmol) en DME (4 mL)/Na2CO3 (2M, 0.9 mL) se
agregó ácido 3-formil-fenilborónico (133 mg, 0.9 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 110°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El aldehído crudo se purificó al tapar a través de SiO2 (20% de EtOAc en Hex) para dar 184 mg de material que se utilizó como estaba. A una solución agitada de aldehído (400 mg, 1.4 mmol) y reactivo de Rupert (483 mg, 3.3 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de agitar durante la noche se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (30% de EtOAc en Hex) para dar 90 mg (41% de rendimiento). 1H RMN (500 MHz, CDCls) 8 = 7.72 (s, 1H), 7.66 (m, 1H), 7.50 (m, 2H), 7.11 (s, 1H), 6.01 (s, 1H), 5.11 (m, 1H), 4.25 (m, 4H), 3.24 (s, 3H), 2.96 (m, 1H)

A una solución agitada de (1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)metanol (427 mg, 1.58 mmol) disuelta en NMP (10 mL) y enfriada a 0°C se agregó NaH (758 mg, 20 mmol). Una vez ha cesado el burbujeo inicial se agregó en forma de gotas Mel (2.24 g, 10 mmol) y la mezcla de reacción se dejó ir a RT. Después de 18 h la reacción se enfrío de nuevo a 0°C y NH4Cl se agregó cuidadosamente. La mezcla de reacción se extrajo con EtOAc (3 X 50 mL), se lavó con solución salina, (1 X 50 mL) y se concentró en vacío. El material crudo luego se purificó mediante cromatografía de columna flash (10% de EtOAc en Hex). 1H RMN (500 MHz, CDCla) 8 = 7.16 (s, 1H), 5.86 (s, 1H), 4.3 (m, 2H), 4.09 (m, 2H), 3.6 (d, J = 6 Hz 2H), 3.40 (s, 3H), 3.08 (m, 1H). A una solución del éter de metilo (245 mg, 0.8 mmol) en DME (5 mL)/Na2CO3 (2M, 1.2.0 mL) se agregó ácido 3-formil-fenilborónico (184 mg, 1.9 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 110°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (DCM) para dar 214 mg de aldehído que se utilizó como estaba. 1H RMN (500 MHz, CDCh) 8 = 10.11 (s, 1H), 8.12 (s, 1H), 7.94 (d, J = 8 Hz, 1H), 7.88 (d, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 7.17 (s, 1H), 6.03 (s, 1H), 4.42 (m, 2H), 4.13 (m, 2H), 3.65 (d, J = 6 Hz, 2H), 3.41 (s, 3H), 3.11 (m, 1H). A una solución agitada del aldehído (214 mg, 1.33 mmol) y reactivo de Rupert (298 mg, 2.1 mmol) en DCM (13 mL) a 0°C se agregó TBAF (0.2 mL, Th F 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con DCM. Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (30% de EtOAc en Hex) para dar 150 mg (94 % de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.71 (m, 1H), 7.64 (m, 1H), 7.51 (m, 2H), 7.13 (s, 1H), 6.02 (s, 1H), 5.11 (m 1H), 4.38 (t, J = 8 Hz, 2H), 4.11 (m, 2H), 3.64 (d, J = 7 Hz, 2H), 3.41 (s, 3H), 3.09 (m, 1H).

Al ácido 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidina-3-carboxílico (250 mg, 0.8 mmol) en DME (5 mL)/Na2CO3 (2M, 1.2 mL) se agregó ácido 3-formil-fenilborónico (181 mg, 1.2 mmol) y Pd(P(Ph)3)4 (50 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 110°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El aldehído crudo se purificó al tapar a través de SiO2 (30% de EtOAc en Hex) para dar 211 mg de material que se utilizó como estaba. A una solución agitada de aldehído (211 mg, 0.63 mmol) y reactivo de Rupert (266 mg, 1.88 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con
DCM. Las fases orgánicas se lavaron con NH4CI saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (20-40% de gradiente EtOAc en Hex) para dar 70 mg (27 % de rendimiento). 1H RMN (500 MHz, CDCls) 8 = 7.71 (s, 1H), 7.64 (m, 1H), 7.49 (m, 2H), 7.13 (s, 1H), 6.04 (s, 1H), 5.12 (s, br, 1H), 4.33 (t, J = 8 Hz, 2H), 4.25 (m, 2H), 2.91 (m, 1H), 2.74 (d, J = 4 Hz, 1H), 1.43 (s, 1H), 1.27 (s, 6H).

A una solución agitada de 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (280 mg, 1.04 mmol) disuelta en DCM (10 mL) se agregó reactivo de Dess-Martin (571 mg, 1.3 mmol) disuelto en DCM (4 mL). Después de 1 hora la reacción se había vuelto turbia y se había formado un precipitado. El material se vertió en NaOH 1 M y se extrajo con TBME, la cetona resultante se utilizó como estaba. 1H RMN (500 MHz, CDCla) 8 = 7.40 (s, 1H), 6.13 (s, 1H), 5.10 (s, 4H). A una solución agitada enfriada a 0°C de cetona (275 mg, 1.03 mmol) disuelta en THF (10 mL) se agregó MeMgBr (1M THF, 3 mL). El baño frío se dejó en su lugar y se le permitió a la reacción llegar a RT durante 8 horas. En ese momento la mezcla se volvió a enfriar y se detuvo con NH4Cl (ac.) y se extrajo con EtOAc (3 X 20 mL), se secó con MgSO4 y se concentró en vacío. El alcohol terciario crudo se pasó a través de un tapón de SiO2 (DCM) para dar un sólido amarillo. A una solución del alcohol terciario (284 mg, 1 mmol) en DME (6 mL)/Na2CO3 (2M, 1.5 mL) se agregó ácido 3-formil-fenilborónico (225 mg, 1.5 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 110°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (10-30% de gradiente EtOAc en Hex) para dar 250 mg de aldehído que se utilizó como estaba. 1H RMN (500 MHz, CDCh) 8 = 10.11 (s, 1H), 8.11 (s, 1H), 7.92 (d, J = 8 Hz, 1H), 7.87 (d, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 7.21 (s, 1H), 6.09 (s, 1H), 4.30 (d, J = 9 Hz, 2H), 4.24 (d, J = 8 Hz, 2H), 2.19 (s, Br, 1H), 1.71 (s, 3H). A una solución agitada de aldehído (250 mg, 0.81 mmol) y reactivo de Rupert (344 mg, 2.4 mmol) en DCM (10 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas se agregó un exceso de TBAF y la reacción se diluyó con d Cm . Las fases orgánicas se lavaron con NH4Cl saturado, solución salina, y luego se secaron con MgSO4 y se concentraron en vacío. El material crudo se purificó mediante cromatografía de columna flash (20-40% de gradiente EtOAc en Hex) para dar 185 mg (60% de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.71 (s, 1H), 7.64 (d, J = 7 Hz, 1H), 7.51 (m, 2H), 7.18 (s, 1H), 6.08 (s, 1H), 5.12 (m, 1H), 4.29 (d, J = 9 Hz, 2H), 4.22 (d, J = 9 Hz, 2H), 2.71 (s, br, 1H). 2.10 (s, br, 1H), 1.70 (s, 3H).

1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (0.500 g, 1.85 mmol) se disolvió en NMP (2 mL) y se enfrió a 0°C. Luego se agrego en forma de porciones NaH (0.096 g, 2.4 mmol) y se continuó la agitación hasta que cesaron todas las burbujas, en cuyo punto se agregó yoduro de etilo (0.16 mL, 2.0 mmol). La reacción se dejó calentar a temperatura ambiente durante la noche. La mezcla luego se volvió a enfriar a 0°C y se detuvo con NH4Cl saturado (ac.). Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x) solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un éter de etilo crudo. Este éter y ácido 3-formilfenilborónico (0.2908 g, 1.94 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (4.1 mL) y Na2CO3 2M (2.8 mL, 5.6 mmol) seguidos por Pd(Pph3)4 (0.1075 g, 0.093 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5.6 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.41 mL, 2.8 mmol) seguido por TBAF (0.1 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C. Se agregó TBAF (5.6 mL, solución 1.0 M en THF) y la reacción se dejó
calentar a temperatura ambiente durante la noche. La mezcla se detuvo con solución salina y luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (20% de EtOAc/hexano) para producir 0.3755 g (51% de rendimiento durante 3 etapas) de TRV-1437. 1H RMN (CDC13, 500 MHz) 8 = 7.72 (s, 1H), 7.66-7.64 (m, 1H), 7.55-7.50 (m, 2H), 7.17 (s, 1H), 6.06 (s, 1H), 5.14-5.11 (m, 1H), 4.56-4.54 (m, 3H), 4.18 (d, J = 5 Hz, 2H), 3.54 (q, J = 5 Hz, 2H), 2.67 (d, J = 5 Hz, 1H), 1.26 (t, J = 5 Hz, 3H).
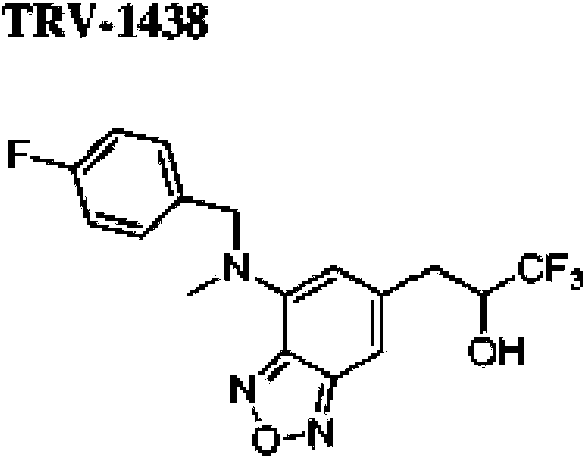
Se agregó 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (2.085 g, 6.20 mmol) a un tubo, que luego se evacuó y purgó con argón (3 ciclos). A este frasco luego se agrego dietilmalonato (1.9 mL, 12.4 mmol), P(tBu)3 (4 mL, 1.98 mmol) y tolueno (18 mL) antes de agregar Pd2(dba)3 (0.4542 g, 0.496 mmol) y K3PO4 (4.6063 g, 21.7 mmol). El tubo luego se selló y se calentó a 100°C durante 16 horas. La reacción luego se enfrío y se filtró a través de un tapón de Celite y luego se concentró. El residuo se purificó a través de cromatografía instantánea (15% de EtOAc/hexano) para producir 1.257 g (49% de rendimiento) de malonato sustituido. Este material (1.2572 g, 3.03 mmol) se disolvió en d Ms O (30 mL) y luego se agregaron NaCl (0.3536 g, 6.05 mmol) y H2O (1.8 mL, 97 mmol). Esta mezcla se calentó a 150°C durante 8 horas. Luego de enfriar a temperatura ambiente la mezcla se diluyó con EtOAc y agua. La capa orgánica luego se lavó con H2O (63), solución salina, se secó (Na2SO4), se filtró y se concentró para dar el éster de etilo crudo. Este material se purificó a través de cromatografía instantánea (10% de EtOAc/hexano) para producir 0.7641 g (73% de rendimiento) de sólido naranja. El éster de etilo (0.5873 g, 1.71 mmol) luego se disolvió en DCM (20 mL) y se enfrió a -78°C. Se agregó en forma de gotas DIBAL (4.0 mL, solución 1.0 M en hexano). La reacción se agitó a -78°C durante 5 minutos y luego se calentó a -30°C. Después de agitar a esta temperatura durante 3 horas se detuvo con metanol y se dejó calentar a temperatura ambiente. Se agregaron agua (5 mL) y Na2SO4, la mezcla se agitó durante 30 minutos y luego se filtró para dar una mezcla del aldehído y alcohol. Esta mezcla se tomó en DCM (50 mL) y luego se agregó DMP (0.7253 g, 1.71 mmol) con agitación vigorosa. Después de 60 minutos la reacción se detuvo con NaHCO3 acuoso saturado y exceso de Na2S2O3, se continuó la agitación hasta que todos los sólidos se disolvieron. La mezcla luego se extrajo con DCM. Las capas orgánicas combinadas se secaron con Na2SO4, se filtraron y se concentraron. El residuo luego se purificó a través de cromatografía instantánea (20% de EtOAc/hexano) para producir 0.0726 g (14 % de rendimiento, 2 etapas) de aldehído 9. Este aldehído (0.0726 g, 0.243 mmol) se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.05 mL) seguido por TBAF (0.03 mL, solución 1.0 M en THF). La mezcla se agitó durante 60 minutos y se agregó luego TBAF (0.46 mL, solución 1.0 M en THF) y la reacción se agitó durante la noche. La reacción luego se detuvo con solución salina y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (20% de EtOAc/hexano) para producir 0.0349 g (39% de rendimiento) de TRV-1438. 1H RMN (CDC13, 500 MHz) 8 = 7.23-7.21 (m, 2H), 7.03-6.99 (m, 3H), 6.01 (s, 1H), 5.08 (s, 2H), 4.23 (br s, 1H), 3.11 (s, 3H), 3.03 (dd, J = 14, 2.5 Hz, 1H), 2.87 (dd, J = 14, 10 Hz, 1H), 2.21 (s, 1H).

Se agregó 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (2.085 g, 6.20 mmol) a un tubo, que luego se evacuó y purgó con argón (3 ciclos). A este frasco luego se agrego dietilmalonato (1.9 mL, 12.4 mmol), P(tBu)3 (4 mL, 1.98 mmol) y tolueno (18 mL) antes de agregar Pd2(dba)3 (0.4542 g, 0.496 mmol) y K3PO4 (4.6063 g, 21.7 mmol). El tubo luego se selló y se calentó a 100°C durante 16 horas. La reacción luego se enfrío y se filtró a través de un tapón de Celite y luego se concentró. El residuo se purificó a través de cromatografía instantánea (15%
de EtOAc/hexano) para producir 1.257 g (49% de rendimiento) del compuesto 2. Este material (1.2572 g, 3.03 mmol) se disolvió en d MSo (30 mL) y NaCl (0.3536 g, 6.05 mmol) y H2O (1.8 mL, 97 mmol) luego se agregaron. Esta mezcla se calentó a 150°C durante 8 horas. Luego de enfriar a temperatura ambiente la mezcla se diluyó con EtOAc y agua. La capa orgánica luego se lavó con H2O (63), solución salina, se secó (Na2SO4), se filtró y se concentró para dar el éster de etilo crudo. Este material se purificó a través de cromatografía instantánea (10% de EtOAc/hexano) para producir 0.7641 g (73% de rendimiento) de sólido naranja. Este material (0.1736 g, 0.506 mmol) se disolvió en THF (10 mL) y se enfrió a -78°C. Se agregó en forma de gotas MeLi (0.70 mL, solución 1.6 M en Et2O), y la reacción se dejó calentar lentamente a temperatura ambiente. Luego se volvió a enfriar a 0°C y se detuvo con cloruro de amonio. Esta mezcla se extrajo con EtOAc y los extractos orgánicos combinados se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un residuo crudo. El material crudo se purificó a través de cromatografía instantánea (20% de EtOAc/hexano) para producir 0.0288 g (17 % de rendimiento) del alcohol terciario TRV-1439. 1H RMN (CDC13, 500 MHz) 8 = 7.24-7.21 (m, 2H), 7.05-6.99 (m, 2H), 6.94 (s, 1H), 6.08 (s, 1H), 5.02 (s, 2H), 3.11 (s, 3H), 2.76 (s, 2H), 1.40 (s, 1H), 1.27 (s, 6H).

A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (480 mg, 0.6 mmol) en DME (11 mL)/Na2CO3 (2M, 2.8 mL) se agregó ácido 3-carboxi-fenilborónico (470 mg, 2.8 mmol) y Pd(P(Ph)3)4 (93 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 100°C O/N. La reacción se sometió a trabajo final al vertir en HCl 1 M (150 mL) y aislar en vacío los sólidos resultantes. El ácido crudo se purificó mediante cromatografía de columna flash (d Cm 1% de AcOH) para dar 330 mg de material que se utilizó como estaba. A una solución agitada de este ácido (110 mg, 0.37 mmol) se disuelto en NMP (2 mL) se agregó DIPEA (130 mL, 0.74 mmol) y piperazina (230 mg, 0.37 mmol). Después de que la mezcla fue homogénea se agregó HATU (140 mg, 0.37 mmol) y la reacción se dejó de agitar. Después de 1 hora a RT la reacción se diluyó con EtOAc (100 mL) y se diluyó con agua. El material crudo se purificó mediante cromatografía de columna flash (50-100% de EtOAc en Hex) para dar (20 mg) 14% de rendimiento. 1H RMN (500 MHz, CDCh) 8 = 7.66 (m, 2H), 7.50 (t, J = 8 Hz, 1H), 7.42 (d, J = 8 Hz, 1H), 7.12 (s, 1H), 6.01 (s, 1H), 4.34 (t, J = 7 Hz, 4H), 3.79 (s, amplio, 2H), 3.43 (s, amplio, 2H), 2.97 (s, amplio, 2H), 2.82 (s, amplio, 2H), 2.61 (s, 1H), 2.53 (p, J = 7 Hz, 2H).
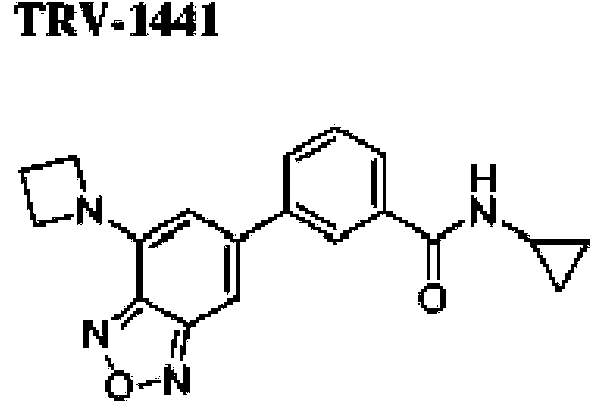
A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (480 mg, 0.6 mmol) en DME (11 mL)/Na2CO3 (2M, 2.8 mL) se agregó ácido 3-carboxi-fenilborónico (470 mg, 2.8 mmol) y Pd(P(Ph)3)4 (93 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 100°C O/N. La reacción se sometió a trabajo final al vertir en HCl 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó mediante cromatografía de columna flash (DCM 1% de AcOH) para dar 330 mg de material que se utilizó como estaba. A una solución agitada de este ácido (165 mg, 0.56 mmol) disuelta en NMP (2 mL) se agregó DIPEA (300 mL, 1.7 mmol) y ciclopropilamina (55 mg, 0.59 mmol). Después de que la mezcla fue homogénea se agregó HATU (213 mg, 0.56 mmol) y la reacción se dejó de agitar. Después de 1 hora a RT la reacción se diluyó con EtOAc (100 mL) y se diluyó con agua. La fase orgánica se lavó con ácido (HCl 1 M, 1 X 100 mL), base (NaOH 1 M, 1 X 100 mL) y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (EtOAc) para dar (120 mg) 64 % de rendimiento. 1H RMN (500 MHz, DMSO-D6) 8 = 7.91 (m, 2H), 7.66 (d, J = 8 Hz, 1H), 7.57 (t, J = 8 Hz, 1H), 7.35 (s, 1H), 6.23 (s, 1H), 4.33 (m, 6H), 4.07 (t, J = 7 Hz, 2H), 2.45 (p, J = 7 Hz, 2 H), 2.27 (p, J = 7 Hz, 2H).

A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (480 mg, 0.6 mmol) en DME (11 mL)/Na2CO3 (2M, 2.8 mL) se agregó ácido 3-carboxi-fenilborónico (470 mg, 2.8 mmol) y Pd(P(Ph)3)4 (93 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 100°C O/N. La reacción se sometió a trabajo final al vertir en HCl 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó mediante cromatografía de columna flash (DCM 1% de AcOH) para dar 330 mg de material que se utilizó como estaba. A una solución agitada de este ácido (110 mg, 0.37 mmol) se disolvió en NMP (2 mL) se agregó DIPEA (130 mL, 0.74 mmol) y morfolina (32 mL, 0.37 mmol). Después de que la mezcla fue homogénea se agregó HATU (140 mg, 0.37 mmol) y la reacción se dejó de agitar. Después de 1 hora a RT la reacción se diluyó con EtOAc (100 mL) y se diluyó con agua. La fase orgánica se lavó con ácido (HCl 1 M, 1 X 100 mL), base (NaOH 1 M, 1 X 100 mL) y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (50-100% de EtOAc en Hex) para dar (40 mg) 29% de rendimiento. 1H RMN (500 MHz, CDCla) 8 = 7.67 (m, 2H), 7.52 (t, J = 8 Hz, 1H), 7.43 (d, J = 7 Hz, 1H), 7.12 (s, 1H), 6.00 (s, 1H), 4.35 (t, J = 7 Hz, 4H), 3.81 (m, br, 4H), 3.65 (m, br, 2H), 3.49 (m, br, 2H), 2.54 (p, J = 7 Hz, 2H).

A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (480 mg, 0.6 mmol) en DME (11 mL)/Na2CO3 (2M, 2.8 mL) se agregó ácido 3-carboxi-fenilborónico (470 mg, 2.8 mmol) y Pd(P(Ph)3)4 (93 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 100°C O/N. La reacción se sometió a trabajo final al vertir en HCl 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó mediante cromatografía de columna flash (DCM 1% de AcOH) para dar 330 mg de material que se utilizó como estaba. A una solución agitada de este ácido (165 mg, 0.56 mmol) disuelta en NMP (2 mL) se agregó DIPEA (300 mL, 1.7 mmol) y clorhidrato de azetidina (55 mg, 0.59 mmol). Después de que la mezcla fue homogénea se agregó HATU (213 mg, 0.56 mmol) y la reacción se dejó de agitar. Después de 1 hora a RT la reacción se diluyó con EtOAc (100 mL) y se diluyó con agua. La fase orgánica se lavó con ácido (HCl 1 M, 1 X 100 mL), base (NaOH 1 M, 1 X 100 mL) y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (EtOAc) para dar (120 mg) 64 % de rendimiento. 1H RMN (500 MHz, DMSO-D6) 8 = 7.91 (m, 2H), 7.66 (d, J = 8 Hz, 1H), 7.57 (t, J = 8 Hz, 1H), 7.35 (s, 1H), 6.23 (s, 1H), 4.33 (m, 6H), 4.07 (t, J = 7 Hz, 2H), 2.45 (p, J = 7 Hz, 2 H), 2.27 (p, J = 7 Hz, 2H).

A una solución de 4-(azetidin-1-il)-6-bromobenzo[c][1,2,5]oxadiazol (480 mg, 0.6 mmol) en DME (11 mL)/Na2CO3 (2M, 2.8 mL) se agregó ácido 3-carboxi-fenilborónico (470 mg, 2.8 mmol) y Pd(P(Ph)3)4 (93 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 100°C O/N. La reacción se sometió a trabajo final al vertir en HCl 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó mediante cromatografía de columna flash (DCM 1% de AcOH) para dar 330 mg de material que se utilizó como estaba. A una solución agitada de este ácido (165 mg, 0.56 mmol) disuelta en NMP (2 mL) se agregó DIPEA (300 mL, 1.7 mmol) y clorhidrato de 3-hidroxiazetidina (64 mg, 0.59 mmol). Después de que la mezcla fue homogénea se
agregó HATU (212 mg, 0.56 mmol) y la reacción se dejó de agitar. Después de 1 hora a RT la reacción se diluyó con EtOAc (100 mL) y se diluyó con agua. La fase orgánica se lavó con ácido (HCl 1 M, 1 X 100 mL), base (NaOH 1 M, 1 X 100 mL) y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (EtOAc) para dar (60 mg) 30% de rendimiento. 1H RMN (500 MHz,, DMSOD6) 8 = 7.89 (m, 2H), 7.67 (d, J = 8 Hz, 1H), 7.58 (t, J = 8 Hz, 1H), 7.35 (s, 1H), 6.22 (s, 1H), 4.50 (m 2H), 4.29 (m, 5H), 4.08 (m, 1H), 3.80 (dd, J = 10 Hz, 2 Hz, 1H), 2.45 (p, J = 7 Hz, 2H).

Se agruparon 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.3010 g, 0.895 mmol) y ácido 3-acetilfenilborónico (0.1542 g, 0.940 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (2.1 mL) y Na2CO3 2M (1.4 mL, 2.69 mmol) seguidos por Pd(PPh3)4 (0.0518 g, 0.0448 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (10 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (1.2 mL, solución 1.0 M en Bu2O) y luego la reacción se dejó calentar lentamente a temperatura ambiente durante la noche. La reacción se enfrió a 0°C y se detuvo con cloruro de amonio saturado. Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El aceite se purificó a través de cromatografía instantánea (20% de EtOAc/hexano) para producir 0.0841 g (24 % de rendimiento, 2 etapas) de TRV-1445. 1H RMN (CDC13, 500 MHz) 8 = 7.75 (d, J = 5 Hz, 1H), 7.54 (d, J = 10 Hz, 1H), 7.49-7.43 (m, 2H), 7.25-7.24 (m, 3H), 7.02 (t, J = 7 Hz, 2H), 6.38 (s, 1H), 5.11 (s, 2H), 3.18 (s, 3H), 1.78 (s, 1H), 1.64 (s, 6H).

Se agruparon 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (0.3207 g, 0.954 mmol) y ácido 3-bromofenilborónico (0.164 g, 1.00 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (2.1 mL) y Na2CO3 2M (1.4 mL, 2.86 mmol) seguidos por Pd(Pph3)4 (0.0551 g, 0.0477 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite crudo se purificó a través de cromatografía instantánea (5% de EtOAc/hexano) para producir 0.3037 g (77 % de rendimiento) del bromuro de arilo correspondiente. Este bromuro de arilo (0.3037 g, 0.74 mmol) se disolvió en THF (7.5 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.39 mL, solución 2.0 M en ciclohexano). La solución se agitó durante 30 minutos a esta temperatura y luego se agregó en forma de gotas oxetan-3-ona (0.0692 g, 0.962 mmol) en THF (1 mL). La solución luego se agitó y se dejó calentar a temperatura ambiente durante la noche. La reacción se enfrió a 0°C y se detuvo con cloruro de amonio saturado. Esta mezcla se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron a un aceite crudo. El aceite luego se purificó a través de cromatografía instantánea (45% de EtOAc/hexano) para producir 0.0587 g (19% de rendimiento) de TRV-1446. 1H RMN (CDC13, 500 MHz) 8 = 7.83 (s, 1H), 7.70 (d, J = 5 Hz, 1H), 7.58 (d, J = 5 Hz, 1H), 7.53 (t, J = 10 Hz, 1H), 7.25 7.24 (m, 3H), 7.02 (t, J = 10 Hz, 2H), 6.36 (s, 1H), 5.12 (s, 2H), 4.96 (s, 4H), 3.18 (s, 3H), 2.62 (s, 1H).

Se disolvió 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (0.500 g, 1.85 mmol) en NMP (2 mL) y se enfrió a 0°C. NaH (0.096 g, 2.4 mmol) luego se agrego en forma de porciones y se continuó la agitación hasta que cesaron todas las burbujas, en cuyo punto se agregó 2-bromopropano (0.19 mL, 2.0 mmol). La reacción se dejó calentar a temperatura ambiente durante la noche. La mezcla luego se volvió a enfriar a 0°C y se agregaron NaH (0.192 g, 4.8 mmol), 2-bromopropano (1.8 mL, 19 mmol) y NaI (1 eq). La reacción se calentó a 50°C durante la noche. La mezcla luego se volvió a enfriar a 0°C y se detuvo con NH4Cl saturado (ac.). Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x) solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el éter de isopropilo crudo. Este éter (0.1942 g, 0.622 mmol) y ácido 3-formilfenilborónico (0.0979 g, 1.94 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (1.4 mL) y Na2CO32M (0.93 mL, 1.87 mmol) seguidos por Pd(PPh3)4 (0.0359 g, 0.0311 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo (0.1047 g, 0.31 mmol) luego se disolvió en THF (1.0 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.092 mL, 0.62 mmol) seguido por t Ba F (0.03 mL, solución 1.0 M en t Hf ). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C. Se agregó TBAF (1.0 mL, solución 1.0 M en THF) y la reacción se dejó calentar a temperatura ambiente durante la noche. La mezcla se detuvo con solución salina y luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (20% de EtOAc/hexano) para producir 0.073 g (58 % de rendimiento durante 3 etapas) de TRV-1447. 1H RMN (CDC13, 500 MHz) 8 = 7.71 (s, 1H), 7.65 (d, J = 5 Hz, 1H), 7.54-7.50 (m, 2H), 7.16 (s, 1H), 6.06 (s, 1H), 5.13-5.12 (m, 1H), 4.60-4.55 (m, 3H), 4.16-4.14 (m, 2H), 3.70 (sept, J = 5 Hz, 1H), 2.71 (s, 1H), 1.21 (d, J = 5 Hz, 6H).

A una solución de 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (396 mg, 1.7 mmol) en NMP (3 mL) en un frasco de 4 gramos se agregó clorhidrato de 3-hidroxipirrolidina (230 mg, 1.83 mmol) y trietilamina (710 pL, 5.1 mmol). Una tapa se ajustó firmemente y la reacción se calentó a 85°C O/N. La reacción se sometió a tratamiento final al diluir con EtoAc (60 mL) y lavar con HCl 1M (3 X 20 mL) y solución salina (1 X 20 mL). La fase orgánica se secó con MgSO4 se filtró y se concentró en vacío. La anilina cruda se utilizó sin purificación adicional. A una solución de anilina (820 mg, 2.9 mmol) en DME (12 mL)/Na2CO3 (2M, 4.4 mL) se agregó ácido 3-formil-fenilborónico (645 mg, 4.3 mmol) y Pd(P(Ph)3)4 (110 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 110°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (EtOAc) para dar 680 mg de material que se utilizó como estaba. 1H RMN (500 MHz, CDCh) 8 = 10.11 (s, 1H), 8.12 (s, 1H), 7.94 (d, J = 8 Hz, 1H), 7.88 (d, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 7.17 (s, 1H), 6.03 (s, 1H), 4.42 (m, 2H), 4.13 (m, 2H), 3.65 (d, J = 6 Hz, 2H), 3.41 (s, 3H), 3.11 (m, 1H). A una solución agitada del aldehido (680 mg, 2.2 mmol) y reactivo de Rupert (937 mg, 6.6 mmol) en DCM (20 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (50% de EtOAc en Hex) para dar 380 mg (38 % de rendimiento) de TRV-1448, una mezcla de 1:1:1:1 de diastereómeros debido a los dos centros quirales. 1H RMN (500 MHz, CDCh) 8 = 7.73 (s, 1H), 7.66 (dt J = 7 Hz, 2
Hz, 2H), 7.51 (m, 2H), 7.10 (s, 1H), 6.12 (s, 1H), 5.1 (m, 1H), 4.7 (s br, 1H), 3.96 (m, 4H), 2.81 (d, J = 3 Hz, 1H), 2.20 (m, 2H), 1.75 (d, J = 3 Hz, 1H).

Se disolvieron 4,6-dibromobenzo[c][1,2,5]oxadiazol (0.3765 g, 1.35 mmol), clorhidrato de pirrolidina-2-carboxilato de etilo (0.2677 g, 1.49 mmol) y DIPEA (0.59 mL, 3.38 mmol) en NMP (1.8 mL) bajo argón y se agitaron en un tubo sellado a 100°C durante la noche. Luego de enfriar a temperatura ambiente, la reacción se diluyó con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (5X), HCl 1 N (ac.), NaHCO3 saturado (ac.) y solución salina antes de secar con Na2SO4, filtrar y concentrar para dar 0.1756 g (38 % de rendimiento) de material crudo. La anilina (0.4346 g, 1.28 mmol) luego se disolvió en THF (12 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (3.2 mL, solución 1.0 M en Bu2O) y la reacción se dejó calentar lentamente a temperatura ambiente durante la noche. La reacción se detuvo con NH4Cl (ac.) y luego se extrajo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite luego se purificó a través de cromatografía instantánea (15% de EtOAc/hexano) para producir 0.1293 g (31% de rendimiento) de alcohol terciario. El alcohol terciario (0.1293 g, 0.316 mmol) y ácido 3-formilfenilborónico (0.0624 g, 0.416 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (1.0 mL) y Na2CO3 2M (0.6 mL, 1.19 mmol) seguidos por Pd(PPh3)4 (0.0229 g, 0.0198 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (3 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.12 mL, 0.792 mmol) seguido por TBAF (0.05 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C. Se agregó TBAF (1.4 mL, solución 1.0 M en THF) y la reacción se dejó calentar a temperatura ambiente durante la noche. La mezcla se detuvo con solución salina y luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (2x), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (30% de EtOAc/hexano) para producir 0.0748 g (45% de rendimiento durante 2 etapas) de TRV-1449. 1H RMN (CDC13, 500 MHz) 8 = 7.73 (s, 1H), 7.67-7.65 (m, 1H), 7.54-7.49 (m, 2H), 7.14 (s, 1H), 6.46 (s, 1H), 5.12-5.10 (m, 1H), 4.92 (d, J = 5 Hz, 1H), 3.99-3.95 (m, 1H), 3.73-3.68 (m, 1H), 2.83 (s, 1H), 2.77-2.21 (m, 1H), 2.11 2.02 (m, 3H), 1.82 (s, 1H), 1.31 (s, 3H), 1.24 (s, 3H).

4,6-dibromobenzo[c][1,2,5]oxadiazol (2.013 g, 7.24 mmol) y sal de clorhidrato de pirrolidina-2-carboxilato de (S)-etilo (1.43 g, 7.96 mmol) se agruparon en un tubo. El tubo se evacuó y se enjuagó con argón durante tres ciclos. Luego se agregaron NMP (10 mL) y DIPEA (3.5 mL, 19.9 mmol) y el tubo se selló y calentó a 50°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite crudo luego se purificó a través de cromatografía de columna flash (15% de EtOAc/hexano) para producir 0.6105 g (25% de rendimiento) de anilina. La anilina (1.696 g, 4.9 mmol) se disolvió en DCM (20 mL) y se enfrió a -78°C. Se agregó en forma de gotas DIBAL (12.5 mL, solución 1.0 M en hexanos) y luego la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción se detuvo con MeOH y luego se agregó Na2SO4 y la mezcla se agitó durante 30 minutos antes de filtrar
a través de Celite. La fase orgánica se diluyó con EtOAc y agua. Las capas se separaron y la capa orgánica se lavó con H2O (3X), solución salina, se secó (Na2SO4), se filtró y se concentró. El material crudo se purificó a través de cromatografía (30% de EtOAc/hexano) para dar 1.294 g (87 % de rendimiento) del alcohol primario. Este alcohol (0.2926 g, 0.98 mmol) y ácido 3-formilfenilborónico (0.1544 g, 1.03 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). DME (2.2 mL) y Na2CO32M (1.5 mL, 2.94 mmol) luego se agregaron seguidos por Pd(PPh3)4 (0.0566 g, 0.049 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (10 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.29 mL, 1.96 mmol) seguido por TBAF (0.1 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (35% de EtOAc/hexano) para producir 0.2158 g (56% de rendimiento, 2 etapas) de TRV-1450. 1H RMN (DMSO, 500 MHz) 8 = 7.87 (s, 1H), 7.79 (d, J = 5 Hz, 1H), 7.57-7.54 (m, 2H), 7.21 (s, 1H), 6.96 (d, J = 5 Hz, 1H), 6.34 (t, J = 5 Hz, 1H), 5.29-5.25 (m, 1H), 4.91-4.89 (m, 1H), 4.55 (br s, 1H), 3.82-3.81 (m, 1H), 3.58-3.57 (m, 3H), 2.12-2.11 (m, 2H), 2.02-1.97 (m, 2H).

Se disolvió ácido 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidina-3-carboxílico (1.5 g, 5 mmol) en THF (50 mL) y se enfrió a 0°C. A este se agregó BH3-THF (10 mL, 10 mmol). Se le permitió a la reacción llegar a la temperatura ambiente durante la noche. Al siguiente día la reacción se detuvo con AcOH y se extrajo en EtOAc. La capa orgánica se lavó con NaOH 1 M hasta que los lavados quedaron azul tornasol y luego se concentraron en vacío. El material crudo se fusionó a SiO2 y se purificó mediante cromatografía de columna flash (3:2 Hex:EtOAc) para dar 800 mg de alcohol primario (56% de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.05 (s, 1H), 6.11 (s, 1H), 5.30 (s, 1H), 4.11 (t, J = 8 Hz, 2H), 3.85 (m, 4H), 3.00 (m, 1H). A una solución agitada del alcohol (500 mg, 1.76 mmol) en DCM (20 mL) se agregó peryodano de Dess-Martin (1.1 g, 2.64 mmol) y la solución se dejó de agitar durante 1 hora. En ese momento la reacción se diluyó con EtOAc (100 mL) y se lavó con carbonato de sodio (sat.). La capa orgánica se secó con MgSO4, y se concentró en vacío. El material crudo se purificó al tapar a través de SiO2 (DCM) para dar 420 mg (1.49 mmol, 84% de rendimiento) de aldehído. 1H RMN (500 MHz, CDCh) 8 = 9.96 (d, J = 2 Hz, 1H), 7.26 (s, 1H), 5.96 (s, 1H), 4.47 (m, 4H), 3.7 (m, 1h ). A una solución agitada a 0°C de este aldehído (420 mg, 1.5 mmol) en THF (10 mL) se agregó bromuro de metilmagnesio (1M, 1.8 mL). Después de 10 min a temperatura reducida el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 1 hora la reacción se enfrío de nuevo a 0°C y se agregó cuidadosamente NH4Cl saturado. La mezcla de reacción luego se diluyó con EtOAc (100 mL) y las fases se separaron. La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (4 g) y el gradiente se destiló (0-2% de MeOH en DCM) para dar 380 mg (1.27 mmol, 85% de rendimiento) del alcohol secundario. 1H RMN (500 MHz, CDCh) 8 = 7.18 (s, 1H), 5.898 (s, 1H), 4.36-4.08 (m, 5H), 2.85 (m, 1H), 1.25 (d, 3H). A una solución de este alcohol secundario (380 mg, 1.28 mmol) en Dm E (8 mL)/Na2CO3 (2M, 1.9 mL) se agregó ácido 3-formil-fenilborónico (286 mg, 1.9 mmol) y Pd(P(Ph)3)4 (70 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (50% de EtOAc en Hex) para dar 356 mg de material que se utilizó como estaba. 1H RMN (500 MHz, CDCl3) 8 = 10.1 (s, 1H), 8.11 (s, 1H), 7.63 (d J = 7 Hz, 1H), 7.88 (d, J = 7 Hz, 1H), 7.65 (t, J = 7 Hz, 1H), 7.17 (s, 1H), 6.05 (s, 1H), 4.40 (m, 2H), 4.27 (m, 1H), 4.15-4.05 (m, 2H), 2.87 (m, 1H), 1.26 (d, J = 6 Hz, 3H). A una solución agitada de este aldehído (356 mg, 1.1 mmol) y reactivo de Rupert (488 mL, 3.3 mmol) en THF (3 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (20-40% de EtOAc en Hex) para dar 100 mg (23% de rendimiento) de TRV-1451, una mezcla de 1:1:1:1 de diastereómeros debido a los dos centros quirales. 1H RMN (500 MHz, CDCh) 8 = 7.71 (s, 1H), 7.64 (m, 1H), 7.51 (m, 2H), 7.14 (s, 1H), 6.04 (s, 1H), 5.11 (m, 1H), 4.39 (m, 2H), 4.24 (m, 1H), 4.10 (m, 2H), 2.85 (m, 1H), 2.74 (m, 1H), 1.26 (d, J = 6 Hz, 3H).

Se agruparon 4,6-dibromobenzo[c][1,2,5]oxadiazol (2.013 g, 7.24 mmol) y sal de clorhidrato de pirrolidina-2-carboxilato de (S)-etilo (1.43 g, 7.96 mmol) en un tubo. El tubo se evacuó y se enjuagó con argón durante tres ciclos. NMP (10 mL) y DIPEA (3.5 mL, 19.9 mmol) luego se agregaron y el tubo se selló y calentó a 50°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite crudo luego se purificó a través de cromatografía de columna flash (15% de EtOAc/hexano) para producir 0.6105 g (25% de rendimiento) de anilina. La anilina (1.696 g, 4.9 mmol) se disolvió en DCM (20 mL) y se enfrió a -78°C. DIbA l (12.5 mL, solución 1.0 M en hexanos) se agregó en forma de gotas y luego la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción se detuvo con MeOH y luego se agregó Na2SO4 y la mezcla se agitó durante 30 minutos antes de filtrar a través de Celite. La fase orgánica se diluyó con EtOAc y agua. Las capas se separaron y la capa orgánica se lavó con H2O (3X), solución salina, se secó (Na2SO4), se filtró y se concentró. El material crudo se purificó a través de cromatografía (30% de EtOAc/hexano) para dar 1.294 g (87 % de rendimiento) del alcohol primario. El alcohol primario (0.9956 g, 3.34 mmol) se disolvió en DCM (100 mL) y se agregó DMP (2.1249 g, 5.0 mmol). La reacción se agitó durante 2 horas y luego se detuvo con NaHCO3 saturado (ac.) y se agregó exceso de Na2SO4 (8.0 g) y la mezcla se agitó hasta que todos los sólidos se disolvieron. Esta mezcla luego se extrajo con DCM y la capa orgánica combinada se secó (Na2SO4), se filtró y se concentró. La purificación (columna de 20% de EtOAc/hexano) produjo 0.6471 g (65% de rendimiento) del aldehído. Este aldehído (0.200 g, 0.675 mmol) y pirrolidina (0.06 mL, 0.743 mmol) se disolvieron en DCM (3.1 mL) y luego se trató con NaBH(OAc)3 (0.2003 g, 0.945 mmol) y la mezcla se agitó durante 2 horas. La solución se enfrió a 0°C y se detuvo con NaOH 1N. Esta mezcla luego se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar la amina cruda. Esta amina y ácido 3-formilfenilborónico (0.1063 g, 0.709 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (1.5 mL) y Na2CO3 2M (1.0 mL, 2.0 mmol) seguidos por Pd(PPh3)4 (0.0389 g, 0.033 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (3 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.20 mL, 1.35 mmol) seguido por TBAF (0.1 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (5% de MeOH/DCM) para producir 0.1537 g (51% de rendimiento) de TRV-1452. 1H RMN (CDC13, 500 MHz) 8 = 7.76-7.74 (m, 2H), 7.68-7.65 (m, 2H), 7.51-7.47 (m, 4H), 7.08 (s, 1H), 7.07 (s, 1H), 6.22 (s, 1H), 6.21 (s, 1H), 5.11-5.05 (m, 2H), 64.65 (br s, 1H), 4.52 (br s, 1H), 3.94-3.89 (m, 2H), 3.65-3.59 (m, 2H), 2.70-2.52 (m, 12 H), 2.28-2.25 (m, 2H), 2.14-2.05 (m, 6H), 1.81 (br s, 8H).

