DE69726841T2 - Heteroatomsubstituierte metallocene für olefipolymerisationkatalysatore un verfahren zu deren herstellung - Google Patents
Heteroatomsubstituierte metallocene für olefipolymerisationkatalysatore un verfahren zu deren herstellung Download PDFInfo
- Publication number
- DE69726841T2 DE69726841T2 DE69726841T DE69726841T DE69726841T2 DE 69726841 T2 DE69726841 T2 DE 69726841T2 DE 69726841 T DE69726841 T DE 69726841T DE 69726841 T DE69726841 T DE 69726841T DE 69726841 T2 DE69726841 T2 DE 69726841T2
- Authority
- DE
- Germany
- Prior art keywords
- group
- indenyl
- same
- tert
- atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003054 catalyst Substances 0.000 title claims description 42
- 238000000034 method Methods 0.000 title claims description 10
- 238000006116 polymerization reaction Methods 0.000 claims description 75
- -1 tetrahydroindenyl Chemical group 0.000 claims description 49
- 238000006243 chemical reaction Methods 0.000 claims description 45
- AQZWEFBJYQSQEH-UHFFFAOYSA-N 2-methyloxaluminane Chemical compound C[Al]1CCCCO1 AQZWEFBJYQSQEH-UHFFFAOYSA-N 0.000 claims description 33
- 239000002904 solvent Substances 0.000 claims description 26
- 239000000460 chlorine Substances 0.000 claims description 23
- 239000003446 ligand Substances 0.000 claims description 23
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 18
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 claims description 16
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 16
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 14
- 229910052710 silicon Inorganic materials 0.000 claims description 13
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 claims description 12
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims description 11
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 11
- 125000005843 halogen group Chemical group 0.000 claims description 11
- 239000010703 silicon Substances 0.000 claims description 11
- 150000001336 alkenes Chemical class 0.000 claims description 10
- 125000003118 aryl group Chemical group 0.000 claims description 10
- 125000004429 atom Chemical group 0.000 claims description 9
- 239000012018 catalyst precursor Substances 0.000 claims description 9
- 125000006657 (C1-C10) hydrocarbyl group Chemical group 0.000 claims description 8
- 229910052732 germanium Inorganic materials 0.000 claims description 7
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 claims description 6
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 6
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 6
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 6
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 6
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 6
- 125000005018 aryl alkenyl group Chemical group 0.000 claims description 6
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 6
- UMJJFEIKYGFCAT-UHFFFAOYSA-N indan-2-one Chemical compound C1=CC=C2CC(=O)CC2=C1 UMJJFEIKYGFCAT-UHFFFAOYSA-N 0.000 claims description 6
- 229910052723 transition metal Inorganic materials 0.000 claims description 6
- 150000003624 transition metals Chemical class 0.000 claims description 6
- 229910052726 zirconium Inorganic materials 0.000 claims description 6
- 238000007334 copolymerization reaction Methods 0.000 claims description 5
- 229910052751 metal Inorganic materials 0.000 claims description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 5
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 claims description 5
- 239000005046 Chlorosilane Substances 0.000 claims description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 4
- 125000004104 aryloxy group Chemical group 0.000 claims description 4
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical compound Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 239000002184 metal Substances 0.000 claims description 4
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 4
- 229910052719 titanium Inorganic materials 0.000 claims description 4
- 239000010936 titanium Substances 0.000 claims description 4
- GBXQPDCOMJJCMJ-UHFFFAOYSA-M trimethyl-[6-(trimethylazaniumyl)hexyl]azanium;bromide Chemical compound [Br-].C[N+](C)(C)CCCCCC[N+](C)(C)C GBXQPDCOMJJCMJ-UHFFFAOYSA-M 0.000 claims description 4
- MEUXNEGJODESOX-UHFFFAOYSA-N chloro-cyclohexyl-dimethylsilane Chemical compound C[Si](C)(Cl)C1CCCCC1 MEUXNEGJODESOX-UHFFFAOYSA-N 0.000 claims description 3
- KAADXUXXXANQKW-UHFFFAOYSA-N chloro-dimethyl-(2-methylpentan-2-yl)silane Chemical compound CCCC(C)(C)[Si](C)(C)Cl KAADXUXXXANQKW-UHFFFAOYSA-N 0.000 claims description 3
- 229910052735 hafnium Inorganic materials 0.000 claims description 3
- VBJZVLUMGGDVMO-UHFFFAOYSA-N hafnium atom Chemical compound [Hf] VBJZVLUMGGDVMO-UHFFFAOYSA-N 0.000 claims description 3
- 238000002360 preparation method Methods 0.000 claims description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 2
- HCFQDMDVMBTQOR-UHFFFAOYSA-N [SiH3]OC1=Cc2ccccc2C1 Chemical compound [SiH3]OC1=Cc2ccccc2C1 HCFQDMDVMBTQOR-UHFFFAOYSA-N 0.000 claims description 2
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 125000000217 alkyl group Chemical group 0.000 claims description 2
- 229910052801 chlorine Inorganic materials 0.000 claims description 2
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 claims description 2
- 125000004639 dihydroindenyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims description 2
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 claims description 2
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 claims description 2
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 claims description 2
- 230000000737 periodic effect Effects 0.000 claims description 2
- 229920006395 saturated elastomer Polymers 0.000 claims description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims 1
- 125000002883 imidazolyl group Chemical group 0.000 claims 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims 1
- 239000002685 polymerization catalyst Substances 0.000 claims 1
- 239000000243 solution Substances 0.000 description 80
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 56
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 42
- 150000001875 compounds Chemical class 0.000 description 40
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 36
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 34
- 239000005977 Ethylene Substances 0.000 description 33
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 32
- VPGLGRNSAYHXPY-UHFFFAOYSA-L zirconium(2+);dichloride Chemical compound Cl[Zr]Cl VPGLGRNSAYHXPY-UHFFFAOYSA-L 0.000 description 31
- 239000011541 reaction mixture Substances 0.000 description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 27
- 229920000642 polymer Polymers 0.000 description 23
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 21
- 229910007926 ZrCl Inorganic materials 0.000 description 20
- 230000000694 effects Effects 0.000 description 20
- 239000000203 mixture Substances 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- 229910052938 sodium sulfate Inorganic materials 0.000 description 13
- 235000011152 sodium sulphate Nutrition 0.000 description 13
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 12
- 238000001704 evaporation Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 230000008020 evaporation Effects 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 239000004698 Polyethylene Substances 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- VVAXKNLRUHHJCZ-UHFFFAOYSA-N tert-butyl-(1h-inden-2-yloxy)-dimethylsilane Chemical compound C1=CC=C2CC(O[Si](C)(C)C(C)(C)C)=CC2=C1 VVAXKNLRUHHJCZ-UHFFFAOYSA-N 0.000 description 8
- APQIUTYORBAGEZ-UHFFFAOYSA-N 1,1-dibromoethane Chemical compound CC(Br)Br APQIUTYORBAGEZ-UHFFFAOYSA-N 0.000 description 7
- PEJJRWHRMLPGDG-UHFFFAOYSA-N 1h-inden-2-yloxy-dimethyl-(2-methylpentan-2-yl)silane Chemical compound C1=CC=C2CC(O[Si](C)(C)C(C)(C)CCC)=CC2=C1 PEJJRWHRMLPGDG-UHFFFAOYSA-N 0.000 description 7
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 7
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 7
- 238000001819 mass spectrum Methods 0.000 description 7
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 7
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 7
- 239000012190 activator Substances 0.000 description 6
- 150000002500 ions Chemical class 0.000 description 6
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Chemical compound [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 6
- DUNKXUFBGCUVQW-UHFFFAOYSA-J zirconium tetrachloride Chemical compound Cl[Zr](Cl)(Cl)Cl DUNKXUFBGCUVQW-UHFFFAOYSA-J 0.000 description 6
- FSLSKEFNMQRNRQ-UHFFFAOYSA-N [1-[1-[2-[dimethyl(2-methylpentan-2-yl)silyl]oxy-1h-inden-1-yl]ethyl]-1h-inden-2-yl]oxy-dimethyl-(2-methylpentan-2-yl)silane Chemical compound CCCC(C)(C)[Si](C)(C)OC1=CC2=CC=CC=C2C1C(C)C1C2=CC=CC=C2C=C1O[Si](C)(C)C(C)(C)CCC FSLSKEFNMQRNRQ-UHFFFAOYSA-N 0.000 description 5
- 230000000052 comparative effect Effects 0.000 description 5
- 125000002524 organometallic group Chemical group 0.000 description 5
- QMYBXECXCNZHMM-UHFFFAOYSA-N tert-butyl-(1h-inden-2-yloxy)-diphenylsilane Chemical compound C=1C2=CC=CC=C2CC=1O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 QMYBXECXCNZHMM-UHFFFAOYSA-N 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 230000008025 crystallization Effects 0.000 description 4
- MIVGNBLEYFDVMG-UHFFFAOYSA-N cyclohexyl-(1h-inden-2-yloxy)-dimethylsilane Chemical compound C=1C2=CC=CC=C2CC=1O[Si](C)(C)C1CCCCC1 MIVGNBLEYFDVMG-UHFFFAOYSA-N 0.000 description 4
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 4
- 238000004821 distillation Methods 0.000 description 4
- ODKBFKLBYDZPQS-UHFFFAOYSA-N tert-butyl-[[1-[1-[2-[tert-butyl(dimethyl)silyl]oxy-1h-inden-1-yl]ethyl]-1h-inden-2-yl]oxy]-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OC1=CC2=CC=CC=C2C1C(C)C1C2=CC=CC=C2C=C1O[Si](C)(C)C(C)(C)C ODKBFKLBYDZPQS-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 235000019502 Orange oil Nutrition 0.000 description 3
- 150000002362 hafnium Chemical class 0.000 description 3
- PDPJQWYGJJBYLF-UHFFFAOYSA-J hafnium tetrachloride Chemical compound Cl[Hf](Cl)(Cl)Cl PDPJQWYGJJBYLF-UHFFFAOYSA-J 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 239000010502 orange oil Substances 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 3
- 239000004711 α-olefin Substances 0.000 description 3
- PWAKRZXJFZFLBA-UHFFFAOYSA-N C[SiH](C)[SiH](C1C(O[Si](C)(C)C(C)(C)C)=Cc2ccccc12)C1C(O[Si](C)(C)C(C)(C)C)=Cc2ccccc12 Chemical compound C[SiH](C)[SiH](C1C(O[Si](C)(C)C(C)(C)C)=Cc2ccccc12)C1C(O[Si](C)(C)C(C)(C)C)=Cc2ccccc12 PWAKRZXJFZFLBA-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 125000005234 alkyl aluminium group Chemical group 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- SZLCRBUXZGPBQK-UHFFFAOYSA-N bis[2-[tert-butyl(dimethyl)silyl]oxy-1h-inden-1-yl]-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OC1=CC2=CC=CC=C2C1[Si](C)(C)C1C2=CC=CC=C2C=C1O[Si](C)(C)C(C)(C)C SZLCRBUXZGPBQK-UHFFFAOYSA-N 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- ODYLMIARNWCFQK-UHFFFAOYSA-N cyclohexyl-[[1-[1-[2-[cyclohexyl(dimethyl)silyl]oxy-1h-inden-1-yl]ethyl]-1h-inden-2-yl]oxy]-dimethylsilane Chemical compound C1CCCCC1[Si](C)(C)OC1=CC2=CC=CC=C2C1C(C)C(C1=CC=CC=C1C=1)C=1O[Si](C)(C)C1CCCCC1 ODYLMIARNWCFQK-UHFFFAOYSA-N 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 238000003306 harvesting Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- CPOFMOWDMVWCLF-UHFFFAOYSA-N methyl(oxo)alumane Chemical compound C[Al]=O CPOFMOWDMVWCLF-UHFFFAOYSA-N 0.000 description 2
- ZGEGCLOFRBLKSE-UHFFFAOYSA-N methylene hexane Natural products CCCCCC=C ZGEGCLOFRBLKSE-UHFFFAOYSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- WCGLAMWRVNHSCL-UHFFFAOYSA-N tert-butyl-[[1-[1-[2-[tert-butyl(diphenyl)silyl]oxy-1h-inden-1-yl]ethyl]-1h-inden-2-yl]oxy]-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](C(C)(C)C)(C=1C=CC=CC=1)OC1=CC2=CC=CC=C2C1C(C)C(C1=CC=CC=C1C=1)C=1O[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 WCGLAMWRVNHSCL-UHFFFAOYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- LSYZRJFMRVUUNC-UHFFFAOYSA-N (2-cyclohexyl-1H-inden-1-yl)oxy-dimethylsilane Chemical compound C1(CCCCC1)C=1C(C2=CC=CC=C2C=1)O[SiH](C)C LSYZRJFMRVUUNC-UHFFFAOYSA-N 0.000 description 1
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 1
- 0 **(*)(*)OC1=C(*)c2c(*)c(*)c(*)c(*)c2C1(BS1C(O*(*)(*)*)=C(*)c2c1c(*)c(*)c(*)c2*)* Chemical compound **(*)(*)OC1=C(*)c2c(*)c(*)c(*)c(*)c2C1(BS1C(O*(*)(*)*)=C(*)c2c1c(*)c(*)c(*)c2*)* 0.000 description 1
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical compound CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 description 1
- KWKAKUADMBZCLK-UHFFFAOYSA-N 1-octene Chemical compound CCCCCCC=C KWKAKUADMBZCLK-UHFFFAOYSA-N 0.000 description 1
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 1
- PUHHRKAPYINUGH-UHFFFAOYSA-N 4,7-dimethyl-1,3-dihydroinden-2-one Chemical compound CC1=CC=C(C)C2=C1CC(=O)C2 PUHHRKAPYINUGH-UHFFFAOYSA-N 0.000 description 1
- DKLQZDIAQKGVTA-UHFFFAOYSA-N 4,7-dimethyl-1h-indene Chemical compound CC1=CC=C(C)C2=C1CC=C2 DKLQZDIAQKGVTA-UHFFFAOYSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- QSZGOMRHQRFORD-UHFFFAOYSA-L [Cl-].[Cl-].C=C.C1=CC2=CC=CC=C2C1[Zr+2]C1C2=CC=CC=C2C=C1 Chemical compound [Cl-].[Cl-].C=C.C1=CC2=CC=CC=C2C1[Zr+2]C1C2=CC=CC=C2C=C1 QSZGOMRHQRFORD-UHFFFAOYSA-L 0.000 description 1
- ATQZKZKLNBGBJQ-UHFFFAOYSA-L [Cl-].[Cl-].CC(C)(C)[Si](C)(C)OC1=Cc2c(C1[Zr++]C1C(O[Si](C)(C)C(C)(C)C)=Cc3c1c1ccccc1c1ccccc31)c1ccccc1c1ccccc21 Chemical compound [Cl-].[Cl-].CC(C)(C)[Si](C)(C)OC1=Cc2c(C1[Zr++]C1C(O[Si](C)(C)C(C)(C)C)=Cc3c1c1ccccc1c1ccccc31)c1ccccc1c1ccccc21 ATQZKZKLNBGBJQ-UHFFFAOYSA-L 0.000 description 1
- JQHPURQXTURPDS-UHFFFAOYSA-L [Cl-].[Cl-].C[Si](C)=[Zr++]([C@H]1C=CC2=C1CCCC2)[C@@H]1C=CC2=C1CCCC2 Chemical compound [Cl-].[Cl-].C[Si](C)=[Zr++]([C@H]1C=CC2=C1CCCC2)[C@@H]1C=CC2=C1CCCC2 JQHPURQXTURPDS-UHFFFAOYSA-L 0.000 description 1
- YIMDFOVCIKFEPU-UHFFFAOYSA-L [Cl-].[Cl-].O([Si](C)(C)C(C)(C)C)C=1C(C=2CCCCC=2C=1)[Zr+2] Chemical compound [Cl-].[Cl-].O([Si](C)(C)C(C)(C)C)C=1C(C=2CCCCC=2C=1)[Zr+2] YIMDFOVCIKFEPU-UHFFFAOYSA-L 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000001555 benzenes Chemical group 0.000 description 1
- UIHFYFJSTRYMGM-UHFFFAOYSA-N bis[2-[dimethyl(2-methylpentan-2-yl)silyl]oxy-1h-inden-1-yl]-dimethylsilane Chemical compound CCCC(C)(C)[Si](C)(C)OC1=CC2=CC=CC=C2C1[Si](C)(C)C1C2=CC=CC=C2C=C1O[Si](C)(C)C(C)(C)CCC UIHFYFJSTRYMGM-UHFFFAOYSA-N 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- IDASTKMEQGPVRR-UHFFFAOYSA-N cyclopenta-1,3-diene;zirconium(2+) Chemical class [Zr+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 IDASTKMEQGPVRR-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 238000004636 glovebox technique Methods 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical group 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QNXSIUBBGPHDDE-UHFFFAOYSA-N indan-1-one Chemical compound C1=CC=C2C(=O)CCC2=C1 QNXSIUBBGPHDDE-UHFFFAOYSA-N 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000012968 metallocene catalyst Substances 0.000 description 1
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 150000002902 organometallic compounds Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- YWAKXRMUMFPDSH-UHFFFAOYSA-N pentene Chemical compound CCCC=C YWAKXRMUMFPDSH-UHFFFAOYSA-N 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 230000037048 polymerization activity Effects 0.000 description 1
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 1
- 229910000105 potassium hydride Inorganic materials 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- BLTUJPISBOAJCN-UHFFFAOYSA-N tert-butyl-(1h-cyclopenta[l]phenanthren-2-yloxy)-dimethylsilane Chemical compound C12=CC=CC=C2C2=CC=CC=C2C2=C1CC(O[Si](C)(C)C(C)(C)C)=C2 BLTUJPISBOAJCN-UHFFFAOYSA-N 0.000 description 1
- PUORDKGMOOZWDP-UHFFFAOYSA-N tert-butyl-[(4,7-dimethyl-1h-inden-2-yl)oxy]-dimethylsilane Chemical compound CC1=CC=C(C)C2=C1CC(O[Si](C)(C)C(C)(C)C)=C2 PUORDKGMOOZWDP-UHFFFAOYSA-N 0.000 description 1
- XBAPTBZSXHMGFX-UHFFFAOYSA-N tert-butyl-[[3-[1-[2-[tert-butyl(dimethyl)silyl]oxy-3-trimethylsilyl-3h-inden-1-yl]ethyl]-1-trimethylsilyl-1h-inden-2-yl]oxy]-dimethylsilane Chemical compound C1=CC=C2C(C(C=3C4=CC=CC=C4C(C=3O[Si](C)(C)C(C)(C)C)[Si](C)(C)C)C)=C(O[Si](C)(C)C(C)(C)C)C([Si](C)(C)C)C2=C1 XBAPTBZSXHMGFX-UHFFFAOYSA-N 0.000 description 1
- FBFPWFZIKYNROP-UHFFFAOYSA-N tert-butyl-[[3-[1-[2-[tert-butyl(dimethyl)silyl]oxy-3h-cyclopenta[l]phenanthren-3-yl]ethyl]-3h-cyclopenta[l]phenanthren-2-yl]oxy]-dimethylsilane Chemical compound C12=CC=CC=C2C2=CC=CC=C2C(C=C2O[Si](C)(C)C(C)(C)C)=C1C2C(C)C1C(C=2C(=CC=CC=2)C=2C3=CC=CC=2)=C3C=C1O[Si](C)(C)C(C)(C)C FBFPWFZIKYNROP-UHFFFAOYSA-N 0.000 description 1
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 1
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 1
- 150000003613 toluenes Chemical class 0.000 description 1
- VOITXYVAKOUIBA-UHFFFAOYSA-N triethylaluminium Chemical compound CC[Al](CC)CC VOITXYVAKOUIBA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F17/00—Metallocenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/04—Monomers containing three or four carbon atoms
- C08F110/06—Propene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65908—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an ionising compound other than alumoxane, e.g. (C6F5)4B-X+
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/65912—Component covered by group C08F4/64 containing a transition metal-carbon bond in combination with an organoaluminium compound
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/659—Component covered by group C08F4/64 containing a transition metal-carbon bond
- C08F4/6592—Component covered by group C08F4/64 containing a transition metal-carbon bond containing at least one cyclopentadienyl ring, condensed or not, e.g. an indenyl or a fluorenyl ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
Description
- Die vorliegende Erfindung betrifft neue Metallocen-Katalysatorsysteme für die Homo- und Copolymerisation von Olefinen, insbesondere Propylen, Ethylen und höheren alpha-Olefinen, in der Gegenwart eines Cokatalysators wie etwa Methylalumoxan (MAO). Insbesondere betrifft die Erfindung Metallocene mit Heteroatomsubstituierten Indenyl- und Indenylderivat-Liganden, ein Verfahren zu deren Herstellung und deren Verwendung bei der Polymerisation von Olefinen, insbesondere Propylen und Ethylen.
- Chirale C2-symmetrische Bis(indenyl)-ansa-Metallocene sind Vorläufer für hoch aktive Katalysatoren für die stereoselektive Polymerisation von alpha-Olefinen. Die Leistungscharakteristika dieser Systeme sind unterschiedlich, wobei die Variationen durch Größe und Position der Substituenten induziert werden. Beispielsweise erzeugen Dimethylsilylen-verbrückte 2,2'-Dimethyl-4,4'-diaryl-substituierte Bis(indenyl)zirkonocene, entwickelt von Brintzinger und Mitarbeitern (Organometallics 1994, 13, 964) und Spaleck et al. (Organometallics 1994, 13, 954) isotaktische Polypropylene, mit Katalysatoraktivitäten und Polymereigenschaften, welche mit den mit heterogenen Ziegler-Natta Katalysatoren erhaltenen vergleichbar sind.
- Das Gebiet der elektronisch veränderten Bis(indenyl)-Metallocene ist relativ unerforscht geblieben. Es war früher berichtet worden, dass Halogen- oder Alkoxysubstitution in den sechsgliedrigen Ringen von Indenen die Aktivität des Katalysatorsystems und das Molekulargewicht des erzeugten Polymers verringert (Consiglio et al., Organometallics 1990, 9, 3098; Collins et al., Organometallics 1992, 11, 2115). Bis(indenyl)zirkonocene mit 2-Amino-funktionalisierten Liganden wurden vor kurzem von mehreren Gruppen berichtet (Luttikhedde et al., Organometallics 1996, 15, 3092; Plenio and Burth, J. Organomet. Chem. 1996, 519, 269; Brintzinger et al., J. Organomet. Chem. 1996, 520, 63). Die verbrückten Komplexe zeigen im Vergleich mit ihren unsubstituierten Bis(indenyl)zirkonocen-Analoga etwas niedrigere Katalysatoraktivitäten. WO 94/28034,
EP 485 822 EP 584 609 - Die vorliegende Erfindung betrifft neue Metallocenkomplexe (I) mit einem Sauerstoffatom direkt an die 2-Position einer Pentahaptoindenyl-Gruppe gebunden, z. B. racemisches [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid, und deren Anwendung bei der Polymerisation von Olefinen. Diese Komplexe (I), in Kombination mit MAO oder anderen Aktivatoren, bilden hoch aktive Katalysatorsysteme für die Homo- und Copolymerisation von Olefinen. Beispielsweise polymerisieren die I/MAO Katalysatorsysteme Propylen zu hoch isotaktischem Polypropylen. Die Propylen- und Ethylenpolymerisationsaktivitäten dieser (I)/MAO-Systeme übersteigen unter ähnlichen Polymerisationsbedingungen die von mehreren herkömmlichen ansa-Metallocen/MAO Katalysatorsystemen, wie etwa von Dimethylsilylenbis(4,5,6,7-tetrahydroindenyl)zirkoniumdichlorid/MAO. Die neuen Katalysatorsysteme sind sehr stabil und behalten ihre hohen Aktivitäten bei außergewöhnlich niedrigen [Al] : [Zr] Verhältnissen.
- Erfindungsgemäß wird ein neuer Katalysatorvorläufer erhalten, bei dem eine Siloxysubstitution in der 2-Position des fünfgliedrigen Rings einer Verbindung vom Indenyl-Typ ausgeführt worden ist. Dadurch ist es möglich, Metallocenverbindungen herzustellen, bei denen ein Sauerstoffatom direkt an die 2-Position einer Pentahaptoindenylgruppe gebunden ist.
- Der erfindungsgemäße Katalysatorvorläufer betrifft somit eine Indenylverbindung mit der Formel (I):worin D Silizium oder Germanium ist; R3, R3' und R4 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe oder eine C1-C10 Hydrocarbyloxygruppe sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C20 Ringstruktur bilden; M ein Übergangsmetall der Gruppe 4 des Periodensystems ist und an den Liganden IndY in mindestens einem η5-Bindungsmodus gebunden ist; jedes R gleich oder verschieden ist und ein Wasserstoffatom, ein Halogenatom, eine C1-C10 Hydrocarbylgruppe, eine C1-C10 Hydrocarbyloxygruppe, eine Trihydrocarbylsilylgruppe ist, oder zwei R zusammen eine C2-C20 Ringstruktur bilden; B ein Brückenatom oder ein Brückengruppe zwischen zwei IndY-Liganden oder zwischen einem IndY-Liganden und dem Übergangsmetall M ist; m gleich 1 oder 2 ist; o gleich 0 oder 1 ist; und n gleich 4-m ist, wenn keine Brücke B vorhanden ist oder B ein Brückenatom oder ein Brückengruppe zwischen zwei IndY-Liganden ist, oder n gleich 4-m-o ist, wenn B ein Brückenatom oder ein Brückengruppe zwischen einem IndY-Liganden und dem Übergangsmetall M ist.

