WO1999005106A1 - Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse - Google Patents
Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse Download PDFInfo
- Publication number
- WO1999005106A1 WO1999005106A1 PCT/FR1998/001640 FR9801640W WO9905106A1 WO 1999005106 A1 WO1999005106 A1 WO 1999005106A1 FR 9801640 W FR9801640 W FR 9801640W WO 9905106 A1 WO9905106 A1 WO 9905106A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl group
- compounds
- day
- compounds according
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- TTWJBBZEZQICBI-UHFFFAOYSA-N CCN(CC)CCNC(c(c(OC)c1)cc(Cl)c1N)=O Chemical compound CCN(CC)CCNC(c(c(OC)c1)cc(Cl)c1N)=O TTWJBBZEZQICBI-UHFFFAOYSA-N 0.000 description 1
- MLIHLJVHWFKPQE-UHFFFAOYSA-N COC(C(C=C1Cl)/C(/NCCN2CNCCC2)=[O]\C)C=C1N Chemical compound COC(C(C=C1Cl)/C(/NCCN2CNCCC2)=[O]\C)C=C1N MLIHLJVHWFKPQE-UHFFFAOYSA-N 0.000 description 1
- LGYPZTNCXCTAGC-UHFFFAOYSA-O C[N+](CCNC(c(cc(c(N)c1)Cl)c1OC)=O)(CC1)CCC1O Chemical compound C[N+](CCNC(c(cc(c(N)c1)Cl)c1OC)=O)(CC1)CCC1O LGYPZTNCXCTAGC-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/20—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by singly bound oxygen or sulphur atoms
- C07D211/22—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by singly bound oxygen or sulphur atoms by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
Definitions
- the present invention relates to the use of benzamide-type compounds as stimulants of gastrointestinal motility, their pharmaceutically acceptable salts and their quaternary ammonium salts, as well as their methods of preparation.
- the compounds according to the present invention are also analgesics Benzamides and in particular 4-am ⁇ no-5-chloro-2-methoxybenzam ⁇ des such as metoclopramide (A)
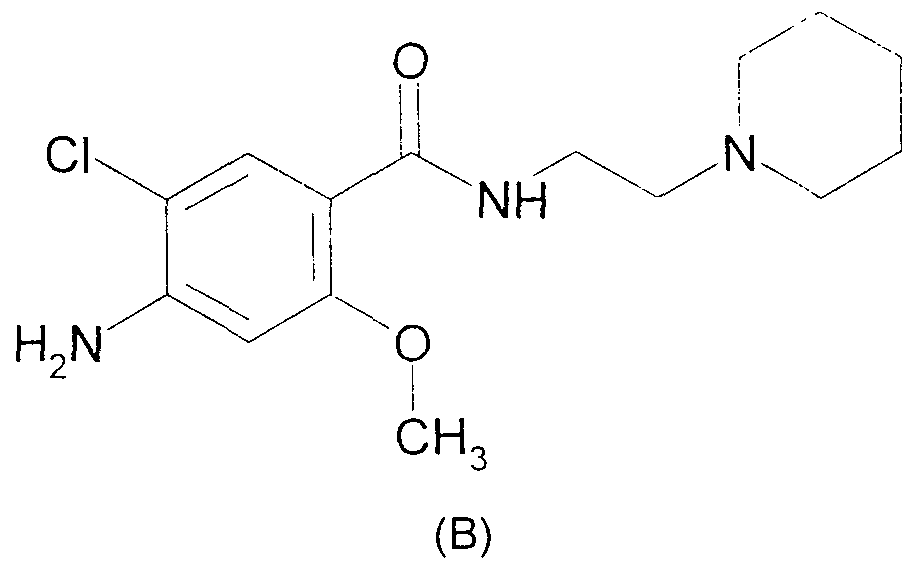
- BE 620543 describes benzamides and in particular the compound of formula (B)
- WO 93/03725 describes compounds close to BE 620 543 and in particular the compounds of formulas (C) and (D);
- piperidinoalkyl amides substituted or not by an alkyl and the 4-hydroxypipe ⁇ dinoalkyle esters therefore have an activity only on the upper intestinal part
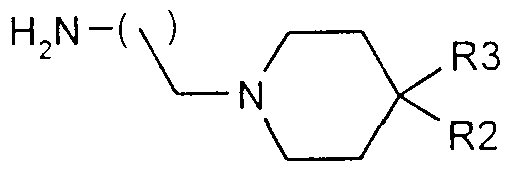
- the present invention therefore relates to compounds of general formula I
- - R-] is a linear alkyl, branched or cyclized C1, Cg or cycloalkyl C3 to Cg, said alkyl group can, if necessary, form a ring of 4 to 5 carbon atoms in the ring, in combination with the carbon in position 3 of the aromatic nucleus, and with that in position 2 carrying the oxygen atom,
- R2 is a hydrogen atom, or a C- alkyl group
- R3 is a hydrogen atom, or a C- alkyl group