Se agruparon 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (1.00 g, 2.97 mmol) y ácido 3-carboxifenilborónico (0.5176 g, 3.12 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (8.9 mL) y Na2CO32M (6.0 mL, 11.9 mmol) seguidos por Pd(PPh3)4 (0.1733 g, 0.15 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas
combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el material crudo. Este se purificó a través de columna de 5% de MeOH/DCM para producir 1.0873 g (97 % de rendimiento, 90% de pureza química) del ácido. Una mezcla del ácido (0.200 g, 0.529 mmol), ciclopropilamina (0.04 mL, 0.529 mmol) y TEA (0.18 mL, 1.32 mmol) se agitó en EtOAc (6 mL) y se enfrió en un baño de hielo. Se agregó en forma de gotas la solución T3P (0.4040 g, 50% p/p en EtOAc). Una vez se completó la adición la reacción se dejó calentar a temperatura ambiente. La reacción luego se detuvo con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó mediante cromatografía (40% de EtOAc/hexano) para dar 0.1109 g (50% de rendimiento) de TRV-1453. 1H RMN (CDC13, 500 MHz) 8 = 8.01 (s, 1H), 7.72 (dd, J = 8, 1.6 Hz, 2H), 7.51 (t, J = 8 Hz, 1H), 7.26-7.23 (m, 3H), 7.02 (t, J = 8 Hz, 2H), 6.35 (s, 1H), 6.32 (br s, 1H), 5.13 (s, 2H), 3.17 (s, 3H), 2.94-2.92 (m, 1H), 0.92-0.85 (m, 2H), 0.67-0.64 (m, 2H).

Se agruparon 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (1.00 g, 2.97 mmol) y ácido 3-carboxifenilborónico (0.5176 g, 3.12 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (8.9 mL) y Na2CO32M (6.0 mL, 11.9 mmol) seguidos por Pd(PPh3)4 (0.1733 g, 0.15 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el material crudo. Este se purificó a través de columna de 5% de MeOH/DCM para producir 1.0873 g (97 % de rendimiento, 90% de pureza química) del ácido. Una mezcla del ácido (0.200 g, 0.529 mmol), morfolina (0.05 mL, 0.529 mmol) y TEA (0.18 mL, 1.32 mmol) se agitó en EtOAc (6 mL) y se enfrió en un baño de hielo. Se agregó en forma de gotas la solución T3P (0.4040 g, 50% p/p en EtOAc). Una vez se completó la adición la reacción se dejó calentar a temperatura ambiente. La reacción luego se detuvo con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó mediante cromatografía (65% de EtOAc/hexano) para dar 0.1331 g (56% de rendimiento) de TRV-1454. 1H RMN (CDC13, 500 MHz) 8 = 7.67 (d, J = 10 Hz, 1H), 7.64 (s, 1H), 7.52 (t, J = 10 Hz, 1H), 7.44-7.41 (m, 2H), 7.26-7.23 (m, 2H), 7.02 (t, J = 10 Hz, 2H), 6.33 (s, 1H), 5.13 (s, 2H), 3.81 (br s, 4H), 3.63 (br s, 2H), 3.48 (br s, 2H), 3.17 (s, 3H).

A una solución agitada de (1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)metanol (500 mg, 1.76 mmol) en DCM (20 mL) se agregó peryodano de Dess-Martin (1.1 g, 2.64 mmol) y la solución se dejó de agitar durante 1 hora. En ese momento la reacción se diluyó con EtOAc (100 mL) y se lavó con carbonato de sodio (sat.). La capa orgánica se secó con MgSO4, y se concentró en vacío. El material crudo se purificó al tapar a través de SiO2 (DCM) para dar 420 mg (1.49 mmol, 84% de rendimiento) de aldehído. 1H RMN (500 MHz, CDCh) 8 = 9.96 (d, J = 2 Hz, 1H), 7.26 (s, 1H), 5.96 (s, 1H), 4.47 (m, 4H), 3.7 (m, 1H). A una solución agitada de este aldehído (316 mg, 1.1 mmol) en DCM (5 mL) se agregó morfolina (105 mL, 1.21 mmol) seguido por triacetoxiborohidruro de sodio (326 mg, 1.54 mmol). Cuando la reacción se consideró completa (se monitorizó mediante TLC), se diluyó con DCM (100 mL) y se lavó con agua (50 mL), hidróxido de sodio (1M, 50 mL), luego se secó con MgSO4 y se concentró en vacío para dar 406 mg de amina. El material se utilizó sin purificación adicional. 1H RMN (500 m Hz , CDCh) 8 = 7.16 (s, 1H), 5.85 (s, 1H), 4.39 (m 2H), 3.97 (m, 2H), 3.70 (m, 4H), 3.05 (m, 1H), 2.68 (d, J = 8 Hz, 2H), 2.45 (m, 4H). A una solución de esta
amina (406 mg, 1.15 mmol) en DME (7 mL)/Na2CO3 (2M, 1.7 mL) se agregó ácido 3-formil-fenilborónico (259 mg, 1.9 mmol) y Pd(P(Ph)3)4 (60 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (50% de Acetona en EtOAc) para dar 380 mg (1 mmol, 85% de rendimiento) de aldehido que se utilizó como estaba. 1H RMN (500 MHz, CDCh) 8 = 10.1 (s, 1H), 8.13 (s, 1H), 7.93 (d, J = 8 Hz, 1H), 7.89 (d, J = 8 Hz, 1H), 7.66 (t, J = 8 Hz, 1H), 7.18 (s, 1H), 6.05 (s, 1H), 4.46 (t, J = 8 Hz, 2H), 4.04 (m, 2H), 3.72 (t, J = 4 Hz, 4H), 3.10 (m, 1H), 2.72 (d, J = 7 Hz, 2H), 2.47 (m, 4H). A una solución agitada de este aldehido (380 mg, 1.0 mmol) y reactivo de Rupert (220 pL, 1.5 mmol) en THF (3 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con, solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (80% de EtOAc, 2% de TEA, Hex en equilibrio) para dar 100 mg (22% de rendimiento). 1H RMN (500 MHz, CDCla) 8 = 7.72 (s, 1H), 7.65 (dm, J = 8 Hz, 1H), 7.52 (m, 2H), 7.15 (s, 1H), 6.03 (s, 1H), 5.13 (q, J = 5 Hz, 1H), 4.44 (t, J = 8 Hz, 2H), 4.01 (m, 2H), 3.72 (t, J = 6 Hz, 4H), 3.09 (m, 1H), 2.92 (s, br, 1H), 2.71 (d, j = 8 Hz, 2H), 2.47 (m, 4H).

A una solución agitada de 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (280 mg, 1.04 mmol) disuelta en DCM (10 mL) se agregó reactivo de Dess-Martin (Oakwood) (571 mg, 1.3 mmol) disuelto en DCM (4 mL). Después de 1 hora la reacción se había vuelto turbia y se había formado un precipitado. El material se vertió en NaOH 1 M y se extrajo con TBME para dar la cetona correspondiente. 1H RMN (500 MHz, CDCla) 8 = 7.40 (s, 1H), 6.13 (s, 1H), 5.10 (s, 4H). A una solución agitada de la cetona (226 mg, 0.8 mmol) en DCM (4 mL) se agregó pirrolidina (75 mL, 0.9 mmol), ácido acético glacial (45 pL, 0.9 mmol) y triacetoxiborohidruro de sodio (250 mg, 1.26 mmol). Cuando la reacción se consideró completa (TLC) se diluyó con EtOAc (100 mL) y se lavó con hidróxido de sodio (1M ac.). La fase orgánica luego se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. La amina cruda se purificó mediante cromatografía instantánea (EtOAc) para dar 216 mg (0.7 mmol, 79% de rendimiento) de material.
1H RMN (500 MHz, CDCls) 8 = 7.18 (s, 1H), 5.89 (s, 1H), 4.28 (m, 2H), 4.21 (m, 2H), 3.53 (m, 1H), 2.57 (m, 4H), 1.58 (m, 4H). A una solución de esta amina (216 mg, 0.7 mmol) en Dm E (4 mL)/Na2CO3 (2M, 1.05 mL) se agregó ácido 3-formil-fenilborónico (158 mg, 1.05 mmol) y Pd(P(Ph)3)4 (60 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (EtOAc 1% de MeOH) para dar 212 mg de aldehído que se utilizó como estaba. 1H RMN (500 MHz, CDCh) 8 = 10.1 (s, 1H), 8.11 (s, 1H), 7.93 (d, J = 8 Hz, 1H), 7.87 (d, J = 8 Hz, 1H), 7.63 (m 1H), 7.17 (s, 1H), 6.05 (s, 1H), 4.43 (m, 2H), 4.24 (m, 2H), 3.53 (m, 1H), 2.58 (m, 4H), 1.85 (m, 4H). A una solución agitada de este aldehído (212 mg, 0.6 mmol) y reactivo de Rupert (266 pL, 1.8 mmol) en THF (3 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (2% de MeOH en DCM) para dar 16 mg (6.2% de rendimiento). 1H RMN (500 MHz, DMSO) 8 = 7.89 (s, 1H), 7.81 (dm, J = 8 Hz, 1H), 7.56 (m, 2H), 7.31 (s, 1H), 6.96 (d, J = 6 Hz, 1H), 6.25 (s, 1H), 5.28 (m 1H), 4.38 (m, 2H), 4.16 (m, 2H), 3.52 (m, 1H), 2.51 (m, 4H), 1.73 (m, 4H).

A una solución agitada de (1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)metanol (500 mg, 1.76 mmol) en DCM (20 mL) se agregó peryodano de Dess-Martin (1.1 g, 2.64 mmol) y la solución se dejó de agitar durante 1 hora. En ese momento la reacción se diluyó con EtOAc (100 mL) y se lavó con carbonato de sodio (sat.). La capa orgánica se secó con MgSO4, y se concentró en vacío. El material crudo se purificó al tapar a través de SO2 (DCM) para dar 420 mg (1.49 mmol, 84% de rendimiento) de aldehido. 1H RMN (500 MHz, CDCls) 8 = 9.96 (d, J = 2 Hz, 1H), 7.26 (s, 1H), 5.96 (s, 1H), 4.47 (m, 4H), 3.7 (m, 1H). A una solución agitada de este aldehido (316 mg, 1.1 mmol) en DCM (5 mL) se agregó pirrolidina (118 pL, 1.45 mmol) seguido por triacetoxiborohidruro de sodio (385 mg, 1.82 mmol). Cuando la reacción se consideró completa (se monitorizó mediante TLC), se diluyó con DCM (100 mL) y se lavó con agua (50 mL), hidróxido de sodio (1M, 50 mL), luego se secó con MgSO4 y se concentró en vacío para dar 425 mg. El material se utilizó sin purificación adicional. 1H RMN (500 MHz, CDCla) 8 = 7.14 (s, 1H), 5.83 (s, 1H), 4.40 (m, 2H), 3.98 (m, 2H), 3.04 (m, 1H), 2.76 (d, J = 8 Hz, 2H), 2.51 (m, 4H), 1.79 (m, 4H). A una solución de amina (425 mg, 1.26 mmol) en DME (8 mL)/Na2CO3 (2m , 1.9 mL) se agregó ácido 3-formil-fenilborónico (283 mg, 1.9 mmol) y Pd(P(Ph)3)4 (66 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SO2 (50% de Acetona en EtOAc) para dar 274 mg (0.75 mmol, 75% de rendimiento) de aldehido que se utilizó como estaba. 1H RMN (500 MHz, CDCl3) 8 = 10.10 (s, 1h ), 8.12 (s, 1H), 7.93 (d, J = 8 Hz, 1H), 7.89 (d, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 717 (s, 1H), 6.03 (s, 1H), 4.48 (t, J = 8 Hz, 2H), 4.05 (m, 2H), 3.10 (m, 1H), 2.81(d, J = 8 Hz, 2H), 2.54 (m, 4H), 1.80 (m, 4H). A una solución agitada de este aldehido (274 mg, 0.75 mmol) y reactivo de Rupert (166 pL, 1.1 mmol) en THF (3 mL) a 0°C se agregó TBAF (0.1 mL, THF 1 M, 0.1 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con t Hf (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (50% de EtOAc, 49% Hex, 1% de TEA) para dar 70 mg (22% de rendimiento). 1H RMN (500 MHz, DMSO-D6) 8 = 7.88 (s, 1H), 7.81 (d, J = 8 Hz, 1H), 7.59 (dm, J = 7 Hz, 1H), 7.54 (t, J = 8 Hz, 1H), 7.30 (s, 1H), 6.96 (d, J = 5 Hz, 1H), 6.24 (s, 1H), 5.28 (m, 1H),4.38 (m, 2H), 3.97 (m, 2H), 3.00 (m, 1H), 2.74 (d, J = 7 Hz, 2H), 2.45 (m, 4H), 1.68 (m, 4H).

Se agruparon 4,6-dibromobenzo[c][1,2,5]oxadiazol (2.013 g, 7.24 mmol) y sal de clorhidrato de pirrolidina-2-carboxilato de (S)-etilo (1.43 g, 7.96 mmol) en un tubo. El tubo se evacuó y se enjuagó con argón durante tres ciclos. Luego se agregaron NMP (10 mL) y DIPEA (3.5 mL, 19.9 mmol) y el tubo se selló y calentó a 50°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite crudo luego se purificó a través de cromatografía de columna flash (15% de EtOAc/hexano) para producir 0.6105 g (25% de rendimiento) de anilina. La anilina (1.696 g, 4.9 mmol) se disolvió en DCM (20 mL) y se enfrió a -78°C. Se agregó en forma de gotas DIBAL (12.5 mL, solución 1.0 M en hexanos) y luego la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción se detuvo con MeOH y luego se agregó Na2SO4 y la mezcla se agitó durante 30 minutos antes de filtrar a través de Celite. La fase orgánica se diluyó con EtOAc y agua. Las capas se separaron y la capa orgánica se lavó con H2O (3X), solución salina, se secó (Na2SO4), se filtró y se concentró. El material crudo se purificó a través de cromatografía (30% de EtOAc/hexano) para dar 1.294 g (87 % de rendimiento) del alcohol primario. El alcohol primario (0.9956 g, 3.34 mmol) se disolvió en DCM (100 mL) y se agregó DMP (2.1249 g, 5.0 mmol). La
reacción se agitó durante 2 horas y luego se detuvo con NaHCO3 saturado (ac.) y se agregó exceso de Na2SO4 (8.0 g) y la mezcla se agitó hasta que todos los sólidos se disolvieron. Esta mezcla luego se extrajo con DCM y la capa orgánica combinada se secó (Na2SO4), se filtró y se concentró. La purificación (columna de 20% de EtOAc/hexano) produjo 0.6471 g (65% de rendimiento) del aldehído. Este aldehído (0.206 g, 0.696 mmol) y clorhidrato de ciclobutilamina (0.0794 g, 0.738 mmol) se disolvieron en metanol (3 mL) y se agregó TEA (0.19 mL, 1.39 mmol). Este material se agitó durante 24 horas y luego se enfrió a 0°C. A esta mezcla luego se agrego en forma de porciones NaBH4 (0.0685 g, 1.81 mmol). La mezcla se agitó durante 60 minutos y luego se detuvo con NaOH 1N (ac.). La mezcla se extrajo con EtOAc (3X) y los orgánicos combinados se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar la amina cruda. A una solución de esta amina en THF (5 mL) a 0°C se agregó TEA (0.19 mL, 1.39 mmol), DMAP (un cristal) y luego BOC2O (0.1822 g, .835 mmol). La reacción se agitó a 0°C mientras que se monitorizó por TLC. Luego se detuvo con H2O y se extrajo con EtOAc. Los orgánicos combinados se lavaron con H2O (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el carbamato crudo. Este carbamato y ácido 3-formilfenilborónico (0.1094 g, 0.73 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (1.6 mL) y Na2CO32M (1.1 mL, 2.09 mmol) seguidos por Pd(PPh3)4 (0.0402 g, 0.0348 mmol). El tubo luego se selló y se calentó a 80°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (4 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.21 mL, 1.39 mmol) seguido por TBAF (0.07 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el trifluorocarbinol, como una mezcla de diastereómeros. Este material crudo luego se disolvió en DCM (15 mL) y se enfrió a 0°C. Se agregó TFA (0.53 mL) y la mezcla se agitó durante 36 horas antes de detenerla con NaHCO3 saturado(ac.). Esta mezcla se extrajo con DCM (5X) y los orgánicos combinados se secaron con NA2SO4, se filtraron y luego se concentraron. Este material crudo se purificó a través de columna de 5% de MeOH/DCM para producir 0.130 g de la amina libre. Este material luego se disolvió en MeOH a 0°C y se trató con un gran exceso de HCl metanólico. La mezcla de reacción se agitó durante 30 minutos y luego se concentró para producir 0.081 g (24 % de rendimiento, 8 etapas) de TRV-1458. 1H RMN (MeOD, 700 MHz) 8 = 7.83 (s, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.52 (t, J = 7.6 Hz, 1H), 7.26 (s, 1H), 6.43 (s, 1H), 5.16 (q, J = 7.1 Hz, 1H), 3.87 (m, 1H), 3.78-3.77 (m, 1H), 3.52-3.50 (m, 1H), 3.30 (s, 2H), 3.25 (dd, J = 12.5, 2.1 Hz, 1H), 3.05 (t, J = 11.0 Hz, 1H), 2.36-2.20 (m, 9H), 1.99-1.90 (m, 2H).

Se agruparon 4,6-dibromobenzo[c][1,2,5]oxadiazol (1.1509 g, 4.14 mmol) y N-metil-1-(tiazol-2-il)metanamina (4.97 mmol) en un tubo. El tubo se evacuó y se enjuagó con argón durante tres ciclos. Luego se agregaron NMP (6 mL) y DIPEA (0.94 mL, 5.38 mmol) y el tubo se selló y calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. El aceite crudo luego se purificó a través de cromatografía de columna flash (30% de EtOAc/hexano) para producir 0.1872 g (14 % de rendimiento) de anilina. Esta anilina (0.1872 g, 0.58 mmol) y ácido 3-formilfenilborónico (0.0899 g, 0.60 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (1.5 mL) y Na2CO3 2M (0.9 mL, 1.74 mmol) seguidos por Pd(PPh3)4 (0.0335 g, 0.029 mmol). El tubo luego se selló y se calentó a 85°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (4 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.17 mL, 1.16 mmol) seguido por TBAF (0.06 mL, solución 1.0 M en t Hf ). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash con etapa-gradiente (100% de DCM a 0.5% de MeOH/DCM) para producir 0.0339 g (14 % de rendimiento, 3 etapas) de TRV-1459. 1H RMN (CDC13, 700 MHz) 8 = 7.77 (d, J = 3.2 Hz, 1H), 7.73 (s, 1H), 7.66 (dt,
J = 7.2, 1.6 Hz, 1H), 7.55-7.51 (m, 2H), 7.31 (s, 1H), 7.30 (d, J = 3.2 Hz, 1H), 6.46 (s, 1H), 5.46 (s, 2H), 5.13 (q, J = 6.6 Hz, 1H), 3.32 (s, 3H).

Se agruparon 6-bromo-N-(4-fluorobencil)-N-metilbenzo[c][1,2,5]oxadiazol-4-amina (1.00 g, 2.97 mmol) y ácido 3-carboxifenilborónico (0.5176 g, 3.12 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3X). Luego se agregaron DME (8.9 mL) y Na2CO32M (6.0 mL, 11.9 mmol) seguidos por Pd(PPh3)4 (0.1733 g, 0.15 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar el material crudo. Este se purificó a través de columna de 5% de MeOH/DCM para producir 1.0873 g (97 % de rendimiento, 90% de pureza química) del ácido. Una mezcla del ácido (0.3774 g, 1.0 mmol), pirrolidina (0.083 mL, 1.0 mmol) y TEA (0.35 mL, 2.5 mmol) se agitaron en EtOAc (10 mL) y se enfrió en un baño de hielo. Se agregó en forma de gotas la solución T3P (0.7636 g, 50% p/p en EtOAc). Una vez se completó la adición la reacción se dejó calentar a temperatura ambiente. La reacción luego se detuvo con agua y se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron. El material crudo luego se purificó mediante dos columnas sucesivas (75% de EtOAc/hexano y luego 70% de EtOAc/hexano) para dar 0.1436 g (33% de rendimiento, 96% c.p.) de TRV-1460. 1H RMN (CDC13, 500 MHz) 8 = 7.75 (s, 1H), 7.65 (d, J = 10 Hz, 1H), 7.55 (d, J = 10 Hz, 1H), 7.52-7.48 (m, 1H), 7.26-7.23 (m, 3H), 7.01 (t, J = 10 Hz, 2H), 6.35 (s, 1H), 5.12 (s, 2H), 3.68 (t, J = 5 Hz, 2H), 3.45 (t, J = 5 Hz, 2H), 2.02-1.95 (m, 2H), 1.92-1.86 (m, 2H).

Se disolvió ácido 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidina-3-carboxílico (1.5 g, 5 mmol) en THF (50 mL) y se enfrió a 0°C. A este se agregó BH3-THF (10 mL, 10 mmol). Se le permitió a la reacción llegar a la temperatura ambiente durante la noche. Al siguiente día la reacción se detuvo con AcOH y se extrajo en EtOAc. La capa orgánica se lavó con NaOH 1 M hasta que los lavados quedaron azul tornasol y luego se concentraron en vacío. El material crudo se fusionó a SiO2 y se purificó mediante cromatografía de columna flash (3:2 Hex:EtOAc) para dar 800 mg de alcohol primario (56% de rendimiento). 1H RMN (500 MHz, CDCh) 8 = 7.05 (s, 1H), 6.11 (s, 1H), 5.30 (s, 1H), 4.11 (t, J = 8 Hz, 2H), 3.85 (m, 4H), 3.00 (m, 1H).A una solución agitada del alcohol (500 mg, 1.76 mmol) en DCM (20 mL) se agregó peryodano de Dess-Martin (1.1 g, 2.64 mmol) y la solución se dejó de agitar durante 1 hora. En ese momento la reacción se diluyó con EtOAc (100 mL) y se lavó con carbonato de sodio (sat.). La capa orgánica se secó con MgSO4, y se concentró en vacío. El material crudo se purificó al tapar a través de SiO2 (DCM) para dar 420 mg (1.49 mmol, 84% de rendimiento) de aldehido. 1H RMN (500 MHz, CDCh) 8 = 9.96 (d, J = 2 Hz, 1H), 7.26 (s, 1H), 5.96 (s, 1H), 4.47 (m, 4H), 3.7 (m, 1h ). A una solución agitada a 0°C de este aldehido (420 mg, 1.5 mmol) en THF (10 mL) se agregó bromuro de metilmagnesio (1M, 1.8 mL). Después de 10 min a temperatura reducida el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 1 hora la reacción se enfrío de nuevo a 0°C y se agregó cuidadosamente NH4Cl saturado. La mezcla de reacción luego se diluyó con EtOAc (100 mL) y las fases se separaron. La fase orgánica se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. El material crudo se fusionó a SiO2 (4 g) y el gradiente se destiló (0-2% de MeOH en DCM) para dar 380 mg (1.27 mmol, 85% de rendimiento) de alcohol secundario. A una solución del alcohol secundario (aproximadamente 190 mg, 0.65
mmol) se disolvió en THF (5 mL) y se enfrió a 0°C se agregó NaH 60% en aceite (100 mg, 3 mmol). Una vez ha cesado el burbujeo inicial MeI (200 j L, 3 mmol) se agregó en forma de gotas la mezcla de reacción se dejó ir a RT. Después de 18 h la reacción se enfrío de nuevo a 0°C y NH4Cl se agregó cuidadosamente. La mezcla de reacción se extrajo con EtOAc (3 X 50 mL), se lavó con solución salina (1 50 mL) y se concentró en vacío. El material crudo se purificó al tapar a través de SiO2 (80% de DCM en Hex) para dar 160 mg (78% de rendimiento) de éter de metilo. A una solución del éter de metilo (160 mg, 0.5 mmol) en DME (4 mL)/Na2CO3 (2 M, 0.75 mL) se agregó ácido 3-formil-fenilborónico (113 mg, 1.5 mmol) y Pd(P(Ph)3)4 (40 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (DCM/5% de EtOAc) para dar 150 mg de aldehido que se utilizó como estaba. A una solución agitada del aldehido (150 mg, 0.4 mmol) y reactivo de Rupert (98 j L, 0.7 mmol) en THF (2 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (30% de EtOAc en Hexano) para dar 37 mg (23% de rendimiento) de TRV-1461, una mezcla de 1:1:1:1 de diastereómeros debido a los dos centros quirales. 1H RMN (500 MHz, DMSO) 8 = 7.72 (s, 1H), 7.65 (d, J = 7 Hz, 1 H), 7.52 (m, 2H), 7.13 (s, 1H), 6.02 (s, 1H), 5.13 (m, 1H), 4.38 (m, 2H), 4.19 (m, 1H), 4.09 (m, 1H), 3.55 (p, J = 6 Hz, 1H), 3.39 (s, 3H), 2.88 (m, 1H), 2.71 (s, br, 1H), 1.78 (d, J = 6 Hz, 3H).

A una solución agitada de 1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-ol (730 mg, 2.7 mmol) disuelta en DCM (10 mL) se agregó reactivo de Dess-Martin (Oakwood) (1.26 g, 2.97 mmol) disuelto en DCM (10 mL). Después de 1 hora la reacción se había vuelto turbia y se había formado un precipitado. El material se vertió en NaOH 1 M y se extrajo con TBME para dar la cetona correspondiente. 1H RMN (500 MHz, CDCh) 8 = 7.40 (s, 1H), 6.13 (s, 1H), 5.10 (s, 4H). A una solución agitada de la cetona (300 mg, 1.1 mmol) en DCM (4 mL) se agregó morfolina (160 j L, 1.23 mmol), ácido acético glacial (65 j L, 1.1 mmol) y triacetoxiborohidruro de sodio (360 mg, 1.68 mmol). Cuando la reacción se consideró completa (TLC) se diluyó con EtOAc (100 mL) y se lavó con hidróxido de sodio (1M ac.). La fase orgánica luego se lavó con solución salina, se secó con MgSO4 y se concentró en vacío. La amina cruda se purificó mediante cromatografía instantánea (EtOAc) para dar 470 mg (1.4 mmol, 52% de rendimiento) de material. A una solución de esta amina (470 mg, 1.4 mmol) en DME (5 mL)/Na2CO3 (2M, 2 mL) se agregó ácido 3-formilfenilborónico (180 mg, 1.05 mmol) y Pd(P(Ph)3)4 (80 mg). El matraz luego se equipó con un condensador de reflujo, se purgó con argón y se calentó a 85°C O/N. La reacción se sometió a trabajo final al vertir en NaOH 1 M (150 mL) y aislar en vacío los sólidos resultantes. El material crudo se purificó al tapar a través de SiO2 (EtOAc 1% de MeOH) para dar 300 mg de aldehído que se utilizó como estaba. A una solución agitada de este aldehído (300 mg, 0.8 mmol) y reactivo de Rupert (240 j L, 1.6 mmol) en THF (3 mL) a 0°C se agregó TBAF (0.2 mL, THF 1 M, 0.2 mmol). Después de 30 min a baja temperatura el baño frío se retiró y se le permitió a la reacción llegar a RT. Después de 3 horas la reacción se concentró en vacío y el matraz se cargó con THF (20 mL). A este se agregó un exceso de TBAF y la reacción se dejó de agitar. Una vez se completo la desprotección se agregó EtOAc (100 mL) y la reacción se lavó con NH4Cl saturado, solución salina, y luego se secó con MgSO4 y se concentró en vacío. El material crudo se purificó mediante cromatografía de columna flash (2% de MeOH en d Cm ) para dar 40 mg (9% de rendimiento). 1H RMN (500 MHz, DMSO) 8 = 7.71 (s, 1H), 7.63 (d, J = 7 Hz, 1H), 7.51 (m, 2H), 7.16 (s, 1H), 6.06 (s, 1H), 5.13 (m, 1H), 4.41 (t, J = 8 Hz, 2H), 4.18 (m, 2H), 3.77 (m, 4H), 3.42 (p, J = 6 Hz, 1H), 2.87 (s, amplio, 1H), 2.48 (m, 4H).

Se agruparon 6-bromo-4-(4-(pirrolidin-1-il)piperidin-1-il)benzo[c][1,2,5]oxadiazol (0.3366 g, 0.958 mmol) y ácido 2-formiltiofeno- 4-borónico (0.1794 g, 1.15 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (2.1 mL) y Na2CO32M (1.4 mL, 2.87 mmol) seguidos por Pd(PPh3)4 (0.0554 g, 0.048 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.21 mL, 1.44 mmol) seguido por TBAF (0.1 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (5% de MeOH/DCM) para producir 0.067 g (15% de rendimiento, 3 etapas) de TRV-1463 como un sólido amarillo. 1H RMN (DMSO, 700 MHz) 8 = 8.29 (d, J = 1.0 Hz, 1H), 7.81 (s, 1H), 7.59 (s, 1H), 7.37 (d, J = 6.0 Hz, 1H), 6.83 (s, 1H), 5.52 5.48 (m, 1H), 4.22 (br s, 2H), 3.13 (t, J = 12 Hz, 2 H), 2.59 (br s, 4 H), 2.32-2.30 (m, 1H), 2.02-2.01 (m, 2H), 1.71 (s, 4H), 1.62-1.61 (m, 2H).

Se agruparon 6-bromo-4-(4-(pirrolidin-1-il)piperidin-1-il)benzo[c][1,2,5]oxadiazol (0.3368 g, 0.958 mmol) y ácido 5-formil-2- tienil-borónico (0.1793 g, 1.15 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (2.1 mL) y Na2CO32M (1.4 mL, 2.87 mmol) seguidos por Pd(PPh3)4 (0.0554 g, 0.048 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.28 mL, 1.92 mmol) seguido por TBAF (0.1 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (10% de MeOH/DCM). La pureza después de la primera columna fue inadecuada de tal manera que se repitió la separación con una columna de 5% de MeOH/DCM para producir 0.060 g (14 % de rendimiento, 3 etapas) de TRV-1464. 1H RMN (DMSO, 700 MHz) 8 = 7.77 (d, J = 5 Hz, 1H), 7.56 (s, 1H), 7.42 (d, J = 5 Hz, 1H), 7.28 (d, J = 5 Hz, 1H), 6.76 (s, 1H), 5.59-5.55 (m, 1H), 4.24 (d, J = 10 Hz, 2H), 3.17 (d, J = 10 Hz, 2H), 2.60 (br s, 4H), 2.37 (br s, 1H), 2.05-2.02 (m, 2H), 1.72 (br s, 4H), 1.64-1.62 (m, 2H).

Una mezcla de dibromobenzofurazan (0.500 g, 1.8 mmol), N1,N1,N3-trimetilpropano-1,3-diamina (0.28 mL, 1.9 mmol) y DIPEA (0.31 mL, 1.8 mmol) en NMP (2 mL) se calentó a 100°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con EtOAc y agua. Después de que capas se separaron, la capa acuosa se basificó y se extrajo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar la anilina cruda. Este material se purificó a través de columna de 10% de MeOH/DCM para producir 0.3988 g de anilina. Esta anilina (0.3988 g, 1.27 mmol) y ácido 3-formilfenilborónico (0.2488 g, 1.66 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3x). DME (2.8 mL) y Na2CO32M (1.9 mL, 3.8 mmol) luego se agregaron seguidos por Pd(PPh3)4 (0.074 g, 0.064 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.38 mL, 2.54 mmol) seguido por TBAF (0.13 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (10% de MeOH/DCM) para producir 0.061 g (12% de rendimiento, 2 etapas) de TRV-1465. 1H RMN (DMSO, 500 MHz) 8 = 7.89 (s, 1H), 7.81 (d, J = 10 Hz, 1H), 7.59-7.53 (m, 2H), 7.29 (s, 1H), 6.95 (d, J = 5 Hz, 1H), 6.40 (s, 1H), 5.31-5.25 (m, 1H), 3.92 (t, J = 10 Hz, 2H), 3.24 (s, 3H), 2.24 (t, J = 10 Hz, 2H), 2.08 (s, 6H), 1.77 1.71 (m, 2H).

Una mezcla de dibromobenzofurazan (0.4669 g, 1.68 mmol), N,N-dimetilpiperidin-4-amina (0.2263 g, 1.76 mmol) y DIPEA (0.29 mL, 1.68 mmol) en NMP (3 mL) se calentó a 95°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con EtOAc y agua. Después de que capas se separaron, la capa acuosa se basificó y se extrajo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar la anilina cruda. Este material se purificó a través de columna de 5% de MeOH/DCM para producir 0.2221 g de anilina. Esta anilina (0.2221 g, 0.68 mmol) y ácido 3-formilfenilborónico (0.1334 g, 0.89 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3x). DME (3.0 mL) y Na2CO32M (2.0 mL, 4 mmol) luego se agregaron seguidos por Pd(PPh3)4 (0.039 g, 0.034 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.20 mL, 1.36 mmol) seguido por TBAF (0.07 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (10% de MeOH/DCM) para producir 0.061 g (24 % de rendimiento, 2 etapas) de TRV-1466. 1H RMN (DMSO, 700 MHz) 8 = 7.90 (s, 1H), 7.83 (d, J = 7 Hz, 1H), 7.59 (d, J = 7 Hz, 1H), 7.55 (t, J = 7 Hz, 1H), 7.51 (d, J = 3 Hz, 1H), 6.96 (d, J = 3
Hz, 1H), 6.73 (s, 1H), 5.30-5.27 (m, 1H), 4.33 (d, J = 7 Hz, 2H), 3.05 (t, J = 14 Hz, 2H), 2.41-2.39 (m, 1H), 2.23 (s, 6H), 1.93 (d, J = 14 Hz, 2H), 1.61-1.54 (m, 2H).

Se agruparon 6-bromo-4-(4-(pirrolidin-1-il)piperidin-1-il)benzo[c][1,2,5]oxadiazol (1 g, 2.8 mmol) y ácido (2-formilfenil)borónico (0.554 g, 3.6 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (6 mL) y Na2CO32M (4.2 mL, 8.4 mmol) seguidos por Pd(PPh3)4 (0.160 g, 0.14 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron, se concentraron y se sometieron a cromatografía de columna flash (5% de MeOH/DCM) para producir 0.95 g de 2-(7-(4-(pirrolidin-1 -il)piperidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzaldehído. Este aldehído luego se disolvió en THF (15 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.738 mL, 5.06 mmol) seguido por TBAF (0.25 mL, solución 1.0 M en THF). El baño de enfriamiento se retiró y la reacción se agitó durante 30 min. La reacción luego se volvió a enfriar y se agregó TBAF (0.30 mL, solución 1.0 M en THF) y el baño frío se retiró. La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (5% de MeOH/DCM) para producir 0.550 g (44 % de rendimiento, 2 etapas) de TRV-1467 como un sólido amarillo. 1H RMN (DMSO, 500 MHz) 8 = 7.72 (d, J = 8.0 Hz, 1H), 7.58-7.48 (m, 2H), 7.36 (d, J = 7.5 Hz, 1H), 7.15 (s, 1H) 6.91 (d, J = 5 Hz, 1H), 6.33 (s, 1H), 5.13-5.11 (m, 1H), 4.16 (br s, 2H), 3.15- 3.09 (m, 2 H), 2.53-2.49 (m, 4 H), 2.26 (br s, 1H), 1.99 1.97 (m, 2H), 1.68 (s, 4H), 1.59-1.57 (m, 2H).

Se agruparon 6-bromo-4-(4-(pirrolidin-1-il)piperidin-1-il)benzo[c][1,2,5]oxadiazol (0.5 g, 1.4 mmol) y ácido (2-formilfenil) borónico (0.277 g, 1.8 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (6 mL) y Na2CO32M (4.2 mL, 8.4 mmol) seguidos por Pd(PPh3)4 (0.080 g, 0.07 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron, se concentraron y se sometieron a cromatografía de columna flash (5% de MeOH/DCM) para producir 0.53 g de 4-(7-(4-(pirrolidin-1-il)piperidin-1 -il)benzo[c][1,2,5] oxadiazol-5-il)benzaldehído. Este aldehído luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.392 mL, 2.8 mmol) seguido por TBAF (0.140 mL, solución 1.0 M en THF). El baño de enfriamiento se retiró y la reacción se agitó durante 30 min. La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (5% de MeOH/DCM) para producir 0.270 g (43% de rendimiento, 2 etapas) de TRV-1468 como un sólido amarillo. 1H RMN (DMSO, 500 MHz) 8 = 7.84 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.56 (s, 1H), 6.94 (d, J = 5.5 Hz, 1H), 6.77 (s, 1H), 5.29-5.23 (m, 1H), 4.23 (br s, 2H), 2.49 (brs, 3 H), 2.25 (br s, 1H), 2.01 -1.99 (m, 2H) 1.69 (br s, 4H), 1.61 1.59 (m, 2H).

Una mezcla de 4,6-dibromobenzo[c][1,2,5]oxadiazol (1.6246 g, 5.8 mmol), N-metil-3-(pirrolidin-1-il)propan-1-amina (aproximadamente 7.6 mmol) y DIPEa (1.1 mL, 5.8 mmol) en NMP (6 mL) se calentó a 90°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con EtOAc y agua. Después de que capas se separaron, la capa acuosa se basificó y se extrajo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar la anilina cruda. Este material se purificó a través de columna de 10% de MeOH/DCM para producir 0.5651 g de anilina. Esta anilina (0.5651 g, 1.67 mmol) y ácido 3-formilfenilborónico (0.3253 g, 2.17 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (3.7 mL) y Na2CO32M (2.5 mL, 5 mmol) seguidos por Pd(PPh3)4 (0.0965 g, 0.084 mmol). El tubo luego se selló y se calentó a 100°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.49 mL, 3.34 mmol) seguido por TBAF (0.17 mL, solución 1.0 M en t Hf ). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (5% de MeOH/DCM con 6 mL de NH4OH) para producir 0.465 g (64 % de rendimiento, 2 etapas) de TRV-1469 como sólido naranja. 1H RMN (DMSO, 700 MHz) 8 = 7.72 (s, 1H), 7.60 (d, J = 7 Hz, 1H), 7.50 (d, J = 7 Hz, 1H), 7.47 (t, J = 7 Hz, 1H), 7.13 (d, J = 3 Hz, 1H), 6.21 (s, 1H), 5.05 (q, J = 7 Hz, 1H), 3.94 (t, J = 7 Hz, 2H), 3.16 (s, 3H), 2.59 (br s, 6 H), 1.95-1.91 (m, 2H), 1.81 (br s, 4H).

Una mezcla de ácido 6-bromo-4-clorobenzo[c][1,2,5]oxadiazol (3.1436 g, 13.4 mmol), 3-azetidinacarboxílico (1.76 g, 17.4 mmol) y DIPEA (7.0 mL, 40.2 mmol) en NMP (15 mL) se calentó a 90°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con EtOAc y HCl 2N. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua (3X), solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar el material crudo. Este material luego se disolvió en EtOAc y se extrajo con NaOH 2 N (3x). Los extractos acuosos combinados se acidificaron a pH 1-2 con HCl concentrado. Esta suspensión luego se extrajo con EtOAc (3x). Las capas orgánicas combinadas luego se lavaron con agua (3X), solución salina, se secaron (MgSO4) se filtraron y se concentraron para producir 3.0657 g de sólido naranja. Esta mezcla cruda (3.066 g, 10.3 mmol) se disolvió en THF y se enfrió a -10°C con un baño de MeOH/hielo. Se agregó en forma de gotas una solución 1 M de BH3-THF en THF (21 mL, 21 mmol), produciendo una solución roja brillante. La solución se agitó vigorosamente durante 10 minutos y luego se dejó calentar a temperatura ambiente durante la noche. La reacción se detuvo con la adición en forma de gotas de 10 mL de AcOH/H2O (1:1 v/v). Después de que se calmó la efervescencia, la mezcla se concentró para eliminar THF. La mezcla acuosa luego se basificó con NaOH 2N y se extrajo con EtOAc (2x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material se purificó a través de cromatografía (40-50% de EtOAc/hexano) para producir 1.942 g del alcohol primario. Este alcohol (0.9217 g, 3.2 mmol) se suspendió en DCM (40 mL) y luego se agregó en forma de porciones reactivo de Dess-Martin (1.789 g, 4.2 mmol) y la mezcla se agitó durante 2.5 horas.
La reacción luego se detuvo con NaHCO3 saturado (ac.) y exceso de Na2S2O3, se continuó la agitación hasta que se disolvieron los sólidos. Esta mezcla luego se extrajo con DCM (3x). Las capas orgánicas combinadas se lavaron con NaHCO3 saturado (ac.) y luego se secaron con MgSO4, antes de filtrar y concentrar la solución. El aldehído crudo luego se purificó a través de cromatografía (40% de EtOAc/hexano) para proporcionar 0.7704 g de aldehído. El aldehído (0.7411 g, 2.63 mmol) se disolvió en DCE (10 mL) y se enfrió en un baño de hielo. Se agregó en forma de gotas morfolina (0.30 mL, 3.42 mmol) y la reacción se agitó durante 5 minutos antes se agregó en forma de porciones NaBH(OAc)3, y la reacción se le permitió agitar durante la noche. La reacción se detuvo con NaHCO3 saturado y luego se extrajo con DCM. Los orgánicos combinados se secaron (MgSO4), se filtraron y se concentraron. El material crudo luego se purificó a través de cromatografía (80% de EtOAc/hexano) para producir 0.7662 g TRV-1470. 1H RMN (CDCl3, 700 MHz) 8 = 7.19 (s, 1H), 5.88 (s, 1H), 4.41 (br s, 2H), 4.00 (br s, 2H), 3.73 (br s, 4H), 3.09 (s, 1H), 2.71 (s, 2H), 2.48 (br s, 4H).

El TRV-1470 (0.4092 g, 1.16 mmol) y ácido 3-formilfenilborónico (0.1829 g, 1.22 mmol) se agruparon en un tubo. El tubo se evacuó y se purgó con argón (3x). Luego se agregaron DME (2.6 mL) y Na2CO32M (1.8 mL, 3.48 mmol) seguidos por Pd(PPh3)4 (0.067 g, 0.058 mmol). El tubo luego se selló y se calentó a 80°C durante la noche. Luego de enfriar a temperatura ambiente la mezcla se diluyó con agua y EtoAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas luego se lavaron con agua (5X), solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite. Este aceite crudo luego se disolvió en THF (10 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.34 mL, 2.32 mmol) seguido por TBAF (0.12 mL, solución 1.0 M en THF). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos. La mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa se basificó. La capa acuosa luego se volvió a extraer con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (Na2SO4), se filtraron y se concentraron para dar un aceite crudo. Este material se purificó a través de cromatografía de columna flash (3% de una solución de NH37 N en MeOH/DCM) para producir 0.125 g de TRV 1471 (24 % de rendimiento, 2 etapas). 1H RMN (CDC13, 700 MHz) 8 = 7.78 (d, J = 7 Hz, 1H), 7.47 (t, J = 7 Hz, 1H), 7.45 (t, J = 7 Hz, 1H), 7.30 (d, J = 1H), 6.90 (s, 1H), 5.73 (s, 1H), 5.21 (br s, 1H), 4.42-4.38 (m, 2H), 3.98 (br s, 2H), 3.71 (br s, 4H), 3.09-3.06 (m, 1H), 2.92 (s, 1H), 2.72 (d, J = 7 Hz, 2H), 2.48 (br s, 4H).

