KR880000112B1 - 메타크릴산의 정제법 - Google Patents
메타크릴산의 정제법 Download PDFInfo
- Publication number
- KR880000112B1 KR880000112B1 KR1019860001812A KR860001812A KR880000112B1 KR 880000112 B1 KR880000112 B1 KR 880000112B1 KR 1019860001812 A KR1019860001812 A KR 1019860001812A KR 860001812 A KR860001812 A KR 860001812A KR 880000112 B1 KR880000112 B1 KR 880000112B1
- Authority
- KR
- South Korea
- Prior art keywords
- acid
- methacrylic acid
- exchange resin
- solution
- maleic acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C57/00—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms
- C07C57/02—Unsaturated compounds having carboxyl groups bound to acyclic carbon atoms with only carbon-to-carbon double bonds as unsaturation
- C07C57/03—Monocarboxylic acids
- C07C57/04—Acrylic acid; Methacrylic acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/47—Separation; Purification; Stabilisation; Use of additives by solid-liquid treatment; by chemisorption
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S203/00—Distillation: processes, separatory
- Y10S203/22—Accessories
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description

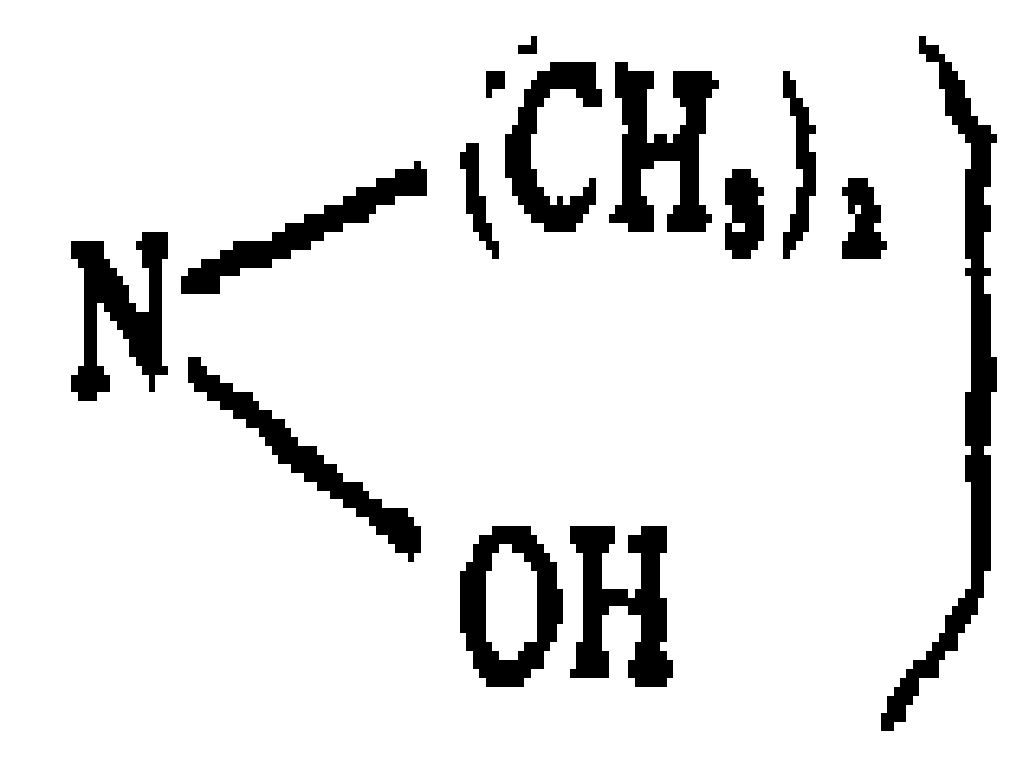







Claims (7)
- 4탄소원자 화합물을 증기의 존재하에 산소분자 함유 기체를 이용하여 촉매산화시켜서 수득한 메타크릴산 함유 용액을 정제하는 방법에 있어서, 그 메타크릴산용액을 염기성-교환수지와 접촉시킴을 특징으로 하는 메타크릴산 함유 용액의 정제방법.
- 제1항에 있어서, 4탄소원자 화합물이 이소부틸렌, t-부탄올, 메타크롤레인 또는 이소부틸알데히드임을 특징으로 하는 정제방법.