- the piperidine nitrogen atom can also be in N-oxide form (N + -0 ⁇ ) or salified with a pharmaceutically acceptable acid such as hydrochloric acid, bromhydnque, sulfu ⁇ que, nitric, phospho ⁇ que, formic, acetic, propionic, glycolic oxalic smoke, lactic, succinic, tart ⁇ que, malic, pamoic
- a pharmaceutically acceptable acid such as hydrochloric acid, bromhydnque, sulfu ⁇ que, nitric, phospho ⁇ que, formic, acetic, propionic, glycolic oxalic smoke, lactic, succinic, tart ⁇ que, malic, pamoic
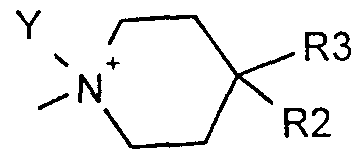
- the piperidine can also be in the form of a quaternary ammonium salt according to the following formula
- CX - or Y is a C- alkyl group
- R2 and R3 are as defined above
- the subject of the invention is the use of one or more compounds of formula I described above, for the preparation of a medicament intended for the treatment of dysfunctions of the upper and lower digestive tract such as
- the subject of the invention is also any pharmaceutical composition comprising one or more compounds of formula I described above, in combination with a pharmaceutically acceptable vehicle.
- the pharmaceutical compositions of the invention can be administered by oral, parenteral or rectal route.
- the pharmaceutical compositions according to the invention are characterized in that the dosage of active principle is approximately 0.1 ⁇ g / kg / day to 20 mg / kg / day by oral and rectal route and approximately 0, 1 ⁇ g / kg / day to 2 mg / kg / day parenterally
- Preferred pharmaceutical compositions according to the invention are in a form which can be administered orally, in a unit dose of 10 ⁇ g to 200 mg of active principle per dose, and preferably from 0.1 mg to 200 mg of active principle per dose, 1 to 4 times a day
- compositions according to the invention are presented in a form which can be administered parenterally, in a unit dose of 10 ⁇ g to 100 mg of active principle per injection, at the rate of 1 to 2 injections per day.
- FIGURE 1 in which R-
- Step 1 consists in condensing diamine III with acid II by one of the coupling methods known in the literature, preferably with CDI in a solvent such as THF at room temperature for 30 minutes to 24 hours to lead to compound V, namely to a compound of the above-mentioned formula I in which R-
- CH3, and R2 and R3 are as defined above.
- Compound V can also be obtained by stage 2 which consists in condensing in the same manner as in stage 1, the hydrochloride of the chloroalkyl amino in the presence of a base such as thethylamine.
- Step 3 consists in reacting the desired piperidine on compound IV in toluene at a temperature between 60 and 110 ° C for 1 h to 48 h.