(1-(6-bromobenzo[c][1,2,5]oxadiazol-4-il)azetidin-3-il)metanol(1.9573 g, 6.89 mmol) se suspendió en DCM (85 mL) y se agregó reactivo de Dess- Martin (3.799 g, 8.9 mmol) todo de una vez y se agitó hasta el consumo completo del material de partida, aproximadamente 90 minutos. La mezcla luego se enfrió en un baño de hielo y se agregaron NaHCO3 saturado (ac.) y Na2S2O3 (4.2 g) y se agitaron hasta que todos los sólidos se disolvieron. La solución luego se extrajo con DCM (3x) y los orgánicos combinados se lavaron con NaHCO3 saturado (ac.), se secaron (MgSO4), se filtraron y se concentraron. El material crudo se purificó a través de columna (20-100% de gradiente EtOAc/hexano) para producir 2.0355 g de aldehído. El aldehído luego se disolvió en DCE (solución 0.28 M) y se enfrió en un baño de hielo. Se agregó pirrolidina (0.20 mL, 2.4 mmol) y se agitó durante 5 minutos, seguido por la adición de NaHB(OAc)3 (0.7630 g, 3.6 mmol) todo de una vez. La mezcla se dejó calentar a temperatura ambiente durante la noche. Después de enfriar de nuevo en un baño de hielo, la reacción se detuvo con NaHCO3 saturado y
se agitó hasta que se detuvo la efervescencia. Esta mezcla luego se extrajo con DCM (3x). Las capas orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron. Por último, este material crudo se purificó a través de cromatografía (63-69% de gradiente MeCN/DCM con 1% de aditivo TEA) para producir TRV-1472. 1H RMN (CDC13, 700 MHz) 8 = 7.18 (s, 1H), 5.88 (s, 1H), 4.44 (s, 2H), 4.03 (s, 2H), 3.13 (s, 1H), 2.87 (br s, 2H), 2.61 (br s, 4H), 1.86 (s, 4H).

El TRV-1472 (0.1350 g, 0.40 mmol) se disolvió en THF (10 mL) bajo argón, y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.23 mL, solución 2.0 M en ciclohexano) y la mezcla se agitó durante 20 minutos. Se agregó metanol (0.16 mL, 4 mmol) a -78°C y la mezcla se dejó calentar a 0°C en donde se detuvo con NH4Cl saturado (ac.). Esta mezcla se extrajo con EtOAc (3x) y las capas orgánicas combinadas se lavaron con H2O, solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (10-100% de gradiente EtOAc/hexano con 5% de aditivo TEA) para producir 0.0585 g (57 % de rendimiento) de TRV-1473 como aceite naranja. 1H RMN (CDC13, 700 MHz) 8 = 7.21 (dd, J = 8.4, 7.7 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 5.84 (d, J = 7.7 Hz, 1H), 4.42 (t, J = 7.7 Hz, 1H), 4.00 (s, 2H), 3.13 (br s, 1H), 2.90 (br s, 2H), 2.62 (br s, 4H), 1.88 (s, 4H).

El TRV-1472 (0.2281 g, 0.68 mmol) y ácido 2-formilbencenoborónico (0.1070 g, 0.71 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.0 mL, solución 2.0 M ac.) junto con DME (1.6 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0393 g, 0.0034 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El aldehido crudo se purificó a través de cromatografía (10-100% de gradiente EtOAc/hexano, 5% de aditivo TEA) para producir 0.0753 g de material puro. El aldehido luego se disolvió en THF (5 mL) y se enfrió en un baño de hielo. A esta solución se agregó CF3TMS (61 pL, 0.415 mmol) y luego TBAF (D20 pL, solución 1.0 M en THF). Después de 5 minutos, se eliminó el baño de hielo y la mezcla se agitó hasta conversión completa del material de partida, la mezcla se enfrió en un baño de hielo y se detuvo con HCl 2N (ac.). Después de 30 minutos, la mezcla luego se hizo básica con NaOH 2N y se extrajo con EtOAc (3x). Los orgánicos combinados luego se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material crudo luego se purificó a través de cromatografía (63% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.0357 g (40% de rendimiento) de TRV-1474. 1H RMN (CDCh, 700 MHz) 8 = 7.80 (d, J = 7.7 Hz, 1H), 7.49 (t, J = 7.7 Hz, 1H), 7.43-7.41 (m, 1H), 7.29-7.27 (m, 1H), 6.93 (s, 1H), 5.78 (s, 1H), 5.24 (q, J = 7.0 Hz, 1H), 4.47 (br s, 1H), 4.38 (s, 1H), 4.13 (br s, 1H), 4.02 (s, 1H), 3.15 (br s, 1H), 2.98 (br s, 2H), 2.74 (br s, 4H), 1.91 (br s, 4H).

El TRV-1472 (0.2750 g, 0.82 mmol) y ácido (2,4-diclorofenil)borónico (0.1643 g, 0.86 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.2 mL, solución 2.0 M ac.) junto con DME (1.8 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0474 g, 0.041 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El aldehído crudo se purificó a través de cromatografía (10-100% de gradiente EtOAc/hexano, 5% de aditivo TEA) para producir 0.1821 g (55% de rendimiento) de TRV-1475 como aceite naranja. 1H RMN (CDCh, 700 MHz) 8 = 7.51 (d, J = 1.4 Hz, 1H), 7.33 (dd, J = 1.4, 8.4 Hz, 1H), 7.31 (d, J = 8.4 Hz, 1H), 6.99 (s, 1H), 5.85 (s, 1H), 4.48 (s, 2H), 4.06 (s, 2H), 3.19 (br s, 1H), 2.95 (br s, 2H), 2.64 (br s, 4H), 1.91 (br s, 4H).

El TRV-1472 (0.2707 g, 0.80 mmol) y ácido piridin-3-ilborónico (0.1033 g, 0.84 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.2 mL, solución 2.0 M ac.) junto con DME (1.8 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0462 g, 0.04 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El aldehído crudo se purificó a través de cromatografía (1-10% de gradiente MeOH/DCM, 1% de aditivo TEA) para producir 0.106 g (40% de rendimiento) de TRV-1476 como aceite naranja. 1H RMN (CDCh, 700 MHz) 8 = 8.87 (d, J = 2.1 Hz, 1H), 8.66 (dd, J = 4.9, 2.1 Hz, 1H), 7.90 (dt, J = 8.4, 2.1 Hz, 1H), 7.40 (dd, J = 8.4, 4.9 Hz, 1H), 7.16 (s, 1H), 6.00 (s, 1H), 4.50 (s, 2H), 4.09 (s, 2H), 3.19 (br s, 1H), 2.93 (br s, 2H), 2.63 (br s, 4H), 1.89 (br s, 4H).

El TRV-1472 (0.3402, 1.0 mmol) se disolvió en THF (10 mL) y se enfrió a -78°C bajo argón. Se agregó en forma de gotas nBuLi (0.60 mL, 1.16 mmol, solución 2.0 M en ciclohexano) y la solución oscura resultante se agitó durante 20 minutos antes se agregó trifluoro Weinreb amida (0.16 mL, 1.26 mmol). Se le permitió a la mezcla agitar durante la noche mientras que se calienta a temperatura ambiente, luego se enfrió a 0°C y se detuvo con HCl 2 N (ac.),
agitación durante 60 minutos. Esta mezcla luego se hizo básica con la adición en forma de gotas de NaOH 2 N (ac.) y luego se extrajo con EtOAc. Las capas orgánicas combinadas luego se lavaron con H2O (3X), solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material se purificó a través de (10-100% de gradiente EtOAc/hexano y luego un gradiente 1-10% de MeOH/EtOAc con 5% de aditivo TEA todo el tiempo) para producir 0.108 g (30% de rendimiento) de TRV-1477 como una mezcla de la cetona y cetona hidratada. 1H Rm N (CDCh, 700 MHz) cetona: 8 = 7.77 (s, 1H), 6.28 (s, 1H), 4.47 (br s, 2H), 4.05 (br s, 2H), 3.13-3.06 (m, 1H), 2.81 (d, J = 7.7 Hz, 2H), 2.54 (br s, 4H), 1.81-1.78 (m, 4H); cetona hidratada: 8 = 7.40 (s, 1H), 6.16 (s, 1H), 4.43-4.38 (m, 2H), 4.01-3.99 (m, 2H), (los picos restantes se superponen con la cetona); 1H RMN (DMSO, 700 MHz) cetona hidratada: 8 = 7.85 (s, 2H), 7.27 (s, 1H), 6.14 (s, 1H), 4.32 (t, J = 8.4 Hz, 2H), 3.88 (t, J = 7.0 Hz, 2H), 3.03-2.95 (m, 1H), 2.69 (d, J = 7.0 Hz, 2H), 2.44-2.43 (m, 4H), 1.68-1.66 (m, 4H); cetona: 8 = 7.83 (s, 1H), 6.25 (s, 1H), 4.42 (br s, 2H), 3.99 (br s, 2H), (los picos restantes se superponen con cetona hidratada).

El TRV-1472 (0.1584 g, 0.47 mmol) y ácido (3,5-diclorofenil)borónico (0.0.0941 g, 0.49 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.71 mL, solución 2.0 M ac.) junto con DME (1.1 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.0272 g, 0.024 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (5% de MeOH/DCM) para producir 0.0348 g (18% de rendimiento) de TRV-1478. 1H RMN (DMSO, 500 MHz) 8 = 7.87 (d, J = 2.0 Hz, 2H), 7.69 (t, J = 2.0 Hz, 1H), 7.45 (s, 1H), 6.28 (s, 1H), 4.38 (t, J = 8.5 Hz, 2H), 3.99-3.96 (m, 2H), 3.03-2.94 (m, 1H), 2.71 (d, J = 7.5 Hz, 2H), 2.46 (br s, 4H), 1.68 (br s, 4H).

El TRV-1472 (0.1987 g, 0.59 mmol) y ácido3-metilsulfonilfenilborónico (0.1240 g, 0.62 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (0.9 mL, solución 2.0 M ac.) junto con DME (1.3 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.034 g, 0.03 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (gradiente 1-10% de MeOH/DCM) para producir 0.1103 g (45% de rendimiento) de TRV-1479. 1H RMN (CDCh, 500 MHz) 8 = 8.17 (s, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.89 (d, J= 8.0 Hz, 1H), 7.68 (t, J = 8.0 Hz, 1H), 7.17 (s, 1H), 6.01 (s, 1H), 4.51 (t, J = 8.5 Hz, 2H), 4.11-4.09 (m, 2H), 3.22 (br s, 1H), 3.12 (s, 3H), 2.98 (br s, 2H), 2.75 (br s, 4H), 1.91 (br s, 4H).

El TRV-1472 (0.2075 g, 0.615 mmol) y ácido 3-acetilbencenoborónico (0.1059 g, 0.65 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.92 mL, solución 2.0 M ac.) junto con DME (1.4 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.036 g, 0.031 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (gradiente 0-10% de MeOH/DCM) para producir 0.1307 g (56% de rendimiento) de TRV-1480. 1H RMN (CDCla, 500 MHz) 8 = 8.19 (t, J = 1.5 Hz, 1H), 7.99 (d, J = 7.5 Hz, 1H), 7.81 (dd, J = 7.5, 1.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.16 (s, 1H), 6.04 (s, 1H), 4.48 (t, J = 8.5 Hz, 2H), 4.07-4.04 (m, 2H), 3.15 (br s, 1H), 2.90 (br s, 2H), 2.67 (s, 3H), 2.64 (br s, 4H), 1.85 (br s, 4H).

El TRV-1472 (0.2009 g, 0.60 mmol) y ácido 2-metilsulfonilfenilborónico (0.1260 g, 0.63 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (0.9 mL, solución 2.0 M ac.) junto con DME (1.3 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.034 g, 0.03 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (0-100% de gradiente EtOAc/hexano con 5% de TEA) para producir 0.0257 g (10% de rendimiento) de TRV-1481. 1H RMN (CDCl3, 500 MHz) 8 = 8.21 (d, J = 7.5 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.39 (d, J = 7.5 Hz, 1H), 6.98 (s, 1H), 5.93 (s, 1H), 4.48 (t, J = 8.0 Hz, 2H), 4.07 (br s, 2H), 3.23 (br s, 1H), 3.05-3.01 (m, 2H), 2.91 (s, 3H), 2.77 (br s, 4H), 1.93 (br s, 4H).

El TRV-1472 (0.2039 g, 0.60 mmol) y ácido (4-acetilaminofenil)borónico (0.1128 g, 0.63 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.9 mL, solución 2.0 M ac.) junto con DME (1.3 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.036 g, 0.03 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (gradiente 0-10% de MeOH/DCM con 1% de TEA) para producir 0.1350 g (57% de rendimiento) de TV-1482. 1H RMN (CDCla, 500 MHz) 8 = 7.61 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H), 7.47 (s, 1H), 7.11 (s, 1H), 6.04 (s, 1H), 4.46 (t, J = 8.0 Hz, 2H), 4.06-4.04 (m, 2H), 3.19 (br s, 1H), 3.00 (br s, 2H), 2.77 (br s, 4H), 2.23 (s, 3H), 1.93 (br s, 4H).

El TRV-1472 (0.1910 g, 0.57 mmol) y ácido 3,4-dimetoxibencenoborónico (0.1092 g, 0.60 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.90 mL, solución 2.0 M ac.) junto con DME (1.3 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.034 g, 0.029 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (0-8% de MeOH/DCM) para producir 0.0471 g (21% de rendimiento) de TRV-1483. 1H RMN (CDCla, 500 MHz) 8 = 7.18 (dd, J = 8.0, 2.0 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 7.11 (s, 1H), 6.95 (d, J = 8.0 Hz, 1H), 6.07 (s, 1H), 4.48 (t, J = 8.0 Hz, 2H), 4.07 (t, J = 6.0 Hz, 2H), 3.96 (s, 3H), 3.94 (s, 3H), 3.20 (br s, 1H), 2.98 (br s, 2H), 2.74 (br s, 4H), 1.92 (br s, 4H).

El TRV-1472 (0.2297 g, 0.68 mmol) y ácido 3-trifluorobencenoborónico (0.1367 g, 0.72 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.0 mL, solución 2.0 M ac.) junto con DME (1.5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.039 g, 0.034 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (0-8% de MeOH/DCM) para producir 0.0377 g (14% de rendimiento) de TRV-1484. 1H RMN (CDCh, 500 MHz) 8 = 7.84 (s, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.16 (s, 1H), 6.02 (s, 1H), 4.51 (t, J = 8.0 Hz, 2H), 4.10 (t, J = 6.0 Hz, 2H), 3.22 (br s, 1H), 3.00 (br s, 2H), 2.75 (br s, 4H), 1.92 (br s, 4H).

El TRV-1472 (0.2067 g, 0.61 mmol) y ácido benzofuran-2-ilborónico (0.1042 g, 0.64 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.92 mL, solución 2.0 M ac.) junto con DME (1.4 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.035 g, 0.031 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (0-8% de MeOH/DCM) para producir 0.1032 g (45% de rendimiento) de TRV-1485. 1H RMN (CDCla, 500 MHz) 6 = 7.62 (d, J = 8.0 Hz, 1H), 7.56 7.54 (m, 2H), 7.37-7.33 (m, 1H), 7.28-7.25 (m, 1H), 7.15 (s, 1H), 6.26 (s, 1H), 4.50 (t, J = 8.0 Hz, 2H), 4.08 (t, J = 6.0 Hz, 2H), 3.16 (br s, 1H), 2.93 (br s, 2H), 2.67 (br s, 4H), 1.88 (br s, 4H).

El TRV-1472 (0.2044 g, 0.61 mmol) y ácido fenilborónico (0.0776 g, 0.63 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (0.92 mL, solución 2.0 M ac.) junto con DME (1.4 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.036 g, 0.031 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (0-8% de MeOH/DCM) para producir 0.0702 g (34% de rendimiento) de TRV-1486. 1H RMN (CDCh, 500 MHz) 6 = 7.62 (d, J = 7.0 Hz, 2H), 7.47 (t, J = 7.0 Hz, 2H), 7.43 (d, J = 7.0 Hz, 1H), 7.16 (s, 1H), 6.08 (s, 1H), 4.47 (t, J = 8.0 Hz, 2H), 4.06-4.03 (m, 2H), 3.16 (br s, 1H), 2.94 (br s, 2H), 2.69 (br s, 4H), 1.88 (br s, 4H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-(tert-butil)fenil) borónico (139 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la
finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 200 mg (85% de rendimiento) de TRV-1487 como un sólido amarillo. 1H RMN (500 MHz, CDCh) 1.42 (s, 4H), 1.95 (br, s, 4H), 2.76 (br, s, 4H), 3.01 (br, s, 2H), 3.23 (br, s, 1H), 4.08-4.11 (m, 2H), 4.52 (t, J=8.2, 2H), 6.16 (s, 1H), 7.21 (s, 1H), 7.55 (d, J=8.4, 2H), 7.62 (d, J=8.4, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-metoxi-3-metilfenil) borónico (130 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 190 mg (84% de rendimiento) de TRV-1488 como aceite rojo. 1H RMN (500 MHz, CDCls) 1.88 (t, J=3.1, 4H), 2.34 (s, 3H), 2.64 (br, s, 4H), 2.90 (d, J=7.4, 2H), 3.13-3.16 (m, 1H), 3.94 (s, 3H), 4.05-4.08 (m, 2H), 4.50 (t, J=8.2, 2H), 6.11 (s, 1H), 6.95 (d, J=8.4, 1H), 7.14 (s, 1H), 7.46 (s, 1H), 7.49 (d-d, J=8.4, J=2.2, 1H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-isopropilfenil) borónico (128 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 165 mg (73% de rendimiento) de TRV-1489 como un sólido amarillo. 1H RMN (500 MHz, CDCla) 1.35 (d, J=7.0, 6H), 1.88 (t, J=3.1, 4H), 2.63 (br, s, 4H), 2.89 (d, J=7.5, 2H), 3.01-3.05 (m, 1H), 3.12-3.18 (m, 1H), 4.05-4.08 (m, 2H), 4.50 (t, J=8.2, 2H), 6.13 (s, 1H), 7.19 (s, 1H), 7.38 (d, J=8.2, 2H), 7.60 (d, J=8.2, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (3-cloro-4-metilfenil) borónico (133 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 165 mg (73% de rendimiento) de TRV-1490 como aceite rojo. 1H RMN (500 MHz, CDCls) 1.88 (t, J=3.1, 4H), 2.48 (s, 3H), 2.64 (br, s, 4H), 2.90 (d, J=7.4, 2H), 3.15-3.18 (m, 1H), 4.06-4.09 (m, 2H), 4.51 (t, J=8.2, 2H), 6.03 (s, 1H), 7.15 (s, 1H), 7.36 (d, J=7.9, 1H), 7.45 (d-d, J=7.9, J=1.7, 1H), 7.64 (d, J=1.7, 1H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-fenoxifenil) borónico (167 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 215 mg (84% de rendimiento) de TRV-1491 como un sólido rojo. 1H RMN (500 MHz, CDCla) 1.97 (br, s, 4H), 2.80 (br, s, 4H), 3.04 (br, s, 2H), 3.5 (br, s, 1H), 4.11-4.13 (m, 2H), 4.54 (t, J=8.2, 2H), 6.12 (s, 1H), 7.12-7.14 (m, 4H), 7.18 (s, 1H), 7.21 (t, J=7.5, 1H),7.44 (t, J=7.8, 2H), 7.63 (d, J=8.6, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-fenoxifenil) borónico (145 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La
fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 178 mg (74% de rendimiento) de TRV-1492 como aceite rojo. 1H RMN (500 MHz, CDCls) 1.98 (br, s, 4H), 2.83 (br, s, 4H), 3.06 (s, 2H), 3.27 (br, s, 1H), 4.02 (s, 3H), 4.12-4.14 (m, 2H), 4.4 (t, J=8.2, 2H), 6.06 (s, 1H), 7.06 (d, J=8.6, 1H), 7.15 (s, 1H), 7.55 (d-d, J=8.6, J=2.3, 1H), 7.69 (d, J=2.3, 1H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (3-cianofenil) borónico (115 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 6 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 159 mg (73% de rendimiento) de TRV-1493 como un sólido rojo. 1H RMN (500 MHz, CDCla) 1.87-1.89 (br, s, 4H), 2.63 (br, s, 4H), 2.90 (d, J=7.5, 2H), 3.15-3.18 (m, 1H), 4.09-4.12 (m, 2H), 4.53 (t, J=8.3, 2H), 5.97 (s, 1H), 7.15 (s, 1H), 7.63 (t, J=7.8, 1H), 7.74 (d, J=7.7, 1H), 7.88 (d, J=7.9, 1H), 7.93 (s, 1H).
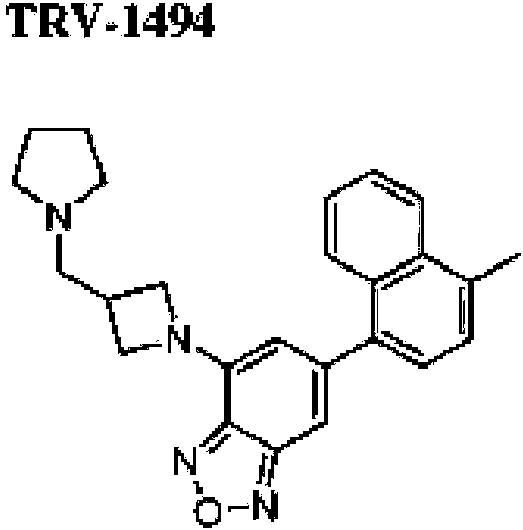
Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-metilnaftalen-1-il) borónico (145 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 204 mg (85% de rendimiento) de TRV-1494 como un sólido rojo. 1H RMN (500 MHz, CDCla) 1.85-1.86 (m, 4H), 2.61 (br, s, 4H), 2.81 (s, 3H), 2.89 (d, J=7.4, 2H), 3.11-3.14 (m, 1H), 4.03-4.06 (m, 2H), 4.45-4.48 (m, 2H), 6.01 (s, 1H), 7.14 (s, 1H), 7.43 (s, 2H), 7.52 (t, J=7.3, J=7.9, 1H), 7.62 (t, J=7.7, J=7.4, 1H), 8.01 (d, J=7.4, 1H), 8.13 (d, J=7.5, 1H).
Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (2-(trifluorometoxi) fenil) borónico (161 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 211 mg (84% de rendimiento) de TRV-1495 como aceite rojo. 1H RMN (500 MHz, CDCla) 2.06 (br, s, 4H), 2.97 (br, s, 4H), 3.19 (br, s, 2H), 3.37(br, s, 1H), 4.12-4.14 (m, 2H), 4.53-4.56 (m, 2H), 6.01 (s, 1H), 7.15 (s, 1H), 7.42-7.46 (m, 2H), 7.49-7.51 (m, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (6-cloropiridin-3-il) borónico (161 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (6:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 167 mg (75% de rendimiento) de TRV-1496 como aceite rojo. 1H RMN (500 MHz, CDCla) 1.92 (br, s, 4H), 2.17 (br, s, 4H), 2.97 (d, J=5.8, 2H), 3.21-3.22 (m, 1H), 4.13 (br, s, 2H), 4.53-4.54 (m, 2H), 5.97 (s, 1H), 7.16 (s, 1H), 7.49 (d, J=8.1, 1H), 7.92 (d, J=7.8, 1H), 8.69 (s, 1H).

Una solución de ácido 3-azeditinecarboxílico (3.4889 g, 34.5 mmol) en agua (60 mL) se enfrió en un baño de hielo y se agregó DIPEA (18 mL, 103.5 mmol). La mezcla se agitó durante 5 minutos y se agregó en forma de gotas luego una solución de 6-bromo-4- fluorobenzo[c][1,2,5]oxadiazol 3 (7.4877 g, 34.5 mmol) en THF (60 mL). La reacción se agitó durante 30 minutos a 0°C, luego se retiró del baño de hielo y se dejó calentar a temperatura ambiente durante la noche. La reacción se diluyó con agua y luego se basificó con NaOH 2n (ac.). La capa acuosa luego se lavó con DCM (3x) y luego se acidificó con HCl 2N (ac.). El precipitado resultante se recolectó mediante filtración para producir 4.8909 gramos (48 % de rendimiento) de un sólido marrón fino. Una mezcla de este material, pirrolidina (1.36 mL, 16.4 mmol) y TEA (5.7 mL, 41 mmol) en THF (140 mL) se enfrió en un baño de hielo. Se agregó en forma de gotas la solución de T3P (12.54 g, 19.7 mmol, 50% p/p de solución en EtOAc) y luego la reacción se dejó calentar a temperatura ambiente. La reacción luego se diluyó con agua y se extrajo con EtOAc (3x). Las capas orgánicas combinadas luego se lavaron con agua (3X), solución salina, se secaron (MgSO4), se filtraron y se concentraron para producir 3.4558 g (60% de rendimiento, < 95% c.p.). 85 mg de este material se purificó a través de columna de 3% de MeOH/DCM. Esto produjo aproximadamente 60 mg (71% de recuperación) de un sólido naranja fino de TRV-1497. 1H RMN (500 MHz, DMSO) 8 = 7.40 (s, 1H), 6.10 (s, 1H), 4.45 (t, J = 8.5 Hz, 2H), 4.35 (t, J = 8.5 Hz, 2H), 3.86-3.81 (m, 1H), 3.35-3.30 (m, 4H), 1.90-1.85 (m, 2H), 1.81-1.75 (m, 2H).

Se agregaron TRV-1497 (0.4031 g, 1.15 mmol) y ácido 3-formilbencenoborónico (0.1810 g, 1.21 mmol) a un tubo y el tubo se evacuó y se purgó con argón (3x). Se agregaron DME (2.8 mL) y Na2CO3 (1.7 mL, 3.4 mmol, solución acuosa 2 N) seguidos por Pd(PPh3)4 (0.069 g, 0.06 mmol). El tubo se selló y luego se calentó a 85°C durante la noche. La reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el aldehído crudo. Este aldehído se disolvió en THF (10 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.19 mL) y luego se agregó el catalizador TBAF (0.1 mL, solución 1.0 M). Después de 30 minutos la reacción se retiró del baño de hielo y se dejó calentar a temperatura ambiente. Una vez se completó mediante TLC, se volvió a enfriar a 0°C y se agregó HCl 2 N (ac.), se agitó durante 40 minutos y luego se basificó con NaOH 2N. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua (2x), solución salina, se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (5% de MeOH/DCM) para producir 0.261 g (51% de rendimiento) de TRV-1498 como un sólido naranja. 1H RMN (500 MHz, DMSO) 8 = 7.91 (s, 1H), 783 (d, J = 7.5 Hz, 1H), 7.60 (d, J = 7.5 Hz, 1H), 7.56 (t, J = 7.5 Hz, 1H), 7.35 (s, 1H), 6.93 (d, J = 5.5 Hz, 1H), 6.32 (s, 1H), 5.32-5.26 (m, 1H), 4.51-4.49 (m, 2H), 4.40 (t, J = 7.0 Hz, 2H), 3.92-3.86 (m, 1H), 3.39 (t, J = 6.5 Hz, 2H), 3.35 (t, J = 6.5 Hz, 2H), 1.94-1.88 (m, 2H), 1.84-1.78 (m, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (2-(hidroximetil) fenil) borónico (119 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 3 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (6:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 142 mg (64% de rendimiento) de TRV-1499 como aceite rojo. 1H RMN (400 MHz, CDCh) 1.78-1.81 (m, 4H), 1.83-1.87 (m, 1H), 2.51 2.55 (m, 4H), 2.81 (d, J=7.5, 2H), 3.02-3.09 (m, 1H),3.97-4.01 (m, 2H), 4.42 (t, J=8.3, 2H), 4.67 (s, 2H), 5.84 (s, 1H), 6.95 (s, 1H), 7.32 (d-d, J=7.6, J=1.5, 1H), 7.38 (t-d, J=7.5, J=1.5, 1H), 7.45 (t-d, J=7.4, J=1.7, 1H), 7.59 (d, J=8.0, 1H).
Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (2,3-dimetoxifenil) borónico (142 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 2 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 141 mg (72% de rendimiento) de TRV-1500 como aceite rojo. 1H RMN (400 MHz, CDCla) 1.79-1.82 (m, 4H), 2.52-2.55 (m, 4H), 2.81 (d, J=7.5, 2H), 3.02-3.09 (m, 1H),3.68 (s, 3H), 3.94 (s, 3H), 3.96-4.00 (m, 2H), 4.42 (t, J=8.3, 2H), 6.08 (s, 1H), 6.98 7.00 (m, 2H), 7.11-7.16 (m, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-(metilsulfonil)fenil) borónico (156 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (4:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 163 mg (66% de rendimiento) de TRV-1501 como un sólido rojo. 1H RMN (400 MHz, CDCh) 1.79-1.83 (m, 4H), 2.53-2.57 (m, 4H), 2.83 (d, J=7.6, 2H), 3.09-3.13 (m, 1H),4.04-4.08 (m, 2H), 4.49 (t, J=8.4, 2H), 5.98 (s, 1H), 7.16 (s, 1H), 7.81 (d, J=8.0, 2H), 8.05 (d, J=8.3, 1H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-(trifluorometil)fenil) borónico (148 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 205 mg (85% de rendimiento) de TRV-1502 como un sólido amarillo. 1H RMN (400 MHz, CDCh) 1.80-1.83 (m, 4H), 2.53-2.57 (m, 4H), 2.83 (d, J=7.5, 2H), 3.07-3.13 (m, 1H), 4.03-4.07 (m, 2H), 4.48 (t, J=8.3, 2H), 6.00 (s, 1H), 7.16 (s, 1H), 7.73 (s, 4H).

Se agregó en forma de gotas LDA (6 ml, 12 mmol, solución 2 M en THF) a una solución agitada de 1-Boc-4-piperidona (1.99 g, 10 mmol) en THF (30 mL) a -78°C bajo atmósfera de nitrógeno. Después de 20 minutos, una solución de THF (10 ml) de N-fenilbis(trifluometanosulfonimida) se agregó a la mezcla a -78°C, y la solución viscosa de color amarillo claro se agitó durante la noche y la temperatura se dejó calentar a RT. Se agregó la mezcla 30 ml de hexanos y se lavó con solución salina. La capa orgánica se secó y se concentró. 4-(((trifluorometil)sulfonil)oxi)-3,6-dihidropiridina-1(2H)-carboxilato de tert-Butilo (2.02 g, 61%) se obtuvo a través de cromatografía (5:95 EtOAc /Hexano).
Un matraz de fondo redondo se cargó con 4-(((trifluorometil)sulfonil)oxi)-3,6-dihidropiridina-1(2H)-carboxilato de tertbutilo (2.02 g, 6.1 mmol), bis(pinacolato)diboro (2.32 g, 9.15 mmol), acetato de potasio (1.79 g, 18.3 mmol), y PdCh(dppf) (223 mg, 0.31 mmol). Después se desgasificó, se agregó dioxano (15 mL). La mezcla de reacción se calentó a 90°C durante 2 h. Después que se completó la reacción, la mezcla se enfrió a rt. Se agregó 40 mL de EtOAc, y la mezcla de reacción se lavó con agua durante 3 veces. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó mediante cromatografía instantánea (1:10 EtOAc/Hexano). 1.55 g (82%) de 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo se obtuvo como un sólido.
Un matraz de fondo redondo se cargó con TRV-1472 (1.422 g, 4.22 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo (1.500 g, 4.85 mmol), y Pd(PPh3)4 (195 mg, 0.169 mmol). Después se desgasificó, dioxano (12 mL) y se agregó carbonato de sodio acuoso (6 mL, 2M). La mezcla de reacción se calentó a 90°C durante 2.5 h. Después de que se verificó la finalización mediante TLC, se agregó 50 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 1.80 g (97% de rendimiento) de 4-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,6-dihidropiridina- 1(2H)-carboxilato de tert-butilo.
Se agitó 4-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo (1.80 g, 4.09 mmol) en la solución de DCM/CF3COOH (2:1, 21 ml) durante 1 hora a 0°C. Después de que se verificó la finalización mediante TLC, la mezcla se neutralizó cuidadosamente con solución de K2CO3 saturado hasta que no haya más gas entregado a 0°C. Se agregó 50 ml de DCM a la solución y la mezcla se lavó con agua. La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El (4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,3,6- tetrahidropiridin-4-il)benzo[c][1,2,5]oxadiazol) crudo se utilizó para la siguiente etapa sin purificación adicional.
Se agregaron en forma de gotas TEA (3.06 ml, 22 mmol) y anhídrido acético (1.04 ml, 11 mmol) a una solución de (4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,3,6-tetrahidropiridin-4-il)benzo[c][1,2,5]oxadiazol) crudo (250 mg, 0.735 mmol) en THF (10 ml) a RT respectivamente. La mezcla se agitó durante 3 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo mediante Hexanos/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (6:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1503 (210 mg 75%) como un sólido amarillo. 1H RMN (400 MHz, CDCl3) 1.77-1.84 (m, 4H), 2.16 (s, 1.28H), 2.19 (s, 1.72H), 2.51-2.56 (m, 4H), 2.57-2.64 (m, 2H), 2.80 (d, J=7.5, 2H), 3.03-3.10 (m, 1H),3.69 (t, J=5.6, 0.85H), 3.83 (t, J=5.8, 1.15H), 3.96-4.00 (m, 2H), 4.17 (d, J=3.0, 0.85H), 4.28 (d, J=2.4, 1.15H), 4.42 (t, J=7.7, 2H), 5.87 (s, 0.43H), 5.90 (s, 0.57H), 6.15 (s, 0.43H), 6.24 (s, 0.57H), 6.90 (s, 0.57H), 6.94 (s, 0.43H).

Un frasco de reacción se cargó con TRV 1472 (0.19 g, 0.56 mmol), ácido (2-fenoxifenil)borónico (0.14 g, 0.67 mmol), y Pd(PPh3)4 (0.032 g, 0.028 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (5 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (2% de trietilamina y 2% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.20 g (84% de rendimiento) de TRV 1504 como un sólido rojo. 1H RMN (CDCla, 400 MHz) 8 = 7.52 (dt, J1 = 1.51 Hz, J2 = 7.53 Hz, 1H), 7.38 (dt, J1 = 1.75 Hz, J2 = 7.80 Hz, 1H), 7.30 (t, J = 8.54 Hz, 2H), 7.24 (t, J = 7.53 Hz, 1H), 7.13 (s, 1H), 7.05 (t, J = 7.40 Hz, 2H), 6.95 (d, J = 7.53 Hz, 2H), 6.03 (s, 1H), 4.32 (t, J = 8.16 Hz, 2H), 3.87 (dd, J1 = 6.12 Hz, J2 = 8.03 Hz, 2H), 3.01 (septeto, J = 7.28 Hz, 1H), 2.76 (d, J = 7.28 Hz, 2H), 2.56 - 2.46 (m, 4H), 1.85 - 1.75 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.16 g, 0.48 mmol), ácido (3-clorofenil)borónico (0.10 g, 0.64 mmol), y Pd(PPh3)4 (0.028 g, 0.024 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (5 mL) y carbonato de sodio ac. (3 mL, 2.0 M, 6.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (2% de trietilamina y 2% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.15 g (85% de rendimiento) de TRV 1505 como aceite rojo. 1H RMN (CDCla, 400 MHz) 8 = 7.60 (s, 1H), 7.52 - 7.48 (m, 1H), 7.43 - 7.38 (m, 2H), 7.13 (s, 1H), 6.99 (s, 1H), 4.48 (t, J = 8.29 Hz, 2H), 4.04 (dd, J1 = 5.90 Hz, J2 = 8.66 Hz, 2H), 3.10 (septeto, J = 6.97 Hz, 1H), 2.82 (d, J = 7.28 Hz, 2H), 2.59 - 2.51 (m, 4H), 1.86 - 1.78 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (3,5-dimetilfenil)borónico (0.15 g, 1.00 mmol), y Pd(PPh3)4 (0.086 g, 0.074 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó
dioxano (5 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (5% de trietilamina y 1.5% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.20 g (75% de rendimiento) de TRV 1506 como un sólido amarillo. 1H RMN (CDCla, 400 MHz) 8 = 7.24 (s, 2H), 7.14 (d, J = 0.75 Hz, 1H), 7.07 (s, 1H), 6.07 (s, 1H), 4.46 (t, J = 8.28 Hz, 2H), 4.02 (dd, J1 = 5.65 Hz, J2 = 8.41 Hz, 2H), 3.09 (septeto, J = 7.66 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.59 -2.51 (m, 4H), 2,41 (s, 6H), 1.86 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (4-propilfenil)borónico (0.16 g, 1.00 mmol), y Pd(PPh3)4 (0.086 g, 0.074 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (5% de trietilamina y 1.5% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.25 g (90% de rendimiento) de TRV 1507 como un sólido amarillo. 1H RMN (CDCla, 400 MHz) 8 = 7.55 (d, J = 8.28 Hz, 2H), 7.29 (d, J = 8.28 Hz, 2H), 7.15 (d, J = 0.75 Hz, 1H), 6.08 (s, 1H), 4.46 (t, J = 8.88 Hz, 2H), 4.02 (dd, J1 = 5.65 Hz, J2 = 8.41 Hz, 2H), 3.08 (septeto, J = 6.78 Hz, 1H), 2.82 (d, J = 7.28 Hz, 2H), 2.66 (t, J = 7.66 Hz, 2H), 2.59 - 2.50 (m, 4H), 1.87 - 1.78 (m, 4H), 1.70 (sexteto, J = 7.28 Hz, 2H), 0.99 (t, J = 7.28 Hz, 3H).
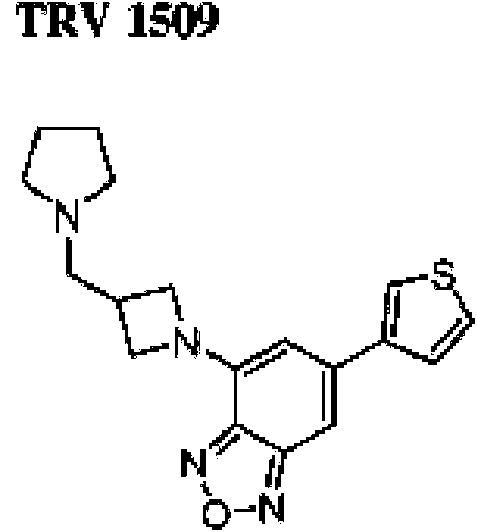
Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido tiofen-3-ilborónico (0.13 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (5% de trietilamina y 1% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.20 g (79% de rendimiento) de TRV 1509 como un sólido naranja. 1H RMN (CDCla, 400 MHz) 8 = 7.58 (t, J = 2.13 Hz, 1H), 7.43 (d, J = 2.01 Hz, 2H), 7.19 (s, 1H), 6.09 (s, 1H), 4.46 (t, J = 8.41 Hz, 2H), 4.03 (dd, J1 = 5.90 Hz, J2 = 8.46 Hz, 2H), 3.09 (septeto, J = 6.59 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.62 - 2.48 (m, 4H), 1.89 - 1.76 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido furan-3-ilborónico (0.15 g, 1.30 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (5% de trietilamina y 1% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.18 g (75% de rendimiento) de TRV 1510 como un sólido naranja. 1H RMN (CDCla, 400 MHz) 8 = 7.81 (s, 1H), 7.52 (t, J = 1.63 Hz, 1H), 7.08 (s, 1H), 6.75 (t, J = 0.87 Hz, 1H), 5.94 (s, 1H), 4.45 (t, J = 8.28 Hz, 2H), 4.01 (dd, J1 = 5.77 Hz, J2 = 8.54 Hz, 2H), 3.08 (septeto, J = 6.72 Hz, 1H), 2.81 (d, J = 7.53 Hz, 2H), 2.59 - 2.51 (m, 4H), 1.87 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (2,6-dimetoxifenil)borónico (0.18 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (5% de trietilamina y 1% de metanol en un solvente de mezcla 5:1 de hexano y acetato de etilo) para producir 0.21 g (72% de rendimiento) de TRV 1511 como un sólido amarillo. 1H RMN (CDCh, 400 MHz) 8 = 7.33 (t, J = 8.26 Hz, 1H), 6.99 (s, 1H), 6.68 (d, J = 8.28 Hz, 1H), 5.82 (s, 1H), 4.39 (t, J = 8.16 Hz, 2H), 3.96 (dd, J1 = 5.65 Hz, J2 = 8.41 Hz, 2H), 3.77 (s, 6H), 3.04 (septeto, J = 6.65 Hz, 1H), 2.79 (d, J = 7.53 Hz, 2H), 2.58 - 2.48 (m, 4H), 1.85 - 1.75 (m, 4H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (4-acetilfenil) borónico (128 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La
fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (3:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 199 mg (88% de rendimiento) de TRV-1512 como un sólido rojo. 1H RMN (400 MHz, CDCl3) 1.80-1.83 (m, 4H), 2.53-2.56 (m, 4H), 2.67 (s, 3H), 2.82 (d, J=7.5, 2H), 3.07-3.13 (m, 1H), 4.03-4.07 (m, 2H), 4.48 (t, J=8.3, 2H), 6.03 (s, 1H), 7.19 (d, J=1.0, 1H), 7.70-7.73 (m, 2H), 8.04-8.07 (m, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (2-(trifluorometil)fenil) borónico (148 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 4 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 190 mg (79% de rendimiento) de TRV-1514 como aceite naranja. 1H RMN (400 MHz, CDCl3) 1.78-1.82 (m, 4H), 2.52-2.55 (m, 4H), 2.81 (d, J=7.3, 2H), 3.03-3.10 (m, 1H),3.98-4.02 (m, 2H), 4.42 (t, J=8.3, 2H), 5.77 (s, 1H), 6.93 (s, 1H), 7.39 (d, J=7.8, 1H), 7.52 (t, J=7.4, 1H), 7.60 (t, J=7.5, 1H), 7.77 (d, J=7.5, 1H).