- Unter mono- oder polysubstituiert wird verstanden, dass zusätzlich zum Substituenten Y gegebenenfalls andere Substituenten an den Ringen des Liganden IndY vorhanden sein können.
- Unter kondensiert oder nicht-kondensiert wird verstanden, dass jeder Ring des Liganden IndY kondensiert oder nicht-kondensiert sein kann, d. h. mit mindestens einem weiteren Ring mindestens zwei Atome gemeinsam haben kann.
- Unter homo- und heterozyklisch wird verstanden, dass jeder Ring des Liganden IndY nur Kohlenstoffringatome (homo- oder isozyklisch) haben kann oder andere Ringatome als Kohlenstoff (heterozyklisch) haben kann.
- Bevorzugt hat die Indenylverbindung nach Formel (I) die nachfolgende Formel (III)worin
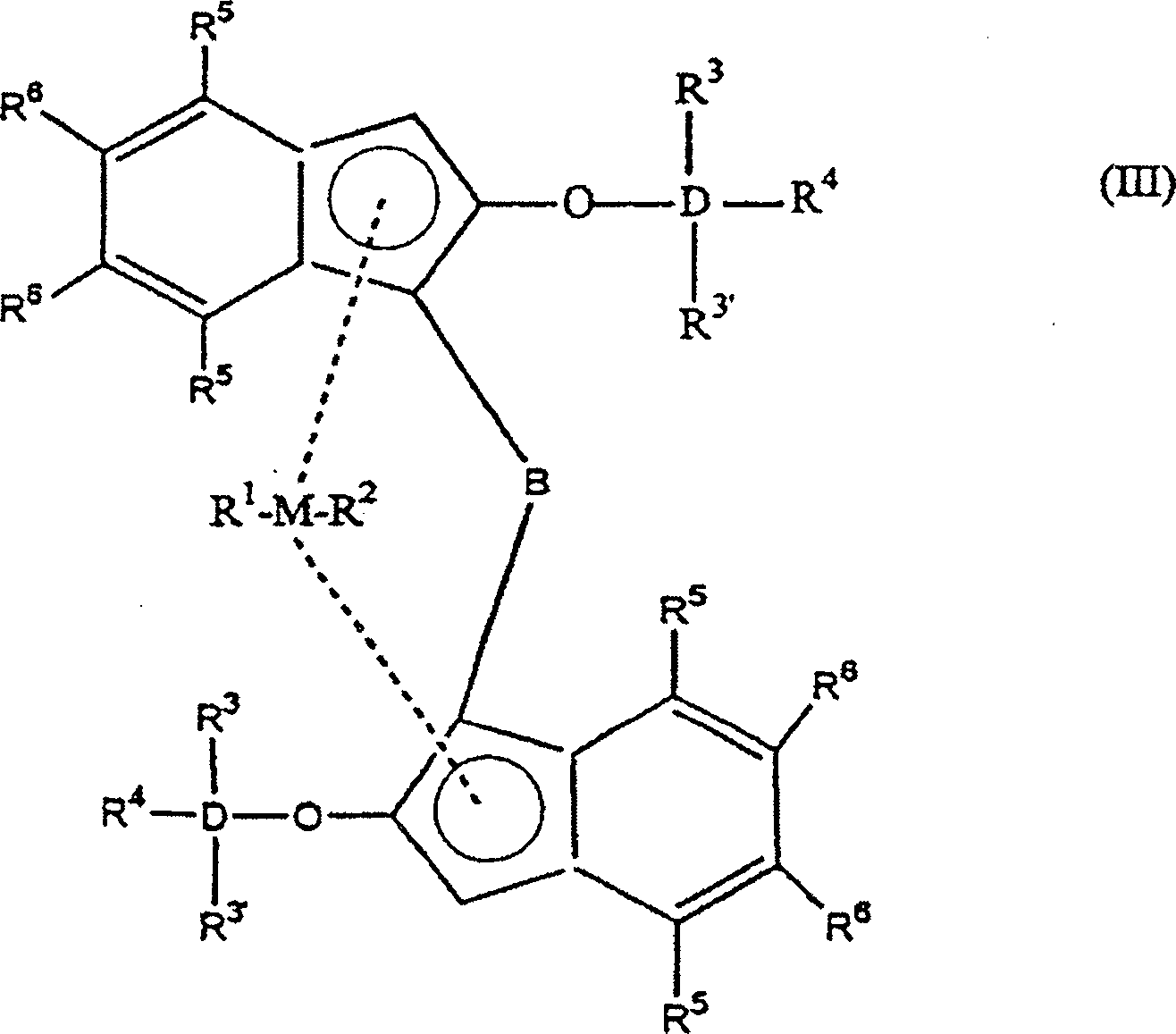
M ein Metall, ausgewählt aus Zirkonium, Titan oder Hafnium ist,
D ein Element, ausgewählt aus Silizium (Si) oder Germanium (Ge) ist,
B eine Brücke ist, umfassend mindestens eine Gruppe aus -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2-,
R1 und R2 die gleichen oder verschiedene Gruppen sind, ausgewählt aus einem Wasserstoffatom, einer C1-C10 Alkylgruppe, C1-C10 Alkoxygruppe, C6-C10 Arylgruppe, C6-C10 Aryloxygruppe, C2-C10 Alkenylgruppe, C2-C10 Arylalkylgruppe, C2-C10 Alkylarylgruppe, C8-C40 Arylalkenylgruppe oder einem Halogenatom, bevorzugt eine C1-C10 Alkylgruppe und/oder ein Halogenatom
R3', R3–R6 die gleichen oder verschiedene Gruppen sind, ausgewählt aus Wasserstoff, einer C1-C10 Alkylgruppe, C1-C10 Alkoxygruppe, C6-C10 Arylgruppe, C6-C10 Aryloxygruppe, C2-C10 Alkenylgruppe, C2-C10 Arylalkylgruppe, C2-C10 Alkylarylgruppe, C8-C40 Arylalkenylgruppe und einem Halogenatom. Die R3'- und R3-Gruppen können auch miteinander verknüpft sein um eine Ringstruktur zu bilden, und R4 kann auch Teil einer Ringstruktur sein. - Bevorzugt ist M Zirkonium.
- R1 und R2 sind bevorzugt gleich und am meisten bevorzugt sind sie Halogenatome, beispielsweise Chloratome.
- R3 ist bevorzugt eine C1-C10 Alkyl- oder Arylgruppe und am meisten bevorzugt ist es eine Methylgruppe. Gleichermaßen ist R4 bevorzugt eine C1-C10 Alkyl- oder Arylgruppe, beispielsweise eine tert.-Butylgruppe, t-Hexylgruppe oder Cyclohexylgruppe.
- Am meisten bevorzugt ist D gleich Si, ist B gleich -CH2CH2-, ist R1 = R2 und gleich Chlor, ist R3 gleich CH3 und ist R5-R6 aromatisch oder kondensiert aromatisch oder Alkyl oder Wasserstoff.
- Die Erfindung betrifft auch 2-Trihydracarbyl- und 2-Trihydrocarbyloxysiloxyinden- und -germyloxyindenverbindungen mit der allgemeinen Formel:worin: D Silizium oder Germanium ist; R3, R3' und R4 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe oder eine C1-C10 Hydrocarbyloxygruppe sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C20 Ringstruktur bilden; R7 eine vieratomige Kette ist, welche einen unsubstituierten oder substituierten, weiter nicht-kondensierten oder weiter kondensierten, homozyklischen (= isozyklischen) oder heterozyklischen, ungesättigten oder gesättigten, aliphatischen oder aromatischen sechsgliedrigen Ring bildet; und R8, R9 und R10 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe, eine C1-C10 Hydrocarbyloxygruppe, eine Tri-C1-C10-hydrocarbylsilylgruppe oder eine Tri-C1-C10-hydrocarbyloxysilylgruppe sind, oder einer aus R8 und R9 eine Gruppe B sein kann, welche ein Brückenatom oder eine Brückengruppe zu einer Cyclopentadienyl-, Fluorenyl- oder Indenylgruppe ist, mit der Maßgabe, dass wenn R7 einen unsubstituierten Benzolring bildet, D Silizium ist, R3, R3' und R4 Methylgruppen sind und R8 und R10 Wasserstoffe sind, R9 kein Wasserstoff ist. Bevorzugt haben sie die Formel:
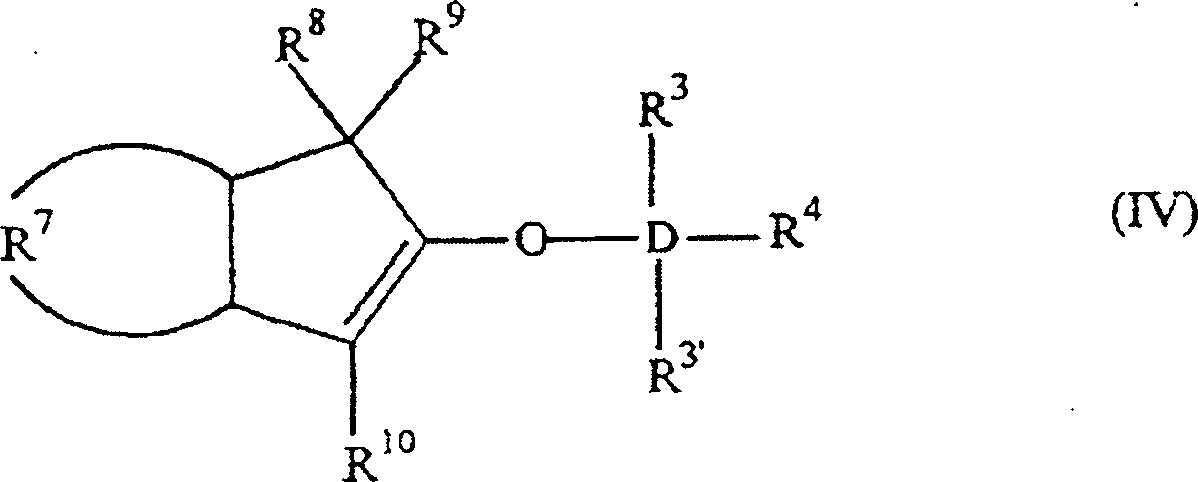 worin: R3', R3–R6 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Alkylgruppe, eine C1-C10 Alkoxygruppe, eine C6-C10 Arylgruppe, eine C2-C10 Alkenylgruppe, eine C7-C22 Arylalkylgruppe, eine C7-C22 Alkylarylgruppe, eine C8-C23 Arylalkenylgruppe oder ein Halogenatom sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C10 Ringstruktur bilden, und R8, R9 und R10 die gleichen wie vorstehend sind, oder alternativ die Formel:
worin: R3', R3–R6 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Alkylgruppe, eine C1-C10 Alkoxygruppe, eine C6-C10 Arylgruppe, eine C2-C10 Alkenylgruppe, eine C7-C22 Arylalkylgruppe, eine C7-C22 Alkylarylgruppe, eine C8-C23 Arylalkenylgruppe oder ein Halogenatom sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C10 Ringstruktur bilden, und R8, R9 und R10 die gleichen wie vorstehend sind, oder alternativ die Formel: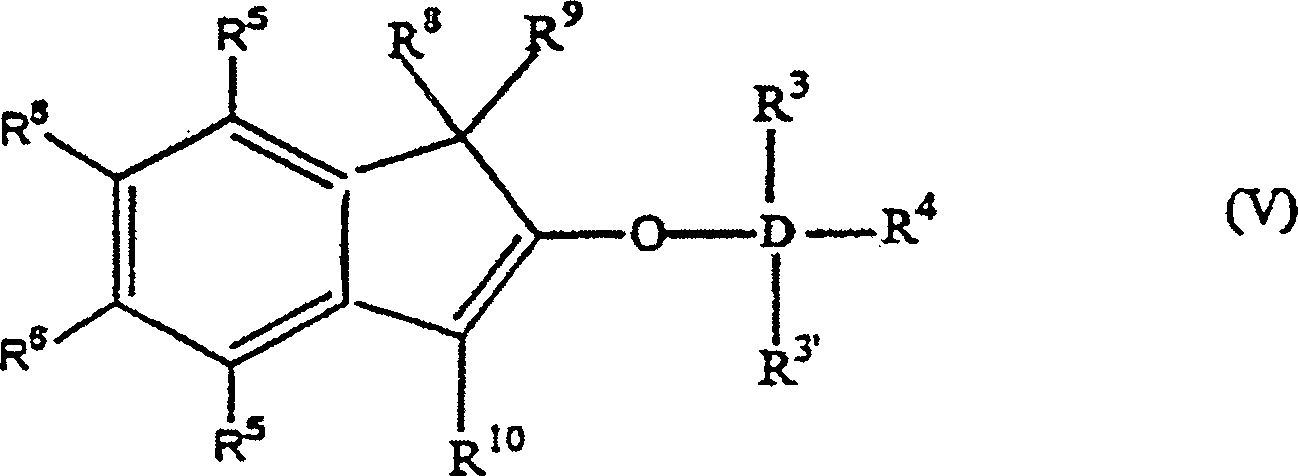 worin: R3, R3', R4-R6, R8, R10 und D die gleichen wie vorstehend sind, und B eine Brücke von mindestens einer der Gruppen -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2- ist, wobei n gleich 1–8 ist und R3 gleich wie vorstehend ist. D ist bevorzugt Si.
worin: R3, R3', R4-R6, R8, R10 und D die gleichen wie vorstehend sind, und B eine Brücke von mindestens einer der Gruppen -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2- ist, wobei n gleich 1–8 ist und R3 gleich wie vorstehend ist. D ist bevorzugt Si.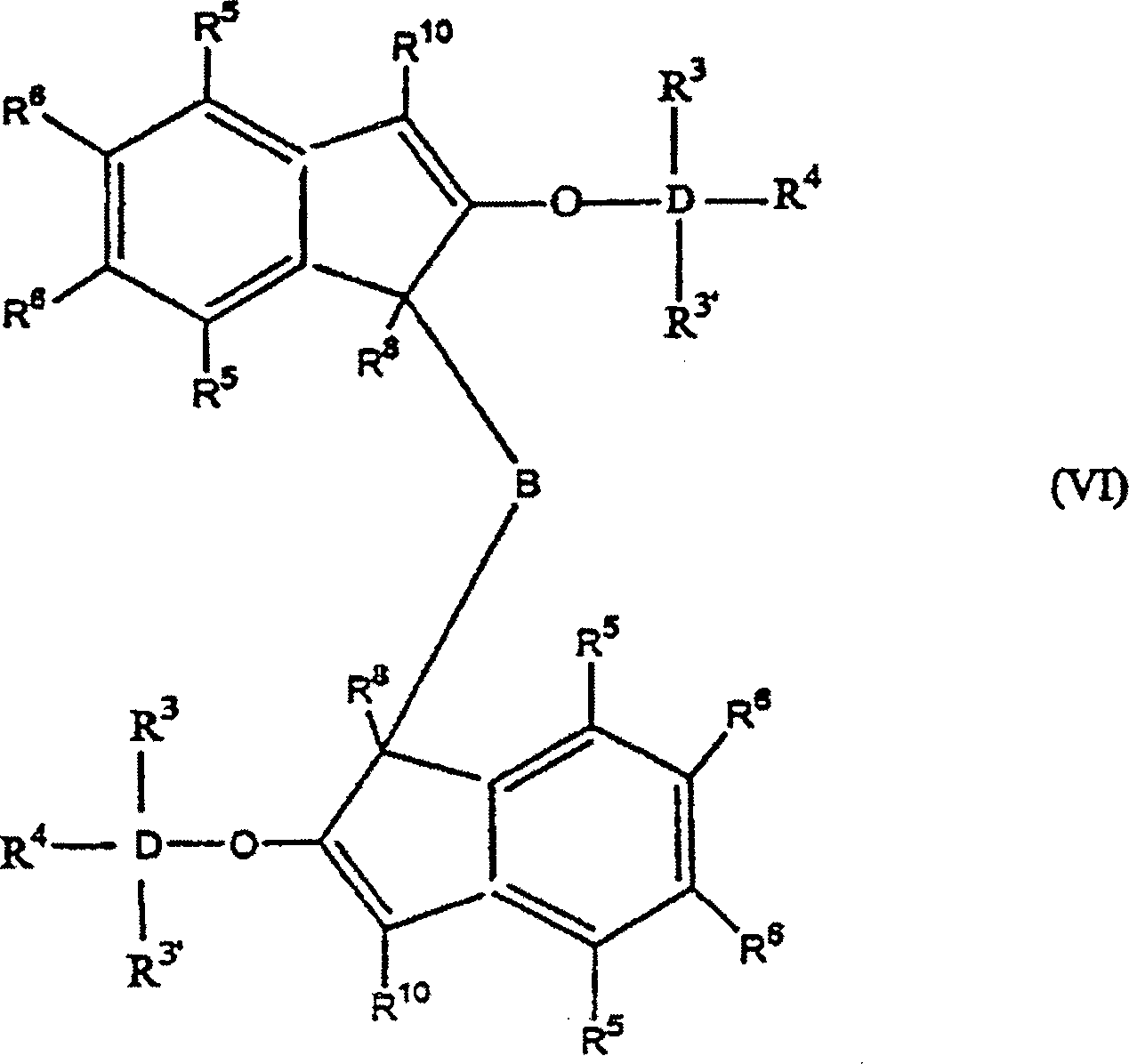
- Die erfindungsgemäßen Katalysatorverbindungen können aus 2-Indanon hergestellt werden. Diese Verbindung kann in einem geeigneten Lösungsmittel mit einer Base und einem Chlorsilan umgesetzt werden, wobei 2-Siloxyinden in einer Ausbeute von über 80% gebildet wird. Geeignete Lösungsmittel sind beispielsweise Dimethylformamid (DMF) und Tetrahydrofuran (THF). Geeignete Basen sind beispielsweise Imidazol, Triethylamin (TEA) und 1,8-Diazabicyclo[5.4.0]undec-7-en. Geeignete Chlorsilane sind beispielsweise tert.-Butyldimethylchlorsilan, t-Hexyldimethylchlorsilan und Cyclohexyldimethylchlorsilan. Die Reaktion verläuft nach dem folgenden Reaktionsschema (VII):
-