- Step 4 consists in demethylating compound V with sodium ethane thiolate (CH3CH2SNa, prepared from ethanethiol and a base such as NaH) in a solvent such that DMF or DMSO at 80-90X for 15 minutes at 2 h and then alkylate the phenate formed with an R-
- Step 5 leading to the compound of the above-mentioned formula V, consists in condensing diamine III on acid VII by one of the coupling methods known in the literature, preferably by an acid chloride prepared with thionyl chloride in toluene at reflux for 1 h then reaction with the corresponding diamine in dichloromethane at room temperature for 30 minutes to 12 h
- the hydrolysis of acetyl is carried out in the presence of a sodium hydroxide-EtOH mixture at 60 ° C for 30 minutes to 24 h
- Step 6 leading to the compound of formula VI above, is identical to step 4 of scheme 1
- Diamines III can be obtained according to scheme 3
- FIGURE 3 in which R-
- Step 7 consists in protecting the amine from chloroalkylamine with, for example, di-tert-butyl dicarbonate in a sodium hydroxide-dioxane mixture at room temperature for 1 h to 20 h which leads to the compound of formula VIII
- Step 8 is identical to step 3 of Scheme 1, and is carried out from the above-mentioned compound VIII, which leads to the compound of formula IX
- Step 9 consists in protecting the amine protecting group from compound IX in an acid medium with, for example, t ⁇ fluroacetic acid in dichloromethane at room temperature for 1 to 5 h, which leads to diamines III Experimental part
- Step A 4-amino-5-chloro-2-methoxy N- (3-chloropropyl) benzamide
- Step B 4-amino-5-chloro-2-methoxy N- [3- (4-hydroxypiperidin-1 -yl) propyl] benzamide
- the organic phase is dried over sodium sulfate and concentrated in vacuo.
- the residue crystallizes from a CH3Cl / MeOH mixture. 3.03 g. YId: 61%.
- the base obtained is transformed into hydrochloride by adding 1 5 eq of hydrochloric ether
- a precipitate is formed which is filtered and treated with an excess of hydrochloric ether After evaporation, the salt is crystallized by adding an acetone / H2 ⁇ mixture (99 1) After filtration, the precipitate is taken up in 1 N sodium hydroxide and extracted twice with a CH2 ⁇ 2 / MeOH mixture After evaporation of the organic phases, the residue is dissolved in MeOH and crystallized by adding ethyl ether then washed with H2O and dried. 0.9 g of a solid yields 35% yield.
- Step B N-tertiobutoxycarbonyl 2- (4-hydroxypiperidin-1-yl) ethylamine
- Step D 4-amino-5-chloro-2-methoxy N- [2- (4-hydroxyp ⁇ peridin-1 -yl) ethyl] benzamide
- Step A 4-amino-5-chloro-2-methoxy N- [3- (4-hydroxy-piperidin-1-yl) propyl] benzamide
- Step B 4-amino-5-chloro-2-ethoxy N- [3- (4-hydroxypiperidin-1-yl) propyl] benzamide
- the crude residue is chromatographed on an alumina flash column with an eluent system CH2Cl2 / AcOEt MeOH / NH4 ⁇ H 90/5/5 / 0.44
- the base obtained is transformed into hydrochloride with an excess of hydrochloric ether
- the salt crystallizes in a mixture EtOH / H2 ⁇ 60 mg are obtained.
- Example 3 To a solution of Example 3 (0.4 g, 1.22 mmol) in 300 ml of CH 2 CI 2 , add 3-chloroperbenzoic acid (0.27 g, 1.59 mmol) at room temperature Stir overnight room temperature then concentrate under vacuum Take up the residue with CH 2 CI 2 and wash with
- the principle of the test consists in recording the electrical activity of the smooth muscle directly responsible for the digestive sinstaltism. The recordings are made in the fasting rabbit
- Electromyographic recordings begin five days after surgery on fasting animals that have been fasting for 12 hours and kept in containment boxes
- the test product is administered intravenously in a volume equal to 0.5 ml at a dose of 0 , 7 mg / kg. It is dissolved in 10 to 20 ⁇ l of pure acetic acid. The volume is completed with physiological serum. The pH is readjusted by 0.1 N NaOH to values between 5 and 7.5
- the product vehicle is used as a placebo II consists of dilute acetic acid (10 to 20 ⁇ l in 0.5 ml of physiological saline), Its pH is also readjusted by 0.1 N NaOH to values between 5 and 7 , 5
- the first intravenous injection (product or vehicle) is carried out 1 hour later (T1 h)
- the second intravenous injection (vehicle or product) is carried out at T2h
- the return to the baseline is checked before each administration
- the activity of the products and the vehicle is evaluated over the 30 minutes of recording following their injection.