- 제1항에 있어서, 용액을 용매로 추출하고, 메타크릴산을 함유하는 추출액을 염기성 음이온-교환수지와 접촉시킴을 특징으로 하는 정제방법.
- 제1항에 있어서, 메탄올을 이용하여 메타크릴산을 에스테르화시켜 수득한, 메틸 메타크릴레이트와 반응하지않는 메타크릴레트를 함유하는 반응 혼합물을 증류하고, 메타크릴산을 함유하는 분획을 염기성-음이온 교환수지와 접촉시킴을 특징으로 하는 정제방법.
- 제1항에 있어서, 용액을 염기성 음이온-교환수지와 접촉시켜 수득한 수지에 말레산을 용리액으로 공급하여 시크라콘산을 분리, 회수한 후 용리액으로 처리한 수지에 강 알칼리를 공급하여 말레산을 용출시킴으로써 수지를 재생시킴을 특징으로 하는 정제방법.
- 제1항에 있어서, 용액을 10∼60℃에서 0.2l∼20l/lhr의 공간 속도로 염기성 음이온-교환수지와 교환시킴을 특징으로 하는 정제방법.
- 말레산 및 시트라콘산이 흡착되어 있는 염기성 음이온-교환수지에 용리액으로서 말레산을 공급하여 시트라콘산을 분리 및 회수한 후 강알칼리를 공급하여 말레산을 용출, 회수함을 특징으로 하는 말레산 및 시트라콘산이 흡착된염기성 음이온-교환수지로 부터 시트라콘산을 분리하는 방법.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP60049248A JPH0669983B2 (ja) | 1985-03-14 | 1985-03-14 | メタクリル酸の精製法 |
| JP49248 | 1985-03-14 | ||
| JP74351 | 1985-04-10 | ||
| JP60074351A JPS61233644A (ja) | 1985-04-10 | 1985-04-10 | シトラコン酸の分離法 |
| JP136882 | 1985-06-25 | ||
| JP13688285A JPH0684330B2 (ja) | 1985-06-25 | 1985-06-25 | メタクリル酸の精製法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR860007190A KR860007190A (ko) | 1986-10-08 |
| KR880000112B1 true KR880000112B1 (ko) | 1988-03-12 |
Family
ID=27293569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1019860001812A Expired KR880000112B1 (ko) | 1985-03-14 | 1986-03-13 | 메타크릴산의 정제법 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US4879412A (ko) |
| EP (1) | EP0194867B1 (ko) |
| KR (1) | KR880000112B1 (ko) |
| CN (1) | CN86102590B (ko) |
| CA (1) | CA1274541A (ko) |
| DE (1) | DE3661909D1 (ko) |
| IN (1) | IN163288B (ko) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5132456A (en) * | 1991-05-07 | 1992-07-21 | The Regents Of The University Of California | Sorption of carboxylic acid from carboxylic salt solutions at PHS close to or above the pKa of the acid, with regeneration with an aqueous solution of ammonia or low-molecular-weight alkylamine |
| JPH08134011A (ja) | 1994-11-04 | 1996-05-28 | Mitsui Toatsu Chem Inc | メタクリル酸の精製方法 |
| US6153791A (en) * | 1999-08-02 | 2000-11-28 | Archer-Daniels-Midland Company | Process for purifying 2-keto-L-gulonic acid |
| GB0403592D0 (en) | 2004-02-18 | 2004-03-24 | Lucite Int Uk Ltd | A catalyst system |
| DE102010062601A1 (de) * | 2010-12-08 | 2012-06-14 | Evonik Stockhausen Gmbh | Verfahren zur Herstellung von (Meth)Acrylsäureestern |
| BR112013019323B1 (pt) | 2011-02-09 | 2019-12-24 | Lucite International Uk Limited | método de extrair ácido (met)acrílico a partir de um meio de reação aquoso, processo para a produção de ácido (met)acrílico, e, método de preparar polímeros ou copolímeros de ácido (met)acrílico ou ésteres de ácido (met)acrílico |
| WO2013163806A1 (en) | 2012-05-03 | 2013-11-07 | Evonik Industries Ag | Process for preparation of highly pure, non-yellowing methacrylic acid |
| CN104203894B (zh) * | 2012-05-03 | 2017-08-04 | 赢创罗姆有限公司 | 高纯度非黄变(甲基)丙烯酸的制备方法 |
| EP3039006B1 (en) * | 2013-08-28 | 2018-10-24 | Archer Daniels Midland Co. | Separation of propionic acid from acrylic acid |
| WO2017188220A1 (ja) * | 2016-04-28 | 2017-11-02 | 三菱ケミカル株式会社 | メタクリル酸の精製方法及び製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1452566A (fr) * | 1964-11-06 | 1966-02-25 | Mitsubishi Petrochemical Co | Procédé perfectionné pour récupérer les esters acryliques et méthacryliques sous forme purifiée à partir de solutions aqueuses diluées d'acide acrylique et méthacrylique |
| AT261561B (de) * | 1966-02-01 | 1968-05-10 | Donau Chemie Ag | Verfahren zur Entfernung insbesondere schwach saurer Verunreinigungen aus alkaliempfindlichen, beschränkt wasserlöslichen Flüssigkeiten |
| JPS502900Y2 (ko) * | 1971-06-11 | 1975-01-25 | ||
| JPS5726253B2 (ko) * | 1973-09-13 | 1982-06-03 | ||
| JPS6054295B2 (ja) * | 1977-01-11 | 1985-11-29 | 三井東圧化学株式会社 | アクリル酸のソ−ダ塩の精製方法 |
| NL7807420A (nl) * | 1977-07-22 | 1979-01-24 | Mitsubishi Chem Ind | Werkwijze voor het afscheiden van methacrylzuur uit een mengsel, dat water, methanol, methacrylzuur en methylmethacrylaat bevat. |
| JPS56108735A (en) * | 1980-02-01 | 1981-08-28 | Sumitomo Chem Co Ltd | Treating method of waste water in preparation of methacrylic acid |
| JPS5777643A (en) * | 1980-10-31 | 1982-05-15 | Asahi Chem Ind Co Ltd | Stabilizing method of methacrylic acid against heat |
-
1986
- 1986-03-03 US US06/835,737 patent/US4879412A/en not_active Expired - Lifetime
- 1986-03-04 IN IN160/CAL/86A patent/IN163288B/en unknown
- 1986-03-12 EP EP86301763A patent/EP0194867B1/en not_active Expired
- 1986-03-12 DE DE8686301763T patent/DE3661909D1/de not_active Expired
- 1986-03-13 KR KR1019860001812A patent/KR880000112B1/ko not_active Expired
- 1986-03-13 CA CA000503986A patent/CA1274541A/en not_active Expired - Lifetime
- 1986-03-14 CN CN86102590A patent/CN86102590B/zh not_active Expired
Also Published As
| Publication number | Publication date |
|---|---|
| EP0194867A1 (en) | 1986-09-17 |
| IN163288B (ko) | 1988-09-03 |
| CN86102590B (zh) | 1988-08-03 |
| KR860007190A (ko) | 1986-10-08 |
| DE3661909D1 (en) | 1989-03-02 |
| EP0194867B1 (en) | 1989-01-25 |
| CA1274541A (en) | 1990-09-25 |
| US4879412A (en) | 1989-11-07 |
| CN86102590A (zh) | 1987-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2102379C1 (ru) | Способ получения уксусной кислоты | |
| US4238294A (en) | Process for recovering heavy metal ions or heavy metal ions and halogen values from solutions comprising a lower aliphatic monocarboxylic acid | |
| CN100522916C (zh) | 从甲醇羰基化工艺流中去除还原高锰酸盐的化合物 | |
| US5414154A (en) | Phenol with low levels of methylbenzofuran | |
| KR880000112B1 (ko) | 메타크릴산의 정제법 | |
| CA2119298A1 (en) | Process for the removal of corrosion metal contaminants from liquid compositions | |
| US20020188151A1 (en) | Process for producing methyl methacrylate | |
| NL8204901A (nl) | Zuivering van carbonyleringsprodukten. | |
| EP0343583B1 (en) | A process for purifying methyl methacrylate | |
| US4039428A (en) | Purification of propionic acid | |
| US5840643A (en) | Recovery of oxidation catalysts used for the production of trimellitic acid | |
| WO2006121081A1 (ja) | 高純度アセトニトリル及びその製造方法 | |
| EP0169254B1 (en) | Process for purification of 1,2-unsaturated carboxylic acids and/or esters thereof | |
| US4061546A (en) | Purification of acetic acid | |
| EP0062653B1 (en) | Vinyl acetate purification process | |
| US4269705A (en) | Method for removing chromium ions from aqueous solutions of organic acids | |
| JPS647064B2 (ko) | ||
| JP3476226B2 (ja) | メタクリルアミド水溶液の精製方法 | |
| JPH02279655A (ja) | アクリル酸エステルの製造方法 | |
