ES2913090T3 - Derivado e intermedio del etomidato, procedimiento de preparación y uso del mismo - Google Patents
Derivado e intermedio del etomidato, procedimiento de preparación y uso del mismo Download PDFInfo
- Publication number
- ES2913090T3 ES2913090T3 ES16853130T ES16853130T ES2913090T3 ES 2913090 T3 ES2913090 T3 ES 2913090T3 ES 16853130 T ES16853130 T ES 16853130T ES 16853130 T ES16853130 T ES 16853130T ES 2913090 T3 ES2913090 T3 ES 2913090T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- etomidate
- present
- formula
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229960001690 etomidate Drugs 0.000 title claims abstract description 26
- NPUKDXXFDDZOKR-LLVKDONJSA-N etomidate Chemical compound CCOC(=O)C1=CN=CN1[C@H](C)C1=CC=CC=C1 NPUKDXXFDDZOKR-LLVKDONJSA-N 0.000 title claims description 72
- 238000000034 method Methods 0.000 title claims description 29
- 238000002360 preparation method Methods 0.000 title description 31
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 46
- 239000001257 hydrogen Substances 0.000 claims abstract description 46
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 45
- 150000002367 halogens Chemical class 0.000 claims abstract description 45
- 150000003839 salts Chemical class 0.000 claims abstract description 31
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 29
- 239000012453 solvate Substances 0.000 claims abstract description 27
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 21
- 150000002431 hydrogen Chemical class 0.000 claims abstract description 20
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims abstract description 19
- -1 etomidate compound Chemical class 0.000 claims abstract description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 18
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims abstract description 14
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims abstract description 14
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 14
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 14
- 239000000460 chlorine Substances 0.000 claims abstract description 14
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims abstract description 13
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 13
- 239000011737 fluorine Substances 0.000 claims abstract description 13
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims abstract description 12
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 12
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 11
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 11
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 10
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims abstract description 10
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims abstract description 10
- 125000001424 substituent group Chemical group 0.000 claims abstract description 10
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 9
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims abstract description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims abstract description 8
- 150000001875 compounds Chemical class 0.000 claims description 88
- 125000003118 aryl group Chemical group 0.000 claims description 18
- 125000000217 alkyl group Chemical group 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 5
- 239000003193 general anesthetic agent Substances 0.000 claims description 4
- 238000001990 intravenous administration Methods 0.000 claims description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract description 3
- 125000000041 C6-C10 aryl group Chemical group 0.000 abstract 2
- 238000006243 chemical reaction Methods 0.000 description 27
- 239000000243 solution Substances 0.000 description 21
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 239000000203 mixture Substances 0.000 description 18
- 230000003444 anaesthetic effect Effects 0.000 description 16
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 14
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 14
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 14
- 239000000543 intermediate Substances 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 239000007858 starting material Substances 0.000 description 13
- 206010002091 Anaesthesia Diseases 0.000 description 12
- 230000037005 anaesthesia Effects 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 230000028327 secretion Effects 0.000 description 11
- 241000699670 Mus sp. Species 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 238000001704 evaporation Methods 0.000 description 9
- 230000008020 evaporation Effects 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 239000012230 colorless oil Substances 0.000 description 7
- 229960000890 hydrocortisone Drugs 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 241000700159 Rattus Species 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000002349 favourable effect Effects 0.000 description 4
- 125000001183 hydrocarbyl group Chemical group 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 230000002085 persistent effect Effects 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical group ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 210000003462 vein Anatomy 0.000 description 4
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 230000003595 spectral effect Effects 0.000 description 3
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 229910004039 HBF4 Inorganic materials 0.000 description 2
- 238000012449 Kunming mouse Methods 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 229940092714 benzenesulfonic acid Drugs 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 210000000748 cardiovascular system Anatomy 0.000 description 2
- 210000001715 carotid artery Anatomy 0.000 description 2
- NEHMKBQYUWJMIP-UHFFFAOYSA-N chloromethane Chemical compound ClC NEHMKBQYUWJMIP-UHFFFAOYSA-N 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 230000008034 disappearance Effects 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 2
- QEWYKACRFQMRMB-UHFFFAOYSA-N fluoroacetic acid Chemical compound OC(=O)CF QEWYKACRFQMRMB-UHFFFAOYSA-N 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 125000000530 1-propynyl group Chemical group [H]C([H])([H])C#C* 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000006020 2-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- WDJHALXBUFZDSR-UHFFFAOYSA-N Acetoacetic acid Natural products CC(=O)CC(O)=O WDJHALXBUFZDSR-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108010091893 Cosyntropin Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000001780 adrenocortical effect Effects 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 150000001718 carbodiimides Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- ZOEFCCMDUURGSE-CQVUSSRSSA-N cortrosyn Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCNC(N)=N)C(=O)N[C@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(O)=O)NC(=O)C(N)CO)C1=CC=C(O)C=C1 ZOEFCCMDUURGSE-CQVUSSRSSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- WOWBFOBYOAGEEA-UHFFFAOYSA-N diafenthiuron Chemical compound CC(C)C1=C(NC(=S)NC(C)(C)C)C(C(C)C)=CC(OC=2C=CC=CC=2)=C1 WOWBFOBYOAGEEA-UHFFFAOYSA-N 0.000 description 1
- PBWZKZYHONABLN-UHFFFAOYSA-N difluoroacetic acid Chemical compound OC(=O)C(F)F PBWZKZYHONABLN-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- VFNGKCDDZUSWLR-UHFFFAOYSA-N disulfuric acid Chemical compound OS(=O)(=O)OS(O)(=O)=O VFNGKCDDZUSWLR-UHFFFAOYSA-N 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 150000002475 indoles Chemical class 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005932 isopentyloxycarbonyl group Chemical group 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000002960 lipid emulsion Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001148 pentyloxycarbonyl group Chemical group 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 150000003233 pyrroles Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 239000013558 reference substance Substances 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000028527 righting reflex Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 125000005930 sec-butyloxycarbonyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- RYYKJJJTJZKILX-UHFFFAOYSA-M sodium octadecanoate Chemical compound [Na+].CCCCCCCCCCCCCCCCCC([O-])=O RYYKJJJTJZKILX-UHFFFAOYSA-M 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/90—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P23/00—Anaesthetics
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Anesthesiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto de etomidato de Fórmula 1: **(Ver fórmula)** en la que, X e Y son independientemente halógeno, preferentemente flúor, cloro o bromo, o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10, preferentemente alquilo C1-6, más preferentemente metilo, etilo, propilo, isopropilo, n-butilo o isobutilo, o una sal farmacéuticamente aceptable, o un solvato del mismo.
Description
DESCRIPCIÓN
Derivado e intermedio del etomidato, procedimiento de preparación y uso del mismo
CAMPO TÉCNICO
La presente invención se relaciona con el campo de la farmacia, específicamente con un derivado del etomidato, o una sal farmacéuticamente aceptable, o un solvato del mismo, una composición farmacéutica que comprende el mismo, así como intermedios y procedimientos para preparar el mismo, y usos del mismo. Se divulga también un polimorfo del derivado de etomidato y un kit que comprende el derivado de etomidato, la sal farmacéuticamente aceptable, el polimorfo o el solvato del mismo.
ANTECEDENTES
El "etomidato", etil R-(+)-1-(1-fenetil)-1H-imidazol-5-carboxilato, es un fármaco anestésico intravenoso con las características de inicio rápido, corta duración de acción, rápida recuperación y ligera inhibición de los sistemas cardiovascular y respiratorio. En la clínica, el etomidato se utiliza principalmente en la inducción de la anestesia y en la anestesia para la cirugía clínica. La estructura del etomidato es la siguiente

Sin embargo, las investigaciones han demostrado que al mismo tiempo que ejerce el efecto anestésico, el etomidato puede tener un efecto inhibidor sobre la 11p-hidroxilasa, y por lo tanto reducir la secreción de cortisol y/o corticosterona. Por lo tanto, existe un riesgo potencialmente mortal en la aplicación a largo plazo de etomidato.
Los químicos farmacéuticos han realizado amplios estudios para superar las desventajas del etomidato y desarrollar novedosos fármacos que posean las ventajas del etomidato sin inhibir la secreción de cortisol y/o corticosterona tan fuertemente como el etomidato. Por ejemplo, Cotten JF et al. (Methoxycarbonyl-etomidate: A novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression, Anesthesiology, 2009, 111: 240-9) informó de un derivado del etomidato de Fórmula 7, que muestra un menor efecto inhibidor sobre la secreción de cortisol y/o corticosterona, pero que tiene un efecto anestésico drásticamente disminuido mientras tanto. Asimismo, Laha, Joydev K et al. (Synthesis of Fused Imidazoles, Pyrroles, and Indoles with a Defined Stereocenter a to Nitrogen Utilizing Mitsunobu Alkylation Followed by Palladium-Catalyzed Cyclization, Journal of Organic Chemistry, 2011, vol. 76, #20, p 8477-8482) divulgó un derivado de etomidato del tipo Carboetomidato de Fórmula 8, que tiene poca inhibición de la secreción de cortisol y/o corticosterona, pero tiene una actividad anestésica disminuida y una duración de acción prolongada mientras tanto