- Gemäß einer Ausführungsform der Erfindung wird 2-(tert-Butyldimethylsiloxy)inden zunächst mit Butyllithium reagiert und danach mit Dimethyldichlorsilan (Me2SiCl2), wobei Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)silan gebildet wird.
- Butyllithium kann durch Methyllithium, Natriumhydrid oder Kaliumhydrid ersetzt werden. Gleichermaßen kann Dimethyldichlorsilan durch ein beliebiges Dialkyl- oder Diarylsilan ersetzt werden. Silizium kann durch Germanium ersetzt werden.
- Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)silan kann mit Butyllithium reagiert werden, was das entsprechende Bislithiumsalz ergibt. Dieses Produkt kann mit Zirkoniumtetrachlorid reagiert werden, wobei [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid als ein Gemisch der racemischen und meso Diastereomere erhalten wird. Butyllithium kann ersetzt werden wie vorstehend beschrieben. Zirkoniumtetrachlorid kann durch Titantetrachlorid oder Hafniumtetrachlorid ersetzt werden, wobei die entsprechenden Titan-. und Hafniumkomplexe erhalten werden. Die Reaktionen verlaufen gemäß den nachfolgenden Reaktionsschemata (VIII) und (IX):
-

- Gemäß einer anderen Ausführungsform der Erfindung wird 2-(tert-Butyldimethylsiloxy)inden zunächst mit Butyllithium und danach mit Dibromethan reagiert, wobei Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan gebildet wird. Diese Verbindung kann mit zwei Äquivalenten Butyllithium reagiert werden, was das entsprechende Bislithiumsalz ergibt. Dieses kann danach mit Zirkoniumtetrachlorid reagiert werden, wobei [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid erhalten wird. Das racemische Diastereomer des letzteren wird in großem Überschuss gebildet und wird vom meso-Isomer durch fraktionierte Kristallisierung leicht abgetrennt. Katalytische Hydrierung von racemischem [Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid ergibt den entsprechenden Tetrahydroindenylkomplex. Diese Reaktionen verlaufen gemäß dem nachfolgenden Reaktionsschema (X):
-

- In den vorstehenden Reaktionen kann Butyllithium ersetzt werden wie vorstehend beschrieben. Zirkoniumtetrachlorid kann durch Titantetrachlorid oder Hafniumtetrachlorid ersetzt werden, wobei die entsprechenden Titan-. und Hafniumkomplexe erhalten werden.
- Gemäß einer nochmals weiteren Ausführungsform der Erfindung wird 2-(t-Hexyldimethylsiloxy)inden zunächst mit Butyllithium und danach mit Dibromethan reagiert, wobei Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan gebildet wird. Diese Verbindung kann mit zwei Äquivalenten Butyllithium reagiert werden, was das entsprechende Bislithiumsalz ergibt. Dieses kann danach mit Zirkoniumtetrachlorid reagiert werden, wobei [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid erhalten wird. Das racemische Diastereomer des letzteren wird in großem Überschuss gebildet und wird vom meso-Isomer durch fraktionierte Kristallisierung leicht abgetrennt. Die Reaktionen verlaufen gemäß dem nachfolgenden Reaktionsschema (XI):
-