Un matraz de fondo redondo se cargó con TRV-1472 (202 mg, 0.6 mmol), ácido (2,4-dimetoxifenil) borónico (142 mg, 0.78 mmol), y Pd(PPh3)4 (35 mg, 0.03 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 2 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 190 mg (61% de rendimiento) de TRV-1515 como un sólido amarillo. 1H RMN (400 MHz, CDCl3): 1.78-1.82 (m, 4H), 2.55 (s, br, 4H), 2.81 (d, J=7.3, 2H), 3.02-3.09 (m, 1H), 3.84 (s, 3H), 3.88 (s, 3H), 3.95-3.99 (m, 2H), 4.41 (t, J=8.2, 2H), 6.04 (s, 1H), 6.58-6.60 (m, 2H), 7.09 (s, 1H), 7.30-7.32 (m, 1H).
Un matraz de fondo redondo se cargó con TRV-1472 (250 mg, 0.742 mmol), ácido (4-(dimetilcarbamoil)fenil) borónico (172 mg, 0.89 mmol), y Pd(PPh3)4 (43 mg, 0.037 mmol). Después se desgasificó, se agregó dioxano (5 mL) y carbonato de sodio acuoso (2.5 mL, 2M). La mezcla de reacción se calentó a 90°C durante 2.5 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de cromatografía de columna flash (5:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 207 mg (85% de rendimiento) de TRV-1516 como un sólido rojo. 1H RMN (400 MHz, CDCls): 1.80-1.84 (m, 4H), 2.54-2.56 (m, 4H), 2.82 (d, J=7.3, 2H), 3.05 (s, 3H), 3.07-3.13 (m, 1H), 3.16 (s, 3H), 4.02-4.06 (m, 2H), 4.47 (t, J=8.2, 2H), 6.03 (s, 1H), 7.15 (s, 1H), 7.27 (s, 1H), 7.53 (d, J=8.3, 1H), 7.66 (d, J=8.3, 2H).

Se agregaron en forma de gotas TEA (2.05 ml, 14.7 mmol) y yoduro de metilo (0.46 ml, 7.35 mmol) a una solución de (4-(3-(pirrolidin-1-ilmetil)azetidin-1 -il)-6-(1,2,3,6-tetrahidropiridin-4-il)benzo[c][1,2,5]oxadiazol) crudo (250 mg, 0.735 mmol) en DCM (10 ml) respectivamente. La mezcla se agitó durante 5 horas. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (7:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1517 (120 mg, 46%) como aceite rojo. 1H RMN (400 MHz, CDCls): 1.79-1.83 (m, 4H), 2.43 (s, 3H), 2.52-2.55 (m, 4H), 2.58-2.63 (m, 2H), 2.67-2.71 (m, 2H), 2.79 (d, J=7.5, 2H), 3.02-3.09 (m, 1H), 3.13-3.17 (m, 2H), 3.94-3.98 (m, 2H), 4.40 (t, J=8.2, 2H), 5.95 (s, 1H), 6.22 (t, J=3.6, 1H), 6.95 (s, 1H).
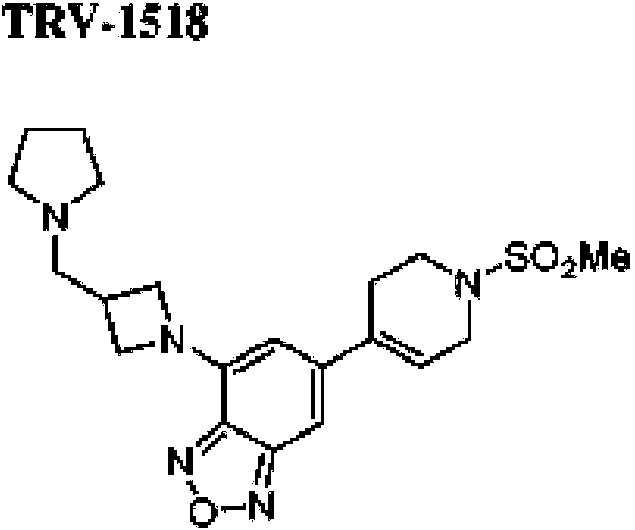
Se agregaron en forma de gotas TEA (0.41 ml, 2.94 mmol) y cloruro de metanosulfonilo (0.11 ml, 1.47 mmol) a una solución de (4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,3,6-tetrahidropiridin-4-il)benzo[c][1,2,5]oxadiazol) crudo (250 mg, 0.735 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 5 horas. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (5:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1518 (250 mg, 81%) como sólido rojo. 1H RMN (400 MHz, CDCla): 1.80-1.82 (m, 4H), 2.52-2.57 (m, 4H), 2.66-2.71 (m, 2H), 2.80 (d, J=7.3, 2H), 2.89 (s, 3H), 3.04-3.11 (m, 1H), 3.54 (t, J=5.6, 2H), 3.97-4.01 (m, 4H), 4.42 (t, J=8.2, 2H), 5.80 (s, 1H), 6.21 (s, br, 1H), 6.93 (s, 1H).

Se agregaron en forma de gotas carbonato de potasio (203 mg, 1.47 mmol) y éter de 2-bromo-etil metilo (0.316 ml, 3.3 mmol) a una solución de (4-(3-(pirrolidin-1-ilmetM)azetidin-1-il)-6-(1,2,3,6-tetrahidropiridin-4-il)benzo[c][1,2,5] oxadiazol) crudo (250 mg, 0.735 mmol) en acetonitrilo (10 ml) a RT respectivamente. La mezcla se agitó durante la noche. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (5:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1519 (180 mg, 61%) como aceite rojo. 1H RMN (400 MHz, CDCla): 1.78-1.82 (m, 4H), 2.52-2.55 (m, 4H), 2.60 (s, br, 2H), 2.73 (d, J=5.6, 2H), 2.77-2.80 (m, 4H), 3.02-3.08 (m, 1H), 3.26-3.29 (m, 2H), 3.58 (s, 3H), 3.60 (t, J=5.6, 2H), 3.94-3.97 (m, 2H), 4.40 (t, J=8.2, 2H), 5.95 (s, 1H), 6.21 (t, J=4.0, 1H), 6.93 (s, 1H).
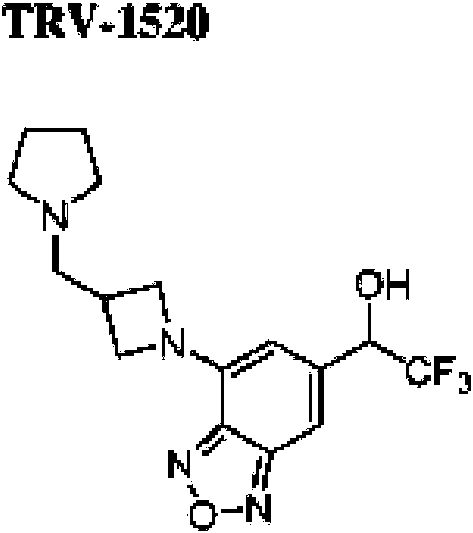
A un matraz de 2 cuellos secado al fuego se agregó TRV-1472 (0.2336 g, 0.69 mmol). El matraz se evacuó y se purgó con argón (3x). Se agregó THF (9.2 mL) y la solución se enfrió a -78°C. Se agregó nBuLi (0.52 mL, solución 1.6 M en hexano) durante 5 minutos, y la reacción se agitó durante unos 15 minutos adicionales. DMF puro (0.08 mL) luego se agregó, se agitó a -78°C durante 60 minutos. En este punto, se agregó MeOH (1 mL) a -78°C y la reacción se dejó calentar a temperatura ambiente. La mezcla se diluyó con agua y EtOAc y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron con MgSO4, se filtraron y se concentraron. El aldehído crudo luego se disolvió en THF (10 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.20 mL, 1.38 mmol) seguido por TBAF (70 mL). La reacción se agitó a 0°C durante 5 minutos y luego se calentó a temperatura ambiente. La reacción se completó después de 90 minutos. Se detuvo con 10 mL de HCl 2 N, luego se diluyó con EtOAc y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron con MgSO4, se filtraron y se concentraron. El material crudo se purificó con columna de 10% de MeOH/DCM para producir 0.0525 g (21% de rendimiento) de TRV-1520. 1H RMN (500 MHz, DMSO) 8 = 7.22 (s, 1H), 7.04 (d, J = 6 Hz, 1H), 6.06 (s, 1H), 5.23-5.18 (m, 1H), 4.33 (t, J = 8 Hz, 2H), 3.91 (t, J = 8 Hz, 2H), 3.02-2.97 (m, 1H), 2.76 (br s, 2H), 2.49-2.48 (m, 4H, se traslapó con señal de DMSO), 1.69 (s, 4H).
Se agregó en forma de gotas LDA (18 ml, 23 mmol, solución 2 M en THF) a una solución agitada de 1-Boc-3-piperidona (5.97 g, 30 mmol) en THF (80 mL) a -78°C bajo atmósfera de nitrógeno. Después de 20 minutos, se agregó una solución de THF (30 ml) de N-fenilbis( trifluometanosulfonimida) a la mezcla a -78°C, y la solución viscosa de color amarillo claro se agitó durante la noche y la temperatura se dejó calentar a RT. Después de adición de NH4Cl saturado (80 ml), la mezcla se diluyó con agua (100 ml). se agregó la mezcla 150 ml de EtOAc y se lavó con solución salina. La capa orgánica se secó y se concentró, 5-(((trifluorometil)sulfonil)oxi)-3,4-dihidropiridina-1(2H)-carboxilato de tert-butilo (4.02 g, 40.5%) y 5-(((trifluorometil)sulfonil)oxi)-3,6-dihidropiridina-1(2H)-carboxilato de tertbutilo (2.5 g, 25.2%) se obtuvieron a través cromatografía (5:95 EtOAc /Hexano).
Un matraz de fondo redondo se cargó con 5-(((trifluorometil)sulfonil)oxi)-3,6-dihidropiridina-1(2H)-carboxilato (2.5 g, 7.55 mmol), bis(pinacolato)diboro (2.88 g, 11.33 mmol), acetato de potasio (2.22 g, 22.7 mmol), y PdCh(dppf) (276 mg, 0.38 mmol). Después se desgasificó, se agregó dioxano (40 mL). La mezcla de reacción se calentó a 90°C durante 3 h. Después que se completó la reacción, la mezcla se enfrió a rt. Se agregó 40 mL de EtOAc, y la mezcla de reacción se lavó con agua durante 3 veces. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó mediante cromatografía instantánea (1:8:12 EtOAc/DCM/Hexano). 1.5 g (64.3%) de 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo se obtuvo como un sólido.
Un matraz de fondo redondo se cargó con TRV-1472 (1.36 g, 4.05 mmol), 5-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo (1.5 g, 4.85 mmol), y Pd(PPh3)4 (280 mg, 0.24 mmol). Después se desgasificó, se agregaron dioxano (16 mL) y carbonato de sodio acuoso (8 mL, 2M). La mezcla de reacción se calentó a 90°C durante 3 h. Después de que se verificó la finalización mediante TLC, se agregó 20 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó mediante cromatografía instantánea (1:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo (1.7 g, 94.4%).
Se agitó 5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,6-dihidropiridina-1(2H)-carboxilato de tert-butilo (1.7 g, 3.86 mmol) en la solución de DCM/CF3COOH (2:1,21 ml) durante 1 hora a 0°C. Después de que se verificó la finalización mediante TLC, la mezcla se neutralizó cuidadosamente con solución de K2CO3 saturado hasta que no haya más gas entregado a 0°C. Se agregó 15 ml de NaOH (2N) a la solución, y se extrajo con de DCM (3 x 30 ml). La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol crudo se utilizó para la siguiente etapa sin purificación adicional.
Se agregaron en forma de gotas TEA (1.02 ml, 7.35 mmol) y anhídrido acético (0.341 ml, 3.62 mmol) a una solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol crudo (250 mg, 0.735 mmol) en THF (10 ml) a RT respectivamente. La mezcla se agitó durante 3 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo mediante Hexano/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (6:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1521 (230 mg 82%) como un sólido amarillo. 1H RMN (400 MHz, CDCl3): 1.81 (s, br, 4H), 2.19 (s, 3H), 2.36-2.43 (m, 2H), 2.54 (s, br, 4H), 2.79-2.81 (m, 2H), 3.03-3.10 (m, 1H), 3.61 (t, J=5.8, 1.2H), 3.77 (t, J=5.8, 0.8H), 3.96-4.02 (m, 2H), 4.31-4.47 (m, 4H), 5.87 (s, 1H), 6.32-6.34 (m, 0.6H), 6.42-6.44 (m, 0.4H), 6.87 (s, 0.4H), 6.97 (s, 0.6H).

Se agregaron en forma de gotas TEA (0.41 ml, 2.94 mmol) y cloruro de metanosulfonilo (0.11 ml, 1.47 mmol) a una solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol crudo (250 mg, 0.735 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 5 horas. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (6:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1522 (240 mg, 78%) como un sólido rojo. 1H RMN (400 MHz, CDCh): 1.79-1.83 (m, 4H), 2.48-2.55 (m, 6H), 2.80 (d, J=7.3, 2H), 2.90 (s, 3H), 3.04-3.11 (m, 1H), 3.47 (t, J=5.8, 2H), 3.97-4.01 (m, 2H), 4.14-4.16 (m, 2H), 4.43 (t, J=8.2, 2H), 5.83 (s, 1H), 6.35-6.37 (m, 1H), 6.89 (s, 1H).

Se agregaron en forma de gotas carbonato de potasio (507 mg, 3.68 mmol) y éter de 2-bromo-etil metilo (0.316 ml, 3.3 mmol) a una solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5] oxadiazol crudo (250 mg, 0.735 mmol) en acetonitrilo (10 ml) a RT respectivamente. La mezcla se agitó durante la noche. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía cromatografía (4:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1523 (210 mg, 72%) como aceite rojo. 1H RMN (400 MHz, CDCl3):_1.80-1.82 (m, 4H), 2.41-2.42 (m, 2H), 2.54 (s, br, 2H), 2.71 (d, J=5.6, 2H), 2.77-2.81 (m, 4H), 3.02-3.08 (m, 1H), 3.40-3.41 (m, 5H), 3.62 (t, J=5.6, 2H), 3.94-3.98 (m, 2H), 4.40 (t, J=8.0, 2H), 5.91 (s, 1H), 6.28 (s, br, 1H), 6.87 (s, 1H).

Se agregaron en forma de gotas TEA (1.23 ml, 8.82 mmol) y yoduro de metilo (0.11 ml, 1.76 mmol) a una solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(1,2,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol crudo (300 mg, 0.882 mmol) en DCM (10 ml) a RT respectivamente. Después la mezcla se agitó durante 2 horas, se agregó yoduro de metilo (0.11 ml, 1.76 mmol) a una solución, y la mezcla se agitó durante otras 3 h. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de cromatografía (4:5:100:500, MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1524 (150 mg, 48%) como aceite rojo. 1H RMN (400 MHz, CDCl3): 1.81-1.86 (m, 4H), 2.42-2.45 (m, 2H), 2.49 (s, 3H), 2.58-2.62 (m, 6H), 2.82 (d, J=7.3, 2H), 3.04-3.11 (m, 1H), 3.29-3.31 (m, 2H), 3.95-3.99 (m, 2H), 4.41 (t, J=8.2, 2H), 5.92 (s, 1H), 6.28-6.30 (m, 1H), 6.88 (s, 1H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (1H-pirazol-3-il)borónico (0.10 g, 0.90 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro
y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 3: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 8: 75: 15) para producir 0.050 g (21% de rendimiento) de TRV 1525 como un sólido rojo. 1H RMN (DMSO, 400 MHz) 8 = l3.11 (amplio, 1H), 8.43 (amplio, 1H), 8.13 (amplio, 1H), 7.31 (s, 1H), 6.32 (s, 1H), 4.34 (t, J = 8.29 Hz, 2H), 3.92 (dd, J1 = 5.53 Hz, J2 = 8.54 Hz, 2H), 2.98 (septeto, J = 6.86 Hz, 1H), 2.71 (d, J = 7.53 Hz, 2H), 2.53 - 2.37 (m, 4H), 1.76 - 1.62 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (0.18 g, 0.90 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 3: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 10: 75: 15) para producir 0.080 g (33% de rendimiento) de TRV 1526 como un sólido rojo. 1H RMN (CDCh, 400 MHz) 8 = 7.66 (d, J = 2.51 Hz, 1H), 7.35 (s, 1H), 6.73 (d, J = 2.26 Hz, 1H), 6.46 (s, 1H), 4.46 (t, J = 8.28 Hz, 2H), 4.02 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.08 (septeto, J = 6.99 Hz, 1H), 2.81 (d, J = 7.28 Hz, 2H), 2.61 - 2.51 (m, 4H), 1.87 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), 4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (0.18 g, 0.90 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 3: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 8: 75: 15) para producir 0.25 g (100% de rendimiento) de TRV 1527 como un sólido rojo. 1H RMN (CDCh, 400 MHz) 87.81 (d, J = 0.75 Hz, 1H), 7.70 (s, 1H), 7.05 (d, J = 1.00 Hz, 1H), 5.95 (s, 1H), 4.44 (t, J = 8.29 Hz, 2H), 4.01 (dd, J1 = 5.65 Hz, J2 = 8.66 Hz, 2H), 3.98 (s, 3H), 3.08 (septeto, J = 7.03 Hz, 1H), 2.81 (d, J = 7.53 Hz, 2H), 2.62 - 2.49 (m, 4H), 1.88 -1.76 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.32 g, 0.95 mmol), ácido piridin-4-ilborónico (0.15 g, 1.20 mmol), y Pd(PPh3)4 (0.055 g, 0.048 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 1: 1: 15: 3) para producir 0.26 g (82% de rendimiento) de TRV 1528 como un sólido rojo. 1H RMN (CDCh, 400 MHz) 8 = 8.71 (dd, J1 = 1.64 Hz, J2 = 6.27 Hz, 1H), 7.52 (dd, J1 = 1.26 Hz, J2 = 6.27 Hz, 1H), 7.21 (s, 1H), 5.99 (s, 1H), 4.49 (t, J = 8.28 Hz, 2H), 4.06 (dd, J1 = 5.66 Hz, J2 = 8.66 Hz, 2H), 3.10 (septeto, J = 6.99 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.60 - 2.50 (m, 4H), 1.87 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido tiofen-2-ilborónico (0.13 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1.2: 75: 15) para producir 0.21 g (83% de rendimiento) de TRV 1529 como un sólido marrón. 8 = 7.54 (dd, J1 = 1.13 Hz, J2 = 3.64 Hz, 1H), 7.38 (dd, J1 = 1.13 Hz, J2 = 5.15 Hz, 1H), 7.22 (d, J = 1.00 Hz, 1H), 7.13 (dd, J1 = 3.64 Hz, J2 = 5.15 Hz, 1H), 6.10 (d, J = 1.00 Hz, 1H), 4.47 (t, J = 8.28 Hz, 2H), 4.03 (dd, J1 = 5.65 Hz, J2 = 8.66 Hz, 2H), 3.09 (septeto, J = 6.90 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.60 - 2.53 (m, 4H), 1.87 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido furan-2-ilborónico (0.11 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno,
y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1.2: 75: 15) para producir 0.18 g (75% de rendimiento) de TRV 1530 como un sólido naranja. 1H RMN (CDCh, 400 MHz) 8 = 7.54 (dd, J-i = 0.75 Hz, J2 = 1.76 Hz, 1H), 7.31 (s, 1H), 7.29 (dd, J1 = 0.75 Hz, J2 = 3.76 Hz, 1H), 6.53 J = 1.76 Hz, J2 = 3.51 Hz, 1H), 6.13 (s, 1H), 4.45 (t, J = 8.28 Hz, 2H), 4.02 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.08 (septeto, J = 6.99 Hz, 1H), 2.81 (d, J = 7.53 Hz, 2H), 2.61 - 2.49 (m, 4H), 1.88 - 1.76 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (3-(dimetilcarbamoil)fenil)borónico (0.18 g, 0.91 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc: DCM 5: 4: 60: 20: 20) para producir 0.24 g (80% de rendimiento) de TRV 1531 como un sólido amarillo. 1H RMN (CDCla, 400 MHz) 8 = 7.69 - 7.65 (m, 2H), 7.51 (dt, J1 = 1.25 Hz, J2 = 7.78 Hz, 1H), 7.46 (dt, J1 = 1.25 Hz, J2 = 7.53 Hz, 1H), 7.15 (s, 1H), 6.04 (s, 1H), 4.47 (t, J = 8.28 Hz, 2H), 4.03 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.16 (s, 3H), 3.09 (septeto, J = 6.78 Hz, 1H), 3.04 (s, 3H), 2.81 (d, J1 = 7.53 Hz, 2H), 2.59 - 2.51 (m, 4H), 1.86 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (2-(metiltio)fenil)borónico (0.11 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15) para producir 0.20 g (71% de rendimiento) de TRV 1532 como un sólido amarillo. 1H RMN (CDCh, 400 MHz) 8 = 7.39 (dt, J1 = 2.01 Hz, J2 = 7.66 Hz, 1H), 7.31 (d, J = 7.78 Hz, 1H), 7.27 (dt, J1 = 2.01 Hz, J2 = 7.53 Hz, 1H), 7.23 (dt, J1 = 1.00 Hz, J2 = 7.03 Hz, 1H), 7.00 (s, 1H), 5.91 (s, 1H), 4.44 (t, J = 8.29 Hz, 2H), 4.01 (dd, J1 = 5.78 Hz, J2 = 8.79 Hz, 2H), 3.06 (septeto, J = 6.78 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.59 - 2.49 (m, 4H), 2.43 (s, 3H), 1.63 - 1.53 (m, 4H).

Un matraz de fondo redondo se cargó con 5-(((trifluorometil)sulfonil)oxi)-3,4-dihidropiridina-1(2H)-carboxilato de tertbutilo (4.02 g, 12.15 mmol), bis(pinacolato)diboro (4.63 g, 18.22 mmol), acetato de potasio (3.57 g, 36.44 mmol), y PdCh(dppf) (445 mg, 0.61 mmol). Después se desgasificó, dioxano (60 mL) se agregó. La mezcla de reacción se calentó a 90°C durante 3 h. Después que se completó la reacción, la mezcla se enfrió a rt. Se agregó 40 mL de EtOAc, y la mezcla de reacción se lavó con agua durante 3 veces. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó mediante cromatografía instantánea (1:19, EtOAc/Hexano). 2.27 g (60.5%) de 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,4-dihidropiridina-1(2H)-carboxilato de tert-butilo se obtuvo como un sólido.
Un matraz de fondo redondo se cargó con TRV-1472 (2.06 g, 6.12 mmol), 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-3,4-dihidropiridina-1(2H)-carboxilato de tert-butilo (2.27 g, 7.35 mmol), y Pd(PPh3)4 (354 mg, 0.306 mmol). Después se desgasificó, se agregaron dioxano (16 mL) y carbonato de sodio acuoso (8 mL, 2M). La mezcla de reacción se calentó a 90°C durante 3 h. Después de que se verificó la finalización mediante TLC, se agregó 20 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó mediante cromatografía instantánea para producir 5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,4-dihidropiridina-1(2H)-carboxilato de tert-butilo (2.65 g, 98.4%). 1H RMN (400 MHz, CDCla): 1.55 (s, 9H), 1.79-1.83 (m, 4H), 1.96-2.02 (m, 2H), 2.47 (t, J=5.9, 2H), 2.54 (s, br, 4H), 2.78-2.82 (m, 2H), 3.02-3.09 (m, 1H), 3.63 (s, br, 2H), 3.94-3.97 (m, 2H), 4.40 (t, J=8.2, 2H), 5.90 (s, 0.44H), 6.03 (s, 0.6H), 6.86 (s, 1H), 7.43 (s, 0.4H), 7.66 (s, 0.6H).
Se agitó 5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-3,4-dihidropiridina-1(2H)-carboxilato de tert-Butilo (440 g, 1.0 mmol) en la solución de DCM/CF3COOH (2:1, 6 ml) durante 6 hora a 0°C. Después de que se verificó la finalización mediante TLC, la mezcla se neutralizó cuidadosamente con solución de K2CO3 saturado hasta que no haya más gas entregado a 0°C. Se agregó 15 ml de NaOH (2N) a la solución, y se extrajo con de DCM (3 x 10 ml). La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)- 6-(1,4,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol crudo se utilizó para la siguiente etapa sin purificación adicional.
Se agregaron en forma de gotas TEA (1.39 ml, 10 mmol) y anhídrido acético (0.471 ml, 5 mmol) a una solución de 4-(3-(pirrolidin-1 -ilmetil)azetidin-1-il)-6-(1,4,5,6-tetrahidropiridin-3-il)benzo[c][1,2,5]oxadiazol (crudo, 1 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 3 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 3:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1533 (65 mg, 17%) como un sólido amarillo. 1H RMN (400 MHz, CDCla): 1.79-1.83 (m, 4H), 1.96-2.02 (m, 1.1H), 2.04-2.10 (m, 0.9H), 2.26 (s, 1.35H), 2.31 (s, 1.65H), 2.50-2.55 (m, m, 6H), 2.79-2.82 (m, 2H), 3.02-3.11 (m, 1H), 3.66-3.69 (m, 0.9H), 3.76-3.78 (m, 1.1H), 3.96-4.01 (m, 2H), 4.42 (t, J=8.2, 2H), 5.84 (s, 0.55H), 6.02 (s, 0.45H), 6.90-6.91 (m, 1H), 7.15 (s, 0.55H), 7.93 (s, 0.45H).
Se agregaron en forma de gotas TEA (0.7 ml, 5 mmol) y cloruro de metanosulfonilo (0.39 ml, 5 mmol) a una solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1 -il)-6-(1,4,5,6-tetrahidropiridin-3-M)benzo[c][1,2,5]oxadiazol (crudo, 1 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 3 horas. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1534 (105 mg, 25%) como un sólido rojo. 1H RMN (400 MHz, CDCh): 1.80-1.83 (m, 4H), 2.06-2.12 (m, 2H), 2.50-2.55 (m, 6H), 2.79 (d, J=7.5, 2H), 2.97 (s, 3H), 3.03-3.09 (m, 1H), 3.64 (t, J=5.5, 2H), 3.96-4.00 (m, 2H), 4.42 (t, J=8.2, 2H), 5.87 (s, 1H), 6.87 (s, 1H), 7.19 (s, 1H).

El TRV-1472 (0.4 g, 1.15 mmol) y ácido 2-formiltiofeno-4-borónico (0.188 g, 1.21 mmol) se agregaron a un tubo y el tubo se evacuó y se purgó con argón (3x). Se agregaron DME (2.8 mL) y Na2CO3 (1.7 mL, 3.4 mmol, solución acuosa 2 N) seguidos por Pd(PPh3)4 (0.069 g, 0.06 mmol). El tubo se selló y luego se calentó a 85°C durante la noche. La reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el aldehído crudo. Se purificó el aldehído utilizando cromatografía de columna y se utilizó directamente en la siguiente etapa. Se disolvió aldehído 4-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)tiofeno-2-carbaldehído en THF (10 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.19 mL) y luego se agregó el catalizador TBAF (0.1 mL, solución 1.0 M). Después de 30 minutos la reacción se retiró del baño de hielo y se dejó calentar a temperatura ambiente. Una vez se completó mediante TLC, se volvió a enfriar a 0°C y se agregó HCl 2 N (ac.), se agitó durante 40 minutos y luego se basificó con NaOH 2N. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua (2x), solución salina, se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (hexano: acetato de etilo: trietilamina: metanol, 5:1:0.3:0.1) para producir 0.210 mg (58%) de TRV 1535 como sólido amarillo. 1H RMN (500 MHz, CDCl3) 87.59 (d, J= 3.6 Hz, 1H), 7.49 (s, 1H), 7.13(d, J = 3.6 Hz, 1H), 5.97 (s, 1H), 5.32-5.28 (m, 1H), 4.44-4.39 (m, 2H), 4.02-3.98 (m, 2H), 3.09-3.03 (m, 1H),2.81-2.83(d, J = 8 Hz, 2H),2.58 (m, 4H), 1.85 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.67 g, 2.00 mmol), 1-metil-5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-1H-pirazol (0.50 g, 2.40 mmol), y Pd(PPh3)4 (0.12 g, 0.10 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (6 mL) y carbonato de sodio ac. (3 mL, 2.0 M, 6.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 5: 75: 15) para producir 0.67 g (99% de rendimiento) de TRV 1536 como un sólido rojo. 1H RMN (CDCh, 400 MHz) 8 = 7.55 (d, J = 2.00 Hz, 2H), 7.01 (d, J = 0.76 Hz, 1H), 6.40 (d, J = 2.01 Hz, 2H), 5.82 (s, 1H), 4.46 (t, J = 8.29 Hz, 2H), 4.03 (dd, J1 = 5.40 Hz, J2 = 8.67 Hz, 2H), 3.97 (s, 3H), 3.09 (septeto, J = 7.09 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.60 - 2.49 (m, 4H), 1.87 - 1.76 (m, 4H).

Una solución de 6-bromo-4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.38 g, 1.13 mmol) en 6 mL de THF anhidro se enfrió a -78°C bajo nitrógeno. A la solución se agregó n-butiMitio (0.55 mL, 2.5 M, 1.38 mmol) en forma de gotas. Después que la adición se completó, la mezcla de reacción se agitó durante 30 minutos a la misma temperatura, y luego se agregó cloruro de tributilestaño (0.46 mL, 1.70 mmol). La reacción se agitó durante 1h a -78°C antes se detuvo con metanol. La mezcla se diluyó con solución salina, se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: hexano: EtOAc 2: 100: 0 a TEA: hexano: EtOAc 2: 95: 5) para producir 0.48 g (77% de rendimiento) de 4-(3-(Pirrolidin-1-ilmetil)azetidin-1 -il)-6-(tri-nbutilestanil)benzo[c][1,2,5]oxadiazol como aceite de color naranja profundo. 1H RMN (CDCh, 400 MHz) 8 = 7.14 (s, 1H), 5.89 (s, 1H), 4.40 (t, J = 8.03 Hz, 2H), 3.96 (dd, J1 = 5.77 Hz, J2 = 8.28 Hz, 2H), 3.06 (septeto, J = 7.53 Hz, 1H), 2.81 (d, J = 7.53 Hz, 2H), 2.61 - 2.49 (m, 4H), 1.88 - 1.76 (m, 4H), 1.61-1.51 (m, 6H), 1.35 (sexteto, J = 7.28 Hz, 6H), 1.09 (t, J = 8.09 Hz, 6H), 0.91 (t, J = 7.28 Hz, 9H).
Un frasco de reacción se cargó con 4-(3-(Pirrolidin-1-ilmetil)azetidin-1-il)-6-(tri-n-butilestanil)benzo[c][1,2,5]oxadiazol (0.33 g, 0.60 mmol), 1-(4-bromotiofen-2-il)-2,2,2-trifluoroetan-1-ol (0.16 g, 0.60 mmol), Pd(PPh3)4 (0.035 g, 0.030 mmol), Cul (0.011 g, 0.06 mmol), y CsF (0.18 g, 1.20 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó DMF (4 mL). La reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 5: 75: 15) para producir 0.080 g (31% de rendimiento) de TRV 1537 como un sólido rojo. 1H RMN (CDCla, 400 MHz) 8 = 8.23 (d, J = 1.51 Hz, 1H), 7.78 (s, 1H), 7.38 (s, 1H), 7.34 (d, J = 5.52 Hz, 1H), 6.35 (s, 1H), 5.50 (penteto, J = 6.52 Hz, 1H), 4.37 (t, J = 8.03 Hz, 2H), 3.96 (dd, J1 = 6.02 Hz, J2 = 8.28 Hz, 2H), 2.99 (septeto, J = 7.53 Hz, 1H), 2.71 (d, J = 7.53 Hz, 2H), 2.48 - 2.41 (m, 4H), 1.73 - 1.63 (m, 4H).

El TRV-1472 (2.0095 g, 5.96 mmol) se disolvió en THF (60 mL) y se enfrió a -78°C. nBuLi (4.5 mL, solución 1.6 M en hexano) se agregó en forma de gotas durante 10 minutos y se agitó durante unos 10 minutos adicionales. Se burbujeó gas CO2 a través de la mezcla de reacción a través de cánula mientras que se calentó gradualmente a temperatura ambiente. La reacción luego se concentró para dar 2.12 gramos de un sólido oscuro. Este material luego se disolvió en NMP (70 mL) y luego se utilizaron 20 mL de alícuotas de esta mezcla en la etapa de acoplamiento HATU. El carboxilato de litio 1 en NMP (20 mL, 0.6068 g de 1) se enfrió a 0°C y se agregó DIPEA (1.0 mL, 5.91 mmol); seguido por HATU (0.8239 g, 2.16 mmol). Esta mezcla se agitó durante 5 minutos antes de agregar morfolina (0.52 mL, 5.91 mmol) y la reacción se agitó hasta que se completó mediante TLC. La reacción se diluyó con solución salina y se extrajo con DCM (3x). Las capas combinadas se secaron con MgSO4, se filtraron y se concentraron. El residuo crudo se purificó a través de cromatografía, el primer intento fue con 10% de MeOH/DCM que falló para dar >95% de pureza química. La purificación se repitió con EtOAc: Hexano:TEA (6:4:0.5) para producir 0.0803 g (11% de rendimiento) de TRV-1538 como un sólido de cera. 1H RMN (500 MHz, DMSO) 8 = 7.06 (s, 1H), 5.88 (s, 1H), 4.34 (t, J = 5 Hz, 2H), 3.92 (t, J = 5 Hz, 2H), 3.65 (br s, 2H), 3.60 (br s, 2H), 3.54 (br s, 2H), 3.35 (br s, 2H), 3.03-2.95 (m, 1H), 2.70 (d, J = 10 Hz, 2H), 2.44 (br s, 4H), 1.67 (br s, 4H).

El TRV-1520 (0.3249 g, 0.91 mmol) se disolvió en NMP (10 mL) y se enfrió en un baño de hielo. Se agregó en forma de porciones NaH (0.0401 g, 1.00 mmol, 60% en aceite mineral) y se agitó durante 20 minutos, después de ese tiempo, se agregó yodometano (0.056 mL, 0.91 mmol) a la reacción y la mezcla se agitó durante la noche. La reacción se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla se extrajo con DCM (3x) y las capas orgánicas combinadas se secaron con MgSO4, se filtraron y se concentraron. El residuo crudo se purificó a través de 8% de MeOH/DCM y luego de nuevo con EtOAc/Hexano/TEA (6:4:0.5) para producir 0.0602 g (18 % de rendimiento) de TRV-1539. 1H RMN (500 MHz, DMSO) 8 = 7.20 (s, 1H), 5.93 (s, 1H), 5.10 (q, J = 7 Hz, 1H), 4.36 4.32 (m, 2H), 3.93-3.89 (m, 2H), 3.39 (s, 3H), 3.01-2.96 (m, 1H), 2.71 (d, J = 5 Hz, 2H), 2.44 (br s, 4 H), 1.70-1.64 (m, 4H).

El TRV-1472 (2.0095 g, 5.96 mmol) se disolvió en THF (60 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (4.5 mL, solución 1.6 M en hexano) durante 10 minutos y se agitó durante unos 10 minutos adicionales. Se burbujeó gas CO2 a través de la mezcla de reacción a través de cánula mientras que se calentó gradualmente a temperatura ambiente. La reacción luego se concentró para dar 2.12 gramos de un sólido oscuro. Este material luego se disolvió en NMP (70 mL) y luego se utilizaron 20 mL de alícuotas de esta mezcla en la etapa de acoplamiento HATU. El carboxilato de litio 1 en NMP (20 mL, 0.6068 g de 1) se enfrió a 0°C y se agregó DIPEA (1.0 mL, 5.91 mmol); seguido por HATU (0.8239 g, 2.16 mmol). Esta mezcla se agitó durante 5 minutos antes de agregar dietilamina (1.0 mL, 5.91 mmol) y la reacción se agitó hasta que se completó mediante TLC. La reacción se diluyó con solución salina y se extrajo con DCM (3x). Las capas combinadas se secaron con MgSO4, se filtraron y se concentraron. El residuo crudo se purificó a través de cromatografía, el primer intento fue con EtOAc: Hexano:TEA (6:4:0.5) que falló para dar >95% de pureza química. La purificación se repitió con 10% de MeOH/DCM para producir 0.0823 g (12% de rendimiento) de TRV-1540 como un sólido de cera. 1H RMN (500 MHz, DMSO) 8 = 6.99 (s, 1H), 5.81 (s, 1H), 4.35 (t, J = 10 Hz, 2H), 3.93 (t, J = 10 Hz, 2H), 3.42 (d, J = 10 Hz, 2H), 3.21 (d, J = 10 Hz, 2H), 3.00 2.97 (m, 1H), 2.72 (br s, 2H), 2.46 (br s, 4H), 1.68 (br s, 4H), 1.19-1.13 (m, 3H), 1.10-1.03 (m, 3H).
El TRV-1472 (2.0095 g, 5.96 mmol) se disolvió en THF (60 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (4.5 mL, solución 1.6 M en hexano) durante 10 minutos y se agitó durante unos 10 minutos adicionales. Se burbujeó gas CO2 a través de la mezcla de reacción a través de cánula mientras que se calentó gradualmente a temperatura ambiente. La reacción luego se concentró para dar 2.12 gramos de un sólido oscuro. Este material luego se disolvió en NMP (70 mL) y luego se utilizaron 20 mL de alícuotas de esta mezcla en la etapa de acoplamiento HATU. El carboxilato de litio 1 en NMP (20 mL, 0.6068 g de 1) se enfrió a 0°C y se agregó DIPEA (1.0 mL, 5.91 mmol); seguido por HATU (0.8239 g, 2.16 mmol). Esta mezcla se agitó durante 5 minutos antes de agregar ciclopropilamina (0.41 mL, 5.91 mmol) y la reacción se agitó hasta que se completó mediante TLC. La reacción se diluyó con solución salina y se extrajo con DCM (3x). Las capas combinadas se secaron con MgSO4, se filtraron y se concentraron. El residuo crudo se purificó a través de cromatografía, el primer intento fue con EtOAc: HexanoTEA (6:4:0.5) que falló para dar >95% de pureza química. La purificación se repitió con 10% de MeOH/DCM para producir 0.0459 g (6.8% de rendimiento) de TRV-1541 como un sólido de cera. 1H RMN (500 MHz, DMSO) 8 = 8.57 (d, J = 5 Hz, 1H), 7.47 (s, 1H), 6.29 (s, 1H), 4.35 (t, J = 10 Hz, 2H), 3.93 (t, J = 10 Hz, 2H), 3.01-2.96 (m, 1H), 2.86-2.81 (m, 1H), 2.71 (d, J = 10 Hz, 2H), 2.45 (br s, 4H), 1.68 (br s, 4 H), 0.72-0.69 (m, 2H), 0.59-0.56 (m, 2H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (2-(dimetilcarbamoil)fenil)borónico (0.18 g, 0.91 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 3: 75: 15) para producir 0.23 g (77 % de rendimiento) de TRV 1542 como aceite rojo. 1H RMN (CDCh, 400 MHz) 8 = 7.52 - 7.38 (m, 4H), 7.02 (s, 1H), 5.98 (s, 1H), 4.42 (t, J = 8.28 Hz, 2H), 3.99 (dd, J1 = 5.65 Hz, J2 = 8.41 Hz, 2H), 3.06 (septeto, J = 7.03 Hz, 1H), 2.94 (s, 3H), 2.79 (d, J = 7.53 Hz, 2H), 2.65 (s, 3H), 2.58 - 2.46 (m, 4H), 1.86 - 1.72 (m, 4H).

Se agregaron TRV-1472 (0.4 g, 1.15 mmol) y ácido 5-formiltiofeno-2-borónico (0.188 g, 1.21 mmol) a un tubo y el tubo se evacuó y se purgó con argón (3x). Se agregaron DME (2.8 mL) y Na2CO3 (1.7 mL, 3.4 mmol, solución acuosa 2 N) seguidos por Pd(PPh3)4 (0.069 g, 0.06 mmol). El tubo se selló y luego se calentó a 80°C durante la noche. La reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el aldehído crudo. Se purificó el aldehído utilizando cromatografía de columna y se utilizó directamente en la siguiente etapa. Aldehído 2 se disolvió en THF (5 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.19 mL) y luego se agregó el catalizador TBAF (0.1 mL, solución 1.0 M). Después de 30 minutos la reacción se retiró del baño de hielo y se dejó calentar a temperatura ambiente. Una vez se completó mediante TLC, se volvió a enfriar a 0°C y se agregó HCl 2 N (ac.), se agitó durante 40 minutos y luego se basificó con NaOH 2N. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua (2x), solución salina, se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (hexano: acetato de etilo: trietilamina: metanol, 5:1:0.3:0.1) para producir 40 mg (28%) de TRV 1543 como sólido amarillo. 1H RMN (400 MHz, CDCh) 8 7.33 (d, J=3.6 Hz, 1H), 7.20-7.20 (m, 2H), 5.99(s, 1H), 5.31-5.25 (m, 1H), 4.45-4.40 (m, 2H), 4.02-3.99 (m, 2H), 3.09-3.03 (m,1H),2.83-2.81 (d,J=8 Hz, 2H),2.57 (m, 4H), 1.83 (m, 4H).

Se cargó 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(tributilestanM)benzo[c][1,2,5]oxadiazol (0.4 g, 0.73 mmol), 1-(5-bromopiridin-3-M)-2,2,2-trifluoroetan-1-ol (0.230 g, 0.9 mmol), CsF (0.45 g, 2.19 mmol), CuI (14 mg, 0.073 mmol), Pd(PPh3)4, (0.042 g, 0.0365 mmol) en tubo de vidrio y se selló, luego se desgasifico y se enjuagó con N2. Se agregó DMF (10 ml) y la mezcla de reacción se calentó a 45°C durante la noche. Después de la finalización de la reacción mediante TLC, la reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el producto crudo que se purificó utilizando cromatografía de columna para dar TRV-1544 (0.120 g, 42%). 1H RMN (400 MHz, DMSO): 89.03 (d, J=3.6 Hz, 1H), 8.75 (s, 1H), 8.26 (s, 1H), 7.41 (s, 1H), 7.19 (d, J=4.0 Hz, 1H), 6.26 (s, 1H), 5.37-5.45 (m, 1H), 4.40 (m, 2H), 3.99 (m, 2H), 2.97-3.04 (m, 1H), 2.72 (d, J=8.0 Hz, 2H), 2.45 (s, 4H), 1.68 (s, 4H).

4-(3-(pirrolidin-1-ilmetil)azetidin-1 -il)-6-(tributilestanil)benzo[c][1,2,5]oxadiazol (0.4 g, 0.73 mmol), 1-(6- bromopiridin-3-il)-2,2,2-trifluoroetan-1-ol (0.230 g, 0.9mmol), CsF (0.45 g, 2.19 mmol), CuI (14 mg, 0.073 mmol), Pd(PPha)4, (0.042 g, 0.0365 mmol) se cargó en tubo de vidrio y se selló, luego se desgasifico y se enjuagó con N2. Se agregó DMF (10 ml) y la mezcla de reacción se calentó a 45°C durante la noche. Después de la finalización de la reacción mediante TLC, la reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el producto crudo que se purificó utilizando cromatografía de columna para dar TRV-1545 (0.108 g, 36%). 1H RMN (400 MHz, DMSO): 88.80 (s, 1H), 8.22 (d, J=8.0 Hz, 1H), 8.03 (d, J=8.0 Hz, 1H), 7.80 (s, 1H), 7.14 (s, 1H), 6.77 (s, 1H), 5.40 (m, 1H), 4.39 (m, 2H), 3.96 (m, 2H), 3.0 (m, 1H), 2.72 (d, J=4.0 Hz, 2H), 2.45 (s, 4H), 1.68 (s, 4H).