- Spontaneous myoelectronic activity is organized on the lair in bursts of potentials which appear in the presence of the vehicle with a frequency equal to 1.1 ⁇ 0.3 bursts / min in 150 rabbits
- the electrical activity is organized in slow waves (17 cycles per minute) randomly superimposed on bursts of potentials responsible for peristalsis
- the results are therefore expressed as a percentage of slow waves overloaded with bursts of potentials
- the molecules tested stimulate significantly (p ⁇ 0.05) digestive pestalism when the frequency on the antrum is greater than or equal to 1.6 bursts / mm and the motor index on the colon is greater than or equal to 35%, whatever the segment recorded (colon life + 5 and colon life + 8)
- the results of the compounds according to the invention are given below (table I) as well as those of the comparators (compounds AE).
- the compounds according to the invention significantly stimulate the proximal colon (Life + 5 and Life + 8) unlike the comparators
- the principle of the test consists in recording the contractile activity of the colon in the fasted and alert animal
- the recording begins ten days after the surgical intervention.
- the animal is alert and fasting since the day before. It is placed in a digestibility cage and connected to the recording device.
- the first three hours constitute the recording during the control period, then the product. tested at a dose of 1 mg / kg or the vehicle (diluted acetic acid) is injected intravenously under a volume equal to 0 8 ml
- the pH is between 5 and 7.5 and the osmolalite is between 100 and 300 mosmol / l
- the activity is quantified over the two hours of recording following the injection of the molecule and compared using the Dunnett test to the recordings obtained under vehicle
- the significance level is equal to 5%
- the parameters used to evaluate the activity of the product are the frequency of contractions and the duration of stimulation of colonic motility
- colonic motility is significantly stimulated (p _. 0.05) when the frequency of contractions per hour is higher. or equal to 2.8
- the compounds of examples 1 and 3 significantly stimulate colonic motility in dogs with a duration greater than 120 minutes
- the visceral analgesic activity was studied on a model of digestive pain in the alert rat This pain is caused by the distention of the colon using a balloon
- the product to be tested or the vehicle (dilute acetic acid) is administered per os in a volume of 1 ml, then the rat is placed under observation in a c ⁇ stallisoir
- Colon distension is performed 2 h 30 min after irritation. It is performed under a fixed volume equal to 1.5 ml of distilled water. Colonic distension causes digestive pain objectified by abdominal cramps, the number of which reflects the intensity of the pain. Colonic distension is maintained for 10 minutes during which the abdominal cramps are counted Statistical analysis is performed with the aid of Dunnett's test comparing the same g r oup animals vehicle (6) to several groups of rats (6 animals per group) receiving the molecules studied The significance is attached seuii at 5 %
- the molecules are tested orally at 1 -10-100 and 1000 ⁇ g / kg They are dissolved in
- the vehicle used as a placebo consists of dilute acetic acid (10 to 20 ⁇ l in 1 ml of physiological saline) Its pH is adjusted (NaOH 0, 1 N) to values between 5 and