- In den vorstehenden Reaktionen kann Butyllithium ersetzt werden wie vorstehend beschrieben. Zirkoniumtetrachlorid kann durch Titantetrachlorid oder Hafniumtetrachlorid ersetzt werden, wobei die entsprechenden Titan-. und Hafniumkomplexe erhalten werden. Hydrierung von [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid ergibt den entsprechenden Tetrahydroindenylkomplex.
- Veranschaulichende, aber nicht einschränkende Beispiele der erfindungsgemäßen Verbindungen sind unter anderem 2-(tert-Butyldimethylsiloxy)inden, 2-(t-Hexyldimethylsiloxy)inden, 2-(Cyclohexyldimethylsiloxy)inden, 2-(tert-Butyldiphenylsiloxy)inden, Dimethylsilylbis(2-(tert-butyldimethylsiloxy)inden), Diphenylsilylbis(2-(tert-butyldimethylsiloxy)inden), Dimethylsilylbis(2-(t- hexyldimethylsiloxy)inden), Diphenylsilylbis(2-(t-hexyldimethylsiloxy)inden), Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)inden), Diphenylsilylbis(2-(cyclohexyldimethylsiloxy)inden), Dimethylsilylbis(2-(tert-butyldiphenylsiloxy)inden), Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)inden), racemisches und meso [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(t-hexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(t-hexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Dimethylsilylbis(2-(tert-butyldiphenylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Diphenylsilylbis(2-(tert-butyldiphenylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan, Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan, Bis(2-(tert-butyldiphenylsiloxy)indenyl)ethan, Bis(2-(cyclohexyldimethylsiloxy)indenyl)ethan, [rac-Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Ethylenbis(2-(cyclohexyldimethylsiloxy)indenyl)]zirkoniumdichlorid, racemisches und meso [Ethylenbis(2-(tert-butyldiphenylsiloxy)indenyl)]zirkoniumdichlorid, [rac-Ethylenbis(2- (tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Ethylenbis(2-(cyclohexyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, racemisches und meso [Ethylenbis(2-(tert-butyldiphenylsiloxy )-4,5,6,7-tetrahydroindenyl)]zirkoniumdichlorid, Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan und [rac-Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid. Titan oder Hafnium können anstelle von Zirkonium in entsprechenden Komplexen verwendet werden.
- Die erfindungsgemäßen Metallocenverbindungen können als Katalysatorkomponenten für die Polymerisation oder Copolymerisation von Olefinen verwendet werden. Die Olefine können ausgewählt werden aus Ethylen, Propylen, Buten, Penten, Hexen, Hepten und Octen oder Gemischen davon. Insbesondere können die Olefine aus Ethylen und Propylen und Gemischen davon oder zusammen mit anderen α-Olefinen ausgewählt werden.
- Die Katalysatoren können entweder trägergestützt oder ungestützt sein. Die trägergestützten Katalysatoren werden hauptsächlich für Suspensions- und Gasphaseverfahren verwendet. Der Träger kann jeder Träger sein, der für Metallocene und andere Typen von Katalysatoren bekannt ist. Bevorzugte Träger sind Siliziumoxid und Aluminiumoxid.
- Die erfindungsgemäßen Verbindungen können in Kombination mit Aktivatorverbindungen, was organometallische Verbindungen sind, verwendet werden oder allen anderen Verbindungen, die in Kombination mit Metallocenverbindungen verwendet werden. Geeignete Cokatalysatoren sind beispielsweise Alkylaluminiumverbindungen, Aluminoxane, Methylaluminoxan oder modifiziertes Methylaluminoxan. Der bevorzugte Cokatalysator ist Methylalumoxan (MAO). Andere Cokatalysatoren, die verwendet werden können, umfassen Lewis-Säuren oder prolische Säuren, wie etwa (B(C6F5)3 oder [PhNMe2H]1B(C6F5)4, welche kationische Metallocene mit kompatiblen nicht-koordinierenden Anionen erzeugen, in der Gegenwart oder Abwesenheit von Alkylaluminiumverbindungen. Besonders geeignete Aktivatoren sind beispielsweise Alumoxanverbindungen mit der Formel R-(Al(R)-O-)m-AlR2, wobei n gleich 1–40 ist, m gleich 3–40 ist und R eine C1-C8 Alkylgruppe ist. Bevorzugt ist R eine Methylgruppe, und der bevorzugte Aktivator ist Methylalumoxan (MAO). Der Aktivator kann gemäß den in der Technik bekannten Methoden angewandt werden.
- Experimenteller Teil
- Alle Arbeitsschritte wurden in Argon- oder Stickstoffatmosphäre unter Verwendung von Schlenk-, Vakuum- oder Handschuhkasten-Standardtechniken durchgeführt. Lösungsmittel wurden vor Verwendung getrocknet und unter Argon destilliert. Die 1H und 13C NMR-Spektren wurden in CDCl3 oder CD2Cl2 Lösung aufgezeichnet, unter Verwendung der NMR-Spektrometer JEOL JNM-LA400 oder JEOL JNM-A500, und gegen Tetramethylsilan (TMS) oder die restlichen Protonen der deuterierten Lösungsmittel abgeglichen. Direkte Elektronenionisation-Massenspektren (EIMS) wurden an einem Varian VG-7070E oder einem Varian-8000 Massenspektrometer erhalten.
- KATALYSATORHERSTELLUNG
- Beispiel 1
- 2-(tert-Butyldimethylsiloxy)inden
- Eine Lösung von tert-Butyldimethylchlorsilan (248,69 g, 1,65 Mol) und Imidazol (112,33 g, 1,65 Mol) in DMF (900 ml) wurde mit 2-Indanon (198,24 g, 1,50 Mol) reagiert und danach über Nacht bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde mit Wasser (800 ml) behandelt und mit Diethylether (3 × 400 ml) extrahiert. Die kombinierten organischen Phasen wurden mit Wasser (2 × 400 ml) gewaschen und über Natriumsulfat getrocknet. Die Lösungsmittel wurden unter verringertem Druck entfernt, wobei ein orangefarbenes Öl zurück blieb. Destillation unter verringertem Druck ergab 331,2 g (89,6%) der Titelverbindung als ein gelbes Öl (Sdp. 105–107°C/0,1 mbar). 1H NMR (CDCl3, δ): 7,19-7,07 (m, 3H); 6,97 (td, 3J = 7,3 Hz, 4J = 1,4 Hz, 1H); 5,72 (dd, 4J = 1,9 Hz, 1,1 Hz, 1H); 3,24 (dd, 4J = 1,7 Hz, 1,1 Hz, 2H); 0,96 (s, 9H); 0,23 (s, 6H). 13C NMR (CDCl3, δ): 162,44; 145,14; 136,53; 126,44; 123,01; 122,39; 118,92; 106,58; 39,46; 25,59; 18,14; –4,68.
- Beispiel 2
- 2-(t-Hexyldimethylsiloxy)inden
- Eine Lösung von t-Hexyldimethylchlorsilan (100,0 g, 559,3 mMol) und Imidazol (38,08 g, 559,3 mMol) in DMF (350 ml) wurde mit 2-Indanon (67,40 g, 510,0 mMol) reagiert und danach zwei Tage bei Raumtemperatur gerührt. Das Reaktionsgemisch wurde mit Wasser (300 ml) behandelt und mit Et2O (3 × 200 ml) extrahiert. Die kombinierten organischen Phasen wurden mit Wasser (2 × 200 ml) gewaschen und über Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein rotes Öl. Destillation unter verringertem Druck ergab 116,89 g (83,5%) der Titelverbindung als ein gelbes Öl (Sdp. 128–130°C/0,4 mbar). 1H NMR (CDCl3, δ): 7,24–7,10 (m, 3H); 7,00 (td, 3J = 7,3 Hz, 4J = 1,3 Hz, 1H); 5,74 (d, 4J = 0,6 Hz, 1H); 3,28 (s, 2H); 1,70 (sept, 3J = 6,8 Hz, 1H); 0,93 (s, 6H); 0,92 (d, 3J = 6,8 Hz, 6H); 0,29 (s, 6H). 13C NMR (CDCl3, δ): 162,82; 145,71; 137,03; 126,86; 123,45; 122,73; 119,29; 106,92; 40,00; 34,49; 25,51; 20,51; 18,89; –2,26.
- Beispiel 3
- 2-(Cyclohexyldimethylsiloxy)inden
- Eine Lösung von Cyclohexyldimethylchlorsilan (84,62 g, 478,7 mMol) und Imidazol (32,59 g, 478,7 mMol) in DMF (300 ml) wurde mit 2-Indanon (57,62 g, 436,0 mMol) reagiert. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt, mit Wasser (300 ml) behandelt und mit Et2O (3 × 200 ml) extrahiert. Die kombinierten organischen Phasen wurden mit Wasser (3 × 300 ml) gewaschen und über Natriumsulfat getrocknet. Die Lösungsmittel wurden entfernt, wobei ein orangefarbenes Öl zurück blieb. Destillation unter verringertem Druck ergab 81,67 g (68,8%) der Titelverbindung als ein gelbes Öl (Sdp. 140–142°C/0,2 mbar). 1H NMR (CDCl3, δ): 7,25–7,11 (m, 3H); 7,01 (td, 3J = 7,3 Hz, 4J = 1,4 Hz, 1H); 5,76 (d, 4J = 0,7 Hz, 1H); 3,29 (s, 2H); 1,77–1,74 (m, 5H); 1,27–1,13 (m, 5H); 0,90 (s, 1H); 0,24 (s, 6H). 13C NMR (CDCl3, δ): 162,44; 145,24; 136,60; 126,47; 123,07; 122,37; 118,91; 106,32; 39,52; 27,69; 26,77; 26,46; 26,33; –3,55.
- Beispiel 4
- 2-(tert-Butyldiphenylsiloxy)inden
- Zu einer Lösung von tert-Butyldiphenylchlorsilan (42,43 g, 154,4 mMol) und DBU (25,64 g, 168,4 mMol) in Benzol (200 ml) wurde 2-Indanon (18,38 g, 139,1 mMol) auf ein Mal zugegeben. Das Reaktionsgemisch wurde über Nacht gerührt, mit Et2O (200 ml) verdünnt, mit 10% HCl (2 × 200 ml) und Wasser (2 × 200 ml) gewaschen und über Natriumsulfat getrocknet. Die Lösungsmittel wurden abgedampft, wobei ein dunkelbraunes Öl zurück blieb. Destillation unter verringertem Druck ergab 38,22 g (74,1%) der Titelverbindung als ein gelbes Öl (Sdp. 172–175°C/0,05 mbar). 1H NMR (CDCl3, δ): 7,77–7,74 (m, 4H); 7,46–7,36 (m, 6H); 7,18–7,16 (m. 1H); 7,11–7,07 (m. 1H); 6,98–6,94 (m. 2H); 5,48 (d, 4J = 0,7 Hz, 1H); 3,29 (s, 2H); 1,09 (s, 9H). 13C NMR (CDCl3, δ): 162,13; 145,17; 136,75; 135,53; 132,43; 130,20; 127,99; 126,51; 123,15; 122,49; 119,24; 107,91; 39,43; 26,60; 19,46.
- Beispiel 5
- 2-(tert-Butyldimethylsiloxy)bisbenz[e,g]inden
- Zu einer Suspension von Bisbenz[e,g]indanon (35,90 g, 154,5 mMol) und tert-Butyldimethylchlorsilan (28,00 g, 185,5 mMol) in Benzol (300 ml) wurde DBU (30,60 g, 201,0 mMol) zugetropft, während das Reaktionsgemisch mit einem Eisbad kalt gehalten wurde. Das Rühren wurde 1 Stunde bei Raumtemperatur fortgesetzt. Die organische Phase wurde mit Wasser (200 ml), 5% HCl (2 × 200 ml), Wasser (200 ml) gewaschen und über Natriumsulfat getrocknet. Nach Verdampfen der Lösungsmittel blieb ein Rückstand zurück, der mit MeOH (3 × 200 ml) gewaschen wurde, wobei die Titelverbindung (38,35 g, 110,7 mMol, 71,6%) als ein leicht rosafarbenes Pulver erhalten wurde. EIMS (berechnet/gefunden): 346,1753/346,1744. 1H NMR (CDCl3, δ): 8,72–8,67 (m, 1H); 8,66–8,62 (m, 1H); 8,02–7,96 (m, 1H); 7,82–7,99 (m, 1H); 7,62–7,57 (m, 2H); 7,56–7,50 (m, 1H); 7,49–7,44 (m, 1H); 6,32 (m, 1H); 3,73 (m, 2H); 1,03 (s, 9H); 0,33 (s, 6H). 13C NMR (CDCl3, δ): 163,16; 139,79; 130,11; 129,41; 128,34; 127,73; 127,21; 126,67; 126,07; 125,53; 124,42; 123,91; 123,38; 123,24; 122,96; 104,55; 39,81; 25,68; 18,29; –4,50.
- Beispiel 6
- 2-(tert-Butyldimethylsiloxy)-4,7-dimethylinden
- Zu einer Lösung von tert-Butyldimethylchlorsilan (2,85 g, 18,9 mMol) und 4,7-Dimethyl-2-indanon (2,53 g, 15,8 mMol, erhalten durch Oxidation von 4,7-Dimethylinden) in Benzol (30 ml) wurde DBU (3,13 g, 20,5 mMol) zugegeben und das Reaktionsgemisch wurde 2 Stunden bei Raumtemperatur gerührt. Das Gemisch wurde mit Et2O (50 ml) verdünnt, mit Wasser (2 × 50 ml), 5% HCl (50 ml), Wasser (2 × 50 ml) gewaschen und über Natriumsulfat getrocknet. Nach Verdampfen der Lösungsmittel blieb ein dunkles Öl zurück, das in Pentan (30 ml) aufgelöst wurde. Die nicht-abreagierten Ausgangsmaterialien wurden bei –15°C kristallisiert und durch Filtration entfernt. Verdampfen des Lösungsmittels ergab 3,03 g (69,9%) der ziemlich reinen Titelverbindung als ein orangefarbenes Öl. 1H NMR (CDCl3, δ): 6,94 (dq, 3J = 7,7 Hz, 4J = 0,3 Hz, 1H); 6,78 (dq, 3J = 7,7 Hz, 4J = 0,3 Hz, 1H); 5,84 (t, 4J = 1,1 Hz, 1H); 3,20 (m, 2H); 2,30 (s, 3H); 2,25 (s, 3H); 1,00 (s, 9H); 0,27 (s, 6H). 13C NMR (CDCl3, δ): 162,08; 143,39; 134,89; 129,45; 127,78; 125,49; 123,84; 105,28; 38,80; 25,65; 18,29; 18,21; –4,57.
- Beispiel 7
- Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan
- Zu einer Lösung von 2-(tert-Butyldimethylsiloxy)inden (36,96 g, 150,0 mMol) in THF (150 ml) bei 0°C wurde n-BuLi (60,0 ml einer 2,5 M Lösung in Hexanen, 150,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach auf –80°C gekühlt und tropfenweise mit einer Lösung von Dibromethan (14,09 g, 75,0 mMol) in THF (50 ml) behandelt. Nach Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt und mit gesättigter Ammoniumchloridlösung (300 ml) gewaschen. Lösungsmittel aus der organischen Phase wurden abgedampft und das Produkt wurde in Et2O (300 ml) aufgelöst, mit Wasser (2 × 200 ml) gewaschen und über Natriumsulfat getrocknet. Wiederholte Kristallisationen bei –15°C ergaben 22,54 g (57,9%) der Titelverbindung als einen schmutzig weißen Feststoff. Die erste Kristallfraktion bestand aus diastereomer reinem Material (Smp. 108–110°C). EIMS (berechnet/gefunden): 518,3036/518,3028. 1H NMR (CDCl3, δ, Hauptdiastereomer): 7,18–7,07 (m, 6H); 6,97 (td, 3J = 7,4 Hz, 4J = 1,3 Hz, 2H); 5,66 (s, 2H); 3,17 (m, 2H); 1,89–1,84 (m, AA', 2H); 1,59–1,54 (m, BB', 2H); 0,94 (s, 18H); 0,23 (s, 6H); 0,21 (s, 6H). 13C NMR (CDCl3, δ, Hauptdiastereomer): 164,96; 144,39; 140,62; 126,50; 122,58; 122,41; 118,74; 104,97; 49,18; 25,67; 24,34; 18,12; –4,68; –4,88. 1H NMR (CDCl3, δ, Nebendiastereomer): 7,17–7,05 (m, 6H); 6,97 (td, 3J = 7,4 Hz, 4J = 1,2 Hz, 2H); 5,63 (s, 2H); 3,21 (m, 2H); 1,76–1,75 (m, 4H); 0,94 (s, 18H); 0,22 (s, 6H); 0,20 (s, 6H). 13C NMR (CDCl3, δ, Nebendiastereomer): 165,18; 144,35; 140,68; 126,53; 122,71; 122,35; 118,74; 104,87; 49,04; 25,67; 25,30; 18,14; –4,77.
- Beispiel 8
- Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan
- Zu einer Lösung von 2-(t-Hexyldimethylsiloxy)inden (68,62 g, 250,0 mMol) in THF (250 ml) bei 0°C wurde n-BuLi (100,0 ml einer 2,5 M Lösung in Hexanen, 250,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach auf –80°C gekühlt und tropfenweise mit einer Lösung von Dibromethan (23,48 g, 125,0 mMol) in THF (100 ml) behandelt. Nach Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt und mit gesättigter Ammoniumchloridlösung (350 ml) gewaschen. Lösungsmittel aus der organischen Phase wurden abgedampft und das Produkt wurde in Et2O (350 ml) aufgelöst, mit Wasser (2 × 300 ml) gewaschen und über Natriumsulfat getrocknet. Wiederholte Kristallisationen bei –15°C ergaben 37,48 g (52,2%) der Titelverbindung als schmutzig weiße Kristalle. Die erste Kristallfraktion bestand aus diastereomer reinem Material, das zur spektralen Charakterisierung verwendet wurde. EIMS (berechnet/gefunden): m/e 574,3662/574,3659. 1H NMR (CDCl3, δ): 7,22–7,07 (m, 6H); 6,97 (td, 3J = 7,4 Hz, 4J = 1,2 Hz, 2H); 5,65 (s, 2H); 3,15 (m, 2H); 1,91–1,84 (m, AA', 2H); 1,67 (sept, 3J = 6,8 Hz, 2H); 1,57–1,50 (m, BB', 2H); 0,89 (m, 24H); 0,27 (s, 6H); 0,24 (s, 6H). 13C NMR (CDCl3, δ): 164,75; 144,45; 140,58; 126,46; 122,60; 122,23; 118,68; 104,96; 49,24; 33,93; 25,03; 24,32; 20,14; 20,02; 18,51; 18,47; –2,66; –2,95.
- Beispiel 9
- Bis(2-(cyclohexyldimethylsiloxy)indenyl)ethan
- Zu einer eisgekühlten Lösung von 2-(Cyclohexyldimethylsiloxyinden (13,62 g, 50,0 mMol) in THF (50 ml) wurde n-BuLi (20,0 ml einer 2,5 M Lösung in Hexanen, 50,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach auf –80°C gekühlt und tropfenweise mit einer Lösung von Dibromethan (4,70 g, 25,0 mMol) in THF (30 ml) behandelt. Das Reaktionsgemisch wurde allmählich auf Raumtemperatur erwärmt, über Nacht gerührt und mit gesättigter Ammoniumchloridlösung (150 ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet. Lösungsmittel wurden abgedampft und das zurückbleibende Öl wurde in Et2O (100 ml) aufgelöst. Wiederholte Kristallisationen ergaben eine Gesamtausbeute von 4,03 g (28,2%) der Titelverbindung als schmutzig weiße Kristalle. Die erste Kristallfraktion wurde für die spektrale Charakterisierung verwendet. EIMS (berechnet/gefunden): m/e 570,3349/570,3342. 1H NMR (CDCl3, δ): 7,19–7,07 (m, 6H); 6,99 (td, 3J = 7,3 Hz, 4J = 1,3 Hz, 2H); 5,65 (s, 2H); 3,17 (m, 2H); 1,84–1,79 (m, AA', 2H); 1,73 (m, 10H); 1,59–1,54 (m, BB', 2H); 1,20 (m, 10H); 0,84 (m, 2H); 0,22 (s, 6H); 0,22 (s, 6H). 13C NMR (CDCl3, δ): 164,98; 144,47; 140,65; 126,51; 122,52; 122,33; 118,72; 104,64; 49,09; 27,72; 26,80; 26,48; 26,37; 24,29; –3,60.
- Beispiel 10
- Bis(2-(tert-butyldiphenylsiloxy)indenyl)ethan
- Zu einer Lösung von 2-(tert-Butyldiphenylsiloxy)inden (32,6 g, 88,0 mMol) in THF (200 ml) bei 0°C wurde n-BuLi (35,5 ml einer 2,5 M Lösung in Hexan, 88,8 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach auf –80°C gekühlt und tropfenweise mit einer Lösung von Dibromethan (8,26 g, 44,0 mMol) in THF (50 ml) behandelt. Das Reaktionsgemisch wurde allmählich auf Raumtemperatur erwärmt, über Nacht gerührt und mit gesättigter Ammoniumchloridlösung (2 × 300 ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein braunes Öl, das in siedendem Et2O (300 ml) aufgelöst wurde. Abkühlen auf –15°C ergab 13,7 g (40,7%) der Titelverbindung als ein diastereomer reines, weißes mikrokristallines Pulver. EIMS (berechnet/gefunden): m/e 766,3662/766,3641. 1H NMR (CDCl3, δ): 7,78–7,75 (m, 4H); 7,71–7,68 (m, 4H); 7,48–7,33 (m, 12H); 7,22–7,20 (m, 2H); 7,09–7,05 (m, 2H); 6,93–6,88 (m, 4H); 5,28 (s, 2H); 3,36 (m, 2H); 2,21–2,15 (m, AA', 2H); 1,85–1,79 (m, BB', 2H); 1,06 (s, 18H). 13C NMR (CDCl3, δ): 164,06; 144,19; 140,54; 135,45; 132,28; 131,91; 130,06; 129,99; 127,84; 127,79; 126,45; 122,65; 122,54; 119,06; 106,81; 49,25; 26,61; 25,10; 19,38. Die Mutterlauge wurde zur Trockene eingedampft und in siedendem Pentan (150 ml) aufgelöst. Aufkonzentrieren und Abkühlen auf –15°C ergab die zweite Ernte von 1,62 g (4,8%) der Titelverbindung als ein braunes Pulver, das mit dem Nebendiastereomer angereichert war (Gesamtausbeute 45,5%).
- Beispiel 11
- Bis(2-(tert-butyldimethylsiloxy)bisbenz[e,g]indenyl)ethan
- Zu einer eisgekühlten Lösung von 2-(tert-Butyldimethylsiloxybisbenzyl[e,g]inden (20,00 g, 63,48 mMol) in THF (80 ml) wurde n-BuLi (25,4 ml einer 2,5 M Lösung in Hexanen, 63,50 mMol) zugetropft und das Reaktionsgemisch wurde 1 Stunde bei Raumtemperatur gerührt. Die Lösungsmittel wurden unter Vakuum entfernt und Toluol (200 ml) wurde zugegeben. Die resultierende Lösung wurde auf –80°C gekühlt und Dibromethan (5,95 g, 31,67 mMol) in Toluol (20 ml) wurde zugetropft. Das Reaktionsgemisch wurde allmählich auf Raumtemperatur erwärmt, über Nacht gerührt und mit gesättigter Ammoniumchloridlösung (2 × 100 ml) gewaschen. Die organische Phase wurde über Natriumsulfat getrocknet. Aufkonzentrieren und Abkühlen auf –15°C ergab die Titelverbindung (900 mg, 1,25 mMol, 3,9%) als einen schmutzig weißen Feststoff. EIMS (berechnet/gefunden): m/e 718,3662/718,3659. 1H NMR (CDCl3, δ): 8,73–8,69 (m, 2H); 8,66 (dt, 3J = 8,2 Hz, 4J = 0,6 Hz, 2H); 8,02–7,98 (m, 2H); 7,63–7,58 (m, 6H); 7,42 (ddd, 3J = 8,2 Hz, 3J = 6,9 Hz, 4J = 1,3 Hz, 2H); 7,34 (ddd, 3J = 8,2 Hz, 3J = 6,9 Hz, 4J = 1,2 Hz, 2H); 6,17 (d, 4J = 0,4 Hz, 2H); 3,57–3,54 (m, 2H); 2,09–2,06 (m, 2H); 1,76–1,73 (m, 2H); 0,72 (s, 18H); 0,11 (s, 6H); 0,03 (s, 6H). 13C NMR (CDCl3, δ): 166,68; 139,37; 131,67; 130,32; 129,25; 127,93; 127,04; 126,70; 125,91; 125,46; 124,54; 123,61; 123,43; 123,20; 122,97; 102,67; 49,93; 25,48; 23,68; 18,04; –4,82; –5,03.
- Beispiel 12
- Bis(1-(trimethylsilyl)-2-(tert-butyldimethylsiloxy)-3-indenyl)ethan
- Zu einer Lösung von Bis(2-tert-butyldimethylsiloxy)indenyl)ethan (10,38 g, 20,0 mMol) in THF (100 ml) bei –20°C wurde n-Buli (16,1 ml einer 2,5 M Lösung in Hexan, 40,2 mMol) zugetropft und das Reaktionsgemisch wurde 3 Stunden bei Raumtemperatur gerührt. Zu der resultierenden Lösung wurde Chlortrimethylsilan (6,85 g, 63,0 mMol, 8,0 ml) bei 0°C zugetropft. Nach Beendigung der Zugabe wurde das Reaktionsgemisch über Nacht bei Raumtemperatur gerührt und die Lösungsmittel wurden abgedampft. Der zurückbleibende orangefarbene Feststoff wurde mit CH2Cl2 extrahiert und durch Celite filtriert. Das Lösungsmittel wurde abgedampft und das Produkt wurde in Et2O (150 ml) aufgelöst. Aufkonzentrieren und Abkühlen auf 0°C ergab 10,29 g (77,6%) eines 2 : 1 Diastereomerengemisches der Titelverbindung als schmutzig weiße Kristalle. EIMS (berechnet/gefunden): m/e 662,3827/662,3834. 1H NMR (CD2Cl2, δ, Haupt/Nebendiastereomer): 7,51–7,49 (m, 2H, Haupt); 7,42–7,40 (m, 2H, Neben); 7,29–7,24 (m, 4 + 2H, Haupt/Neben); 7,22–7,17 (m, 2H, Neben); 7,08–7,02 (m, 2 + 2H, Haupt/Neben); 3,29 (s, 1H, Neben); 3,27 (s, 1H, Haupt); 3,00–2,96 (m, AA', 2H, Neben); 2,72–2,65 (m, AA'BB', 4H, Haupt); 2,44–2,40 (m, BB', 2H, Neben); 1,02 (s, 18H, Haupt); 1,01 (s, 18H, Neben); 0,20 (s, 6H, Neben); 0,19 (s, 6H, Haupt); 0,10 (s, 6H, Neben); 0,05 (s, 6H, Haupt); 0,02 (s, 18H, Neben); 0,01 (s, 18H, Haupt). 13C NMR (CD2Cl2, δ, Hauptdiastereomer): 158,32; 144,56; 139,24; 125,09; 122,74; 121,98; 119,89; 118,08; 45,08; 26,14; 24,06; 18,52; –2,39; –3,40; –4,66. 13C NMR (CDCl3, δ, Nebendiastereomer): 158,08; 144,50; 139,14; 124,97; 122,62; 121,95; 119,95; 118,50; 45,13; 26,14; 23,86; 18,47; –2,29; –3,40; –4,24.
- Beispiel 13
- Dimethylbis(2-(tert-butyldimethylsiloxy)indenyl)silan
- Zu einer Lösung von 2-(tert-Butyldimethylsiloxy)inden (12,32 g, 50,0 mMol) in Et2O (50 ml) bei 0°C wurde n-BuLi (20,0 ml einer 2,5 M Lösung in Hexan, 50,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach zu einer Lösung von Dimethyldichlorsilan (3,03 ml, 25,0 mMol) in Et2O (25 ml) bei 0°C zugetropft. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt und mit gesättigter Ammoniumchloridlösung (150 ml), Wasser (3 × 100 ml) gewaschen und über Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein rotes Öl, das in einem 1 : 1 Gemisch von MeOH und Aceton aufgelöst wurde. Aufkonzentrieren und Abkühlen auf –30°C ergab 5,78 g (42,1%) der Titelverbindung als leicht gelbe Kristalle. Die erste Ernte bestand ausschließlich aus dem racemischen Diastereomer. EIMS (berechnet/gefunden): m/e 548,2962/548,2958. Smp. 92–94°C. 1H NMR (CDCl3, δ): 7,22–7,14 (m, 6H); 6,98 (td, 3J = 7,3 Hz, 4J = 1,4 Hz, 2H); 5,85 (s, 2H); 3,99 (s, 2H); 0,97 (s, 18H); 0,34 (s, 6H); 0,28 (s, 6H); –0,23 (s, 6H). 13C NMR (CDCl3, δ): 164,96; 144,44; 137,55; 125,13; 122,75; 121,41; 118,83; 104,00; 43,06; 25,77; 18,20; –4,34; –4,88; –6,99. Der ölige Rückstand wurde ausgiebig mit kaltem MeOH (3 × 50 ml), kaltem Pentan (50 ml) gewaschen und unter Vakuum getrocknet, wobei 3,47 g (24,8%) des ziemlich reinen meso-Diasteromers als ein rotes Öl erhalten wurden (Gesamtausbeute 66,9%).
- Beispiel 14
- Dimethylbis(2-(t-hexyldimethylsiloxy)indenyl)silan
- Zu einer Lösung von 2-(t-Hexyldimethylsiloxy)inden (35,58 g, 129,6 mMol) in Et2O (120 ml) bei 0°C wurde n-BuLi (52,4 ml einer 2,5 M Lösung in Hexan, 130,9 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende Lösung wurde danach zu einer Lösung von Dimethyldichlorsilan (7,9 ml, 64,8 mMol) in Et2O (50 ml) bei 0°C zugetropft. Das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt und mit gesättigter Ammoniumchloridlösung (300 ml), Wasser (2 × 200 ml) gewaschen und über Natriumsulfat getrocknet. Abdampfen der Lösungsmittel ergab ein rotes Öl, das in einem 4 : 1 Gemisch von MeOH und Aceton aufgelöst wurde. Aufkonzentrieren und Abkühlen auf –30°C ergab 11,67 g (29,7%) der Titelverbindung als ein weißes Pulver. Die kristalline erste Ernte bestand ausschließlich aus dem racemischen Diastereomer. EIMS (berechnet/gefunden): m/e 604,3588/604,3585. 1H NMR (CDCl3, δ): 7,22–7,13 (m, 6H); 6,98 (td, 3J = 7,3 Hz, 4J = 1,4 Hz, 2H); 5,85 (s, 2H); 3,99 (s, 2H); 1,72 (sept, 3J = 6,9 Hz, 2H); 0,94 (s, 6H); 0,94 (s, 6H); 0,90 (d, 3J = 6,9 Hz, 6H); 0,89 (d, 3J = 6,9 Hz, 6H); 0,38 (s, 6H); 0,34 (s, 6H); –0,23 (s, 6H). 13C NMR (CDCl3, δ): 164,87; 144,49; 137,53; 125,13; 122,86; 121,38; 118,84; 104,26; 43,15; 33,76; 25,19; 20,17; 20,08; 18,54; 18,45; –2,30; –2,75; –6,73. Der ölige Rückstand wurde mit kaltem MeOH (3 × 100 ml) gewaschen und unter Vakuum getrocknet, wobei 14,02 g (35,8%) des ziemlich reinen meso-Diasteromers als ein rotes Öl erhalten wurden (Gesamtausbeute 65,5%).
- Beispiel 15
- [Bis(2-(tert-butyldimethylsiloxy)bisbenz[e,g]indenyl)]zirkoniumdichlorid
- Zu einer Lösung von 2-tert-Butyldimethylsiloxy)bisbenz[e,g]inden (6,83 g, 19,7 mMol) in THF (50 ml) wurde bei –20°C n-Buli (7,9 ml einer 2,5 M Lösung in Hexanen, 19,7 mMol) zugetropft. Die resultierende rote Lösung wurde auf Raumtemperatur erwärmen gelassen und wurde mittels einer Kanüle einer Lösung von ZrCl4 (2,24 g, 9,6 mMol) in THF (50 ml) zugegeben. Das Reaktionsgemisch wurde 20 h unter Rückfluss erwärmt. Nach Verdampfen des THF blieb ein Feststoff zurück, der mit CH2Cl2 (50 ml) extrahiert wurde und durch Celite filtriert wurde, um Lithiumchlorid zu entfernen. Nach Verdampfen des CH2Cl2 blieb ein roter öliger Feststoff zurück, der mit Et2O (2 × 50 ml) gewaschen wurde, wobei 2,97 g (34,0%) der Titelverbindung als ein blassgrünes Pulver zurück blieben. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C46H50Si2O2ZrCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 850–859 beobachtet. 1H NMR (CDCl3, δ): 8,45 (m, 4H); 7,66 (m, 4H); 7,50–7,41 (m, 8H); 6,24 (s, 4H); 1,08 (s, 18H); 0,34 (s, 12H); 13C NMR (CDCl3, δ): 153,11; 129,88; 129,02; 128,21; 128,13; 126,95; 126,82; 124,25; 123,37; 118,32; 97,22; 25,80; 18,48; –4,12.
- Beispiel 16
- rac-[Ethylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
- Zu einer eisgekühlten Lösung von Bis(2-tert-butyldimethylsiloxy)indenyl)ethan (5,37 g, 10,3 mMol) in THF (50 ml) wurde n-Buli (8,3 ml einer 2,5 M Lösung in Hexanen, 20,7 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende schmutzig gelbe Suspension wurde danach mittels einer Kanüle einer Suspension von ZrCl4 (2,41 g, 10,3 mMol) in THF (20 ml) bei –80°C zugegeben. Das Reaktionsgemisch wurde langsam auf Raumtemperatur erwärmt und über Nacht gerührt. Nach Verdampfen der Lösungsmittel blieb ein gelber Feststoff zurück, der mit CH2Cl2 (150 ml) extrahiert wurde und durch Celite filtriert wurde, um Lithiumchlorid zu entfernen. Aufkonzentrieren und Abkühlen auf –30°C ergab 1,47 g (21,0%) der Titelverbindung als einen gelben mikrokristallinen Feststoff. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C32H44Si2O2ZrCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 676–684 beobachtet. 1H NMR (CD2Cl2, δ): 7,64 (dq, J = 8,6 Hz, 1,9 Hz, 0,9 Hz, 2H); 7,31–7,27 (m, 4H); 7,07–7,03 (m, 2H); 5,93 (d, J = 0,8 Hz, 2H); 4,01–3,90 (m, AA', 2H); 3,58–3,47 (m, BB', 2H); 1,00 (s, 18H); 0,20 (s, 6H); 0,19 (s, 6H). 13C NMR (CD2Cl2, δ): 150,12; 126,17; 125,14; 124,86; 124,79; 123,35; 116,99; 108,54; 98,61; 26,30; 25,80; 18,61; –3,94; –4,27.
- Beispiel 17
- rac-[Ethylenbis(2-(tert-butyldimethylsiloxy)-4,5,6,7-tetrahydroindenyl)1-zirkoniumdichlorid
- Ein Gemisch von rac-[Ethylenbis((2-tert-butyldimethylsiloxy)indenyl)]-zirkoniumdichlorid (1,00 g, 1,47 mMol) und PtO2 (20 mg) in CH2Cl2 (150 ml) wurde 16 h bei 70 bar in einem gerührten Reaktor hydriert. Die hellgrüne Suspension wurde durch Celite filtriert und das Lösungsmittel abgedampft. Der Rückstand wurde in Hexan (50 ml) aufgelöst und auf 0°C abgekühlt, wobei 0,80 g (79,2%) der Titelverbindung als ein hellgrüner mikrokristalliner Feststoff erhalten wurden. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C32H52Si2O2ZrCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 684–692 beobachtet. 1H NMR (CD2Cl2, δ): 5,69 (s, 2H); 3,39–3,29 (m, AA', 2H); 3,04–2,97 (m, 2H); 2,85–2,77 (m, 2H); 2,73–2,64 (m, BB', 2H); 2,48–2,34 (m, 4H); 1,90–1,70 (m, 4H); 1,59–1,42 (m, 4H); 0,92 (s, 18H); 0,20 (s, 6H); 0,16 (s, 6H). 13C NMR (CD2Cl2, δ): 142,24; 127,66; 117,29; 114,18; 106,45; 25,46; 24,46; 23,80; 22,05; 21,92; 21,75; 18,10; –4,09; –4,65.
- Beispiel 18
- rac-Ethylenbis(2-tert-butyldimethylsiloxy-1-indenyl)hafniumdichlorid
- Zu einer Lösung von Bis(2-(tert-butyldimethylsiloxy)indenyl)ethan (10,38 g, 20,0 mMol) in THF (80 ml) bei 0°C wurde n-Buli (16,1 ml einer 2,5 M Lösung in Hexan, 40,2 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die Lösungsmittel wurden unter Vakuum entfernt und das resultierende schmutzig weiße Pulver wurde mit HfCl4 (6,41 g, 20,0 mMol) gemischt. Das Gemisch wurde auf –80°C gekühlt und vorgekühltes CH2Cl2 (150 ml) wurde zugegeben. Die hellgelbe Suspension wurde langsam auf Raumtemperatur erwärmt, über Nacht gerührt und durch Celite filtriert, um Lithiumchlorid zu entfernen. Aufkonzentrieren und Abkühlen auf –30°C ergab 1,96 g (12,8%) der Titelverbindung als einen hellgelben mikrokristallinen Feststoff. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C32H44Si2O2NfCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 760–774 beobachtet. 1H NMR (CD2Cl2, δ): 7,64 (dq, J = 8,5 Hz, 1,8 Hz, 1,0 Hz, 2H); 7,34–7,25 (m, 4H); 7,05–7,01 (m, 2H); 5,86 (d, 4J = 0,7 Hz, 2H); 4,02–3,92 (AA', 2H); 3,71–3,61 (BB', 2H); 1,02 (s, 18H); 0,22 (s, 6H); 0,21 (s, 6H). 13C NMR (CD2Cl2, δ): 148,64; 126,19; 124,97; 124,35; 124,30; 123,49; 115,47; 105,37; 96,46; 25,84; 25,52; 18,62; –3,92; –4,26.
- Beispiel 19
- rac-[Ethylenbis(2-(t-hexyldimethylsiloxy)indenyl)]zirkoniumdichlorid
- Zu einer eisgekühlten Lösung von Bis(2-(t-hexyldimethylsiloxy)indenyl)ethan (11,50 g, 20,0 mMol) in THF (100 ml) wurde n-Buli (16,0 ml einer 2,5 M Lösung in Hexan, 40,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die resultierende schmutzig gelbe Suspension wurde danach mittels einer Kanüle einer Suspension von ZrCl4 (4,66 g, 20,0 mMol) in THF (25 ml) bei –80°C zugegeben. Das Reaktionsgemisch wurde langsam auf Raumtemperatur erwärmt und über Nacht gerührt. Nach Verdampfen der Lösungsmittel blieb ein gelber Feststoff zurück, der mit CH2Cl2 (150 ml) extrahiert wurde und durch Celite filtriert wurde, um Lithiumchlorid zu entfernen. Aufkonzentrieren und Abkühlen auf
–30°C ergab 3,25 g (22,1%) der Titelverbindung als einen gelben mikrokristallinen Feststoff. Einkristalle für Röntgenbeugung wurden aus einer gesättigten Toluollösung bei Raumtemperatur erhalten. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C36H52Si2O2ZrCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 732–740 beobachtet. 1H NMR (CD2Cl2, δ): 7,66 (dq, J = 8,6 Hz, 1,8 Hz, 1,0 Hz, 2H); 7,35–7,29 (m, 4H); 7,08–7,04 (m, 2H); 5,95 (d, 4J = 0,6 Hz, 2H); 4,04–3,94 (m, AA', 2H); 3,56–3,46 (m, BB', 2H); 1,74 (sept, 3J = 6,8 Hz, 2H); 0,99 (s, 6H); 0,97 (s, 6H); 0,95 (d, 3J = 6,8 Hz, 6H); 0,94 (d, 3J = 6,8 Hz, 6H); 0,26 (s, 6H); 0,25 (s, 6H). 13C NMR (CD2Cl2, δ): 150,48; 126,13; 125,18; 125,10; 124,85; 123,32; 116,94; 98,53; 34,22; 26,41; 25,84; 20,35; 20,14; 18,80; 18,64; –1,62; –2,01. - Beispiel 20
- rac- und meso-[Dimethylsilylenbis(2-(tert-butyldimethylsiloxy)indenyl)]zirkoniumdichlorid
- Zu einer Lösung von Dimethylbis(2-(tert-butyldimethylsiloxy)indenyl)silan (10,98 g, 20,0 mMol) in Et2O (40 ml) bei 0°C wurde n-Buli (16,0 ml einer 2,5 M Lösung in Hexan, 40,0 mMol) zugetropft und das Reaktionsgemisch wurde über Nacht bei Raumtemperatur gerührt. Die Lösungsmittel wurden unter Vakuum entfernt, wobei ein schmutzig weißes Pulver zurück blieb, das mit ZrCl4 (4,66 g, 20 mMol) gemischt wurde. Das Gemisch wurde auf –80°C gekühlt und vorgekühltes CH2Cl2 (150 ml) wurde zugegeben. Die hellgelbe Suspension wurde langsam auf Raumtemperatur erwärmt, über Nacht gerührt und durch Celite filtriert, um Lithiumchlorid zu entfernen. Aufkonzentrieren und Abkühlen auf –30°C ergab 7,95 g (56,0%) eines annähernd 2 : 1 rac/meso-Gemisches der ziemlich reinen Titelverbindung als ein hellgelbes Pulver. Im EIMS Massenspektrum der Titelverbindung wurden Stammionen der Zusammensetzung C32H46Si3O2ZrCl2 + in den entsprechenden Isotopenverhältnissen bei m/e = 706–714 beobachtet. Kleine Mengen des ziemlich reinen rac-Diastereomers der Titelverbindung wurden erhalten durch Ausführen der Reaktion in THF-Lösung, gefolgt von Entfernen von LiCl und Kristallisation aus Et2O. 1H NMR (CD2Cl2, δ, rac): 7,47–7,44 (m, 2H); 7,37–7,27 (m, 4H); 6,91–6,87 (m, 2H); 6,22 (d, 4J = 0,8 Hz, 2H); 1,19 (s, 6H); 0,92 (s, 18H); 0,27 (s, 6H); 0,21 (s, 6H). 13C NMR (CD2Cl2, δ, rac): 155,89; 128,69; 127,13; 126,84; 125,07; 124,94; 119,69; 104,50; 73,22; 26,12; 19,23; 1,04; –3,36; –3,87.
- POLYMERISATIONSTESTS
- Beispiel 21
- Die Polymerisationen wurden in einem 0,5 l Büchi-Glasautoklav in 200 ml Toluol durchgeführt. In einem typischen Lauf wurde die Hälfte der MAO/Toluol-Lösung dem Reaktor zugegeben und 5 Minuten gerührt, um Verunreinigungen im Reaktor zu reduzieren. In einer parallelen Prozedur wurden 10–15 μmMol des Metallocens in der restlichen Hälfte der MAO/Toluol-Lösung aufgelöst und 5 min bei 25°C präaktiviert. Das Katalysator/Aktivator-Gemisch wurde in den Reaktor gegeben und die Polymerisation wurde durch Einleiten des Olefinmonomers gestartet. Die Polymerisation wurde nach 20 oder 60 Minuten durch Zugabe von Ethanol oder Methanol unterbrochen. Das Polymer wurde nach Waschen mit Ethanol/HCl oder Methanol/HCl analysiert.
- Methylalumoxan (MAO) wurde als eine 29,3 Gew.-% Toluollösung verwendet (Al gesamt 13,1% w/w, Al als TMA 3,50% w/w). Ethylen und Propylen mit hoher Reinheit (99,5%) wurden als Monomere verwendet.
- Die Ergebnisse sind nachstehend in Tabelle 1 angegeben.
- Tabelle 1