Un frasco de reacción se cargó con 4-(3-(Pirrolidin-1-ilmetil)azetidin-1-il)-6-(tri-n-butilestanil)benzo[c][1,2,5]oxadiazol (0.33 g, 0.60 mmol), 1-(4-bromotiofen-2-il)-2,2,2-trifluoroetan-1-ol (0.16 g, 0.60 mmol), Pd(PPh3)4 (0.035 g, 0.030 mmol), Cul (0.011 g, 0.06 mmol), y CsF (0.18 g, 1.20 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó DMF (4 mL). La reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se
extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 5: 75: 15) para producir 0.14 g (42% de rendimiento) de 2-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol- 5-il)tiazol-5-carbaldehído como un sólido amarillo.
Se disolvió 2-(7-(3-(Pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)tiazol-5-carbaldehído (0.14 g, 0.38 mmol) en THF (5 mL) y se enfrió a 0°C. Se agregó CF3TMS (0.085 mL, 0.57 mmol) seguido por TbAf (0.050 mL, solución 1.0 M en THF, 0.050 mmol). La reacción luego se agitó durante 60 minutos antes de volver a enfriarse a 0°C y se agregó HCl 4N (ac.) y se agitó durante 60 minutos, y luego se basificó con 3N NaOH ac. y se extrajo con EtOAc. La capa orgánica se secó sobre Na2SO4 anhidro, se filtró y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 5: 75: 15) para producir 0.090 g (54% de rendimiento) de TRV 1546 como un sólido rojo. 1H RMN (CDCl3, 400 MHz) 8 = 8.54 (s, 1H), 7.48 (s, 1H), 6.42 (s, 1H), 5.20 (q, J = 6.53 Hz, 1H), 4.49 (t, J = 8.28 Hz, 2H), 4.30 (amplio, 1H), 4.11 -4.03 (m, 2H), 3.10 (septeto, J = 7.19 Hz, 1H), 2.82 (d, J = 7.28 Hz, 2H), 2.60 -2.50 (m, 4H), 1.87 - 1.77 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (4-fluorofenil)borónico (0.14 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15) para producir 0.20 g (77% de rendimiento) de TRV 1547 como aceite rojo. 1H RMN (CDCh, 400 MHz) 8 = 7.63 - 7.56 (m, 2H), 7.20 - 7.13 (m, 2H), 7.10 (s, 1H), 6.00 (s, 1H), 4.47 (t, J = 8.28 Hz, 2H), 4.03 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.09 (septeto, J = 6.65 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.60 - 2.50 (m, 4H), 1.86 - 1.77 (m, 4H).

Se disolvió 7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carboxilato de metilo (0.2146 g, 0.68 mmol) en metanol (2 mL) y NH37N en metanol (12 mL) se agregó al tubo. El tubo se selló y calentó a 50°C durante 48 horas detrás de un escudo explosivo. La mezcla luego se concentró para producir la amida cruda. La amida cruda se purificó a través de cromatografía (10% de MeOH/DCM con capa de NH4OH) para producir 0.1439 g (70% de rendimiento) de TRV-1548 1H RMN (500 MHz, DMSO) 8 = 8.10 (s, 1H), 7.58 (s, 1H), 7.57 (s, 1H), 6.36 (s, 1H), 4.35 (t, J = 10 Hz, 2H), 3.92 (q, J = 5 Hz, 2H), 3.02-2.94 (m, 1H), 2.70 (d, J = 10 Hz, 2H), 2.44 (br s, 4H), 1.68-1.65 (m, 4H).

El TRV-1551 (0.1830 g, 0.54 mmol) se disolvió en DCM (10 mL) y se enfrió a 0°C. Se agregó TEA (0.75 mL, 5.4 mmol) seguido por la adición en forma de gotas de MeSO2Cl (0.21 mL, 2.7 mmol). La reacción se dejó calentar a temperatura ambiente y se agitó hasta que se completó mediante TLC. La reacción luego se concentró y el residuo se hizo básico mediante la adición de NaOH 2N(ac.). Este residuo básico luego se extrajo con DCM (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para producir la amida cruda. El material se purificó a través de cromatografía EtOAc:hexano:TEA (9:1:0.5) para producir 0.1417 g (63% de rendimiento) de TRV-1549. 1H RMN (500 MHz, CDCls) 8 = 6.98 (s, 1H), 5.95 (s, 1H), 5.17 (q, J = 10 Hz, 1H), 4.39 (q, J = 10 Hz, 2H), 3.99-3.95 (m, 2H), 3.05 (br s, 1H), 2.97 (s, 3H), 2.81 (br s, 2H), 2.54 (br s, 4H), 2.30-2.26 (m, 1H), 1.81 (br s, 4H), 1.73 (d, J = 10 Hz, 3H), 1.02-0.96 (m, 1H), 0.81-0.75 (m, 1H), 0.57-0.52 (m, 2H).

El TRV-1551 (0.2133 g, 0.62 mmol) se disolvió en DCM (10 mL) y se enfrió a 0°C. TEA (0.86 mL, 6.2 mmol) se agregó seguido por la adición en forma de gotas de AcCl (0.22 mL, 3.1 mmol). La reacción se dejó calentar a temperatura ambiente y se agitó hasta que se completó mediante TLC. La reacción luego se concentró y el residuo se hizo básico mediante la adición de NaOH 2N(ac.). Este residuo básico luego se extrajo con DCM (3x). Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para producir la amida cruda. El material se purificó a través de cromatografía EtOAc:hexano:TEA (9:1:0.5) para producir 0.1858 g (78% de rendimiento) de TRV-1550. 1H RMN (500 MHz, CDCla) 8 = 6.87 (s, 1H), 5.70 (s, 1H), 5.68-5.67 (m, 1H), 4.37 (t, J = 10 Hz, 1H), 4.32 (t, J = 10 Hz, 1H), 3.95-3.89 (m, 2H), 3.05-2.97 (m, 1H), 2.78-2.73 (m, 2H), 2.56 2.52 (m, 5H), 2031 (s, 3H), 1.79 (s, 4H), 1.67 (d, J = 10 Hz, 3H), 0.82-0.75 (m, 1H), 0.69-0.60 (m, 3H).

El TRV-1472 (1.7789 g, 5.28 mmol) se disolvió en tolueno (25 mL) en un tubo y se agregó Tributil(1-etoxivinil)estaño (2.4789 g, 6.86 mmol). La solución se desgasificó durante 10 minutos al burbujear argón a través de la solución. Luego se agregó Pd(PPh3)4 (0.6125 g, 0.53 mmol), el tubo se selló y la mezcla se calentó a 110°C durante 16 horas. Una alícuota durante 1H RMN indicó que la reacción había alcanzado 100% de conversión. El material se filtró a través de Celite y luego se concentró. El residuo se disolvió en DCM y se lavó con solución de KF acuosa, lo que
resulta en un precipitado. Este material se retiró mediante filtración. La capa orgánica luego se lavó con agua, solución salina, se secó (MgSO4), se filtró y se concentró. El material crudo se disolvió parcialmente en THF (80 mL) y se agregó EtOH (6 mL) para alcanzar solución completa. La solución luego se enfrió a 0°C se agregó en forma de gotas y HCl 2N (ac.) (11 mL). Se agitaron a 0°C durante 5 minutos antes de eliminar el baño de hielo. Se agitó hasta que se completó mediante TLC (60 minutos) y luego se concentró para eliminar el THF. El residuo se enfrió a 0°C y luego se hizo básico con NaOH 2N (ac.). Esta mezcla básica se extrajo con DCM (3x) y los orgánicos combinados se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para producir la cetona cruda 8. Esta cetona luego se disolvió en metanol (50 mL) y ciclopropil amina (0.76 mL, 11 mmol) y Ti(OiPr)4 (2.1 mL, 6.9 mmol) se agregaron produciendo un precipitado marrón. Esta mezcla luego se enfrió en un baño de hielo y se agregó NaBH4 (0.2989 g, 7.9 mmol) en forma de porciones y luego se agitó hasta que se completó mediante TLC. La reacción luego se detuvo con la adición de NH4Cl y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc. Las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4) se filtraron y se concentraron. La amina cruda se purificó a través de cromatografía (10% de MeOH/DCM) para producir 0.8689 g (48% de rendimiento de TRV-1472) de TRV-1551. 1H RMN (500 MHz, DMSO) 8 = 6.99 (s, 1H), 6.09 (s, 1H), 4.29 (t, J = 10 Hz, 2H), 3.86 (t, J = 10 Hz, 2H), 3.73 (s, 1H), 3.00-2.91 (m, 1H), 2.75 (s, 1H), 2.69 (d, J = 10 Hz, 2H), 2.44 (s, 4H), 1.88-1.84 (m, 1H), 1.70-1.64 (m, 4H), 1.23 (d, J = 5 Hz, 3H), 0.29-0.19 (m, 4H).

El TRV-1472 (0.7014 g, 2.08 mmol) se disolvió en THF (20 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (1.8 mL, solución 1.6 M en hexano) durante 10 minutos y se agitó durante unos 10 minutos adicionales. Se burbujeó gas CO2 a través de la mezcla de reacción a través de cánula mientras que se calentó gradualmente a temperatura ambiente. La reacción luego se concentró para dar 800 mg de un sólido oscuro. Este material luego se disolvió en NMP (30 mL) y luego 15 mL de alícuotas de esta mezcla se utilizaron en la etapa de acoplamiento HATU. El carboxilato de litio 1 en NMP (15 mL, 1.04 mmol de 1) se enfrió a 0°C y se agregó DlpEA (0.54 mL, 3.12 mmol); seguido por HATU (0.4349 g, 1.14 mmol). Esta mezcla se agitó durante 5 minutos antes de agregar piperidina (0.31 mL, 3.13 mmol) y la reacción se agitó hasta que se completó mediante TLC. La reacción se diluyó con solución salina y se extrajo con DCM (3x). Las capas combinadas se secaron con MgSO4, se filtraron y se concentraron. El residuo crudo se purificó a través de cromatografía, el primer intento fue con EtOAc: Hexano:TEA (6:4:0.5) que falló para dar >95% de pureza química. La purificación se repitió con 10% de MeOH/DCM para producir 0.081 g (21% de rendimiento) de TRV-1552 como un sólido de cera. 1H RMN (500 MHz, DMSO) 8 = 7.00 (s, 1H), 5.84 (s, 1H), 4.34 (t, J = 10 Hz, 2H), 3.92 (t, J = 10 Hz, 2H), 3.56 (br s, 2H), 3.29-3.24 (m, 2H), 2.98-2.97 (m, 1H), 2.72 (br s, 2H), 2.49 (se traslapó con señal de DMSO, br s, 4H), 1.68 (s, 4H), 1.61-1.59 (m, 2H), 1.56 (s, 2H), 1.46 (s, 2H).

Un matraz de fondo redondo se cargó con TRV-1472 (600 mg, 1.78 mmol), benciltrietilamonio (406 mg, 1.78 mmol) y Pd(OAc)2 (40 mg, 0.178 mmol). El matraz se purgó con nitrógeno durante varias veces, y luego se agregaron DMF seco (8 ml) alcohol alílico (0.181 ml, 2.67 mmol) y TEA (2.47 ml, 17.8 mmol) a la mezcla por separado. La mezcla resultante luego se calentó a 55-60°C, y se verificó con TLC. Después que se completó la reacción, se agregó 50 ml de EtOAc a la mezcla. La mezcla se lavó con solución salina durante 3 veces. La capa orgánica se secó y se concentró. Se obtuvo 3-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)propanal (340 mg, 60.7%) a
través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 4:5:100:500 MeOH/TEA/EtOAc/Hexano). 1H RMN (400 MHz, CDCl3): 1.78-1.83 (m, 4H), 2.52 (s, br, 4H), 2.78-2.84 (m, 4H), 2.91-2.94 (m, 2H), 3.01-3.07 (m, 1H), 3.95 3.97 (m, 2H), 4.38 (t, J=8.2, 2H), 5.63 (s, 1H), 6.77 (s, 1H), 9.85 (s, 1H).
El aldehído anterior (250 mg, 0.79 mmol) se enfrío a 0°C en solución de THF seco (6 ml) bajo atmósfera de nitrógeno. Se agregaron lentamente TMSCF3 (140 ul, 0.95 mmol) y TBAF (0.08 ml, 0.08 mmol, solución 1 M) a la mezcla por separado. El color se cambió de rojo a púrpura y de nuevo a rojo durante la adición. La solución se agitó durante 2 horas a 0°C. Después de que se verificó la finalización mediante TLC, se agregó la mezcla con 5 ml de agua y se agitó durante 1 h. Esta mezcla se agregó con 50 ml de EtOAc y se lavó con NaOH 1 N y solución salina. La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El producto (TRV-1553, 220 mg, 72.6%) se obtuvo a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 4:5:100:500 MeOH/TEA/EtOAc/Hexano). 1H RMN (400 MHz, CDCl3): 1.85 (s, br, 4H), 1.92-2.08 (m, 2H), 2.54 (s, br, 4H), 2.70 2.78 (m, 1H), 2.80 (d, J=7.6, 2H), 2.86-2.93 (m, 1H), 3.02-3.09 (m, 1H), 3.92-3.97 (m, 3H), 4.40 (t, J=8.2, 2H), 5.67 (s, 1H), 6.82 (s, 1H).

Se agregó en forma de gotas n-BuLi (0.96 ml, 2.4 mmol, solución 4 M) a una solución agitada de TRV-1472 (674 mg, 2 mmol) en THF (10 mL) a -78°C bajo atmósfera de nitrógeno. Después de 20 minutos, una solución de THF (5 ml) de 1-Boc-2-piperidona (440 mg, 2.2 mmol) se agregó a la mezcla a -78°C, y la solución negra se agitó durante 1 hora y luego se detuvo con MeOH. Después de adición de EtOAc (50 ml), la mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró, se obtuvo (5-oxo-5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)pentil)carbamato de tert-butilo (360 mg, 39.3%) a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano). 1H RMN (400 MHz, CDCl3): 1.45 (s, 9H), 1.56-1.60 (m, 2H), 1.74-1.82 (m, 6H), 2.53 (s, br, 4H), 2.79 (d, J=7.6, 2H), 3.00-3.09 (m, 3H), 3.17-3.20 (m, 2H), 4.00 4.04 (m, 2H), 4.45 (t, J=8.2, 2H), 4.63 (s, br, 1H), 6.34 (s, 1H), 7.61 (s, 1H).
(5-oxo-5-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5] oxadiazol-5-il)pentil) carbamato de tert-butilo (280 mg, 1.0 mmol) se agitó en la solución de DCM/CF3COOH (2:1, 3 ml) durante 1 hora a 0°C. Después de que se verificó la finalización mediante TLC, la mezcla se neutralizó cuidadosamente con solución de K2CO3 saturado hasta que no haya más gas entregado a 0°C. Se agregó 15 ml de NaOH (2N) a la solución, y se extrajo con de DCM (3 x 10 ml). La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo (4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(3,4,5,6- tetrahidropiridin-2-il)benzo[c][1,2,5]oxadiazol) se utilizó para la siguiente etapa sin purificación adicional. NaBH4 (114 mg, 1.83 mmol) se agregó a la solución agitada en la solución de 4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)-6-(3,4,5,6-tetrahidropiridin-2-il)benzo[c][1,2,5]oxadiazol (210 mg, 0.61 mmol) en MeOH (8 ml) durante 2 horas a rt. Después de que se verificó la finalización mediante TLC, la mezcla se concentró. 15 ml de NaOH (2N) se agregó al residuo, y se extrajo con de DCM (3 x 10 ml). La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo (compuesto 3) se utilizó para la siguiente etapa sin purificación adicional. Se agregaron en forma de gotas TEA (0.51 ml, 3.66 mmol) y cloruro de metanosulfonilo (0.14 ml, 1.83 mmol) a una solución de 6-(piperidin-2-il)-4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (crudo, 0.61 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 3 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 4:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir el Compuesto TRV-1554 (192 mg 75%) como sólido amarillo. 1H RMN (400 MHz, CDCl3): 1.56-1.76 (m, 4H), 1.81 (s, br, 4H), 1.95-2.02 (m, 1H), 2.34 (d, J=8.0, 1H), 2.54 (s, br, 4H), 2.80 (d, J=7.6, 2H), 2.9 (s, 3H), 3.02-3.16 (m, 2H), 3.85 (d, J=7.2, 1H), 3.97 4.00 (m, 2H), 4.39-4.43 (m, 2H), 5.01 (d, J=4.8, 1H), 5.88 (s, 1H), 7.00 (s, 1H).

Se agregaron TEA (0.81 ml, 5.84 mmol) y anhídrido acético (0.276 ml, 2.92 mmol) a una solución de 6-(piperidin- 2-il)-4-(3-(pirrolidin-1 -ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (200 mg, 0.584 mmol) en DCM (10 ml) a RT respectivamente. La mezcla se agitó durante 2 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 3:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1555 (205 mg 91%) como sólido amarillo. 1H RMN (400 MHz, CDCla): 1.52-1.89 (m, 9H), 2.13-2.34 (m, 4H), 2.53 (s, br, 4H), 2.73-2.80 (m, 2.31H), 3.04-3.12 (m, 1.69H), 3.69-3.72 (m, 0.69H), 3.95 (s, br, 2H), 4.34-4.41 (m, 2H), 4.63-4.65 (m, 0.31H), 4.95 (s, br, 0.31H), 5.61-5.67 (m, 1H), 5.88 (s, 0.69H), 6.88 (s, 1H).

Se agregaron carbonato de potasio (828 mg, 6 mmol) y éter de 2-bromo-etil metilo (0.432 ml, 4.5 mmol) a una solución de 6-(piperidin-2-il)-4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (513 mg, 1.5 mmol) en acetonitrilo (10 ml) respectivamente. Luego la mezcla se calentó a 50°C durante la noche, se agregó otra tanda de éter de 2-bromo-etil metilo (0.144 ml, 1.5 mmol) y se calentó durante 8 h a 50°C. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1556 (280 mg, 46%) como aceite rojo. 1H RMN (400 MHz, CDCla): 1.31-1.42 (m, 1H), 1.59-1.82 (m, 9H), 2.09-2.18 (m, 2H), 2.54 (s, br, 4H), 2.71-2.78 (m, 1H), 2.80 (d, J=7.6, 2H), 3.03 3.07 (m, 2H), 3.23 (d, J=7.6, 1H), 3.28 (s, 3H), 3.34-3.39 (m, 1H), 3.46-3.52 (m, 1H), 3.95-3.99 (m, 2H), 4.41 (q, J=8.0, J=6.0, 2H), 6.04 (s, 1H), 6.91 (s, 1H).

Se agregó en forma de gotas n-BuLi (0.48 ml, 2.5 M, 1.2 mmol) a una solución de 6-(piperidin-2-il)-4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (342 mg, 1.0 mmol) en THF (10 ml) a -78°C. Después la mezcla se agitó durante 5 minutos, se agregó yoduro de metilo (0.125 ml, 2.0 mmol) a una solución, y la mezcla se agitó durante 1 h
a -78°C. La reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 2 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 4:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1557 (156 mg, 44%) como aceite rojo. 1H RMN (400 MHz, CDCla): 1.31-1.41 (m, 1H), 1.55-1.72 (m, 4H), 1.77-1.82 (m, 5H), 2.03-2.13 (m, 4H), 2.54 (s, br, 4H), 2.75-2.81 (m, 3H), 3.00-3.08 (m, 2H), 3.94-4.00 (m, 2H), 4.39-4.44 (m, 2H), 5.99 (s, 1H), 6.90 (s, 1H).

Un frasco de reacción se cargó con TRV 1472 (0.25 g, 0.74 mmol), ácido (2,4,6-trimetilfenil)borónico (0.16 g, 1.00 mmol), y Pd(PPh3)4 (0.043 g, 0.037 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15) para producir 0.21 g (75% de rendimiento) de TRV 1558 como aceite rojo. 1H RMN (CDCh, 400 MHz) 8 = 6.96 (d, J = 0.50 Hz, 2H), 6.79 (d, J = 0.75 Hz, 1H), 5.62 (s, 1H), 4.40 (t, J = 8.28 Hz, 2H), 3.97 (dd, J1 = 5.90 Hz, J2 = 8.16 Hz, 2H), 3.05 (septeto, J = 7.06 Hz, 1H), 2.82 (d, J = 7.53 Hz, 2H), 2.58 - 2.49 (m, 4H), 2.35 (s, 3H), 2.10 (s, 6H), 1.85 -1.76 (m, 4H).

Un frasco de reacción se cargó con TRV 1472 (0.16 g, 0.47 mmol), ácido (2,5-dimetilfenil)borónico (0.085 g, 0.57 mmol), y Pd(PPh3)4 (0.030 g, 0.026 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y carbonato de sodio ac. (2 mL, 2.0 M, 4.0 mmol). La reacción se volvió a desgasificar, se rellenó con nitrógeno, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (TEA: MeOH: hexano: EtOAc 5: 1: 75: 15) para producir 0.17 g (100% de rendimiento) de TRV 1559 como aceite rojo. 1H RMN (CDCh, 400 MHz) 8 = 7.18 (d, J = 7.78 Hz, 1H), 7.12 (d, J = 7.78 Hz, 1H), 7.09 (s, 1H), 6.90 (d, J = 0.75 Hz, 1H), 5.79 (s, 1H), 4.42 (t, J = 8.28 Hz, 2H), 3.99 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.06 (septeto, J = 7.15 Hz, 1H), 2.81 (d, J = 7.28 Hz, 2H), 2.60 - 2.48 (m, 4H), 2.37 (s, 3H), 2.27 (s, 3H), 1.87 - 1.75 (m, 4H).

Un frasco de reacción se cargó con 4-(3-(Pirrolidin-1 -ilmetil)azetidin-1-M)-6-(tri-n-butilestanil)benzo[c][1,2,5]oxadiazol (0.33 g, 0.60 mmol), 2-bromotiazol (0.081 mL, 0.90 mmol), Pd(PPh3)4 (0.035 g, 0.030 mmol), Cul (0.011 g, 0.06 mmol), y CsF (0.18 g, 1.20 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó NMP (4 mL). La reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 0: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 3: 75: 15) para producir 0.086 g (42% de rendimiento) de TRV 1560 como un sólido rojo. 1H RMN (CDCla, 400 MHz) 8 = 7.93 (d, J = 3.26 Hz, 1H), 7.56 (s, 1H), 7.44 (d, J = 3.26 Hz, 1H), 6.56 (s, 1H), 4.50 (t, J = 8.53 Hz, 2H), 4.07 (dd, J1 = 6.02 Hz, J2 = 8.53 Hz, 2H), 3.09 (septeto, J = 7.53 Hz, 1H), 2.81 (d, J = 7.28 Hz, 2H), 2.58 -2.50 (m, 4H), 1.85 - 1.77 (m, 4H).

Un frasco de reacción se cargó con 4-(3-(Pirrolidin-1-ilmetil)azetidin-1-il)-6-(tri-n-butilestanil)benzo[c][1,2,5]oxadiazol (0.41 g, 0.75 mmol), 4-bromotiazol (0.10 mL, 1.13 mmol), Pd(PPha)4 (0.043 g, 0.037 mmol), Cul (0.014 g, 0.076 mmol), y CsF (0.28 g, 1.86 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó DMF (4 mL). La reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: hexano: EtOAc 5: 0: 75: 15 a TEA: MeOH: hexano: EtOAc 5: 3: 75: 15) para producir 0.11 g (43% de rendimiento) de TRV 1561 como un sólido rojo. 1H RMN (CDCla, 400 MHz) 8 = 8.91 (d, J = 2.01 Hz, 1H), 7.68 (d, J = 2.01 Hz, 1H), 7.59 (s, 1H), 6.44 (s, 1H), 4.48 (t, J = 8.28 Hz, 2H), 4.05 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.09 (septeto, J = 7.00 Hz, 1H), 2.81 (d, J = 7.28 Hz, 2H), 2.59 -2.49 (m, 4H), 1.85 - 1.75 (m, 4H).

Se agregaron 6-bromo-4-fluorobenzo[c][1,2,5]oxadiazol (0.427 g, 2.0 mmol) y ácido 5-formiltiofeno-2-borónico (0.340 g, 2.2 mmol) a un tubo de vidrio y el tubo se evacuó y se purgó con argón (3x). Se agregaron DME (2.8 mL) y Na2CO3 (3.0 mL, 6 mmol, solución acuosa 2 N) seguidos por Pd(PPh3)4 (0.115 g, 0.1 mmol). El tubo se selló y luego se calentó a 80°C durante la noche. La reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar 5-(7-fluorobenzo[c][1,2,5] oxadiazol-5-il)tiofeno-2-carbaldehído que se purificó utilizando cromatografía de columna y se utilizó en la siguiente etapa.
Se disolvió 5-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiofeno-2-carbaldehído (0.2 g) en THF (5 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.19 mL) y luego se agregó el catalizador TBAF (0.1 mL, solución 1.0 M). Después de 30 minutos la reacción se retiró del baño de hielo y se dejó calentar a temperatura ambiente. Una vez se completó mediante TLC, se volvió a enfriar a 0°C y se agregó HCl 2 N (ac.), se agitó durante 40 minutos y luego se basificó con NaOH 2N. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua (2x), solución salina, se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (hexano: acetato de etilo, 80:20) para producir 0.2 mg (80%) de 2,2,2-trifluoro-1-(5-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiofen-2-il)etan-1-ol como sólido amarillo. 1H RMN (400 MHz, CDCh) 87.82(s1H), 7.42(d,J= 4.0 Hz, 1H), 7.39(d,J= 8.0 Hz, 1H), 7.26(d,J= 4.0 Hz, 1H), 5.33-5.38 (m, 1H), 2.88(d,J=8.0 Hz, 1H).
2, 2, 2-trifluoro-1-(5-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiofen-2-il)etan-1-ol (0.172 g, 0.54 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil)morfolina (0.185 g, 0.81 mmol) se disolvieron en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.3 ml, 2.16 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 2,2,2-trifluoro-1-(5-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)tiofen-2-il)etan-1-ol TRV1562.1H RMN (DMSO, 700 MHz) 8 = 7.77 (d, J = 5 Hz, 1H), 7.56 (s, 1H), 7.42 (d, J = 5 Hz, 1H), 7.28 (d, J = 5 Hz, 1H), 6.76 (s, 1H), 5.59-5.55 (m, 1H), 4.38 (m, 2H), 3.96 (m, 2H), 3.54 (m, 4H), 3.08 (m, 1H), 2.64 (d, J=8.0 Hz, 2H), 2.39 (m, 4H)

2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin- 3-ilmetil)morfolina (0.110 g, 0.47 mmol) se disolvieron en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 2,2,2-trifluoro-1-(2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol TRV-1563. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener el compuesto del título (0.105 g, 80%) como sólido color naranja. 1H RMN (DMSO-D6, 400 MHz): 88.70 (d, J=4.0 Hz, 1H), 8.13 (d,J=8.0 Hz, 1H), 7.60 (m, 1H), 7.12 (s, 1H), 7.05 (s, 1H), 5.93 (s, 1H), 5.28 (m, 1H), 4.37 (m, 2H), 3.94 (m, 2H), 3.56 (m, 4H), 3.05 (m, 1H), 2.63 (d, J=8.0 Hz, 2H), 2.37 (m, 4H).

Se disolvieron 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 1-(azetidin-3-ilmetil)pirrolidina (0.102 g, 0.48 mmol) en acetonitrilo (5 ml) a temperatura ambiente, se
agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2 h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 2,2,2-trifluoro-1-(2-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1 -ol TRV1564. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener el compuesto del título (0.103 g, 78%) como sólido color naranja.1H RMN (DMSO-D6, 400 MHz): 88.70 (d, J=4.0 Hz, 1H), 8.13(d,J=8.0 Hz, 1H), 7.60 (m, 1H), 7.12 (s, 1H), 7.04 (s, 1H), 5.93 (s, 1H), 5.27 (m, 1H), 4.37 (m, 2H), 3.94 (m, 2H), 3.02 (m, 1H), 2.72 (d, J=8.0 Hz, 2H), 2.43 (m, 4H), 1.67 (m, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.33 g, 1.25 mmol), 2-bromotiazol-4-carbaldehído (0.19 g, 1.00 mmol), Pd(PPh3)4 (0.058 g, 0.050 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y Na2CO3 ac. (2 mL, 2.0 M, 5.0 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0: 100 a 10: 90) para producir 0.13 g (51% de rendimiento) de 2-(7-fluorobenzo[c][1,2,5] oxadiazol-5-il)tiazol-4-carbaldehído como un sólido incoloro.
A una solución de 2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-carbaldehído (0.13 g, 0.51 mmol) en se agregó THF (5 mL) borohidruro de sodio (0.15 g, 4.07 mmol). La mezcla de reacción se agitó a rt hasta que se completó. La reacción se enfrió en un baño de hielo-agua, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 0: 100 a 20: 80) para producir 0.070 g (53% de rendimiento) de (2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-il)metanol como un sólido incoloro.
A una solución de (2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-il)metanol (0.070 g, 0.27 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)morfolina (0.093 g, 0.41 mmol) seguido por TEA (0.14 mL, 1.03 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 25: 75 a 5: 10: 25: 75) para producir 0.086 g (80% de rendimiento) de TRV 1565 como un sólido rojo. 1H RMN (CDCl3, 400 MHz) 8 = 7.55 (s, 1H), 7.30 (s, 1H), 6.31 (s, 1H), 4.87 (s, 2H), 4.48 (t, J = 8.28 Hz, 2H), 4.07 (dd, J1 = 5.52 Hz, J2 = 8.53 Hz, 2H), 3.74 (t, J = 4.65 Hz, 4H), 3.10 (septeto, J = 6.65 Hz, 1H), 2.72 (d, J = 7.28 Hz, 2H), 2.49 (t, J = 4.39 Hz, 4H), 2.40 (amplio, 1H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.33 g, 1.25 mmol), 4-bromotiazol-2-carbaldehído (0.19 g, 1.00 mmol), cloro(2-diciclohexilfosfino-2',4',6'-triisopropil-1,1'-bifenil)[2-(2'-ammo-1,1'-bifenil)]paladio(II) (0.016 g, 0.020 mmol). El frasco se desgasificó y se rellenó con
nitrógeno. Al frasco se agregó dioxano (3 mL) y K3PO4 ac. (3 mL, 0.68 M, 2.0 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 80°C durante la noche. La mezcla se diluyó con agua, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0: 100 a 10: 90) para producir 0.15 g (59% de rendimiento) de 4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-carbaldehído como un sólido incoloro.
A una solución de 4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-carbaldehído (0.15 g, 0.59 mmol) en se agregó THF (5 mL) borohidruro de sodio (0.067 g, 1.77 mmol). La mezcla de reacción se agitó a rt hasta que se completó. La reacción se enfrió en un baño de hielo-agua, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se disolvió en acetonitrilo (5 mL). A la solución se agregó diclorhidrato de 1 -(azetidin-3-ilmetil) morfolina (0.17 g, 0.74 mmol) seguido por TEA (0.26 mL, 1.87 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 1: 25: 75 a 5: 10: 25: 75) para producir 0.15 g (66% de rendimiento para dos etapas) TRV1566 como un sólido rojo. 1H RMN (CDCla, 400 MHz) 8 = 7.60 (s, 1H), 7.55 (s, 1H), 6.37 (s, 1H), 5.04 (s, 2H), 4.45 (t, J = 8.16 Hz, 2H), 4.07 (dd, J1 = 5.52 Hz, J2 = 8.28 Hz, 2H), 3.74 (t, J = 4.65 Hz, 4H), 3.09 (septeto, J = 6.86 Hz, 1H), 2.78 (amplio, 1H), 2.72 (d, J = 7.53 Hz, 2H), 2.39 (t, J = 4.39 Hz, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.29 g, 1.08 mmol), 1-(4-bromotiazol-2-il)-2,2,2-trifluoroetan-1-ol (0.22 g, 0.83 mmol), Pd(PPha)4 (0.048 g, 0.042 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y Na2CO3 ac. (2 mL, 2.0 M, 5.0 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0: 100 a 15: 85) para producir 0.050 g (19% de rendimiento) de 2,2,2- trifluoro-1-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-il)etan-1-ol como un sólido incoloro.
A una solución de 2,2,2-trifluoro-1-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-il)etan-1-ol (0.050 g, 0.16 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)pirrolidina (0.050 g, 0.24 mmol) seguido por TEA (0.09 mL, 0.62 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 1: 25: 75 a 5: 5: 25: 75) para producir 0.050 g (72% de rendimiento) TRV 1567 como un sólido amarillo. 1H RMN (CDCla, 400 MHz) 8 = 7.73 (s, 1H), 7.58 (s, 1H), 6.29 (s, 1H), 5.38 (q, J = 7.27 Hz, 1H), 4.47 (t, J = 8.28 Hz, 2H), 4.07 - 4.00 (m, 2H), 3.09 (septeto, J = 7.03 Hz, 1H), 2.83 (d, J = 7.28 Hz, 2H), 2.61 - 2.52 (m, 4H), 1.87 - 1.78 (m, 4H).

Se disolvieron 2,2,2-trifluoro-1-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiofen-2-il)etan-1-ol (0.1, 0.31 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil)morfolina (0.108 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.25 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 2,2,2-trifluoro-1-(4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il) tiofen-2-il)etan-1-ol TRV 1568. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener el compuesto del título (0.118 g, 83%) como sólido color naranja. 1H RMN (DMSO-D6, 400 MHz): 88.23 (s, 1H), 7.78 (s, 1H), 7.38 (s, 1H), 7.35 (d, J=8.0 Hz, 1H), 6.36 (s, 1H), 5.50 (m, 1H), 4.37 (m, 2H), 3.95 (m, 2H), 3.57 (m, 4H), 3.06 (m, 1H), 2.64 (d, J=8.0 Hz, 2H), 2.39(m,2H).

Se disolvieron 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil)morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 2,2,2-trifluoro-1-(3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-2-il) etan-1-ol TRV1569. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener el compuesto del título (0.105 g, 80%) como sólido color naranja.1H RMN (DMSO-D6, 400 MHz): 88.72 (d, J=4.0 Hz, 1H), 7.73 (d, J=8.0 Hz, 1H), 7.48 (m, 1H), 6.93 (s, 1H), 5.67 (s, 1H), 5.33 (m, 1H), 4.98 (d, J=8.0 Hz, 1H), 4.43 (m, 2H), 4.02 (m, 2H), 3.72 (m, 4H), 3.10 (m, 1H), 2.73 (d, J=4.0 Hz, 2H), 2.48 (m, 4H).

Se agregaron CuCN (594 mg, 6.6 mmol) y TRV1470 (1.06 g, 3.0 mmol) a NMP seco (8 ml). El frasco se purgó con nitrógeno durante varias veces. La mezcla se calentó a 150°C y se agitó durante 10 horas. Después de que se
verificó la finalización mediante TLC, la reacción se detuvo con hidróxido de amoniaco acuoso (5 ml), y se agregó EtOAc (50 ml) a la solución. La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carbonitrilo TRV1570 (708 mg, 79%) como un sólido rojo. 1H RMN (400 MHz, CDCls): 2.44-2.49 (m, 4H), 2.72 (d, J=7.6, 2H), 3.08-3.15 (m, 1H), 3.69-3.76 (m, 4H), 4.05-4.11 (m, 2H), 4.45-4.49 (m, 2H), 5.83 (s, 1H), 7.40 (s, 1H).

Se agregó en forma de gotas n-BuLi (2.22 ml, 2.5 M, 5.56 mmol) a una solución de TRV1470 (1.51 mg, 4.28 mmol) en THF (20 ml) a -78°C bajo atmósfera de nitrógeno. Después la mezcla se agitó durante 10 minutos, DMF (0.46 ml, 6.0 mmol) se agregó a una solución, y la mezcla se agitó durante 1 h a -78°C. La reacción se detuvo mediante MeOH (1 ml), y se agregó EtOAc (50 ml) a la solución. La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carbaldehído (980 mg, 76%) como sólido rojo.
Se agregó NaBH4 (370 mg, 9.73 mmol) a una solución de 7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5] oxadiazol-5-carbaldehído (980 mg, 3.24 mmol) en MeOH (10 ml) a temperatura ambiente. Después la mezcla se agitó durante 1 hora, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 10:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir (7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)metanol TRV1571 (560 mg, 56%) como un sólido amarillo. 1H RMN (400 MHz, CDCla): 1.94 (s, br, 1H), 2.46-2.48 (m, 4H), 2.70 (d, J=7.6, 2H), 3.03-3.10 (m, 1H), 3.72-3.74 (m, 4H), 3.95-3.98 (m, 2H), 4.39 (t, J=8.2, 2H), 4.68 (s, 2H), 5.81 (s, 1H), 7.01 (s, 1H).
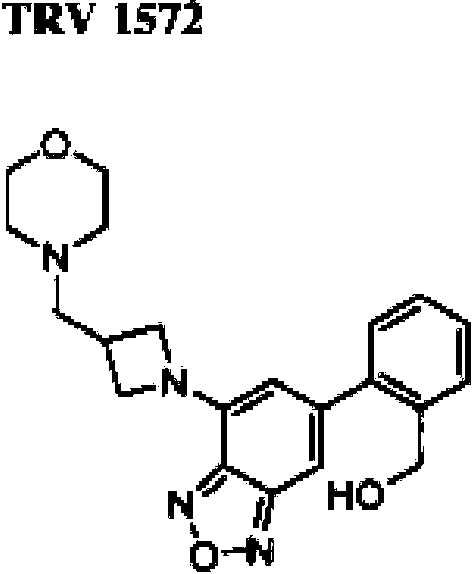
Un frasco de reacción se cargó con 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (1.20 g, 3.40 mmol), ácido (2-formilfenil)borónico (0.76 g, 5.07 mmol), Pd(PPh3)4 (0.20 g, 0.17 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (15 mL) y Na2CO3 ac. (6 mL, 2.0 M, 12.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 100°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 5 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 15: 75 a 5: 2: 15: 75) para producir 0.93 g (73% de rendimiento) de 2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzaldehído como un semisólido amarillo.
Una solución de 2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzaldehído (0.20 g, 0.53 mmol) en THF (5 mL) se enfrió en un baño de hielo-agua. A la solución enfriada se agregó en forma de porción borohidruro de sodio (0.060 g, 1.60 mmol). La mezcla de reacción se agitó hasta que se completó, luego se detuvo con HCl ac. 1 N, se basificó con NaOH 1N, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 25: 75 a 5: 5: 25: 75) para producir 0.12 g (60% de rendimiento) de el TRV1572 como aceite rojo. 1H RMN (CDCla, 400 MHz) 8 = 7.59 (d, J = 7.53 Hz, 1H), 7.45 (dt, J = 1.25 Hz, J2 = 7.53 Hz, 1H), 7.39 (dt, J1 = 1.25 Hz, J2 = 7.53 Hz, 1H), 7.33 (dd, J1 = 1.51 Hz, J2 = 7.53 Hz, 1H), 6.97 (s, 1H), 5.86 (s, 1H), 4.68 (d, J = 4.27 Hz, 2H), 4.41 (t, J = 8.28 Hz, 2H), 3.99 (dd, J1 = 5.77 Hz, J2 = 8.53 Hz, 2H), 3.72 (t, J = 4.65 Hz, 4H), 3.07 (septeto, J = 6.59 Hz, 1H), 2.72 (d, J = 7.53 Hz, 2H), 2.47 (t, J = 4.39 Hz, 4H), 1.67 (amplio, 1H).

Se sellaron TRV1470 (0.250 g, 0.7 mmol) y ácido 3,4-dimetoxibencenoborónico (0.155 g, 0.85 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego Pd(PPh3)4 (0.041 g, 0.035 mmol) se agregó todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante la noche. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (3% de MeOH/DCM) para producir 0.238 g (82% de rendimiento) de 6-(3,4-dimetoxifenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1573.. 1H RMN (CDCfj, 500 MHz) 8 = 7.22 (dd, J = 4.0 Hz, 1H), 7.14 (d, J = 2.0 Hz, 1H), 7.12 (s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.06 (s, 1H), 4.47 (t, J = 8.0 Hz, 2H), 4.07 (m, 2H), 3.98 (s, 3H), 3.96 (s, 3H), 3.74 (m, 4H), 3.10-3.06 (m, 1H), 2.74 (d,J=8.0 Hz, 2H), 2.49 (m, 4H).

Un matraz de fondo redondo se cargó con TRV1470 (250 mg, 0.7 mmol), ácido (4-cianofenil) borónico (125 mg, 0.85 mmol), y Pd(PPh3)4 (42 mg, 0.035 mmol). Después se desgasificó, dioxano (5 mL) y carbonato de sodio acuoso (1.5 mL, 2M) se agregó. La mezcla de reacción se calentó a 80°C durante 3 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó sobre sistema de cromatografía de columna ISCO (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 219 mg (83% de rendimiento) de 4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzonitrilo TRV1574 como un sólido rojo. 1H RMN (400 MHz, CDCfj): 8 7.78(d,J=8.0 Hz, 2H), 7.73(d,J=8.0 Hz, 2H), 7.16 (s, 1H), 5.97 (s, 1H), 4.49(t,J=8.0 Hz, 2H), 4.07(t,J=8.0 Hz, 2H), 3.74 (m, 4H), 3.15 (m, 1H), 2.74 (d, J=8.0 Hz, 2H), 2.48 (m, 4H).

El TRV-1470 (0.250 g, 0.7 mmol) y ácido (3,5-diclorofenil) borónico (0.162 g, 0.85 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.041 g, 0.035 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante 3 h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (2% de MeOH/DCM) para producir 0.255 g (86% de rendimiento) de 6-(3,5-diclorofenN)-4-(3-(morfoNnometN)azetidin-1-il)benzo[c][1,2, 5]oxadiazol TRV1575. 1H RMN (CDCla, 400 MHz) 8 = 7.48 (m, 2H), 7.42 (m, 1H), 7.11 (s, 1H), 5.99 (s, 1H), 4.49 (t, J = 8.0 Hz, 2H), 4.06-4.03 (m, 2H),3.74 (m, 4H), 3.14-3.08 (m, 1H), 2.74 (d, J = 8.0 Hz, 2H), 2.49 (m, 4H).