- the compounds according to the invention significantly reduce digestive pain
- the ch i ffers parentheses is the number of animals studied TABLE III
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Nutrition Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description






















Claims

Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU88678/98A AU8867898A (en) | 1997-07-25 | 1998-07-24 | Novel benzamide derivatives stimulating high and low gastrointestinal motricity |
| EP98940329A EP0998458A1 (fr) | 1997-07-25 | 1998-07-24 | Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR97/09477 | 1997-07-25 | ||
| FR9709477A FR2766484B1 (fr) | 1997-07-25 | 1997-07-25 | Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1999005106A1 true WO1999005106A1 (fr) | 1999-02-04 |
| WO1999005106A8 WO1999005106A8 (fr) | 1999-04-15 |
Family
ID=9509623
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR1998/001640 Ceased WO1999005106A1 (fr) | 1997-07-25 | 1998-07-24 | Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse |
Country Status (8)
| Country | Link |
|---|---|
| EP (1) | EP0998458A1 (fr) |
| AR (1) | AR014900A1 (fr) |
| AU (1) | AU8867898A (fr) |
| CO (1) | CO4990994A1 (fr) |
| FR (1) | FR2766484B1 (fr) |
| TN (1) | TNSN98138A1 (fr) |
| WO (1) | WO1999005106A1 (fr) |
| ZA (1) | ZA986633B (fr) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE620543A (fr) * | ||||
| WO1993003725A1 (fr) * | 1991-08-20 | 1993-03-04 | Smithkline Beecham Plc | Antagonistes du recepteur 5-ht4 |
| WO1995025100A1 (fr) * | 1994-03-14 | 1995-09-21 | Sanofi | Utilisation d'esters de l'acide 4-amino-5-chloro-2-methoxybenzoique comme 5-ht4 agonistes |
| FR2735693A1 (fr) * | 1995-06-23 | 1996-12-27 | Logeais Labor Jacques | Nouvelles applications therapeutiques de n-cyclohexyl benzamides |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1015921A (en) * | 1961-10-06 | 1966-01-05 | Benger Lab Ltd | Benzamides |
| AR216043A1 (es) * | 1974-03-21 | 1979-11-30 | Anphar Sa | Procedimiento para la preparacion de 1-derivados de benzoilamino-4-piperidina y sus sales fisiologicamente aceptables |
| EP0222533A1 (fr) * | 1985-10-25 | 1987-05-20 | The Upjohn Company | Cis-N-[(2-aminocycloaliphatique)-benzèneacétamide et -benzamides anticonvulsifs |
| FR2674849B1 (fr) * | 1991-04-02 | 1994-12-23 | Logeais Labor Jacques | Nouveaux derives de n-cyclohexyl benzamides ou thiobenzamides, leurs preparations et leurs applications en therapeutique. |
-
1997
- 1997-07-25 FR FR9709477A patent/FR2766484B1/fr not_active Expired - Fee Related
-
1998
- 1998-07-15 TN TNTNSN98138A patent/TNSN98138A1/fr unknown
- 1998-07-24 EP EP98940329A patent/EP0998458A1/fr not_active Withdrawn
- 1998-07-24 WO PCT/FR1998/001640 patent/WO1999005106A1/fr not_active Ceased
- 1998-07-24 ZA ZA986633A patent/ZA986633B/xx unknown
- 1998-07-24 AR ARP980103645A patent/AR014900A1/es unknown
- 1998-07-24 CO CO98042280A patent/CO4990994A1/es unknown
- 1998-07-24 AU AU88678/98A patent/AU8867898A/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE620543A (fr) * | ||||
| WO1993003725A1 (fr) * | 1991-08-20 | 1993-03-04 | Smithkline Beecham Plc | Antagonistes du recepteur 5-ht4 |
| WO1995025100A1 (fr) * | 1994-03-14 | 1995-09-21 | Sanofi | Utilisation d'esters de l'acide 4-amino-5-chloro-2-methoxybenzoique comme 5-ht4 agonistes |