- Beispiel 22
- Propylen wurde gemäß Beispiel 21 polymerisiert. Die Bedingungen und die Ergebnisse sind nachstehend in Tabelle 2 angegeben. Tabelle 2

- Tpol = 20°C; p = 2,0 bar; Polymerisationsdauer = 60 min; Al : Zr = 3000 : 1
- Beispiel 23
- Die Polymerisationen mit dem Katalysatorsystem [Metallocen]+[B(C6F6)4]– wurden bei –20°C und 2,0 bar Propylendruck ausgeführt, unter Verwendung von Triethylaluminium (TEA) als einen Abfänger für Verunreinigungen. In einem typischen Lauf wurden 0,3 g TEA mit 50 ml Toluol gerührt, gefolgt von der Zugabe von 5 μMol des Metallocens. Das Kation-bildende Mittel (14 μMol) wurde über eine Spritze zugegeben. Die Polymerisation wurde nach 60 Minuten durch Zugabe von Ethanol oder Methanol unterbrochen. Das Polymer wurde nach Waschen mit Ethanol/HCl oder Methanol/HCl analysiert.
- Die Bedingungen und die Ergebnisse sind nachstehend in Tabelle 3 angegeben. Tabelle 3
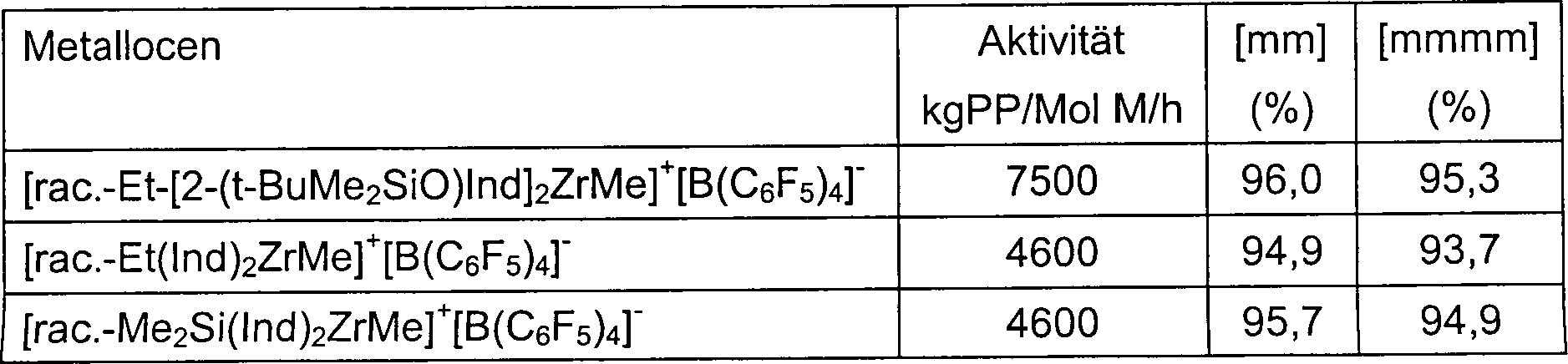
- Temperatur = –20°C; Polymerisationsdauer = 20 min
- Beispiel 24
- Ethylen wurde wie vorstehend polymerisiert. Die Ergebnisse sind nachstehend in Tabelle 4 angegeben.
- Tabelle 4: Ethylenpolymerisation mit den Katalysatorsystemen Et[IndOSiMe2tBu]2ZrCl2/MAO und Me2Si[IndH4]2ZrCl2/MAO/(Referenz) in Toluol (Al : Zr = 3000 : 1; Monomerdruck = 2 bar)
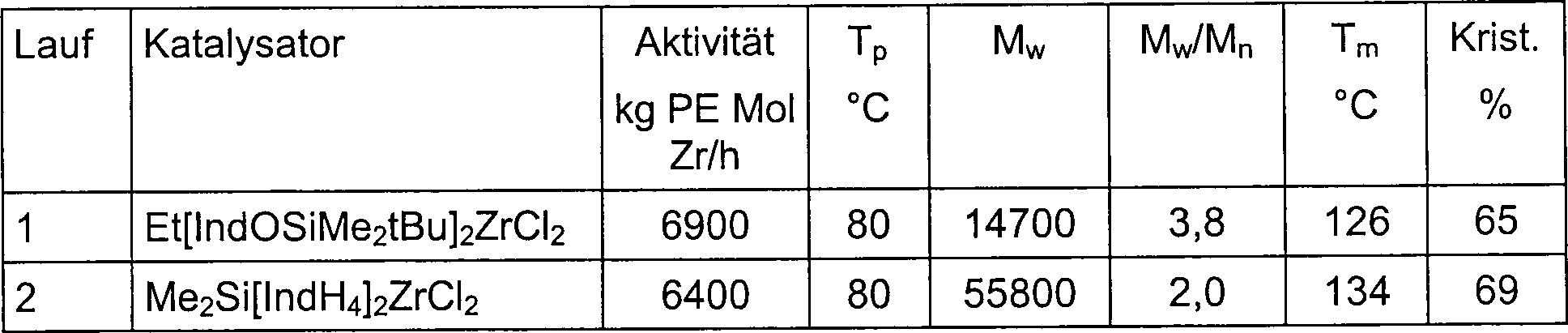
- Beispiel 25
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 2,5 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 120 g Polymer, was eine Katalysatoraktivität von 526 kgHD-PE/g·Zr·h ergibt.
- Beispiel 26
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 1,25 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 103 g Polymer, was eine Katalysatoraktivität von 903 kgHD-PE/g·Zr·h ergibt.
- Beispiel 27
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 100, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,63 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 96 g Polymer, was eine Katalysatoraktivität von 1670 kgHD-PE/g·Zr·h ergibt.
- Beispiel 28
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 100, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 12 g Polymer, was eine Katalysatoraktivität von 435 kgHD-PE/g·Zr·h ergibt.
- Beispiel 29
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 27 g Polymer, was eine Katalysatoraktivität von 936 kgHD-PE/g·Zr·h ergibt.
- Beispiel 30
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von ÅBO3 (rac-Et[2-(t-BuMe2SiO)Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 500, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,6 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 73 g Polymer, was eine Katalysatoraktivität von 2534 kgHD-PE/g·Zr·h ergibt.
- Vergleichsbeispiel 1
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von TA2677 (Et[1-Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 100, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,65 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 9 g Polymer, was eine Katalysatoraktivität von 151 kgHD-PE/g·Zr·h ergibt.
- Vergleichsbeispiel 2
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von TA2677 (Et[1-Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 200, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,65 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 27 g Polymer, was eine Katalysatoraktivität von 455 kgHD-PE/g·Zr·h ergibt.
- Vergleichsbeispiel 3
- Die Polymerisationen wurden in einem 2,0 l Büchi-Autoklav in 1300 ml Pentan durchgeführt. Eine Komplexlösung von TA2677 (Et[1-Ind]2ZrCl2) und MAO in Toluol, mit Al/Zr = 500, wurde in den Reaktor eingebracht. Die Metallocenmenge betrug 0,65 μMol. Nach dem Zuführen der Katalysatorkomplexlösung wurde der Polymerisationsreaktor auf die Polymerisationstemperatur von 70°C erwärmt. Bei Beginn der Reaktion wurde ein Ethylenpartialdruck von 5 bar in den Polymerisationsreaktor eingeführt und die Reaktion wurde gestartet. Nach 30 min Polymerisation wurde die Reaktion durch Schließen des Ethylenventils und Freisetzen von Überdruck aus dem Reaktor gestoppt.
- Nach 30 min Polymerisation betrug die Polymerausbeute 25 g Polymer, was eine Katalysatoraktivität von 421 kgHD-PE/g·Zr·h ergibt. Erklärungen von Chemikalien die in den Experimenten verwendet wurden

- MAO
- 30 Gew.-% Lösung von Methylalumoxan in Toluol, hergestellt von Albemarle
- Pentan
- Ultrareines Pentan in Polymerisationsgüte, H2O und O2 << 1,0 ppm
- Ethylen
- Polymerisationsgüte, H2O und 02 << 1,0 ppm
- Die Polymerisationen 28–30 wurden mit den Vergleichspolymerisationen 1–3 gemäß dem bekannten Stand der Technik verglichen. Die Ergebnisse sind in Tabelle 5 angegeben.
- Tabelle 5 Vergleich von ÅBO3 und TA2677

- Figur 1: Vergleich der Wirkung einer Siloxysubstitution auf die Aktivität des Metallzentrums

- Beschreibung von Beispielen
- Die Beispiele 25–27 beschreiben den Einfluss der Metallocenkonzentration auf die Polymerisation. Bei Verwendung einer zu hohen Konzentration wird die Metallaktivität zu niedrig. Aus diesen Beispielen ist ersichtlich, dass die Metallaktivität erhöht wird, wenn die Katalysatormenge von 2,5 μMol herab auf 0,6 μMol erniedrigt wird.
- Die Beispiele 28–30 und die Vergleichsbeispiele 1–3 zeigen die Bedeutung eines 2-Siloxysubstituenten auf die Katalysatoraktivität. Wenn Ethylenbisindenylzirkoniumdichlorid einen Siloxysubstituenten aufweist, wird die Katalysatoraktivität insbesondere bei niedrigen Al/Zr-Verhältnissen erhöht, was auf eine leichte Komplexbildung zwischen Metallocen und MAO und auch auf eine erhöhte Aktivität des Zirkoniums hinweist. Der Unterschied kann am besten in der vorstehenden
1 erkannt werden.
Claims (17)
- Katalysatorvorläufer mit der Formel (I):worin D Silizium oder Germanium ist; R3, R3' und R4 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe oder eine C1-C10 Hydrocarbyloxygruppe sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C20 Ringstruktur bilden; M ein Übergangsmetall der Gruppe 4 des Periodensystems ist und an den Liganden IndY in mindestens einem η5-Bindungsmodus gebunden ist; jedes R gleich oder verschieden ist und ein Wasserstoffatom, ein Halogenatom, eine C1-C10 Hydrocarbylgruppe, eine C1-C10 Hydrocarbyloxygruppe, eine Trihydrocarbylsilylgruppe ist, oder zwei R zusammen eine C2-C20 Ringstruktur bilden; B ein Brückenatom oder ein Brückengruppe zwischen zwei IndY-Liganden oder zwischen einem IndY-Liganden und dem Übergangsmetall M ist; m gleich 1 oder 2 ist; o gleich 0 oder 1 ist; und n gleich 4-m ist, wenn keine Brücke vorhanden ist oder B ein Brückenatom oder ein Brückengruppe zwischen zwei IndY-Liganden ist, oder n gleich 4-m-o ist, wenn B ein Brückenatom oder ein Brückengruppe zwischen einem IndY-Liganden und dem Übergangsmetall M ist.

- Katalysatorvorläufer mit der Formel (I):worin M ein Metall, ausgewählt aus Zirkonium, Titan oder Hafnium ist, D ein Element, ausgewählt aus Silizium (Si) oder Germanium (Ge) ist, B eine Brücke ist, umfassend mindestens eine Gruppe aus -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2 –, wobei n = 1–8, R1 und R2 die gleichen oder verschiedene Gruppen sind, ausgewählt aus Wasserstoff, einer C1-C10 Alkylgruppe, C1-C10 Alkoxygruppe, C6-C10 Arylgruppe, C6-C10 Aryloxygruppe, C2-C10 Alkenylgruppe, C2-C10 Arylalkylgruppe, C2-C10 Alkylarylgruppe, C8-C40 Arylalkenylgruppe oder einem Halogenatom, R3', R3-R6 die gleichen oder eine verschiedene Gruppen sind, ausgewählt aus Wasserstoff, einer C1-C10 Alkylgruppe, C1-C10 Alkoxygruppe, C6-C10 Arylgruppe, C6-C10 Aryloxygruppe, C2-C10 Alkenylgruppe, C7-C22 Arylalkylgruppe, C7-C22 Alkylarylgruppe, C8-C40 Arylalkenylgruppe und einem Halogenatom, die R3'- und R3-Gruppen können auch miteinander verknüpft sein um eine Ringstruktur zu bilden und R4 kann auch eine Ringstruktur sein.