| JPS6160623A (ja) | t−ブチルスチレンの精製法 | |
| JPS6124533A (ja) | 有機溶媒の脱アルコ−ル方法 | |
| JPH1135542A (ja) | 蒸留回収/イオン交換樹脂処理プロセスによるアセトニトリルの精製 | |
| JPH05255155A (ja) | tert−ブチルアルコールの精製法 | |
| JPS60130546A (ja) | 1,2−不飽和カルボン酸の精製方法 | |
| JPH0722714B2 (ja) | イオン交換樹脂の再生方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| PA0109 | Patent application |
Patent event code: PA01091R01D Comment text: Patent Application Patent event date: 19860313 |
|
| PA0201 | Request for examination | ||
| PG1501 | Laying open of application | ||
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
Comment text: Notification of reason for refusal Patent event date: 19870429 Patent event code: PE09021S01D |
|
| G160 | Decision to publish patent application | ||
| PG1605 | Publication of application before grant of patent |
Comment text: Decision on Publication of Application Patent event code: PG16051S01I Patent event date: 19871229 |
|
| E701 | Decision to grant or registration of patent right | ||
| PE0701 | Decision of registration |
Patent event code: PE07011S01D Comment text: Decision to Grant Registration Patent event date: 19880527 |
|
| GRNT | Written decision to grant | ||
| PR0701 | Registration of establishment |
Comment text: Registration of Establishment Patent event date: 19880718 Patent event code: PR07011E01D |
|
| PR1002 | Payment of registration fee |
Payment date: 19880718 End annual number: 3 Start annual number: 1 |
|
| PR1001 | Payment of annual fee |
Payment date: 19910206 Start annual number: 4 End annual number: 4 |
|
| PR1001 | Payment of annual fee |
Payment date: 19920207 Start annual number: 5 End annual number: 5 |
|
| PR1001 | Payment of annual fee |
Payment date: 19930303 Start annual number: 6 End annual number: 6 |
|
| PR1001 | Payment of annual fee |
Payment date: 19940215 Start annual number: 7 End annual number: 7 |
|
| PR1001 | Payment of annual fee |
Payment date: 19950209 Start annual number: 8 End annual number: 8 |
|
| PR1001 | Payment of annual fee |
Payment date: 19960309 Start annual number: 9 End annual number: 9 |
|
| PR1001 | Payment of annual fee |
Payment date: 19970307 Start annual number: 10 End annual number: 10 |
|
| PR1001 | Payment of annual fee |
Payment date: 19980303 Start annual number: 11 End annual number: 11 |
|
| PR1001 | Payment of annual fee |
Payment date: 19990227 Start annual number: 12 End annual number: 12 |
|
| PR1001 | Payment of annual fee |
Payment date: 20000229 Start annual number: 13 End annual number: 13 |
|
| PR1001 | Payment of annual fee |
Payment date: 20010307 Start annual number: 14 End annual number: 14 |
|
| PR1001 | Payment of annual fee |
Payment date: 20020307 Start annual number: 15 End annual number: 15 |
|
| PR1001 | Payment of annual fee |
Payment date: 20030224 Start annual number: 16 End annual number: 16 |
|
| PR1001 | Payment of annual fee |
Payment date: 20040219 Start annual number: 17 End annual number: 17 |
|
| FPAY | Annual fee payment |
Payment date: 20050225 Year of fee payment: 18 |
|
| PR1001 | Payment of annual fee |
Payment date: 20050225 Start annual number: 18 End annual number: 18 |
|
| EXPY | Expiration of term | ||
| PC1801 | Expiration of term |