| FR2735693A1 (fr) * | 1995-06-23 | 1996-12-27 | Logeais Labor Jacques | Nouvelles applications therapeutiques de n-cyclohexyl benzamides |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2766484B1 (fr) | 1999-09-17 |
| CO4990994A1 (es) | 2000-12-26 |
| TNSN98138A1 (fr) | 2005-03-15 |
| EP0998458A1 (fr) | 2000-05-10 |
| AU8867898A (en) | 1999-02-16 |
| FR2766484A1 (fr) | 1999-01-29 |
| WO1999005106A8 (fr) | 1999-04-15 |
| ZA986633B (en) | 1999-06-15 |
| AR014900A1 (es) | 2001-04-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FI63220B (fi) | Foerfarande foer framstaellning av nya benshydrylsulfinyler me terapeutisk verkan pao det centrala nervsystemet | |
| JP3231042B2 (ja) | 2−(2,6−ジ−ハロ−フェニルアミノ)フェニル酢酸の誘導体の硝酸エステルおよびその製造法 | |
| JPS6049192B2 (ja) | 新規置換ベンズアミド、その製造方法及びこれを有効成分とする向精神薬 | |
| NO144211B (no) | Analogifremgangsmaate til fremstilling av nye tyrosinderivater med farmasoeytisk virkning paa glatte muskler | |
| EP0022118A1 (fr) | Nouveaux dérivés de sulfonyl-aniline, leur procédé de préparation et leur application en thérapeutique | |
| CH641438A5 (fr) | Derives du 3-(aminoethyl)phenol et leurs sels, leur procede de preparation et medicaments les renfermant. | |
| KR840001836B1 (ko) | 9-아미노알킬플루오렌의 제조방법 | |
| FR2584713A1 (fr) | Nouveaux derives de l'indole carboxamide, leurs sels, procede et intermediaires de preparation, application a titre de medicaments et compositions les renfermant | |
| US5834495A (en) | Crystalline xamoneline tartrate | |
| EP0246126A1 (fr) | Benzimidazoles et imidazopyridines sulfonamides, leur préparation et leur application en tant que médicaments | |
| JP3383668B2 (ja) | 5−htアゴニストまたはアンタゴニストとしての1−アザアダマンタン誘導体 | |
| EP0077427A1 (fr) | Dérivés de pipéridine, leur procédé de préparation et leur application en thérapeutique | |
| JPH03866B2 (fr) | ||
| WO2000046201A1 (fr) | Dérivés de benzamide et médicaments les contenant | |
| EP0998458A1 (fr) | Nouveaux derives benzamides stimulants de la motricite gastrointestinale haute et basse | |
| CH655721A5 (fr) | Derives du furanne et sels d'addition de ces derives leurs procedes de preparation et composition pharmaceutique les contenant. | |
| CA1093582A (fr) | Procede d'obtention de nouvelles propylenediamines | |
| EP0998469A1 (fr) | Cycloalkyles benzamides stimulants de la motricite gastrointestinale haute et basse | |
| CH674845A5 (fr) | ||
| CH658786A5 (fr) | Medicaments qui contiennent d'alpha-(n-pyrrolyl)-acides ou de leurs sels ou esters. | |
| JPS62108863A (ja) | 2−ピリジル酢酸誘導体、その製法およびそれを含む医薬 | |
| FR2617164A1 (fr) | Nouveaux derives de la decahydroquinoleine, leur procede de preparation, leur application comme medicaments et les compositions pharmaceutiques les renfermant | |
| EP0010030A1 (fr) | Nouveaux dérivés d'ortho chloro benzoyl-2 chloro-4 glycylanilide, leur préparation et leur application en tant que médicaments | |
| CA1064489A (fr) | Procede d'obtention de nouveaux benzene sulfonamides | |
| CA1092136A (fr) | Procede d'obtention de nouveaux acetamides et les produits ainsi obtenus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| AK | Designated states |
Kind code of ref document: C1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: PAT. BUL. 05/99 UNDER (30) REPLACE "97/19477" BY "97/09477" |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998940329 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998940329 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998940329 Country of ref document: EP |