- Katalysatorvorläufer nach Anspruch 1 oder 2, dadurch gekennzeichnet, daß M Zirkonium ist und D Silizium ist.
- Katalysatorvorläufer nach Anspruch 2, dadurch gekennzeichnet, daß R1 und R2 bevorzugt gleich sind und am meisten bevorzugt Halogenatome sind, beispielsweise Chloratome.
- Katalysatorvorläufer nach einem der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß R3' und/oder R3 eine C1-C10 Alkyl- oder Arylgruppe und am meisten bevorzugt eine Methylgruppe ist, und R4 eine C1-C10 Alkyl- oder Arylgruppe, bevorzugt eine tert.-Butylgruppe, t-Hexylgruppe oder Cyclohexylgruppe ist.
- Katalysatorvorläufer nach einem der vorhergehenden Ansprüche, dadurch gekennzeichnet, daß D gleich Si ist, B gleich -CH2CH2- ist, R = R1 = R2 und Chlor ist, R3 gleich CH3 ist und R5-R6 aromatisch oder kondensiert aromatisch oder Alkyl oder Wasserstoff ist.
- 2-Trihydrocarbylsiloxyinden-, 2-Trihydrocarbyloxysiloxyinden-, 2-Trihydrocarbylgermyloxyinden- oder 2-Trihydrocarbyloxygermyloxyindenverbindung mit der allgemeinen Formel:worin: D Silizium oder Germanium ist; R3, R3' und R4 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe oder eine C1-C10 Hydrocarbyloxygruppe sind, oder mindestens zwei aus R3, R3' und R4 zusammen eine C2-C20 Ringstruktur bilden; R7 eine vieratomige Kette ist, welche einen unsubstituierten oder substituierten, weiter nicht-kondensierten oder weiter kondensierten, homozyklischen (= isozyklischen) oder heterozyklischen, ungesättigten odergesättigten, aliphatischen oder aromatischen sechsgliedrigen Ring bildet; R8, R9 und R10 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Hydrocarbylgruppe, eine C1-C10 Hydrocarbyloxygruppe, eine Tri-C1-C10-hydrocarbylsilylgruppe oder eine Tri-C1-C10-hydrocarbyloxysilylgruppe sind, oder einer aus R8 und R9 eine Gruppe B sein kann, welche ein Brückenatom oder eine Brückengruppe zu einer Cyclopentadienyl-, Fluorenyl- oder Indenylgruppe ist, mit der Maßgabe, daß wenn R7 einen unsubstituierten Benzolring bildet, D Silizium ist, R3, R3' und R4 Methylgruppen sind, R8 und R10 Wasserstoffe sind, R9 kein Wasserstoff ist.
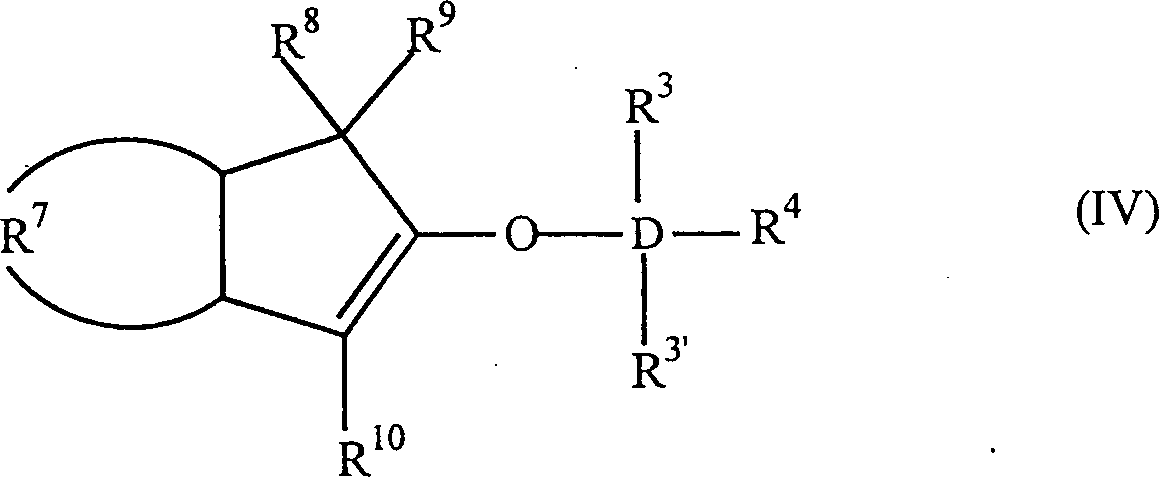
- 2-Trihydrocarbylsiloxyinden oder 2-Trihydrocarbylgermyloxyinden nach Anspruch 7, dadurch gekennzeichnet, daß es die nachfolgende Formel hat:worin: R3', R3-R5 gleich oder verschieden sind und ein Wasserstoffatom, eine C1-C10 Alkylgruppe, eine C1-C10 Alkoxygruppe, eine C6-C10 Arylgruppe, eine C2-C10 Alkenylgruppe, eine C7-C22 Arylalkylgruppe, eine C7-C22 Alkylarylgruppe, eine C8-C23 Arylalkenylgruppe oder ein Halogenatom sind, oder mindestens zwei aus R3, R3'- und R4 zusammen eine C2-C10 Ringstruktur bilden, und R8, R9 und R10 die gleichen wie vorstehend sind.
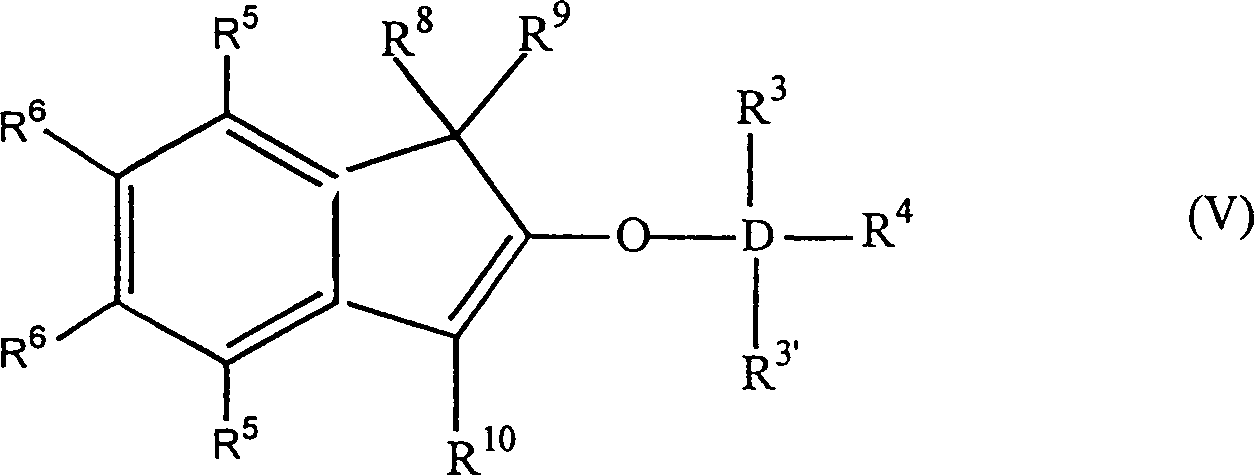
- 2-Trihydrocarbylsiloxyinden oder 2-Trihydrocarbylgermyloxyinden nach Anspruch 7 oder 8, dadurch gekennzeichnet, daß es die allgemeine Formel (VI) hat:worin: R3, R3', R4-R6, R8, R10 und D die gleichen wie vorstehend sind, und B eine Brücke von mindestens einer der Gruppen -(CH2)n-, -Si(R3)2-, oder -Ge(R3)2- ist, wobei n gleich 1–8 ist und R3 gleich wie vorstehend ist.

- 2-Trihydrocarbylsiloxyinden oder 2-Trihydrocarbylgermyloxyinden nach einem der Ansprüche 7 bis 9, dadurch gekennzeichnet, daß D Silizium ist.
- Verfahren zur Herstellung von Katalysatorkomponenten für die Olefinpolymerisation und -copolymerisation, dadurch gekennzeichnet, daß 2-Indanon in einem Lösungsmittel mit einer Base und einem Chlorsilan umgesetzt wird, wobei 2-Siloxyinden nach dem folgenden Reaktionsschema gebildet wird:wobei R' eine tert.-Butyl- oder Hexylgruppe ist.

- Verfahren nach Anspruch 11, dadurch gekennzeichnet, daß die Base aus Imidazol und Triethylamin (TEA) ausgewählt wird, und Chlorsilane aus tert.-Butyldimethylchlorsilan, t-Hexyldimethylchlorsilan und Cyclohexyldimethylchlorsilan ausgewählt werden.
- Verfahren nach einem der Ansprüche 11 oder 12, dadurch gekennzeichnet, daß die Reaktion nach dem folgenden Reaktionsschema weitergeführt wird:
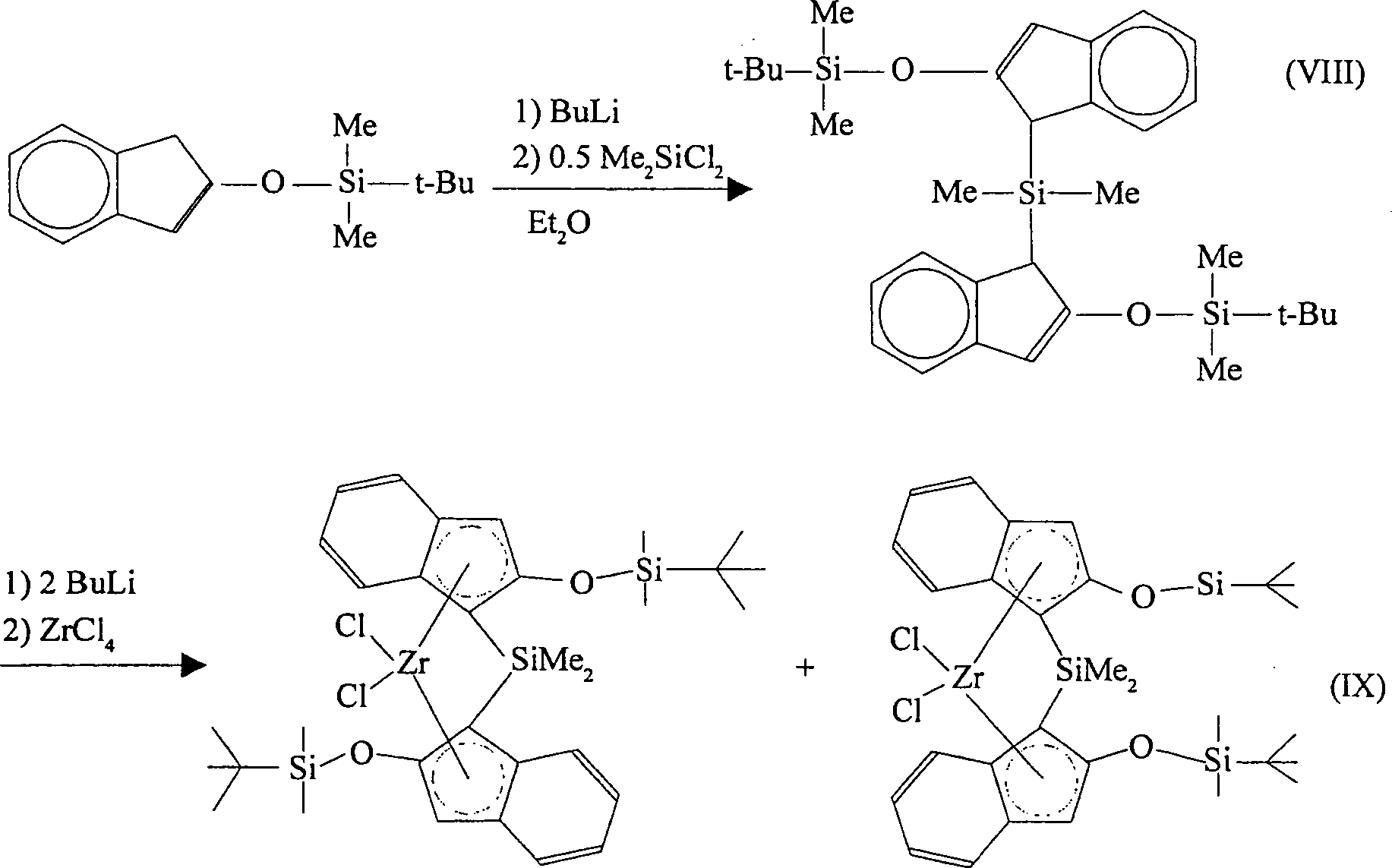
- Verfahren nach einem der Ansprüche dadurch gekennzeichnet, daß die Reaktion nach dem folgenden Reaktionsschema (X) weitergeführt wird:

- Verfahren nach einem der Ansprüche dadurch gekennzeichnet, daß die Reaktion nach dem folgenden Reaktionsschema weitergeführt wird:
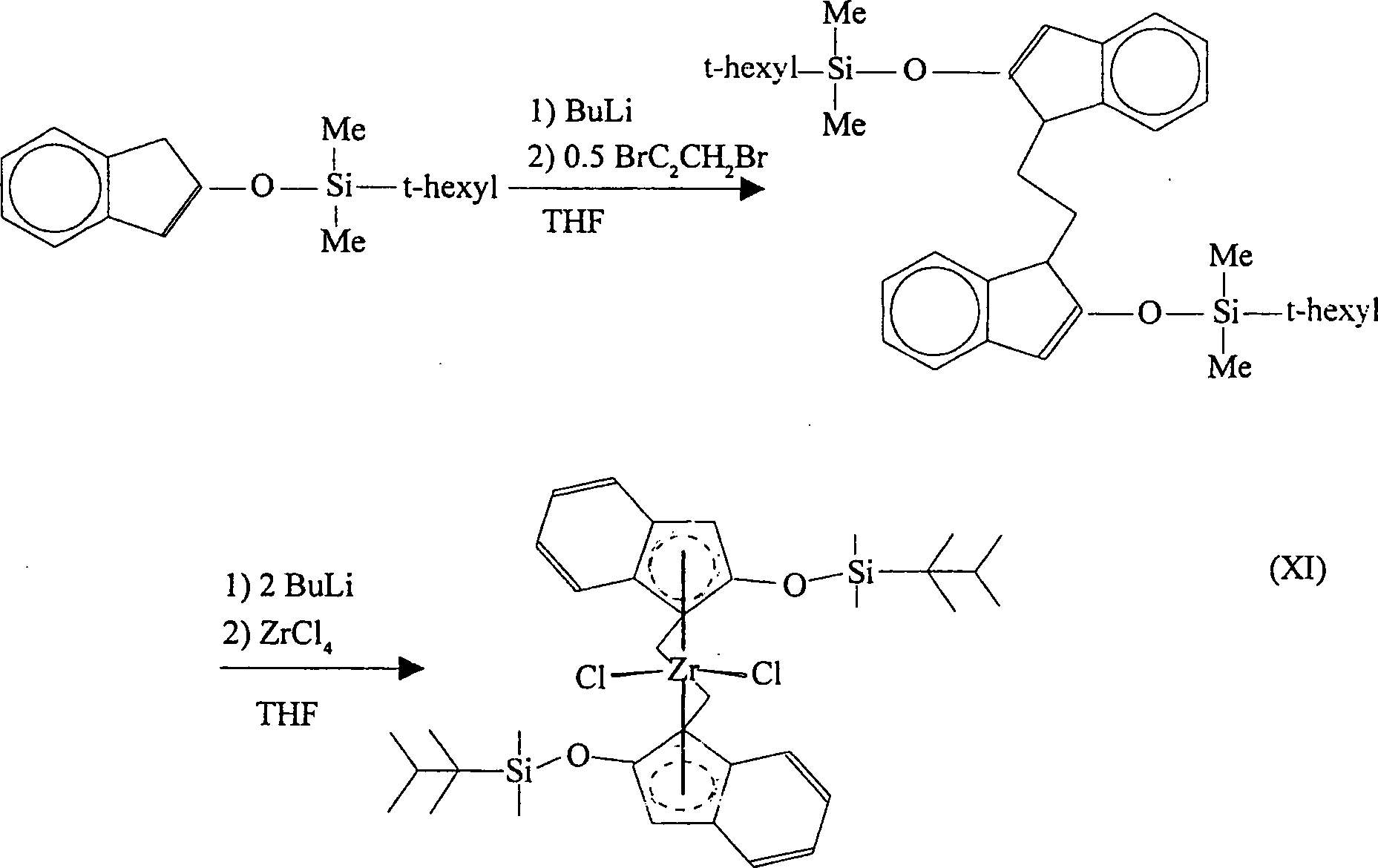
- Verwendung von Katalysatorvorläufern nach den Ansprüchen 1 bis 5 als Polymerisationskatalysatorkomponenten für die Polymerisierung oder Copolymerisierung von Olefinen.
- Verwendung nach Anspruch 16, dadurch gekennzeichnet, daß Methylalumoxan (MAO) als Cokatalysator verwendet wird.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FI960437A FI104826B (fi) | 1996-01-30 | 1996-01-30 | Heteroatomilla substituoituja metalloseeniyhdisteitä olefiinipolymerointikatalyytti-systeemejä varten ja menetelmä niiden valmistamiseksi |
| FI960437 | 1996-01-30 | ||
| PCT/FI1997/000049 WO1997028170A1 (en) | 1996-01-30 | 1997-01-30 | Heteroatom substituted metallocene compounds for olefin polymerization catalyst systems and methods for preparing them |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69726841D1 DE69726841D1 (de) | 2004-01-29 |
| DE69726841T2 true DE69726841T2 (de) | 2004-11-04 |
Family
ID=8545178
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69726841T Expired - Lifetime DE69726841T2 (de) | 1996-01-30 | 1997-01-30 | Heteroatomsubstituierte metallocene für olefipolymerisationkatalysatore un verfahren zu deren herstellung |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US6277778B1 (de) |
| EP (1) | EP0880534B1 (de) |
| JP (1) | JP4051089B2 (de) |
| KR (1) | KR19990082158A (de) |
| CN (1) | CN1089342C (de) |
| AR (1) | AR005612A1 (de) |
| AT (1) | ATE256691T1 (de) |
| AU (1) | AU722731B2 (de) |
| BR (1) | BR9707236A (de) |
| CZ (1) | CZ297674B6 (de) |
| DE (1) | DE69726841T2 (de) |
| ES (1) | ES2212070T3 (de) |
| FI (1) | FI104826B (de) |
| HU (1) | HUP9902106A3 (de) |
| IL (1) | IL125542A (de) |
| MY (1) | MY120110A (de) |
| SK (1) | SK285145B6 (de) |
| TW (1) | TW442509B (de) |
| WO (1) | WO1997028170A1 (de) |
| ZA (1) | ZA97766B (de) |
Families Citing this family (235)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1270253B (it) | 1994-06-20 | 1997-04-29 | Spherilene Srl | Copolimeri dell'etilene e procedimento per la preparazione di polimeri dell'etilene |
| FI104826B (fi) | 1996-01-30 | 2000-04-14 | Borealis As | Heteroatomilla substituoituja metalloseeniyhdisteitä olefiinipolymerointikatalyytti-systeemejä varten ja menetelmä niiden valmistamiseksi |
| US20030195109A1 (en) * | 1996-10-31 | 2003-10-16 | Jose Sancho Royo | Catalytic systems for the polimerisation and copolimerisation of alpha-olefins |
| FI971565A7 (fi) | 1997-04-14 | 1998-10-15 | Borealis As | Olefiinien polymerointiin tarkoitettujen katalysaattorisysteemien substituoituja metalloseeniyhdisteitä, niiden välituotteet ja valmistusmenetelmä |
| EP0953581B1 (de) * | 1998-04-27 | 2004-01-07 | Repsol Quimica S.A. | Katalysatorsysteme für die Polymerisation und Copolymerisation von Alpha-Olefinen |
| FI981148A7 (fi) * | 1998-05-25 | 1999-11-26 | Borealis As | Uusia aktivaattorijärjestelmä metalloseeniyhdisteitä varten |
| KR100367463B1 (ko) * | 1999-03-03 | 2003-01-14 | 주식회사 엘지화학 | 메탈로센 화합물 및 이를 이용한 올레핀 중합 |
| FI111954B (fi) | 2000-02-21 | 2003-10-15 | Borealis Tech Oy | Menetelmä polyeteenipäällysteen valmistamiseksi substraatille |
| US7074863B2 (en) | 2001-06-22 | 2006-07-11 | Borealis Technology Oy | Metallocene catalysts containing an idenyl moiety substituted at the 4,-5,-6- or 7-position by a siloxy or germyloxy group |
| GB0118010D0 (en) * | 2001-07-24 | 2001-09-19 | Borealis Tech Oy | Catalysts |
| EP1323747A1 (de) | 2001-12-19 | 2003-07-02 | Borealis Technology Oy | Herstellung von Katalysatoren für die Olefinpolymerisation |
| ATE422508T1 (de) | 2001-12-19 | 2009-02-15 | Borealis Tech Oy | Herstellung von geträgerten katalysatoren für die olefinpolymerisation |
| AU2003261617A1 (en) | 2002-08-29 | 2004-03-19 | Lg Chem, Ltd. | Fulvene, metallocene catalysts and preparation method thereof, and preparation of polyolefines copolymer using the same |
| SE0300195D0 (sv) | 2003-01-28 | 2003-01-28 | Borealis Tech Oy | Coating composition, method of preparation thereofand substrate coated therewith |
| ATE541868T1 (de) | 2003-06-20 | 2012-02-15 | Borealis Polymers Oy | Verfahren zur herstellung eines katalysators für die olefinpolymerisation |
| EP1616887A1 (de) * | 2004-07-13 | 2006-01-18 | Total Petrochemicals Research Feluy | Verfahren zur Extrusionsbeschichtung mit Polyethylen |
| GB0423212D0 (en) | 2004-10-19 | 2004-11-24 | Borealis Tech Oy | Polymer |
| GB0423555D0 (en) | 2004-10-22 | 2004-11-24 | Borealis Tech Oy | Process |
| EP1739103A1 (de) | 2005-06-30 | 2007-01-03 | Borealis Technology Oy | Katalysator |
| DE102005035477A1 (de) | 2005-07-26 | 2007-02-01 | Basell Polyolefine Gmbh | Verfahren zur Steuerung der relativen Aktivität der unterschiedlichen aktiven Zentren von Hybridkatalysatoren |
| DE602005013376D1 (de) | 2005-08-09 | 2009-04-30 | Borealis Tech Oy | Siloxy substituierte Metallocenkatalysatoren |
| WO2007045415A1 (en) | 2005-10-21 | 2007-04-26 | Borealis Technology Oy | Composition |
| EP1795542A1 (de) | 2005-12-07 | 2007-06-13 | Borealis Technology Oy | Polymer |
| EP1803747A1 (de) | 2005-12-30 | 2007-07-04 | Borealis Technology Oy | Oberflächemodifizierte Polymerisationskatalysatoren zur Herstellung von Polyolefinfilmen mit niedrigem Gelgehalt |
| EP1803743B1 (de) | 2005-12-30 | 2016-08-10 | Borealis Technology Oy | Katalysatorpartikel |
| ATE480586T1 (de) | 2006-07-14 | 2010-09-15 | Borealis Tech Oy | Polyethylen hoher dichte |
| ATE461973T1 (de) | 2007-01-22 | 2010-04-15 | Borealis Tech Oy | Polypropylenzusammensetzung mit niedriger oberflächenenergie |
| US20100280206A1 (en) | 2007-11-09 | 2010-11-04 | Borealis Technology Oy | Polyethylene copolymer |
| PL2072587T3 (pl) | 2007-12-20 | 2020-11-02 | Borealis Technology Oy | Powlekane rury o ulepszonych właściwościach mechanicznych w wysokich temperaturach i sposób ich wytwarzania |
| EP2072588B1 (de) | 2007-12-20 | 2012-10-10 | Borealis Technology Oy | Verfahren zum Beschichten eines Rohres mit hohem Durchsatz unter Verwendung von multimodalem Ethylen-Copolymer und daraus gewonnene beschichtete Rohre |
| PL2072586T3 (pl) | 2007-12-20 | 2021-05-31 | Borealis Technology Oy | Powlekane rury o ulepszonych właściwościach mechanicznych i sposób ich wytwarzania |
| EP2072589A1 (de) | 2007-12-20 | 2009-06-24 | Borealis Technology Oy | Verfahren zum Beschichten eines Rohres mit hohem Durchsatz unter Verwendung von multimodalem Ethylen-Copolymer und daraus gewonnene beschichtete Rohre |
| EP2130863A1 (de) | 2008-06-02 | 2009-12-09 | Borealis AG | Hochdichte Polymerzusammensetzungen, Verfahren für ihre Herstellung und daraus hergestellte druckfeste Rohre |
| EP2130859A1 (de) | 2008-06-02 | 2009-12-09 | Borealis AG | Polymerzusammensetzungen mit verbesserter Homogenität und verbessertem Geruch, Verfahren zu deren Herstellung und daraus hergestellte Rohre |
| EP2130862A1 (de) | 2008-06-02 | 2009-12-09 | Borealis AG | Polymerzusammensetzung und daraus hergestellte druckfeste Rohre |
| EP2130860A1 (de) | 2008-06-06 | 2009-12-09 | Borealis Technology Oy | Polymer |
| EP2133389A1 (de) | 2008-06-12 | 2009-12-16 | Borealis AG | Polypropylenzusammensetzung |
| EP2182524A1 (de) | 2008-10-31 | 2010-05-05 | Borealis AG | Kabel und Polymerzusammensetzung enthaltend ein multimodales Ethylen-Copolymer |
| EP2182526A1 (de) | 2008-10-31 | 2010-05-05 | Borealis AG | Kabel und Polymerzusammensetzung enthaltend ein multimodales Ethylen-Copolymer |
| EP2182525A1 (de) | 2008-10-31 | 2010-05-05 | Borealis AG | Kabel und Polymerzusammensetzung enthaltend ein multimodales Ethylen-Copolymer |
| WO2010052264A1 (en) | 2008-11-07 | 2010-05-14 | Borealis Ag | Solid catalyst composition |
| EP2186832B1 (de) | 2008-11-10 | 2012-09-12 | Borealis AG | Verfahren zur Herstellung eines nicht geträgerten, festen Metallocenkatalysatorsystems und dessen Verwendung bei der Polymerisation von Olefinen |
| EP2186831B1 (de) | 2008-11-10 | 2013-01-02 | Borealis AG | Verfahren zur Herstellung eines nicht geträgerten, festen Olefinpolymerisationskatalysators und Verwendung bei der Polymerisation von Olefinen |
| EP2367854B1 (de) | 2008-12-17 | 2012-11-28 | Basell Polyolefine GmbH | Katalysatorsystem für die olefinpolymerisation und herstellung und verwendung davon |
| EP2204410A1 (de) | 2008-12-31 | 2010-07-07 | Borealis AG | Mit einer Zusammensetzung aus mittels eines Single-Site-Katalysators hergestelltem Polyethylen beschichteter Artikel |
| EP2275476A1 (de) | 2009-06-09 | 2011-01-19 | Borealis AG | Automobilmaterial mit ausgezeichneter Strömung, hoher Steifheit, ausgezeichneter Leitfähigkeit und geringem CLTE |
| ES2433646T5 (es) | 2009-08-26 | 2024-04-26 | Borealis Ag | Cable y composición polimérica |
| PL2308923T3 (pl) | 2009-10-09 | 2012-11-30 | Borealis Ag | Kompozyt włókna szklanego o ulepszonej przetwarzalności |
| US8501881B2 (en) | 2009-11-13 | 2013-08-06 | Borealis Ag | Process for olefin polymerization |
| WO2011058089A1 (en) | 2009-11-13 | 2011-05-19 | Borealis Ag | Process for producing a polymerization catalyst |
| EP2322568B1 (de) | 2009-11-13 | 2013-05-15 | Borealis AG | Verfahren zur Herstellung eines Olefin-Polymerisationskatalysatoren |
| WO2011058088A1 (en) | 2009-11-13 | 2011-05-19 | Borealis Ag | Process for recovering a transition metal compound |
| EP2330135B1 (de) | 2009-12-02 | 2012-11-07 | Borealis AG | Verfahren zur Herstellung von Polyolefinen |
| EP2330136B1 (de) | 2009-12-07 | 2013-08-28 | Borealis AG | Verfahren zur Herstellung eines nicht geträgerten, festen Metallocenkatalysatorsystems und dessen Verwendung bei der Polymerisation von Olefinen |
| US8859451B2 (en) | 2009-12-18 | 2014-10-14 | Basell Polyolefine Gmbh | Process for the preparation of supported catalysts |
| EP2338920A1 (de) | 2009-12-22 | 2011-06-29 | Borealis AG | Katalysatoren |
| ES2535323T3 (es) | 2009-12-22 | 2015-05-08 | Borealis Ag | Preparación de catalizadores de sitio único |
| EP2516484A1 (de) | 2009-12-22 | 2012-10-31 | Borealis AG | Herstellung von polypropylen in gegenwart eines einseitigen katalysators |
| CN102712715B (zh) | 2010-01-21 | 2014-10-29 | 巴塞尔聚烯烃股份有限公司 | 在低聚催化剂存在下制备乙烯共聚物组合物的方法 |
| ES2564189T3 (es) | 2010-05-07 | 2016-03-18 | Borealis Ag | Preparación de un sistema de catalizador sólido |
| EP2738183A1 (de) | 2010-05-07 | 2014-06-04 | Borealis AG | Herstellung eines Feststoffkatalysatorsystems |
| EP2386583A1 (de) | 2010-05-07 | 2011-11-16 | Borealis AG | Herstellung eines Feststoffkatalysatorsystems |
| EP2385073A1 (de) | 2010-05-07 | 2011-11-09 | Borealis AG | Katalysatoren |
| WO2011147573A2 (en) | 2010-05-28 | 2011-12-01 | Basell Polyolefine Gmbh | Process for preparing a supported catalyst system for olefin polymerization, the catalyst system and its use |
| EP2415598B1 (de) | 2010-08-06 | 2014-02-26 | Borealis AG | Mehrschichtige Folie |
| EP2632980B1 (de) | 2010-10-28 | 2018-05-16 | Borealis AG | Single-site-polymer |
| PL2495037T3 (pl) | 2011-03-02 | 2021-01-11 | Borealis Ag | Zespół reaktora o wysokiej przepustowości do polimeryzacji olefin |
| PT2495038T (pt) | 2011-03-02 | 2020-11-06 | Borealis Ag | Conjunto de reator flexível para polimerização de olefinas |
| EP2495264B1 (de) | 2011-03-04 | 2013-05-08 | Borealis AG | Äußere Kraftfahrzeugartikel mit verringerter Anstreichbarkeitsstörung |
| EP2520625A1 (de) | 2011-05-06 | 2012-11-07 | Borealis AG | Beschichtungszusammensetzung |
| ES2462544T3 (es) | 2011-08-03 | 2014-05-23 | Borealis Ag | Película |
| EP2570195B1 (de) | 2011-09-15 | 2014-12-03 | Borealis AG | Schutzpolymerschicht |
| ES2462166T3 (es) | 2011-12-19 | 2014-05-22 | Borealis Ag | Reactor de bucle que proporciona un avanzado control de la división de la producción |
| WO2013149915A1 (en) | 2012-04-04 | 2013-10-10 | Borealis Ag | High-flow fiber reinforced polypropylene composition |
| US9382359B2 (en) | 2012-08-29 | 2016-07-05 | Borealis Ag | Reactor assembly and method for polymerization of olefins |
| EP2722344B1 (de) | 2012-10-18 | 2017-03-22 | Borealis AG | Polymerisationsverfahren |
| EP2722346A1 (de) | 2012-10-18 | 2014-04-23 | Borealis AG | Polymerisationsverfahren und Katalysator |
| ES2711081T3 (es) | 2012-10-18 | 2019-04-30 | Borealis Ag | Catalizador para la polimerización de olefinas |
| EP2740748B1 (de) | 2012-12-07 | 2015-06-10 | Borealis AG | Verfahren zur Polymerisierung von Olefinen in Suspensionsreaktoren |
| EP2745926A1 (de) | 2012-12-21 | 2014-06-25 | Borealis AG | Gasphasenpolymerisation und Reaktoranordnung mit einem Fließbettreaktor und einem externen Wanderbettreaktor |
| EP2745927A1 (de) | 2012-12-21 | 2014-06-25 | Borealis AG | Fließbettreaktor mit interner Wanderbettreaktionseinheit |
| EP2829558B1 (de) | 2013-07-24 | 2016-12-14 | Borealis AG | Verfahren |
| ES2607378T3 (es) | 2013-07-24 | 2017-03-31 | Borealis Ag | Proceso |
| JP6216887B2 (ja) | 2013-08-14 | 2017-10-18 | ボレアリス・アクチェンゲゼルシャフトBorealis Ag | 低温における耐衝撃性が改善されたプロピレン組成物 |
| WO2015024887A1 (en) | 2013-08-21 | 2015-02-26 | Borealis Ag | High flow polyolefin composition with high stiffness and toughness |
| MX2016001705A (es) | 2013-08-21 | 2016-05-18 | Borealis Ag | Composicion de poliolefina de alto flujo con alta rigidez y tenacidad. |
| EP2853563B1 (de) | 2013-09-27 | 2016-06-15 | Borealis AG | Filme zur BOPP-Verarbeitung aus Polymeren mit hohem XS und hoher Schmelztemperatur |
| ES2568615T3 (es) | 2013-10-11 | 2016-05-03 | Borealis Ag | Película para etiquetas orientada en la dirección de la máquina |
| ES2574428T3 (es) | 2013-10-24 | 2016-06-17 | Borealis Ag | Artículo moldeado por soplado basado en copolímero al azar bimodal |
| US10519259B2 (en) | 2013-10-24 | 2019-12-31 | Borealis Ag | Low melting PP homopolymer with high content of regioerrors and high molecular weight |
| EP3063185B9 (de) | 2013-10-29 | 2017-11-15 | Borealis AG | Feste single-site-katalysatoren mit hoher polymerisationsaktivität |
| CA2927448C (en) | 2013-11-22 | 2017-01-17 | Borealis Ag | Low emission propylene homopolymer with high melt flow |
| EP3077426B1 (de) | 2013-12-04 | 2022-10-05 | Borealis AG | Phthalatfreie pp-homopolymere für schmelzgeblasene fasern |
| KR101873134B1 (ko) | 2013-12-18 | 2018-06-29 | 보레알리스 아게 | 향상된 강성/인성 균형을 가진 bopp 필름 |
| CN105829364B (zh) | 2014-01-17 | 2017-11-10 | 博里利斯股份公司 | 用于制备丙烯/1‑丁烯共聚物的方法 |
| JP2017508032A (ja) | 2014-02-06 | 2017-03-23 | ボレアリス エージー | 高衝撃強さを有する軟質コポリマー |
| BR112016017227B1 (pt) | 2014-02-06 | 2021-06-29 | Borealis Ag | Copolímero de propileno heterofásico, película não orientada, recipiente, e uso de um copolímero de propileno heterofásico |
| EP2907841A1 (de) | 2014-02-14 | 2015-08-19 | Borealis AG | Polypropylenverbundstoff |
| EP2913346B1 (de) | 2014-02-28 | 2016-11-02 | Borealis AG | Verfahren zur Polymerisation von Olefinen in einem Fließbett |
| EP2913345B1 (de) | 2014-02-28 | 2016-11-02 | Borealis AG | Gasphasenpolymerisationsverfahren |
| EP2947118B1 (de) | 2014-05-20 | 2017-11-29 | Borealis AG | Polypropylenzusammensetzung für Anwendungen im Fahrzeuginnenraum |
| EP2995631A1 (de) | 2014-09-12 | 2016-03-16 | Borealis AG | Verfahren zur Herstellung von Pfropfcopolymeren auf einem Polyolefinrückgrat |
| EP3023450B1 (de) | 2014-11-21 | 2017-07-19 | Borealis AG | Verfahren zur Herstellung von Pellets aus weichen Copolymeren |
| KR101907331B1 (ko) | 2014-11-26 | 2018-10-11 | 보레알리스 아게 | 필름 층을 위한 폴리에틸렌 조성물 |
| KR102006091B1 (ko) | 2014-11-26 | 2019-07-31 | 보레알리스 아게 | 필름 층 |
| CN107112071A (zh) | 2014-12-19 | 2017-08-29 | 博里利斯股份公司 | 包含热塑性塑料并且具有有利性质的电力缆线聚合物组合物 |
| CN107207658B (zh) | 2014-12-19 | 2022-11-01 | 博里利斯股份公司 | 用于w&c应用的具有有利电性能的聚合物组合物 |
| EP3053976A1 (de) | 2015-02-09 | 2016-08-10 | Borealis AG | Klebstoffzusammensetzung |
| ES2724011T3 (es) | 2015-02-27 | 2019-09-05 | Borealis Ag | Estructura de película laminada basada solo en polietileno |
| WO2016184812A1 (en) | 2015-05-20 | 2016-11-24 | Borealis Ag | Process for producing polyethylene composition |
| KR101926833B1 (ko) | 2015-06-05 | 2019-03-07 | 주식회사 엘지화학 | 메탈로센 화합물 |
| KR101973191B1 (ko) | 2015-06-05 | 2019-04-26 | 주식회사 엘지화학 | 메탈로센 담지 촉매 및 이를 이용하는 폴리올레핀의 제조 방법 |
| HUE039059T2 (hu) | 2015-06-12 | 2018-12-28 | Borealis Ag | Eljárás és berendezés olefinek gázfázisban történõ polimerizálására |
| EP3173443A1 (de) | 2015-11-27 | 2017-05-31 | Borealis AG | Halbleitende polyethylenzusammensetzung |
| EP3173442A1 (de) | 2015-11-27 | 2017-05-31 | Borealis AG | Halbleitende polyethylenzusammensetzung |
| EP3178853B1 (de) | 2015-12-07 | 2018-07-25 | Borealis AG | Verfahren zur polymerisierung von alpha-olefin-monomeren |
| US11535012B2 (en) | 2015-12-15 | 2022-12-27 | Borealis Ag | Polyethylene based laminated film structure with barrier properties |
| EP3184166A1 (de) | 2015-12-22 | 2017-06-28 | Borealis AG | Verfahren zum entnehmen von agglomeraten aus einem fliessbettreaktor |
| EP3184167B8 (de) | 2015-12-22 | 2022-03-30 | Borealis AG | Verfahren zur rückführung von polymer zu einem fliessbettreaktor |
| EP3241611B1 (de) | 2016-05-02 | 2020-03-04 | Borealis AG | Verfahren zum zuführen eines polymerisations-katalysators |
| EP3243622B1 (de) | 2016-05-13 | 2020-09-09 | Borealis AG | Verfahren zum hydraulischen transportieren von polyolefinpellets |
| EP3463859B2 (de) | 2016-06-03 | 2025-03-26 | Borealis AG | Mehrschichtige struktur |
| EP3257879A1 (de) | 2016-06-17 | 2017-12-20 | Borealis AG | Bi- oder multimodales polyethylen mit niedrigem ungesättigtheitsgrad |
| KR20190021324A (ko) | 2016-06-17 | 2019-03-05 | 보레알리스 아게 | 향상된 유동학적 특성을 갖는 바이모달 또는 멀티모달 폴리에틸렌 삼원공중합체 |
| EP3257895A1 (de) | 2016-06-17 | 2017-12-20 | Borealis AG | Bi- oder multimodales polyethylenterpolymer mit verbesserten rheologischen eigenschaften |
| US20190330397A1 (en) | 2016-06-17 | 2019-10-31 | Borealis Ag | Bi- or multimodal polyethylene with low unsaturation level |
| CN109415545A (zh) | 2016-06-17 | 2019-03-01 | 博里利斯股份公司 | 具有增强的流变性能的双峰或多峰聚乙烯 |
| CN109328211B (zh) | 2016-06-21 | 2024-08-06 | 博里利斯股份公司 | 具有有利的热机械性能和电气特性的用于电线和电缆应用的聚合物组合物 |
| EP3261093B1 (de) | 2016-06-21 | 2023-08-02 | Borealis AG | Kabel mit vorteilhaften elektrischen eigenschaften |
| EP3261096A1 (de) | 2016-06-21 | 2017-12-27 | Borealis AG | Kabel und zusammensetzung |
| FI3261095T3 (fi) | 2016-06-21 | 2026-03-25 | Borealis Gmbh | Kaapeli, jolla on parannetut sähköiset ominaisuudet |
| CN109328200B (zh) | 2016-06-23 | 2022-08-19 | 博里利斯股份公司 | 催化剂失活的方法 |
| WO2018091653A1 (en) | 2016-11-18 | 2018-05-24 | Borealis Ag | Catalyst |
| ES2786756T3 (es) | 2017-06-20 | 2020-10-13 | Borealis Ag | Un método, una disposición y uso de una disposición para la polimerización de olefinas |