En vista de los problemas anteriores, tiene gran importancia práctica desarrollar un derivado del etomidato que tenga tanto un efecto anestésico favorable como perfiles de seguridad.
El documento CN103739553 A se refiere a un compuesto quiral de éster de ácido imidazolecarboxílico N-sustituido que contiene una cadena lateral de éter, un procedimiento de preparación y una aplicación del mismo. En el compuesto divulgado en CN103739553 Ax e Y en la Fórmula 1 son ambos H, y R1 es metoxietilo.
SUMARIO DE LA INVENCIÓN
La presente invención se define por el objeto de las reivindicaciones independientes.
En un primer aspecto se proporciona un derivado de etomidato, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo. El derivado de etomidato de la presente invención no sólo tiene una buena actividad anestésica, un rápido inicio y una corta duración de la acción, sino que también muestra poca inhibición de la secreción de cortisol y/o corticosterona, y por lo tanto tiene tanto un efecto anestésico favorable como perfiles de seguridad.
El derivado de etomidato de la presente invención tiene la estructura de la Fórmula I:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10.
En un segundo aspecto se proporciona una composición farmacéutica que comprende el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo, y uno o más portadores farmacéuticamente aceptables.
En un tercer aspecto se proporciona un kit que comprende el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo, o la composición farmacéutica de la presente invención.
En un cuarto aspecto se proporciona el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo, para su uso en anestesia.
En un quinto aspecto se proporciona el uso del derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo, o un solvato del mismo en la fabricación de un medicamento anestésico.
En un sexto aspecto se proporciona un uso anestésico, que comprende la administración de una cantidad eficaz del derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo.
En un séptimo aspecto se proporciona un procedimiento para preparar el derivado de etomidato de la presente invención, que incluye: preparar el derivado de etomidato a partir de un compuesto de Fórmula 2:

en la que X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
En un octavo aspecto se proporciona un intermedio para la preparación del derivado de etomidato de la presente invención, o una sal, un polimorfo o un solvato del mismo. El intermedio tiene la estructura de la Fórmula 2:

en la que X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
En un noveno aspecto se proporciona el uso del compuesto de Fórmula 2, o una sal, un polimorfo, o un solvato del mismo en la preparación del derivado de etomidato de la presente invención.
En un décimo aspecto se proporciona otro procedimiento para preparar el derivado de etomidato de la presente invención, que incluye: preparar el derivado de etomidato a partir de un compuesto de Fórmula 3:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10.
En un undécimo aspecto se proporciona otro intermedio para la preparación del derivado de etomidato de la presente invención, o una sal, un polimorfo o un solvato del mismo. El intermedio tiene la estructura de la Fórmula 3:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10.
En un duodécimo aspecto se proporciona el uso del compuesto de Fórmula 3, o una sal, un polimorfo, o un solvato del mismo en la preparación del derivado de etomidato de la presente invención.
DEFINICIONES
A menos que se definan de otro modo, todos los términos técnicos y científicos utilizados en el presente documento tienen el mismo significado que los entendidos comúnmente por un experto en la materia. Las referencias a las técnicas empleadas en el presente documento pretenden referirse a las técnicas tal y como se entienden comúnmente en la técnica, incluidas las variaciones de dichas técnicas o las sustituciones de técnicas equivalentes que serían evidentes para un experto en la materia. Aunque se cree que la mayoría de los siguientes términos serán fácilmente comprendidos por un experto en la materia, las siguientes definiciones se presentan para ilustrar mejor la presente invención.
Los términos "incluir", "comprender", "tener", "contener" o "involucrar", así como otras variaciones utilizadas en el presente documento, son inclusivos o abiertos, y no excluyen elementos o pasos del procedimiento adicionales y no citados.
El término "sustituido", tal como se utiliza aquí, significa que uno o más (por ejemplo, 1,2, 3 o 4) átomos de hidrógeno del átomo designado se sustituyen por grupos específicos, siempre que no se supere la valencia normal del átomo designado en las circunstancias existentes, y que la sustitución dé lugar a un compuesto estable. Las combinaciones de sustituyentes y/o variables sólo son admisibles si dichas combinaciones dan lugar a compuestos estables.
El término "opcionalmente sustituido", tal y como se utiliza en este documento, significa que la estructura descrita puede estar sin sustituir o sustituida con un grupo, radical o fracción específica.
El término "halógeno", tal y como se utiliza aquí, se refiere al flúor, al cloro, al bromo o al yodo, preferentemente flúor, cloro o bromo.
El término "alquilo C-i-a", tal como se utiliza en este documento, se refiere a un grupo hidrocarburo saturado, lineal o ramificado, que tiene de 1 a 6 átomos de carbono, como el metilo, el etilo, el propilo, el isopropilo, el n-butilo, el isobutilo, el sec-butilo, el tert-butilo, el n-pentilo, el isopentilo, el neo-pentilo, el n-hexilo, el isohexilo y similares, preferentemente metilo, etilo, propilo, isopropilo, n-butilo o isobutilo.
El término "alquenilo C2-6", tal como se utiliza en el presente documento, se refiere a un grupo de hidrocarburo insaturado, lineal o ramificado, que tiene al menos un doble enlace carbono-carbono y 2-6 átomos de carbono, como el etenilo, el 1-propenilo, el 2-propenilo (alilo), el isopropenilo, el 2-metil-1-propenilo, el 1 -butenilo, el 2-butenilo y similares.
El término "alquinilo C2-6", tal y como se utiliza en este documento, se refiere a un grupo de hidrocarburo insaturado, lineal o ramificado, que tiene al menos un triple enlace carbono-carbono y 2-6 átomos de carbono, como el etilo, el 1-propinilo, el 2-propinilo, el 1 -butilo, el 2-butilo y similares.
El término "cicloalquilo C3-6", tal como se utiliza en el presente documento, se refiere a un grupo de hidrocarburos monocíclicos saturados que tienen de 3 a 6 átomos de carbono, como el ciclopropilo, el ciclobutilo, el ciclopentilo, el ciclohexilo y similares.
El término "arilo C6-10", tal como se utiliza aquí, se refiere a un grupo aromático que tiene de 6 a 10 átomos de carbono, como el fenilo o el naftilo.
El término "alcoxilo C1 -6 ", tal como se utiliza en el presente documento, se refiere a un grupo hidrocarburo monovalente saturado de la fórmula -O-alquilo C1-6, en el que el término "alquilo C1-6" se define como anteriormente, por ejemplo, metoxi, etoxi, n-propoxi, isopropoxi, n-butoxi, isobutoxi, tert-butoxi, sec-butoxi, pentiloxi, isopentiloxi, n-hexiloxi y similares.
El término "alcoxicarbonilo C2-7", tal y como se utiliza en el presente documento, se refiere a un alcoxilo C1-6 unido al resto de la molécula a través de un enlace carbonilo, en el que el término "alcoxilo C1-6" se define como anteriormente, por ejemplo metoxicarbonilo, etoxicarbonilo, n-propoxicarbonilo, isopropoxicarbonilo, n-butoxicarbonilo, isobutoxicarbonilo, tert-butoxicarbonilo, sec-butoxicarbonilo, pentiloxicarbonilo, isopentiloxicarbonilo, n-hexilocarbonilo y similares.
El término "sal", tal y como se utiliza en el presente documento, se refiere a una sal formada a partir de un ácido y el átomo de nitrógeno del anillo de imidazol, donde el ácido se refiere a un ácido inorgánico o a un ácido orgánico comúnmente utilizado en el campo de la química orgánica que puede reaccionar con el átomo de nitrógeno del anillo de imidazol para formar una sal. Los ejemplos de ácido inorgánico incluyen el ácido clorhídrico, el ácido bromhídrico, el ácido yodhídrico, el ácido sulfúrico, el ácido pirosulfúrico, el ácido fosfórico, el ácido nítrico, etc. Los ejemplos de ácido orgánico incluyen el ácido fórmico, el ácido acético, el ácido propiónico, el ácido butírico, el ácido piválico, el ácido trifluoroacético, el ácido difluoroacético, el ácido fluoroacético, el ácido acetoacético, el ácido benzoico, el ácido metanosulfónico, el ácido etanosulfónico, el ácido trifluorometanosulfónico, el ácido bencenosulfónico, el ácido ptoluenosulfónico, el ácido naftalenosulfónico, el ácido canforsulfónico, etc.
El término "sal farmacéuticamente aceptable", tal como se utiliza aquí, se refiere a una sal formada a partir de un ácido farmacéuticamente aceptable y el átomo de nitrógeno del anillo de imidazol. Entre los ejemplos del ácido farmacéuticamente aceptable se encuentran el ácido clorhídrico, el ácido bromhídrico, el ácido sulfúrico, el ácido
fosfórico, el ácido acético, el ácido propiónico, el ácido metanosulfónico, el ácido etanosulfónico, el ácido bencenosulfónico, el ácido p-toluenosulfónico, el ácido naftalenosulfónico, el ácido canforsulfónico, etc.
El compuesto aquí divulgado puede existir en forma de cristal o polimorfo, y puede estar en forma de un solo polimorfo o una mezcla de más de un polimorfo en cualquier relación.
El compuesto de la presente invención puede existir en forma de un solvato, especialmente un hidrato, en el que el compuesto de la presente invención contiene un disolvente polar, en particular agua, etanol, isopropanol, acetato de etilo o acetona, por ejemplo, como elemento estructural de la red cristalina del compuesto. Los disolventes polares, en particular el agua, pueden existir en una cantidad estequiométrica o no estequiométrica.
COMPUESTOS
Es un objeto de la presente invención proporcionar un derivado de etomidato, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo. El derivado de etomidato de la presente invención no sólo tiene una buena actividad anestésica, un rápido inicio y una corta duración de la acción, sino que también muestra poca inhibición de la secreción de cortisol y/o corticosterona en el animal, y por lo tanto tiene tanto un efecto anestésico favorable como perfiles de seguridad.
Específicamente, se proporciona un derivado de etomidato de Fórmula 1 como se indica a continuación:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10,
o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo.
De acuerdo con una realización de la presente invención, uno de X e Y es halógeno, y el otro es hidrógeno.
De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno.
De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
De acuerdo con otra realización de la presente invención, X e Y son halógenos diferentes.
De acuerdo con una realización de la presente invención, X e Y son independientemente flúor, cloro, bromo o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, R1 es alquilo C1-6, por ejemplo, metilo, etilo, propilo, isopropilo, n-butilo o isobutilo.
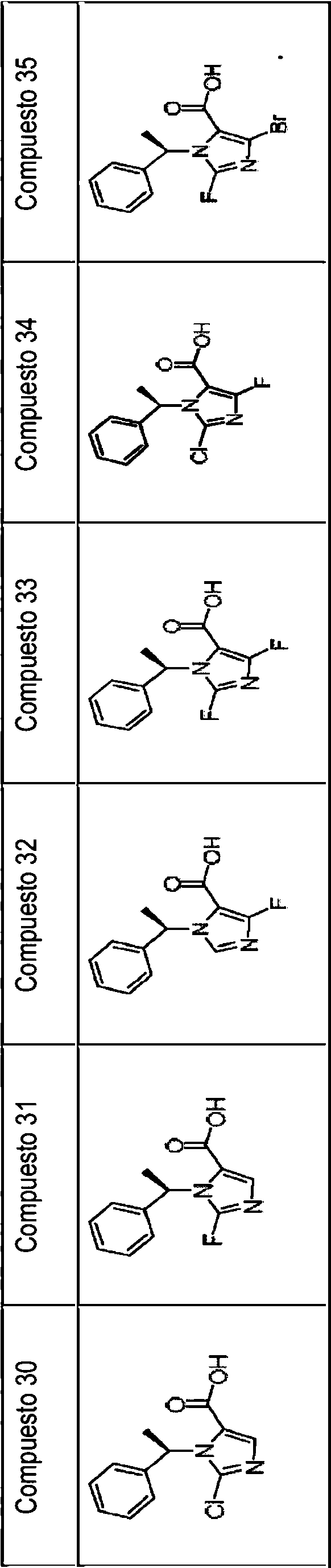
De acuerdo con una realización de la presente invención, el derivado de etomidato de la presente invención se selecciona del grupo que consiste en:

COMPOSICIÓN FARMACÉUTICA Y KIT
Es otro objeto de la presente invención proporcionar una composición farmacéutica que comprenda el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo, o un solvato del mismo, y uno o más portadores farmacéuticamente aceptables.
El término "portador farmacéuticamente aceptable" utilizado en la presente invención se refiere a un diluyente, adyuvante, excipiente o vehículo administrado junto con el agente terapéutico, que, según el buen criterio médico, es adecuado para entrar en contacto con los tejidos del ser humano y/u otros animales sin una toxicidad, irritación, reacción alérgica u otros problemas o complicaciones indebidos más allá de una relación beneficio/riesgo razonable.
El portador farmacéuticamente aceptable que puede emplearse en la composición farmacéutica de la presente invención puede incluir líquidos estériles, como el agua y los aceites, incluidos los aceites de petróleo, de origen animal, vegetal o sintético, como el aceite de cacahuete, el aceite de soja, el aceite mineral, el aceite de sésamo y similares. El agua es un portador ejemplar cuando la composición farmacéutica se administra por vía intravenosa. Las soluciones salinas fisiológicas, así como las soluciones acuosas de dextrosa y glicerol, también pueden emplearse como soportes líquidos, especialmente para soluciones inyectables. Entre los excipientes farmacéuticos adecuados se encuentran el almidón, la glucosa, la lactosa, la sacarosa, la gelatina, la maltosa, la tiza, el gel de sílice, el estearato de sodio, el monoestearato de glicerol, el talco, el cloruro de sodio, la leche desnatada en polvo, el glicerol, el propilenglicol, el agua, el etanol y otros similares. La composición, si se desea, puede contener también pequeñas cantidades de agentes humectantes, emulsionantes o agentes amortiguadores del pH.
La composición farmacéutica de la presente invención puede administrarse por vías adecuadas. Preferentemente, la composición farmacéutica de la presente invención se administra por vía parenteral, por ejemplo, por vía intravenosa, intraarterial, subcutánea, intraperitoneal, intramuscular o percutánea. Más preferentemente, la composición farmacéutica de la presente invención se administra por vía intravenosa.
Es otro objeto de la presente invención proporcionar un kit que comprenda el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo, o un solvato del mismo, o la composición farmacéutica de la presente invención.
TRATAMIENTO Y USO
Es otro objeto de la presente invención proporcionar el derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo, o un solvato del mismo, para su uso en anestesia, particularmente en anestesia intravenosa.
Es otro objeto de la presente invención proporcionar el uso del derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo, o un solvato del mismo en la fabricación de un medicamento anestésico. Preferentemente, el medicamento se administra por vía parenteral, por ejemplo, por vía intravenosa, intraarterial, subcutánea, intraperitoneal, intramuscular o percutánea. Más preferentemente, el medicamento se administra por vía intravenosa.
Es otro objeto de la presente invención proporcionar un uso anestésico, que comprende la administración de una cantidad eficaz del derivado de etomidato de la presente invención, o una sal farmacéuticamente aceptable, un polimorfo o un solvato del mismo. Preferentemente, la administración se realiza por vía parenteral, por ejemplo, por vía intravenosa, intraarterial, subcutánea, intraperitoneal, intramuscular o percutánea. Más preferentemente, la administración se realiza por vía intravenosa.
PROCEDIMIENTO DE PREPARACIÓN
Es otro objeto de la presente invención proporcionar un procedimiento para preparar el derivado de etomidato de la presente invención, que incluye: preparar el derivado de etomidato a partir de un compuesto de Fórmula 2:

en la que X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, uno de X e Y es halógeno, y el otro es hidrógeno.
De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno.
De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
De acuerdo con otra realización de la presente invención, X e Y son halógenos diferentes.
De acuerdo con una realización de la presente invención, X e Y son independientemente flúor, cloro, bromo o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, el compuesto de Fórmula 2 se selecciona del grupo que consiste en:
De acuerdo con una realización de la presente invención, el derivado de etomidato de Fórmula 1 puede prepararse según el siguiente esquema A (condensación con un alcohol) a partir del compuesto de Fórmula 2:

donde X e Y y R1 se definen como arriba.
De acuerdo con una realización de la presente invención, el agente condensador es un compuesto que comprende una estructura de carbodiimida, por ejemplo, EDCI o DCC, etc.
Preferentemente, la reacción descrita anteriormente se realiza a una temperatura de 10°C a 50°C.
Preferentemente, la relación molar del compuesto de Fórmula 2 con R1OH y el agente condensador es 1:(1-10):(1-15).
De acuerdo con otra realización de la presente invención, el derivado de etomidato de Fórmula 1 puede prepararse según el siguiente esquema B a partir del compuesto de Fórmula 2:

donde X, Y y R1 se definen como arriba.
De acuerdo con una realización de la presente invención, el agente halogenante es cloruro de tionilo, cloruro de oxalilo o un reactivo químico similar.
De acuerdo con una realización de la presente invención, el reactivo alcalino es un reactivo orgánico que contiene nitrógeno, por ejemplo, trietilamina, DMAP, etc.
Preferentemente, la reacción descrita anteriormente se realiza a una temperatura de -20°C a 50°C.
Preferentemente, la relación molar del compuesto de Fórmula 2 con respecto a R1OH es de 1 :(1-10).
De acuerdo con otra realización de la presente invención, el derivado de etomidato de Fórmula 1 puede prepararse según el siguiente esquema C a partir del compuesto de Fórmula 2:

donde X, Y y R1 se definen como arriba.
De acuerdo con una realización de la presente invención, H+ significa ácido sulfúrico, HCl, HBr, etc.
Preferentemente, la reacción descrita anteriormente se realiza a una temperatura de -10°C a 60°C.
Preferentemente, la relación molar del compuesto de Fórmula 2 con respecto a R1OH es de 1 :(5-20).
De acuerdo con otra realización de la presente invención, el derivado de etomidato de Fórmula 1 puede prepararse según el siguiente esquema D a partir del compuesto de Fórmula 2:

donde X, Y y R1 se definen como arriba.
El agente activador es un agente que puede activar el grupo acilo para facilitar la esterificación, por ejemplo, cloroformiato de etilo, cloroformiato de bencilo, etc.
Preferentemente, la reacción descrita anteriormente se realiza a una temperatura de -20°C a 50°C.
Preferentemente, la relación molar del compuesto de Fórmula 2 con respecto a R1OH es de 1 :(1-10).
Es otro objeto de la presente invención proporcionar otro procedimiento para preparar el derivado de etomidato de la presente invención, incluyendo: preparar el derivado de etomidato a partir de un compuesto de Fórmula 3:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10.
De acuerdo con una realización de la presente invención, uno de X e Y es halógeno, y el otro es hidrógeno.
De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno.
De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
De acuerdo con una realización de la presente invención, X e Y son halógenos diferentes.
De acuerdo con una realización de la presente invención, X e Y son independientemente flúor, cloro, bromo o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, R1 es alquilo C1-6, por ejemplo, metilo, etilo, propilo, isopropilo, n-butilo o isobutilo.
De acuerdo con una realización de la presente invención, el compuesto de Fórmula 3 se selecciona del grupo que consiste en:

De acuerdo con una realización de la presente invención, el derivado de etomidato de Fórmula 1 puede prepararse según el siguiente esquema E a partir del compuesto de Fórmula 3:

donde X, Y y R1 se definen como arriba.
De acuerdo con una realización de la presente invención, la reacción del esquema E se realiza en presencia de Ph^P y DEAD.
Preferentemente, la reacción descrita anteriormente se realiza a una temperatura de -20°C a 60°C.
Preferentemente, la relación molar del compuesto de Fórmula 3 con el S-fenetanol es de 1:(1-10).
Es otro objeto de la presente invención proporcionar otros procedimientos para preparar el derivado de etomidato de la presente invención. Por ejemplo, el derivado de etomidato de la Fórmula 1 puede prepararse según los siguientes esquemas F y G a partir de compuestos de las Fórmulas 4 y 5, respectivamente

Por ejemplo, cuando X e Y son simultáneamente cloro o bromo, el compuesto de Fórmula 1 puede prepararse haciendo reaccionar el compuesto de Fórmula 4 con NCS o NBS

Por ejemplo, cuando uno de X e Y es flúor o cloro, y el otro es bromo, el compuesto de Fórmula 1 puede prepararse haciendo reaccionar el compuesto de Fórmula 5 con NCS o NBS.
COMPUESTOS INTERMEDIOS
Es otro objeto de la presente invención proporcionar un compuesto intermedio para la preparación del derivado de etomidato de la presente invención, donde el compuesto intermedio tiene la estructura de la Fórmula 2:

en la que X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, uno de X e Y es halógeno, y el otro es hidrógeno.
De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno.
De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
De acuerdo con otra realización de la presente invención, X e Y son halógenos diferentes.
De acuerdo con una realización de la presente invención, X e Y son independientemente flúor, cloro, bromo o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, el compuesto de Fórmula 2 se selecciona del grupo que consiste en:

El compuesto de Fórmula 2 puede obtenerse por hidrólisis del derivado de etomidato de Fórmula 1. Así, el compuesto de Fórmula 2 también puede utilizarse como sustancia de referencia de impurezas para el compuesto de Fórmula 1

Es otro objeto de la presente invención proporcionar otro compuesto intermedio para la preparación del derivado de etomidato de la presente invención, donde el compuesto intermedio tiene la estructura de la Fórmula 3:

en la que,
X e Y son independientemente halógeno o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo formado por halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10.
De acuerdo con una realización de la presente invención,  uno de X e Y es halógeno, y el otro es hidrógeno. De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno. De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
uno de X e Y es halógeno, y el otro es hidrógeno. De acuerdo con otra realización de la presente invención, X e Y son independientemente halógeno. De acuerdo con una realización de la presente invención, X e Y son el mismo halógeno.
De acuerdo con otra realización de la presente invención, X e Y son halógenos diferentes.
De acuerdo con una realización de la presente invención, X e Y son independientemente flúor, cloro, bromo o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
De acuerdo con una realización de la presente invención, R1 es alquilo C1-6, por ejemplo, metilo, etilo, propilo, isopropilo, n-butilo o isobutilo.
De acuerdo con una realización de la presente invención, el compuesto de Fórmula 3 se selecciona del grupo que consiste en:

El compuesto de Fórmula 3 puede prepararse de acuerdo con procedimientos convencionales a partir de materias primas comercialmente disponibles, por ejemplo, refiriéndose a los procedimientos descritos en las siguientes literaturas: [1] Journal of Organic Chemistry, 2011 (20), 76, 8477-8482; [2] Journal of Medicine Chemistry, 2008 (7), 51, 2244-2253; [3] Journal of American Chemical Society, 1973, 95, 4619; y [4] Journal of Organic Chemistry, 1984 (11), 49, 1951-1954.
EJEMPLOS
La presente invención se ha descrito más detalladamente con referencia a los siguientes ejemplos para que se entienda el propósito y la solución técnica de la presente invención. Además, los procedimientos experimentales específicos que no se mencionan en los ejemplos siguientes se llevan a cabo de acuerdo con los procedimientos experimentales convencionales.
Las abreviaturas utilizadas en este documento tienen el siguiente significado:

Preparación del compuesto de Fórmula 3
Ejemplo 1
Preparación del 1H-4-cloroimidazol-5-carboxilato de etilo
A temperatura ambiente, se añadió el ácido 1H-4-cloroimidazol-5-carboxílico disponible en el mercado (300 mg) al etanol (5 ml), después se añadió cloruro de tionilo (0,5 ml) y se dejó reaccionar la mezcla a reflujo durante 1 hora. El disolvente se eliminó por evaporación bajo presión reducida para dar clorhidrato de 1H-4-cloroimidazol-5-carboxilato de etilo como un sólido blanco. Se añadió una solución acuosa saturada de NaHCO3 para neutralizar en un baño de agua helada, y la mezcla resultante se extrajo con acetato de etilo. El disolvente se eliminó por evaporación bajo presión reducida para dar el compuesto del título (290 mg).
MS-ESI [M+H]+ = 175,53
Ejemplo 2
Preparación del 1H-4-fluoroimidazol-5-carboxilato de metilo
En un baño de hielo, se disolvió el 1H-imidazol-4-amino-5-carboxilato de metilo (0,5 g) en una solución de HBF4 al 50% (12,5 ml), y luego se añadió una solución acuosa (1 ml) de NaNO2 (0,28 g). La mezcla se dejó reaccionar continuamente bajo la irradiación de una lámpara de mercurio (234 nm) hasta que no se produjera más evolución de gas nitrógeno. Una vez completada la reacción, se añadió una solución de agua helada 1 N de NaOH a la mezcla en un baño de hielo para ajustar el pH a 6. La capa acuosa se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas se combinaron, se lavaron con salmuera saturada, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron bajo presión reducida. El residuo se separó por cromatografía en columna (EA:PE = 1:1) para dar el compuesto del título (0,23 g) como un aceite incoloro.
MS-ESI [M+H]+ = 145,08
Los compuestos 36-50 se prepararon de acuerdo con el procedimiento descrito en el Ejemplo 1 o 2, utilizando las correspondientes materias primas disponibles en el mercado. Los datos espectrales de masas del compuesto 36-50 se enumeran en la tabla siguiente:

Preparación del compuesto de Fórmula 2
Ejemplo 3
Preparación del ácido R-1-(1-fenil)-1H-4-fluoroimidazol-5-carboxílico (compuesto 32)
El R-1-(1-fenil)-1H-4-fluoroimidazol-5-carboxilato de metilo (2,48 g, 10 mmol) se disolvió en metanol (30 ml) mientras se agitaba en un baño de hielo, y luego se añadió gota a gota una solución de NaOH 1 N. La reacción se controló por TLC. Una vez completada la reacción, el metanol se eliminó por evaporación bajo presión reducida. La capa de agua se ajustó a pH 5 utilizando HCl 1 N, y luego se extrajo con acetato de etilo (25 ml * 3). Las capas orgánicas se combinaron, se lavaron con salmuera saturada, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron para dar el compuesto del título (2,22 g, rendimiento 94,8%) como un sólido blanco.
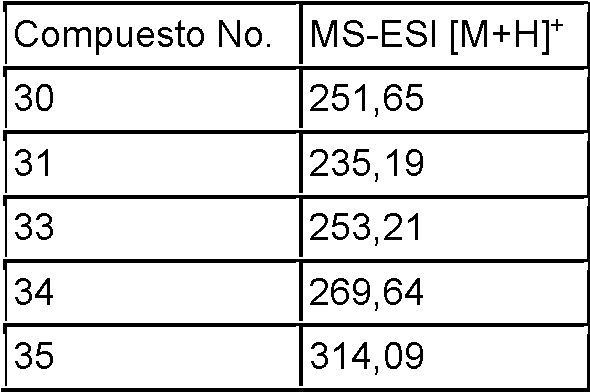
MS-ESI [M+H]+ = 235,18
Los compuestos 30-31 y 33-35 se prepararon de acuerdo con el procedimiento descrito en el Ejemplo 3, utilizando las materias primas correspondientes. Los datos de los espectros de masas de los compuestos 30-31 y 33-35 se enumeran en la siguiente tabla:
Preparación del derivado de etomidato de Fórmula 1
Ejemplo 4
Preparación del R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxilato de metilo (compuesto 13)
A una mezcla de 1H-2-fluoroimidazol-5-carboxilato de metilo (144 mg) y Ph3P (325 mg) en THF (3 ml) se añadió S-fenetanol (122 mg) en THF (2 ml) gota a gota a -30° C. A continuación se añadió DEAD (220 mg) en THF (2 ml) a la solución de reacción. Después de la adición, la solución de reacción se calentó a 0° C. La reacción se monitorizó por TLC. Una vez completada la reacción, el disolvente se eliminó por evaporación bajo presión reducida. El residuo se separó por cromatografía en columna (EA:PE = 1:3) para dar el compuesto del título (73 mg) como un aceite incoloro.
MS-ESI [M+H]+ = 249,21
Ejemplo 5
Preparación del R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxilato de etilo (compuesto 14)
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 4, utilizando 1H-2-fluoroimidazol-5-carboxilato de etilo como materia prima.
MS-ESI [M+H]+ = 263,24
Ejemplo 6
Preparación del R-1-(1-fenetil)-1H-2,4-difluoroimidazol-5-carboxilato de etilo (compuesto 18)
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 4, utilizando 1H-2,4-difluoroimidazol-5-carboxilato de etilo como materia prima.
MS-ESI [M+H]+ = 281,23
Ejemplo 7
Preparación del R-1-(1-fenetil)-1H-4-cloroimidazol-5-carboxilato de etilo (compuesto 4)
1) 1H-4-cloroimidazol-5-carboxilato de etilo: A temperatura ambiente, se añadió el ácido 1H-4-cloroimidazol-5-carboxílico (300 mg) al etanol (5 ml) mientras se agitaba, y a continuación se añadió cloruro de tionilo (0,5 ml). La mezcla se dejó reaccionar a reflujo durante 1 hora. El disolvente se eliminó por evaporación bajo presión reducida para dar clorhidrato de 1H-4-cloroimidazol-5-carboxilato de etilo como un sólido blanco. Se añadió lentamente una solución de agua helada de bicarbonato de sodio para neutralizar en un baño de agua helada, y la mezcla resultante se extrajo con acetato de etilo. El disolvente se eliminó por evaporación bajo presión reducida para dar 1H-4-cloroimidazol-5-carboxilato de etilo (280 mg).
2) El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 4, utilizando como materia prima 1H-4-cloroimidazol-5-carboxilato de etilo.
MS-ESI [M+H]+ = 279,69
Ejemplo 8
Preparación del R-1-(1-fenetil)-1H-4-fluoroimidazol-5-carboxilato de metilo (compuesto 15)
1) 1H-imidazol-4-fluoro-5-carboxilato de metilo: En un baño de hielo, se disolvió el 1H-imidazol-4-amino-5-carboxilato de metilo (0,5 g) en una solución de HBF4 al 50% (12,5 ml) en un baño de hielo, y luego se añadió una solución acuosa (1 ml) de NaNO2 (0,28 g). La mezcla se dejó reaccionar continuamente bajo la irradiación de una lámpara de mercurio (234 nm) hasta que no se produjera más evolución de gas nitrógeno. Una vez completada la reacción, se añadió una solución de agua helada 1 N de NaOH a la mezcla en un baño de hielo para ajustar el pH por encima de 6. La capa acuosa se extrajo con acetato de etilo (20 ml * 3). Las capas orgánicas se combinaron, se lavaron con salmuera saturada, se secaron sobre sulfato sódico anhidro, se filtraron y se concentraron bajo presión reducida. El residuo se separó por cromatografía en columna (EA:PE = 1:1) para dar 1H-4-fluoroimidazol-5-carboxilato de metilo (0,23 g) como un aceite incoloro.
2) El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 4, utilizando como materia prima 1H-4-fluoroimidazol-5-carboxilato de metilo.
MS-ESI [M+H]+ = 249,22
1H-RMN (DMSO, 400MHz): 1,73-1,84 (t, 3H); 3,68 (s, 3H); 6,13 (q, 1H), 7,1-7,3 (m, 5H).
F-RMN 8: -113,40 (s).
Ejemplo 9
Preparación del R-1-(1-fenetil)-1H-4-fluoroimidazol-5-carboxilato de etilo (compuesto 16)
A una mezcla de 1H-4-fluoroimidazol-5-carboxilato de etilo (158 mg, 1,1 mmol) y Ph3P (340 mg, 1,3 mmol) en THF seco (3 ml) se añadió S-fenetanol (134 mg, 1,1 mmol) en THF seco (2 ml) gota a gota. A continuación, se añadió gota a gota DEAD (230 mg, 1,32 mmol) en THF seco (2 ml) a la solución de reacción. Tras la adición, la solución de reacción se calentó lentamente hasta 0° C. La reacción se monitorizó por TLC. Una vez completada la reacción, el disolvente se eliminó por evaporación bajo presión reducida. El residuo se separó por cromatografía en columna (EA:PE = 1:3) para dar el compuesto del título (81 mg, rendimiento del 28%) como un aceite incoloro.
MS-ESI [M+H]+ = 263,24
1H-RMN (CDaCl, 400MHz): 1,31 (t, J = 7,1 Hz, 3H); 1,96 (d, J = 7,2 Hz, 3H); 4,20-4,33 (q, J = 7,1 Hz, 2H); 6,22-6,31 (m, 1H), 7,16-7,38 (m, 5H).
F-RMN 8: -113,40 (s).
Los compuestos 1-3, 5-12, 17 y 19-29 se prepararon de acuerdo con el procedimiento descrito en el Ejemplo 4-9, utilizando los correspondientes compuestos de Fórmula 3 como materias primas.
Ejemplo 10
Preparación de R-1-(1-fenil)-1H-2-cloroimidazol-5-carboxilato de etilo (compuesto 2), R-1-(1-fenil)-1H-4-cloroim idazol-5-carboxilato de etilo (compuesto 4) y R-1-(1-fenil)-1H-2,4-dicloroimidazol-5-carboxilato de etilo (compuesto 6)
El R-1-(1-fenetil)-1H-imidazol-5-carboxilato de etilo (488 mg, 2 mmol) y el NCS (280 mg, 2,1 mmol) se disolvieron en acetonitrilo (20 ml) a temperatura ambiente. La mezcla se calentó a reflujo y se dejó reaccionar durante 12 horas. Una vez completada la reacción, el disolvente se eliminó por evaporación bajo presión reducida. El residuo se separó por cromatografía en columna para dar R-1-(1-fenil)-1H-2-cloroimidazol-5-carboxilato de etilo (80 mg), R-1-(1-fenil)-1H-4-cloroimidazol-5-carboxilato de etilo (120 mg) y R-1-(1-fenil)-1H-2,4-dicloroimidazol-5-carboxilato de etilo (50 mg), respectivamente.
R-1-(1-fenetil)-1H-2-cloroimidazol-5-carboxilato de etilo
1H-RMN <CHaCl, 400 MHz) 8: 1,30 (t, J = 7,1 Hz, 3H); 2,00 (d, J = 7,2 Hz, 3H); 4,22-4,38 (q, J = 7,1 Hz, 2H); 6,25-6,38 (m, 1H), 7,15-7,37 (m, 5H); 7,55 (s, 1H).
MS-ESI [M+H]+ = 279,71.
R-1-(1-fenetil)-1H-2-cloroimidazol-5-carboxilato de etilo
1H-RMN<CH3Cl, 400 MHz) 8: 1,33 (t, J = 7,1 Hz, 3H); 1,97 (d, J = 7,2 Hz, 3H); 4,24-4,31 (q, J = 7,1 Hz, 2H); 6,65-6,77 (m, 1H); 7,15-7,35 (m, 5H); 7,70 (s, 1H).
MS-ESI [M+H]+ = 279,70.
R-1-(1-fenetil)-1H-2,4-dicloroimidazol-5-carboxilato de etilo:
1H-RMN <CHaCl, 400 MHz) 8: 1,31 (t, J = 7,1 Hz, 3H); 1,96 (d, J = 7,2 Hz, 3H); 4,26-4,36 (q, J = 7,1 Hz, 2H); 6,60-6,69 (m, 1H); 7,16-7,36 (m, 5H).
MS-ESI [M+H]+ = 341,15.
Ejemplo 11
Preparación de R-1-(1-fenetil)-1H-2-bromoimidazol-5-carboxilato de etilo (compuesto 7), R-1-(1-fenetil)-1H-4-bromoimidazol-5-carboxilato de etilo (compuesto 10) y R-1-(1-fenetil)-1-hidrógeno-2,4-dibromoimidazol-5-carboxilato de etilo (compuesto 12)
Los compuestos del título se prepararon de acuerdo con el procedimiento descrito en el Ejemplo 10, utilizando R-1-(1-fenil)-1H-imidazol-5-carboxilato de etilo como materia prima y NBS como agente bromante.
R-1-(1-fenetil)-1H-2-bromoimidazol-5-carboxilato de etilo:
1H-RMN (C^C l, 400 MHz) 8: 1,30 (t, J = 7,1 Hz, 3H); 2,00 (d, J = 7,2 Hz, 3H); 4,22-4,38 (q, J = 7,1 Hz, 2H); 6,26-6,40 (m, 1H), 7,09-7,39 (m, 5H); 7,58 (s, 1H).
MS-ESI [M+H]+ = 324,17.
R-1-(1-fenetil)-1H-2-bromoimidazol-5-carboxilato de etilo:
1H-RMN (C^C l, 400 MHz) 8: 1,30 (t, J = 7,1 Hz, 3H); 2,00 (d, J = 7,2 Hz, 3H); 4,20-4,27 (q, J = 7,1 Hz, 2H); 6,65-6,71 (m, 1H), 7,36-7,12 (m, 5H); 7,75 (s, 1H).
MS-ESI [M+H]+ = 324,18.
R-1-(1-fenetil)-1H-2,4-dibromoimidazol-5-carboxilato de etilo:
1H-RMN (CH3Cl, 400 MHz) 8: 1,31 (t, J = 7,1 Hz, 3H); 1,99 (d, J = 7,1 Hz, 3H); 4,35-4,16 (q, J = 7,1 Hz, 2H); 6,49-6,6 (m, 1H); 7,42-7,09 (m, 5H).
MS-ESI [M+H]+ = 403,07.
Ejemplo 12
Preparación del R-1-(1-fenetil)-1H-2-cloro-4-bromo-imidazol-5-carboxilato de etilo (compuesto 19)
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 10, utilizando R-1 -(1-fenetil)-1H-2-cloroimidazol-5-carboxilato de etilo como materia prima y NBS como agente bromante.
MS-ESI [M+H]+ = 358,61.
Ejemplo 13
Preparación del R-1-(1-fenetil)-1H-2-fluoro-4-bromo-imidazol-5-carboxilato de isopropilo (compuesto 24) El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 10, utilizando R-1 -(1-fenetil)-1H-2-fluoroimidazol-5-carboxilato de isopropilo como materia prima y NBS como agente bromante.
MS-ESI [M+H]+ = 356,19.
Ejemplo 14
Preparación del R-1-(1-fenetil)-1H-2-fluoro-4-cloro-imidazol-5-carboxilato de isopropilo (compuesto 25) El ácido R-1-(1-fenil)-1H-2-fluoro-4-cloro-imidazol-5-carboxílico (270 mg, 1 mmol) y la trietilamina (111 mg, 1,1 mmol) se disolvieron en diclorometano seco (15 ml) a -20°C. A continuación, se añadió gota a gota una solución de cloroformiato de etilo (110 mg, 1 mmol) en diclorometano seco (2 ml). La mezcla de reacción se agitó durante 1 hora a -20° C y luego se filtró. Se añadió alcohol isopropílico (0,5 ml) gota a gota al filtrado en un baño de hielo, y se dejó reaccionar la mezcla durante 4 horas en un baño de hielo. Una vez completada la reacción, la solución de reacción se lavó en secuencia con HCl 0,1 N (5 ml * 2), solución saturada de NaHCO3 (10 ml * 2) y salmuera saturada (15 ml * 1). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró bajo presión reducida. El residuo se purificó por cromatografía en columna para dar el compuesto del título (280 mg, rendimiento del 90%) como un aceite incoloro.
MS-ESI [M+H]+ = 311,68.
Ejemplo 15
Preparación del R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxilato de isopropilo (compuesto 26)
A temperatura ambiente, se añadieron el ácido R-1-(1-fenil)-1H-2-fluoroimidazol-5-carboxílico (234 mg, 1 mmol), EDCI (250 mg, 1,3 mmol) y DMAP (12 mg, 0,1 mmol) a diclorometano seco (10 ml) mientras se agitaba. A continuación, se añadió gota a gota alcohol isopropílico (12 mg, 2 mmol). La mezcla se dejó reaccionar mientras se agitaba a temperatura ambiente durante 6 horas. Una vez completada la reacción, la solución de reacción se lavó en secuencia con HCl 0,1 N (5 ml * 2), NaHCO3 saturado (10 ml * 2) y salmuera saturada (15 ml * 1). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró bajo presión reducida. El residuo se purificó por cromatografía en columna para dar el compuesto del título (248 mg, rendimiento del 90%) como un aceite incoloro.
MS-ESI [M+H]+ = 277,28.
Ejemplo 16
Preparación del R-1-(1-fenetil)-1H-4-fluoroimidazol-5-carboxilato de n-butilo (compuesto 27)
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 15, utilizando el ácido R-1-(1-fenil)-1H-4-fluoroimidazol-5-carboxílico y n-butanol como materias primas.
MS-ESI [M+H]+ = 291,31.
Ejemplo 17
Preparación del R-1-(1-fenetil)-1H-2,4-dibromoimidazol-5-carboxilato de isobutilo (compuesto 28)
El compuesto del título se preparó de acuerdo con el procedimiento descrito en el Ejemplo 15, utilizando el ácido R-1-(1-fenil)-1H-2,4-dibromoimidazol-5-carboxílico y el isobutanol como materias primas.
MS-ESI [M+H]+ = 431,09.
Preparación del R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxilato de n-butilo (compuesto 29)
En un baño de hielo, se añadió el ácido R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxílico (470 mg, 2 mmol) a diclorometano seco (10 ml) mientras se agitaba, y luego se añadió cloruro de oxalilo (0,5 ml) y una cantidad catalítica de DMF. Se retiró el baño de hielo y se dejó reaccionar la mezcla a temperatura ambiente durante 12 horas. El disolvente y el exceso de cloruro de oxalilo se eliminaron por evaporación bajo presión reducida para dar cloruro de R-1-(1-fenil)-1H-2-fluoroimidazol-5-carboxilo.
Una solución de cloruro de R-1-(1-fenetil)-1H-2-fluoroimidazol-5-carboxilo preparada en el último paso en diclorometano seco (5 ml) se añadió gota a gota lentamente a una solución de n-butanol (0,2 ml) y DMAP (300 mg, 2,5 mmol) en diclorometano seco (10 ml) en un baño de hielo mientras se agitaba. Se retiró el baño de hielo y se dejó reaccionar la mezcla a temperatura ambiente durante 6 horas. Una vez completada la reacción, la solución de reacción se lavó en secuencia con HCl 0,1 N (5 ml * 2), NaHCO3 saturado (10 ml * 2) y salmuera saturada (15 ml * 1). La capa orgánica se secó sobre sulfato sódico anhidro, se filtró y se concentró bajo presión reducida. El residuo se purificó por cromatografía en columna para dar el compuesto del título (550 mg, rendimiento del 95%) como un aceite incoloro.
MS-ESI [M+H]+ = 291,30.
Los compuestos 1-24 se prepararon de acuerdo con el procedimiento descrito en el Ejemplo 14-18, utilizando los correspondientes compuestos de Fórmula 2 como materias primas.
Los datos espectrales de masas del compuesto 1, 3, 5, 8, 9, 11, 17, 20-23 se enumeran en la tabla siguiente:

Ejemplos experimentales biológicos
Ejemplo experimental 1
Prueba de ED50 y LD50 del derivado de etomidato de la presente invención en ratones
Fármacos de prueba y administración:
Los compuestos 14, 15, 16 y 18 pesados con precisión (5 mg para cada uno de los compuestos) se colocaron en tubos de centrífuga de 10 ml, respectivamente. A continuación, se añadió 1 ml de emulsión en blanco (emulsión en blanco de aceite de soja al 20%) a cada tubo, y la mezcla se sometió a sonicación en un aparato de ultrasonidos durante varios minutos para obtener una emulsión homogénea de 5 mg/ml, que se extrajo con una jeringa de inyección justo antes de su uso. Se utilizó como control una inyección de emulsión grasa de etomidato (disponible en el mercado, 2 mg/ml). La concentración de administración en los ratones se fijó, y el volumen de administración varió de acuerdo con la situación real.
Procedimiento de prueba:
Los valores ED50 y LD50 relativos a la anestesia se determinaron mediante un procedimiento secuencial. Las administraciones de este ensayo se realizaron en ratones KM calificados sanos (machos) mediante una inyección a una tasa constante por la vena caudal durante 10 segundos. Antes de la prueba, se realizó un ensayo preliminar para determinar una dosis aproximada (volumen de administración) que conduce a la anestesia (o muerte) de los animales, que luego se fijó como dosis intermedia en la prueba formal. se establecieron 2-3 grupos de dosis por encima y por debajo del grupo de dosis intermedia con un intervalo de 0,8. La desaparición del reflejo de enderezamiento o la muerte se utilizaron como indicadores de la eficacia farmacológica o de la toxicidad, respectivamente. La prueba formal comenzó con la administración de la dosis intermedia. Si los animales estaban anestesiados, se les administraba una dosis menor; en caso contrario, si los animales no estaban anestesiados, se les administraba una dosis mayor, hasta que el ciclo se repetía por 3-4 veces. Los valores LD50 y ED50 de la anestesia se calcularon mediante un procedimiento secuencial en el software aot425. El IT se calculó de acuerdo con la siguiente ecuación: TI = LD50/ ED50.
Resultados de la prueba:
Los resultados de las pruebas de LD50/ED50 e índice TI de los compuestos en ratones se enumeran en la Tabla 1:
Tabla 1. Resultados de las pruebas de LD50/ED50 e índice TI de los compuestos en ratones (n = 10-20)

Conclusión: el derivado de etomidato de la presente invención logra un efecto anestésico en ratones, y la eficacia anestésica es cercana al etomidato.
Ejemplo experimental 2:
Prueba del derivado de etomidato de la presente invención para el período latente y el período persistente de anestesia en ratones
Los ratones Kunming macho se dividieron en grupos, 5 por cada grupo. El fármaco de prueba se administró por inyección a un ritmo constante a través de la vena caudal durante 10 segundos. Se registró el tiempo de desaparición del reflejo de giro a la derecha (período latente) y el tiempo de recuperación (período persistente).
Los resultados de la prueba del período latente y del período persistente de anestesia de los compuestos en ratones se enumeran en la Tabla 2:
Tabla 2. Resultados de la prueba del período latente y del período persistente de anestesia de los compuestos (ratones, mg/kg, n = 5)

Conclusión: el derivado de etomidato de la presente invención consigue un efecto anestésico cercano al etomidato, junto con un inicio rápido y una corta duración de acción.
Además, el inventor también encontró que en la prueba de anestesia en ratones, los compuestos de la presente invención tenían una influencia menor en la presión sanguínea y en las reacciones de seguimiento después de la recuperación en ratones. Los datos figuran en la Tabla 3:
Tabla 3. Influencia de los compuestos en la presión arterial y reacciones de seguimiento tras la recuperación en ratones

Ejemplo experimental 3
Prueba del grado de inhibición de la secreción de corticosterona en ratas por el derivado de etomidato de la presente invención
El grado de inhibición de la secreción de corticosterona en ratas por el derivado de etomidato de la presente invención se evaluó de acuerdo con el procedimiento descrito en Anesthesiolgy 2010; 112 (3): 637-644 utilizando etomidato como control positivo.
Procedimiento experimental:
Las ratas se recuperaron durante 1 día con cateterismo, y se dividieron en grupos (6 para el grupo de control negativo, 8 para cada uno de los grupos restantes). A continuación, se administraron 0,2 mg/kg de dexametasona (concentración 0,04 mg/mL, volumen de administración 5 mL/kg) por inyección a través de la vena caudal. Después de 2 horas, se tomaron muestras de sangre a través de las arterias carótidas y se determinaron los valores de referencia de la corticosterona sérica (Cümin de corticosterona, control negativo). A continuación, se administraron 0,2 mg/kg de dexametasona, el compuesto 15, el compuesto 16 y el etomidato (control positivo), respectivamente, seguidos inmediatamente por la administración de 25 pg/kg de ACTH1-24 (concentración 5 pg/mL, volumen de administración 5 mL/kg) mediante inyección por vena caudal. Después de 15 minutos, se tomaron muestras de sangre a través de las arterias carótidas y se determinaron las concentraciones de corticosterona (C15min de corticosterona). Los resultados de las pruebas figuran en la Tabla 4:
Tabla 4. El grado de inhibición de la secreción de corticosterona en ratas por el derivado de etomidato de la presente invención

De los resultados anteriores, se puede ver que el derivado de etomidato de la presente invención no muestra ninguna inhibición de la secreción de corticosteroides en la rata.
En resumen, el compuesto de la presente invención no sólo tiene ventajas similares a las del etomidato (es decir, tiene una buena actividad anestésica, un inicio rápido y una corta duración de la acción, con poca influencia en el sistema cardiovascular), sino que también no muestra ninguna inhibición de la secreción de cortisol y/o corticosterona, y por lo tanto tiene tanto un efecto anestésico favorable como perfiles de seguridad.
Claims (10)
1. Un compuesto de etomidato de Fórmula 1:
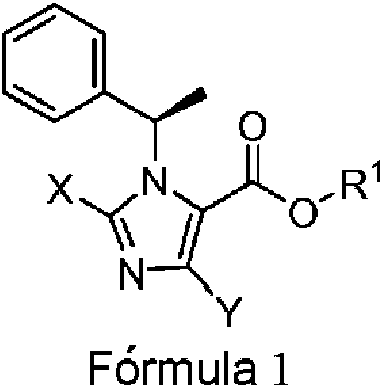
en la que,
X e Y son independientemente halógeno, preferentemente flúor, cloro o bromo, o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10, preferentemente alquilo C1-6, más preferentemente metilo, etilo, propilo, isopropilo, n-butilo o isobutilo,
o una sal farmacéuticamente aceptable, o un solvato del mismo.
2. El compuesto de etomidato de acuerdo con la reivindicación 1, seleccionado del grupo que consiste en:

o una sal farmacéuticamente aceptable, o un solvato del mismo.
3. Una composición farmacéutica que comprende el compuesto de etomidato de acuerdo con la reivindicación 1 o 2, o una sal farmacéuticamente aceptable, o un solvato del mismo, y uno o más portadores farmacéuticamente aceptables.
4. El compuesto de etomidato de acuerdo con la reivindicación 1 o 2, o una sal farmacéuticamente aceptable, o un solvato del mismo, para su uso como fármaco anestésico, especialmente como fármaco anestésico intravenoso.
5. Un procedimiento para preparar el compuesto de etomidato de acuerdo con la reivindicación 1, que incluye: preparar el compuesto de etomidato a partir de un compuesto de Fórmula 2:

en la que X e Y son independientemente halógeno, preferentemente flúor, cloro o bromo, o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo.
6. El procedimiento de acuerdo con la reivindicación 5, en el que el compuesto de Fórmula 2 se selecciona del grupo que consiste en:

7. Un compuesto de Fórmula 2:

en la que,
X e Y son independientemente halógeno, preferentemente flúor, cloro o bromo, o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo,
o una sal, o un solvato del mismo.
8. El compuesto de Fórmula 2 de acuerdo con la reivindicación 7, seleccionado del grupo que consiste en:

o una sal, o un solvato del mismo.
9. Un procedimiento para preparar el compuesto de etomidato de acuerdo con la reivindicación 1, que incluye: preparar el derivado de etomidato a partir de un compuesto de Fórmula 3:

en la que,
X e Y son independientemente halógeno, preferentemente flúor, cloro o bromo, o hidrógeno, con la condición de que X e Y no sean hidrógeno al mismo tiempo; y
R1 es alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo C3-6 o arilo C6-10, opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en halógeno, hidroxilo, amino, ciano, alcoxilo C1-6, alcoxicarbonilo C2-7, cicloalquilo C3-6 y arilo C6-10, preferentemente alquilo C1-6, más preferentemente metilo, etilo, propilo, isopropilo, n-butilo o isobutilo.
10. El procedimiento de acuerdo con la reivindicación 9, en el que el compuesto de Fórmula 3 se selecciona del grupo que consiste en:
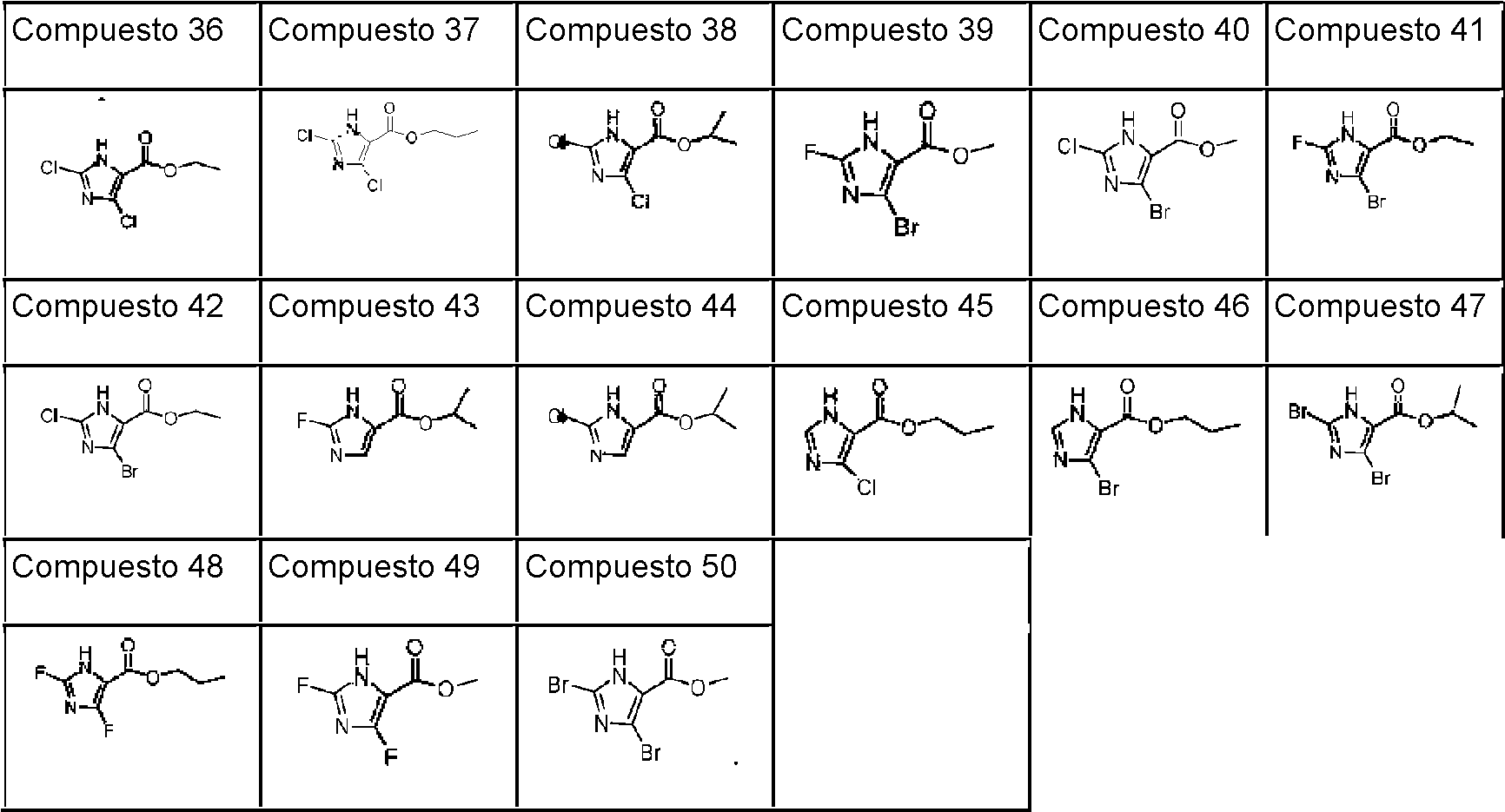
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201510650913 | 2015-10-10 | ||
| PCT/CN2016/101696 WO2017059827A1 (zh) | 2015-10-10 | 2016-10-10 | 依托咪酯衍生物及其中间体、制备方法和用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2913090T3 true ES2913090T3 (es) | 2022-05-31 |
Family
ID=58487268
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16853130T Active ES2913090T3 (es) | 2015-10-10 | 2016-10-10 | Derivado e intermedio del etomidato, procedimiento de preparación y uso del mismo |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US10392352B2 (es) |
| EP (1) | EP3360863B1 (es) |
| JP (1) | JP6983416B2 (es) |
| CN (2) | CN108349906B (es) |
| CA (1) | CA3001431C (es) |
| DK (1) | DK3360863T3 (es) |
| ES (1) | ES2913090T3 (es) |
| HU (1) | HUE058909T2 (es) |
| PL (1) | PL3360863T3 (es) |
| WO (1) | WO2017059827A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109748876A (zh) * | 2017-11-02 | 2019-05-14 | 北京蓝丹医药科技有限公司 | 依托咪酯衍生物及其制备方法 |
| CN109824656B (zh) * | 2018-01-30 | 2023-11-10 | 成都麻沸散医药科技有限公司 | 多取代咪唑甲酸酯类衍生物及其用途 |
| US12570612B2 (en) | 2019-10-11 | 2026-03-10 | Chengdu Mfs Pharma. Co., Ltd. | Substituted nitrogen heterocyclic compound and anesthetic effect thereof |
| CN116262729A (zh) * | 2021-12-15 | 2023-06-16 | 燃点(南京)生物医药科技有限公司 | 一类依托咪酯杂质化合物、制备方法及其应用 |
| CN116265442B (zh) * | 2021-12-16 | 2024-08-06 | 燃点(南京)生物医药科技有限公司 | 一种依托咪酯的制备方法 |
| CN120769745A (zh) * | 2024-05-27 | 2025-10-10 | 江苏恩华药业股份有限公司 | 含有依托咪酯衍生物与环泊酚的药物组合及其用途 |
| WO2025247238A1 (zh) * | 2024-05-27 | 2025-12-04 | 江苏恩华药业股份有限公司 | 含有依托咪酯衍生物与环泊酚的药物组合及其用途 |
| CN119462514B (zh) * | 2025-01-16 | 2025-03-14 | 四川大学华西医院 | 依托咪酯衍生物及其制备方法和在制备麻醉药物中的用途 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8557856B2 (en) * | 2008-03-31 | 2013-10-15 | The General Hospital Corporation | Etomidate analogues with improved pharmacokinetic and pharmacodynamic properties |
| WO2011005969A2 (en) * | 2009-07-10 | 2011-01-13 | The General Hospital Corporation | Etomidate analogues that do not inhibit adrenocortical steroid synthesis |
| DK2802325T3 (en) * | 2012-01-13 | 2017-03-13 | Massachusetts Gen Hospital | ANESTHETIC RELATIONS AND RELATED APPLICATIONS THEREOF |
| CN104706587A (zh) * | 2013-12-16 | 2015-06-17 | 天津迈迪瑞康生物医药科技有限公司 | 一种依托咪酯脂肪乳浓缩液、其制备方法及用途 |
| CN103739553B (zh) * | 2013-12-23 | 2014-11-12 | 四川大学华西医院 | 含有醚侧链的n-取代咪唑羧酸酯手性化合物、制备方法和用途 |
-
2016
- 2016-10-10 EP EP16853130.9A patent/EP3360863B1/en active Active
- 2016-10-10 CN CN201680059075.4A patent/CN108349906B/zh active Active
- 2016-10-10 WO PCT/CN2016/101696 patent/WO2017059827A1/zh not_active Ceased
- 2016-10-10 ES ES16853130T patent/ES2913090T3/es active Active
- 2016-10-10 CN CN202111196263.0A patent/CN113880770A/zh active Pending
- 2016-10-10 CA CA3001431A patent/CA3001431C/en active Active
- 2016-10-10 US US15/767,009 patent/US10392352B2/en active Active
- 2016-10-10 HU HUE16853130A patent/HUE058909T2/hu unknown
- 2016-10-10 JP JP2018537709A patent/JP6983416B2/ja active Active
- 2016-10-10 DK DK16853130.9T patent/DK3360863T3/da active
- 2016-10-10 PL PL16853130.9T patent/PL3360863T3/pl unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3360863A1 (en) | 2018-08-15 |
| JP6983416B2 (ja) | 2021-12-17 |
| CN108349906A (zh) | 2018-07-31 |
| EP3360863B1 (en) | 2022-04-20 |
| WO2017059827A1 (zh) | 2017-04-13 |
| CN108349906B (zh) | 2021-11-02 |
| US10392352B2 (en) | 2019-08-27 |
| CN113880770A (zh) | 2022-01-04 |
| CA3001431C (en) | 2023-09-26 |
| DK3360863T3 (da) | 2022-06-13 |
| US20180297958A1 (en) | 2018-10-18 |
| EP3360863A4 (en) | 2019-06-19 |
| HUE058909T2 (hu) | 2022-09-28 |
| CA3001431A1 (en) | 2017-04-13 |
| PL3360863T3 (pl) | 2022-08-08 |
| JP2018530611A (ja) | 2018-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2913090T3 (es) | Derivado e intermedio del etomidato, procedimiento de preparación y uso del mismo | |
| JP7850537B2 (ja) | トレプロスチニル誘導体ならびにその組成物および使用 | |
| DD273833A5 (de) | N 9 - cyclopentylsubstituierte adeninderivate | |
| CN106478502B (zh) | 1,2,3,4-四氢异喹啉衍生物、其制备方法和应用 | |
| JP6905597B2 (ja) | N−置換イミダゾールカルボン酸エステル系化合物とその調製方法及び用途 | |
| CN103588757B (zh) | 超短效麻醉效应的n-取代咪唑羧酸酯类化合物、制备方法和用途 | |
| ES2909438T3 (es) | Enantiómero (S) de mepacina como inhibidor de paracaspasa (MALT1) para tratar cáncer | |
| KR20130040180A (ko) | 피리피로펜 유도체의 제조방법 | |
| WO2019156074A1 (ja) | アミノアルキル化合物 | |
| EP0136198A1 (fr) | Dérivés de triazolo pyrimidine, leur procédé de préparation et leur application thérapeutique en tant que toni-cardiaques | |
| JP2980749B2 (ja) | フラン誘導体 | |
| PT1221445E (pt) | Compostos pentacíclicos derivados de taxamos | |
| JPS637549B2 (es) | ||
| SU1634134A3 (ru) | Способ получени 4-оксо-4-(замещенный фенил)-бутеноилсалицилатов Е-конфигурации | |
| KR910002838B1 (ko) | 4.7-디히드로피라졸로[3,4-b]피리딘 유도체의 제조방법 | |
| IL29707A (en) | Phenthiazine derivatives | |
| SU793404A3 (ru) | Способ получени производных винциновой кислоты или их солей или их четвертичных солей | |
| BRPI0822432B1 (pt) | "processo para a preparação de composto intermediário para sintetizar um antiulcerante" | |
| SU48311A1 (ru) | Способ получени ацетильных производных альфа и альфа' аминоникотина | |
| KR950014459B1 (ko) | 코르티코스테롤-21-알칸올 에테르 | |
| KR810001958B1 (ko) | 모라노린 유도체의 제조법 | |
| PT93725A (pt) | Processo para a preparacao de derivados de benzopirano anti-hipertensivos | |
| JPS60246376A (ja) | N置換3,4−ジヒドロピリミジン誘導体、その製造方法および循環系疾患治療剤 | |
| JPS60255778A (ja) | 2位置換ジヒドロピリミジン誘導体およびその製造法 | |
| JPS6213359B2 (es) |