Se disolvieron 3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)benzamida (0.102 g,0.4 mmol) y sal de clorhidrato de 4 (azetidin-3-ilmetil)morfolina (0.137g, 0.6 mmol) en NMP (5 ml) a temperatura ambiente, trietilamina (0.22 ml, 1.6 mmol) se agregó y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 3-(7-(3-(morfolinometil)zetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il) benzamida TRV1576. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener el compuesto del título (0.128 g, 80%) como sólido color naranja. 1H RMN (DMSO-D6, 400 MHz): 88.22 (s, 1H), 8.16 (s, 1H), 7.94 (s, 1H), 7.92 (s, 1H), 7.59(t, J=8.0 Hz, 1H),7.51 (s, 1H), 7.41 (s, 1H), 6.31 (s, 1H), 4.40(t, J=8.0 Hz, 2H), 3.98 (m, 2H), 3.58 (m, 4H), 3.09-3.02 (m, 1H), 2.63 (d, J=4.0 Hz, 2H), 2.38(m,2H).

Un matraz de fondo redondo se cargó con TRV-1470 (250 mg, 0.7 mmol), ácido (3-cianofenil) borónico (125 mg, 0.85 mmol), y se agregó Pd(PPh3)4 (42 mg, 0.035 mmol). Después se desgasificó, dioxano (5 mL) y carbonato de sodio acuoso (1.5 mL, 2M). La mezcla de reacción se calentó a 80°C durante 3 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó sobre sistema de cromatografía de columna ISCO (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 250 mg (94% de rendimiento) de 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5] oxadiazol-5-il)benzonitrilo TRV1577 como un sólido rojo. 1H RMN (400 MHz, CDCla): 87.90 (s, 1H), 7.87(d,J=8.0 Hz, 1H), 7.73(d,J=8.0 Hz, 1H), 7.61(t,J=8.0 Hz, 1H), 7.13 (s, 1H), 5.95 (s, 1H), 4.49(t,J=8.0 Hz, 2H), 4.07(t,J=8.0 Hz, 2H), 3.74 (m, 4H), 3.12 (m, 1H), 2.74 (d, J=4.0 Hz, 2H), 2.50 (m, 4H),

El TRV1470 (0.250 g, 0.7 mmol) y ácido (3-cloro-4-metoxifenil) borónico (0.158 g, 0.85 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.041 g, 0.035 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante 3h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (2% de MeOH/DCM) para producir 0.225 g (78% de rendimiento) de 6-(3- cloro-4-metoxifenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1578. 1H RMN (CDCh, 400 MHz) 8 = 7.65 (d, J=4.0 Hz, 1H), 7.52-7.49(dd,J=4.0 Hz, 1H), 7.09 (s, 1H), 7.03 (d, J=8 Hz, 1H), 5.99 (s, 1H), 4.47 (t, J = 8.0 Hz, 2H), 4.04-4.01 (m, 2H), 3.97(s,3H), 3.74 (m, 4H), 3.13-3.06 (m, 1H), 2.74 (d, J = 8.0 Hz, 2H), 2.49 (m, 4H).
Se disolvieron 4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)benzamida (0.102 g, 0.4 mmol) y sal de clorhidrato de 1-(azetidin-3-il metil)pirrolidina (0.128 g, 0.6 mmol) en NMP (5 ml) a temperatura ambiente, se agregó trietilamina (0.22 ml, 1.6 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar 4-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzamida TRV1579. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener el compuesto del título (0. 110 g, 73%) como sólido color naranja. 1H RMN (DMSO-D6, 400 MHz): 68.08(bs, 1H), 7.99 (d, J=8.0 Hz, 2H), 7.88 (d,J=8.0 Hz, 2H), 7.46(bs, 1H), 7.40 (s, 1H), 6.30 (s, 1H), 4.41 (t,J=8.0 Hz, 2H), 4.0 (t,J=8.0 Hz, 2H), 3.04-2.99 (m, 1H), 2.80 (m, 2H), 2.40 (m, 2H), 1.69 (m, 4H).

Un frasco de reacción se cargó con 4-(3-(Pirrolidin-1-ilmetil)azetidin-1-il)-6-(tri-n-butilestanil)benzo[c][1,2,5]oxadiazol (0.25 g, 0.74 mmol), 2-bromopiridina (0.90 mmol), Pd(PPh3)4 (35 mg, 0.030 mmol), Cul (11.4 mg, 0.06 mmol), y Cs2COa (182 mg, 1.20 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó NMP (4 mL). La reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea (sistema Combi-Flash RF200) para conseguir 6-(Piridin-2-il)-4-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1580. 1H RMN (CDCh, 400 MHz) 6 = 8.73 (d, J = 4.52 Hz, 1H), 7.83 - 7.78 (m, 2H), 7.52 (s, 1H), 7.35 - 7.30 (m, 1H), 6.65 (s, 1H), 4.50 (t, J = 8.28 Hz, 2H), 4.07 (dd, J1 = 5.90 Hz, J2 = 8.41 Hz, 2H), 3.09 (septeto, J = 6.90 Hz, 1H), 2.81 (d, J = 7.28 Hz, 2H), 2.58 - 2.50 (m, 4H), 1.85 - 1.77 (m, 4H).

El TRV1470 (0.250 g, 0.7 mmol) y ácido (4-fluoro-3-metoxifenil) borónico (0.145 g, 0.85 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego se agregó todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante 3h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (2% de MeOH/DCM) para producir 0.248 g (88% de rendimiento) de 6-(4-fluoro-3-metoxifenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1584. 1H RMN (CDCh, 400 MHz) 6 = 7.20-7.15 (m, 3H), 7.10 (s, 1H), 5.99 (s, 1H), 4.47 (t, J = 8.0 Hz, 2H), 4.04 4.01 (m, 2H), 3.98(s,3H), 3.74 (m, 4H), 3.13-3.07 (m, 1H), 2.74 (d, J = 8.0 Hz, 2H), 2.48 (m, 4H).

El TRV1470 (0.250 g, 0.7 mmol) y ácido (3-fluoro-4-metoxifenil) borónico (0.145 g, 0.85 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.041 g, 0.035 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante 3h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (2% de MeOH/DCM) para producir 0.260 g (92% de rendimiento) de 6-(3-fluoro-4-metoxifenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1585. 1H RMN (CDCla, 400 MHz) 8 = 7.38-7.36 (m, 2H), 7.10 (s, 1H), 7.07-7.03(t, J=8 Hz, 1H), 5.99 (s, 1H), 4.46 (t, J = 8.0 Hz, 2H), 4.04-4.02 (m, 2H), 3.96(s,3H), 3.75 (m, 4H), 3.13-3.06 (m, 1H), 2.73 (d, J = 8.0 Hz, 2H), 2.49 (m, 4H).

Un matraz de fondo redondo se cargó con TRV1470 (250 mg, 0.7 mmol), ácido (2-cianofenil) borónico (125 mg, 0.85 mmol), y Pd(PPh3)4 (42 mg, 0.035 mmol). Después se desgasificó, dioxano (5 mL) y carbonato de sodio acuoso (1.5 mL, 2M) se agregó. La mezcla de reacción se calentó a 80°C durante 6 h. Después de que se verificó la finalización mediante TLC, se agregaron 10 mL de agua, y la mezcla de reacción se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó sobre sistema de cromatografía de columna ISCO (2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 150 mg (57% de rendimiento) de 2-(7-(3-(morfolinometil)azetidin-l-il)benzo[c][1,2,5]oxadiazol-5-il)benzonitrilo TRV1586 como un sólido rojo. 1H RMN (400 MHz, CDCla): 87.84(t, J=8.0 Hz, 1H), 7.71(t, J=8.0 Hz, 1H), 7.49-7.59 (m, 2H), 7.10 (s, 1H), 5.96 (s, 1H), 4.49(t,J=8.0 Hz, 2H), 4.07(t,J=8.0 Hz, 2H), 3.74 (m, 4H), 3.12 (m, 1H), 2.74 (d, J=8.0 Hz, 2H), 2.48 (m, 4H).

4-(3-(pirrolidin-1-ilmetil)azetidin-1 -il)-6-(tributilestanil)benzo[c][1,2,5]oxadiazol (0.4 g, 0.73 mmol), 1-(2-bromopiridin-4-il)-2,2,2-trifluoroetan-1-ol (0.230 g, 0.9 mmol), CsF (0.45 g, 2.19 mmol), CuI (14 mg, 0.073 mmol), Pd(PPh3)4, (0.042 g, 0.0365 mmol) se cargó en tubo de vidrio y se selló, luego se desgasifico y se enjuagó con N2. Se agregó DMF (10 ml) y la mezcla de reacción se calentó a 45°C durante la noche. Después de la finalización de la reacción mediante TLC, la reacción se enfrío y luego se diluyó con EtOAc y agua. La capa orgánica se lavó con agua (3x), solución salina, se secó (MgSO4), se filtró y se concentró para dar el producto crudo que se purificó utilizando cromatografía de columna para dar 2,2,2-trifluoro-1-(2-(7-(3-(pirrolidin-1 -ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-4-il)etan-1 -ol TRV1587 (0.08 g, 26%). 1H RMN (400 MHz, CDCl3): 88.74(d,J=4.0 Hz, 1H), 7.86 (s, 1H), 7.47 (s, 1H), 7.43(d,J=4.0 Hz, 1H), 6.39 (s, 1H), 5.09 (m, 1H), 4.42 (m, 2H), 3.99 (m, 2H), 3.05 (m, 1H), 2.83(d,J=8.0 Hz, 2H), 2.57 (m, 4H), 1.84 (m, 4H).

El TRV1470 (0.250 g, 0.7 mmol) y ácido (3,5-difluorofenil) borónico (0.135 g, 0.85 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO3 2M (1.2 mL, solución 2.0 M ac.) junto con Dioxano (5 mL). La solución se desgasificó durante 10 minutos y luego se agregó Pd(PPh3)4 (0.041 g, 0.035 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 80°C durante 3h. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía sobre ISCO (2% de MeOH/DCM) para producir 0.240 g (88% de rendimiento) de 6-(3,5-difluorofenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1588. 1H RMN (CDCl3, 400 MHz) 8 = 7.15-7.13 (m, 3H), 6.89(t, J=8.0 Hz, 1H), 5.94 (s, 1H), 4.48 (t, J = 8.0 Hz, 2H), 4.06-4.02 (m, 2H), 3.74 (m, 4H), 3.14-3.08 (m, 1H), 2.74 (d, J = 8.0 Hz, 2H), 2.49 (m, 4H).

El TRV-1470 (0.5571 g, 1.58 mmol) y ácido 4-formilfenilborónico (0.2606 g, 1.74 mmol) se pesaron en un tubo. El tubo se evacuó y se purgó con argón (3x). Se agregó DME (3.5 mL) y Na2CO3 acuoso (2.4 mL, 2N solución ac.) al tubo. El tubo se desgasificó durante 5 minutos al burbujear gas argón a través de la solución. Se agregó Pd(PPh3)4 (0.0924 g, 0.08 mmol) todo de una vez, el tubo se selló y calentó a 95°C durante 4 horas. La reacción luego se enfrió a temperatura ambiente y se diluyó con DCM y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con DCM (2x). Las capas orgánicas combinadas se lavaron con agua, se secaron (MgSO4), se filtraron y se concentraron para dar un sólido marrón crudo. Este material crudo se purificó a través de cromatografía instantánea (60% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.5709 g (95% de rendimiento) de aldehído 3 como un sólido naranja. Se disolvió aldehído 3 (0.2852 g, 0.75 mmol) en una mezcla de MeOH (12.5 mL) y THF (5 mL) y luego se enfrió en un baño de hielo. Luego se agrego NaBH4 (0.0567 g, 1.5 mmol) todo de una vez, y la mezcla se agitó durante la noche mientras que se calienta a temperatura ambiente. La reacción luego se volvió a enfriar a 0°C y se detuvo con cloruro de amonio saturado. La mezcla acuosa luego se hizo básica con NaOH 2N (ac.) antes de extraer con DCM. Los extractos orgánicos combinados se secaron (MgSO4), se filtraron y se concentraron para dar el alcohol bencílico crudo. Este material se purificó a través de cromatografía instantánea (5% de MeOH/DCM) para producir 0.1551 g (54% de rendimiento) de TRV1592 como un sólido naranja. 1H RMN (500 MHz, DMSO) 8 = 7.74 (d, J = 8 Hz, 2H), 7.42 (d, J = 8 Hz, 2H), 7.30 (s, 1H), 6.27 (s, 1H), 5.24 (t, J = 5.5 Hz, 1H), 4.55 (d, J = 5.5 Hz, 2H), 4.37 (t, J = 8.5 Hz, 2H), 3.97-3.94 (m, 2H), 3.57 (t, J = 4.5 Hz, 4H), 3.09-3.01 (m, 1H), 2.63 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.40 g, 1.50 mmol), (2-bromotiazol-5-il)metanol (0.23 g, 1.19 mmol), Pd(PPh3)4 (0.068 g, 0.059 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (4 mL) y Na2CO3 ac. (2 mL, 2.0 M, 5.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 100°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0: 100 a 15: 85) para producir 0.20 g (68% de rendimiento) de (2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)metanol como un sólido incoloro.
A una solución de (2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)metanol (0.20 g, 0.80 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)pirrolidina (0.28 g, 1.20 mmol) seguido por TEA (0.42 mL, 3.00 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 25: 75 a 5: 10: 25: 75) para producir 0.050 g (72% de rendimiento) de TRV1594 como un sólido naranja. 1H RMN (DMSO, 400 MHz) 8 = 7.82 (s, 1H), 7.59 (s, 1H), 6.51 (s, 1H), 5.72 (t, J = 5.65 Hz, 1H), 4.74 (d, J = 5.52 Hz, 2H),
4.39 (t, J = 8.28 Hz, 2H), 4.00 - 3.91 (m, 2H), 3.57 (t, J = 74.52 Hz, 4H), 3.06 (septeto, J = 6.71 Hz, 1H), 2.63 (d, J = 7.53 Hz, 2H), 2.42 - 2.34 (m, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametiM,3,2-dioxaborolan-2-N)benzo[c][1,2,5]oxadiazol (0.16 g, 0.61 mmol), 1-(2-bromotiazol-5-il)-2,2,2-trifluoroetan-1-ol (0.16 g, 0.61 mmol), Pd(PPh3)4 (35 mg, 0.030 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (4 mL) y Na2CO3 ac. (2 mL, 2.0 M, 4.0 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0: 100 a 15: 85) para producir 0.082 g (42% de rendimiento) de 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)etan-1-ol como un sólido incoloro.
A una solución de 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)etan-1-ol (0.082 g, 0.26 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)pirrolidina (0.082 g, 0.39 mmol) seguido por TEA (0.14 mL, 1.0 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 1: 25: 75 a 5: 10: 25: 75) para producir 0.10 g (88% de rendimiento) de TRV1597 como un sólido amarillo. 1H RMN (DMSO, 400 MHz) 8 = 8.06 (s, 1H), 7.67 (s, 1H), 7.54 (d, J = 5.77 Hz, 1H), 6.51 (s, 1H), 5.74 (penteto, J = 6.40 Hz, 1H), 4.41 (t, J = 8.28 Hz, 2H), 3.98 (dd, J1 = 5.77 Hz, J2 = 8.28 Hz, 2H), 3.00 (septeto, J = 6.71 Hz, 1H), 2.72 (d, J = 7.53 Hz, 2H), 2.48 - 2.38 (m, 4H), 1.74 -1.61 (m, 4H).

Un frasco de reacción se cargó con 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (1.20 g, 3.40 mmol), ácido (3-formilfenil)borónico (0.63 g, 4.25 mmol), Pd(PPh3)4 (0.20 g, 0.17 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (10 mL) y Na2CO3 ac. (6 mL, 2.0 M, 12.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 100°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 5 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 15: 75 a 5: 2: 15: 75) para producir 1.20 g (93% de rendimiento) de 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzaldehído como aceite rojo.
Una solución de 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzaldehído (0.26 g, 0.69 mmol) en THF (5 mL) se enfrió en un baño de hielo-agua. A la solución enfriada se agregó borohidruro de sodio (0.040 g se disolvió en 2 mL de NaOH 1N ac., 1.06 mmol) lentamente. La mezcla de reacción se agitó hasta que se completó, luego se detuvo con HCl ac. 1 N, se basificó con NaOH 1N, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 5: 0: 25: 75 a 5: 5: 25: 75) para producir 0.086
g (80% de rendimiento) de TRV1598 como un sólido marrón. 1H RMN (CDCI3, 400 MHz) 5 = 7.64 (s, 1H), 7.56 (d, J = 7.53 Hz, 1H), 7.47 (t, J = 7.53 Hz, 1H), 7.43 (d, J = 7.53 Hz, 1H), 7.16 (s, 1H), 6.07 (s, 1H), 4.80 (s, 2H), 4.44 (t, J = 8.16 Hz, 2H), 4.02 (dd, J1 = 5.52 Hz, J2 = 8.53 Hz, 2H), 3.73 (t, J = 4.65 Hz, 4H), 3.09 (septeto, J = 6.59 Hz, 1H), 2.72 (d, J = 7.53 Hz, 2H), 2.47 (t, J = 4.27 Hz, 4H), 1.90 (amplio, 1H).

Se agregó en forma de gotas n-BuLi (0.64 ml, 2.5 M, 1.6 mmol) a una solución de TRV1470 (435 mg, 1.23 mmol) en THF (10 ml) a -78°C bajo atmósfera de nitrógeno. Después la mezcla se agitó durante 10 minutos, yoduro de metilo (0.153 ml, 2.46 mmol) se agregó a una solución, y la mezcla se agitó durante 1 h a -78°C. La reacción se detuvo mediante MeOH (1 ml), y EtOAc (50 ml) se agregó a la solución. La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 6-metil-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1599
(205 mg, 57%) como un sólido rojo. 1H RMN (400 MHz, CDCla): 2.35 (s, 1H), 2.46-2.49 (m, 4H), 2.70 (d, J=7.6, 2H), 3.02-3.09 (m, 1H), 3.72-3.74 (m, 4H), 3.94 (d-d, J=5.6, J=8.4, 2H), 4.37 (t, J=8.2, 2H), 5.68 (s, 1H), 6.79 (s, 1H).

Se agregó NaH (52 mg, 1.32 mmol) a una solución de TRV1571 (200 mg, 0.658 mmol) en THF (10 ml) a 0°C. Luego se agregó yoduro de metilo (0.122 ml, 3.0 mmol) a una solución, y la mezcla se agitó durante 5 h de 0°C a temperatura ambiente. Después de la finalización, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtoAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 6-(metoximetil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1600 (170 mg, 81%) como un sólido amarillo. 1H RMN (400 MHz, CDCla): 2.46-2.48 (m, 4H), 2.70 (d, J=7.6, 2H), 3.03-3.09 (m, 1H), 3.44 (s, 3H), 3.72-3.74 (m, 4H), 3.97 (d-d, J=5.6, J=8.3, 2H), 4.38-4.42 (m, 4H), 5.83 (s, 1H), 6.97 (s, 1H).

(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)oxazol-4-il)metanol (0.150 g, 0.64 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.218 g, 0.95 mmol) se disolvieron en acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.3 ml, 1.92 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar (2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5] oxadiazol-5-il)oxazol-4-il)metanol TRV1606. La purificación se realizó sobre cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener TRV 1606 (0.210 g, 86%) como sólido color naranja. 1H RMN (CDCla, 400 MHz): 87.72 (s, 1H), 7.67 (s, 1H), 6.52 (s, 1H), 4.71 (s, 2H), 4.48(t,J=8.0 Hz, 2H), 4.06(t, J=4.0,8.0 Hz, 2H), 3.75 (m, 4H), 3.11 (m, 1H), 2.73 (d, J=8.0 Hz, 2H), 2.48 (m, 4H)

Se disolvieron (2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)oxazol-5-il)metanol (0.236 g, 1.0 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil)morfolina (0.34 3 g, 1.5 mmol) en acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.4 ml, 3.0 mmol) y la mezcla se calentó a 80°C durante 6 h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar el producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener (2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)oxazol-5-il)metanol TRV1607 (0.260 g, 70%) como sólido color naranja. 1H RMN (CDCh, 400 MHz): 87.65 (s, 1H), 7.18 (s, 1H), 6.48 (s, 1H), 4.79 (s, 2H), 4.47 (t,J=8.0 Hz, 2H), 4.05 (t, J=8.0 Hz, 2H), 3.75 (m, 4H), 3.12 (m, 1H), 2.73 (d, J=8.0 Hz, 2H), 2.48 (m, 4H).

Se disolvió 2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2, 5]oxadiazol-5-il) oxazol-4-carbaldehído (0.08 g, 0.217 mmol) en THF (5 mL) y se enfrió en un baño de hielo. Se agregó CF3TMS (0.062 g, 0.434 mmol) y luego se agregó el catalizador TBAF (0.05 mL, solución 1.0 M en THF). Después de 30 minutos la reacción se retiró del baño de hielo y se dejó calentar a temperatura ambiente. Una vez se completó mediante TLC, se volvió a enfriar a 0°C y se agregó
HCl 2 N (ac.), se agitó durante 40 minutos y luego se basificó con NaOH 2N. Esta mezcla se extrajo con EtOAc. Los extractos combinados se lavaron con agua (2x), solución salina, se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (hexano: acetato de etilo: MeOH: Et3N, 5:1:0.3:0.3) para producir 0.07 mg (75%) de 2,2,2-trifluoro-1-(2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)oxazol-4-il)etan-1-ol TRV 1608 como sólido rojo. 1H RMN (CDCla, 400 MHz): 87.85 (s, 1H), 7.69 (s, 1H), 6.50 (s, 1H), 5.16 (m, 1H), 4.51 (t, J=8.0 Hz, 2H), 4.09(t,J=8.0 Hz, 2H), 3.75 (m, 4H), 3.15 (m, 1H), 2.74 (d, J=4.0 Hz, 2H), 2.49 (m, 4H).

Se disolvieron 3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)benzamida(0.102 g, 0.4 mmol) y sal de clorhidrato de 1-(azetidin-3-ilmetil) pirrolidina (0.128 g, 0.6 mmol) en NMP (5 ml) a temperatura ambiente, trietilamina (0.22 ml, 1.6 mmol) se agregó y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (90:10) para obtener 3-(7-(3-(pirrolidin-1-ilmetil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzamida TRV1609 (0.118 g, 78%) como sólido color naranja. 1H RMN (DMSO-D6, 400 MHz): 88.22 (s, 1H), 8.16 (s, 1H), 7.94 (m, 2H), 7.60(t,J=8.0 Hz, 1H), 7.51 (s, 1H), 7.41 (s, 1H), 6.32 (s, 1H), 4.42 (t,J=8.0 Hz, 2H), 4.0(t,J=8.0 Hz, 2H), 3.02 (m, 1H), 2.73 (d, J=8.0 Hz, 2H), 2.45 (m, 4H), 1.68 (m, 4H).
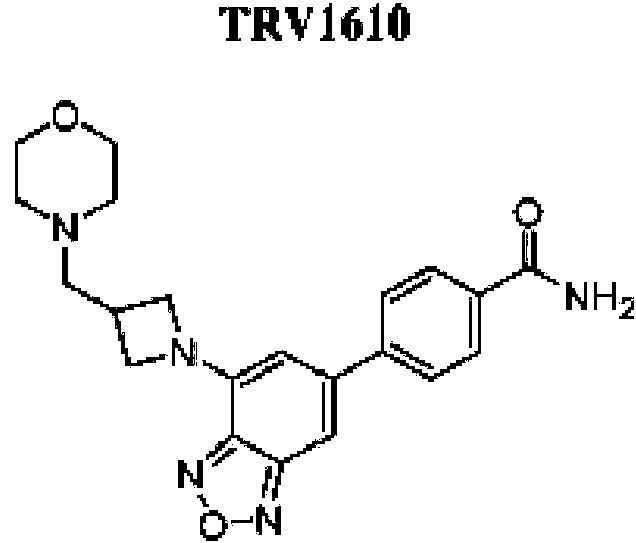
4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)benzamida (0.102 g, 0.4 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.137 g, 0.6 mmol) se disolvieron en NMP (5 ml) a temperatura ambiente, trietilamina (0.22 ml, 1.6 mmol) se agregó y la mezcla se calentó a 80°C durante 2 h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando sistema de solventes diclorometano: metanol (95:5) para obtener 4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)benzamida TRV1610 (0.068 g, 51%) como sólido amarillo.
1H RMN (DMSO-D6, 400 MHz): 88.08 (s, 1H), 7.99 (s, 1H), 7.97 (s, 1H), 7.89 (s, 1H), 7.87 (s, 1H), 7.46 (s, 1H), 7.41 (s, 1H), 6.31 (s, 1H), 4.41 (t, J=8.0 Hz, 2H), 3.99 (m, 2H), 3.58 (m, 4H), 308 (m, 1H), 2.64 (d, J=8.0 Hz, 2H), 2.39 (m, 4H).

Se agregó en forma de gotas H2O2 (30% en H2O, 2.65 ml) a una solución de 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (700 mg, 2.65 mmol) y NaOH (1 N, 2.65 ml) en EtOH (10 ml) a temperatura ambiente. Después la mezcla se agitó durante 20 minutos, la reacción se detuvo con HCl (1 N) y el pH se ajustó a 2. La mezcla de reacción se concentró, y se agregó 20 ml de EtOAc al residuo. La mezcla luego se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (1:10, EtOAc/Hexano a 1:5 EtOAc/Hexano) para producir 7-fluorobenzo[c][1,2,5]oxadiazol-5-ol (330 mg, 81%) como un sólido amarillo pálido.
Se agregó MeI (0.25 ml, 4 mmol) a una solución en suspensión de 7-fluorobenzo[c][1,2,5]oxadiazol-5-ol (154 mg, 1.0 mmol) y K2CO3 (276 mg, 2.0 mmol) en MeCN (10 ml) a temperatura ambiente. Después la mezcla se agitó durante la noche, la reacción se concentró hasta secado. Al residuo se agregó otra tanda de K2CO3 (518 mg, 3.75 mmol), sal de 2HCl de 4-(azetidin-3-ilmetil)morfolina (344 mg, 1.5 mmol) y 10 ml de MeCN. La mezcla se calentó a 55°C durante 6 horas. Después la mezcla se enfrió a temperatura ambiente, EtOAc (30 ml) se agregó a la solución. La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 3:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir 6-metoxi-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1611 (210 mg, 69%) como un sólido amarillo.
1H RMN (400 MHz, CDCls): 2.44-2.46 (m, 4H), 2.68 (d, J=7.5, 2H), 2.98-3.06 (m, 1H), 3.69-3.72 (m, 4H), 3.84 (s, 3H), 3.89 (d-d, J=5.5, J=8.3, 2H), 4.33 (t, J=8.2, 2H), 5.51 (d, J=1.3, 1H), 6.20 (d, J=1.8, 1H).
y 5-metoxi-4-metil-7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1613 (45 mg, 14%) como un sólido rojo. 1H RMN (400 MHz, CDCla): 2.32 (s, 3H), 2.46-2.48 (m, 4H), 2.70 (d, J=7.5, 2H), 3.03-3.09 (m, 1H), 3.72-3.74 (m, 4H), 3.90 (s, 3H), 3.93 (d-d, J=5.5, J=8.0, 2H), 4.36 (t, J=8.0, 2H), 5.73 (s, 1H).

Se agregó en forma de gotas n-BuLi (1.0 ml, 2.5 M, 2.5 mmol) a una solución de TRV1470 (706 mg, 2.0 mmol) en THF (20 ml) a -78°C bajo atmósfera de nitrógeno. Después la mezcla se agitó durante 10 minutos, se agregó disulfuro de dimetilo (0.233 ml, 2.5 mmol) a una solución, y la mezcla se agitó durante 1 h a -78°C. La reacción se detuvo mediante MeOH (1 ml), y se agregó EtOAc (50 ml) a la solución. La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 6-(metiltio)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5] oxadiazol TRV1612
(550 mg, 86%) as a red sólido marrón. 1H RMN (400 MHz, CDCls): 2.46-2.49 (m, 4H), 2.53 (s, 3H), 2.69 (d, J=7.5, 2H), 3.02-3.09 (m, 1H), 3.72-3.74 (m, 4H), 3.95 (d-d, J=5.5, J=8.4, 2H), 4.38 (t, J=8.2, 2H), 5.66 (s, 1H), 6.61 (s, 1H).

Para detalles experimentales véase [00822] y [00823]

Un frasco de reacción se cargó con 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.44 g, 1.25 mmol), ácido (2-(dimetilcarbamoil)fenil)borónico (0.30 g, 1.55 mmol), Pd(PPh3)4 (0.072 g, 0.063 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (7.5 mL) y Na2CO3 ac. (3 mL, 2.0 M, 6.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: MeOH: EtOAc: Hex 2: 0: 15: 75 a 2: 5: 15: 75) para producir 0.52 g (100% de rendimiento) de TRV1615 como un sólido naranja. 1H RMN (CDCla, 400 MHz) 8 = 7.52 - 7.37 (m, 4H), 7.03 (s, 1H), 6.01 (s, 1H), 4.40 (t, J = 8.16 Hz, 2H), 3.98 (dd, J1 = 5.65 Hz, J2 = 8.41 Hz, 2H), 3.72 (t, J = 4.65 Hz, 4H), 3.06 (septeto, J = 6.65 Hz, 1H), 2.94 (s, 3H), 2.70 (d, J = 7.53 Hz, 2H), 2.65 (s, 3H), 2.46 (t, J = 4.27 Hz, 4H).

Se agruparon TRV1470 (0.4179 g, 1.18 mmol) y ácido (4-(metoxicarbonil) fenil) borónico (0.2772 g, 1.54 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). DME (2.7 mL) y Na2CO3 acuoso (1.8 mL, 2N solución ac.) se agregó al tubo. El tubo se desgasificó durante 5 minutos al burbujear gas argón a través de la solución. se agregó Pd(PPh3)4 (0.069 g, 0.06 mmol) todo de una vez, el tubo se selló y calentó a 95°C durante 16 horas. La reacción luego se enfrió a temperatura ambiente y se diluyó con DCM y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con DCM (2x). Las capas orgánicas combinadas se lavaron con agua, se secaron (MgSO4), se filtraron y se concentraron para dar un sólido marrón crudo. Este material crudo se purificó a través de cromatografía instantánea (60% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.4752 g (99% de rendimiento) éster de
metilo 5. Este material se disolvió en THF (15 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (1.2 mL, solución 3.0 M en Et2O) y la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción luego se volvió a enfriar y se detuvo con cloruro de amonio saturado. Este se extrajo con EtOAc (3x) y las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar el alcohol crudo. El material crudo se purificó a través de cromatografía (60% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.2202 g (46% de rendimiento) de TRV1616. 1H RMN (500 MHz, DMSO) 8 = 7.70 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.29 (s, 1H), 6.26 (s, 1H), 5.06 (s, 1H), 4.37 (t, J = 8.0 Hz, 2H), 3.97-3.94 (m, 2H), 3.57 (t, J = 4.5 Hz, 4H), 3.09-3.01 (m, 1H), 2.62 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H), 1.45 (s, 6H).

Se agruparon TRV1470 (0.4044 g, 1.14 mmol) y (3-(metoxicarbonil) fenil) ácido borónico (0.2664 g, 1.48 mmol) en un tubo. El tubo se evacuó y se purgó con argón (3x). DME (2.6 mL) y Na2CO3 acuoso (1.7 mL, 2N solución ac.) se agregó al tubo. El tubo se desgasificó durante 5 minutos al burbujear gas argón a través de la solución. Pd(PPh3)4 (0.069 g, 0.06 mmol) se agregó todo de una vez, el tubo se selló y calentó a 95°C durante 16 horas. La reacción luego se enfrió a temperatura ambiente y se diluyó con DCM y agua. Las capas se separaron y la capa acuosa se extrajo de nuevo con DCM (2x). Las capas orgánicas combinadas se lavaron con agua, se secaron (MgSO4), se filtraron y se concentraron para dar un sólido marrón crudo. Este material crudo se purificó a través de cromatografía instantánea (70% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.4769 g (99% de rendimiento) éster de metilo. Este material se disolvió en THF (15 mL) y se enfrió a 0°C. Se agregó en forma de gotas MeMgBr (1.2 mL, solución 3.0 M en Et2O) y la reacción se dejó calentar a temperatura ambiente durante la noche. La reacción luego se volvió a enfriar y se detuvo con cloruro de amonio saturado. Este se extrajo con EtOAc (3x) y las capas orgánicas combinadas se lavaron con agua, solución salina, se secaron (MgSO4), se filtraron y se concentraron para dar el alcohol crudo. El material crudo se purificó a través de cromatografía (60% de EtOAc/hexano con 5% de aditivo TEA) para producir 0.2507 g (53% de rendimiento) de TRV1617. 1H RMN (500 MHz, DMSO) 8 = 7.82 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 7.27 (s, 1H), 6.23 (s, 1H), 5.08 (s, 1H), 4.38 (t, J = 8.0 Hz, 2H), 3.97-3.94 (m, 2H), 3.57 (t, J = 4.5 Hz, 4H), 3.10-3.01 (m, 1H), 2.63 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H), 1.48 (s, 6H).

Se disolvieron 2,2,2-trifluoro-1-(5-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-2-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1618 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(5-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-2-il)etan-1-ol TRV1618 (0.115 g, 85%) como sólido color naranja. 1H RMN(DMSO-D6, 400 MHz): 89.0 (s, 1H), 8.32 (dd, J=4.0 Hz, 1H), 7.74 (d, J=8.0 Hz, 1H), 7.47 (m, 1H), 7.15 (d,J=8.0 Hz, 1H), 6.33 (s, 1H), 5.24 (m, 1H), 4.41 (t,J=8.0 Hz, 2H), 3.98 (t,J=8.0 Hz, 2H), 3.57 (m, 4H), 3.10 (m, 1H), 2.64 (d, J=4.0 Hz, 2H), 2.38 (m, 4H).

Se disolvieron 2,2,2-trifluoro-1-(6-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-2-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1569 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(6-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-2-il)etan-1-ol TRV1619 (0.105 g, 80%) como sólido color naranja. 1H RMN(DMSO-Da, 400 MHz): 88.18 (d, J=8.0 Hz, 1H), 8.06(t, J=8.0 Hz, 1H), 7.84 (s, 1H), 7.68(d,J=8.0 Hz, 1H), 7.12(d,J=4.0 Hz, 1H), 6.78 (s, 1H), 5.33 (m, 1H), 4.38(t, J=8.0 Hz, 2H), 3.97(t, J=8.0 Hz, 2H), 3.58 (m, 4H), 3.10 (m, 1H), 2.64 (d, J=4.0 Hz, 2H), 2.38 (m, 4H).

Se disolvieron 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-4-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1569 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-4-il)etan-1-ol TRV1620 (0.110 g, 82%) como sólido color naranja. 1H RMN(DMSO-Da, 400 MHz): 88.78 (d, J=4.0 Hz, 1H), 8.23 (s, 1H), 7.75 (s, 1H), 7.58 (d, J=4.0 Hz, 1H),7.23 (d, J=4.0 Hz, 1H), 6.74 (s, 1H), 5.39 (m, 1H), 4.42(t,J=8.0 Hz, 2H),3.99(t,J=8.0 Hz,2H,), 3.58 (m, 4H), 3.09 (m, 1H), 2.65 (d, J=4.0 Hz, 2H), 2.39 (m, 4H).
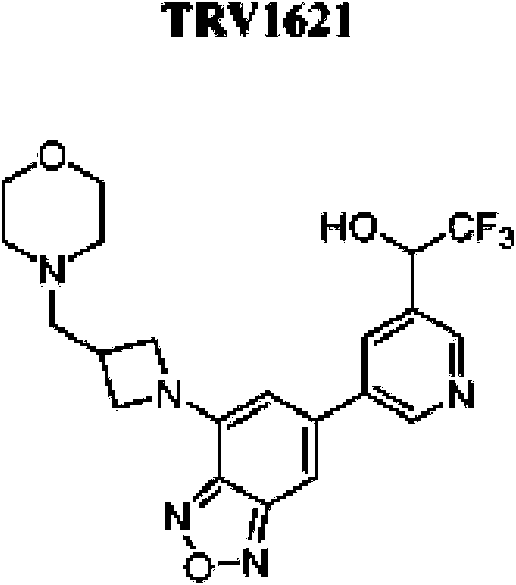
Se disolvieron 2,2,2-trifluoro-1-(5-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se
agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1569 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(5-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)pindin-3-il)etan-1-ol TRV-1621 (0.099g, 75%) como sólido color naranja. 1H RMN(DMSO-Da, 400 MHz): 89.04 (d, J=4.0 Hz, 1H), 8.75 (s, 1H), 8.26 (s, 1H),7.42(s,1H) 7.21(d,J=8.0 Hz,1H),6.27 (s, 1H), 5.43 (m, 1H), 4.98 (d, J=8.0 Hz, 1H), 4.43(t,J=8.0 Hz, 2H),4.01(t,J=8.0 Hz,2H,), 3.57 (m, 4H), 3.11 (m, 1H), 2.64 (d, J=4.0 Hz, 2H), 2.39 (m, 4H).
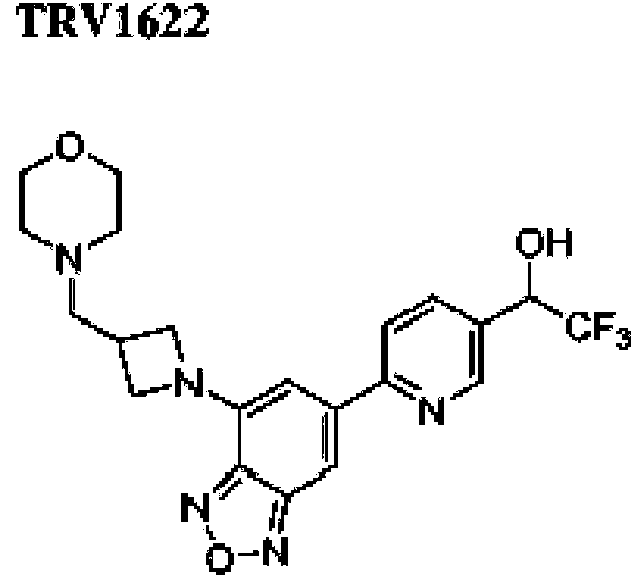
Se disolvieron 2,2,2-trifluoro-1-(6-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-3-il)etan-1-ol (0.1, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1569 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(6-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)pindin-3-il)etan-1-ol TRV1622 (0.095 g, 73%) como sólido color naranja. 1H RMN(DMSO-Da, 400 MHz): 8 8.80 (s, 1H), 8.22(d,J=8.0 Hz, 1H), 8.03(d,J=8.0 Hz, 1H), 7.81 (s, 1H), 7.19 (s, 1H), 6.77 (s, 1H), 5.41 (m, 1H), 4.43 (m, 2H), 4.02 (m, 2H), 3.96 (m, 4H), 3.10 (m, 1H), 2.64 (d, J=4.0 Hz, 2H), 2.38 (m, 4H).
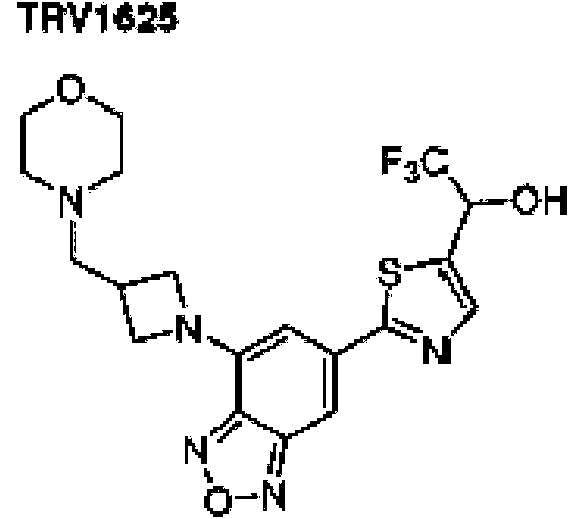
Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.35 g, 1.34 mmol), 1-(2-bromotiazol-5-il)-2,2,2-trifluoroetan-1-ol (0.28 g, 1.07 mmol), Pd(PPh3)4 (0.090 g, 0.080 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (5 mL) y Na2CO3 ac. (3 mL, 2.0 M, 6.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 0: 100 a 20: 80) para producir 0.20 g (60% de rendimiento) de 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)etan-1-ol como un sólido incoloro.
El 2,2,2-Trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-5-il)etan-1-ol (0.20 g, 0.64 mmol) obtenido anteriormente y Dihidrocloruro de 4-(azetidin-3-ilmetil) morfolina (0.23 g, 1.00 mmol) se suspendió en MeCN (5 mL). A la suspensión se agregó TEA (0.35 mL, 2.55 mmol). La solución resultante se calentó a 50 °C durante la noche, se enfrió a temperatura ambiente, se diluyó con acetato de etilo, y se lavó con solución salina. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: d Cm 0: 100 a 5: 95) para producir 0.25 g (86% de rendimiento) de TRV1625 como un sólido naranja. 1H RMN (CDCh, 400 MHz) 8 = 8.06 (s, 1H), 7.67 (s, 1H), 7.55 (d, J = 5.77 Hz, 1H), 6.51 (s, 1H), 5.74 (penteto, J = 6.60 Hz, 1H), 4.40 (t, J = 8.15 Hz, 2H), 3.97 (t, J = 6.90 Hz, 2H), 3.57 (t, J = 4.27 Hz, 4H), 3.07 (septeto, J = 6.65 Hz, 1H), 2.63 (d, J = 7.53 Hz, 2H), 2.44 - 2.33 (m, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.29 g, 1.10 mmol), 2-bromotiazol-4-carbaldehído (0.19 g, 1.00 mmol), XPhos Pd G2 (0.024 g, 0.030 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (3 mL) y K3PO4 ac. (3.3 mL, 0.62 M, 2.05 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 60°C durante 24 h. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc:Hex 0:100 a 20:80) para producir 0.120 g (48% de rendimiento) de 2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-carbaldehído como un sólido incoloro.
2-(7-Fluorobenzo[c][1,2,5] oxadiazol-5-il) tiazol-4-carbaldehído (0.22 g, 0.88 mmol) y trimetil (trifluorometil) silano (0.20 mL, 1.30 mmol) se disolvió en THF anhidro (7.5 mL). La solución se enfrió en un baño de hielo-agua antes se agregó en forma de gotas TBAF (0.080 mL, 1M, 0.080 mmol). La solución oscura resultante se agitó a temperatura de baño de hielo-agua hasta que se completó, y luego se detuvo con HCl ac- 2 N. La mezcla de reacción se calentó a temperatura ambiente, se agitó durante 0.5h, luego se diluyó con agua, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: DCM: Hex 0: 30: 50 a 20: 30: 50) para producir 0.060 g (21% de rendimiento) de 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-il)etan-1-ol como un sólido blanco.
A una solución de 2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-4-il)etan-1-ol (0.060 g, 0.19 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)morfolina (0.060 g, 0.26 mmol) seguido por TEA (0.10 mL, 0.72 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: DCM 0: 100 a 5: 100) para producir 0.065 g (76% de rendimiento) de TRV1626 como un sólido naranja. 1H RMN (DMSO, 400 MHz) 8 = 7.98 (s, 1H), 7.66 (s, 1H), 7.09 (d, J = 6.27 Hz, 1H), 6.49 (s, 1H), 5.40 (penteto, J = 6.90 Hz, 1H), 4.41 (t, J = 8.15 Hz, 2H), 3.98 (dd, J1 = 6.02 Hz, J2 = 8.03 Hz, 2H), 3.57 (t, J = 4.40 Hz, 4H), 3.07 (septeto, J = 6.65 Hz, 1H), 2.64 (d, J = 7.53 Hz, 2H), 2.44 - 2.34 (m, 4H).

Un frasco de reacción se cargó con 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (0.58 g, 2.20 mmol), 4-bromotiazol-2-carbaldehído (0.38 g, 2.00 mmol), XPhos Pd G2 (0.047 g, 0.060 mmol). El frasco se desgasificó y se rellenó con nitrógeno. Al frasco se agregó dioxano (6.5 mL) y K3PO4 ac. (6.5 mL, 0.62 M, 4.00 mmol). El frasco de reacción se volvió a desgasificar, se rellenó con nitrógeno, se selló, y luego se calentó a 60°C durante 24 h. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 0: 100 a 10: 90) para producir 0.36 g (72% de rendimiento) de 4-(7-fluorobenzo[c][1,2, 5]oxadiazol-5-il)tiazol-2-carbaldehído como un sólido incoloro.
4-(7-Fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-carbaldehído (0.36 g, 1.44 mmol) y trimetil(trifluorometil)silano (0.35 mL, 2.28 mmol) se disolvió en THF anhidro (10 mL). La solución se enfrió en un baño de hielo-agua antes se agregó en forma de gotas TBAF (0.15 mL, 1M, 0.15 mmol). La solución oscura resultante se agitó a temperatura de baño de hielo-agua hasta que se completó, y luego se detuvo con HCl ac. 2 N. La mezcla de reacción se calentó a temperatura ambiente, se agitó durante 0.5h, luego se diluyó con agua, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: DCM: Hex 0: 30: 50 a 20: 30: 50) para producir 0.050 g (11% de rendimiento) de 2,2,2-trifluoro-1-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-il)etan-1-ol como un sólido blanco.
A una solución de 2,2,2-trifluoro-1-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)tiazol-2-il)etan-1-ol (0.050 g, 0.16 mmol) en acetonitrilo (5 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)morfolina (0.045 g, 0.20 mmol) seguido por TEA (0.080 mL, 0.58 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: DCM 0: 100 a 5: 100) para producir 0.060 g (84% de rendimiento) de TRV1627 como un sólido naranja. 1H RMN (DMSO, 400 MHz) 8 = 8.57 (s, 1H), 7.94 (d, J = 6.02 Hz, 1H), 7.65 (s, 1H), 6.60 (s, 1H), 5.67 (penteto, J = 6.65 Hz, 1H), 4.38 (t, J = 7.78 Hz, 2H), 3.96 (dd, J1 = 6.27 Hz, J2 = 8.03 Hz, 2H), 3.58 (t, J = 4.15 Hz, 4H), 3.07 (septeto, J = 6.75 Hz, 1H), 2.64 (d, J = 7.53 Hz, 2H), 2.44 - 2.34 (m, 4H).

Se agregaron CuCN (594 mg, 6.6 mmol) y TRV1470 (1.06 g, 3.0 mmol) a NMP seco (8 ml). El frasco se purgó con nitrógeno durante varias veces. La mezcla se calentó a 150°C y se agitó durante 10 horas. Después de que se verificó la finalización mediante TLC, la reacción se detuvo con hidróxido de amoniaco acuoso (5 ml), y se agregó EtOAc (50 ml) a la solución. La mezcla se lavó con NaOH 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 2:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV-1570 (708 mg, 79%) como un sólido rojo.
Se agregaron clorhidrato de hidroxilamina (167 mg, 2.4 mmol) y hidróxido de sodio (96 mg, 2.4 mmol) a una solución de TRV-1570 (598 mg, 2 mmol) en metanol a temperatura ambiente. La mezcla de reacción se agitó durante dos días a temperatura ambiente. El solvente se evaporó y el residuo se utilizó para la siguiente etapa sin purificación.
N,N-Diisopropiletilamina (0.9 mL, 5.2 mol) se agregó a N-hidroxi-7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carboximidamida (664 mg, 2.0 mol) en DMF (15 mL) a temperatura ambiente bajo nitrógeno. La mezcla se enfrió a 0°C y cloruro de oxalilo etilo (0.29 mL, 2.6 mmol) se agregó en forma de gotas. Después de agitar a 0°C durante 10 min, la mezcla se calentó a temperatura ambiente y luego a 50°C y se agitó durante 3 h. La mezcla se diluyó con EtOAc (50 mL) y se lavó con solución salina (3 X 50 mL). La solución se secó (Na2SO4) y se evaporó El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 2:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1,2,4-oxadiazol-5-carboxilato de etilo (397 mg, 48%) como aceite rojo.
Se agregó MeMgBr (0.50 ml, 1.5 mmol) a una solución de 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1,2,4-oxadiazol-5-carboxilato de etilo (200 mg, 0.48 mmol) THF (10 ml) a 0°C. Después la mezcla se agitó 2 h, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 3:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir TRV1628 (130 mg, 67%) como un sólido rojo. 1H RMN (400 MHz, CDCla): 1.71 (s, 6H), 2.47-2.49 (m, 4H), 2.72 (d, J=7.5, 2H), 3.05-3.11 (m, 1H), 3.72-3.75 (m, 4H), 4.04 (d-d, J=5.5, J=8.5, 2H), 4.46 (t, J=8.3, 2H), 6.44 (s, 1H), 7.81 (s, 1H).

Se agregó NaBH4 (46 mg, 1.21 mmol) a una solución de 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1,2,4-oxadiazol-5-carboxilato de etilo (250 mg, 0.604 mmol) en THF (10 ml) a 0°C. Después la mezcla se agitó 2 h, la reacción se detuvo mediante EtOAc/H2O (1:1, 50 ml). La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 3:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir 3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1,2,4-oxadiazol-5-il)metanol TRV1629 (40 mg, 18%) como un sólido rojo. 1H RMN (400 MHz, CDCla): 2.48-2.50 (m, 4H), 2.73 (d, J=7.3, 2H), 3.06-3.11 (m, 1H), 3.73-3.75 (m, 4H), 4.03 (d-d, J=5.6, J=8.4, 2H), 4.46 (t, J=8.3, 2H), 4.99 (s, 2H), 6.44 (s, 1H), 7.79 (s, 1H).

Se disolvieron 2,2,2-trifluoro-1-(3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)piridin-2-il)etan-1-ol (0.1g, 0.32 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.110 g, 0.47 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.18 ml, 1.28 mmol) y la mezcla se calentó a 80°C durante 2h. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(3-(7-(3-(morfolinometil)azetidin-l-il)benzo[c][1,2,5]oxadiazol-5-il)piridin-4-il)etan-1-ol TRV1636 (0.080 g, 75%) como sólido color naranja. 1H RMN(DMSO-D6, 400 MHz): 88.74(d,J=8.0 Hz, 1H), 8.57 (s, 1H), 7.70 (d, J=8.0 Hz, 1H), 7.19 (d, J=8.0 Hz, 1H), 7.03 (s, 1H), 5.85 (s, 1H), 5.24 (m, 1H), 4.40 (m, 2H), 3.95 (m, 2H), 3.56 (m, 4H), 3.09 (m, 1H), 2.64 (d, J=8.0 Hz, 2H), 2.37 (m, 4H).

El TRV1470 (0.4197 g, 1.19 mmol) y ácido 3-bromofenilborónico (0.2510 g, 1.25 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). se agregó Na2CO32M (1.8 mL, solución 2.0 M ac.) junto con DME (2.7 mL). Luego se agregó Pd(PPh3)4 (0.0690 g, 0.059 mmol) todo de una vez. El tubo se volvió a sellar y se calentó
a 95°C durante 5 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (50% de EtOAc/hexano 5% de TEA) para producir 0.3918 g (75% de rendimiento) de bromuro de arilo 2. El bromuro de arilo (0.3881 g, 0.90 mmol) se disolvió en THF (10 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.74 mL, solución 1.6 M en hexano) durante 5 minutos. La reacción se le permitió agitar durante 15 minutos y luego se agregó en forma de gotas una solución de 3-oxetanona (0.1042 g, 1.45 mmol) en THF (1 mL). La reacción luego se agitó durante la noche mientras que se calentó lentamente a temperatura ambiente. Se volvió a enfriar en un baño de hielo y se detuvo con cloruro de amonio saturado y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas combinadas luego se lavaron con agua (2x), solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material crudo por último se purificó con cromatografía de columna (70% de EtOAc/hexano 5% de TEA) para producir TRV1638 (0.1406 g, 37% de rendimiento). 1H RMN (DMSO, 500 MHz) 8 = 7.90 (s, 1H), 7.69 (d, J = 7.5 Hz, 2H), 7.52 (t, J = 7.5 Hz, 1H), 7.32 (s, 1H), 6.41 (s, 1H), 6.25 (s, 1H), 4.81-4.77 (m, 4H), 4.38 (t, J = 8.5 Hz, 2H), 3.98 3.95 (m, 2H), 3.57 (t, J = 4.5 Hz, 4H), 3.09-3.01 (m, 1H), 2.64 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H).

El TRV1470 (0.4054 g, 1.15 mmol) y ácido 4-bromofenilborónico (0.2430 g, 1.21 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.8 mL, solución 2.0 M ac.) junto con DME (2.7 mL). Luego se agregó Pd(PPh3)4 (0.0670 g, 0.058 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 95°C durante 5 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (40% de EtOAc/hexano 5% de TEA) para producir 0.3273 g (66% de rendimiento) de bromuro de arilo 1. El bromuro de arilo (0.3054 g, 0.71 mmol) se disolvió en THF (10 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.50 mL, solución 1.6 M en hexano) durante 5 minutos. La reacción se le permitió agitar durante 15 minutos y luego se agregó en forma de gotas una solución de 3-oxetanona (0.0793 g, 1.1 mmol) en THF (1 mL). La reacción luego se agitó durante la noche mientras que se calentó lentamente a temperatura ambiente. Se volvió a enfriar en un baño de hielo y se detuvo con cloruro de amonio saturado y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas combinadas luego se lavaron con agua (2x), solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material crudo por último se purificó con cromatografía de columna (85% de EtOAc/hexano 5% de TEA) para producir TRV1639 (0.1084 g, 36% de rendimiento). 1H RMN (DMSO, 500 MHz) 8 = 7.82 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz, 2H), 7.33 (s, 1H), 6.42 (s, 1H), 6.28 (s, 1H), 4.80 (d, J = 6.5 Hz, 2H), 4.71 (d, J = 6.5 Hz, 2H), 4.38 (t, J = 8.0 Hz, 2H), 3.97 (t, J = 8.0 Hz, 2H), 3.57 (t, J = 4.5 Hz, 4H)3.09-3.01 (m, 1H), 2.63 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H).
Se agregaron Diisopropiletilamina (1.043 ml, 6.0 mmol) y anhídrido trifluoroacético (0.42 ml, 3.0 mmol) por separado a una solución de N-hidroxi-7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carboximidamida (500 mg, 1.50 mmol) en THF (20 ml) y DMF (30 ml) a temperatura ambiente. Después la mezcla se agitó 2 h, la reacción se agregó con EtOAc (60 ml). La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:100:500, TEA/EtOAc/Hexano a 3:5:100:500 MeOH/TEA/EtOAc/Hexano) para producir 4-(3-(morfolinometil)azetidin-1-il)-6-(5-(trifluorometil)-1,2,4-oxadiazol-3-il)benzo[c][1,2,5]oxadiazol TRV1643 (350 mg, 57%) como un sólido rojo. 1H RMN (400 MHz, CDCh): 2.48-2.50 (m, 4H), 2.73 (d, J=7.5, 2H), 3.07-3.16 (m, 1H), 3.73-3.75 (m, 4H), 4.09 (d-d, J=5.3, J=8.5, 2H), 4.51 (t, J=8.2, 2H), 6.45 (s, 1H), 7.88 (s, 1H).

Se agregó DCC (618 mg, 3.0 mmol) a una solución de N-hidroxi-7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-carboximidamida (500 mg, 1.5 mmol) y ácido 3,3,3-trifluoro-láctico (432 mg, 3.0 mmol) en THF (20 ml) y DMF (30 ml) a temperatura ambiente. Después la mezcla se agitó 3 h, la reacción se calentó a 70°C durante la noche. Uno o más equivalentes de DCC (309 mg, 1.5 mmol) y ácido 3,3,3-trifluoro-láctico (216 mg, 1.5 mmol) se agregaron a la mezcla de reacción y la reacción se agitó durante 8 horas a esta temperatura. La reacción se agregó con EtOAc (60 ml). La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (1:99, MeOH/DCM a 3:97, MeOH/DCM) para producir 2,2,2-trifluoro-1-(3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1,2,4-oxadiazol-5-il)etan-1 -ol TRV1644 (350 mg, 53%) como un sólido naranja. 1H RMN (400 MHz, CDCla): 2.50-2.52 (m, 4H), 2.74 (d, J=7.5, 2H), 3.07-3.14 (m, 1H), 3.74-3.77 (m, 4H), 4.05 (d-d, J=5.5, J=8.5, 2H), 4.47 (t, J=8.3, 2H), 5.43 (q, J=5.9, 1H), 6.41 (s, 1H), 7.82 (s, 1H).

Un matraz de fondo redondo se cargó con 6-bromo-4-fluorobenzo[c][1,2,5]oxadiazol (3.255 g, 15.0 mmol), dppp (371 mg, 0.9 mmol) y Pd(OAc)2 (101 mg, 0.45 mmol). El matraz se purgó con nitrógeno durante varias veces, y luego se agregaron DMF seco (30 ml), acrilato de etilo (4.9 ml, 45.0 mmol) y TEA (6.26 ml, 45 mmol) a la mezcla por separado. La mezcla resultante luego se calentó a 80°C. Después que se completó la reacción durante 2 h, 100 ml de EtOAc se agregó a la mezcla. La mezcla se lavó con solución salina durante 3 veces. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:95, EtOAc/Hexano a 6:85 EtOAc/Hexano) para producir (E)-3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il) acrilato de etilo (3.3 g, 93%) como un sólido naranja.
Se agregó MeMgBr (10 mg, 30 mmol) a una solución para producir (E)-3-(7-fluorobenzo[c][1,2, 5]oxadiazol-5-il) acrilato de etilo (1.652 g, 7.0 mmol) en THF (35 ml) a -5°C. Después la mezcla se agitó 4 h, la reacción se detuvo mediante 3 ml de MeOH. Se agregó EtOAc (80 ml) a la mezcla. La mezcla se lavó con NH4Cl saturado y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (5:95, EtOAc/Hexano a 15:85 EtOAc/Hexano) para producir (E)-4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)-2-metilbut-3-en-2-ol (1.0 g, 64%) como un sólido amarillo claro.
Se agregó TEA (0.50 ml, 3.6 mmol) a una solución de (E)-4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)-2-metilbut-3-en-2-ol (200 mg, 0.90 mmol) y sal de 2HCl de 4-(azetidin-3-ilmetil)morfolina (309 mg, 1.35 mmol) en MeCN (10 ml). La mezcla se calentó a 70°C durante 36 horas. Después la mezcla se enfrió a temperatura ambiente, se agregó EtOAc (30 ml) a la solución. La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 4:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir (E)-2-metil-4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)but-3-en-2-ol TRV1645 (230 mg, 72%) como un sólido amarillo. 1H RMN (400 MHz, CDCla): 1.46 (s, 6H), 2.47-2.49 (m, 4H), 2.71 (d, J=7.3, 2H), 3.05-3.11 (m, 1H), 3.72-3.75 (m, 4H), 3.97 (d-d, J=5.8, J=8.2, 2H), 4.41 (t, J=8.2, 2H), 5.96 (s, 1H), 5.43 (d, J=16.1, 1H), 6.59 (d, J=16.1, 1H), 6.92 (s, 1H).

Un frasco de reacción se cargó con 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.71 g, 2.00 mmol), 1-metil-4-(3,3,4,4-tetrametilborolan-1-il)-1H-pirazol (0.47 g, 2.30 mmol), Pd(PPh3)4 (0.070 g, 0.061 mmol). Después se desgasificó y se rellenó con argón, se agregó dioxano (6 mL) y Na2CO3 ac. (4.0 mL, 2.0 M, 8.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con argón, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, se agregó 3 mL de NaOH 2N, y se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 5: 2: 25: 75) para producir 0.70 g 6-(1 -metil-1H-pirazol-4-il)-4-(3-(morfolinometil)azetidin-1 -il) benzo[c][1,2,5]oxadiazol como un sólido naranja (99% de rendimiento).
Un frasco de reacción se cargó con 6-(1-Metil-1H-pirazol-4-il)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.29 g, 0.82 mmol). Después se desgasificó y se rellenó con argón, se agregó THF (5 mL). La solución resultante se enfrió a -78°C en un baño de hielo seco. Se agregó en forma de gotas n-BuLi (0.40 mL, 2.5 M, 1.00 mmol). Después se agitaron durante 1h -78°C, se agregó 2,2,2-trifluoroacetato de etilo (0.36 mL, 3.00 mmol). La reacción se agitó a -78°C durante 2h antes se detuvo con metanol. La mezcla se diluyó con agua, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 5: 2: 25: 75) para producir 0.20 g (55% de rendimiento) de 2,2,2-trifluoro-1-(1-metil-4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1H-pirazol-5-il)etan-1-ona como un sólido naranja.
Una solución de 2,2,2-trifluoro-1-(1-metil-4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1H-pirazol-5-il)etan-1-ona (0.050 g, 0.16 mmol) en metanol (5 mL) se enfrió en un baño de hielo-agua. Se agregó borohidruro de sodio (0.025 g, 0.66 mmol). La mezcla de reacción se calentó a rt, y se agitó hasta que se completó. La reacción se detuvo con HCl ac. 1 N, se diluyó con agua, se basificó con NaOH ac., y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 5: 2: 25: 75) para producir 0.18 g (90% de rendimiento) de TRV1647 como un sólido naranja. 1H RMN (CDCl3, 400 MHz) 8 = 7.55 (s, 1H), 6.83 (s, 1H), 5.70 (s, 1H), 5.44 (q, J = 7.27 Hz, 1H), 5.08 (amplio, 1H), 4.34 (t, J = 8.03 Hz, 1H), 4.27 (t, J = 8.16 Hz, 1H), 4.14 (s, 3H), 3.96 - 3.86 (m, 2H), 3.71 (t, J = 4.52 Hz, 4H), 3.01 (septeto, J = 6.90 Hz, 1H), 2.68 (d, J = 7.53 Hz, 2H), 2.45 (t, J = 4.27 Hz, 4H).

Un frasco de reacción se cargó con 6-bromo-4-fluorobenzo[c][1,2,5]oxadiazol (0.65 g, 3.00 mmol), 1-metil-5-(3,3,4,4-tetrametilborolan-1-il)-1H-pirazol (0.72 g, 3.46 mmol), Pd(PPh3)4 (0.10 g, 0.090 mmol). Después se desgasificó y se rellenó con argón, dioxano (8 mL) y se agregó Na2CO3 ac. (6.0 mL, 2.0 M, 12.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con argón, se selló, y luego se calentó a 90°C hasta que se completo la reacción. La mezcla se diluyó con agua, y se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex: DCM 0: 50: 30 a 8: 50: 30) para producir 0.45 g de 4-fluoro-6-(1-metil-1H-pirazol-5-il)benzo[c][1,2,5]oxadiazol (69% de rendimiento) como un sólido incoloro.
Una solución de 4-fluoro-6-(1-metil-1H-pirazol-5-il)benzo[c][1,2,5]oxadiazol (0.23 g, 1.04 mmol) en DCM (5.0 mL) se enfrió en un baño de hielo-agua. Se agregó lentamente bromo (0.65 mL, 2.0 M en DCM, 1.30 mmol). La mezcla de reacción se calentó lentamente a temperatura ambiente y se agitó hasta que se completó. La reacción se concentró y el producto crudo se utilizó directamente en la siguiente etapa sin purificación adicional.
Una solución de 6-(4-bromo-1-metil-1H-pirazol-5-il)-4-fluorobenzo[c][1,2,5]oxadiazol (0.42 g, 1.41 mmol) en acetonitrilo (10 mL) se agregó diclorhidrato de 1-(azetidin-3-ilmetil)morfolina (0.40 g, 1.75 mmol) seguido por TEA (1.10 mL, 7.91 mmol). La reacción luego se calentó a 50°C hasta que se completo la reacción. La mezcla se diluyó con agua y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se utilizó directamente en la siguiente etapa sin purificación adicional.
Un frasco de reacción se cargó con 6-(4-bromo-1-metil-1H-pirazol-5-il)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.11 g, 0.25 mmol). Después se desgasificó y se rellenó con argón, Se agregó THF (3 mL). La solución resultante se enfrió a -78°C en un baño de hielo seco. Se agregó en forma de gotas n-BuLi (0.13 mL, 2.5 M, 0.31 mmol). Después se agitó durante 1h -78°C, se agregó 2,2,2-trifluoroacetato de etilo (0.12 mL, 1.00 mmol). La reacción se agitó a -78°C durante 2h antes se detuvo con metanol. La mezcla se diluyó con agua, y se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 5: 2: 25: 75) para producir 0.040 g de 2,2,2-trifluoro-1-(1-metil-5-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-1H-pirazol-4-il)etan-1-ona.
Una solución de 2,2,2-trifluoro-1-(1-metil-5-(7-(3-(morfolinometil)azetidin-1-il) benzo[c][1,2,5]oxadiazol-5-il)-1H-pirazol-4-il)etan-1-ona (0.13 g, 0.28 mmol) en THF (5 mL) se enfrió en un baño de hielo-agua. Borohidruro de sodio (0.016 g, 0.42 mmol) en NaOH ac. Se agregó (0.80 mL, 1 M, 0.80 mmol). La mezcla de reacción se agitó a temperatura de baño de hielo-agua hasta que se completó. La reacción se diluyó con acetato de etilo y se lavó con solución salina. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: DCM 0: 100 a 5: 95) para producir 0.11 g (88% de rendimiento) de TRV1651 como un sólido naranja. 1H RMN (CDCh, 400 MHz) 8 = 7.75 (s, 1H), 6.98 (s, 1H), 5.68 (s, 1H), 4.84 (q, J = 7.03 Hz, 1H), 4.54 - 4.44 (m, 2H), 4.08 - 3.98 (m, 2H), 3.86 -3.82 (m, 3H), 3.76 -3.70 (m, 4H), 3.11 (septeto, J = 6.53 Hz, 1H), 2.74 (d, J = 7.28 Hz, 2H), 2.52 -2.45 (m, 4H).

6-(2-metoxipiridin-3-il)-4-fluorobenzo[c][1,2,5]oxadiazol (0.150 g,0.612 mmol) y sal de clorhidrato de 4-(azetidin-3-il metil) morfolina (0.140 g, 0.612 mmol) se disolvieron en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.3 ml, 1.85 mmol) y la mezcla se calentó a 50°C durante la noche. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 6-(2-metoxipiridin-3-il)-4-(3-(morfolinometil)azetidin-l-il)benzo[c][1,2, 5]oxadiazol TRV1658 (0.208 g, 90%) como sólido de color rojo. 1H RMN (CDCla, 400 MHz): 88.23 (d, J=4.0 Hz, 1H), 7.68 (d, J=8.0 Hz, 1H) 7.14 (s, 1H), 7.02 (m, 1H), 6.02 (s, 1H), 4.44(t, J=8.0 Hz, 2H), 4.05(s,3H), 3.98 (m, 2H), 3.74 (m, 4H), 3.11 (m, 1H), 2.73 (d, J=8.0 Hz, 2H), 2.48 (m, 4H)

Se disolvieron 6-(2-metoxipiridin-4-il)-4-fluorobenzo[c][1,2,5]oxadiazol (0.150 g,0.612 mmol) y sal de clorhidrato de 4-(azetidin-3-il metil) morfolina (0.140 g, 0.612 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.3 ml, 1.85 mmol) y la mezcla se calentó a 50°C durante la noche. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 6-(2-metoxipiridin-4-il)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1659 (0.2180 g, 92%) como sólido de color rojo. 1H RMN (CDCla, 400 MHz): 88.26 (d, J=8.0 Hz, 1H), 7.20 (s, 1H), 7.12 (d, J=8.0 Hz, 1H), 6.96 (s, 1H), 5.99 (s, 1H), 4.47(t, J=8.0 Hz, 2H), 4.05 (m, 2H), 4.01(s,3H), 3.74 (m, 4H), 3.14 (m, 1H), 2.73 (d, J=4.0 Hz, 2H), 2.48 (m, 4H).
Se disolvieron 6-(4-ciclopropil-3-metoxifenil)-4-fluorobenzo[c][1,2,5]oxadiazol (0.142g, 0.5 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.114 g, 0.5 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.2 ml, 1.5 mmol) y la mezcla se calentó a 50°C durante la noche. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 6-(4-ciclopropil-3-metoxifenil)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1660 (0.1780 g, 85%) como sólido color naranja. 1H RMN(CDCh,400 MHz): 87.30 (s, 1H), 7.08 (s, 1H), 7.05 (d, J=8.0 Hz, 1H), 7.0 (s, 1H), 5.96 (s, 1H), 4.42(t,J=8.0 Hz, 2H), 3.99 (m, 2H), 3.85(s,3H), 3.74 (m, 4H), 3.10 (m, 1H), 2.72 (d, J=8.0 Hz, 2H), 2.47 (m, 4H) 1.85 (m, 1H), 1.51 (m, 2H), 1.26(m,2H).

El TRV1470 (0.4480 g, 1.27 mmol) y ácido 2-bromofenilborónico (0.2678 g, 1.33 mmol) se sellaron en un tubo. El tubo se evacuó y se purgó con argón (3 ciclos). Se agregó Na2CO32M (1.9 mL, solución 2.0 M ac.) junto con DME (2.8 mL). Luego se agregó Pd(PPh3)4 (0.0734 g, 0.064 mmol) todo de una vez. El tubo se volvió a sellar y se calentó a 95°C durante 5 horas. Después de enfriar a temperatura ambiente, la mezcla se diluyó con agua y EtOAc. Las capas se separaron y la capa acuosa luego se extrajo de nuevo. Los extractos orgánicos combinados luego se lavaron con H2O (3X), solución salina y luego se secaron (Na2SO4), se filtraron y se concentraron. El material crudo se purificó a través de cromatografía (40% de EtOAc/hexano 5% de TEA) para producir 0.4642 g (85% de rendimiento) de bromuro de arilo. Este bromuro de arilo (0.4642 g, 1.08 mmol) se disolvió en THF (10 mL) y se enfrió a -78°C. Se agregó en forma de gotas nBuLi (0.90 mL, solución 1.6 M en hexano) durante 5 minutos. La reacción se le permitió agitar durante 15 minutos y luego se agregó en forma de gotas una solución de 3-oxetanona (0.1247 g, 1.73 mmol) en THF (1 mL). La reacción luego se agitó durante la noche mientras que se calentó lentamente a temperatura ambiente. Se volvió a enfriar en un baño de hielo y se detuvo con cloruro de amonio saturado y luego se diluyó con EtOAc. Las capas se separaron y la capa acuosa se extrajo de nuevo con EtOAc (3x). Las capas combinadas luego se lavaron con agua (2x), solución salina, se secaron (MgSO4), se filtraron y se concentraron. El material crudo se purificó con cromatografía de columna (85% de EtOAc/hexano 5% de TEA) para producir TRV1663 (0.3337 g, 73% de rendimiento). 1H RMN (DMSO, 500 MHz) 8 = 7.46-7.38 (m, 3H), 7.30 (d, J = 7.5 Hz, 1H), 7.19 (s, 1H), 6.61 (br s, 1H), 6.15 (s, 1H), 4.62 (d, J = 7.0 Hz, 2H), 4.41 (d, J = 7.0 Hz, 2H), 4.34 (t, J = 8.0 Hz, 2H), 3.94-3.91 (m, 2H), 3.57 (t, J = 4.5 Hz, 4H), 3.07-3.01 (m, 1H), 2.63 (d, J = 7.5 Hz, 2H), 2.38 (br s, 4H).

Se agregó DIBAL (25.76 ml, 25.76 mmol) a una solución para producir (E)-3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)acrilato de etilo (1.52 g, 6.44 mmol) en THF (35 ml) a -5°C. Después la mezcla se agitó durante la noche a temperatura ambiente, la reacción se detuvo mediante 3 ml de MeOH. EtOAc (40 ml) se agregó a la mezcla. La mezcla se lavó con HCl 1 N y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (10:90, EtOAc/Hexano a 15:85 EtOAc/Hexano) para producir (E)-3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)prop-2-en-1-ol (340 mg, 28%) como un sólido amarillo claro.
Se agregó TEA (0.98 ml, 7.08 mmol) a una solución de (E)-3-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)prop-2-en-1-ol (340 mg, 1.77 mmol) y sal de 2 HCl de 4-(azetidin-3-ilmetil)morfolina (608 mg, 2.66 mmol) en MeCN (10 ml). La mezcla se calentó a 70°C durante 48 horas. Después la mezcla se enfrió a temperatura ambiente, EtOAc (30 ml) se agregó a la solución. La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 4:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir (E)-3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)prop-2-en-1-ol (450 mg, 77%) como un sólido amarillo.
Se agregó periodinano de Dess-Martin (694 mg, 1.64 mmol) a una solución de (E)-3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)prop-2-en-1-ol (450 mg, 1.36 mmol) en DCM (15 ml) a temperatura ambiente. Después la mezcla se agitó 1 h, la reacción se detuvo mediante tiosulfato de sodio saturado (10 ml). La mezcla se lavó con bicarbonato de sodio saturado y solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 3:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir (E)-3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)acrilaldehído (360 mg, 81%) como un sólido naranja.
(E)-3-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)acrilaldehído (360 mg, 1.1 mmol) se enfrío a 0°C en solución de THF seco (10 ml) bajo atmósfera de nitrógeno. TMSCF3 (0.195 ml, 1.32 mmol) y TbAf (0.22 ml, 0.22 mmol, solución 1 M) se agregaron lentamente a la mezcla por separado. La solución se agitó durante 2 horas a 0°C. Después de que se verificó la finalización mediante TLC, se agregó la mezcla con 5 ml de agua. Esta mezcla se agregó con 50 ml de EtOAc y se lavó con solución salina. La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de elución de gradiente (2:100:500, TEA/EtOAc/Hexano a 4:2:100:500 MeOH/TEA/EtOAc/Hexano) para producir (E)-1,1,1-trifluoro-4-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)but-3-en-2-ol (280 mg, 64%) TRV1665 como un sólido naranja. 1H RMN (400 MHz, CDC13): 2.48-2.50 (m, 4H), 2.72 (d, J=7.5, 2H), 3.06-3.12 (m, 1H), 3.73-3.75 (m, 4H), 3.99 (d-d, J=5.6, J=8.2, 2H), 4.42 (t, J=8.2, 2H), 4.67-4.74 (m, 1H), 5.92 (s, 1H), 6.26 (d-d, J=6.0, J=16.1, 1H), 6.86 (d, J=16.1, 1H), 6.98 (s, 1H).

Un matraz de fondo redondo de un cuello, 250 mL se cargó con 45 mL de diclorometano seco y 1.60 mL (15.0 mmol) de 1,2-dimetoxietano (DME). La solución se enfrió a -30°C y 1.50 mL (15.0 mmol) de dietilzinc se agregó. A este se agregó solución agitada 2.40 mL (30.0 mmol) de diyodometano durante un periodo de 20 min mientras que mantiene la temperatura entre -20°C y -30°C. Después de que se completa la adición, la solución clara resultante se agitó durante 1 hora a -10°C. Una solución de 500 mg (2.25 mmol) de (E)-4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)-2-metilbut-3-en-2- ol en 10 mL de diclorometano se agregó a través de cánula bajo argón. La mezcla de reacción se dejó calentar a temperatura ambiente y se agitó durante 24 horas. Después de que se verificó la finalización mediante TLC, se agregó la mezcla con 5 ml de agua. Esta mezcla se lavó con HCl 1 M y solución salina. La capa orgánica se secó sobre sulfato de sodio anhidro y luego se concentró. El residuo se purificó a través de elución de gradiente (10:90, EtOAc/Hexano a 20:80 EtOAc/Hexano) para producir 2-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)ciclopropil)propan-2-ol (120 mg, 23%).
Se agregó TEA (0.28 ml, 2.0 mmol) a una solución de 2-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)ciclopropil)propan-2-ol (120 mg, 0.51 mmol) y sal de 2HCl 4-(azetidin-3-ilmetil)morfolina (175 mg, 0.76 mmol) en MeCN (10 ml). La mezcla se calentó a 70°C durante 30 horas. Después la mezcla se enfrió a temperatura ambiente, se agregó EtOAc (30 ml) a la solución. La mezcla se lavó con solución salina. La capa orgánica se secó y se concentró. El residuo se purificó a través de elución de gradiente (2:25:75, TEA/EtOAc/Hexano a 4:2:25:75 MeOH/TEA/EtOAc/Hexano) para producir 2-(2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)ciclopropil)propan-2-ol TRV1666 (150 mg, 79%) como un sólido amarillo. 1H RMN (400 MHz, CDCl3): 1H RMN (400 MHz, CDCl3): 0.93-0.98 (m, 1H), 1.07 1.24 (m, 1H), 1.32 (s, 6H), 1.42-1.47 (m, 1H), 1.92-1.97 (m, 1H), 2.47 (s, br, 4H), 2.70 (d, J=7.3, 2H), 3.01-3.08 (m, 1H), 3.72-3.74 (m, 4H), 3.92-3.95 (m, 2H), 4.36 (t, J=8.0, 2H), 5.59 (s, 1H), 6.67 (s, 1H).

Se disolvió Oxazol (0.50 mL, 7.60 mmol) en THF anhidro (20 mL). La solución se enfrió en un baño de hielo/acetona seco. A la solución se agregó en forma de gotas n-BuLi (3.5 mL, 2.5 M, 8.8 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó triflato de trimetilsililo (3.0 mL, 11.2 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 0:100 a 5: 95) para dar 1.36 g de 2-triisopropilsililoxazol (79%).
Se disolvió 2-Triisopropilsililoxazol (0.45 g, 2.0 mmol) en THF anhidro (10 mL). La solución se enfrió en un baño de hielo seco/acetona. A la solución se agregó en forma de gotas n-BuLi (1.0 mL, 2.5 M, 2.5 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó 2-isopropoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (0.6 mL, 2.94 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se utilizó directamente en la siguiente etapa con purificación adicional.
Un frasco de reacción se cargó con 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2-(triisopropilsilil)oxazol anterior, 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.70 g, 2.0 mmol), Pd(Pph3)4 (0.070 g, 0.060 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (7.5 mL) y Na2CO3 ac. (4.0 mL, 2.0 M, 8.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 95°C hasta que se completo la reacción. La mezcla se diluyó con agua, se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 1.5: 2: 25: 75) para producir 0.60 g (61% de rendimiento para dos etapas) de 4-(3-(morfolinometil)azetidin-1-il)-6-(2-(triisopropilsilil)oxazol-5-il)benzo [c][1,2,5]oxadiazol como un sólido rojo.
Se disolvió 4-(3-(morfolinometil)azetidin-1-il)-6-(2-(triisopropilsilil)oxazol-5-il)benzo[c][1,2,5] oxadiazol (0.48 g, 0.97 mmol) en THF anhidro (7.5 mL). La solución se enfrió en un baño de hielo seco/acetona. A la solución se agregó en forma de gotas LDA (0.65 mL, 2.0 M, 1.30 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó trifluoroacetato de etilo (0.24 mL, 2.0 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (TEA: EtOAc: Hex 0.5: 10: 90 a 0.5: 80: 20) para producir 0.26 g del producto como un sólido rojo.
Se disolvió 2,2,2-Trifluoro-1-(5-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)-2-(triisopropilsilil)oxazol-4-il)etan-1-ona (0.26 g, 0.44 mmol) en THF (5 mL). La solución se enfrió en un baño de hieloagua. Se agregó borohidruro de sodio (0.025 g, 0.66 mmol) en 0.5 mL de NaOH 0.5 N acuoso NaOH. La solución se agitó durante 10 min, y luego se calentó a temperatura ambiente. La reacción se agitó hasta finalización. La mezcla de reacción se enfrió en un baño de hielo-agua, se detuvo con HCl acuoso 1 N, se agitó durante 1 h, se basificó con bicarbonato de sodio saturado, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TeA: EtOAc: Hex 0: 2: 25: 75 a 5: 2: 25: 75) para producir 0.20 g de TRV1667 como un sólido amarillo. 1H RMN (CDCla, 400 MHz) 8 = 8.03 (s, 1H), 7.24 (s, 1H), 6.01 (s, 1H), 5.27 (q, J = 6.19 Hz, 1H), 4.46 (t, J = 7.53 Hz, 2H), 4.05 (t, J = 6.78 Hz, 2H), 3.79 (amplio, 1H), 3.74 (t, J = 4.52 Hz, 4H), 3.12 (septeto, J = 6.90 Hz, 1H), 2.73 (t, J = 7.53 Hz, 2H), 2.49 (t, J = 4.14 Hz, 4H).

Se disolvieron 6-(6-metoxipiridin-3-il)-4-fluorobenzo[c][1,2,5]oxadiazol (0.150 g,0.612 mmol) y sal de clorhidrato de 4-(azetidin-3-il metil) morfolina (0.140 g, 0.612 mmol) en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.3 ml, 1.85 mmol) y la mezcla se calentó a 80°C durante la noche. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar producto crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 6-(6-metoxipiridin-3-il)-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol TRV1670 (0.218 g, 93%) como sólido de color rojo. 1H RMN (CDCla, 400 MHz): 88.45 (d, J=4.0 Hz, 1H), 7.84 (d, J=4.0 Hz, 1H), 7.82 (d, J=4.0 Hz, 1H), 7.10 (s, 1H), 6.86 (d, J=8.0 Hz, 1H), 5.98 (s, 1H), 4.47(t, J=8.0 Hz, 2H), 4.04 (m, 2H), 4.01(s,3H), 3.75 (m, 4H), 3.13 (m, 1H), 2.74 (d, J=8.0 Hz, 2H), 2.48 (m, 4H).

Se disolvió Oxazol (0.50 mL, 7.60 mmol) en THF anhidro (20 mL). La solución se enfrió en un baño de hielo seco/acetona. A la solución se agregó en forma de gotas n-BuLi (3.5 mL, 2.5 M, 8.8 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó triflato de trimetilsililo (3.0 mL, 11.2 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 0:100 a 5: 95) para dar 1.36 g de 2-triisopropilsililoxazol (79%).
Se disolvió 2-Triisopropilsililoxazol (0.45 g, 2.0 mmol) en THF anhidro (10 mL). La solución se enfrió en un baño de hielo seco/acetona. A la solución se agregó en forma de gotas n-BuLi (1.0 mL, 2.5 M, 2.5 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó 2-isopropoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (0.6 mL, 2.94 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con cloruro de amonio saturado, y se extrajo con acetato de etilo. La capa orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se utilizó directamente en la siguiente etapa con purificación adicional.
Un frasco de reacción se cargó con 5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-2-(triisopropilsilil)oxazol anterior, 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5] oxadiazol (0.76 g, 2.0 mmol), Pd(PPh3)4 (0.070 g, 0.060 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (7.5 mL) y Na2CO3 ac. (4.0 mL, 2.0 M, 8.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 95°C hasta que se completo la reacción. La mezcla se enfrió a rt, se sometió a tratamiento final con HCl 1N, se agitó durante 1h, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 7.5: 2: 25: 75) para producir 0.55 g (62% de rendimiento para dos etapas) de 4-(3-(morfolinometil)azetidin-1-il)-6-(oxazol-5-il)benzo[c][1,2,5] oxadiazol como un sólido rojo.
Se disolvió 4-(3-(morfolinometil)azetidin-1-il)-6-(oxazol-5-il)benzo[c][1,2,5]oxadiazol (0.36 g, 1.06 mmol) en THF anhidro (7.5 mL). La solución se enfrió en un baño de hielo seco/acetona. A la solución se agregó en forma de gotas
LDA (1.60 mL, 2.0 M, 3.20 mmol). Después que la adición se completó, la solución se maduró durante 1h a -78°C, y luego se agregó trifluoroacetato de etilo (0.54 mL, 4.54 mmol). La mezcla de reacción se calentó naturalmente a temperatura ambiente, se detuvo con agua, y se volvió a enfriar en un baño de hielo-agua antes se agregó borohidruro de sodio (0.17 g, 4.54 mmol). La solución se agitó durante 10 min, y luego se calentó a temperatura ambiente y se agitó hasta finalización. La mezcla de reacción se enfrió en un baño de hielo-agua, se detuvo con HCl acuoso 1N, se agitó durante 1 h, se basificó con bicarbonato de sodio saturado, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: EtOAc: Hex 0: 2: 25: 75 a 7.5: 2: 25: 75) para producir 0.27 g (un rendimiento de 58%) de TRV 1677 como un sólido naranja. 1H RMN (CDCh, 400 MHz) 8 = 8.07 (s, 1H), 7.58 (d, J = 7.03 Hz, 1H), 7.33 (s, 1H), 6.32 (s, 1H), 5.62 - 5.52 (m, 1H), 4.38 (t, J = 8.28 Hz, 2H), 4.03 - 3.88 (m, 2H), 3.58 (t, J = 8.28 Hz, 4H), 3.07 (septeto, J = 6.78 Hz, 1H), 2.63 (t, J = 7.53 Hz, 2H), 2.43 -2.34 (m, 4H).

2,2,2-trifluoro-1-(2-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)oxazol-5-il)etan-1-ol (0.152g, 0.5 mmol) y sal de clorhidrato de 4-(azetidin-3-ilmetil) morfolina (0.114 g, 0.5 mmol) se disolvieron en Acetonitrilo (5 ml) a temperatura ambiente, se agregó trietilamina (0.2 ml, 1.5 mmol) y la mezcla se calentó a 50°C durante la noche. Después de la finalización de la reacción mediante TLC se detuvo con Na2CO3 (2M) y se extrajo con EtOAc para dar TRV-1660 crudo. La purificación se realizó sobre sistema de cromatografía instantánea ISCO utilizando diclorometano: metanol (95:5) como solvente para obtener 2,2,2-trifluoro-1-(2-(7-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol-5-il)oxazol-5-il)etan-1-ol TRV1686 (0.1400 g, 68%) como sólido color naranja. 1H RMN (DMSO, 400 MHz): 87.59 (s, 1H), 7.53 (s, 1H), 7.36 (d, J=4.0 Hz, 1H), 6.45 (s, 1H), 5.57 (m, 1H), 4.41(t, J=8.0 Hz, 2H), 3.98 (m, 2H), 3.57 (m, 4H), 3.08 (m, 1H), 2.65 (d, J=8.0 Hz, 2H), 2.38 (m, 4H).

Se disolvió en 6-bromo-4-(3-(morfolinometil)azetidin-1-il)benzo[c][1,2,5]oxadiazol (0.353 g, 1.0 mmol) THF seco (5 ml) y se enfrió a -78°C bajo argón atm. Se agregó en forma de gotas n-BuLi 2.5 M (0.48 ml, 1.2 mmol) y la mezcla se le permitió agitar durante 20 min a - 78°C. 3-(benciloxi)ciclobutan-1-ona luego se agrego a la mezcla de reacción y se agitó durante 30 min adicionales y se llevó a rt seguido por agitación durante otros 30 min luego se detuvo con cloruro de amonio y se extrajo con diclorometano. La evaporación del solvente dio el producto crudo que se purificó sobre ISCO para conseguir de 3-(benciloxi)-1-(7-(3-(morfolinometil)azetidin-l-il)benzo[c][1,2,5]oxadiazol-5-il)ciclobutan-1-ol TRV1692 (47%). 1H RMN (CDCla, 400 MHz): 87.36(m,5H), 7.01 (s, 1H), 5.98 (s, 1H), 4.49 (s, 2H), 4.40(t, J=8.0 Hz, 2H), 3.98 (m, 2H), 3.94 (m, 1H), 3.73 (m, 4H), 3.08 (m, 1H), 2.94 (m, 2H), 2.71 (d, J=8.0 Hz, 2H), 2.47(m,6H).

1,4-Dibromobenceno (4.72 g, 20.00 mmol) se disolvió en THF anhidro (50 mL) bajo argón. La solución se enfrió a -78°C y n-BuLi se agregó en forma de gotas (2.5 M, 8.8 mL, 20.5 mmol). La solución se agitó a -78°C durante 0.5 h antes se agregó oxetan-3-ona (1.2 mL, 20.5 mmol). La solución se agitó a -78°C durante 1 h, luego se detuvo con cloruro de amonio acuoso saturado , y se extrajo con acetato de etilo. La capa orgánica se lavó con solución salina, se secó con sulfato de sodio anhidro, y luego se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 10:90 a 50: 50) para producir 3.94 g de 3-(4-bromofenil) oxetan-3-ol como un sólido blanco (86% de rendimiento).
Un frasco de reacción se cargó con 3-(4-bromofenil)oxetan-3-ol (1.72 g, 7.5 mmol), 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (2.28 g, 8.64 mmol), Pd(PPh3)4 (0.26 g, 0.23 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (15 mL) y Na2CO3 ac. (10.0 mL, 2.0 M, 20.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 95°C hasta que se completó. La mezcla se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 10: 90 a 50: 50) para producir 1.95 g (91% de rendimiento) de 3-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol como un sólido blanco.
3-(4-(7-Fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol (0.43 g, 1.50 mmol), 4-(piperidin-4-il)morfolina (0.30 g, 1.80 mmol), base de Hunig (0.78 mL, 4.49 mmol) en MeCN (10 mL) se calentó a 80°C durante la noche. La mezcla de reacción se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: t Ea : EtOAc: Hex: 0: 2: 25: 75 a 5: 2: 25: 75) para producir a total de 0.63 g (96% de rendimiento) de TRV1717 como un sólido naranja. 1H RMN (CDCla, 400 MHz) 8 = 7.75 (d, J = 8.53 Hz, 2H), 7.69 (d, J = 8.53 Hz, 2H), 7.35 (s, 1H), 6.59 (s, 1H), 4.98 (d, J = 7.03 Hz, 2H), 4.96 (d, J = 7.03 Hz, 2H), 4.43 (d, J = 12.80 Hz, 2H), 3.76 (t, J = 4.65 Hz, 4H), 3.05 (dt, J1 = 1.76 Hz, J2 = 12.30 Hz, 2H), 2.71 (amplio, 1H), 2.62 (t, J = 4.52 Hz, 4H), 2.49 (tt, J1 = 1.76 Hz, J2= 11.04 Hz, 1H), 2.05 (d, J = 12.80 Hz, 2H), 1.78 (dq, J1 = 3.51 Hz, J2 = 12.05 Hz, 2H).

3-(4-(7-Fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol (0.29 g, 1.00 mmol), 4-(pirrolidin-3-il) morfolina (0.19 g, 1.20 mmol), base de Hunig (0.52 mL, 3.00 mmol) en MeCN (7.5 mL) se calentó a 80°C durante la noche. La mezcla de reacción se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: t Ea : THF: Hex: 0: 2: 25: 75 a 5: 2: 25: 75) para producir un total de 0.36 g (85% de rendimiento) de TRV1719 como un sólido naranja. 1H RMN (DMSO, 400 MHz) 8 = 7.84 (d, J = 8.53 Hz, 2H), 7.71 (d, J = 8.53 Hz, 2H), 7.26 (s, 1H), 6.46 (s, 1H), 6.33 (s, 1H), 4.81 (d, J = 6.53 Hz, 2H), 4.71 (d, J = 6.53 Hz, 2H), 4.05 -3.92 (m, 2H), 3.75 (q, J = 9.20 Hz, 1H), 3.61 (t, J = 4.40 Hz, 4H), 3.52 (t, J = 9.29 Hz, 1H), 3.00 (penteto, J = 7.91 Hz, 1H), 2.55 - 2.45 (m, 4H, traslapo con picos de DMSO), 2.31 - 2.22 (m, 1H), 1.91 (penteto, J = 9.98 Hz, 1H).

Se disolvió 1,4-Dibromobenceno (4.72 g, 20.00 mmol) en THF anhidro (50 mL) bajo argón. La solución se enfrió a -78°C y se agregó en forma de gotas n-BuLi (2.5 M, 8.8 mL, 20.5 mmol). La solución se agitó a -78°C durante 0.5 h antes se agregó oxetan-3-ona (1.2 mL, 20.5 mmol). La solución se agitó a -78°C durante 1 h, luego se detuvo con cloruro de amonio acuoso saturado, y se extrajo con acetato de etilo. La capa orgánica se lavó con solución salina, se secó con sulfato de sodio anhidro, y luego se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 10:90 a 50: 50) para producir 3.94 g de 3-(4-bromofenil)oxetan-3-ol como un sólido blanco (86% de rendimiento).
Un frasco de reacción se cargó con 3-(4-bromofenil)oxetan-3-ol (1.72 g, 7.5 mmol), 4-fluoro-6-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)benzo[c][1,2,5]oxadiazol (2.28 g, 8.64 mmol), Pd(PPh3)4 (0.26 g, 0.23 mmol). Después se desgasificó y se rellenó con nitrógeno, el frasco se cargó con dioxano (15 mL) y Na2CO3 ac. (10.0 mL, 2.0 M, 20.0 mmol). El frasco de reacción se volvió a desgasificar adicionalmente, se rellenó con nitrógeno, se selló, y luego se calentó a 95°C hasta que se completó. La mezcla se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase de acetato de etilo se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (EtOAc: Hex 10: 90 a 50: 50) para producir 1.95 g (91% de rendimiento) de 3-(4-(7-fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol como un sólido blanco.
3-(4-(7-Fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol (0.29 g, 1.00 mmol), clorhidrato de N-(4-piperidinil)acetamida (0.22 g, 1.25 mmol), base de Hunig (0.80 mL, 4.60 mmol) en MeCN (7.5 mL) se calentó a 100°C durante 48 h. La mezcla de reacción se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: THF: Hex: 0: 2: 25: 75 a 7.5: 2: 25: 75) para producir a total de 0.25 g (82% de rendimiento) de TRV1735 como un sólido amarillo. 1H RMN (DMSO, 400 MHz) 8 = 7.89 (d, J = 7.78 Hz, 1H), 7.86 (d, J = 8.28 Hz, 2H), 7.73 (d, J = 8.28 Hz, 2H), 7.56 (s, 1H), 6.80 (s, 1H), 6.47 (s, 1H), 4.81 (d, J = 6.78 Hz, 2H), 4.71 (d, J = 6.53 Hz, 2H), 4.31 -4.21 (m, 2H), 3.90 -3.78 (m, 1H), 3.24 - 3.11 (m, 2H), 1.96 - 1.86 (s, 2H), 1.80 (s, 3H), 1.63 -1.49 (m, 2H).
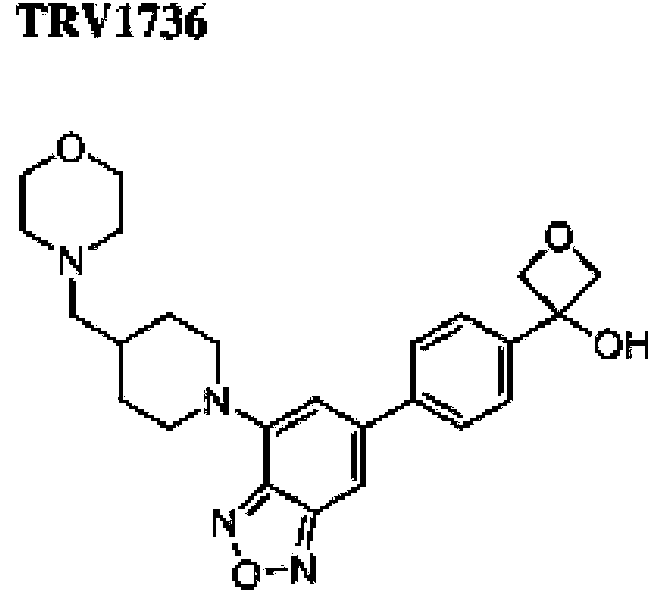
3-(4-(7-Fluorobenzo[c][1,2,5]oxadiazol-5-il)fenil)oxetan-3-ol (0.21 g, 0.75 mmol), diclorhidrato de 4-(piperidin-4-ilmetil) morfolina (0.26 g, 1.00 mmol), base de Hunig (0.80 mL, 4.60 mmol) en MeCN (7.5 mL) se calentó a 100°C durante 48h. La mezcla de reacción se enfrió a rt, se diluyó con agua, y luego se extrajo con acetato de etilo. La fase orgánica se secó sobre sulfato de sodio anhidro y se concentró. El residuo se purificó mediante cromatografía instantánea utilizando elución de gradiente (MeOH: TEA: THF: Hex: 0: 2: 25: 75 a 5: 2: 25: 75) para producir un total de 0.30 g (88% de rendimiento) de TRV1736 como un sólido naranja. 1H RMN (DMSO, 400 Mh z ) 8 = 7.85 (d, J = 8.28 Hz, 2H), 7.73 (d, J = 8.28 Hz, 2H), 7.53 (s, 1H), 6.76 (s, 1H), 6.47 (s, 1H), 4.81 (d, J = 6.53 Hz, 2H), 4.71 (d, J =
6.53 Hz, 2H), 4.38 - 4.28 (m, 2H), 3.57 (t, J = 4.40 Hz, 4H), 3.02 (t, J = 12.30 Hz, 2H), 2.43 - 2.25 (m, 4H), 2.18 (d, J = 6.78 Hz, 2H), 1.92 - 1.77 (m, 3H), 1.37 - 1.25 (m, 2H).
Datos biológicos
Se utilizaron las siguientes metodologías
Preparación de soluciones madre de Ap40
Se trató previamente Ap40 (1.0 mg) en un tubo de microcentrífuga de 1.5 ml con HFIP (1 mL) y se trató con ultrasonidos durante 20 minutos para desensamblar cualquier agregado de Ap preformado. E1HFIP se eliminó con una corriente de argón y el Ap se disolvió en base Tris (5.8 mL, 20 mM, pH ~10). El pH se ajustó a 7.4 con HCl concentrado (~10 |iL) y la solución se filtró con un filtro de jeringa (0.2 |im) antes de uso.
Las proteínas Ap42 y tau se prepararon de manera análoga como en el procedimiento anterior.
Ensayo de agregación de ThT Ap
El ensayo cinético de ThT para la agregación de Ap es similar al de Chalifour et al (Chalifour et al, 2003. J. Biol. Chem.278: 34874-81). Brevemente, se diluyó Ap40 o Ap42 pretratado (40 |jM en Tris 20 mM, pH 7.4) con un volumen igual de tioflavina T(ThT) 8 j M en Tris (20 mM, pH 7.4. NaCl 300 mM). Se agregaron alícuotas de Ap/ThT (200 j l ) a los pozos de una placa de 96 pozos de poliestireno negro, seguido de 2 j L de un compuesto en DMSO (concentración variable), o DMSO solo (controles). Las incubaciones se realizaron por triplicado y se tomó para contener 20 j M de Ap, diversas concentraciones de compuesto en Tris 20 mM, pH 7.4. NaCl 150 mM, DMSO al 1%. Las placas se cubrieron con tapas de poliestireno transparentes y se incubaron a 37°C en un lector de microplacas Tecan Genios.
Ensayo de agregación de ThS Tau
El ensayo cinético de ThS para la agregación de tau generalmente sigue el procedimiento anterior, excepto que se utiliza tioflavina S (ThS) en lugar de ThT y se utiliza proteína tau en lugar de Ap.
Análisis de los ensayos de agregación de ThT y ThS
Se tomaron lecturas de fluorescencia (Aex=450 nm, Aem=480 nm) cada 15 minutos, después de la primera agitación a alta intensidad durante 15 s y se dejó reposar durante 10 s antes de cada lectura. Los compuestos activos atenuaron el aumento de la fluorescencia en el tiempo que se produjo en los controles. En las Figuras 1-7. el tiempo en que se realizó este procedimiento se extendió a 80 horas (la parte más a la derecha del eje X), momento en el que el aumento de la fluorescencia generalmente alcanzó una asíntota. Aplicando una definición de control DMSO a las 80 horas como 100% de agregación (0% de inhibición) y control DMSO a las 0 horas como 0% de agregación (100% de inhibición), se puede calcular un % de puntuación de inhibición en el que más alto es mejor para una concentración dada de compuesto. Al repetir este procedimiento en varias concentraciones, se puede medir una concentración inhibitoria media (IC50), como se muestra en la siguiente tabla para algunos compuestos.
Ensayo biotinilado Ap42
Se disolvió beta-amiloide biotinilado (biotina-AB) en HFIP a la concentración de 1 mg/ml y se dividió en partes alícuotas a 50 jl/tubo en tubos de microcentrífuga coloreados de 0.65 ml (Fisher 02-681-248). Las alícuotas se diluyeron inmediatamente al agregar 450 |jl de HFIP a 0.1 mg/ml. Se tomaron aproximadamente 46 ul (que contenían aproximadamente 4.6 ug de biotina-AB) de un tubo, se agregaron 100 j l de HFIP, se agitaron y se secaron hasta obtener una película delgada bajo una corriente de N2. Se agregaron 100 j l de ácido trifluoroacético (TFA) y se agitaron en vórtex. Después de 10 minutos de incubación a temperatura ambiente en una campana, dando como resultado la desagregación de las semillas, la muestra se agitó en vórtice nuevamente y se secó nuevamente hasta obtener una película delgada bajo una corriente de N2. Se agregaron 100 j l de HFIP, se mezclaron y la muestra de biotina-AB se secó por tercera vez bajo una corriente de N2 para eliminar el TFA residual. La biotina-AB tratada se disolvió en DMSO hasta una concentración final de péptido de 2.3 jg/m l (50X).
Se recubrió una placa de “captura” de ELISA (Costar 9018) con 50 j l de 1 jg/m l de NeutrAvidin™ (NA) en un tampón de fosfato de sodio 10 mM, pH 7.5. (NeutrAvidin se puede preparar como 1 mg/ml (1000X) en agua milliQ/glicerol al 10% y almacenarse a -80°C antes de su uso). La placa se selló con una placa adhesiva y se almacenó a 4°C durante la noche. luego se bloqueó durante horas (sin sellar) a temperatura ambiente con 200 jl/pozo OFB 0.1% v/v Tween 20. Se dejó que la placa de “captura” alcanzara la temperatura ambiente.
Se prepararon varias diluciones del compuesto de ensayo con DMSO al 100%, de acuerdo con la concentración deseada a ensayar. Se pipetearon 2.5 j l de compuesto de ensayo diluido en el fondo de cada pozo de una placa de polipropileno de “dilución” de 96 pozos (Costar 3365), a la que se agregaron 250 ul de tampón de formación de oligómeros (OFB: fosfato de sodio 20 mM, pH 7.5). 150 mM de NaCl). Para comenzar la oligomerización, se agregaron 2 jl/pozo de biotina-AB al fondo de cada pozo de una placa de polipropileno de “reacción” de 96 pozos (Costar 3365) y se transfirieron 100 j l de cada pozo de la placa de “dilución” a la placa de “reacción” (la
concentración final de DMSO es de hasta el 1%). La placa de reacción se selló y se incubó durante 1 hora a temperatura ambiente sin agitar. Luego se detuvo la reacción mediante la adición de 50 ul de Tween 20 al 0.3% en agua.
La solución de bloqueo de la placa de “captura” se eliminó, y se transfirieron 50 ul de cada pozo de la placa de “reacción” a cada pozo de la placa de “captura”. La placa de “captura” se selló e incubó durante 2 horas a temperatura ambiente con agitación a 150 rpm. La placa de “captura” se lavó luego en una lavadora de placas (3x) con 200 ul/pozo de TBST (Tris-HCl 20 mM, pH 7.5/150 mM NaCl/Tween 20 al 0.1%). Se agregaron 50 ul de proteína de rábano picante de estreptavidina (dilución 1:20.000) en OFB Tween 20 al 0.1% a cada pozo, la placa de “captura” se selló y se incubó durante 1 hora con agitación a 150 rpm. Como antes, se lavó la placa y luego se agregaron a cada pozo 100 |jl de solución de sustrato TMB/H2O2. Después de 5-10 minutos, se agregaron 100 ul de ácido sulfúrico al 1% v/v y se leyó la absorbancia de cada pozo a 450 nm utilizando un lector de placas estándar.
Los datos se resumen en la siguiente tabla; los números dados entre paréntesis para los ensayos Ap40 de tioflavina T son los valores en ese experimento particular para el compuesto ID 1027, dados como la primera entrada en esta tabla para comparación estructural. La columna “ IC50 Ap42-bio” se refiere al valor aproximado del ensayo de biotina Ap42 IC50 en micromoles/L.




































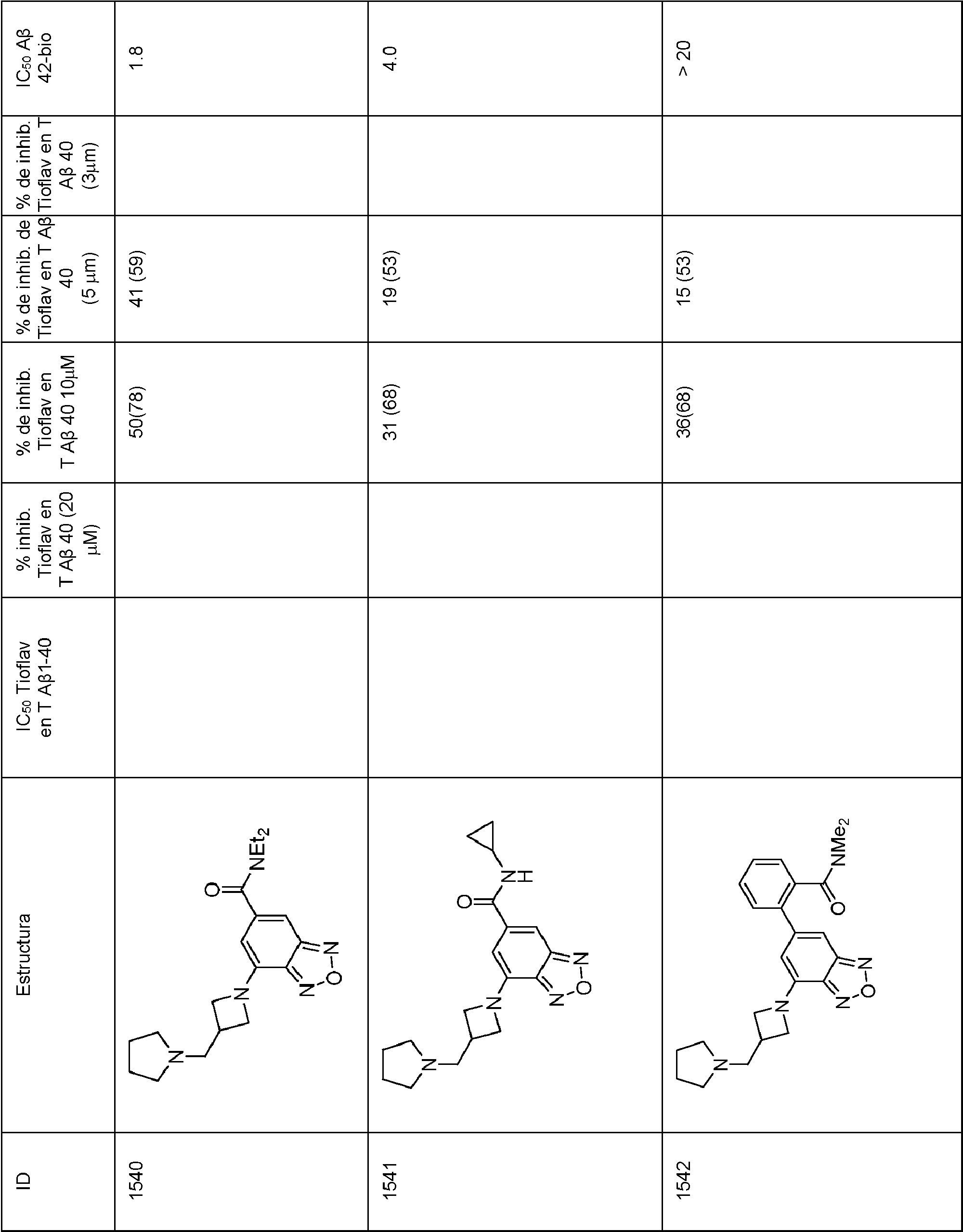
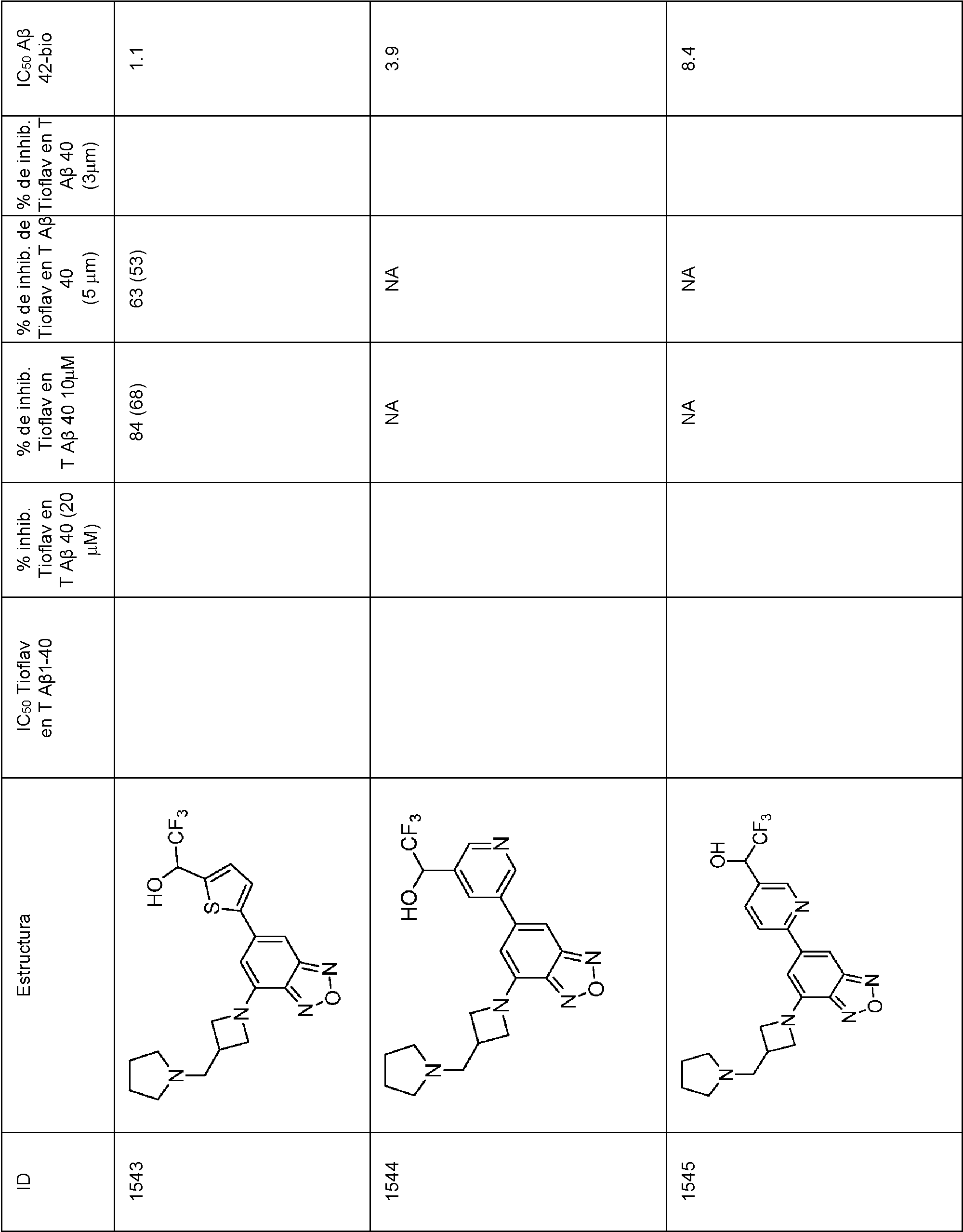














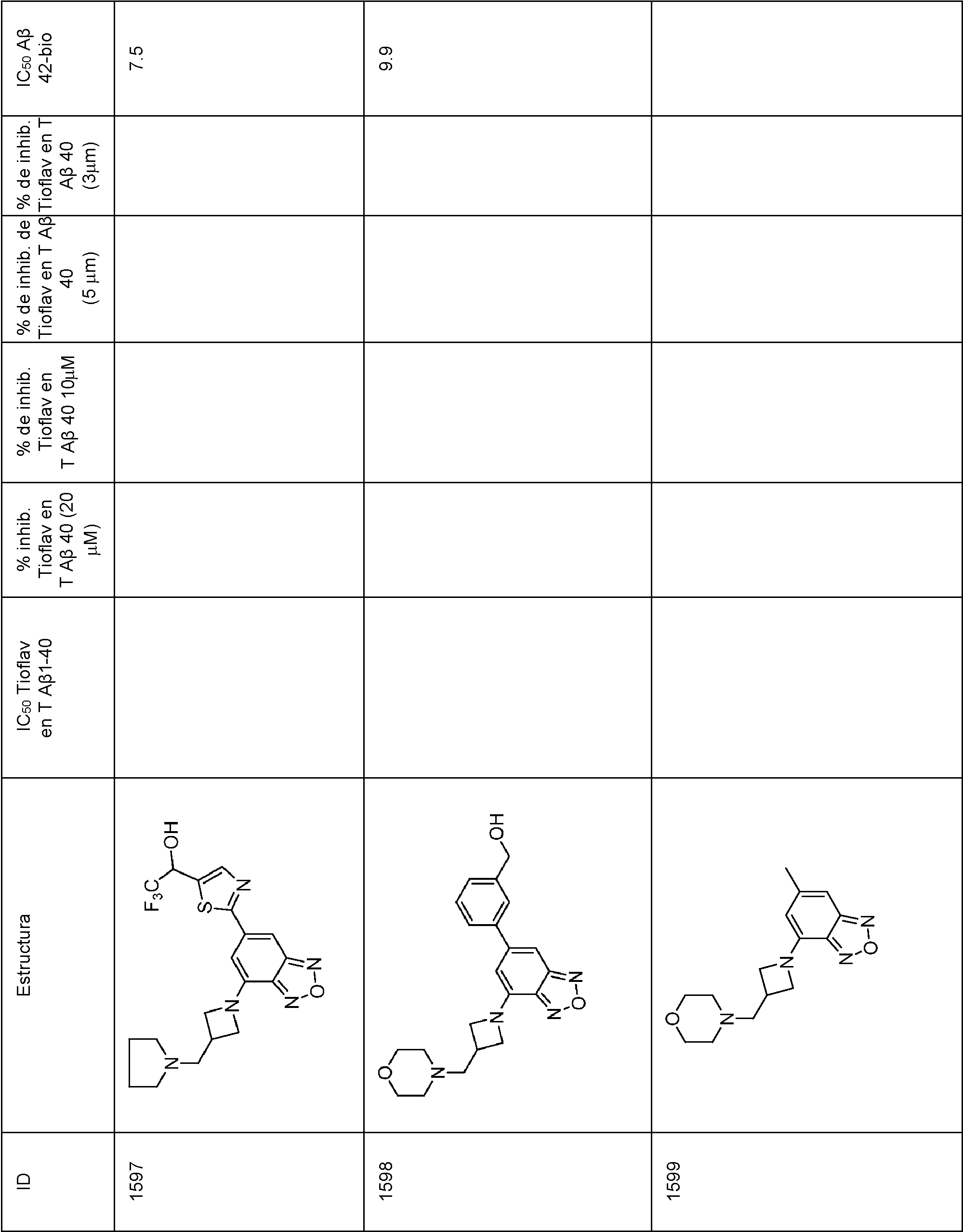














Claims (16)
1. Un compuesto de fórmula I:

o una de sus sales farmacéuticamente aceptables,
en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1-amino, N1,N1,N3 trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1-ilo, 3-(pirrolidin-1-ilmetanon)azetidin-1-ilo, 3-(morfolina)-1-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-4-il)pirrolidin-1-1-ilo, 3-(morfolin-4-ilmetil)pirrolidin-1-ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolin-4-il)piperidin-1-ilo, 4-(morfolin-4-ilmetil)piperidin-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N-metil)azetidin-1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo;
R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más sustituyentes A, oxadiazol opcionalmente sustituido con uno o más sustituyentes A, tiofeno opcionalmente sustituido con uno o más sustituyentes A, tiazol opcionalmente sustituido con uno o más sustituyentes A, furano opcionalmente sustituido con uno o más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más de los sustituyentes A, N-metilpirazol opcionalmente sustituido con uno o más sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectado en cualquier dirección) opcionalmente sustituido con uno o más alquilo, -CH2OH, 1-ol-2,2,2-trifluoroetan-1-ilo, 2-ol-propan-2-ilo, y ciclopropilo; y
R12 y R14 son cada uno independientemente hidrógeno o alquilo, con la condición de que si A es 1-ol-2,2,2-trifluoroetan-1-ilo, entonces no puede conectarse a tiofeno.
2. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo,
en el que R11 se selecciona del grupo que consiste en 4-(pirrolidin-1-il)piperidin-1-1-ilo, N-metil-3-(pirrolidin-1-il)propan-1-amino, N1,N1N3,-trimetilpropano-1,3-diamino, N,N-dimetilpiperidin-4-amino, 3-(pirrolidin-1-ilmetil)azetidin-1-ilo, 3-(pirrolidin-1-ilmetanon)azetidin-1-1-ilo, 3-(morfolin-4-ilmetil)pirrolidin-1-ilo, y 3-(morfolin-4-ilmetil)azetidin-1-ilo; y
R13 se selecciona del grupo que consiste en tiofeno opcionalmente sustituido con uno o más de los sustituyentes A, tiazol opcionalmente sustituido con uno o más de los sustituyentes A, furano opcionalmente sustituido con uno o más de los sustituyentes A, pirazol opcionalmente sustituido con uno o más sustituyentes A, N-metilpirazol opcionalmente sustituido con uno o más de los sustituyentes A, 2-piridina opcionalmente sustituido con uno o más de los sustituyentes A, 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, y 4-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona del grupo que consiste en alquilo, alcoxi, alquiltilo, isopropilo, t-butilo, trifluorometilo, cloro, ciano, CF3O-, metanonilo, metilsulfonilo, trifluorometilsulfonilo, amida (conectada en cualquier dirección) opcionalmente sustituida con uno o más alquilo, y -CH2OH.
3. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en el que R11 se selecciona del grupo que consiste en 3-(morfolin-4-ilmetil)azetidin-1-ilo, 3-(2-etanolamino-N-metil)azetidin-1-ilo, 3-(morfolin-4-il) pirrolidin-1-ilo, 4-(etilamido)piperidin-1-ilo, 3-(aminometil)azetidin-1-ilo, 4-(morfolin-4-il)piperidin-1-il, 4-(morfolin-4-ilmetil) piperidin-1-ilo, 3-amidoazetidin-1-ilo, 3-(propan-2-ol-2-il)azetidin-1-ilo, 3-(metilsulfonamida-N-metil)azetidin-1-ilo, 3-(metilamido-N metil)azetidin-1-ilo, y 3-(metilamino-N-metil)azetidin-1-ilo
y
R13 se selecciona del grupo que consiste en oxazol opcionalmente sustituido con uno o más de los sustituyentes A, oxadiazol opcionalmente sustituido con uno o más de los sustituyentes A, y 3-piridina opcionalmente sustituido con uno o más de los sustituyentes A, donde A se selecciona de grupo que consiste en 1-ol-2,2,2-trifluoroetan-1-ilo, 2-olpropan-2-ilo y ciclopropilo.
4. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en el que R13 se selecciona del grupo que consiste en 5-(2-ol-propan-2-il)oxadizol-3-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-5-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)piridin-2-ilo, 4-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo, 5-(1-ol-2,2,2-trifluoroetan-1-il)oxazol-2-ilo, 5-(1-ol-2,2,2 trifluoroetan-1-il)oxazol-3-ilo, 3-(1-ol-2,2,2-trifluoroetan-1-il)piridin-2-ilo, y 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo.
5. El compuesto de la reivindicación 4 o una sal farmacéuticamente aceptable del mismo, en el que R11 es 3-(morfolin-4-ilmetil)azetidin-1-ilo y R13 se selecciona del grupo que consiste en 2-(1-ol-2,2,2-trifluoroetan-1-il)piridin-3-ilo y 5-(2-olpropan-2-il)oxadizol-3-ilo.
6. Un compuesto para uso en un método de tratamiento de una enfermedad amiloide en un sujeto que comprende administrar una cantidad terapéuticamente eficaz de un compuesto de cualquiera de las reivindicaciones 1-5 al sujeto.
7. El compuesto para uso de la reivindicación 6 en el que la enfermedad amiloide es la enfermedad de Alzheimer.
8. El compuesto para el uso de la reivindicación 6 en el que  la enfermedad amiloide es la enfermedad de Parkinson.
la enfermedad amiloide es la enfermedad de Parkinson.
9. El compuesto para el uso de la reivindicación 6 en el que  la enfermedad amiloide es la demencia frontotemporal.
la enfermedad amiloide es la demencia frontotemporal.
10. El compuesto para el uso de la reivindicación 6 en el que la enfermedad amiloide es una parálisis supranuclear progresiva.
11. El compuesto para el uso de la reivindicación 6 en el que la enfermedad amiloide es la diabetes mellitus tipo 2.
12. Una composición farmacéutica que comprende un compuesto de cualquiera de las reivindicaciones 1-5 y un portador farmacéuticamente aceptable.
13. Un compuesto para uso en un método para tratar una afección que es un miembro seleccionado de pérdida de memoria, pérdida de cognición y una combinación de los mismos, dicho método comprende administrar a un sujeto que lo necesite una cantidad terapéuticamente eficaz de un compuesto de cualquier de reclamaciones 1-5.
14. El compuesto para su uso de acuerdo con la reivindicación 13, en el que dicha condición está asociada con la enfermedad de Alzheimer.
15. El compuesto para uso de acuerdo con la reivindicación 13, en el que se administra una dosis diaria total de 0,0003 a mg/kg de peso corporal.
16. El compuesto de la reivindicación 3 o una sal farmacéuticamente aceptable del mismo, en el que R13 es 4-(1-ol-2,2,2-trifluoroetan1-il)tiazol-2-ilo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461945697P | 2014-02-27 | 2014-02-27 | |
| PCT/US2015/017939 WO2015131021A2 (en) | 2014-02-27 | 2015-02-27 | Anti-amyloid compounds containing benzofurazan |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2734432T3 true ES2734432T3 (es) | 2019-12-05 |
Family
ID=54009787
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15755943T Active ES2734432T3 (es) | 2014-02-27 | 2015-02-27 | Compuestos antiamiloides que contienen benzofurazano |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US9938249B2 (es) |
| EP (1) | EP3110812B1 (es) |
| JP (1) | JP6663361B2 (es) |
| AU (1) | AU2015222903B2 (es) |
| CA (1) | CA2977360C (es) |
| CY (1) | CY1121827T1 (es) |
| DK (1) | DK3110812T3 (es) |
| ES (1) | ES2734432T3 (es) |
| HR (1) | HRP20191150T1 (es) |
| HU (1) | HUE044999T2 (es) |
| IL (1) | IL247428B (es) |
| LT (1) | LT3110812T (es) |
| PL (1) | PL3110812T3 (es) |
| PT (1) | PT3110812T (es) |
| RS (1) | RS58976B1 (es) |
| SI (1) | SI3110812T1 (es) |
| SM (1) | SMT201900387T1 (es) |
| TR (1) | TR201910291T4 (es) |
| WO (1) | WO2015131021A2 (es) |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19643037A1 (de) | 1996-10-18 | 1998-04-23 | Boehringer Ingelheim Kg | Neue Oxadiazole, Verfahren zu ihrer Herstellung sowie deren Verwendung als Arzneimittel |
| MXPA04004443A (es) | 2001-11-14 | 2004-08-11 | Merck Patent Gmbh | Derivados de pirazol. |
| US20050282818A1 (en) * | 2004-06-22 | 2005-12-22 | Rigel Pharmaceuticals, Inc. | Ubiquitin ligase inhibitors |
| WO2012080729A2 (en) * | 2010-12-14 | 2012-06-21 | Electrophoretics Limited | CASEIN KINASE 1δ (CK1δ) INHIBITORS |
| MX365851B (es) * | 2012-08-24 | 2019-06-17 | Treventis Corp | Compuestos de benzofurazano anti-amiloides y metodos. |
-
2015
- 2015-02-27 RS RS20190884A patent/RS58976B1/sr unknown
- 2015-02-27 JP JP2016572360A patent/JP6663361B2/ja active Active
- 2015-02-27 ES ES15755943T patent/ES2734432T3/es active Active
- 2015-02-27 EP EP15755943.6A patent/EP3110812B1/en active Active
- 2015-02-27 LT LTEP15755943.6T patent/LT3110812T/lt unknown
- 2015-02-27 CA CA2977360A patent/CA2977360C/en active Active
- 2015-02-27 HR HRP20191150TT patent/HRP20191150T1/hr unknown
- 2015-02-27 PL PL15755943T patent/PL3110812T3/pl unknown
- 2015-02-27 US US15/118,310 patent/US9938249B2/en active Active
- 2015-02-27 AU AU2015222903A patent/AU2015222903B2/en active Active
- 2015-02-27 SM SM20190387T patent/SMT201900387T1/it unknown
- 2015-02-27 HU HUE15755943A patent/HUE044999T2/hu unknown
- 2015-02-27 TR TR2019/10291T patent/TR201910291T4/tr unknown
- 2015-02-27 WO PCT/US2015/017939 patent/WO2015131021A2/en not_active Ceased
- 2015-02-27 SI SI201530798T patent/SI3110812T1/sl unknown
- 2015-02-27 PT PT15755943T patent/PT3110812T/pt unknown
- 2015-02-27 DK DK15755943.6T patent/DK3110812T3/da active
-
2016
- 2016-08-22 IL IL247428A patent/IL247428B/en active IP Right Grant
-
2019
- 2019-07-10 CY CY20191100740T patent/CY1121827T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| WO2015131021A3 (en) | 2015-11-19 |
| US20170174641A1 (en) | 2017-06-22 |
| IL247428A0 (en) | 2016-11-30 |
| LT3110812T (lt) | 2019-07-10 |
| HUE044999T2 (hu) | 2019-12-30 |
| CA2977360C (en) | 2022-09-27 |
| US9938249B2 (en) | 2018-04-10 |
| EP3110812A4 (en) | 2017-07-05 |
| EP3110812A2 (en) | 2017-01-04 |
| PL3110812T3 (pl) | 2019-10-31 |
| IL247428B (en) | 2020-01-30 |
| EP3110812B1 (en) | 2019-04-10 |
| TR201910291T4 (tr) | 2019-07-22 |
| CA2977360A1 (en) | 2015-09-03 |
| HRP20191150T1 (hr) | 2019-10-04 |
| PT3110812T (pt) | 2019-07-10 |
| WO2015131021A2 (en) | 2015-09-03 |
| SI3110812T1 (sl) | 2019-08-30 |
| JP6663361B2 (ja) | 2020-03-11 |
| JP2017506674A (ja) | 2017-03-09 |
| AU2015222903A1 (en) | 2016-10-06 |
| RS58976B1 (sr) | 2019-08-30 |
| AU2015222903B2 (en) | 2018-02-01 |
| CY1121827T1 (el) | 2020-07-31 |
| DK3110812T3 (da) | 2019-07-08 |
| SMT201900387T1 (it) | 2019-09-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2021202065B2 (en) | Hepatitis B core protein modulators | |
| AU2015371294B2 (en) | Compounds, compositions, and methods for increasing CFTR activity | |
| ES2901418T3 (es) | Difluoropirrolidinas como moduladores del receptor de orexina | |
| ES2858503T3 (es) | Alcoholes trifluorometílicos como moduladores de RORgammat | |
| ES2767303T3 (es) | Tiazoles sustituidos por amida como moduladores de Rorgamma T | |
| ES2616084T3 (es) | Compuestos de pirazol como inhibidores del receptor sigma | |
| ES2742843T3 (es) | Moduladores de quinolinilo de ROR(gamma)t | |
| BR112020001825A2 (pt) | compostos e métodos para a degradação direcionada de receptor de androgênio | |
| WO2015123424A1 (en) | Cyclopropylamines as lsd1 inhibitors | |
| ES2714705T3 (es) | Inhibidor de PDE4 novedoso | |
| BR112021007602A2 (pt) | compostos de heteroaril carboxamida de 5 membros para tratamento de hbv | |
| ES2923790T3 (es) | Piperidinas halosustituidas como moduladores de receptores de orexina | |
| ES2906258T3 (es) | 6-aminopiridin-3-ilo tiazoles como moduladores de ROR gamma t | |
| KR102046499B1 (ko) | 벤조푸라잔 항아밀로이드 화합물 및 방법 | |
| ES2314612T3 (es) | Derivados de heteroarilo condensados y su uso como inhibidores de la quinasa p38. | |
| CA3085097A1 (en) | Small molecule modulators of the btb domain of keap1 | |
| RS99503A (sr) | Derivati pirolidin oksadiazola i tiadiazola | |
| ES2734432T3 (es) | Compuestos antiamiloides que contienen benzofurazano |