| EP3418309A1 (de) | 2017-06-20 | 2018-12-26 | Borealis AG | Verfahren, anordnung und verwendung einer anordnung zur herstellung von polymer |
| EP3418310B1 (de) | 2017-06-23 | 2020-04-08 | Borealis AG | Verfahren und vorrichtung zur entfernung von polymermaterial aus einem gas-festkörper-olefin-polymerisationsreaktor |
| WO2019081611A1 (en) | 2017-10-24 | 2019-05-02 | Borealis Ag | MULTILAYER POLYMER FILM |
| EP3479896A1 (de) | 2017-11-03 | 2019-05-08 | Borealis AG | Polymerisationsreaktorsystem mit mindestens einer enthnahmeventil |
| EP3483189A1 (de) | 2017-11-14 | 2019-05-15 | Borealis AG | Automatisiertes verfahren zur beendigung einer olefinpolymerisierungsreaktion unter notbedingungen |
| ES2806646T3 (es) | 2017-11-17 | 2021-02-18 | Borealis Ag | Procedimiento para mejorar la capacidad de enfriamiento de un reactor de polimerización de olefinas de gas-sólidos |
| EP3486260B1 (de) | 2017-11-17 | 2020-04-01 | Borealis AG | Verfahren zur spaltung des rücklauffluidisierungsgases in einem gas-feststoff-olefin-polymerisationsreaktor |
| EP3728342A1 (de) | 2017-12-21 | 2020-10-28 | Borealis AG | Verfahren zur herstellung eines festen katalysators |
| JP7249349B2 (ja) | 2017-12-27 | 2023-03-30 | ボレアリス エージー | チーグラー・ナッタ触媒及びその調製 |
| EA202091168A1 (ru) | 2017-12-28 | 2020-11-24 | Бореалис Аг | Катализатор и его получение |
| WO2019166652A1 (en) | 2018-03-02 | 2019-09-06 | Borealis Ag | Process |
| WO2019180166A1 (en) | 2018-03-21 | 2019-09-26 | Borealis Ag | Bi- or multimodal polyethylene composition |
| WO2019238428A1 (en) | 2018-06-14 | 2019-12-19 | Borealis Ag | Process for polymerizing olefin in a gas phase reactor with improved thermal homogeneity |
| EP3830140A1 (de) | 2018-08-02 | 2021-06-09 | Borealis AG | Verfahren zur polymerisation von ethylen in einem mehrstufigen polymerisationsverfahren |
| WO2020064313A1 (en) | 2018-09-26 | 2020-04-02 | Borealis Ag | Propylene random copolymer for use in film applications |
| CN112638959B (zh) | 2018-09-26 | 2023-05-02 | 博里利斯股份公司 | 具有优异光学性能的丙烯共聚物 |
| US12404393B2 (en) | 2018-09-26 | 2025-09-02 | Borealis Ag | Propylene copolymer composition with excellent optical and mechanical properties |
| US20210363314A1 (en) | 2018-11-07 | 2021-11-25 | Borealis Ag | Polyolefin composition with improved impact and whitening resistance |
| EP3880430B1 (de) | 2018-11-15 | 2024-09-25 | ABU DHABI POLYMERS CO. LTD (BOROUGE) - Sole Proprietorship L.L.C. | Polymerzusammensetzungen für blasformanwendungen |
| EP3883989B1 (de) | 2018-11-23 | 2024-02-28 | Borealis AG | Polypropylenzusammensetzung mit verbesserten optischen eigenschaften und aufhellungsbeständigkeit |
| CN120157797A (zh) | 2018-11-29 | 2025-06-17 | 博里利斯股份公司 | 聚合物生产工艺和聚合物 |
| WO2020109452A1 (en) | 2018-11-30 | 2020-06-04 | Borealis Ag | Washing process |
| ES2945963T3 (es) | 2018-12-14 | 2023-07-11 | Borealis Ag | Composición de polipropileno con combinación favorable de óptica, suavidad y bajo sellado |
| WO2020156989A1 (en) | 2019-01-28 | 2020-08-06 | Borealis Ag | Polymer composition |
| CN113329858B (zh) | 2019-01-28 | 2024-01-12 | 博里利斯股份公司 | 用于生产聚合物组合物的方法 |
| WO2020242921A1 (en) | 2019-05-24 | 2020-12-03 | Eastman Chemical Company | Recycle content cellulose ester |
| WO2020247192A1 (en) | 2019-05-24 | 2020-12-10 | Eastman Chemical Company | Recycle content cracked effluent |
| CN113993977B (zh) | 2019-05-24 | 2024-09-13 | 伊士曼化工公司 | 进入气体裂化器中加工的液体流中混入少量热解油 |
| EP3976734A4 (de) | 2019-05-24 | 2023-03-15 | Eastman Chemical Company | Cracken einer c4-c7-fraktion von pyrolyseöl |
| CN113993616B (zh) | 2019-06-04 | 2023-06-02 | 北欧化工股份公司 | 用于增强气固流化床反应器中流体动力学的方法和反应器组件 |
| EP3980170A1 (de) | 2019-06-04 | 2022-04-13 | Borealis AG | Verfahren und mehrstufige reaktoranordnung zur herstellung von polyolefinen |
| US12291594B2 (en) | 2019-06-24 | 2025-05-06 | Borealis Ag | Process for preparing polypropylene with improved recovery |
| JP7438324B2 (ja) | 2019-07-22 | 2024-02-26 | アブ・ダビ・ポリマーズ・カンパニー・リミテッド・(ブルージュ)・リミテッド・ライアビリティ・カンパニー | シングルサイト触媒によるマルチモーダルポリエチレン組成物 |
| US12338211B2 (en) | 2019-07-29 | 2025-06-24 | Eastman Chemical Company | Recycle content (C4)alkanal |
| US12534590B2 (en) | 2019-07-29 | 2026-01-27 | Eastman Chemical Company | Recycle content cyclobutane diol polyester |
| US11945998B2 (en) | 2019-10-31 | 2024-04-02 | Eastman Chemical Company | Processes and systems for making recycle content hydrocarbons |
| WO2021087057A1 (en) | 2019-10-31 | 2021-05-06 | Eastman Chemical Company | Pyrolysis method and system for recycled waste |
| EP4054996A4 (de) | 2019-11-07 | 2024-03-27 | Eastman Chemical Company | Oxoalkohole und oxoweichmacher mit recyceltem inhalt |
| EP4055001A4 (de) | 2019-11-07 | 2024-02-14 | Eastman Chemical Company | Gemischte ester und lösungsmittel mit recyceltem inhalt |
| KR20220093368A (ko) | 2019-11-07 | 2022-07-05 | 이스트만 케미칼 컴파니 | 재활용물 알파 올레핀 및 지방 알코올 |
| CN118851872A (zh) | 2019-11-07 | 2024-10-29 | 伊士曼化工公司 | 回收成分丙醇 |
| EP3825357B1 (de) | 2019-11-25 | 2025-04-09 | Borealis AG | Propylenzusammensetzung zur aufschäumung mit verbesserten mechanischen eigenschaften |
| EP3838984A1 (de) | 2019-12-20 | 2021-06-23 | Borealis AG | Polymerzusammensetzung und -artikel |
| EP3868793B1 (de) | 2020-02-24 | 2026-01-07 | Borealis GmbH | Verfahren zur herstellung von alpha-olefinpolymeren in einem mehrstufigen polymerisationsverfahren |
| WO2021191018A1 (en) | 2020-03-24 | 2021-09-30 | Borealis Ag | Polyethylene composition for a film layer |
| EP4126994A1 (de) | 2020-03-24 | 2023-02-08 | Borealis AG | Polyethylenzusammensetzung für eine folienschicht |
| MX2022012464A (es) | 2020-04-13 | 2022-10-27 | Eastman Chem Co | Polietileno de contenido de reciclado. |
| AU2021275475B2 (en) | 2020-05-20 | 2024-02-08 | Borealis Ag | Polymer for cable jacket |
| EP4153676B1 (de) | 2020-05-20 | 2025-09-17 | Borealis GmbH | Polymer zur stromkabelisolierung |
| CN115803411A (zh) | 2020-07-13 | 2023-03-14 | 博里利斯股份公司 | 粘合剂聚乙烯组合物 |
| KR102926344B1 (ko) | 2020-07-13 | 2026-02-11 | 보레알리스 게엠베하 | 접착성 폴리에틸렌 조성물 |
| US20230227637A1 (en) | 2020-07-23 | 2023-07-20 | Borealis Ag | Multimodal ethylene copolymer |
| CA3187544A1 (en) | 2020-08-05 | 2022-02-10 | Mohamed Esseghir | Thermoplastic compositions comprising bimodal polyethylene and articles manufactured therefrom |
| EP4263631A1 (de) | 2020-12-18 | 2023-10-25 | Borealis AG | Modifizierung von polyethylenterpolymer |
| EP4019582B1 (de) | 2020-12-23 | 2025-07-30 | Borealis GmbH | Polyethylenfilm |
| KR20230130052A (ko) | 2021-01-08 | 2023-09-11 | 보레알리스 아게 | 조성물 |
| EP4029914B1 (de) | 2021-01-14 | 2025-03-05 | Borealis AG | Heterophasische polyolefinzusammensetzung |
| EP4036130B1 (de) | 2021-01-29 | 2023-03-22 | Borealis AG | Modifizierung von polyethylencopolymer |
| EP4053194B1 (de) | 2021-03-03 | 2023-08-02 | Borealis AG | Einschichtige blasfolie |
| EP4305075A1 (de) | 2021-03-09 | 2024-01-17 | Borealis AG | Polyethylenzusammensetzung für eine folienschicht |
| EP4056598B1 (de) | 2021-03-09 | 2023-09-13 | Borealis AG | Polyethylenzusammensetzung für eine folienschicht |
| EP4056599B1 (de) | 2021-03-09 | 2023-07-26 | Borealis AG | Polyethylenzusammensetzung für eine folienschicht |
| CA3223017A1 (en) | 2021-06-24 | 2022-12-29 | Pascal Castro | Method for determining compressive character of olefin polymerisation catalysts |
| EP4359449A1 (de) | 2021-06-24 | 2024-05-01 | Borealis AG | Verwendung eines quellmittels bei der mehrstufigen polyolefinherstellung |
| US20240294681A1 (en) | 2021-06-24 | 2024-09-05 | Borealis Ag | Improving catalyst performance in multi-stage polyolefin production |
| CA3223212A1 (en) | 2021-06-24 | 2022-12-29 | Borealis Ag | Utilization of 1-hexene in multi-stage polyolefin production |
| EP4116091A1 (de) | 2021-07-07 | 2023-01-11 | Borealis AG | Mehrschichtige folie |
| IL309627A (en) | 2021-07-08 | 2024-02-01 | Borealis Ag | Polyethylene for use in the production of crosslinked polyethylene (pex) |
| EP4144435A1 (de) | 2021-09-01 | 2023-03-08 | Borealis AG | Gasphasenpolymerisationsverfahren mit verbesserter gasrückführung |
| US12195674B2 (en) | 2021-09-21 | 2025-01-14 | Eastman Chemical Company | Using spent caustic solution from pygas treatment to neutralize halogens from liquified waste plastic |
| EP4163309A1 (de) | 2021-10-07 | 2023-04-12 | Borealis AG | Hdpe |
| EP4163332B1 (de) | 2021-10-10 | 2026-04-01 | Borealis GmbH | Polyethylenzusammensetzung für eine folienschicht |
| EP4163334A1 (de) | 2021-10-10 | 2023-04-12 | Borealis AG | Polyethylenzusammensetzung für eine folienschicht |
| EP4166581A1 (de) | 2021-10-12 | 2023-04-19 | Borealis AG | Propylenzusammensetzung zum schäumen mit hoher schmelzfliessfähigkeit |
| EP4257640B1 (de) | 2022-04-04 | 2024-08-28 | Borealis AG | Rohr aus einer polypropylenzusammensetzung |
| EP4507896A1 (de) | 2022-04-11 | 2025-02-19 | Borealis AG | Mehrschichtige folie |
| EP4317216A1 (de) | 2022-08-03 | 2024-02-07 | Abu Dhabi Polymers Co. Ltd (Borouge) LLC | Äthylenterpolymerzusammensetzung mit niedriger dichte |
| EP4389418A1 (de) | 2022-12-19 | 2024-06-26 | Abu Dhabi Polymers Co. Ltd (Borouge) - Sole Proprietorship L.L.C. | Mehrlagige kollationsschrumpffolie |
| EP4389414A1 (de) | 2022-12-19 | 2024-06-26 | Abu Dhabi Polymers Co. Ltd (Borouge) - Sole Proprietorship L.L.C. | Mehrlagige kollationsschrumpffolie |
| EP4389783A1 (de) | 2022-12-20 | 2024-06-26 | Borealis AG | Katalysatorübergangsverfahren |
| EP4389776A1 (de) | 2022-12-20 | 2024-06-26 | Borealis AG | Verfahren |
| TW202440666A (zh) | 2022-12-23 | 2024-10-16 | 奧地利商柏列利斯股份公司 | 聚丙烯均聚物或共聚物的製造方法及由該製造方法獲得的聚丙烯均聚物或共聚物 |
| CN120380039A (zh) | 2022-12-23 | 2025-07-25 | 北欧化工股份公司 | 聚丙烯共聚物的生产方法 |
| WO2024133045A1 (en) | 2022-12-23 | 2024-06-27 | Borealis Ag | Process for producing a high-flow polypropylene homopolymer |
| EP4417629A1 (de) | 2023-02-14 | 2024-08-21 | Borealis AG | Polyethylenmischung für eine folienschicht |
| WO2024223777A1 (en) | 2023-04-26 | 2024-10-31 | Borealis Ag | Layer element suitable as integrated backsheet for a bifacial photovoltaic module |
| EP4701858A1 (de) | 2023-04-26 | 2026-03-04 | Borealis GmbH | Als integrierte rückseitenfolie für ein zweiseitiges fotovoltaisches modul geeignetes schichtelement |
| EP4455170A1 (de) | 2023-04-28 | 2024-10-30 | Borealis AG | Kabelzusammensetzungen mit einem multimodalen ethylen-buten-hexen-terpolymer |
| WO2025003435A1 (en) | 2023-06-30 | 2025-01-02 | Borealis Ag | Process |
| EP4538321A1 (de) | 2023-10-11 | 2025-04-16 | Borealis AG | Verfahren zur erhöhung der nukleierungseffizienz von ssc-materialien |
| EP4541583A1 (de) | 2023-10-18 | 2025-04-23 | Abu Dhabi Polymers Co. Ltd (Borouge) - Sole Proprietorship L.L.C. | Mehrschichtige folie |
| WO2025125458A1 (en) | 2023-12-12 | 2025-06-19 | Abu Dhabi Polymers Co. Ltd (Borouge) - Sole Proprietorship L.L.C. | Modified polyethylene having improved hydrostatic pressure resistance and slow crack growth resistance |
| EP4574848B1 (de) | 2023-12-19 | 2026-04-29 | Borealis GmbH | Polypropylencopolymerzusammensetzung mit hohem molekulargewicht |
| WO2025219533A1 (en) | 2024-04-18 | 2025-10-23 | Borealis Gmbh | Process for the preparation of a propylene homopolymer |
| WO2025219499A1 (en) | 2024-04-18 | 2025-10-23 | Borealis Gmbh | Processes for polymerising olefins |
| WO2025219537A1 (en) | 2024-04-18 | 2025-10-23 | Borealis Gmbh | Process for propylene polymerization with optimized prepolymerization conditions |
| WO2025262147A1 (en) | 2024-06-19 | 2025-12-26 | Borealis Gmbh | Processes for polymerising olefins |
| WO2026002813A1 (en) | 2024-06-24 | 2026-01-02 | Borealis Gmbh | Processes for polymerising olefins |
| EP4674614A1 (de) | 2024-07-04 | 2026-01-07 | Borealis GmbH | Polyethylen-mdo-film mit hoher steifigkeit, haftung und verbesserten sperrbildungsfähigkeiten |
| EP4685183A1 (de) | 2024-07-25 | 2026-01-28 | Borealis GmbH | Polyethylenzusammensetzung aus gemischten kunststoffen für filmanwendungen |
| WO2026022206A1 (en) | 2024-07-25 | 2026-01-29 | Borealis Gmbh | Mixed-plastics polyethylene composition suitable for film applications |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5166216A (en) * | 1988-10-27 | 1992-11-24 | Basf Aktiengesellschaft | Methyl α-arylacrylates substituted by a heterocyclic radical and their use |
| ES2091273T3 (es) * | 1990-11-12 | 1996-11-01 | Hoechst Ag | Procedimiento para la preparacion de un polimero olefinico de alto peso molecular. |
| DE4120009A1 (de) * | 1991-06-18 | 1992-12-24 | Basf Ag | Loesliche katalysatorsysteme zur herstellung von polyalk-1-enen mit hohen molmassen |
| ES2128371T3 (es) * | 1992-08-15 | 1999-05-16 | Targor Gmbh | Procedimiento para la obtencion de poliolefinas. |
| KR100314971B1 (ko) | 1993-05-25 | 2002-07-03 | 엑손모빌 케미칼 패턴츠 인코포레이티드 | 올레핀중합을위한지지된메탈로센촉매시스템,그의제조방법및용도 |
| ES2143636T3 (es) | 1994-06-24 | 2000-05-16 | Exxon Chemical Patents Inc | Sistemas de catalizadores de polimerizacion, su produccion y uso. |
| US5635437A (en) * | 1995-07-28 | 1997-06-03 | Exxon Chemicals Patents, Inc. | Method for preparing metallocene catalyst systems |
| FI104826B (fi) | 1996-01-30 | 2000-04-14 | Borealis As | Heteroatomilla substituoituja metalloseeniyhdisteitä olefiinipolymerointikatalyytti-systeemejä varten ja menetelmä niiden valmistamiseksi |
| ATE217318T1 (de) * | 1998-08-11 | 2002-05-15 | Dow Chemical Co | Ansa bis(.mu.-aluminium)-substituierte gruppe-4- metallkomplexe |
-
1996
- 1996-01-30 FI FI960437A patent/FI104826B/fi active
-
1997
- 1997-01-30 IL IL12554297A patent/IL125542A/xx not_active IP Right Cessation
- 1997-01-30 BR BR9707236A patent/BR9707236A/pt not_active IP Right Cessation
- 1997-01-30 DE DE69726841T patent/DE69726841T2/de not_active Expired - Lifetime
- 1997-01-30 CN CN97193331A patent/CN1089342C/zh not_active Expired - Fee Related
- 1997-01-30 ZA ZA9700766A patent/ZA97766B/xx unknown
- 1997-01-30 US US09/117,439 patent/US6277778B1/en not_active Expired - Fee Related
- 1997-01-30 SK SK1008-98A patent/SK285145B6/sk unknown
- 1997-01-30 HU HU9902106A patent/HUP9902106A3/hu unknown
- 1997-01-30 KR KR1019980705881A patent/KR19990082158A/ko not_active Withdrawn
- 1997-01-30 AU AU15485/97A patent/AU722731B2/en not_active Ceased
- 1997-01-30 WO PCT/FI1997/000049 patent/WO1997028170A1/en not_active Ceased
- 1997-01-30 CZ CZ0229498A patent/CZ297674B6/cs not_active IP Right Cessation
- 1997-01-30 AR ARP970100376A patent/AR005612A1/es active IP Right Grant
- 1997-01-30 EP EP97901652A patent/EP0880534B1/de not_active Expired - Lifetime
- 1997-01-30 ES ES97901652T patent/ES2212070T3/es not_active Expired - Lifetime
- 1997-01-30 JP JP52733197A patent/JP4051089B2/ja not_active Expired - Fee Related
- 1997-01-30 AT AT97901652T patent/ATE256691T1/de not_active IP Right Cessation
- 1997-01-30 MY MYPI97000351A patent/MY120110A/en unknown
- 1997-05-19 TW TW086106655A patent/TW442509B/zh not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000505794A (ja) | 2000-05-16 |
| FI104826B (fi) | 2000-04-14 |
| CZ229498A3 (cs) | 1998-12-16 |
| ATE256691T1 (de) | 2004-01-15 |
| US6277778B1 (en) | 2001-08-21 |
| JP4051089B2 (ja) | 2008-02-20 |
| HUP9902106A2 (hu) | 1999-11-29 |
| EP0880534A1 (de) | 1998-12-02 |
| IL125542A (en) | 2003-03-12 |
| KR19990082158A (ko) | 1999-11-25 |
| CN1214687A (zh) | 1999-04-21 |
| CN1089342C (zh) | 2002-08-21 |
| AU722731B2 (en) | 2000-08-10 |
| MY120110A (en) | 2005-09-30 |
| AR005612A1 (es) | 1999-06-23 |
| ES2212070T3 (es) | 2004-07-16 |
| ZA97766B (en) | 1997-08-18 |
| BR9707236A (pt) | 1999-07-20 |
| FI960437L (fi) | 1997-07-31 |
| SK285145B6 (sk) | 2006-07-07 |
| AU1548597A (en) | 1997-08-22 |
| SK100898A3 (en) | 1999-03-12 |
| CZ297674B6 (cs) | 2007-02-28 |
| IL125542A0 (en) | 1999-03-12 |
| FI960437A0 (fi) | 1996-01-30 |
| TW442509B (en) | 2001-06-23 |
| EP0880534B1 (de) | 2003-12-17 |
| HUP9902106A3 (en) | 2000-04-28 |
| WO1997028170A1 (en) | 1997-08-07 |
| DE69726841D1 (de) | 2004-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69726841T2 (de) | Heteroatomsubstituierte metallocene für olefipolymerisationkatalysatore un verfahren zu deren herstellung | |
| DE69829365T2 (de) | Substituierte metallocenverbindungen für olefinpolymerisationkatalysatorsysteme, deren zwischenverbindungen und verfahren zu ihrer herstellung | |
| DE60109680T2 (de) | Verfahren zur herstellung von im wesentlichen amorphen polymeren auf propylen-basis | |
| US5585509A (en) | Metallocene complexes having heterofunctional groups in the cyclopentadienyl system | |
| DE60003936T2 (de) | Katalysatorsystem unf verfahren zur polymerisierung von olefine | |
| DE69810894T2 (de) | Metallocene und katalysatoren zur olefinpolymerisation | |
| DE69903388T2 (de) | Verfahren zur herstellung von metallocenen | |
| DE69710838T2 (de) | Katalysator zur polyolefinherstellung und polyolefinherstellungsverfahren | |
| DE69416721T2 (de) | Silylverbrückte Metallocene, und ihre Verwendung | |
| DE69612331T2 (de) | Metallocenkatalysator für die Homo- oder Copolymerisation von Alpha-Olefinen | |
| EP0990003A1 (de) | Neue katalysatorsysteme für (co-)polymerisationsreaktionen, metallocenamidhalogenide, ihre herstellung und verwendung | |
| JPH03179006A (ja) | シンジオタクチツク重合体の製造方法および製造用触媒 | |
| EP0528287A1 (de) | Verbrückte, chirale Metallocene, Verfahren zu ihrer Herstellung und ihre Verwendung als Katalysatoren | |
| EP1074557A2 (de) | Übergangsmetallverbindung, Ligandensystem, Katalysatorsystem und seine Verwendung zur Polymerisation von Olefinen | |
| DE69915626T2 (de) | Durch ein Kohlenstoffatom verbundene Biscyclopentadienylverbindungen und darmit hergestellte Metallocene | |
| DE69905011T2 (de) | Bis(tetrahydro-indenyl) metallocene, und diesen enthaltende olefinpolymerisationskatalysatoren | |
| EP0670336B1 (de) | Geträgerte Metallocenkomplexe mit heterofunktionellen Gruppen am Cyclopentadienylsystem als Katalysatorsysteme | |
| EP0836608B1 (de) | Metallocene mit silylsubstituierten brücken und deren einsatz für die olefinpolymerisation | |
| EP0704454B1 (de) | Verfahren zur Herstellung verbrückter Metallocene | |
| EP1000073A1 (de) | Verfahren zur herstellung von metallocenen | |
| EP1003757B1 (de) | Verfahren zur herstellung von metallocenen | |
| DE60210089T2 (de) | Metallocene, verwendung in katalysatoren für olefinpolymerisation | |
| DE60001902T2 (de) | Verfahren zur herstellung titankomplexen | |
| DE69519217T2 (de) | Organische Übergangsmetall-Verbindung und Verfahren zur Herstellung von Olefinen unter Gebrauch derselben | |
| DE69914012T2 (de) | Katalysatorsysteme für die Polymerisation und Copolymerisation von Alpha-Olefinen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |